 Suna Huang
Suna Huang Su Li
Su Li Hua Feng
Hua Feng Yujie Chen
Yujie Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 16 March 2021
Sec. Neurodegeneration
Volume 15 - 2021 | https://doi.org/10.3389/fnins.2021.587197
This article is part of the Research Topic Ferroptosis in Stroke, Neurotrauma and Neurodegeneration View all 17 articles
Traumatic brain injury (TBI) is one of the most harmful forms of acute brain injury and predicted to be one of the three major neurological diseases that cause neurological disabilities by 2030. A series of secondary injury cascades often cause cognitive dysfunction of TBI patients leading to poor prognosis. However, there are still no effective intervention measures, which drive us to explore new therapeutic targets. In this process, the most part of mild traumatic brain injury (mTBI) is ignored because its initial symptoms seemed not serious. Unfortunately, the ignored mTBI accounts for 80% of the total TBI, and a large part of the patients have long-term cognitive dysfunction. Iron deposition has been observed in mTBI patients and accompanies the whole pathological process. Iron accumulation may affect long-term cognitive dysfunction from three pathways: local injury, iron deposition induces tau phosphorylation, the formation of neurofibrillary tangles; neural cells death; and neural network damage, iron deposition leads to axonal injury by utilizing the iron sensibility of oligodendrocytes. Thus, iron overload and metabolism dysfunction was thought to play a pivotal role in mTBI pathophysiology. Cerebrospinal fluid-contacting neurons (CSF-cNs) located in the ependyma have bidirectional communication function between cerebral–spinal fluid and brain parenchyma, and may participate in the pathway of iron-induced cognitive dysfunction through projected nerve fibers and transmitted factor, such as 5-hydroxytryptamine, etc. The present review provides an overview of the metabolism and function of iron in mTBI, and to seek a potential new treatment target for mTBI with a novel perspective through combined iron and CSF-cNs.
Traumatic brain injury (TBI) is a general term for pathological or structural changes in the brain caused by impact, bumps, penetrating injuries, or blasting under external forces (Menon et al., 2010; Cook and Hawley, 2014; Pavlovic et al., 2019), which involves at least one of the following symptoms: loss or a decreased level of consciousness, loss of memory for events before or after the injury, neurological deficits (weakness, loss of balance, changes in vision, etc.), or alterations in mental state (confusion, disorientation, slowed thinking, etc.). Based on the clinical awareness level of the Glasgow Coma Scale (GCS), the severity of TBI is usually classified as mild (mTBI) (13–15 points), moderate (9–12 points), or severe (3–8 points). The primary and secondary injuries of trauma may cause severe and long-lasting damage to TBI patients.
In the acute phase, moderate and severe TBI often cause intracranial hemorrhage, and half of the TBI patients may suffer from traumatic cerebral hemorrhage in addition to the primitive hematoma that causes compressive damage to brain tissue (Kurland et al., 2012). Some acute clinical symptoms, like sharply increased intracranial pressure and cerebral hernia, lead to the mortality of sever TBI as much as 30–40% (Rosenfeld et al., 2012). Due to the severe clinical symptoms of this part of TBI patients, neurosurgeons always administrate posttraumatic intracranial hematoma (TICH) removal to prevent increased intracranial pressure and cerebral hernia (Tao et al., 2017). Basing on the secondary injury mechanism, a proportion of severe TBI survivors have long-term physical and cognitive disorder, but after high quality care in the emergency or ICU, the consequence may be attenuated.
Reversely, mTBI, accounting for more than 80% of all TBI patients, is often ignored, as it is generally non-fatal (0.1% lethality) (Af Geijerstam and Britton, 2003). MTBI mainly consists of sport-related TBI and military-related TBI. Sport-related mTBI has the highest incidence (approximately 20%) among all types of TBI (Theadom et al., 2014). Except the professional athletes, due to the benefits for physical and mental health, more and more people participating in sports, and the incidence of global sports-related TBI is continuously rising (Daneshvar et al., 2011). From 2007 to 2011, approximately 84% of military-related TBIs were classified as mTBIs (Mondello et al., 2014); military personnel exposed to explosive device blasts on the battlefield are especially prone to TBI, and this incidence is closely related to international military situations. Many single-event mTBI patients experience symptoms or disability even for more than 1 year (Nelson et al., 2019), including chronic dizziness, fatigue, and headache (Silver et al., 2009), which are collectively referred to as postconcussive syndrome (Sterr et al., 2006; Dikmen et al., 2010).
Many professional athletes or veterans have a history of repetitive head impacts and are likely to develop chronic traumatic encephalopathy (CTE) due to multiple episodes of mTBI (McKee et al., 2015; Stein et al., 2015). Compared with single-event mTBI, repeat mTBI will induce more serious neurological dysfunction (Ayubcha et al., 2021). With passage of time, CTE patients gradually present with cognitive impairment and memory loss, and at last, dementia is virtually inevitable (McKee et al., 2013). Cognitive dysfunction is often observed after TBI and is the major complication of TBI during the subacute or chronic phase. Cognitive dysfunction in patients with mTBI can usually last for 3–6 months or longer (Whiteneck et al., 2004; Boake et al., 2005; Roe et al., 2009). Due to the inconsistency of research methods and the different subtypes of patients included, conflicting results on cognitive dysfunction may be reported. However, it was consistently reported in all studies that the impact of post-TBI cognitive dysfunction on patients’ work, daily life, and social functions as well as the burden on families and society cannot be ignored (Gustavsson et al., 2011). To date, the mechanism of early cognitive dysfunction after TBI is still unclear (Wolf and Koch, 2016; Zhao et al., 2017).
Chronic traumatic encephalopathy is a condition that significantly shortens the life span (Wortzel et al., 2013). Autopsy revealed that the most typical pathological feature of CTE is excessive deposition of tau protein and formation of neurofibrillary tangles (McKee et al., 2015; Kanaan et al., 2016). Excessive iron deposits were observed in these nerve fiber tangles, where free iron can induce tau phosphorylation through oxidative stress (Bouras et al., 1997; Hui et al., 2011). Coincidently, earlier reports revealed the deposition of non-heme iron in the deep cortex and hippocampus in the postmortem brain tissues of mTBI patients (Bouras et al., 1997). Thus, iron gradually becomes the intervention target in TBI.
After TBI, the primary injury leads to neuronal death around the injury site. A series of secondary injury cascades, such as the release of blood metabolites, microglial activation, thrombin activity, the release of excitatory amino acids, and proinflammatory factors, are initiated (Figure 1). Patients with various degrees of TBI may suffer from a long period of secondary injury, which plays a key role in determining the final severity and recovery of nerve injury after TBI (Lee et al., 2015).
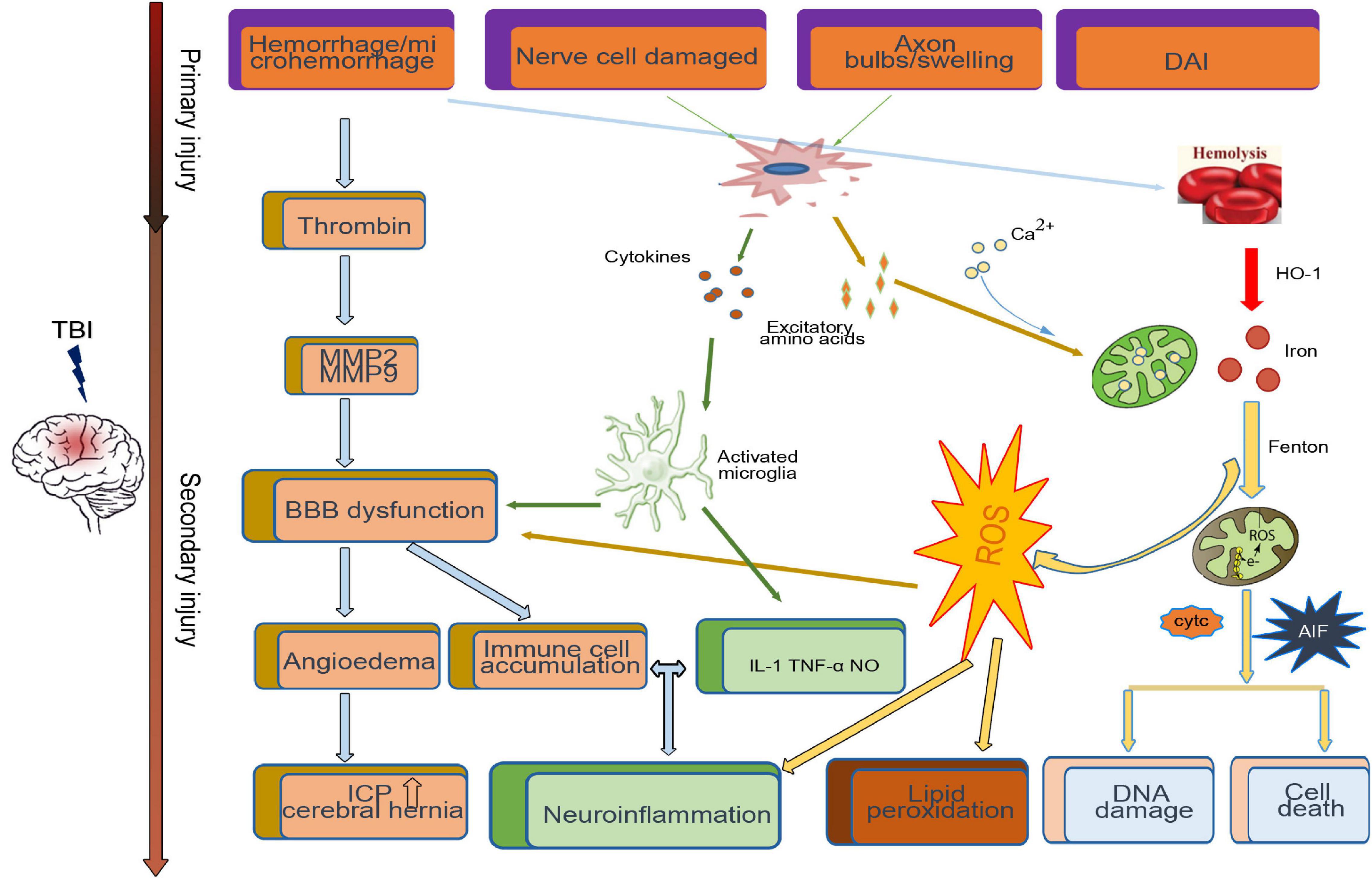
Figure 1. Primary and secondary pathophysiology in traumatic brain injury. Immediately at the time of impact, the brain suffers a direct mechanical trauma, which induced TBI primary injury including hemorrhage/microhemorrhage, cell death, diffuse axonal injury, etc. Hours to days after impact, the secondary injury initiated by the primary injury, such as the microglia activated by cytokines and excitatory amino acids from damaged cell, cooperates with the dysfunctional blood–brain barrier (BBB)-induced immune cell accumulation and neuroinflammation. Excessive iron and calcium-mediated mitochondrial dysfunction increase the generation of free radical and oxidative species, which induce lipid peroxidation, DNA damage, and cell death.
Neuroinflammation is a key pathophysiological mechanism of secondary injury, as it can produce neurotoxic effects on neurons through oxidative stress, apoptosis, and excitotoxicity (Faden et al., 2016). After TBI, there is an activation of neuroinflammation response pathway mediated by microglia cells that were not initially damaged, and the activated microglial cells may promote the release of proinflammatory factors such as interleukin 1, tumor necrosis factor, nitric oxide, etc., (Kumar and Loane, 2012), inducing a progressive cycle of chronic inflammation (Borlongan et al., 2015). Except microglia activation, the ROS generated after TBI and peripheral immune cells like macrophages, neutrophils, and lymphocytes recruit to the injured site through the disrupted blood–brain barrier (BBB), which are key components to exacerbate the inflammation (Sulhan et al., 2020).
The BBB is crucial in maintaining homeostasis within the brain’s biochemical environment. After TBI, activation of microglia and the cytokines released by damaged cells destroy tight junctions through impairment of BBB endothelial cells, pericytes, astrocytes, and through the synthesis of tight junction proteins; this leads to an increase in BBB permeability, subsequent edema, and the accumulation of plasma-derived factors and immune cells in brain tissue (Shetty et al., 2014; Sulhan et al., 2020), leading to a sharp increase in intracranial pressure and eventually cerebral hernia (Winkler et al., 2016).
