
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Neurosci. , 25 February 2021
Sec. Neurodegeneration
Volume 15 - 2021 | https://doi.org/10.3389/fnins.2021.614855
This article is part of the Research Topic Deciphering the biomarkers of Alzheimer’s disease View all 20 articles
 Kunie Ando1*†
Kunie Ando1*† Christophe Erneux2†
Christophe Erneux2† Mégane Homa1
Mégane Homa1 Sarah Houben1Marie-Ange de Fisenne1
Sarah Houben1Marie-Ange de Fisenne1 Jean-Pierre Brion1
Jean-Pierre Brion1 Karelle Leroy1
Karelle Leroy1Alzheimer's disease (AD) is the most common type of dementia and its prevalence is expected to rise in response to an aging human population. Yet, there is no disease-modifying drug currently available. The neuropathological hallmarks of AD are amyloid plaques composed of amyloid ß (Aß) peptides derived from successive cleavages of Amyloid Precursor Protein (APP) and neurofibrillary tangles (NFTs) constituted of the microtubule-associated protein tau (Brion, 2006). In AD brains, tau is hyperphosphorylated and aggregated to form paired helical filaments (PHF-tau). The disease pathogenesis precedes the overt clinical symptoms by 10–15 years. Early diagnosis and biomarkers are thus crucial for future clinical trials of AD. However, current standard biomarkers such as amyloid-PET scans are highly expensive and the patients are exposed to a considerable amount of ionizing radiation at each test. Cerebrospinal-fluid analyses for Aß and tau are highly invasive due to lumbar punctures (Dolgin, 2018). We need to search for additional biomarkers that are less expensive and less invasive.
Emerging evidence suggests that Aß modifies the metabolism of phosphoinositides (PIs) (Berman et al., 2008; Kam et al., 2016). PIs control major signaling pathways and cell processes in eukaryotic cells. Ten enzymes of the inositol and phosphoinositide 5-phosphatases (hereafter, PI 5-phosphatases) have been identified in the human genome i.e., INPP5A, INPP5D (SHIP1), INPPL1 (SHIP2), INPP5G (SYNJ1), INPP5H (SYNJ2), OCRL, INPP5E (Pharbin), INPP5B, INPP5J (PIPP) and INPP5K (SKIP) (Figure 1). Except for INPP5A, PI 5-phosphatases essentially dephosphorylate PI(4,5)P2 and PI(3,4,5)P3 at the 5-position of the inositol ring with different degrees of catalytic efficiency and selectivity for each isoenzyme. PI 5-phosphatases are involved in fine-tuning regulation of PI(4,5)P2 and PI(3,4,5)P3, key intracellular signaling molecules known to be present in different subcellular compartments of the cells. Recent genetic and epigenetic studies have unequivocally suggested that some of the PI 5-phosphatases are implicated in AD, in addition to several other human diseases (Ramos et al., 2019). In this opinion article, we review recent findings on the PI 5-phosphatases in relation to AD, aging and cognitive functions. Such information could be potentially useful for developing novel biomarkers for AD in the future.
