 Kelly A. Rich
Kelly A. Rich Jennifer Roggenbuck1
Jennifer Roggenbuck1 Stephen J. Kolb
Stephen J. Kolb- 1Department of Neurology, The Ohio State University Wexner Medical Center, Columbus, OH, United States
- 2Department of Biological Chemistry and Pharmacology, The Ohio State University Wexner Medical Center, Columbus, OH, United States
Genome-wide association studies (GWAS) and rare variant association studies (RVAS) are applied across many areas of complex disease to analyze variation in whole genomes of thousands of unrelated patients. These approaches are able to identify variants and/or biological pathways which are associated with disease status and, in contrast to traditional linkage studies or candidate gene approaches, do so without requiring multigenerational affected families, prior hypotheses, or known genes of interest. However, the novel associations identified by these methods typically have lower effect sizes than those found in classical family studies. In the motor neuron disease amyotrophic lateral sclerosis (ALS), GWAS, and RVAS have been used to identify multiple disease-associated genes but have not yet resulted in novel therapeutic interventions. There is significant urgency within the ALS community to identify additional genetic markers of disease to uncover novel biological mechanisms, stratify genetic subgroups of disease, and drive drug development. Given the widespread and increasing application of genetic association studies of complex disease, it is important to recognize the strengths and limitations of these approaches. Here, we review ALS gene discovery via GWAS and RVAS.
Introduction
In the timeline of gene discovery for hereditary disease, high penetrance genes are historically identified by linkage analysis in multi-generational family studies and subsequently replicated in high-risk case-control studies of independent disease cohorts. These Mendelian genes, with highly significant (or moderately significant) effect sizes, generally represent the “low-hanging fruit” of gene discovery. Identifying the genetic underpinnings of complex diseases requires an approach to assess variation in many genes simultaneously. Genome-wide association studies (GWAS) were developed using single nucleotide variant (SNV) array technology to identify disease-associated variation in large cohorts of cases and controls and became widely adopted in the late 2000s. GWAS are able to interrogate millions of common genetic variants [minor allele frequency (MAF) > 5%] in thousands of unrelated individuals to identify associations with disease that potentially explain some percentage of disease heritability within a population (Tam et al., 2019).
Despite the impact of GWAS in identifying disease-associated genetic changes, the majority of genetic contribution to many complex diseases remains unexplained. Rare variant association studies (RVAS) extend the genome-wide approach by using massively parallel sequencing to identify less-common variants (MAF < 0.5 or 0.1%) that would be missed by GWAS (Lee et al., 2014). This has been made possible by increasing sample sizes in disease cohorts as well as advances in sequencing technology, leading to greater genomic resolution. Next generation sequencing approaches such as whole exome sequencing (WES) and whole genome sequencing (WGS), sequence the coding regions and the entirety of the genome, respectively, allowing for inclusion of rare variants into large association studies of complex disease (Kosmicki et al., 2016).
Rare variant studies extend the reach of traditional association studies by identifying rare and potentially more clinically significant variants using powerful sequencing technologies. Variants identified via GWAS only explain a fraction of missing heritability in most diseases, limiting the clinical relevance of GWAS findings (Manolio et al., 2009). Targeted candidate gene studies have revealed that rare coding variants may produce large effect sizes in complex disease, motivating further investigation into rare variant contribution (Kosmicki et al., 2016). Rare variants are known to play important roles in human disease (Rivas et al., 2011; Gudmundsson et al., 2012) and explain phenotypic differences across the disease spectrum (Cohen, 2004; Cohen et al., 2005).
While GWAS can be performed on WGS or WES, it is most commonly conducted using SNV array to maximize sample size. The associations evaluated via GWAS often do not include variants of less than 0.1% allele frequency. High-depth WGS offers the greatest opportunity for assessing low-frequency or rare variants using an RVAS approach. RVAS is able to assess both single-variants or the cumulative effects of multiple variants on a gene or region (Lee et al., 2014). The latter includes approaches such as burden tests, variance-component tests, and exponential-combination tests (Lee et al., 2014). Further, RVAS can also be used to confirm candidate associations identified via GWAS or screen a known disease-associated gene in a separate cohort (Auer and Lettre, 2015).
The typical association study includes four components; (1) accrual of a large group of individuals with the disease of interest as well as a carefully matched control group for comparison; (2) genotyping of hundreds of thousands to millions of variants in disease and control groups, traditionally via SNV arrays in GWAS and sequencing in RVAS; (3) statistical analyses to test for common- or rare-variant association with disease; and (4) prioritizing and replicating significant findings in a non-overlapping, independent cohort or performing functional experiments to examine variant consequences (Pearson, 2008). Data from association studies can be easily visualized via a Manhattan plot in which significant regions or variants appear as “skyscrapers,” an example of which is provided in Figure 1.
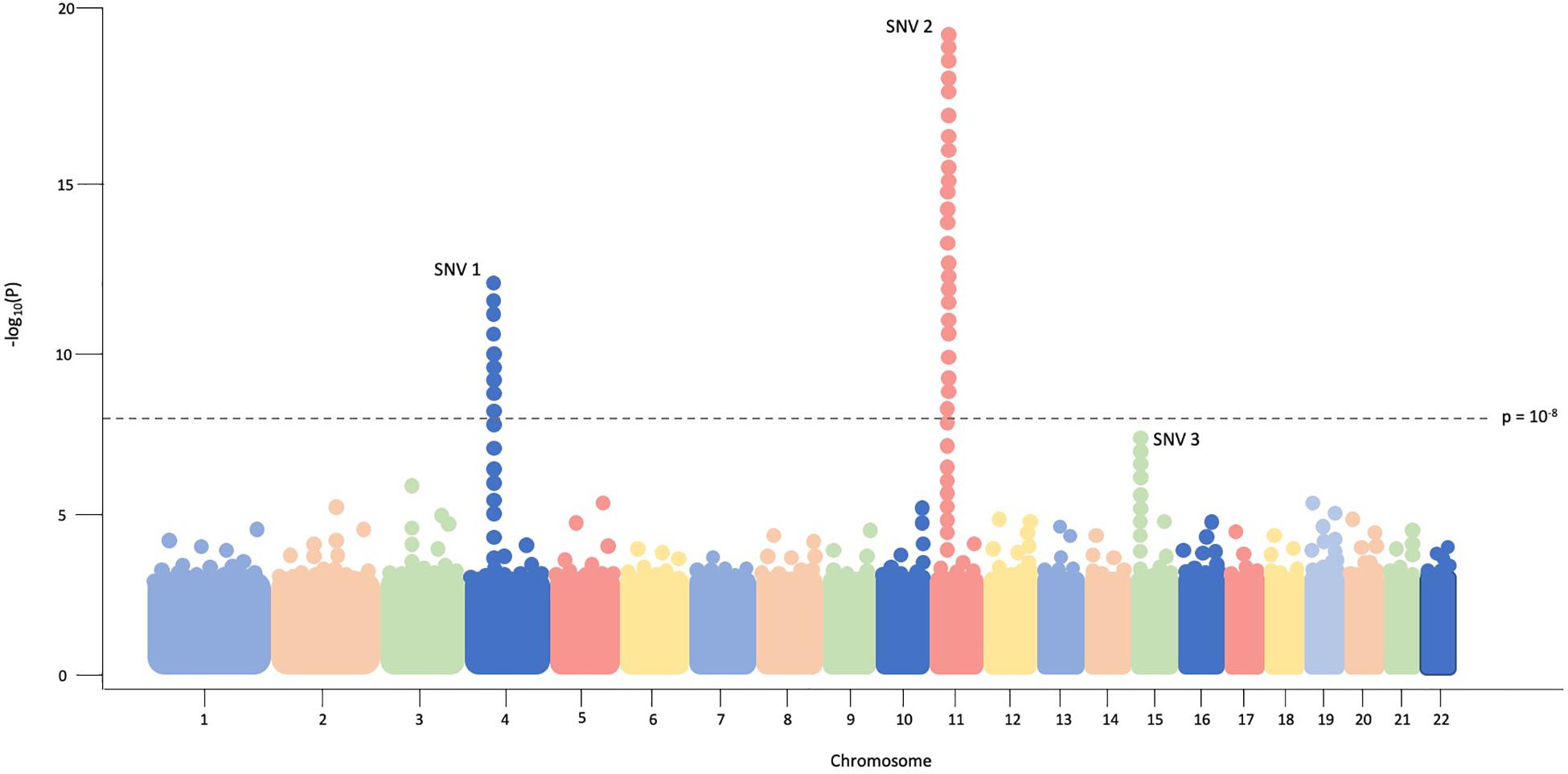
Figure 1. In this example Manhattan plot, each dot represents a single SNV assessed in every individual in the cohort. Genomic coordinates are displayed in ascending fashion on the X-axis, beginning with chromosome 1 on the left. The negative logarithm of the association p-value for each SNV is shown on the Y-axis. The SNVs with the strongest association will have the most negative p-value, so the negative logarithm will be greatest for these SNVs. In this example plot, SNV1 and SNV2 exceed the threshold of significance (p = 10– 8) while SNV3 does not.
