 Ji Wang
Ji Wang Fushun Wang4,5
Fushun Wang4,5 Dongmei Mai
Dongmei Mai Shaogang Qu
Shaogang Qu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 26 November 2020
Sec. Neurodegeneration
Volume 14 - 2020 | https://doi.org/10.3389/fnins.2020.585584
Parkinson’s disease (PD) is a common neurodegenerative disease, the pathological features of which include the presence of Lewy bodies and the neurodegeneration of dopaminergic neurons in the substantia nigra pars compacta. However, until recently, research on the pathogenesis and treatment of PD have progressed slowly. Glutamate and dopamine are both important central neurotransmitters in mammals. A lack of enzymatic decomposition of extracellular glutamate results in glutamate accumulating at synapses, which is mainly absorbed by excitatory amino acid transporters (EAATs). Glutamate exerts its physiological effects by binding to and activating ligand-gated ion channels [ionotropic glutamate receptors (iGluRs)] and a class of G-protein-coupled receptors [metabotropic glutamate receptors (mGluRs)]. Timely clearance of glutamate from the synaptic cleft is necessary because high levels of extracellular glutamate overactivate glutamate receptors, resulting in excitotoxic effects in the central nervous system. Additionally, increased concentrations of extracellular glutamate inhibit cystine uptake, leading to glutathione depletion and oxidative glutamate toxicity. Studies have shown that oxidative glutamate toxicity in neurons lacking functional N-methyl-D-aspartate (NMDA) receptors may represent a component of the cellular death pathway induced by excitotoxicity. The association between inflammation and excitotoxicity (i.e., immunoexcitotoxicity) has received increased attention in recent years. Glial activation induces neuroinflammation and can stimulate excessive release of glutamate, which can induce excitotoxicity and, additionally, further exacerbate neuroinflammation. Glutamate, as an important central neurotransmitter, is closely related to the occurrence and development of PD. In this review, we discuss recent progress on elucidating glutamate as a relevant neurotransmitter in PD. Additionally, we summarize the relationship and commonality among glutamate excitotoxicity, oxidative toxicity, and immunoexcitotoxicity in order to posit a holistic view and molecular mechanism of glutamate toxicity in PD.
Glutamate is the most abundant excitatory neurotransmitter in the mammalian brain. It is widely distributed and involved in a variety of functions and metabolic processes in the central nervous system (CNS) (Murrough et al., 2017; Madji Hounoum et al., 2018; Wang et al., 2018). In the CNS, glutamate is present in more than one type of cell and distributed in many subcellular compartments, including the cytoplasm and mitochondria (Nedergaard et al., 2002). In mammals, there is 5–10 mmol glutamate per kilogram of CNS tissue (Butcher et al., 2010), which is higher than levels of virtually all other neurotransmitters. These levels are a thousand times greater than many other important neurotransmitters, such as dopamine, serotonin, and norepinephrine (Sheldon and Robinson, 2007).
PD is a chronic neurodegenerative disease that affects 2–3% of the population over age 65 (Poewe et al., 2017). The main pathological features of PD are the appearance of Lewy bodies and the death of neurons in the basal ganglia [about 70% of dopaminergic neurons in the substantia nigra pars compacta (SNpc)]. This loss of neurons is accompanied by the death of astrocytes, and a significant increase and activation of microglia in the SNpc. PD is characterized by movement symptoms, such as tremors, bradykinesia, rigidity, and postural instability (Jankovic, 2008). Although many factors have been linked to an increased risk of PD—environmental and genetic factors, aging, and exposure to certain pesticides and fungicides—the exact cause of death of dopaminergic neurons remains unknown.