Similarly, increased release of excitatory amino acids is also involved in nerve function damage after TBI. Increased release of glutamate from presynaptic terminals activates N-methyl-D-aspartate (NMDA) receptors on postsynaptic terminals to lead to a widespread neuronal depolarization or spreading depression, further disrupting the sodium ion, potassium ion, and calcium ion equilibria (Bouley et al., 2019). Then, the function of the mitochondria, which is the calium regulator, is hampered by abnormally elevated intracellular calium (Langlois et al., 2006). As a result of such abnormalities, the membrane pump is continuously activated to correct the example level of imbalance, consuming a large amount of glucose and generating a large amount of lactic acid, leading to acidosis and edema (Zetterberg et al., 2013; Barkhoudarian et al., 2016).
Basing on the secondary injury mechanism, anti-oxidation and anti-excitatory amino have been used in releasing the neurological dysfunction, not showing effect, and currently, there are no effective treatments approved by clinical trials for TBI patients (Pearn et al., 2017).
Imaging examination found a subgroup of mTBI patients with microbleeding (Nisenbaum et al., 2014). Long time, continuous, or discontinuous microbleeding can induce red blood cells, and thrombin enters the brain continuously. Iron is released into the brain tissue 24 h after the onset of bleeding (Xiong et al., 2013); on the seventh day, the non-heme iron content is tripled and can remain at high levels for 28 days in the brain (Xiong et al., 2013). The evidence suggested that increased iron accumulation was negatively associated with cognitive outcomes in chronic TBI patients (Lu et al., 2015). With the formation of a membrane attack complex, the complementary cascade reaction may be a mechanism of thrombin-induced brain damage, resulting in the lysis of red blood cells (Gong et al., 2005). Catalyzed by HO-1, heme-bound iron is released as free iron and contributes to the nerve injury process (Beschorner et al., 2000). Therefore, due to iron deposition accompany with the whole pathological process, iron is considered an important pathophysiological factor involved in these secondary injuries after mTBI (Nisenbaum et al., 2014).
Iron is necessary for brain metabolism and is the most abundant metal in the brain. Iron plays an essential role in myelination, oxygen transport, neurotransmitter transmission, and mitochondrial energy production (Salvador, 2010; Ward et al., 2014).
The BBB plays a vital role in brain iron uptake and protects the brain from fluctuations in iron levels throughout the body. Iron in the circulatory system enters the brain through the BBB mainly in two forms: a transferrin-bound iron-transferrin receptor complex and a non-transferrin-bound iron (Singh et al., 2014). Studies have reported that choroid plexus endothelial cells also regulate the entry of iron into the central nervous system (Rouault and Cooperman, 2006).
Astrocytes redistribute iron, which have gotten through the BBB to other cells in the central nervous system with the assistance of ceruloplasmin (Dringen et al., 2007). Ceruloplasmin can oxidize ferrous iron to Fe3+, which binds to transferrin in the cerebral spinal fluid (CSF) (Singh et al., 2014). In addition, there is a large amount of non-transferrin-bound iron circulating in the CSF together with ATP, citrate, and ascorbic acid in cells that do not express transferrin receptors (Malecki et al., 1999; Ke and Qian, 2007). Oligodendrocytes have the highest iron content in the brain (Connor and Menzies, 1996), which may explain their high sensitivity to oxidative stress (Smith et al., 1999). Although oligodendrocytes are mainly responsible for transferrin expression and secretion, they take non-transferrin-bound iron in the form of ferritin via the Tim-2 receptor; astrocytes take non-transferrin-bound iron via DMT1 (Rouault and Cooperman, 2006; Todorich et al., 2008; Lane et al., 2010). Neurons take iron from the CSF and interstitial fluid by transferrin-bound iron.
Part of the cytoplasmic iron is involved in the normal metabolic activities of the cell, such as the activity of mitoferrin expressed in the mitochondrial inner membrane, which transports iron from the cytoplasm to the mitochondria to participate in the synthesis of heme and iron–sulfur clusters (Benarroch, 2009). Some iron in the form of a complex is directly transferred to ferritin through the PCBP1 protein–protein effect (Philpott et al., 2017). A large portion of cytoplasmic iron is contained in the lysosome (Yu et al., 2003). Apart from the iron serving as co-factors and the iron stored in ferritin, the remaining divalent iron in the cytoplasm (a concentration of approximately 0.5–5 μm) is called the free iron pool (Espósito et al., 2002; Ma et al., 2006; Bayır et al., 2020). Under normal circumstances, >95% of the ferrous iron in the cytoplasm is bound with GSH at a ratio of 1:1 and forms a stable iron complex to prevent free Fe2+ from generating ROS by the Fenton reaction to damage cells (Patel et al., 2019).
The uptake and storage of cellular iron mainly depend on the regulatory effects of the cytoplasmic protein iron regulatory protein (IRP)1 and IRP2 (Ponka, 1999; Styś et al., 2011). When cells experience iron deficiency, IRPs bind to the iron-responsive element (IRE) of ferritin mRNA and inhibit the translation of ferritin to reduce iron storage. Under the mediation of nuclear receptor coactivator 4 (NCOA4), ferritin is transported to the lysosome and degraded through selective autophagy to release free iron for cell utilization (Quiles del Rey and Mancias, 2019). On the other hand, IRPs combine with the IRE on the transferrin receptor mRNA to stabilize its mRNA, promote the expression of transferrin receptor, and increase iron uptake (Recalcati et al., 2010; Wagner et al., 2016). Meanwhile, excessive cellular iron may trigger the opposite regulation mechanism. Similar to peripheral tissues, brain tissues also transport iron out of cells under the synergistic effect of Fpn/hephaestin and/or Fpn/CP (Li and Qian, 2002; Rouault and Cooperman, 2006). Brain iron is reabsorbed from the subarachnoid space back into the blood through the CSF.
Studies have confirmed that iron homeostasis in the brain is disrupted after TBI (Figure 2). The possible causes of brain iron metabolism disorder may include iron accumulation caused by red blood cell lysis, iron metabolism-related protein changes, mitochondria, and lysosome dysfunction.
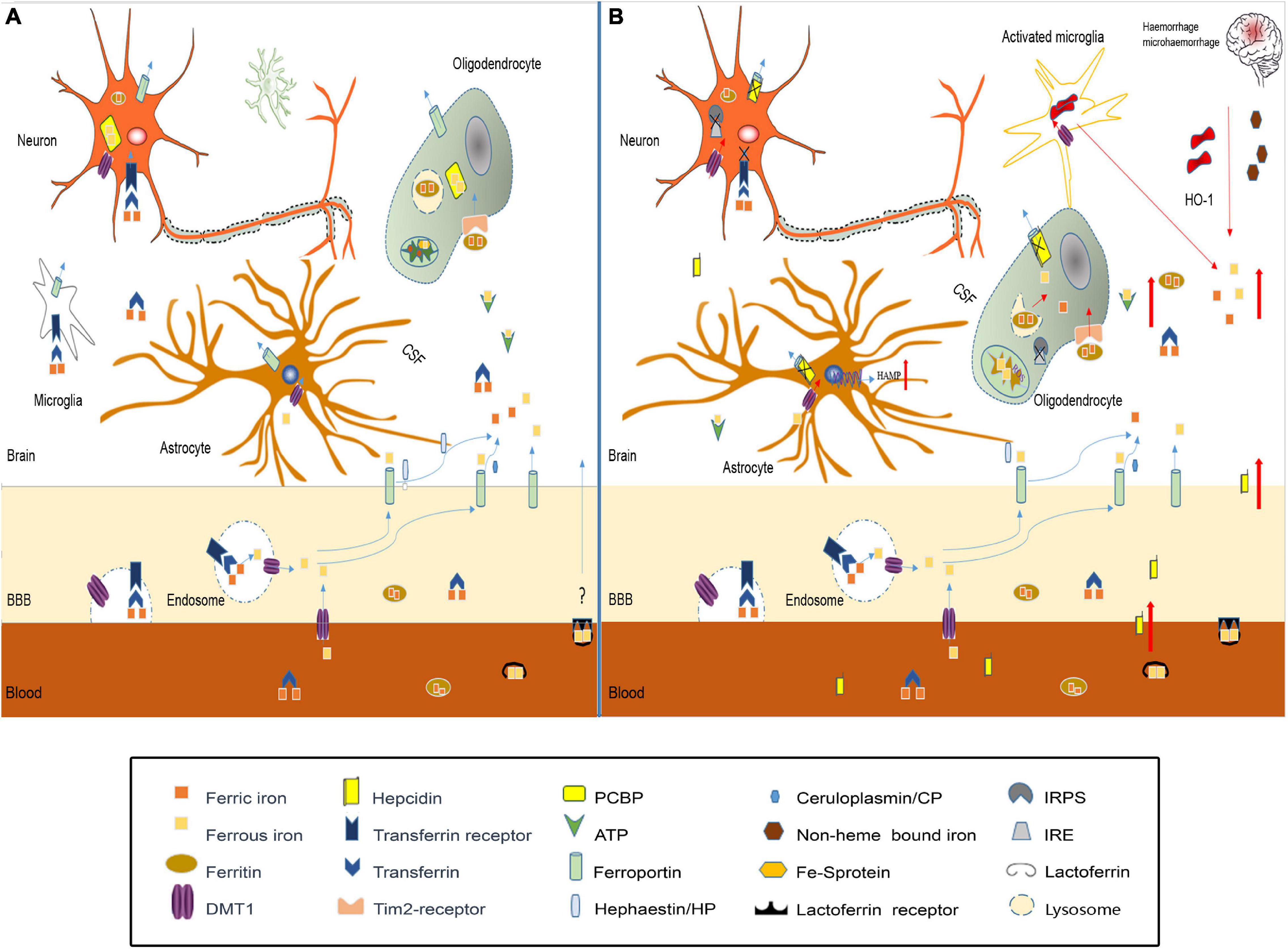
Figure 2. Iron metabolism disorder in traumatic brain injury. (A) In normal brain, iron transport across the luminal membrane of the BBB mainly relies on the transferrin/transferrin receptor (Tf/TfR) pathway, Tf-TfR-bounding iron complex under the help of divalent metal transporter 1 (DMT1) released from the endosome then transported across the abluminal membrane by ferroportin 1/hephaestin (Fpn1/Heph) and/or Fpn1/ceruloplasmin (CP). Non-transferrin-bound iron can be transported across the BBB by DMT1, etc. After release from the microvascular endothelial cells through Fpn1, iron mainly in the forms of Tf-fe3+, ferritin-Fe3+, and ATP-Fe2+, circulates in the cerebral spinal fluid (CSF), which is very convenient to be utilized by nerve cells. Astrocytes take iron via DMT1, and oligodendrocytes take iron in the form of ferritin via the Tim-2 receptor. Neurons and microglias expressing TfR can take iron from the CSF through TfR and DMT1. Cytoplasm iron: (1) Participates in cell metabolic activities, such as the synthesis of heme in mitochondria. (2) Contained in poly(rC)-binding protein (PCBP), ferritin, lysosome to prevent free iron from generating ROS. (B) In traumatic brain injury, iron accumulation occurs in the situation of hemorrhage/microhemorrhage. Excessive iron suppresses iron regulatory proteins (IRPS) combined with TfR and Fpn1, ferritin iron regulatory elements (IRES) induce TfR decrease and Fpn1, and ferritin increased to prevent neuron iron overload, but brain hepcidin expression is increased including local and peripheral hepcidin, which is transported into the brain through the dysfunctional BBB. Hepcidin internalizes Fpn1 to suppress the output of iron. At the same time, increased DMT1 enhances iron uptake in all nerve cells except oligodenrocytes, but increased ferritin promotes oligodenrocytes and takes iron through the Tim2 receptor. A large amount of increased cytoplasm iron is transported into the mitochondria generating ROS-induced organelle damage including lysosome and cell death.
Iron accumulation is observed after TBI (Raz et al., 2011; Liu et al., 2013), which is consistent with the fact that moderate to severe TBI and many cases of mTBI have cerebral hemorrhage. Under the effect of HO-1, iron is released from heme as free iron. Microglia, as brain-specific phagocytic cells, will engulf red blood cells, and then release degraded iron into the interstitium of the brain (Andersen et al., 2013); these cells are all a source of excessive iron in the brain after TBI. The increased iron in the interstitium and CSF will be transported to various types of nerve cells in various forms, eventually leading to iron accumulation in nerve cells, which can produce large amounts of ROS to intensify oxidative stress and aggravate secondary damage after TBI (Nunez et al., 2012).