As opposed to candidate gene studies assessing variation in specific genes, neither approach requires prior hypotheses of associations between genetic variants and disease. Given the widespread and increasing application of GWAS and RVAS to uncover genetic associations in complex diseases, it is important to recognize the strengths and limitations of each approach. Here we will consider the contribution of association studies in unraveling the genetic etiologies of the motor neuron disease amyotrophic lateral sclerosis (ALS).
Strengths and Successes in Association Studies
Association studies have been used to identify significant risk loci in conditions such as type 2 diabetes (Zhao et al., 2017), schizophrenia (Li et al., 2017), hypocholesterolemia (Cohen, 2004; Cohen et al., 2005) and coronary artery disease (Nikpay et al., 2015). Beyond identifying novel disease associations, GWAS and RVAS may also serve as a first step in uncovering biological mechanisms and/or pathways for therapeutic intervention. In schizophrenia, GWAS identified a significant association signal within the major histocompatibility complex (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014) and sparked investigation into complement factor haplotypes including C4 (Sekar et al., 2016). C4 is a known marker of synaptic pruning which was later found to be overexpressed in the brain tissue of individuals with schizophrenia. This biological context supports the theory of excessive synaptic elimination (“pruning”) as a mechanism of disease. Similarly, a GWAS approach to type 2 diabetes identified the risk locus SLC30A8, and follow-up investigation discovered that loss-of-function variants at this locus are protective against disease. This discovery led to the development of several drugs which aim to antagonize the product of SLC30A8, a zinc transporter in pancreatic islet cells (Flannick and Florez, 2016). RVAS identified rare variants in PCSK9 as a key component of low-density lipoprotein metabolism and individuals with loss-of-function variants in this gene had consistently low cholesterol levels throughout their lifetimes (Cohen et al., 2005). Since then, three PCSK9 inhibitors have been tested in human trials, two approved in the United States (Shapiro et al., 2018). This association finding is among the most compelling examples of translation from genetic findings to therapeutic intervention.
Association studies may also provide an avenue for disease subgroup stratification, where a subgroup may have a particular clinical course (Ridker et al., 2008; Reiner et al., 2009; Owen et al., 2010; Thanabalasingham et al., 2011) or may be more likely to benefit from a certain intervention (Nelson et al., 2015). These approaches can provide insight into the impact of geoancestry in disease (Choquet et al., 2013; Wen et al., 2014; Liu et al., 2015; Minster et al., 2016; Visscher et al., 2017). GWAS and RVAS may uncover modifier genes and shed light on the contribution of multiple variants (Pigeyre et al., 2016; Whitacre et al., 2017; Tam et al., 2019). Polygenic risk scores (PRS), which predict an individual’s risk for disease based on the combination of multiple risk alleles, can be calculated using tens of thousands of association “hits” together (Wray et al., 2007). Individuals in the top 1–5% of risk profile may face a disease risk that approaches that of individuals who inherit a single monogenic pathogenic mutation (Khera et al., 2018). Informed by association results, PRS have shown modest but reliable prediction capability in a number of disease areas (Barrett et al., 2009; Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014; Hoffmann et al., 2017; Seibert et al., 2018) as well as the ability to modify risk prediction for monogenic variants (Fahed et al., 2020).
Limitations of Association Studies
Association studies require large datasets and a stringent threshold for significance to avoid false positives. Given the nature of studying rare variants, even larger sample sizes are required with RVAS so that patients with such rare variants will be included. For less-common diseases such as ALS, the sample size required to identify risk variants of low effect size may not be feasible. Further, the overwhelming majority of association studies have been performed in European cohorts (Haga, 2010; Duncan et al., 2019). Since variant frequencies as well as linkage disequilibrium vary between ethnic groups, findings may not be applicable across racial and ethnic groups (Need and Goldstein, 2009; Gravel et al., 2011).
Perhaps most importantly, association studies, by their nature, only measure the association of a risk loci with a disease but cannot determine the impact of a SNV on lifetime risk nor the mechanism by which it confers such risk (Altshuler et al., 2008). Specifically, GWAS findings often highlight non-coding SNVs in linkage disequilibrium with several other genes or regions, making it difficult to specifically identify causal genes. Variants that are associated with disease may in fact act as direct drivers of disease progression, or such a link to disease or phenotype may not be understood, potentially because the true causal variant at that locus has not yet been identified or multiple variants at a locus must work together. Even though association to a particular variant may be statistically significant with cases compared to controls, causality cannot necessarily be assigned by the GWAS approach. Thus, GWAS may reveal synthetic associations (Dickson et al., 2010). Typical GWAS reveal multiple variants associated with disease due to linkage disequilibrium, and functional studies are necessary to determine which are truly meaningful in the context of disease (Pearson, 2008).
Additionally, in such large cohorts the cost of uniform, deep sequencing approaches such as WGS can be a prohibiting factor, so other testing approaches may be considered, each with important caveats. A number of statistical methods have been developed to increase the power of RVAS in the context of sample size limitations.
Gene Discovery in Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis is a progressive neurodegenerative disorder affecting 1–2 per 100,000, involving selective loss of upper and lower motor neurons and typically resulting in death in 2–5 years (van Es et al., 2017). The discovery of multiple genes associated with ALS has led to an era of targeted gene therapies and multiple lines of mechanistic inquiry. As such, the story of gene discovery in ALS provides a useful context in which to understand the significance of GWAS- and RVAS-identified variants in a genetically heterogeneous disease population.