In 1994, Bergman et al. proposed that excessive activation of glutamate receptors in nigrostriatal neurons might lead to cellular death via glutamate excitotoxicity (Bergman et al., 1994; Hassani et al., 1996; Benazzouz et al., 2002; Hutchison et al., 2010). Previous studies have shown that glutamate excitotoxicity may induce the degeneration of dopaminergic neurons and concomitant motor dysfunction in PD (Sonsalla et al., 1998; Meredith et al., 2009). Under pathological conditions, when excessive glutamate is released from the presynaptic membrane or the reuptake function of glutamate is impaired, the extracellular glutamate concentration increases. Activated microglia and reactive astrocytes release large amounts of glutamate (Takeuchi et al., 2006; Wetherington et al., 2008). Too much glutamate induces excessive stimulation of glutamate receptors and increases the concentration of Na+ and Ca2+ in the cell, which may directly cause neuronal damage and cell death. In this way, increased extracellular glutamate concentration is the basis for the effects of glutamate excitotoxicity.
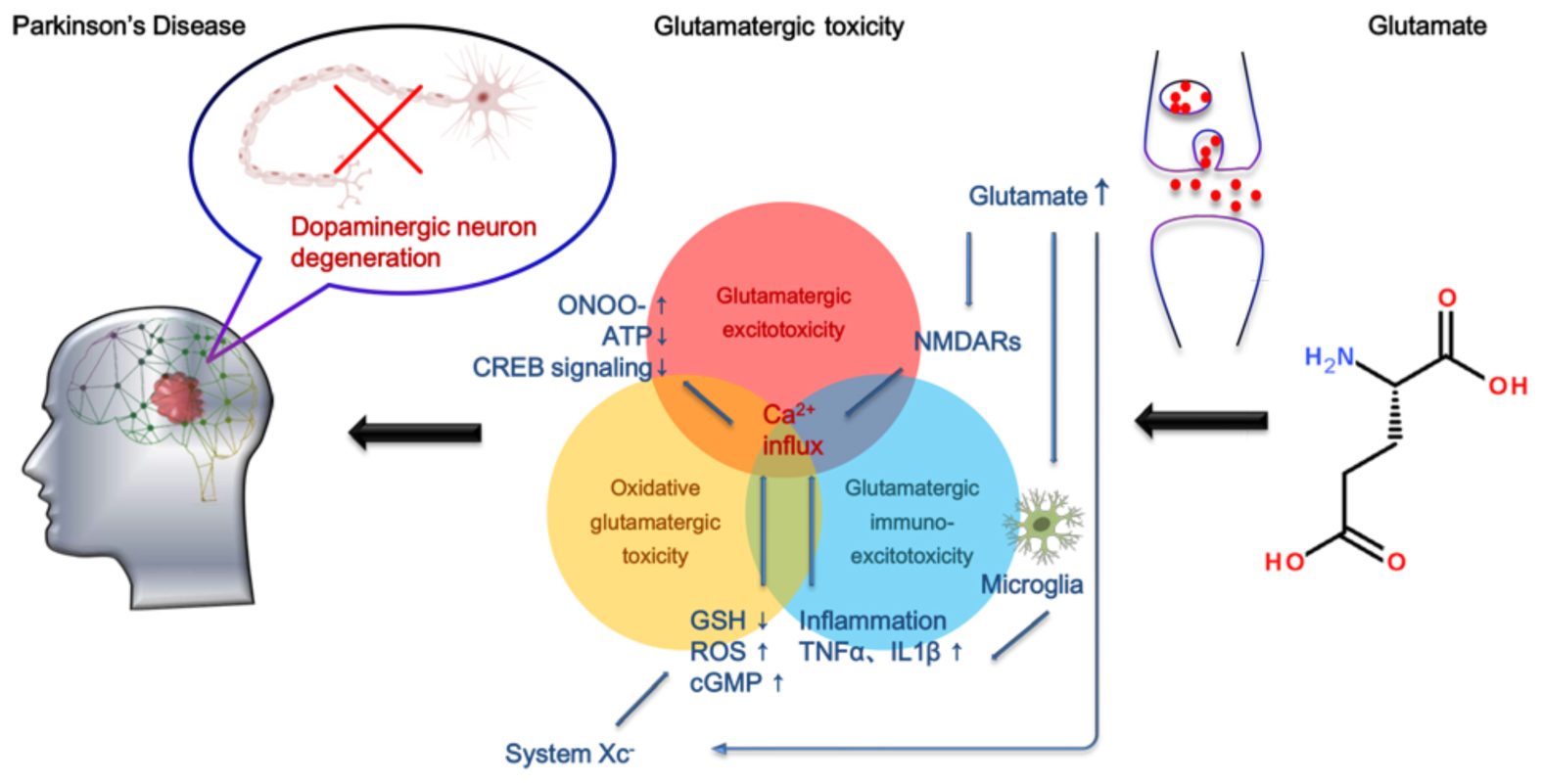
Graphical Abstract Molecular mechanisms of glutamate toxicity in PD.