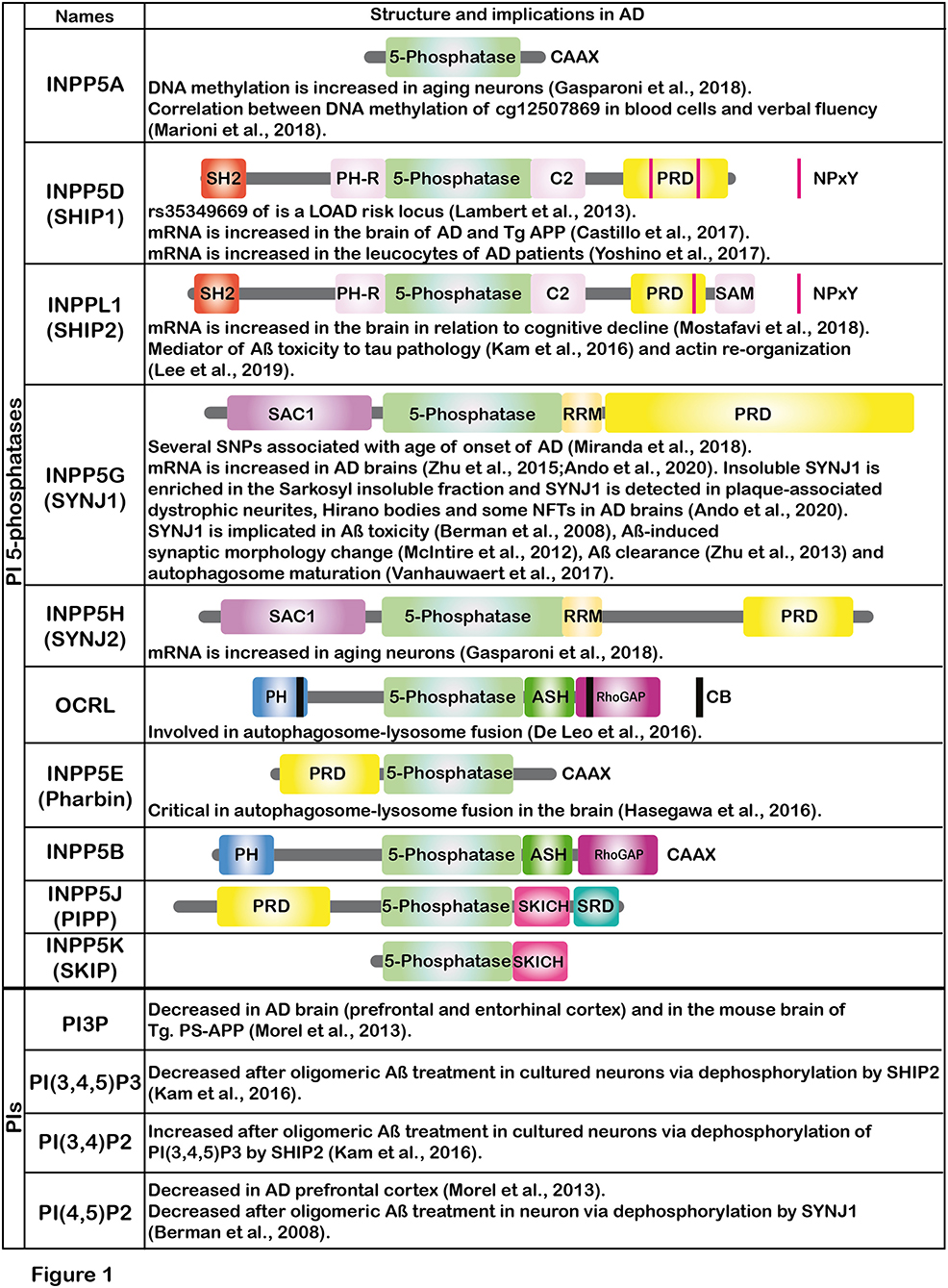
Figure 1. The figure shows the schematic illustrations of the major domains of PI 5-phosphatases and summarizes the implications of PI 5-phosphatases and PIs in AD. Each PI 5-phosphatase contains a highly conserved 5-phosphatase domain shown in green. PI, phosphoinositide; SHIP1, SH2 domain-containing inositol polyphosphate 5-phosphatase-1; SHIP2, SH2 domain-containing inositol polyphosphate 5-phosphatase-2; SYNJ1, Synaptojanin 1; SYNJ2, Synaptojanin 2; OCRL, oculocerebrorenal syndrome of Lowe, PIPP, proline-rich inositol polyphosphate 5-phosphatase; SKIP, skeletal muscle and kidney enriched inositol phosphatase; CAAX, CAAX motif; SH2, Src homology 2; PH, Pleckstrin-homology; PRD, proline-rich domain; NPxY, a conserved tyrosine phosphorylation motif (Asn-Pro-x-Tyr) for binding to a phospho-tyrosine binding (PTB) domain; SAM, sterile alpha motif; SAC1, suppressor of actin 1; RRM, RNA recognition motif; ASH, ASPM-SPD2-Hydin domain; RhoGAP, Rho GTPase-activating protein domain; CB, clathrin binding domain; SRD, serine rich domain; SKICH, SKIP COOH terminal homology domain.