The first gene identified to cause familial ALS (fALS, defined as having a history of ALS in a first-, second- or third-degree relative), SOD1, was identified via linkage studies in 1993 (Rosen, 1993). Since then, variants in over 50 genes have been identified in individuals with both familial and sporadic ALS (an up-to-date list of these genes can be found at alsod.ac.uk). Many of these genes were identified via linkage analysis in high-penetrance fALS families and confirmed in follow-up case-control studies that utilized either Sanger sequencing, SNV arrays, or exome sequencing (Siddique et al., 1989; Hosler et al., 1998; Nishimura, 2004). Other studies utilized prior biological knowledge to identify candidate genes and then conducted case-control sequencing studies (Kwiatkowski et al., 2009; Fecto, 2011). Currently, a monogenic etiology can be identified in up to two-thirds of fALS and 10% of sporadic ALS (sALS) cases (Chia et al., 2018). As with other genetically complex diseases, traditional linkage or candidate gene approaches were responsible for the discovery of the most highly penetrant ALS genes, including C9orf72 (DeJesus-Hernandez et al., 2011; Renton et al., 2011), SOD1 (Rosen, 1993), and FUS (Kwiatkowski et al., 2009).
Association Studies in Amyotrophic Lateral Sclerosis: Discovery and Reproducibility
The primary goal of most association studies in ALS is to identify new ALS-associated genes, either common (via GWAS) or rare (via RVAS), and this has been successful in recent years as evidenced by the identification of multiple disease-associated ALS genes (summarized in Table 1).

Table 1. A chronological overview of ALS-related genes initially identified via association studies.
Rare variation appears to play an important role in explaining missing heritability within ALS (van Rheenen et al., 2016). As such, the field has made significant strides in applying RVAS using large, international collaborations (Smith et al., 2014; van Rheenen et al., 2016; Nicolas et al., 2018), the most recent of which identified KIF5A as a Mendelian ALS gene (Nicolas et al., 2018). Some GWAS studies (using SNV genotyping data) have performed RVAS (using sequencing data on smaller cohorts) as a follow-on validation step. Other approaches leverage additional sources of genetic data, such as gene expression data, to prioritize GWAS findings (Diekstra et al., 2012).
Association studies may be used to confirm previously identified findings, resulting either from early linkage studies or from other association studies in separate human ALS cohorts (Table 1). For example, SOD1 was originally identified via linkage (Rosen, 1993) and in subsequent GWAS a clear signal was found at the SOD1 locus (Laaksovirta et al., 2010). Additionally, association studies can help to more completely characterize a linkage finding. A 9p21.2 locus causing dominant ALS was originally discovered via linkage analysis, with multiple reports defining a minimum linkage region of 3.7 Mb including only five known genes (Luty et al., 2008; Le Ber et al., 2009; Boxer et al., 2011). This region was ultimately pinpointed to C9orf72 in part by association studies which condensed the locus to a few genes (van Es et al., 2009b; Laaksovirta et al., 2010; Shatunov et al., 2010), providing avenues for targeted repeat-mapping in C9orf72 (DeJesus-Hernandez et al., 2011).
Positive replication studies add support that the original finding was in fact a true association. If the replication cohort differs in geographical origin and/or phenotypic features to the original cohort, the findings may be more applicable in additional disease populations. Cross-ethnic analyses have uncovered such genes (Benyamin et al., 2017). Identifying genes and variants that are robustly replicated over time and across populations is a critical first step in characterizing the biological mechanisms underlying ALS. For example, association studies in ALS have replicated C9orf72 as a disease-associated gene across ethnic groups (Laaksovirta et al., 2010; Shatunov et al., 2010). Additional examples of gene replication exist in small cohorts (Cronin et al., 2007; Li et al., 2009) and in larger meta-analyses (van Rheenen et al., 2016; Benyamin et al., 2017). In addition, RVAS studies have also lent support to prior GWAS findings (Kenna et al., 2016).
Nevertheless, across the board, many gene-specific replication studies have failed to replicate association findings (Chiò et al., 2009; Cronin et al., 2009; van Es et al., 2009a; Daoud et al., 2010; Fernández-Santiago et al., 2011; Fogh et al., 2011; Chen et al., 2012; Cai et al., 2014). Lack of reproducibility of association findings is common and may reflect several issues common in human genetic studies. As the risk variants often confer very small increases in risk, small sample sizes of less than 10,000 individuals are frequently underpowered to detect these risk variants. Larger study sizes (>50,000), which are most commonly assembled via large, international collaborations, are much more likely to reproduce association findings (Feliciano et al., 2018; Nishino et al., 2018; Zhang et al., 2020). The sample sizes in ALS, GWAS, and RVAS have grown steadily over time but are much smaller than those in other disease areas (Michailidou et al., 2017). Inherent population stratification also influences varying allele frequencies between individuals from different geographical regions and/or different ancestral backgrounds (Tam et al., 2019). Thus, positive associations may not be found in a subsequent study if population differences exist. Selection criteria for each cohort are not always consistent and may be subject to bias based on clinical or demographic standards (McClellan and King, 2010). Phenotypic variation in different cohorts may influence diagnosis and inclusion in genomic studies. Finally, variation in genetic testing technology and analysis as well as genotyping errors may occur between cohorts.
Genetically homogenous ethnic populations are typically selected for GWAS and RVAS because they introduce the least amount of genetic diversity and maximize the chances of identifying variants that are disease-related rather than geoancestry-related. However, the results from such cohorts are often not replicable in subsequent analyses and/or generalizable in other populations, often due to varying allele prevalence and unequal representation of different populations in case and control groups. For decades, Caucasian individuals have made up the vast majority of people studied in association studies across all diseases, including ALS (Popejoy and Fullerton, 2016). In ALS association studies, there is notable lack of replication between Asian and European ALS cohorts, which may reflect inherent population differences in SNV frequencies and disease phenotypes (Gravel et al., 2011). For example, ALS onset occurs at a younger age in Han Chinese patients and is more likely to present with bulbar-onset, as compared to limb-onset (Deng et al., 2013).
GWAS and RVAS Studies Result in Appreciation of Relevant Mechanistic Pathways
Experiments to determine the functional consequences of ALS-associated variants in genes identified via GWAS and RVAS have further characterized the pathology underlying disease either via a specific gene product itself or the network or pathway in which it operates. Such ALS genes play roles in glutamate-mediated neurotransmission and excitability (ITPR2 and UNC13A; Varoqueaux et al., 2005; Gambardella et al., 2020), regulation of neuronal excitability (DPP6; Lin et al., 2018), autophagy (TBK1; Duan et al., 2019), cytoskeletal organization (TUBA4A; Smith et al., 2014), and axonal transport (KIF5A; Nicolas et al., 2018).