Glucose is a key chemical molecule involved in cell metabolism in the brain; it cannot penetrate the blood–brain barrier (Sibson et al., 1998; Danbolt, 2001), so local synthesis is essential. In the brain, glutamate can be directly synthesized de novo by astrocytes or indirectly produced from glucose molecules through the actions of pyruvate dehydrogenase and astrocyte-specific enzyme pyruvate carboxylase (Krzyżanowska Pomierny et al., 2014; Schousboe et al., 2014). Once inside the cell, glucose breaks down through intracellular glucose enzymes to yield acetic acid. Acetic acid enters the tricarboyxlic acid cycle, and then the transfer of ammonia yields alpha-ketoglutarate, which receives an ammonia donor from a chain of amino acids—such as leucine, isoleucine, and valine—and a variety of amino donors, such as aspartic acid, gamma-aminobutyric acid (GABA), and alanine (Luc and Magistretti, 2004). It should be noted that, in addition to acting as a neurotransmitter, glutamic acid also acts as a metabolic precursor of GABA and various amino acid derivatives (Shen et al., 1999). Glutamate taken up by perisynaptic astrocytes is then converted to glutamine via glutamine synthetase (Danbolt, 2001). Astrocytes and neurons contain glutamine transporters, which—under appropriate electrophysiological conditions—result in a net exchange of glutamine between astrocytes and neurons. In neurons, the mitochondrial phosphate-specific enzyme, glutaminase, converts inactive glutamine to glutamate and repackages it into synaptic vesicles. The circulation of glutamate/glutamine in astrocytes and neurons is called the glutamate–glutamine cycle (Niciu et al., 2012). In summary, neuronal glutamate is produced/obtained in two ways: (1) from glucose and amino acid derivatives through energy metabolism; and (2) recovery of glutamate from glutamine via glutamate reuptake (Erecinska and Silver, 1990).
In axons, glutamate is stored in synaptic vesicles; upon sufficient depolarization, glutamate is released via synaptic calcium influx from voltage-gated calcium channels. Glutamate release leads to a significant rise in synaptic glutamate concentrations (∼1,000-fold). Release of presynaptic glutamate and activation of postsynaptic glutamate receptors on the postsynaptic neuron stimulate influx of Na+ and Ca2+ ions into the postsynaptic neuron, which contributes to depolarization and action potentials (Danbolt, 2001). Astrocytes expressing high levels of excitatory amino acid transporters (EAATs) sequester excess glutamate at synapses (Chaudhry et al., 1995). Glutamate is quickly cleared from the synaptic cleft via glutamate transporters to prevent excessive glutamate-receptor stimulation.