The possible causes of brain iron accumulation may also include the expression of iron metabolism-related proteins changes. Studies have shown that the expression of ferritin, transferrin, and transferrin receptors in the brain is increased after intracranial hemorrhage (Wu et al., 2003), which may increase iron absorption in neurons and oligodendrocytes, which is consistent with previous reports that iron accumulates in brain cells after TBI. In the case of inflammation, McCarthy et al. (2018) found that an acidic environment increased the expression of DMT1 in microglia and increased the uptake of non-transferrin-conjugated iron. Additionally, excessive iron can inhibit IRPs in cells, then regulating the increased expression of ferritin and Fpn1 to enhance the storage capacity and iron output capacity for the increased iron pool (Thirupathi and Chang, 2019; Wang et al., 2019). However, when inflammation and iron overload in cells occur, the expression of hepcidin is also increased (Lieblein-Boff et al., 2013; Rochette et al., 2015). Studies have shown that hepcidin can enter the central nervous system through the BBB (Raha-Chowdhury et al., 2015), which, with increased permeability after TBI, may allow a large amount of hepcidin to the brain tissue; thus, hepcidin internalizes Fpn1, which affects the iron output. As a result, iron uptake capacity is enhanced, while iron output capacity is weakened, aggravating the accumulation of iron in cells.
In addition, the possible causes of brain iron metabolism disorder may include cell mitochondrial and lysosomal dysfunction. Under pathological conditions of TBI, the MCU protein on the inner mitochondrial membrane can transport a large amount of calcium ions into the mitochondria, disrupting the normal function of the mitochondria (Peng and Jou, 2010; Zhang et al., 2019). When MCUs rapidly mediate calcium ions into the mitochondria, they also simultaneously mediate a large amount of iron into the mitochondrial matrix (Zhang et al., 2019), which can cause iron homeostasis disorder. Excessive iron generates ROS through the Fenton reaction, which further aggravates mitochondrial dysfunction and forms a progressive vicious cycle. Using MCU inhibitors can promote mitochondrial dysfunction recovery caused by iron overload by reducing ROS production and mitochondrial depolarization (Sripetchwandee et al., 2013). In normal cells, a large portion of iron is contained in lysosomes in the form of divalent iron (Yu et al., 2003). After TBI, oxidative stress in cells can cause damage to certain lysosomes. The sensitivity to oxidative stress is related to the amount of iron contained in lysosomes (Terman and Kurz, 2013). The damaged lysosome ruptures and releases large amounts of iron into the cytoplasm.
Iron deposition in the brain could induce local pathological appearance. Previous studies have observed iron deposition in CTE patients. The main pathological feature of CTE is the abnormal aggregation of tau protein in neurons, astrocytes, and cell synapses in the deep cortical sulci. In this process, accumulated iron promotes the phosphorylation of tau protein in the brains of CTE patients through oxidative stress and aggregates to form nerve fiber tangles (Shen, 2015). Studies have shown that the sustained occurrence of mild TBI can increase the risk of Parkinson’s disease in patients over 55 years old in 5–7 years (Gardner et al., 2015), which may also be related to excessive iron deposition after TBI.
There is a hypothesis that damage to the brain network, mainly axons, may be the basis of cognitive dysfunction after TBI (Wolf and Koch, 2016), and iron may play an important role in the process. Despite the primary injury due to mechanical force, diffuse and multifocal damage often appears, leading to diffuse axonal injury by blunt force of shearing, tearing, and stretching of the axon and further damage of the long-distance white matter connection, with large-scale network disconnection as the core mechanism of cognitive impairment. Diffuse axonal injury induced by initial mechanical force could lead to cognitive decline immediately when the TBI occurs and is always irreversible. Besides the initial mechanical force, the complex secondary cascade always induces long-term diffuse axonal injury. This characteristic is consistent with TBI especially that mTBI patients have long-term cognitive dysfunction (Mu et al., 2019).
The mixed and intertwined axon and myelin is one pathology nature in diffuse axonal injury. Oligodendrocytes provide an important biological basis for neural network integration and high-level functions. Oligodendrocyte loss following TBI takes place. Demyelinated axons are prone to damage, so the loss of oligodendrocytes may lead to secondary axonal injury (Armstrong et al., 2016). Oligodendrocytes have the highest iron content in the brain and are highly sensitive to oxidative stress and iron accumulation. A recent study demonstrated that iron homeostasis disorder and ferroptosis can induce oligodendrocyte loss and demyelination, and iron chelation can rescue iron-mediated oligodendrocyte death (Nobuta et al., 2019). Thus, iron accumulation and metabolism disorder after TBI may lead to secondary axonal injury and cognitive dysfunction through inducing oligodendrocyte apoptosis.
Excessive iron can lead to cell death. After cell death, the capsule ruptures, and the contents are released into the tissue, activating the surrounding immune cells that then promote inflammation (Tonnus et al., 2018).
Autophagy is an evolutionarily and conservatively important process for the turnover of intracellular materials in eukaryotes. Autophagy is divided into macroautophagy (usually called autophagy), microautophagy, and molecular chaperone-mediated autophagy (Jiang and Mizushima, 2013), which generally refers to macroautophagy. The autophagy mentioned in this article refers to macroautophagy. Under normal circumstances, nerve cells continuously maintain a very low level of autophagy (Rubinsztein et al., 2005), but in situations such as starvation, ischemia, hypoxia, protein misfolding, or damage to organelles, autophagy will be activated to maintain cell homeostasis (Yorimitsu and Klionsky, 2005; Yang and Klionsky, 2010).
After TBI, the expression of several autophagy flux indicators increased, such as Beclin1, LC3-II/LC3-I ratio, and Atg12–Atg5 conjugates, suggesting an upregulation of autophagy formation (Diskin et al., 2005; Clark et al., 2008; Liu et al., 2008; Sadasivan et al., 2008). Au et al. (2016) performed LC3 immunohistochemistry on damaged brain tissue from patients, showing that the neurons, oligodendrocytes, and microglia in the damaged area of patients undergoing decompression of the cranial flap after TBI had autophagosome formation, and the increase in P62 was correlated with adverse results 6 months after TBI. This result is consistent with the results of autopsy on TBI patients by Sakai. The findings by Sakai demonstrated that the expression of P62 in neurons and glial cells increased within 1 h after TBI, P62 and LC3 immunohistochemical staining was higher in the contused site than in the non-contused site, and the expression of autophagy markers increased for a long time—or even months after injury (Sakai et al., 2014).
The mechanism of autophagy flux change after TBI is not very clear. One mechanism is brain iron accumulation. Autophagy occurs after cerebral hemorrhage and that using an iron chelator significantly reduces cerebral hemorrhage-induced autophagy. It is suggested that iron plays an important role in inducing autophagic cell death after cerebral hemorrhage, and this effect may be one of the secondary injury mechanisms of the brain (He et al., 2007). Iron overload changes the size and number of lysosomes and affects autophagic flux (Fernández et al., 2016). The increased free iron pool generates ROS through the Fenton reaction (Dowdle et al., 2014). H2O2 produced by oxidative stress can bind to the cysteine 81 site of ATG4 to regulate the activity of ATG4 (Scherz-Shouval et al., 2007), then enhance autophagy and cause autophagic cell death (Asnaghi et al., 2004; Koike et al., 2008). At the same time, increased autophagy can degrade ferritin by lysosome and increase intracellular iron levels (Dong et al., 2008), forming a negative feedback.
Ferroptosis is a form of regulated cell death that occurs as a consequence of lethal lipid peroxidation (Stockwell et al., 2017). It is due to the inactivation of the glutathione (GSH)-dependent antioxidant defense mechanism in cells, and the ability of GPX4 to suppress lipid ROS accumulation is inhibited, which eventually leads to lipid peroxidation and cell death (Dixon et al., 2012).
Three factors are closely related to ferroptosis: iron, polyunsaturated fatty acids, and amino acids. Several studies have shown that iron is necessary for ferroptosis, and excessive iron deposition has been reported after TBI (Raz et al., 2011). The free iron pool in the cytoplasm is the most powerful producer of ROS. The brain is the most abundant source of polyunsaturated fatty acids and susceptible to lipoxygenase catalysis and ROS oxidation (Cheng and Li, 2007; Skouta et al., 2014). After TBI, the release of glutamate at the terminals of presynaptic neurons increases. High concentrations of extracellular glutamate can inhibit cystine, which is necessary for the synthesis of glutathione entering the cytoplasm by Sxc–; pertinently, insufficient GSH inhibits GPX4 activity and causes ferroptosis. At last, iron-induced oxidative stress exceeds the ability of anti-oxidative stress, and ferroptosis occurs.
To date, almost all genes related to iron-induced cell death are regulated by Nrf2 transcription (Abdalkader et al., 2018). Nrf2 is a central regulator that maintains redox homeostasis and regulates the redox state of cells in a harmful state (Villeneuve et al., 2010); it can be transferred to the nucleus under oxidative stress to activate a variety of antioxidant enzymes, such as GPX (de Vries et al., 2008). In the acute stage of cerebral hemorrhage, the expression of GPX4 is significantly reduced, and the increase in GPX4 can improve iron-dependent neuron death in rats and improve the recovery of cerebral hemorrhage (Zhang et al., 2018).
Cerebral spinal fluid comprises the inherent contents of the skull mainly produced by the choroid plexus in the ventricle and subarachnoid space. It surrounds and supports brain tissue and the spinal cord in their entirety and plays a role in supporting and removing metabolites. CSF contains a variety of signals that participate in the regulation of central nervous system activities and exchange substances with blood through a variety of ion channels. Brain metabolites are secreted and infiltrated into CSF to regulate the entire central nervous system and even organs throughout the entire body (Illes, 2017).
Recent research suggests that there is a class of neurons in contact with CSF on the ependyma called CSF-contacting neurons (CSF-cNs), and the location of CSF-cNs and ciliary morphology suggests that they sense physical and chemical signals in the CSF (Orts-Del’Immagine and Wyart, 2017). The hippocampus is closely related to learning, memory, and cognitive function. Studies have found that 5-HT can help hippocampal learning and memory (Oliveira-Silva et al., 2007). Through the horseradish peroxidase tracing method, it has been confirmed that CSF-cNs project nerve fibers to the dCA1 area and transmit 5-HT to the hippocampus; they can also transport 5-HT from adjacent cells or CSF to hippocampal neurons. When a CSF-cN is impaired, 5-HT transmission decreases, and cognitive function declines (Li et al., 2020).
After TBI, CSF-cN iron-induced damage may be part of the reason why cognitive dysfunction develops. In addition to contact with CSF, CSF-cNs also connect with other neurons, glial cells, and blood vessels in the brain parenchyma. As mentioned above, oxidative stress is the main secondary injury in TBI caused by iron overload. Savman et al. (2001) compared CSF non-protein-bound iron in 20 premature infants with ventricular hemorrhage and 10 normal premature infants and found that 75% of intraventricular hemorrhage (IVH) infants had CSF non-protein-bound iron, but none of the infants in the control group had CSF non-protein-bound iron, and all children with CSF non-protein-bound iron suffered white matter damage and subsequent disability. CSF-cNs are highly likely to take a large amount of iron from CSF to generate ROS. Under oxidative stress, CSF-cNs upregulated the activity of ATG4 by combining H2O2 with the cysteine 81 sites of ATG4 and activated ATG4-promoted LC3 lipidation, which is essential for autophagy initiation, and promoted the formation of autophagosomes (Scherz-Shouval et al., 2007; Lin and Kuang, 2014). TRPML1 (also known as MCOLN1) belongs to the mucoprotein subfamily of transient receptor potential channels and co-express with the CSF-cNs special marker TRPP2 channel in vestibular (Takumida and Anniko, 2010). Recently, the Xu Haoxing team in the United States confirmed that TRPML1 is a Fe2+ transport channel in late endosomes and lysosomes. MCOLN1−/− cells show obvious Fe2+ deposition in lysosomes and cause cellular iron metabolism disorder and neuronal degeneration (Dong et al., 2008). In TRPML1-deficient mice, an increase in p62 and a clearance defect in LC3-II were found, which shows that the complete autophagy flux was destroyed (Curcio-Morelli et al., 2010). Thus, the abnormal autophagy of CSF-cNs may be part of the pathophysiological mechanism for neurological dysfunction after TBI (Figure 3).