Some association findings have led to promising results in in vitro and in vivo models of ALS. For example, conditional knockout of TBK1 was reported to result in motor and cognitive defects in mice as well as pathological features typical of autophagy dysfunction (Duan et al., 2019). In SOD1G93A-transfected cells, TBK1 overexpression reduced the number and size of SOD1 aggregates. SOD1G93A transgenic mice demonstrating an ALS phenotype show increased survival and decreased protein aggregates after intracerebroventricular injection of AAV vectors encoding TBK1 (Duan et al., 2019). TBK1 expression may have the therapeutic potential to promote autophagy even in the absence of TBK1 variants.
Other disease pathway studies for GWAS- and RVAS-identified genes have lent support to the approaches of current therapeutic options for ALS. For example, UNC13A functions to regulate the release of neurotransmitters, such as glutamate at neuromuscular synapses (Rossner et al., 2004; Engel et al., 2016). In mice, UNC13A acts in synaptic vesicle priming, and mice lacking UNC13A demonstrate altered glutamatergic neurotransmission (Varoqueaux et al., 2005; Gambardella et al., 2020). UNC13A variants may therefore promote disease via glutamate-mediated excitotoxicity. Riluzole, one of two FDA-approved treatments for ALS, is a glutamate release inhibitor and can lead to a 2-3 month increase in survival for some patients (Bellingham, 2011; Dharmadasa and Kiernan, 2018). However, in clinical trials, other treatments aimed at decreasing glutamate neurotransmission have demonstrated limited or negative results (Bedlack, 2019).
Current Impact of GWAS and RVAS on Preclinical and Clinical Therapy
Efforts to translate genetic discoveries into therapeutic clinical trials in ALS have thus far been et with limited success, in contrast to other diseases in which GWAS had led to new drugs currently in clinical trials or clinical practice (Visscher et al., 2017). The high degree of clinical and genetic heterogeneity, unknown influence of endogenous and exogenous factors on disease susceptibility, and unknown reasons for selective vulnerability of certain cell types present significant challenges to therapeutic development (Katyal and Govindarajan, 2017).
Gene-targeted clinical trials for ALS patients with variants in three genes (FUS, C9orf72, and SOD1) are underway. Antisense oligonucleotides targeted at C9orf72 mutant transcripts have shown promising results in ALS models and are currently in development for patients with C9orf72-related ALS (Riboldi et al., 2014; Martier et al., 2019). Recently, two approaches to down-regulate SOD1 expression in patients with SOD1-ALS [one utilizing an antisense oligonucleotide (Miller et al., 2020) and the other an adeno-associated viral vector (Mueller et al., 2020)] have been reported. The genes targeted in ALS genomic therapies were each originally identified in high-penetrance families demonstrating Mendelian inheritance, not via GWAS or RVAS. They also represent the most common, known genetic causes of ALS and were discovered prior to the widespread application of association-based technology.
There is potential utility of association studies in identifying subgroups of medication responders. This has been demonstrated in a survival analysis of patients with a particular UNC13A genotype who were treated with lithium carbonate (van Eijk et al., 2017). UNC13A was originally identified via an association study (van Es et al., 2009b). This finding suggests that GWAS and RVAS may lead to more targeted studies in the future and/or improved interpretation of clinical trial results.
Currently, results from ALS association studies are used broadly in several ways, such as improving understanding of the genetic architecture of ALS, illuminating tissue- or cell-specific pathways involving ALS-associated genes, and informing variable expressivity and penetrance of disease. Moving forward, GWAS and RVAS findings may assist in the design of combinatorial therapies that target multiple gene products and disease pathways, reflecting the proposed oligogenic nature of disease (Nguyen et al., 2018). Finally, larger, more powerful association studies may one day enable the calculation of clinical PRS to identify healthy individuals at highest risk of disease, who may be candidates for neuroprotective interventions.
Discussion
Advances in genetic testing and identification of genetic subtypes of disease have been the cornerstone of ALS research in recent years, marked by widespread genetic testing in larger and more diverse cohorts, bioinformatic and molecular characterization of identified variants, and progress toward clinical trials for genetic subtypes of disease. Gene discovery has been driven by linkage analysis of families with high-penetrance genes, candidate gene approaches and more recently, association studies such as GWAS and RVAS. Association studies represent an attractive option for novel gene discovery because they do not require prior knowledge or hypotheses, compared with hypothesis-confirming sequencing studies.
Currently, there are no effective treatment options to halt progression of ALS and only two FDA-approved medications. Significant urgency exists within the ALS community to identify additional genetic markers of disease in order to uncover novel biological mechanisms, stratify genetic subgroups of disease, and drive drug development. Lower-penetrance genes and risk factors identified via association studies may serve as important components of combinatorial gene-targeted therapies in the future. Gene-targeted clinical trials are currently underway, though to date, no ALS genes initially identified via GWAS or RVAS have been developed for gene therapy approaches. In general, consideration of the potential of a GWAS or RVAS finding must be approached with measured expectations, particularly when such genes are quickly added to ALS clinical genetic testing panels. Association studies of common and rare genetic variation, when critically evaluated and contextualized properly, are a powerful tool in understanding the genetic basis of complex diseases such as ALS.
Author Contributions
KR: background research. All authors: manuscript writing and development.
Funding
We are grateful for support from the Julie Bonasera Fund for ALS Research.
Conflict of Interest
SK has received compensation for consulting from Biogen Idec, and AveXis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge Amanda E. Toland, Ph.D. for her contributions to manuscript development.