Increased levels of extracellular glutamate, particularly asynchronistic glutamate levels, cause cellular damage when excitatory neurotransmission is sufficiently aberrant (Pitt et al., 2000; Ouardouz et al., 2009). However, the uptake of glutamate at synapses, due to a lack of extracellular enzymes, is achieved primarily through EAATs located in the plasma membranes of neurons and glia (Zhang et al., 2016). Five different EAATs have been identified, and the amino acid sequence homology of these transporters is approximately 50–60% (Sheldon and Robinson, 2007). Based on the crystalline structure of bacterial glutamate transporters (Dinesh et al., 2004), EAATs have eight cross-membrane structures, with a carboxylic acid end and an amino end. It most likely exists as a tripolymer (Yernool et al., 2003; Koch and Peter, 2005). The specific EAAT glutamate aspartate transporter (GLAST) (Storck et al., 1992) is also known as excitatory amino acid transporter 1 (EAAT1) (Arriza et al., 1994), and the EAAT glutamate transporter 1 (GLT-1) (Pines et al., 1992) is also known as excitatory amino acid transporter 2 (EAAT2) (Arriza et al., 1994). GLT-1 and GLAST are mainly found in astrocytes (Rothstein et al., 1994; Lehre et al., 1995). These transporters are also found in many other cells, such as oligodendrocytes and macrophages (Domercq et al., 2010; Gras et al., 2010). Each EAAT has a specific expression pattern, and some of the functional characteristics of EAATs can be attributed to their different spatiotemporal localizations (Furuta et al., 1997b). GLT-1 is not only expressed in astrocytes; its splice variant GLT-1a is also expressed in the axons of hippocampal neurons, which may contribute significantly to glutamate uptake into the axonal terminal (Schmitt et al., 1996; Chen et al., 2004; Berger et al., 2005). Two members of the EAAT family, EAAC1 (also referred to as EAAT3) and EAAT4, are neuronal transporters (Rothstein et al., 1994; Furuta et al., 1997a); in many neurons, however, they appear to be located in postsynaptic elements, indicating that they do not participate in the delivery cycle as monoamine transporters do (Blakely and Bauman, 2000; Torres and Caron, 2003). EAAC1 has been shown to be expressed in hippocampal and cortical pyramidal neurons, as well as in GABAergic neurons (Rothstein et al., 1994), oligodendrocytes (Conti et al., 1998), and other glutamate neurons. The expression of EAAT4 is thought to be limited to cerebellar GABAergic Purkinje cells, but it has also been observed in astrocytes (Wen-Hui et al., 2003). The expression of EAAT5 is confined to retinal rod photoreceptors and bipolar cells (Arriza et al., 1997). About 90% of glutamate transport is mediated by EAAT2. These transporters cooperate with each glutamate (or asparaginase) molecule in the co-transport of two to three Na+ ions and a proton and cooperate with the reverse transport of one K+ ion (Zerangue and Kavanaugh, 1996). Our previous work has shown that EAATs contribute to the removal of synaptic glutamate, and the protein structures and functions of these transporters are closely related (Shaogang and Kanner, 2008; Crisman et al., 2009; Teichman et al., 2009; Zhang and Qu, 2012; Yunlong et al., 2014; Rong et al., 2015). These findings have helped elucidate the physiological functions of EAATs and their contributions to regulating extracellular glutamate concentrations. However, the role of EAATs in the pathogenesis of various neurodegenerative diseases remains unclear.
Glutamate is released in the synaptic cleft and binds to glutamate receptors, which can lead to action potentials. Ionotropic glutamate receptors (iGluRs) are ligand-gated ion channels that are activated by glutamate neurotransmitters, which mediate excitatory synaptic transmission in the CNS; they are the key to synaptic plasticity and play an important role in learning and memory.
According to the selective nomenclature of agonists, the three types of ionotropic receptors are identified as the following: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), N-methyl-D-aspartic acid receptors (NMDARs), and kainate receptors (KARs). NMDARs consist of a mandatory subunit GluN1 in a combination with GluN2A–2D subunits (Paoletti and Neyton, 2007). AMPARs consist of a combination of GluR1–4 subunits, and KARs are made up of a combination of GluR5–7, KA1, and KA2 subunits (Rosenmund et al., 1998). AMPARs and KARs mainly mediate influx of Na+, while NMDARs are doubly gated channels and have high calcium conductivity. Compared with that of other iGluRs, the activity of NMDARs is inhibited via a so-called Mg2+ block, which is removed via membrane depolarization (Vargas-Caballero and Robinson, 2004).
NMDARs have the highest affinity for glutamate and are substantially regulated in the mammalian CNS. Activation of synaptic NMDARs can induce neuronal survival through Ca2+-mediated signal transduction pathways, whereas overactivation of extrasynaptic pathways can induce neuronal death (Stefanie et al., 2010). Synaptic and extrasynaptic NMDAR activation exert opposing neural protective and neurotoxic effects, respectively, and these processes involve a variety of corresponding neuroprotective and apoptotic pathways (Chai et al., 2001).