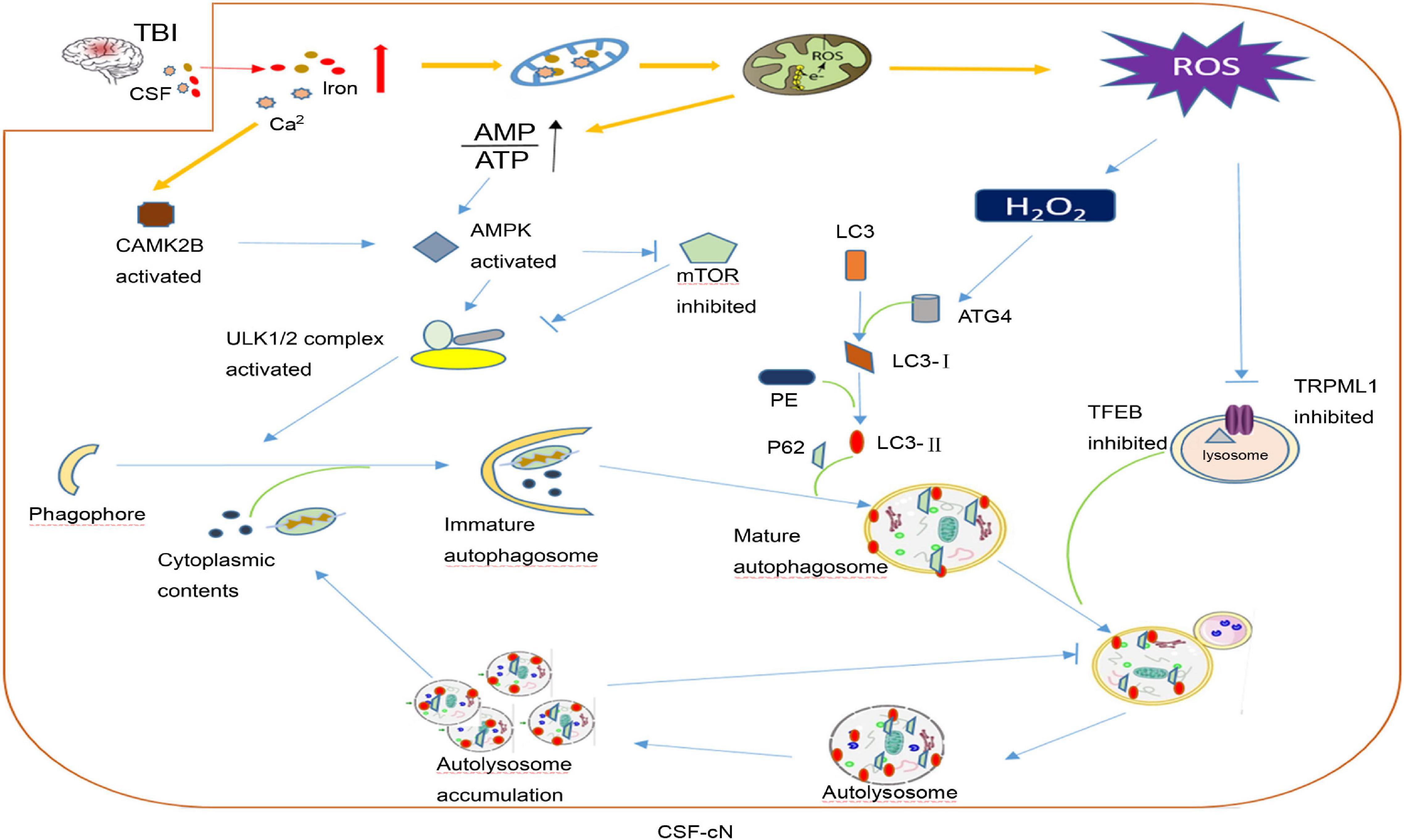
Figure 3. A hypothesis of cerebrospinal fluid-contacting neuron (CSF-cN) autophagy abnormality in traumatic brain injury (TBI). After traumatic brain injury, both iron and calcium are increased in cerebral spinal fluid, which are largely taken by CSF-cNs. Excessive Ca2+ activates calcium/calmodulin-dependent protein kinase IIβ (CAMK2B) and increases the permeability of mitochondria resulting in respiratory chain destruction and AMP/ATP ratio increase. Both activated CAMK2B and increased AMP/ATP ratio could activate AMPK to promote the formation of immature autophagosomes by directly activating ULK1 or inhibiting mTOR. Excessive iron in the mitochondria increases the generation of ROS including H2O2. H2O2 could combine with cysteine 81 sites of ATG4 to promote lipidation of LC3 to LC3-II, which participates in mature autophagosome formation. In addition, excessive ROS may inhibit lysosomal ion channel TRPML1, which may be one of the mechanisms that mediate the abnormality of CSF-cN autophagy after TBI and transcription factor EB (TFEB) to affect lysosome biogenesis and suppress the hydrolysis ability of the lysosome. At last, autophagosome formation increases and autolysosome hydrolysis is abnormal, both together leading to autophagy flux abnormality and cytoplasmic content accumulation including damaged mitochondria, misfolded protein, etc.
Some studies have shown that serum ferritin is independently associated with severe cerebral edema and poor prognosis after cerebral hemorrhage. Serum iron and transferrin levels were negatively correlated with hematoma volume; serum ferritin levels in the experimental group were significantly higher than those in the control group, and the worse prognosis is when the serum ferritin level is higher. There was no significant difference in serum CP and serum transferrin (Yang et al., 2016). In a 3-year randomized double-blind experiment including 818 elderly individuals, an increase in serum ferritin was significantly correlated with a decline in cognitive function (Schiepers et al., 2010). The research by Gao et al. (2017) also indicated that severe cognitive impairment is associated with elevated serum ferritin. Ferritin is an acute phase protein of the inflammatory response (Marshall, 2001), and the increase in ferritin in inflammation causes a decrease in serum iron (Drakesmith and Prentice, 2012). At the same time, hepcidin is also regulated by inflammation, and the expression of serum hepcidin in patients with cerebral hemorrhage is increased (Ganz and Nemeth, 2012). Mecklenburg confirmed the correlation between serum ferritin and serum hepcidin in the case of inflammation (Mecklenburg et al., 2014). In general, the combined application of serum ferritin, hepcidin, and iron indexes may be more clinically meaningful for patients with hematoma after TBI.
For patients who meet the surgical standards, posttraumatic intracranial hematoma (TICH) removal can reduce the neurotoxic effect of iron released by lysis from red blood cells (Tao et al., 2017) and prevent the occurrence of cerebral hernia caused by the increase in intracranial pressure (Arboix et al., 2002). The hematoma removal technique used for TICH varies around the world. Although there are clinical trials for spontaneous cerebral hemorrhage removal, there are still no clinical trials specifically for TICH hematoma removal (Table 1). As early as the 1990s, Zumkeller et al. (1992) found that the incidence of undesirable prognosis in the hematoma removal group of TICH patients was better than that in the non-surgical group of TICH patients. Similarly, in a single-center retrospective study, Choksey et al. (2009) found that in patients with a low GCS score whose TICH hematoma volume was greater than 16 ml, the proportion of patients with undesirable prognosis in the hematoma removal group was 38% less than that in the non-surgical group (56%). Open surgery may cause damage to offset the benefits of clearing the hematoma; thus, research to determine minimally invasive hematoma removal strategies are currently underway (Wilkinson et al., 2018), such as that evaluated in the MISTIE III experiment, endoscopic hematoma removal, and the stereotactic underwater blood aspiration (sCUBa) (Turner et al., 2015; Kellner et al., 2018).
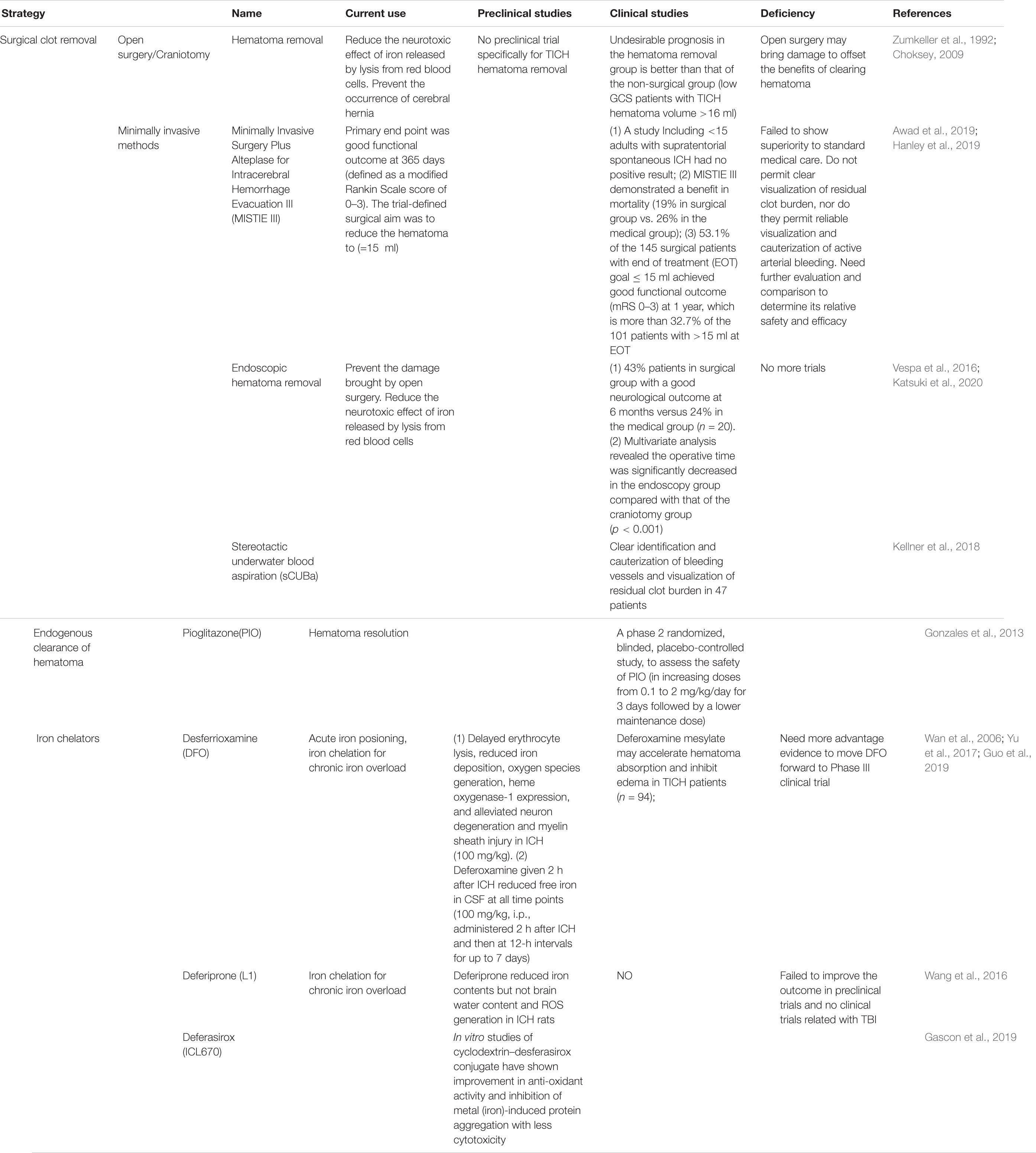
Table 1. Intervention strategies and clinical trials for iron overload in traumatic brain injury (TBI).