References
Altshuler, D., Daly, M. J., and Lander, E. S. (2008). Genetic mapping in human disease. Science (New York, N.Y.) 322, 881–888. doi: 10.1126/science.1156409
Auer, P. L., and Lettre, G. (2015). Rare variant association studies: considerations, challenges and opportunities. Genome Med. 7:16. doi: 10.1186/s13073-015-0138-2
Barrett, J. C., Clayton, D. G., Concannon, P., Akolkar, B., Cooper, J. D., Erlich, H. A., et al. (2009). Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 41, 703–707. doi: 10.1038/ng.381
Bedlack, R. (2019). ALSUntangled 48: perampanel (Fycompa). Amyotroph. Lateral Scler. Frontotemporal Degener. 20, 453–456. doi: 10.1080/21678421.2019.1573850
Bellingham, M. C. (2011). A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade?: neural mechanisms of action and clinical efficiency of riluzole in treating ALS. CNS Neurosci. Ther. 17, 4–31. doi: 10.1111/j.1755-5949.2009.00116.x
Benyamin, B., He, J., Zhao, Q., Gratten, J., Garton, F., Leo, P. J., et al. (2017). Cross-ethnic meta-analysis identifies association of the GPX3-TNIP1 locus with amyotrophic lateral sclerosis. Nat. Commun. 8:611. doi: 10.1038/s41467-017-00471-1
Boxer, A. L., Mackenzie, I. R., Boeve, B. F., Baker, M., Seeley, W. W., Crook, R., et al. (2011). Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J. Neurol. Neurosurg. Psychiatry 82, 196–203. doi: 10.1136/jnnp.2009.204081
Cai, B., Tang, L., Zhang, N., and Fan, D. (2014). The single-nucleotide polymorphism rs6690993 in FGGY is not associated with amyotrophic lateral sclerosisin a large Chinese cohort. Neurobiol. Aging 35, 1512.e3–1512.e4. doi: 10.1016/j.neurobiolaging.2013.12.018
Chen, Y., Zeng, Y., Huang, R., Yang, Y., Chen, K., Song, W., et al. (2012). No association of five candidate genetic variants with amyotrophic lateral sclerosis in a Chinese population. Neurobiol. Aging 33, 2721.e3–5. doi: 10.1016/j.neurobiolaging.2012.06.004
Chi, L., Ke, Y., Luo, C., Gozal, D., and Liu, R. (2007). Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience 144, 991–1003. doi: 10.1016/j.neuroscience.2006.09.064
Chia, R., Chiò, A., and Traynor, B. J. (2018). Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 17, 94–102. doi: 10.1016/S1474-4422(17)30401-5
Chiò, A., Schymick, J. C., Restagno, G., Scholz, S. W., Lombardo, F., Lai, S.-L., et al. (2009). A two-stage genome-wide association study of sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 18, 1524–1532. doi: 10.1093/hmg/ddp059
Choquet, H., Kasberger, J., Hamidovic, A., and Jorgenson, E. (2013). Contribution of common PCSK1 genetic variants to obesity in 8,359 subjects from multi-ethnic American population. PLoS One 8:e57857. doi: 10.1371/journal.pone.0057857
Cirulli, E. T., Lasseigne, B. N., Petrovski, S., Sapp, P. C., Dion, P. A., Leblond, C. S., et al. (2015). Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441. doi: 10.1126/science.aaa3650
Cohen, J., Pertsemlidis, A., Kotowski, I. K., Graham, R., Garcia, C. K., and Hobbs, H. H. (2005). Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 37, 161–165. doi: 10.1038/ng1509
Cohen, J. C. (2004). Multiple rare alleles contribute to low plasma levels of HDL Cholesterol. Science 305, 869–872. doi: 10.1126/science.1099870
Cronin, S., Berger, S., Ding, J., Schymick, J. C., Washecka, N., Hernandez, D. G., et al. (2007). A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum. Mol. Genet. 17, 768–774. doi: 10.1093/hmg/ddm361
Cronin, S., Tomik, B., Bradley, D. G., Slowik, A., and Hardiman, O. (2009). Screening for replication of genome-wide SNP associations in sporadic ALS. Eur. J. Hum. Genet. 17, 213–218. doi: 10.1038/ejhg.2008.194
Daoud, H., Valdmanis, P. N., Dion, P. A., and Rouleau, G. A. (2010). Analysis of DPP6 and FGGY as candidate genes for amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 11, 389–391. doi: 10.3109/17482960903358857
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Deng, M., Wei, L., Zuo, X., Tian, Y., Xie, F., Hu, P., et al. (2013). Genome-wide association analyses in Han Chinese identify two new susceptibility loci for amyotrophic lateral sclerosis. Nat. Genet. 45, 697–700. doi: 10.1038/ng.2627
Dharmadasa, T., and Kiernan, M. C. (2018). Riluzole, disease stage and survival in ALS. Lancet Neurol. 17, 385–386. doi: 10.1016/S1474-4422(18)30091-7
Dickson, S. P., Wang, K., Krantz, I., Hakonarson, H., and Goldstein, D. B. (2010). Rare variants create synthetic genome-wide associations. PLoS Biol. 8:e1000294. doi: 10.1371/journal.pbio.1000294
Diekstra, F. P., Saris, C. G. J., van Rheenen, W., Franke, L., Jansen, R. C., van Es, M. A., et al. (2012). Mapping of Gene Expression Reveals CYP27A1 as a susceptibility gene for sporadic ALS. PLoS One 7:e35333. doi: 10.1371/journal.pone.0035333
Duan, W., Guo, M., Yi, L., Zhang, J., Bi, Y., Liu, Y., et al. (2019). Deletion of Tbk1 disrupts autophagy and reproduces behavioral and locomotor symptoms of FTD-ALS in mice. Aging (Albany NY) 11, 2457–2476. doi: 10.18632/aging.101936
Duncan, L., Shen, H., Gelaye, B., Meijsen, J., Ressler, K., Feldman, M., et al. (2019). Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun. 10:3328. doi: 10.1038/s41467-019-11112-0
Dunckley, T., Huentelman, M. J., Craig, D. W., Pearson, J. V., Szelinger, S., Joshipura, K., et al. (2007). Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N. Engl. J. Med. 357, 775–788. doi: 10.1056/NEJMoa070174
Engel, A. G., Selcen, D., Shen, X.-M., Milone, M., and Harper, C. M. (2016). Loss of MUNC13-1 function causes microcephaly, cortical hyperexcitability, and fatal myasthenia. Neurol. Genet. 2:e105. doi: 10.1212/NXG.0000000000000105
Fahed, A. C., Wang, M., Homburger, J. R., Patel, A. P., Bick, A. G., Neben, C. L., et al. (2020). Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat. Commun. 11:3635. doi: 10.1038/s41467-020-17374-3
Fecto, F. (2011). SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 68:1440. doi: 10.1001/archneurol.2011.250
Feliciano, P., Daniels, A. M., Green Snyder, L., Beaumont, A., Camba, A., Esler, A., et al. (2018). SPARK: a US cohort of 50,000 families to accelerate autism research. Neuron 97, 488–493. doi: 10.1016/j.neuron.2018.01.015
Fernández-Santiago, R., Sharma, M., Berg, D., Illig, T., Anneser, J., Meyer, T., et al. (2011). No evidence of association of FLJ10986 and ITPR2 with ALS in a large German cohort. Neurobiol. Aging 32, 551.e1–4. doi: 10.1016/j.neurobiolaging.2009.04.018
Flannick, J., and Florez, J. C. (2016). Type 2 diabetes: genetic data sharing to advance complex disease research. Nat. Rev. Genet. 17, 535–549. doi: 10.1038/nrg.2016.56