Glutamate can also activate a type of G-protein-coupled receptors known as metabotropic glutamate receptors (mGluRs). The mGluR family consists of eight subtypes (mGluR1 to mGluR8) that are divided into three groups according to their amino-acid sequences, G-protein coupling, and pharmacological profiles. Together, they participate in the formation of extracellular ligand-binding domains and are responsible for the activation of G-proteins in the hepatic helical transmembrane domain (Rondard and Pin, 2015). Subtypes of Group I include mGluR1 and mGluR5 receptors conjugated with Gq/G11 (Abdulghani et al., 1996; Nicoletti et al., 2011). Group-I mGluRs activate phospholipase C (PLC) to produce inositol 1,4,5 triphosphate (IP3) and diacylglycerol, each of which has multiple second-messenger roles (Nakanishi, 1994; Joly et al., 1995; Pin and Duvoisin, 1995). Group-I mGluRs are mainly located in the post-synaptic density near ionotropic receptors, the function of which is to regulate the excitability of neurons (Shigemoto et al., 1993, 1997). Subtypes of group II (mGluR2, mGluR3) and group III (mGluR4, mGluR6, mGluR7, and mGluR8) are coupled with Gi/Go to activate MAP kinase and PI-3-kinase pathways and negatively regulate adenylate cyclase (Iacovelli et al., 2010; Niswender and Conn, 2010; Nicoletti et al., 2011). These subtypes are located presynaptically and act as self-receptors to inhibit the release of glutamate and/or GABA (Schoepp, 2001). mGluRs have seven transmembrane domains (Platt, 2007), as do other metabotropic receptors, but they are not ion channels. Nevertheless, they activate biochemical cascades that lead to modifications of other proteins. This may lead to changes in synaptic excitability, such as presynaptic inhibition of neurotransmission (Sladeczek et al., 1993) or induction of postsynaptic responses (Chu and Hablitz, 2000; Endoh, 2004; Bonsi et al., 2005; Platt, 2007).
The term “excitatory toxicity” was first proposed in 1986 to describe the ability of excessive extracellular glutamate to kill neurons by activating NMDARs (Choi, 1988; Meldrum and Garthwaite, 1990). Although the mechanisms of excitotoxicity have long been studied, there is still a lack of understanding of the intracellular mechanisms of excitotoxicity leading to neuronal death. When glutamate is released from presynaptic terminals or when glutamate reuptake is impaired, extracellular levels of glutamate increase (Lin et al., 2012). Elevated levels of extracellular glutamate lead to the overactivation of Ca2+-permeable NMDARs, subsequent Ca2+ overload, and excitotoxicity (Figure 1; Choi, 1988; Bano et al., 2005). In contrast, iGluRs do not have significant permeability to Ca2+ (Arundine and Tymianski, 2003; Pinheiro and Mulle, 2006). Under pathological conditions, activated microglia and reactive astrocytes release large amounts of glutamate (Hideyuki et al., 2006; Wetherington et al., 2008). Aberrant high levels of intracellular Ca2+ activate catalytic enzymes, produce toxic radicals, and impair production of cellular energy, which ultimately induces cell death (acute necrosis and/or delayed apoptosis; Figure 1; Dutta and Trapp, 2011). Previous studies have shown that this process may activate a variety of enzymes, including kinases, phospholipases, nitric-oxide synthases, and proteases. However, the role of proteases in excitotoxicity remains controversial. The use of calpain inhibitors in hippocampal ischemic neurons was found to reduce the severity of injury (Manev et al., 1990, 2010), but the use of calpain inhibitors did not yield any toxic or protective effect (Manev et al., 2010). In addition, Ca2+ influx is not the only factor of excitation-induced cell death. Studies have shown that glutamate exposure or hypoxia/ischemia can trigger the activation of extrasynaptic NMDARs, activate the shutdown of cAMP response element-binding protein (CREB), and lead to mitochondrial membrane-potential loss and cell death (Figure 1). However, the activation of synaptic NMDARs activates only the CREB pathway and does not activate apoptosis (Hardingham and Bading, 2010).