Unlike other PI 5-phosphatase family members, INPP5A recognizes only soluble inositol 1,4,5-trisphosphate [Ins(1,4,5)P3] and inositol 1,3,4,5-tetrakisphosphate [Ins(1,3,4,5)P4] as substrates. INPP5A is ubiquitously expressed including in the hippocampus and prefrontal cortex, the brain regions highly affected in AD, and is abundantly detected in cerebellum (Liu et al., 2020). INPP5A negatively controls the mobilization of intracellular calcium by decreasing Ins(1,4,5)P3 levels (De Smedt et al., 1997). DNA methylation of the INPP5A gene is increased in association with aging in neurons (Gasparoni et al., 2018). Meta-analysis of blood-based DNA methylation has shown that the methylation of cg12507869 located in the INPP5A gene had a significant negative correlation with phonemic verbal fluency and was associated with logical memory and vocabulary (Marioni et al., 2018). Blood-based DNA methylation of cg12507869 in the INPP5A could be thus considered as a potential biomarker for aging and cognitive functions.
SHIP1 is a hematopoietic-specific PI 5-phosphatase activated downstream of a multitude of receptors for growth factors, cytokines, antigens, immunoglobulin and toll-like receptor agonists. Once activated and correctly localized, SHIP1 generally acts as a negative regulator of signaling processes in hematopoietic cells, for example on the B cell receptor activation signaling pathway (Ramos et al., 2019). SHIP1 is detected in the brain, primarily in microglia reflecting its myeloid origin. Genome-wide association studies (GWAS) have identified the risk variant rs35349669 in INPP5D, the gene encoding human SHIP1 for late-onset AD (Lambert et al., 2013). INPP5D mRNA is significantly upregulated in human AD brains and in transgenic mouse brains with knock-in mutations of APPNL−G−F/NL−G−F (Castillo et al., 2017). INPP5D mRNA expression in peripheral leucocytes is elevated in early AD but is decreased with cognitive decline (Yoshino et al., 2017). Further long-time follow-up of the participants would be necessary to decipher the correlation between the level of INPP5D mRNA and cognitive decline. Since SHIP1 converts PI(3,4,5)P3 to PI(3,4)P2, the amounts of these PIs in the blood leucocytes may also be altered and needs to be further investigated (as discussed in section PI Metabolism and Autophagic-Endosomal-Lysosomal Abnormalities). Taken together, the level of INPP5D mRNA in leucocytes could be an interesting target to develop a blood-based biomarker in the early stages of AD.
SHIP2, encoded by INPPL1, is ubiquitously expressed including in the brain (Muraille et al., 1999). By using PI(3,4,5)P3 as substrate, SHIP2 controls PI(3,4)P2 content, a major SHIP2 product (Ghosh et al., 2018). SHIP2 can also dephosphorylate PI(4,5)P2, another albeit less potent substrate (Elong Edimo et al., 2016). PI(3,4)P2 is scarce under normal conditions but increases through signaling following PI 3-kinase activation. This lipid plays critical roles as a second messenger in cell migration, polarity, feedback control of PI(3,4,5)P3 generation, and basal mTORC1 activity (Ramos et al., 2019). SHIP2 is directly implicated in several human diseases: mutations in INPPL1 cause opsismodysplasia, a rare autosomal recessive disease characterized by delayed bone maturation (Fradet and Fitzgerald, 2017). SHIP2 is also upregulated in some cancer cells, particularly in aggressive human breast cancer cells (Ghosh et al., 2018). SHIP2 negatively regulates insulin/IGF-I actions and is implicated in type 2 diabetes and metabolic syndrome (Marion et al., 2002). Recent network-based approach has unraveled that SHIP2 is also linked to AD and cognitive decline: upregulation of INPPL1 transcript in the brain significantly correlates with cognitive decline in human AD patients (Mostafavi et al., 2018). The same study also reported that SHIP2 immunoreactivity was detected in astrocytes and neurons in the post-mortem human brain tissues of AD patients and that lentivirus-mediated down regulation of SHIP2 in cultured astrocytes significantly reduced Aß production (Mostafavi et al., 2018). Other independent studies have reported SHIP2 functions as a mediator of amyloid toxicity via tau hyperphosphorylation (Kam et al., 2016) and actin-cytoskeleton reorganization (Lee et al., 2019). Kam