Fogh, I., D’Alfonso, S., Gellera, C., Ratti, A., Cereda, C., Penco, S., et al. (2011). No association of DPP6 with amyotrophic lateral sclerosis in an Italian population. Neurobiol. Aging 32, 966–967. doi: 10.1016/j.neurobiolaging.2009.05.014
Fogh, I., Ratti, A., Gellera, C., Lin, K., Tiloca, C., Moskvina, V., et al. (2014). A genome-wide association meta-analysis identifies a novel locus at 17q11.2 associated with sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 23, 2220–2231. doi: 10.1093/hmg/ddt587
Gallus, G. N., Dotti, M. T., and Federico, A. (2006). Clinical and molecular diagnosis of cerebrotendinous xanthomatosis with a review of the mutations in the CYP27A1 gene. Neurol. Sci. 27, 143–149. doi: 10.1007/s10072-006-0618-7
Gambardella, J., Lombardi, A., Morelli, M. B., Ferrara, J., and Santulli, G. (2020). Inositol 1,4,5-Trisphosphate receptors in human disease: a comprehensive update. J. Clin. Med. 9:1096. doi: 10.3390/jcm9041096
Gateva, V., Sandling, J. K., Hom, G., Taylor, K. E., Chung, S. A., Sun, X., et al. (2009). A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 41, 1228–1233. doi: 10.1038/ng.468
Gravel, S., Henn, B. M., Gutenkunst, R. N., Indap, A. R., Marth, G. T., Clark, A. G., et al. (2011). Demographic history and rare allele sharing among human populations. Proc. Natl. Acad. Sci. U. S. A. 108, 11983–11988. doi: 10.1073/pnas.1019276108
Gudmundsson, J., Sulem, P., Gudbjartsson, D. F., Masson, G., Agnarsson, B. A., Benediktsdottir, K. R., et al. (2012). A study based on whole-genome sequencing yields a rare variant at 8q24 associated with prostate cancer. Nat. Genet. 44, 1326–1329. doi: 10.1038/ng.2437
Haga, S. B. (2010). Impact of limited population diversity of genome-wide association studies. Genet. Med. 12, 81–84. doi: 10.1097/GIM.0b013e3181ca2bbf
Hergovich, A. (2011). MOB control: reviewing a conserved family of kinase regulators. Cell. Signal. 23, 1433–1440. doi: 10.1016/j.cellsig.2011.04.007
Hoffmann, T. J., Ehret, G. B., Nandakumar, P., Ranatunga, D., Schaefer, C., Kwok, P.-Y., et al. (2017). Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nat. Genet. 49, 54–64. doi: 10.1038/ng.3715
Hosler, B. A., Sapp, P. C., Berger, R., O’Neill, G., Bejaoui, K., Hamida, M. B., et al. (1998). Refined mapping and characterization of the recessive familial amyotrophic lateral sclerosis locus (ALS2) on chromosome 2q33. Neurogenetics 2, 34–42. doi: 10.1007/s100480050049
Iida, A., Takahashi, A., Kubo, M., Saito, S., Hosono, N., Ohnishi, Y., et al. (2011). A functional variant in ZNF512B is associated with susceptibility to amyotrophic lateral sclerosis in Japanese. Hum. Mol. Genet. 20, 3684–3692. doi: 10.1093/hmg/ddr268
Kanai, Y., Dohmae, N., and Hirokawa, N. (2004). Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43, 513–525. doi: 10.1016/j.neuron.2004.07.022
Katyal, N., and Govindarajan, R. (2017). Shortcomings in the current amyotrophic lateral sclerosis trials and potential solutions for improvement. Front. Neurol. 8:521. doi: 10.3389/fneur.2017.00521
Kenna, K. P., van Doormaal, P. T. C., Dekker, A. M., Ticozzi, N., Kenna, B. J., Diekstra, F. P., et al. (2016). NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 48, 1037–1042. doi: 10.1038/ng.3626
Khan, A. O., Eisenberger, T., Nagel-Wolfrum, K., Wolfrum, U., and Bolz, H. J. (2015). C21orf2 is mutated in recessive early-onset retinal dystrophy with macular staphyloma and encodes a protein that localises to the photoreceptor primary cilium. Br. J. Ophthalmol. 99, 1725–1731. doi: 10.1136/bjophthalmol-2015-307277
Khera, A. V., Chaffin, M., Aragam, K. G., Haas, M. E., Roselli, C., Choi, S. H., et al. (2018). Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224. doi: 10.1038/s41588-018-0183-z
Kosmicki, J. A., Churchhouse, C. L., Rivas, M. A., and Neale, B. M. (2016). Discovery of rare variants for complex phenotypes. Hum. Genet. 135, 625–634. doi: 10.1007/s00439-016-1679-1
Kwiatkowski, T. J., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Laaksovirta, H., Peuralinna, T., Schymick, J. C., Scholz, S. W., Lai, S.-L., Myllykangas, L., et al. (2010). Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet. Neurol. 9, 978–985. doi: 10.1016/S1474-4422(10)70184-8
Landers, J. E., Melki, J., Meininger, V., Glass, J. D., van den Berg, L. H., van Es, M. A., et al. (2009). Reduced expression of the Kinesin-Associated Protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U. S. A. 106, 9004–9009. doi: 10.1073/pnas.0812937106
Laufman, O., Kedan, A., Hong, W., and Lev, S. (2009). Direct interaction between the COG complex and the SM protein, Sly1, is required for Golgi SNARE pairing. EMBO J. 28, 2006–2017. doi: 10.1038/emboj.2009.168
Le Ber, I., Camuzat, A., Berger, E., Hannequin, D., Laquerriere, A., Golfier, V., et al. (2009). Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology 72, 1669–1676. doi: 10.1212/WNL.0b013e3181a55f1c
Lee, S., Abecasis, G. R., Boehnke, M., and Lin, X. (2014). Rare-Variant Association analysis: study designs and statistical tests. Am. J. Hum. Genet. 95, 5–23. doi: 10.1016/j.ajhg.2014.06.009
Li, X.-G., Zhang, J.-H., Xie, M.-Q., Liu, M.-S., Li, B.-H., Zhao, Y.-H., et al. (2009). Association between DPP6 polymorphism and the risk of sporadic amyotrophic lateral sclerosis in Chinese patients. Chin. Med. J. 122, 2989–2992.
Li, Z., Chen, J., Yu, H., He, L., Xu, Y., Zhang, D., et al. (2017). Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat. Genet. 49, 1576–1583. doi: 10.1038/ng.3973
Lin, L., Murphy, J. G., Karlsson, R.-M., Petralia, R. S., Gutzmann, J. J., Abebe, D., et al. (2018). DPP6 loss impacts hippocampal synaptic development and induces behavioral impairments in recognition, learning and memory. Front. Cell Neurosci. 12:84. doi: 10.3389/fncel.2018.00084
Liu, J. Z., van Sommeren, S., Huang, H., Ng, S. C., Alberts, R., Takahashi, A., et al. (2015). Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986. doi: 10.1038/ng.3359
Luty, A. A., Kwok, J. B., Thompson, E. M., Blumbergs, P., Brooks, W. S., Loy, C. T., et al. (2008). Pedigree with frontotemporal lobar degeneration – motor neuron disease and Tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9. BMC Neurol. 8:32. doi: 10.1186/1471-2377-8-32
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi: 10.1038/nature08494