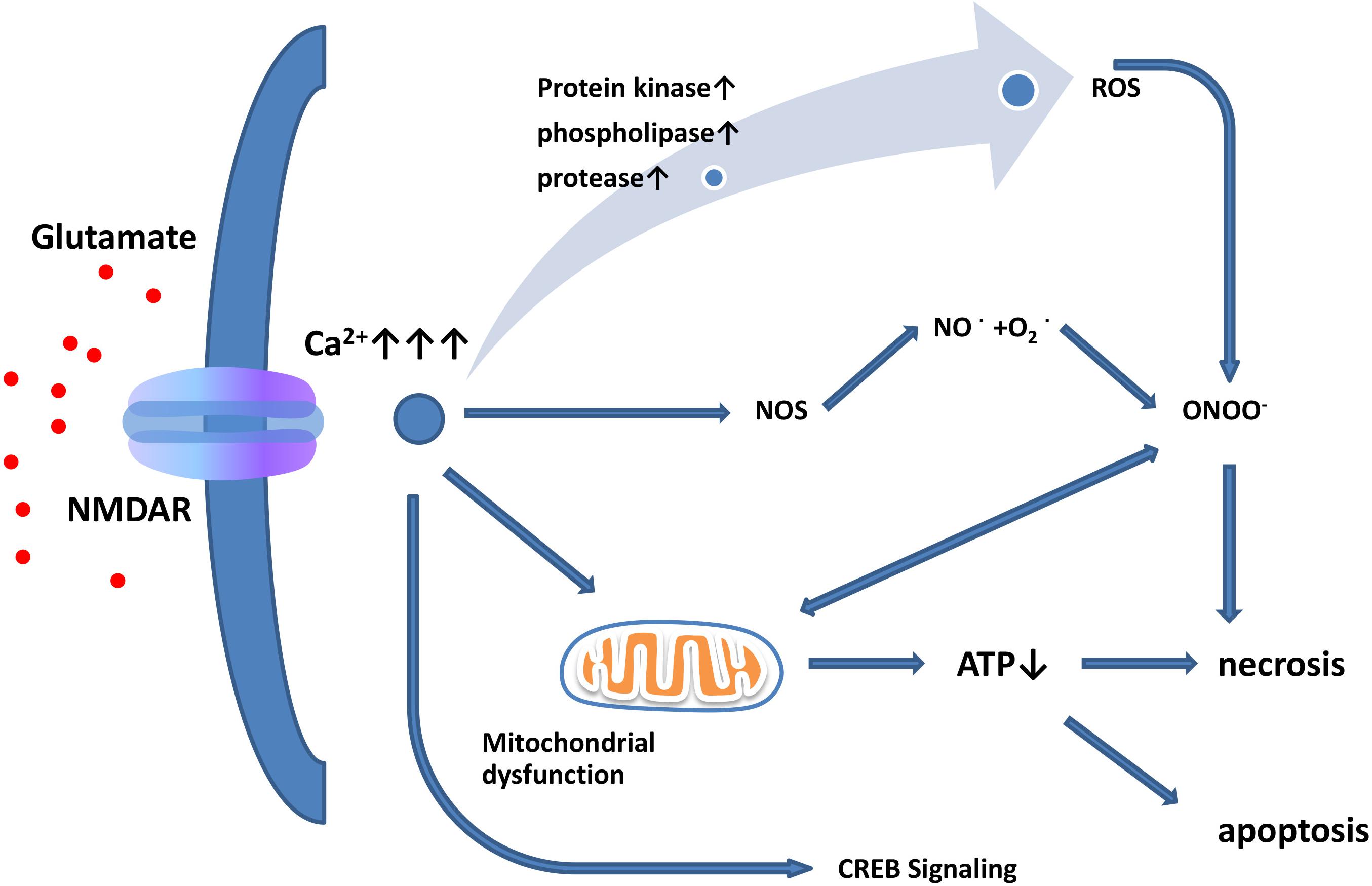
Figure 1. Glutamate excitotoxicity leads to neuronal necrosis and apoptosis. Increased extracellular glutamate levels lead to overactivation of NMDARs and induce Ca2+ influx. Ca2+ influx increases nitric oxide synthase (NOS) activity. Through this enzyme, NO can react with superoxide radicals to generate ONOO–, thus causing serious oxidative damage to cellular contents. In addition, mitochondria are damaged by oxidation, leading to ATP depletion and cellular death. Excessive activation of extrasynaptic NMDARs leads to reduced CREB signaling, resulting in mitochondrial membrane-potential loss and cell death, while synaptic NMDARs affect the CREB pathway but do not induce apoptosis.