et al. reported that the interaction between Aß and the FcγRIIb immuno-receptor leads to a translocation of SHIP2 to the plasma membrane to form a protein complex in which SHIP2 dephosphorylates PI(3,4,5)P3 into PI(3,4)P2. Increased amounts of PI(3,4)P2 lead to decreased inhibitory phosphorylation of GSK3ß at Ser9 via endoplasmic reticulum (ER) stress in cultured neurons (Kam et al., 2016). Consequently, tau phosphorylation by GSK3ß is increased by Aß via FcγRIIb-SHIP2 complex (Kam et al., 2016). SHIP2 inhibitors are thus under active scrutiny as a novel therapeutic target for AD. Actually, SHIP2 inhibitors represent new treatments for several diseases: SHIP2 inhibition has been reported to partially rescue memory deficits in transgenic mouse models of diabetes and AD (Soeda et al., 2010; Kam et al., 2016) and to prevent metastasis in breast cancer cells (Ghosh et al., 2018). Since both SHIP1 and SHIP2 play critical roles in antagonizing microglial proliferation and phagocytosis, the use of both SHIP1 and SHIP2 inhibitors has been proposed in AD to enhance basal microglial homeostatic functions for therapeutic purposes (Pedicone et al., 2020). Although SHIP2 could be a potential biomarker and a valuable therapeutic target for AD, it remains largely elusive whether SHIP2 undergoes a significant alteration in subcellular localization and post-translational modifications during the progression of the disease. SHIP2 has more than 20 putative phosphorylation sites and its phosphatase activity and substrate recognition are, at least partially, regulated by phosphorylation, protein-protein interaction and subcellular localization (Elong Edimo et al., 2011). Given that SHIP2 is translocated to plasma membranes upon Aß-FcγRIIb interaction (Kam et al., 2016), subcellular localization of SHIP2 should be significantly altered in AD brains. Since FcγRIIb activation leads to tyrosine phosphorylation of SHIP2 (Muraille et al., 1999), the post-translational modifications of SHIP2 could be altered in the affected areas of AD brains. It remains to be carefully determined in post-mortem brain tissues of AD patients whether there are changes in SHIP2 subcellular localizations, post-translational modifications and the impact of SHIP2 upregulation in AD on PI amounts, particularly PI(3,4,5)P3 and PI(3,4)P2.
SYNJ1 and SYNJ2 are both highly conserved and their genetic variants are associated with cognitive abilities in a cohort with a mean age of 70 (Lopez et al., 2012). SYNJ1 is a brain-enriched presynaptic phosphatase involved in synaptic vesicle recycling, clathrin-coated vesicle uncoating at synapse (Cremona et al., 1999) and autophagosomal maturation within presynaptic terminals (Vanhauwaert et al., 2017). SYNJ1, whose gene is located in chromosome 21, is linked to endolysosomal abnormalities in Down syndrome (Cossec et al., 2012). Several mutations in SYNJ1 gene are associated with early-onset Parkinsonism (Tran et al., 2020). Some of the polymorphisms in SYNJ1 are also linked with age of onset in familial AD, late-onset AD and Down syndrome with AD (Miranda et al., 2018). SYNJ1 is expressed in neurons and is implicated in Aß toxicity (Berman et al., 2008), synaptic toxicity (McIntire et al., 2012) and Aß clearance (Zhu et al., 2013). The mRNA level of SYNJ1 is significantly upregulated in post-mortem AD brains in association with APOE genotype (Zhu et al., 2015; Ando et al., 2020). SYNJ1 protein undergoes a significant solubility change and is co-enriched with PHF-tau in the sarkosyl-insoluble fraction (Ando et al., 2020). SYNJ1 immunoreactivity is detected in actin-positive Hirano bodies, some NFTs and plaque-associated dystrophic neurites in post-mortem human AD brains (Ando et al., 2020). Such aberrant alteration of mRNA levels, protein localization, and protein solubility of SYNJ1 could be applied to establish a valid biomarker for AD. While SYNJ1 is brain specific, its paralog SYNJ2 is ubiquitously expressed, but is also abundantly expressed in the synapse. In the temporal cortex from patients with depressive disorder, SYNJ2 transcript expression is significantly decreased (Aston et al., 2005). Furthermore, differential methylation in the gene of SYNJ2 has been also reported in association with aging in neuronal cells (Gasparoni et al., 2018).