Martier, R., Liefhebber, J. M., García-Osta, A., Miniarikova, J., Cuadrado-Tejedor, M., Espelosin, M., et al. (2019). Targeting RNA-Mediated Toxicity in C9orf72 ALS and/or FTD by RNAi-based gene therapy. Mol. Ther. Nucleic Acids 16, 26–37. doi: 10.1016/j.omtn.2019.02.001
McClellan, J., and King, M.-C. (2010). Genetic heterogeneity in human disease. Cell 141, 210–217. doi: 10.1016/j.cell.2010.03.032
Michailidou, K., Lindström, S., Dennis, J., Beesley, J., Hui, S., Kar, S., et al. (2017). Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94. doi: 10.1038/nature24284
Miller, T., Cudkowicz, M., Shaw, P. J., Andersen, P. M., Atassi, N., Bucelli, R. C., et al. (2020). Phase 1–2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N. Engl. J. Med. 383, 109–119. doi: 10.1056/NEJMoa2003715
Minster, R. L., Hawley, N. L., Su, C.-T., Sun, G., Kershaw, E. E., Cheng, H., et al. (2016). A thrifty variant in CREBRF strongly influences body mass index in Samoans. Nat. Genet. 48, 1049–1054. doi: 10.1038/ng.3620
Mueller, C., Berry, J. D., McKenna-Yasek, D. M., Gernoux, G., Owegi, M. A., Pothier, L. M., et al. (2020). SOD1 suppression with adeno-associated virus and MicroRNA in Familial ALS. N. Engl. J. Med. 383, 151–158. doi: 10.1056/NEJMoa2005056
Nair, R. P., Duffin, K. C., Helms, C., Ding, J., Stuart, P. E., Goldgar, D., et al. (2009). Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet. 41, 199–204. doi: 10.1038/ng.311
Need, A. C., and Goldstein, D. B. (2009). Next generation disparities in human genomics: concerns and remedies. Trends Genet. TIG 25, 489–494. doi: 10.1016/j.tig.2009.09.012
Nelson, M. R., Tipney, H., Painter, J. L., Shen, J., Nicoletti, P., Shen, Y., et al. (2015). The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860. doi: 10.1038/ng.3314
Nguyen, H. P., Van Broeckhoven, C., and van der Zee, J. (2018). ALS genes in the genomic era and their implications for FTD. Trends Genet. 34, 404–423. doi: 10.1016/j.tig.2018.03.001
Nicolas, A., Kenna, K. P., Renton, A. E., Ticozzi, N., Faghri, F., Chia, R., et al. (2018). Genome-wide analyses identify KIF5A as a Novel ALS Gene. Neuron 97, e1268–e1283. doi: 10.1016/j.neuron.2018.02.027
Nikpay, M., Goel, A., Won, H.-H., Hall, L. M., Willenborg, C., Kanoni, S., et al. (2015). A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 47, 1121–1130. doi: 10.1038/ng.3396
Nishimura, A. L. (2004). A novel locus for late onset amyotrophic lateral sclerosis/motor neurone disease variant at 20q13. J. Med. Genet. 41, 315–320. doi: 10.1136/jmg.2003.013029
Nishino, J., Ochi, H., Kochi, Y., Tsunoda, T., and Matsui, S. (2018). Sample size for successful genome-wide association study of major depressive disorder. Front. Genet. 9:227. doi: 10.3389/fgene.2018.00227
Osterloh, J. M., Yang, J., Rooney, T. M., Fox, A. N., Adalbert, R., Powell, E. H., et al. (2012). dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484. doi: 10.1126/science.1223899
Owen, K. R., Thanabalasingham, G., James, T. J., Karpe, F., Farmer, A. J., McCarthy, M. I., et al. (2010). Assessment of high-sensitivity C-reactive protein levels as diagnostic discriminator of maturity-onset diabetes of the young due to HNF1A mutations. Diabetes Care 33, 1919–1924. doi: 10.2337/dc10-0288
Ozeki, Y., Tomoda, T., Kleiderlein, J., Kamiya, A., Bord, L., Fujii, K., et al. (2003). Disrupted-in-Schizophrenia-1 (DISC-1): mutant truncation prevents binding to NudE-like (NUDEL) and inhibits neurite outgrowth. Proc. Natl. Acad. Sci. U. S. A. 100, 289–294. doi: 10.1073/pnas.0136913100
Pearson, T. A. (2008). How to interpret a genome-wide association study. JAMA 299:1335. doi: 10.1001/jama.299.11.1335
Pigeyre, M., Yazdi, F. T., Kaur, Y., and Meyre, D. (2016). Recent progress in genetics, epigenetics and metagenomics unveils the pathophysiology of human obesity. Clin. Sci. (London, England: 1979) 130, 943–986. doi: 10.1042/CS20160136
Popejoy, A. B., and Fullerton, S. M. (2016). Genomics is failing on diversity. Nature 538, 161–164. doi: 10.1038/538161a
Reiner, A. P., Gross, M. D., Carlson, C. S., Bielinski, S. J., Lange, L. A., Fornage, M., et al. (2009). Common coding variants of the HNF1A gene are associated with multiple cardiovascular risk phenotypes in community-based samples of younger and older European-American adults: the Coronary Artery Risk Development in Young Adults Study and The Cardiovascular Health Study. Circ. Cardiovasc. Genet. 2, 244–254. doi: 10.1161/CIRCGENETICS.108.839506
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 Is the cause of chromosome 9p21-Linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Riboldi, G., Zanetta, C., Ranieri, M., Nizzardo, M., Simone, C., Magri, F., et al. (2014). Antisense oligonucleotide therapy for the treatment of C9ORF72 ALS/FTD Diseases. Mol. Neurobiol. 50, 721–732. doi: 10.1007/s12035-014-8724-7
Ridker, P. M., Pare, G., Parker, A., Zee, R. Y. L., Danik, J. S., Buring, J. E., et al. (2008). Loci related to metabolic-syndrome pathways including LEPR, HNF1A, IL6R, and GCKR associate with plasma C-reactive protein: the Women’s Genome Health Study. Am. J. Hum. Genet. 82, 1185–1192. doi: 10.1016/j.ajhg.2008.03.015
Rivas, M. A., Beaudoin, M., Gardet, A., Stevens, C., Sharma, Y., Zhang, C. K., et al. (2011). Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 43, 1066–1073. doi: 10.1038/ng.952
Rosen, D. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364, 362–362. doi: 10.1038/364362c0
Rossner, S., Fuchsbrunner, K., Lange-Dohna, C., Hartlage-Rübsamen, M., Bigl, V., Betz, A., et al. (2004). Munc13-1-mediated vesicle priming contributes to secretory amyloid precursor protein processing. J. Biol. Chem. 279, 27841–27844. doi: 10.1074/jbc.C400122200
Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Seibert, T. M., Fan, C. C., Wang, Y., Zuber, V., Karunamuni, R., Parsons, J. K., et al. (2018). Polygenic hazard score to guide screening for aggressive prostate cancer: development and validation in large scale cohorts. BMJ 360:j5757. doi: 10.1136/bmj.j5757
Sekar, A., Bialas, A. R., de Rivera, H., Davis, A., Hammond, T. R., Kamitaki, N., et al. (2016). Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183. doi: 10.1038/nature16549
Shapiro, M. D., Tavori, H., and Fazio, S. (2018). PCSK9: from basic science discoveries to clinical trials. Circ. Res. 122, 1420–1438. doi: 10.1161/CIRCRESAHA.118.311227
Shatunov, A., Mok, K., Newhouse, S., Weale, M. E., Smith, B., Vance, C., et al. (2010). Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet. Neurol. 9, 986–994. doi: 10.1016/S1474-4422(10)70197-6