Twenty-four hours after bleeding, iron begins to be released into the brain; on the seventh day, the non-heme iron content of the brain triples and remains at a high level for at least 28 days. This is an important time window for the treatment of secondary damage caused by blood catabolites. The first step is to promote the absorption of the endogenous hematoma and reduce the continuous decomposition of red blood cells to prevent the release of toxic substances such as iron. In a clinical trial, the use of the FDA-approved peroxisome proliferator-activated receptor (PPAR)-g agonist pioglitazone can promote the endogenous absorption of hematoma (Gonzales et al., 2013). In addition, in animal model studies, the enhancement of the activity of low-density lipoprotein receptor-related protein-1 (LRP1) and the inhibition of the expression of erythrocyte integrin-related protein CD-47 can enhance endogenous hematoma clearance (Cao et al., 2016). These elements may all become potential therapeutic targets for clinical endogenous hematoma removal. Although a previous study has confirmed that hematoma removal is beneficial for improving cognitive function of cerebral hemorrhage patients (Tyler et al., 1999), not all hematoma removal show outcome benefit. In the clear III trial administrated by the Daniel F. Hanley team, the outcome shows a reduced mortality of IVH patients after alteplase treatment, but does not improve the secondary neurological dysfunction significantly (Hanley et al., 2017). In view of the uncertain benefit of hematoma removal, researchers and neurosurgeons then put the treatment targets on overload iron and the secondary injury induced by iron. So, the second step is the use of an iron chelator. For this step, deferoxamine has been used for more than 40 years. It mainly forms stable compounds with ferric iron and hemosiderin to reduce the oxidative stress caused by iron to play a neuroprotective role (Zaman et al., 1999). It has the characteristics of fewer side effects and can quickly concentrate in the brain tissue through the BBB (Palmer et al., 1994). An injection dose of 62 mg/kg/day for three consecutive days to treat patients with cerebral hemorrhage proved to be safe and tolerated (Selim et al., 2011). Magdy found that the use of deferoxamine can reduce serum oxidative stress markers and increase antioxidant capacity (Selim, 2008). In a controlled experiment by Yu et al. (2017) on 94 posttraumatic cerebral hemorrhage patients, the size of edema on days 3, 7, and 14 in the deferoxamine group was significantly smaller than that in the unused group, confirming that deferoxamine can promote the absorption of hematoma in the short term. Currently, deferoxamine is undergoing a second phase clinical trial (Yeatts et al., 2013). There are three iron chelators, desferrioxamine, deferiprone, and desferasirox, for the treatment of β-thalassemia (Ward et al., 2015). At present, there are also a large number of experiments to study its application in neurodegenerative diseases. Due to oxidative stress caused by iron and ferroptosis caused by excitatory amino acid synergy, a large number of clinical trials on anti-amino acid toxicity and antioxidative stress treatment have been conducted. Importantly, selfotel is a glutamate antagonist. In the second phase of human experiments, we found that the mortality caused by its protective treatment was higher than that of the control group (Davis et al., 2000). Additionally, cerestat is a non-competitive glutamate antagonist with good drug tolerance in human experiments because the number of patients in most experimental centers is too small, greatly reducing the validity of the experimental data (Narayan et al., 2002). Some endogenous and synthetic antioxidants, such as polyethylene glycol absorbed (PEGSOD), tirilizad (Upjohn), and vitamin E, have been studied for antioxidant effects, but none have shown therapeutic effects (Hawryluk and Bullock, 2015).
In consideration of the relevance between CSF-cNs and cognitive function (mentioned above), although there is no clinical trial, some rodent animal model and cellular-level studies give us a preliminary understanding. The Li Qing team used the cholera toxin subunit (CB)-saporin (SAP) into the lateral ventricle of rats to exclusively damage the CSF-cNs. As a result, learning and memory abilities in animals decreased significantly after the destruction of CSF-cNs (Li et al., 2020). We have demonstrated that the brain is with acidosis after TBI in the Current Understanding of Secondary Brain Injury After Traumatic Brain Injury section. Actually, the unpublished data of our team indicates that acidosis can induce neural stem cell damage through excessive activated acid-sensing ion channels (ASICS), which are abundantly located on the dendritic spine of CSF-cNs. As a specific ASIC1a blocker, Psalmotoxin 1 (PcTX1) injected in the ventricular could reduce the focal infarct size up to 60% forever or temporarily in the cerebral ischemia model (Xiong et al., 2004). In general, CSF-cNs could be a new treatment target after TBI.
It is very clear that iron overload and iron metabolism-related protein disorder is closely related to mTBI. Iron overload can produce autophagic death and ferroptosis, and promote the phosphorylation of tau protein to form nerve fiber tangles, increasing the risk of TBI patients suffering from neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease; In addition, it also can induce neural network damage.
However, there is little research about iron accumulation in the TBI injury mechanism. A single research strategy resulted in few effective treatments for nerve damage caused by iron toxicity. A large number of studies found that cell damage caused by iron overload cannot be alleviated by antioxidant or anti-excitatory amino acid treatment, and it was not very helpful for the improvement of cognitive function and the long-term recovery of the patient. Many related agents have played a role in preclinical animal experiments but rarely can be converted into clinical applications. In the early stage, some effects of iron chelators, such as deferoxamine, have been found in anti-iron therapy, but they have not yet been formally investigated for the clinical treatment of TBI patients. In mechanistic research, various research centers around the world have adopted a variety of experimental animal models to simulate the pathophysiological characteristics of human TBI. A large number of neuroprotective agents have been found in animal and cell brain injury models, and none of them have shown positive long-term results in clinical trials (Menon, 2009; Agoston et al., 2012). Most preclinical studies focus on the acute and short-term pathological effects of TBI (Agoston et al., 2012), but ignoring mTBI, which is the large proportion of TBI, and mTBI’s clinical characteristics are always with long-term emotional and cognitive dysfunction. The clinical characteristics of TBI are extremely heterogeneous, and all animal models possessed only a few pathological features (DeWitt et al., 2018). For example, the chronic traumatic encephalopathy experimental animal model cannot replicate the typical pathological features of human chronic traumatic encephalopathy: phosphorylation of tau protein and the formation of nerve fiber tangles. Most of the current multicenter preclinical trials on TBI are not effective. In future research, we should seek better methods for making animal models that are characterized by pathophysiological mechanisms closer to human TBI patients and should be replicated in large experimental animals.
Iron overload has an extremely important role in the secondary injury of mTBI. Autophagy exists in various nerve cells, and the effect of autophagy inhibition or activation on the nervous system after brain trauma is not yet clear, but it has been confirmed that the expression of autophagy formation-related proteins increases. We assume that CSF-cNs suffer from iron accumulation, and the autophagy flux changed, and maybe an important mechanism of cognitive dysfunction after mTBI. In future research, we should further explore the development of autophagy and the interaction mechanism with iron after mTBI and seek new targets for intervention. The CSF-cN neurotransmitter transmission can affect the learning and memory ability of the hippocampus. Our research team has confirmed that iron deposition around the ventricle and ependymal cilia damage occur after IVH, potentially representing a new TBI treatment target, and may be related to the long-term recovery of cognitive function.
SH, SL, and YC drafted the manuscript and figures. HF and YC contributed to proofread and revised the manuscript. YC gave the final proof for this submission. All the authors contributed to the article and approved the submitted version.
This study was supported by the State Key Laboratory of Trauma, Burn and Combined Injury (SKLYQ202002 to YC), Southwest Hospital (SWH2018BJKJ-05 to YC), National Natural Science Foundation of China (82030036 to HF), and Chongqing Talent Program (4139Z2391 to HF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdalkader, M., Lampinen, R., Kanninen, K. M., Malm, T. M., and Liddell, J. R. (2018). Targeting Nrf2 to suppress ferroptosis and mitochondrial dysfunction in neurodegeneration. Front. Neurosci. 12:466. doi: 10.3389/fnins.2018.00466.
Af Geijerstam, J. L., and Britton, M. (2003). Mild head injury - mortality and complication rate: meta-analysis of findings in a systematic literature review. Acta Neurochir (Wien) 145, 843–850 doi: 10.1007/s00701-003-0115-1
Agoston, D. V., Risling, M., and Bellander, B. -M. (2012). Bench-To-Bedside and bedside back to the bench; coordinating clinical and experimental traumatic brain injury studies. Front. Neurol. 3:3. doi: 10.3389/fneur.2012.00003.
Andersen, H. H., Johnsen, K. B., and Moos, T. (2013). Iron deposits in the chronically inflamed central nervous system and contributes to neurodegeneration. Cell. Mol. Life Sci. 71, 1607–1622. doi: 10.1007/s00018-013-1509-8
Arboix, A., Comes, E., García-Eroles, L., Massons, J., Oliveres, M., Balcells, M., et al. (2002). Site of bleeding and early outcome in primary intracerebral hemorrhage. Acta Neurol. Scand 105, 282–288. doi: 10.1034/j.1600-0404.2002.1o170.x
Armstrong, R. C., Mierzwa, A. J., Sullivan, G. M., and Sanchez, M. A. (2016). Myelin and oligodendrocyte lineage cells in white matter pathology and plasticity after traumatic brain injury. Neuropharmacology 110, 654–659. doi: 10.1016/j.neuropharm.2015.04.029
Asnaghi, L., Bruno, P., Priulla, M., and Nicolin, A. (2004). mTOR: a protein kinase switching between life and death. Pharmacol. Res. 50, 545–549. doi: 10.1016/j.phrs.2004.03.007
Au, A. K., Aneja, R. K., Bayır, H., Bell, M. J., Janesko-Feldman, K., Kochanek, P. M., et al. (2016). Autophagy biomarkers beclin 1 and p62 are increased in cerebrospinal fluid after traumatic brain injury. Neurocrit. Care 26, 348–355. doi: 10.1007/s12028-016-0351-x
Awad, I. A., Polster, S. P., Carrion-Penagos, J., Thompson, R. E., Cao, Y., Stadnik, A., et al. (2019). Surgical performance determines functional outcome benefit in the minimally invasive surgery plus recombinant tissue plasminogen activator for intracerebral hemorrhage evacuation (MISTIE) procedure. Neurosurgery 84, 1157–1168.
Ayubcha, C., Revheim, M. E., Newberg, A., Moghbel, M., Rojulpote, C., Werner, T. J., et al. (2021). A critical review of radiotracers in the positron emission tomography imaging of traumatic brain injury: FDG, tau, and amyloid imaging in mild traumatic brain injury and chronic traumatic encephalopathy. Eur. J. Nucl. Med. Mol. Imag. 48, 623–641. doi: 10.1007/s00259-020-04926-4
Barkhoudarian, G., Hovda, D. A., and Giza, C. C. (2016). The molecular pathophysiology of concussive brain injury - an update. Phys. Med. Rehabil. Clin. N. Am. 27, 373–393. doi: 10.1016/j.pmr.2016.01.003
Bayır, H., Anthonymuthu, T. S., Tyurina, Y. Y., Patel, S. J., Amoscato, A. A., Lamade, A. M., et al. (2020). Achieving life through death: redox biology of lipid peroxidation in ferroptosis. Cell Chem. Biol. 27, 387–408. doi: 10.1016/j.chembiol.2020.03.014
Benarroch, E. E. (2009). Brain iron homeostasis and neurodegenerative disease. Neurology 72, 1436–1440. doi: 10.1212/wnl.0b013e3181a26b30
Beschorner, R., Adjodah, D., Schwab, J. M., Mittelbronn, M., Pedal, I., Mattern, R., et al. (2000). Long-term expression of heme oxygenase-1 (HO-1, HSP-32) following focal cerebral infarctions and traumatic brain injury in humans. Acta Neuropathol. 100, 377–384. doi: 10.1007/s004010000202
Boake, C., Mccauley, S. R., Levin, H. S., Pedroza, C., Contant, C. F., Song, J. X., et al. (2005). Diagnostic criteria for postconcussional syndrome after mild to moderate traumatic brain injury. J. Neuropsychiatry Clin. Neurosci. 17, 350–356. doi: 10.1176/jnp.17.3.350
Borlongan, C., Acosta, S., De La Pena, I., Tajiri, N., Kaneko, Y., Lozano, D., et al. (2015). Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatric Dis. Treatment 11:97–106.