The implication of the other members of PI 5-phosphatase family in AD remains largely unknown. Given that AD is associated with autophagic-endosomal-lysosomal dysfunction (Nixon et al., 2008), we speculate that INPP5E and OCRL, highly expressed in the brain and critical in autophagosome-lysosome fusion (De Leo et al., 2016; Hasegawa et al., 2016), might be involved in dysregulation of autophagy in AD brains.
Consistent with alterations of some PI 5-phosphatases observed in AD brains, there are substantial findings suggesting that PIs undergo dysregulation during the disease progression in AD brains (Stokes and Hawthorne, 1987) and in the AD blood plasma (Mapstone et al., 2014). In the AD prefrontal cortex where both amyloid and tau pathologies are abundant, the amounts of PI 3-phosphate (PI3P) and PI(4,5)P2 are significantly decreased (Morel et al., 2013). Deficiency of PIs in AD brains may be linked to autophagic-endosomal-lysosomal abnormalities observed in neurons of the AD patients even at an early stage (Nixon et al., 2008). Considering that PIs regulate membrane dynamics, we hypothesize that autophagic-endosomal-lysosomal abnormalities could be a potential target for developing AD biomarkers. For instance, endosomal morphology alteration has been observed in iPSC-neurons derived from AD fibroblasts (Israel et al., 2012) and AD blood monocytes (Corlier et al., 2015). Whereas the precise mechanisms underlying endosomal abnormalities remain to be determined, such endosomal alterations in peripheral cells could be considered as a novel potential approach to develop AD biomarkers.
Upregulation of some PI 5-phosphatases and PI dysregulations have been evidenced in AD and such alterations could be useful to develop new biomarkers for AD. Careful investigations will be needed to assess if these alterations are AD-specific or also associated with other diseases. Blood-based analyses of some PI 5-phosphatases, PI metabolism, transcriptomic and epigenetic changes have demonstrated alterations in AD and are conceivable strategies toward development of new biomarkers. Further studies will also be needed to evaluate the sensitivity and the specificity of these alterations during the progression of AD compared to currently available other markers such as those of PET and CSF analyses. These studies will be critical for deciphering the most reliable biomarkers and their complementarity for the diagnosis and the prognosis of this devastating disease.
All the coauthors participated in constructing the concept and writing the manuscript. KA, CE, and J-PB contributed conception and design of this article. All authors contributed to manuscript revision, read, and approved the submitted version.
This study was supported by grants from the Belgian Fonds de la Recherche Scientifique Médicale (T.0027.19 to J-PB and J.0078.18 to CE), the Fund Aline (King Baudouin Foundation) to J-PB, the Foundation for Alzheimer Research (FRA/SAO) to KL, and the Génicot Fund of ULB to J-PB and KL.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize that many interesting studies had to be omitted due to the word limitations.