Glutamate-mediated excitotoxicity is involved in many types of neurodegenerative diseases. Currently, there is no safe and effective drug to prevent excitotoxicity. Since overactivation of NMDARs is considered to be the main factor causing glutamate excitotoxicity (Choi, 1987), blocking these receptors is a potential strategy to prevent excitotoxicity. However, most NMDAR antagonists are competitive antagonists that can affect the physiological function of the brain and have negative side effects (Lipton, 2006). Another potential way to prevent excitotoxicity is by enhancing glutamate reuptake. The glial glutamate transporters, EAAT1 and EAAT2, are mainly present in the peripharyngeal processes of astrocytes closely related to excitatory synaptic contact and are responsible for maintaining low levels of extracellular glutamate. EAAT2-knockout mice exhibit fatal spontaneous epilepsy and increased sensitivity to acute cortical injury (Tanaka et al., 1997). In cases of chronic neurodegenerative diseases, such as like PD and Alzheimer’s disease (AD), aberrant EAAT2 function may contribute to excitotoxicity. In other neurodegenerative diseases—such as epilepsy, stroke, and brain trauma—a rapid rise in the levels of extracellular glutamic acid can cause severe neuronal damage. Increasing EAAT2 expression immediately reduces the levels of extracellular glutamate to prevent neuronal damage. This suggests that the up-regulation of EAAT2 may be a potential means of preventing neuroexcitatory toxicity. The expression of EAAT2 in PD animal models has been studied, and the down-regulation of EAAT2 has been found in both 6-hydroxydopamine and acute 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine models in the mouse striatum (Holmer et al., 2005; Chung et al., 2010). One study reported that thalidomide can heal the functional damage of nigrostriatal cell substratum by inhibiting excitotoxicity in MPTP mouse models (Palencia et al., 2015). Another study showed that NMDAR antagonists have anti-exercise and anti-dyskinesia effects in Parkinson’s disease (PD) rat models, and they have therapeutic potential in the treatment of PD (Löschmann et al., 2004).
In the pathogenesis of PD, regulation of the glutamate transporter gene is considered to be affected by epigenetic modifications. Epigenetics is currently defined as the study of stable and heritable patterns of gene expression that do not require any changes to the original DNA sequence (Egger et al., 2004; Bird, 2007). Epigenetic modifications mainly include changes in histone modification, DNA methylation, and non-coding RNA. Studies have reported that valproic acid (VPA), which is a histone deacetylase (HDAC) inhibitor can increase the levels of GLAST, GLT-1, and EAAC1 mRNA/protein in astrocytes and oligodendrocytes, thereby combating neurotoxicity (Hassel et al., 2001; Bianchi et al., 2012; Johnson et al., 2018). VPA has different effects on the expression of GLT-1 in different regions of the brain. VPA can increase the expression of GLT-1 in the cortex and hippocampus but decrease expression in the cerebellum (Perisic et al., 2012). This effect may be affected by promoter methylation. Research has indicated that the GLT-1 promoter has multiple methylated CpG islands (Zschocke et al., 2007; Yang et al., 2010), and transcription activation requires demethylation of the GLT-1 promoter. By co-culturing neurons and astrocytes, the methylation of the GLT-1 promoter is reduced (Yang et al., 2010). In the forebrain region, the promoter is hypomethylated and up-regulates the expression of GLT-1, while in the brainstem/cerebellum, the promoter is hypermethylated, while the expression of GLT-1 is not affected (Zschocke et al., 2007). Therefore, GLT-1 expression may be jointly regulated by acetylation and methylation. It has been reported that the increase of miR-543-3p and miR-342-3p in the PD model is associated with decreased expression and function of GLT-1 and increased accumulation of extracellular glutamate and that inhibition of either microRNAs can reverse the effect on the expression and function of GLT-1 and reduce dyskinesia (Wu et al., 2019; Wu et al., 2019).