Bouley, J., Chung, D. Y., Ayata, C., Brown, R. H., Jr., and Henninger, N. (2019). Cortical spreading depression denotes concussion injury. J. Neurotrauma 36, 1008–1017. doi: 10.1089/neu.2018.5844
Bouras, C., Giannakopoulos, P., Good, P. F., Hsu, A., Hof, P. R., and Perl, D. P. (1997). A laser microprobe mass analysis of brain aluminum and iron in dementia pugilistica: comparison with Alzheimer’s disease. Eur. Neurol. 38, 53–58. doi: 10.1159/000112903
Cao, S., Zheng, M., Hua, Y., Chen, G., Keep, R. F., and Xi, G. (2016). Hematoma changes during clot resolution after experimental intracerebral hemorrhage. Stroke 47, 1626–1631. doi: 10.1161/strokeaha.116.013146
Cheng, Z., and Li, Y. (2007). What is responsible for the initiating chemistry of iron-mediated lipid peroxidation: an update. Chem. Rev. 107, 748–766. doi: 10.1021/cr040077w
Choksey, M. S. (2009). Peri-operative lumbar epidural bleeding controlled by dural tube reflation. Br. J. Neurosurg. 23, 437–438. doi: 10.1080/02688690902887556
Choksey, M., Crockard, H. A., and Sandilands, M. (2009). Acute traumatic intracerebral haematomas: determinants of outcome in a retrospective series of 202 cases. Br. J. Neurosurg. 7, 611–622. doi: 10.3109/02688699308995090
Clark, R. S., Bayir, H., Chu, C. T., Alber, S. M., Kochanek, P. M., and Watkins, S. C. (2008). Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy 4, 88–90. doi: 10.4161/auto.5173
Connor, J. R., and Menzies, S. L. (1996). Relationship of iron to oligodendrocytes and myelination. Glia 17, 83–93. doi: 10.1002/(sici)1098-1136(199606)17:2<83::aid-glia1>3.0.co;2-7
Cook, G. A., and Hawley, J. S. (2014). A review of mild traumatic brain injury diagnostics: current perspectives, limitations, and emerging technology. Mil. Med. 179, 1083–1089. doi: 10.7205/milmed-d-13-00435
Curcio-Morelli, C., Charles, F. A., Micsenyi, M. C., Cao, Y., Venugopal, B., Browning, M. F., et al. (2010). Macroautophagy is defective in mucolipin-1-deficient mouse neurons. Neurobiol. Dis. 40, 370–377. doi: 10.1016/j.nbd.2010.06.010
Daneshvar, D. H., Nowinski, C. J., Mckee, A. C., and Cantu, R. C. (2011). The epidemiology of sport-related concussion. Clin. Sports Med. 30, 1–17. doi: 10.1016/j.csm.2020.08.013
Davis, S. M., Lees, K. R., Albers, G. W., Diener, H. C., Markabi, S., Karlsson, G., et al. (2000). Selfotel in acute ischemic stroke : possible neurotoxic effects of an NMDA antagonist. Stroke 31, 347–354. doi: 10.1161/01.str.31.2.347
de Vries, H. E., Witte, M., Hondius, D., Rozemuller, A. J., Drukarch, B., Hoozemans, J., et al. (2008). Nrf2-induced antioxidant protection: a promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic Biol Med 45, 1375–1383. doi: 10.1016/j.freeradbiomed.2008.09.001
DeWitt, D. S., Hawkins, B. E., Dixon, C. E., Kochanek, P. M., Armstead, W., Bass, C. R., et al. (2018). Pre-Clinical testing of therapies for traumatic brain injury. J. Neurotrauma 35, 2737–2754.
Dikmen, S., Machamer, J., Fann, J. R., and Temkin, N. R. (2010). Rates of symptom reporting following traumatic brain injury. J. Int. Neuropsychol. Soc. 16, 401–411. doi: 10.1017/s1355617710000196
Diskin, T., Tal-Or, P., Erlich, S., Mizrachy, L., Alexandrovich, A., Shohami, E., et al. (2005). Closed head injury induces upregulation of Beclin 1 at the cortical site of injury. J. Neurotrauma 22, 750–762. doi: 10.1089/neu.2005.22.750
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Dong, X. P., Cheng, X., Mills, E., Delling, M., Wang, F., Kurz, T., et al. (2008). The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 455, 992–996. doi: 10.1038/nature07311
Dowdle, W. E., Nyfeler, B., Nagel, J., Elling, R. A., Liu, S., Triantafellow, E., et al. (2014). Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 16, 1069–1079. doi: 10.1038/ncb3053
Drakesmith, H., and Prentice, A. M. (2012). Hepcidin and the iron-infection axis. Science 338, 768–772. doi: 10.1126/science.1224577
Dringen, R., Bishop, G. M., Koeppe, M., Dang, T. N., and Robinson, S. R. (2007). The pivotal role of astrocytes in the metabolism of iron in the brain. Neurochem. Res. 32, 1884–1890. doi: 10.1007/s11064-007-9375-0
Espósito, B. P., Epsztejn, S., Breuer, W., and Cabantchik, Z. I. (2002). A review of fluorescence methods for assessing labile iron in cells and biological fluids. Anal. Biochem. 304, 1–18. doi: 10.1006/abio.2002.5611
Faden, A. I., Wu, J., Stoica, B. A., and Loane, D. J. (2016). Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br. J. Pharmacol. 173, 681–691. doi: 10.1111/bph.13179
Fernández, B., Fdez, E., Gómez-Suaga, P., Gil, F., Molina-Villalba, I., Ferrer, I., et al. (2016). Iron overload causes endolysosomal deficits modulated by NAADP-regulated 2-pore channels and RAB7A. Autophagy 12, 1487–1506. doi: 10.1080/15548627.2016.1190072
Ganz, T., and Nemeth, E. (2012). Hepcidin and iron homeostasis. Biochim. Biophys. Acta (BBA) - Mol. Cell Res. 1823, 1434–1443.
Gao, L., Jiang, Z., Cai, Z., Cai, M., Zhang, Q., Ma, Y., et al. (2017). Brain iron deposition analysis using susceptibility weighted imaging and its association with body iron level in patients with mild cognitive impairment. Mol. Med. Rep. 16, 8209–8215. doi: 10.3892/mmr.2017.7668
Gardner, R. C., Burke, J. F., Nettiksimmons, J., Goldman, S., Tanner, C. M., and Yaffe, K. (2015). Traumatic brain injury in later life increases risk for Parkinson disease. Ann. Neurol. 77, 987–995. doi: 10.1002/ana.24396
Gascon, J. M., Oliveri, V., Mcgown, A., Kaya, E., Chen, Y. L., Austin, C., et al. (2019). Synthesis and study of multifunctional cyclodextrin-deferasirox hybrids. ChemMedChem 14, 1484–1492. doi: 10.1002/cmdc.201900334
Gong, Y., Xi, G. H., Keep, R. F., Hoff, J. T., and Hua, Y. (2005). Complement inhibition attenuates brain edema and neurological deficits induced by thrombin. Acta Neurochir Suppl. 95, 389–392. doi: 10.1007/3-211-32318-x_79
Gonzales, N. R., Shah, J., Sangha, N., Sosa, L., Martinez, R., Shen, L., et al. (2013). Design of a prospective, dose-escalation study evaluating the Safety of Pioglitazone for Hematoma Resolution in Intracerebral Hemorrhage (SHRINC). Int. J. Stroke 8, 388–396. doi: 10.1111/j.1747-4949.2011.00761.x
Guo, X., Qi, X., Li, H., Duan, Z., Wei, Y., Zhang, F., et al. (2019). Deferoxamine alleviates iron overload and brain injury in a rat model of brainstem hemorrhage. World Neurosurg. 128, e895–e904.
Gustavsson, A., Svensson, M., Jacobi, F., Allgulander, C., Alonso, J., Beghi, E., et al. (2011). Cost of disorders of the brain in Europe 2010. Eur. Neuropsychopharmacol. 21, 718–779.
Hanley, D. F., Thompson, R. E., Rosenblum, M., Yenokyan, G., Lane, K., Mcbee, N., et al. (2019). Efficacy and safety of minimally invasive surgery with thrombolysis in intracerebral haemorrhage evacuation (MISTIE III): a randomised, controlled, open-label, blinded endpoint phase 3 trial. Lancet 393, 1021–1032.
Hanley, D., Lane, K., Mcbee, N., Ziai, W., Tuhrim, S., Lees, K., et al. (2017). Thrombolytic removal of intraventricular haemorrhage in treatment of severe stroke: results of the randomised, multicentre, multiregion, placebo-controlled CLEAR III trial. Lancet 389, 603–611. doi: 10.1016/s0140-6736(16)32410-2
Hawryluk, G. W. J., and Bullock, M. R. (2015). Design of acute neuroprotection studies. Handb. Clin. Neurol. 128, 761–778. doi: 10.1016/b978-0-444-63521-1.00047-9
He, Y., Wan, S., Hua, Y., Keep, R. F., and Xi, G. (2007). Autophagy after experimental intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 28, 897–905. doi: 10.1038/sj.jcbfm.9600578
Hui, Y., Wang, D., Li, W., Zhang, L., Jin, J., Ma, N., et al. (2011). Long-term overexpression of heme oxygenase 1 promotes tau aggregation in mouse brain by inducing tau phosphorylation. J. Alzheimers Dis. 26, 299–313. doi: 10.3233/jad-2011-102061
Illes, S. (2017). More than a drainage fluid: the role of CSF in signaling in the brain and other effects on brain tissue. Handb. Clin. Neurol. 146, 33–46. doi: 10.1016/b978-0-12-804279-3.00003-4
Kanaan, N. M., Cox, K., Alvarez, V. E., Stein, T. D., Poncil, S., and Mckee, A. C. (2016). Characterization of early pathological tau conformations and phosphorylation in chronic traumatic encephalopathy. J. Neuropathol. Exp. Neurol. 75, 19–34. doi: 10.1093/jnen/nlv001
Katsuki, M., Kakizawa, Y., Nishikawa, A., Yamamoto, Y., and Uchiyama, T. (2020). Endoscopic hematoma removal of supratentorial intracerebral hemorrhage under local anesthesia reduces operative time compared to craniotomy. Sci. Rep. 10:10389.
Ke, Y., and Qian, Z. M. (2007). Brain iron metabolism: neurobiology and neurochemistry. Prog. Neurobiol. 83, 149–173. doi: 10.1016/j.pneurobio.2007.07.009
Kellner, C. P., Chartrain, A. G., Nistal, D. A., Scaggiante, J., Hom, D., Ghatan, S., et al. (2018). The stereotactic intracerebral hemorrhage underwater blood aspiration (SCUBA) technique for minimally invasive endoscopic intracerebral hemorrhage evacuation. J. Neurointerv. Surg. 10, 771–776. doi: 10.1136/neurintsurg-2017-013719
Koike, M., Shibata, M., Tadakoshi, M., Gotoh, K., Komatsu, M., Waguri, S., et al. (2008). Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am. J. Pathol. 172, 454–469. doi: 10.2353/ajpath.2008.070876
Kumar, A., and Loane, D. J. (2012). Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav. Immunity 26, 1191–1201. doi: 10.1016/j.bbi.2012.06.008
Kurland, D., Hong, C., Aarabi, B., Gerzanich, V., and Simard, J. M. (2012). Hemorrhagic progression of a contusion after traumatic brain injury: a review. J. Neurotrauma 29, 19–31. doi: 10.1089/neu.2011.2122
Lane, D. J., Robinson, S. R., Czerwinska, H., Bishop, G. M., and Lawen, A. (2010). Two routes of iron accumulation in astrocytes: ascorbate-dependent ferrous iron uptake via the divalent metal transporter (DMT1) plus an independent route for ferric iron. Biochem. J. 432, 123–132. doi: 10.1042/bj20101317
Langlois, J. A., Rutland-Brown, W., and Wald, M. M. (2006). The epidemiology and impact of traumatic brain injury: a brief overview. J. Head Trauma Rehabil. 21, 375–378. doi: 10.1097/00001199-200609000-00001
Lee, S., Park, S., Won, J., Lee, S. -R., Chang, K. -T., and Hong, Y. (2015). The incremental induction of neuroprotective properties by multiple therapeutic strategies for primary and secondary neural injury. Int. J. Mol. Sci. 16, 19657–19670. doi: 10.3390/ijms160819657
Li, H., and Qian, Z. M. (2002). Transferrin/transferrin receptor-mediated drug delivery. Med. Res. Rev. 22, 225–250. doi: 10.1002/med.10008
Li, Q., Liu, H., Zhang, L., Zhang, H., and Zhou, F. (2020). The CSF-contacting nucleus regulates learning and memory through synaptic connections with the hippocampus. Neurosci. Lett. 728:134972. doi: 10.1016/j.neulet.2020.134972
Lieblein-Boff, J. C., Mckim, D. B., Shea, D. T., Wei, P., Deng, Z., Sawicki, C., et al. (2013). Neonatal E. coli infection causes neuro-behavioral deficits associated with hypomyelination and neuronal sequestration of iron. J. Neurosci. 33, 16334–16345. doi: 10.1523/jneurosci.0708-13.2013
Lin, W. J., and Kuang, H. Y. (2014). Oxidative stress induces autophagy in response to multiple noxious stimuli in retinal ganglion cells. Autophagy 10, 1692–1701. doi: 10.4161/auto.36076
Liu, C. L., Chen, S., Dietrich, D., and Hu, B. R. (2008). Changes in autophagy after traumatic brain injury. J. Cereb. Blood Flow Metab. 28, 674–683.