Ando, K., Ndjim, M., Turbant, S., Fontaine, G., Pregoni, G., Dauphinot, L., et al. (2020). The lipid phosphatase synaptojanin 1 undergoes a significant alteration in expression and solubility and is associated with brain lesions in Alzheimer's disease. Acta Neuropathol. Commun. 8:79. doi: 10.1186/s40478-020-00954-1
Aston, C., Jiang, L., and Sokolov, B. P. (2005). Transcriptional profiling reveals evidence for signaling and oligodendroglial abnormalities in the temporal cortex from patients with major depressive disorder. Mol. Psychiatry 10, 309–322. doi: 10.1038/sj.mp.4001565
Berman, D. E., Dall'armi, C., Voronov, S. V., Mcintire, L. B., Zhang, H., Moore, A. Z., et al. (2008). Oligomeric amyloid-beta peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat. Neurosci. 11, 547–554. doi: 10.1038/nn.2100
Brion, J. P. (2006). “Cellular changes in Alzheimer's disease,” in Principles and Practice of Geriatric Medicine, 4th Edn, eds M. S. John Pathy, A. J. Sinclair, and J. E. Morley (Chicester: John Wiley and Sons), 1073–1081. doi: 10.1002/047009057X.ch92
Castillo, E., Leon, J., Mazzei, G., Abolhassani, N., Haruyama, N., Saito, T., et al. (2017). Comparative profiling of cortical gene expression in Alzheimer's disease patients and mouse models demonstrates a link between amyloidosis and neuroinflammation. Sci. Rep. 7:17762. doi: 10.1038/s41598-017-17999-3
Corlier, F., Rivals, I., Lagarde, J., Hamelin, L., Corne, H., Dauphinot, L., et al. (2015). Modifications of the endosomal compartment in peripheral blood mononuclear cells and fibroblasts from Alzheimer's disease patients. Transl. Psychiatry 5:e595. doi: 10.1038/tp.2015.87
Cossec, J. C., Lavaur, J., Berman, D. E., Rivals, I., Hoischen, A., Stora, S., et al. (2012). Trisomy for synaptojanin1 in down syndrome is functionally linked to the enlargement of early endosomes. Hum. Mol. Genet. 21, 3156–3172. doi: 10.1093/hmg/dds142
Cremona, O., Di Paolo, G., Wenk, M. R., Luthi, A., Kim, W. T., Takei, K., et al. (1999). Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99, 179–188. doi: 10.1016/S0092-8674(00)81649-9
De Leo, M. G., Staiano, L., Vicinanza, M., Luciani, A., Carissimo, A., Mutarelli, M., et al. (2016). Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat. Cell Biol. 18, 839–850. doi: 10.1038/ncb3386
De Smedt, F., Missiaen, L., Parys, J. B., Vanweyenberg, V., De Smedt, H., and Erneux, C. (1997). Isoprenylated human brain type I inositol 1,4,5-trisphosphate 5-phosphatase controls Ca2+ oscillations induced by ATP in Chinese hamster ovary cells. J. Biol. Chem. 272, 17367–17375. doi: 10.1074/jbc.272.28.17367
Dolgin, E. (2018). Alzheimer's disease is getting easier to spot. Nature 559, S10–S12. doi: 10.1038/d41586-018-05721-w
Elong Edimo, W., Derua, R., Janssens, V., Nakamura, T., Vanderwinden, J. M., Waelkens, E., et al. (2011). Evidence of SHIP2 Ser132 phosphorylation, its nuclear localization and stability. Biochem. J. 439, 391–401. doi: 10.1042/BJ20110173
Elong Edimo, W., Ghosh, S., Derua, R., Janssens, V., Waelkens, E., Vanderwinden, J. M., et al. (2016). SHIP2 controls plasma membrane PI(4,5)P2 thereby participating in the control of cell migration in 1321 N1 glioblastoma cells. J. Cell Sci. 129, 1101–1114. doi: 10.1242/jcs.179663
Fradet, A., and Fitzgerald, J. (2017). INPPL1 gene mutations in opsismodysplasia. J. Hum. Genet. 62, 135–140. doi: 10.1038/jhg.2016.119