Shimizu, K., Kawabe, H., Minami, S., Honda, T., Takaishi, K., Shirataki, H., et al. (1996). SMAP, an Smg GDS-associating protein having arm repeats and phosphorylated by Src tyrosine kinase. J. Biol. Chem. 271, 27013–27017. doi: 10.1074/jbc.271.43.27013
Siddique, T., Pericak-Vance, M. A., Brooks, B. R., Roos, R. P., Hung, W. Y., Antel, J. P., et al. (1989). Linkage analysis in familial amyotrophic lateral sclerosis. Neurology 39, 919–925. doi: 10.1212/wnl.39.7.919
Singh, C., Glaab, E., and Linster, C. L. (2017). Molecular identification of d-ribulokinase in budding yeast and mammals. J. Biol. Chem. 292, 1005–1028. doi: 10.1074/jbc.M116.760744
Smith, B. N., Ticozzi, N., Fallini, C., Gkazi, A. S., Topp, S., Kenna, K. P., et al. (2014). Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84, 324–331. doi: 10.1016/j.neuron.2014.09.027
Sun, L., Youn, H. D., Loh, C., Stolow, M., He, W., and Liu, J. O. (1998). Cabin 1, a negative regulator for calcineurin signaling in T lymphocytes. Immunity 8, 703–711. doi: 10.1016/s1074-7613(00)80575-0
Takemoto-Kimura, S., Terai, H., Takamoto, M., Ohmae, S., Kikumura, S., Segi, E., et al. (2003). Molecular cloning and characterization of CLICK-III/CaMKIgamma, a novel membrane-anchored neuronal Ca2+/calmodulin-dependent protein kinase (CaMK). J. Biol. Chem. 278, 18597–18605. doi: 10.1074/jbc.M300578200
Tam, V., Patel, N., Turcotte, M., Bossé, Y., Paré, G., and Meyre, D. (2019). Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484. doi: 10.1038/s41576-019-0127-1
Thanabalasingham, G., Shah, N., Vaxillaire, M., Hansen, T., Tuomi, T., Gašperíková, D., et al. (2011). A large multi-centre European study validates high-sensitivity C-reactive protein (hsCRP) as a clinical biomarker for the diagnosis of diabetes subtypes. Diabetologia 54, 2801–2810. doi: 10.1007/s00125-011-2261-y
Thiel, C., Kessler, K., Giessl, A., Dimmler, A., Shalev, S. A., der Haar, S., et al. (2011). NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am. J. Hum. Genet. 88, 106–114. doi: 10.1016/j.ajhg.2010.12.004
van Eijk, R. P. A., Jones, A. R., Sproviero, W., Shatunov, A., Shaw, P. J., Leigh, P. N., et al. (2017). Meta-analysis of pharmacogenetic interactions in amyotrophic lateral sclerosis clinical trials. Neurology 89, 1915–1922. doi: 10.1212/WNL.0000000000004606
van Es, M. A., Van Vught, P. W., Blauw, H. M., Franke, L., Saris, C. G., Andersen, P. M., et al. (2007). ITPR2 as a susceptibility gene in sporadic amyotrophic lateral sclerosis: a genome-wide association study. Lancet Neurol. 6, 869–877. doi: 10.1016/S1474-4422(07)70222-3
van Es, M. A., van Vught, P. W., Blauw, H. M., Franke, L., Saris, C. G., Van Den Bosch, L., et al. (2008). Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 40, 29–31. doi: 10.1038/ng.2007.52
van Es, M. A., Van Vught, P. W. J., Veldink, J. H., Andersen, P. M., Birve, A., Lemmens, R., et al. (2009a). Analysis of FGGY as a risk factor for sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 10, 441–447. doi: 10.3109/17482960802673042
van Es, M. A., Veldink, J. H., Saris, C. G. J., Blauw, H. M., van Vught, P. W. J., Birve, A., et al. (2009b). Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat. Genet. 41, 1083–1087. doi: 10.1038/ng.442
van Es, M. A., Hardiman, O., Chio, A., Al-Chalabi, A., Pasterkamp, R. J., Veldink, J. H., et al. (2017). Amyotrophic lateral sclerosis. Lancet 390, 2084–2098. doi: 10.1016/S0140-6736(17)31287-4
van Rheenen, W., Shatunov, A., Dekker, A. M., McLaughlin, R. L., Diekstra, F. P., Pulit, S. L., et al. (2016). Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 48, 1043–1048. doi: 10.1038/ng.3622
Varoqueaux, F., Sons, M. S., Plomp, J. J., and Brose, N. (2005). Aberrant morphology and residual transmitter release at the Munc13-deficient mouse neuromuscular synapse. Mol. Cell. Biol. 25, 5973–5984. doi: 10.1128/MCB.25.14.5973-5984.2005
Visscher, P. M., Wray, N. R., Zhang, Q., Sklar, P., McCarthy, M. I., Brown, M. A., et al. (2017). 10 Years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22. doi: 10.1016/j.ajhg.2017.06.005
Wang, Z., Iida, A., Miyake, N., Nishiguchi, K. M., Fujita, K., Nakazawa, T., et al. (2016). Axial spondylometaphyseal dysplasia is caused by C21orf2 Mutations. PLoS One 11:e0150555. doi: 10.1371/journal.pone.0150555
Watson, A. P., Evans, R. L., and Egland, K. A. (2013). Multiple functions of sushi domain containing 2 (SUSD2) in breast tumorigenesis. Mol. Cancer Res. 11, 74–85. doi: 10.1158/1541-7786.MCR-12-0501-T
Wen, W., Zheng, W., Okada, Y., Takeuchi, F., Tabara, Y., Hwang, J.-Y., et al. (2014). Meta-analysis of genome-wide association studies in East Asian-ancestry populations identifies four new loci for body mass index. Hum. Mol. Genet. 23, 5492–5504. doi: 10.1093/hmg/ddu248
Whitacre, L. K., Hoff, J. L., Schnabel, R. D., Albarella, S., Ciotola, F., Peretti, V., et al. (2017). Elucidating the genetic basis of an oligogenic birth defect using whole genome sequence data in a non-model organism. Bubalus bubalis. Sci. Rep. 7:39719. doi: 10.1038/srep39719
Wray, N. R., Goddard, M. E., and Visscher, P. M. (2007). Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 17, 1520–1528. doi: 10.1101/gr.6665407
Yamamoto, Y., Mizuno, R., Nishimura, T., Ogawa, Y., Yoshikawa, H., Fujimura, H., et al. (1994). Cloning and expression of myelin-associated oligodendrocytic basic protein. A novel basic protein constituting the central nervous system myelin. J. Biol. Chem. 269, 31725–31730.
Zhang, Y. D., Hurson, A. N., Zhang, H., Choudhury, P. P., Easton, D. F., Milne, R. L., et al. (2020). Assessment of polygenic architecture and risk prediction based on common variants across fourteen cancers. Nat. Commun. 11:3353. doi: 10.1038/s41467-020-16483-3
Keywords: amyotrophic lateral sclerosis, genetic testing, gene therapy, genome, genetic variants, rare variant association study (RVAS), genome wide association studies (GWAS)
Citation: Rich KA, Roggenbuck J and Kolb SJ (2021) Searching Far and Genome-Wide: The Relevance of Association Studies in Amyotrophic Lateral Sclerosis. Front. Neurosci. 14:603023. doi: 10.3389/fnins.2020.603023
Received: 04 September 2020; Accepted: 03 December 2020;
Published: 14 January 2021.
Edited by:
Henry Houlden, University College London, United KingdomReviewed by:
Wouter Van Rheenen, University Medical Center Utrecht, NetherlandsDavid G. Ashbrook, University of Tennessee Health Science Center (UTHSC), United States
Copyright © 2021 Rich, Roggenbuck and Kolb. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelly A. Rich, S2VsbHkuUmljaEBvc3VtYy5lZHU=