Liu, H. D., Li, W., Chen, Z. R., Zhou, M. L., Zhuang, Z., Zhang, D. D., et al. (2013). Increased expression of ferritin in cerebral cortex after human traumatic brain injury. Neurol. Sci. 34, 1173–1180. doi: 10.1007/s10072-012-1214-7
Lu, L., Cao, H., Wei, X., Li, Y., and Li, W. (2015). Iron deposition is positively related to cognitive impairment in patients with chronic mild traumatic brain injury: assessment with susceptibility weighted imaging. Biomed. Res. Int. 2015:470676.
Ma, Y., De groot, H., Liu, Z., Hider, R C., and Petrat, F. (2006). Chelation and determination of labile iron in primary hepatocytes by pyridinone fluorescent probes. Biochem. J. 395, 49–55. doi: 10.1042/bj20051496
Malecki, E. A., Devenyi, A. G., Beard, J. L., and Connor, J. R. (1999). Existing and emerging mechanisms for transport of iron and manganese to the brain. J. Neurosci. Res. 56, 113–122. doi: 10.1002/(sici)1097-4547(19990415)56:2<113::aid-jnr1>3.0.co;2-k
Marshall, J. C. (2001). Inflammation, coagulopathy, and the pathogenesis of multiple organ dysfunction syndrome. Crit. Care Med. 29, S99–106.
McCarthy, R. C., Sosa, J. C., Gardeck, A. M., Baez, A. S., Lee, C. -H., and Wessling-Resnick, M. (2018). Inflammation-induced iron transport and metabolism by brain microglia. J. Biol. Chem. 293, 7853–7863. doi: 10.1074/jbc.ra118.001949
McKee, A. C., Stein, T. D., Kiernan, P. T., and Alvarez, V. E. (2015). The neuropathology of chronic traumatic encephalopathy. Brain Pathol. 25, 350–364.
McKee, A. C., Stern, R. A., Nowinski, C. J., Stein, T. D., Alvarez, V. E., Daneshvar, D. H., et al. (2013). The spectrum of disease in chronic traumatic encephalopathy. Brain 136, 43–64.
Mecklenburg, I., Reznik, D., Fasler-Kan, E., Drewe, J., Beglinger, C., and Hruz, P. (2014). Serum hepcidin concentrations correlate with ferritin in patients with inflammatory bowel disease. J. Crohn’s Colitis 8, 1392–1397. doi: 10.1016/j.crohns.2014.04.008
Menon, D. K. (2009). Unique challenges in clinical trials in traumatic brain injury. Crit. Care Med. 37, S129–S135.
Menon, D. K., Schwab, K., Wright, D. W., and Maas, A. I. (2010). position statement: definition of traumatic brain injury. Arch. Phys. Med. Rehabil. 91, 1637–1640. doi: 10.1016/j.apmr.2010.05.017
Mondello, S., Schmid, K., Berger, R. P., Kobeissy, F., Italiano, D., Jeromin, A., et al. (2014). The challenge of mild traumatic brain injury: role of biochemical markers in diagnosis of brain damage. Med. Res. Rev. 34, 503–531. doi: 10.1002/med.21295
Mu, J., Li, M., Wang, T., Li, X., Bai, M., Zhang, G., et al. (2019). Myelin damage in diffuse axonal injury. Front. Neurosci. 13:217. doi: 10.3389/fnins.2019.00217.
Narayan, R. K., Michel, M. E., Ansell, B., Baethmann, A., Biegon, A., Bracken, M. B., et al. (2002). Clinical trials in head injury. J. Neurotrauma 19, 503–557.
Nelson, L. D., Temkin, N. R., Dikmen, S., Barber, J., Giacino, J. T., Yuh, E., et al. (2019). Recovery after mild traumatic brain injury in patients presenting to US Level I trauma centers. JAMA Neurol. 76:1049.
Nisenbaum, E. J., Novikov, D. S., and Lui, Y. W. (2014). The presence and role of iron in mild traumatic brain injury: an imaging perspective. J. Neurotrauma 31, 301–307. doi: 10.1089/neu.2013.3102
Nobuta, H., Yang, N., Ng, Y. H., Marro, S. G., Sabeur, K., Chavali, M., et al. (2019). Oligodendrocyte death in pelizaeus-merzbacher disease is rescued by iron chelation. Cell Stem Cell 25, 531–541.e6.
Nunez, M. T., Urrutia, P., Mena, N., Aguirre, P., Tapia, V., and Salazar, J. (2012). Iron toxicity in neurodegeneration. Biometals 25, 761–776. doi: 10.1007/s10534-012-9523-0
Oliveira-Silva, I. F., Pinto, L., Pereira, S. R., Ferraz, V. P., Barbosa, A. J., Coelho, V. A., et al. (2007). Age-related deficit in behavioural extinction is counteracted by long-term ethanol consumption: correlation between 5-HIAA/5HT ratio in dorsal raphe nucleus and cognitive parameters. Behav. Brain Res. 180, 226–234. doi: 10.1016/j.bbr.2007.03.012
Orts-Del’Immagine, A., and Wyart, C. (2017). Cerebrospinal-fluid-contacting neurons. Curr. Biol. 27, R1198–R1200.
Palmer, C., Roberts, R. L., and Bero, C. (1994). Deferoxamine posttreatment reduces ischemic brain injury in neonatal rats. Stroke 25, 1039–1045. doi: 10.1161/01.str.25.5.1039
Patel, S. J., Frey, A. G., Palenchar, D. J., Achar, S., Bullough, K. Z., Vashisht, A., et al. (2019). A PCBP1-BolA2 chaperone complex delivers iron for cytosolic [2Fe-2S] cluster assembly. Nat. Chem. Biol. 15, 872–881. doi: 10.1038/s41589-019-0330-6
Pavlovic, D., Pekic, S., Stojanovic, M., and Popovic, V. (2019). Traumatic brain injury: neuropathological, neurocognitive and neurobehavioral sequelae. Pituitary 22, 270–282. doi: 10.1007/s11102-019-00957-9
Pearn, M. L., Niesman, I. R., Egawa, J., Sawada, A., Almenar-Queralt, A., Shah, S. B., et al. (2017). Pathophysiology associated with traumatic brain injury: current treatments and potential novel therapeutics. Cell. Mol. Neurobiol. 37, 571–585. doi: 10.1007/s10571-016-0400-1
Peng, T. -I., and Jou, M. -J. (2010). Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 1201, 183–188. doi: 10.1111/j.1749-6632.2010.05634.x
Philpott, C. C., Ryu, M. -S., Frey, A., and Patel, S. (2017). Cytosolic iron chaperones: proteins delivering iron cofactors in the cytosol of mammalian cells. J. Biol. Chem. 292, 12764–12771. doi: 10.1074/jbc.r117.791962
Quiles del Rey, M., and Mancias, J. D. (2019). NCOA4-Mediated ferritinophagy: a potential link to neurodegeneration. Front. Neurosci. 13:238. doi: 10.3389/fnins.2019.00238.
Raha-Chowdhury, R., Raha, A. A., Forostyak, S., Zhao, J. -W., Stott, S. R. W., and Bomford, A. (2015). Expression and cellular localization of hepcidin mRNA and protein in normal rat brain. BMC Neurosci. 16:24. doi: 10.1186/s12868-015-0161-7.
Raz, E., Jensen, J. H., Ge, Y., Babb, J. S., Miles, L., Reaume, J., et al. (2011). Brain Iron quantification in mild traumatic brain injury: a magnetic field correlation study. Am. J. Neuroradiol. 32, 1851–1856. doi: 10.3174/ajnr.a2637
Recalcati, S., Minotti, G., and Cairo, G. (2010). Iron regulatory proteins: from molecular mechanisms to drug development. Antioxid Redox Signal 13, 1593–1616. doi: 10.1089/ars.2009.2983
Rochette, L., Gudjoncik, A., Guenancia, C., Zeller, M., Cottin, Y., and Vergely, C. (2015). The iron-regulatory hormone hepcidin: a possible therapeutic target? Pharmacol. Ther. 146, 35–52. doi: 10.1016/j.pharmthera.2014.09.004
Roe, C., Sveen, U., Alvsaker, K., and Bautz-Holter, E. (2009). Post-concussion symptoms after mild traumatic brain injury: influence of demographic factors and injury severity in a 1-year cohort study. Disabil. Rehabil. 31, 1235–1243. doi: 10.1080/09638280802532720
Rosenfeld, J. V., Maas, A. I., Bragge, P., Morganti-Kossmann, M. C., Manley, G. T., and Gruen, R. L. (2012). Early management of severe traumatic brain injury. Lancet 380, 1088–1098.
Rouault, T. A., and Cooperman, S. (2006). Brain iron metabolism. Sem. Pediatric Neurol. 13, 142–148.
Rubinsztein, D. C., Difiglia, M., Heintz, N., Nixon, R. A., Qin, Z. H., Ravikumar, B., et al. (2005). Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 1, 11–22. doi: 10.4161/auto.1.1.1513
Sadasivan, S., Dunn, W. A., Hayes, R. L., and Wang, K. K. W. (2008). Changes in autophagy proteins in a rat model of controlled cortical impact induced brain injury. Biochem. Biophys. Res. Commun. 373, 478–481. doi: 10.1016/j.bbrc.2008.05.031
Sakai, K., Fukuda, T., and Iwadate, K. (2014). Immunohistochemical analysis of the ubiquitin proteasome system and autophagy lysosome system induced after traumatic intracranial injury: association with time between the injury and death. Am. J. Forensic Med. Pathol. 35, 38–44. doi: 10.1097/paf.0000000000000067
Salvador, G. A. (2010). Iron in neuronal function and dysfunction. BioFactors 36, 103–110. doi: 10.1002/biof.80
Savman, K., Nilsson, U. A., Blennow, M., Kjellmer, I., and Whitelaw, A. (2001). Non-protein-bound iron is elevated in cerebrospinal fluid from preterm infants with posthemorrhagic ventricular dilatation. Pediatr. Res. 49, 208–212. doi: 10.1203/00006450-200102000-00013
Scherz-Shouval, R., Shvets, E., Fass, E., Shorer, H., Gil, L., and Elazar, Z. (2007). Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Embo J. 26, 1749–1760. doi: 10.1038/sj.emboj.7601623
Schiepers, O. J. G., Van Boxtel, M. P. J., De Groot, R. H. M., Jolles, J., De Kort, W. L. A. M., Swinkels, D. W., et al. (2010). Serum iron parameters, HFE C282Y genotype, and cognitive performance in older adults: results from the FACIT study. J. Gerontol.: Ser. A 65A, 1312–1321. doi: 10.1093/gerona/glq149
Selim, M. (2008). Deferoxamine mesylate: a new hope for intracerebral hemorrhage: from bench to clinical trials. Stroke 40, S90–S91.
Selim, M., Yeatts, S., Goldstein, J. N., Gomes, J., Greenberg, S., Morgenstern, L. B., et al. (2011). Safety and tolerability of deferoxamine mesylate in patients with acute intracerebral hemorrhage. Stroke 42, 3067–3074. doi: 10.1161/strokeaha.111.617589
Shen, H. (2015). Researchers seek definition of head-trauma disorder. Nature 518, 466–467. doi: 10.1038/518466a
Shetty, A. K., Mishra, V., Kodali, M., and Hattiangady, B. (2014). Blood brain barrier dysfunction and delayed neurological deficits in mild traumatic brain injury induced by blast shock waves. Front. Cell. Neurosci. 8. 232. doi: 10.3389/fncel.2014.00232.