Gasparoni, G., Bultmann, S., Lutsik, P., Kraus, T. F. J., Sordon, S., Vlcek, J., et al. (2018). DNA methylation analysis on purified neurons and glia dissects age and Alzheimer's disease-specific changes in the human cortex. Epigenetics Chromatin 11:41. doi: 10.1186/s13072-018-0211-3
Ghosh, S., Scozzaro, S., Ramos, A. R., Delcambre, S., Chevalier, C., Krejci, P., et al. (2018). Inhibition of SHIP2 activity inhibits cell migration and could prevent metastasis in breast cancer cells. J. Cell Sci. 131:jcs216408. doi: 10.1242/jcs.216408
Hasegawa, J., Iwamoto, R., Otomo, T., Nezu, A., Hamasaki, M., and Yoshimori, T. (2016). Autophagosome-lysosome fusion in neurons requires INPP5E, a protein associated with Joubert syndrome. EMBO J. 35, 1853–1867. doi: 10.15252/embj.201593148
Israel, M. A., Yuan, S. H., Bardy, C., Reyna, S. M., Mu, Y., Herrera, C., et al. (2012). Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature 482, 216–220. doi: 10.1038/nature10821
Kam, T. I., Park, H., Gwon, Y., Song, S., Kim, S. H., Moon, S. W., et al. (2016). FcgammaRIIb-SHIP2 axis links Abeta to tau pathology by disrupting phosphoinositide metabolism in Alzheimer's disease model. eLife 5:e18691. doi: 10.7554/eLife.18691
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Lee, H. N., Sim, K. M., Kim, H., Ju, J., Pae, A. N., Park, J. B., et al. (2019). Abeta modulates actin cytoskeleton via SHIP2-mediated phosphoinositide metabolism. Sci. Rep. 9:15557. doi: 10.1038/s41598-019-51914-2
Liu, Q., Huang, S., Yin, P., Yang, S., Zhang, J., Jing, L., et al. (2020). Cerebellum-enriched protein INPP5A contributes to selective neuropathology in mouse model of spinocerebellar ataxias type 17. Nat. Commun. 11:1101. doi: 10.1038/s41467-020-14931-8
Lopez, L. M., Harris, S. E., Luciano, M., Liewald, D., Davies, G., Gow, A. J., et al. (2012). Evolutionary conserved longevity genes and human cognitive abilities in elderly cohorts. Eur. J. Hum. Genet. 20, 341–347. doi: 10.1038/ejhg.2011.201
Mapstone, M., Cheema, A. K., Fiandaca, M. S., Zhong, X., Mhyre, T. R., Macarthur, L. H., et al. (2014). Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 20, 415–418. doi: 10.1038/nm.3466
Marion, E., Kaisaki, P. J., Pouillon, V., Gueydan, C., Levy, J. C., Bodson, A., et al. (2002). The gene INPPL1, encoding the lipid phosphatase SHIP2, is a candidate for type 2 diabetes in rat and man. Diabetes 51, 2012–2017. doi: 10.2337/diabetes.51.7.2012
Marioni, R. E., Mcrae, A. F., Bressler, J., Colicino, E., Hannon, E., Li, S., et al. (2018). Meta-analysis of epigenome-wide association studies of cognitive abilities. Mol. Psychiatry 23, 2133–2144. doi: 10.1038/s41380-017-0008-y
McIntire, L. B., Berman, D. E., Myaeng, J., Staniszewski, A., Arancio, O., Di Paolo, G., et al. (2012). Reduction of synaptojanin 1 ameliorates synaptic and behavioral impairments in a mouse model of Alzheimer's disease. J. Neurosci. 32, 15271–15276. doi: 10.1523/JNEUROSCI.2034-12.2012
Miranda, A. M., Herman, M., Cheng, R., Nahmani, E., Barrett, G., Micevska, E., et al. (2018). Excess synaptojanin 1 contributes to place cell dysfunction and memory deficits in the aging hippocampus in three types of Alzheimer's disease. Cell Rep. 23, 2967–2975. doi: 10.1016/j.celrep.2018.05.011
Morel, E., Lasiecka, Z. M., Chan, R. B., Williamson, R. L., Vetanovetz, C., et al. (2013). Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun. 4:2250. doi: 10.1038/ncomms3250