Silver, J. M., Mcallister, T. W., and Arciniegas, D. B. (2009). Depression and cognitive complaints following mild traumatic brain injury. Am. J. Psychiatry 166, 653–661. doi: 10.1176/appi.ajp.2009.08111676
Singh, N., Haldar, S., Tripathi, A. K., Horback, K., Wong, J., Sharma, D., et al. (2014). Brain iron homeostasis: from molecular mechanisms to clinical significance and therapeutic opportunities. Antiox. Redox Signal. 20, 1324–1363. doi: 10.1089/ars.2012.4931
Skouta, R., Dixon, S. J., Wang, J., Dunn, D. E., Orman, M., Shimada, K., et al. (2014). Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 136, 4551–4556. doi: 10.1021/ja411006a
Smith, K. J., Kapoor, R., and Felts, P. A. (1999). Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol. 9, 69–92. doi: 10.1111/j.1750-3639.1999.tb00212.x
Sripetchwandee, J., Sanit, J., Chattipakorn, N., and Chattipakorn, S. C. (2013). Mitochondrial calcium uniporter blocker effectively prevents brain mitochondrial dysfunction caused by iron overload. Life Sci. 92, 298–304. doi: 10.1016/j.lfs.2013.01.004
Stein, T. D., Alvarez, V. E., and Mckee, A. C. (2015). Concussion in chronic traumatic encephalopathy. Curr. Pain Headache Rep. 19:47.
Sterr, A., Herron, K. A., Hayward, C., and Montaldi, D. (2006). Are mild head injuries as mild as we think? neurobehavioral concomitants of chronic post-concussion syndrome. BMC Neurol. 6:7. doi: 10.1186/1471-2377-6-7.
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Styś, A., Galy, B., Starzyński, R. R., Smuda, E., Drapier, J. -C., Lipiński, P., et al. (2011). Iron regulatory protein 1 outcompetes iron regulatory protein 2 in regulating cellular iron homeostasis in response to nitric oxide. J. Biol. Chem. 286, 22846–22854. doi: 10.1074/jbc.m111.231902
Sulhan, S., Lyon, K. A., Shapiro, L. A., and Huang, J. H. (2020). Neuroinflammation and blood-brain barrier disruption following traumatic brain injury: pathophysiology and potential therapeutic targets. J. Neurosci. Res. 98, 19–28. doi: 10.1002/jnr.24331
Takumida, M., and Anniko, M. (2010). Expression of transient receptor potential channel mucolipin (TRPML) and polycystine (TRPP) in the mouse inner ear. Acta Otolaryngol. 130, 196–203. doi: 10.3109/00016480903013593
Tao, C., Hu, X., Li, H., and You, C. (2017). White matter injury after intracerebral hemorrhage: pathophysiology and therapeutic strategies. Front. Hum. Neurosci. 11:422. doi: 10.3389/fnhum.2017.00422.
Terman, A., and Kurz, T. (2013). Lysosomal iron, iron chelation, and cell death. Antiox. Redox. Signal. 18, 888–898. doi: 10.1089/ars.2012.4885
Theadom, A., Starkey, N. J., Dowell, T., Hume, P. A., Kahan, M., Mcpherson, K., et al. (2014). Sports-related brain injury in the general population: an epidemiological study. J. Sci. Med. Sport 17, 591–596. doi: 10.1016/j.jsams.2014.02.001
Thirupathi, A., and Chang, Y. Z. (2019). Brain iron metabolism and CNS diseases. Adv. Exp. Med. Biol. 1173, 1–19. doi: 10.1007/978-981-13-9589-5_1
Todorich, B., Zhang, X., Slagle-Webb, B., Seaman, W. E., and Connor, J. R. (2008). Tim-2 is the receptor for H-ferritin on oligodendrocytes. J. Neurochem. 107, 1495–1505. doi: 10.1111/j.1471-4159.2008.05678.x
Tonnus, W., Gembardt, F., Latk, M., Parmentier, S., Hugo, C., Bornstein, S. R., et al. (2018). The clinical relevance of necroinflammation—highlighting the importance of acute kidney injury and the adrenal glands. Cell Death Differ. 26, 68–82. doi: 10.1038/s41418-018-0193-5
Turner, R. D., Chesler, D. A., Davis, R., Gutman, F., Chaudry, M. I., Turk, A. S., et al. (2015). Initial multicenter technical experience with the apollo device for minimally invasive intracerebral hematoma evacuation. Operat. Neurosurg. 11, 243–251. doi: 10.1227/neu.0000000000000698
Tyler, D., Mandybur, G. J. S., and Neurosurgery, F. (1999). Interventional MRI-guided stereotactic aspiration of acute/subacute intracerebral hematomas. Stereotact. Funct. Neurosurg. 72, 129–135. doi: 10.1159/000029712
Vespa, P., Hanley, D., Betz, J., Hoffer, A., Engh, J., Carter, R., et al. (2016). ICES (Intraoperative Stereotactic Computed Tomography-Guided Endoscopic Surgery) for brain hemorrhage: a multicenter randomized controlled trial. Stroke 47, 2749–2755. doi: 10.1161/strokeaha.116.013837
Villeneuve, N. F., Lau, A., and Zhang, D. D. (2010). Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: an insight into cullin-ring ubiquitin ligases. Antioxid. Redox. Signal. 13, 1699–1712. doi: 10.1089/ars.2010.3211
Wagner, K. R., Sharp, F. R., Ardizzone, T. D., Lu, A., and Clark, J. F. (2016). Heme and iron metabolism: role in cerebral hemorrhage. J. Cereb. Blood Flow Metab. 23, 629–652. doi: 10.1097/01.wcb.0000073905.87928.6d
Wan, S., Hua, Y., Keep, R. F., Hoff, J. T., and Xi, G. (2006). Deferoxamine reduces CSF free iron levels following intracerebral hemorrhage. Acta Neurochir. Suppl. 96, 199–202. doi: 10.1007/3-211-30714-1_43
Wang, G., Hu, W., Tang, Q., Wang, L., Sun, X. G., Chen, Y., et al. (2016). Effect comparison of both iron chelators on outcomes, iron deposit, and iron transporters after intracerebral hemorrhage in rats. Mol. Neurobiol. 53, 3576–3585. doi: 10.1007/s12035-015-9302-3
Wang, L., Liu, X., You, L. H., Ci, Y. Z., Chang, S., Yu, P., et al. (2019). Hepcidin and iron regulatory proteins coordinately regulate ferroportin 1 expression in the brain of mice. J. Cell Physiol. 234, 7600–7607. doi: 10.1002/jcp.27522
Ward, R. J., Dexter, D. T., and Crichton, R. R. (2015). Neurodegenerative diseases and therapeutic strategies using iron chelators. J. Trace Elements Med. Biol. 31, 267–273. doi: 10.1016/j.jtemb.2014.12.012
Ward, R. J., Zucca, F. A., Duyn, J. H., Crichton, R. R., and Zecca, L. (2014). The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 13, 1045–1060. doi: 10.1016/s1474-4422(14)70117-6
Whiteneck, G. G., Gerhart, K. A., and Cusick, C. P. (2004). Identifying environmental factors that influence the outcomes of people with traumatic brain injury. J. Head. Trauma Rehabil. 19, 191–204. doi: 10.1097/00001199-200405000-00001
Wilkinson, D. A., Keep, R. F., Hua, Y., and Xi, G. (2018). Hematoma clearance as a therapeutic target in intracerebral hemorrhage: from macro to micro. J. Cereb. Blood Flow Metab. 38, 741–745. doi: 10.1177/0271678x17753590
Winkler, E. A., Minter, D., Yue, J. K., and Manley, G. T. (2016). Cerebral edema in traumatic brain injury: pathophysiology and prospective therapeutic targets. Neurosurg. Clin. N. Am. 27, 473–488.
Wolf, J. A., and Koch, P. F. (2016). Disruption of network synchrony and cognitive dysfunction after traumatic brain injury. Front. Syst. Neurosci. 10:43. doi: 10.3389/fnsys.2016.00043
Wortzel, H. S., Shura, R. D., and Brenner, L. A. (2013). Chronic traumatic encephalopathy and suicide: a systematic review. Biomed. Res. Int. 2013:424280.
Wu, J., Hua, Y., Keep, R. F., Nakamura, T., Hoff, J. T., and Xi, G. (2003). Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke 34, 2964–2969. doi: 10.1161/01.str.0000103140.52838.45
Xiong, X. -Y., Wang, J., Qian, Z. -M., and Yang, Q. -W. (2013). Iron and intracerebral hemorrhage: from mechanism to translation. Trans. Stroke Res. 5, 429–441. doi: 10.1007/s12975-013-0317-7
Xiong, Z. G., Zhu, X. M., Chu, X. P., Minami, M., Hey, J., Wei, W. L., et al. (2004). Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell 118, 687–698.
Yang, G., Hu, R., Zhang, C., Qian, C., Luo, Q. -Q., Yung, W. -H., et al. (2016). A combination of serum iron, ferritin and transferrin predicts outcome in patients with intracerebral hemorrhage. Sci. Rep. 6:21970.
Yang, Z., and Klionsky, D. J. (2010). Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22, 124–131. doi: 10.1016/j.ceb.2009.11.014
Yeatts, S. D., Palesch, Y. Y., Moy, C. S., and Selim, M. (2013). High dose deferoxamine in intracerebral hemorrhage (Hi-Def) trial: rationale, design, and methods. Neurocrit. Care 19, 257–266. doi: 10.1007/s12028-013-9861-y
Yorimitsu, T., and Klionsky, D. J. (2005). Autophagy: molecular machinery for self-eating. Cell Death Differ. 12, 1542–1552. doi: 10.1038/sj.cdd.4401765
Yu, J., Yuan, Q., Sun, Y. R., Wu, X., Du, Z. Y., Li, Z. Q., et al. (2017). Effects of deferoxamine mesylate on hematoma and perihematoma edema after traumatic intracerebral hemorrhage. J. Neurotrauma 34, 2753–2759. doi: 10.1089/neu.2017.5033
Yu, Z., Persson, H. L., Eaton, J. W., and Brunk, U. T. (2003). Intralysosomal iron: a major determinant of oxidant-induced cell death. Free Radical. Biol. Med. 34, 1243–1252. doi: 10.1016/s0891-5849(03)00109-6
Zaman, K., Ryu, H., Hall, D., O’donovan, K., Lin, K. I., Miller, M. P., et al. (1999). Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21(waf1/cip1), and erythropoietin. J. Neurosci. 19, 9821–9830. doi: 10.1523/jneurosci.19-22-09821.1999
Zetterberg, H., Smith, D. H., and Blennow, K. (2013). Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nat. Rev. Neurol. 9, 201–210. doi: 10.1038/nrneurol.2013.9
Zhang, L., Wang, H., Zhou, X., Mao, L., Ding, K., and Hu, Z. (2019). Role of mitochondrial calcium uniporter-mediated Ca(2+) and iron accumulation in traumatic brain injury. J. Cell Mol. Med. 23, 2995–3009. doi: 10.1111/jcmm.14206
Zhang, Z., Wu, Y., Yuan, S., Zhang, P., Zhang, J., Li, H., et al. (2018). Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res. 1701, 112–125. doi: 10.1016/j.brainres.2018.09.012
Zhao, Z. A., Zhao, Y., Ning, Y. L., Yang, N., Peng, Y., Li, P., et al. (2017). Adenosine A2A receptor inactivation alleviates early-onset cognitive dysfunction after traumatic brain injury involving an inhibition of tau hyperphosphorylation. Transl. Psychiatry 7:e1123. doi: 10.1038/tp.2017.98
Keywords: traumatic brain injury, cognitive dysfunction, cerebrospinal-fluid contacting neuron, autophagy, iron metabolism
Citation: Huang S, Li S, Feng H and Chen Y (2021) Iron Metabolism Disorders for Cognitive Dysfunction After Mild Traumatic Brain Injury. Front. Neurosci. 15:587197. doi: 10.3389/fnins.2021.587197
Received: 25 July 2020; Accepted: 10 February 2021;
Published: 16 March 2021.
Edited by:
Anwen Shao, Zhejiang University, ChinaReviewed by:
Yi Yang, First Affiliated Hospital of Jilin University, ChinaCopyright © 2021 Huang, Li, Feng and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yujie Chen, eXVqaWVjaGVuNjg4NkBmb3htYWlsLmNvbQ==; orcid.org/0000-0002-9905-9138
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.