Mostafavi, S., Gaiteri, C., Sullivan, S. E., White, C. C., Tasaki, S., Xu, J., et al. (2018). A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer's disease. Nat. Neurosci. 21, 811–819. doi: 10.1038/s41593-018-0154-9
Muraille, E., Pesesse, X., Kuntz, C., and Erneux, C. (1999). Distribution of the src-homology-2-domain-containing inositol 5-phosphatase SHIP-2 in both non-haemopoietic and haemopoietic cells and possible involvement of SHIP-2 in negative signalling of B-cells. Biochem J. 342(Pt 3), 697–705. doi: 10.1042/bj3420697
Nixon, R. A., Yang, D. S., and Lee, J. H. (2008). Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy 4, 590–599. doi: 10.4161/auto.6259
Pedicone, C., Fernandes, S., Dungan, O. M., Dormann, S. M., Viernes, D. R., Adhikari, A. A., et al. (2020). Pan-SHIP1/2 inhibitors promote microglia effector functions essential for CNS homeostasis. J. Cell Sci. 133:jcs238030. doi: 10.1242/jcs.238030
Ramos, A. R., Ghosh, S., and Erneux, C. (2019). The impact of phosphoinositide 5-phosphatases on phosphoinositides in cell function and human disease. J. Lipid Res. 60, 276–286. doi: 10.1194/jlr.R087908
Soeda, Y., Tsuneki, H., Muranaka, H., Mori, N., Hosoh, S., Ichihara, Y., et al. (2010). The inositol phosphatase SHIP2 negatively regulates insulin/IGF-I actions implicated in neuroprotection and memory function in mouse brain. Mol. Endocrinol. 24, 1965–1977. doi: 10.1210/me.2010-0163
Stokes, C. E., and Hawthorne, J. N. (1987). Reduced phosphoinositide concentrations in anterior temporal cortex of Alzheimer-diseased brains. J. Neurochem. 48, 1018–1021. doi: 10.1111/j.1471-4159.1987.tb05619.x
Tran, J., Anastacio, H., and Bardy, C. (2020). Genetic predispositions of Parkinson's disease revealed in patient-derived brain cells. NPJ Parkinsons Dis. 6:8. doi: 10.1038/s41531-020-0110-8
Vanhauwaert, R., Kuenen, S., Masius, R., Bademosi, A., Manetsberger, J., Schoovaerts, N., et al. (2017). The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J. 36, 1392–1411. doi: 10.15252/embj.201695773
Yoshino, Y., Yamazaki, K., Ozaki, Y., Sao, T., Yoshida, T., Mori, T., et al. (2017). INPP5D mRNA expression and cognitive decline in Japanese Alzheimer's disease subjects. J. Alzheimers. Dis. 58, 687–694. doi: 10.3233/JAD-161211
Zhu, L., Zhong, M., Elder, G. A., Sano, M., Holtzman, D. M., Gandy, S., et al. (2015). Phospholipid dysregulation contributes to ApoE4-associated cognitive deficits in Alzheimer's disease pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 112, 11965–11970. doi: 10.1073/pnas.1510011112
Keywords: synaptojanin, microglia, SHIP2, Alzheimer's disease, phosphoinositide phosphatases, phosphoinositides
Citation: Ando K, Erneux C, Homa M, Houben S, de Fisenne M-A, Brion J-P and Leroy K (2021) Dysregulation of Phosphoinositide 5-Phosphatases and Phosphoinositides in Alzheimer's Disease. Front. Neurosci. 15:614855. doi: 10.3389/fnins.2021.614855
Received: 07 October 2020; Accepted: 26 January 2021;
Published: 25 February 2021.
Edited by:
Kin Ying Mok, University College London, United KingdomReviewed by:
Tamas Balla, National Institutes of Health (NIH), United StatesCopyright © 2021 Ando, Erneux, Homa, Houben, de Fisenne, Brion and Leroy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kunie Ando, S3VuaWUuQW5kb0B1bGIuYWMuYmU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.