In addition to the receptor-mediated excitotoxicity, an increase in extracellular glutamate can also lead to oxidative stress in the form of the non-receptor-mediated oxidative glutamate toxicity (Tan et al., 2001); this is also the pathway that is induced by glutamate interacting with the Xc– system to produce toxicity. The Xc– system consists of heavy-chain 4F2hc (or CD98/SLC3A2) and light-chain xCT (or SLC7A11), which are connected by disulfide bonds. The light chain of the xCT protein and histone acetyltransferases (HATs) are distinctly homogeneous, and the HATs are a family of amino acid transporters that are linked by the light chain and the heavy chain through disulfide bonds (François et al., 2004). xCT and 4F2hc are both highly expressed in the brain (Kanai et al., 1998). However, it has been shown that the substitution of rBAT for 4F2hc heavy chain does not affect the function of the transporter (Wang et al., 2003; Fernández et al., 2006). The Xc– system is a sodium- and chloride-dependent amino acid antiporter. It transports non-essential sulfur-containing amino acid cysteine anionic compounds into cells and glutamate in a ratio of 1:1 in the opposite direction (Sato et al., 1999; Makowske and Christensen, 2012). Cystine-mediated uptake is critical to cellular antioxidant stress, and xCT plays a major role in maintaining low concentrations of extracellular glutamate and cystine-mediated uptake.
The cell death pathway, which works via oxidative glutamate toxicity, is as follows. The first step in oxidative glutamate toxicity is the inhibition of cystine uptake through the Xc– system leading to GSH deficiency in cells. Although it has been found that some cells can synthesize cystathionine via cystathionine beta-lyase, neurons do not normally express this enzyme and, thus, rely primarily on extracellular cystine. When cystine absorption is blocked, intracellular GSH consumption is much faster than protein synthesis (Tan et al., 1998). In addition, the inhibition of the Xc– system, the subsequent accumulation of ROS, and the decrease of GSH levels create a void in the antioxidant defenses of the cell, which may ultimately lead to ferroptosis (Dixon et al., 2012; Xie et al., 2016). The second step in oxidative glutamate toxicity occurs when the concentration of GSH falls below 20% (about 6 h after glutamate exposure). The continuous generation of free radicals via mitochondrial complex I activity increases exponentially. At this time, the activation of the lipid-oxidizing enzyme, 12/15-lipoxygenase (12/15-LOX), produces 12/15-hydroxyeicosatetraenoic acid (Li et al., 1997a). Third, 12/15-lox can directly damage mitochondria, leading to mitochondrial depolarization and increased the production of reactive oxygen species (ROS) (Stefanie et al., 2010). The eicosanoids produced by 12-LOX are the activators of soluble functional acid cyclase, which can increase intracellular guanosine monophosphate (GMP) (Li et al., 1997b). Elevated cyclic GMP (cGMP) eventually opens calcium channels, leading to harmful influx of calcium (Li et al., 1997b). Finally, oxidative glutamate toxicity induces oxidation of glutamate for approximately 10–12 h. At this time, both ROS and intracellular calcium levels reach maximal values, apoptotic inducing factor (AIF) translocates from mitochondria to nuclei, and intracellular induction of nuclear condensation and caspase-independent cell death occur within a few minutes (Landshamer et al., 2008). This important process is depicted in Figure 2.
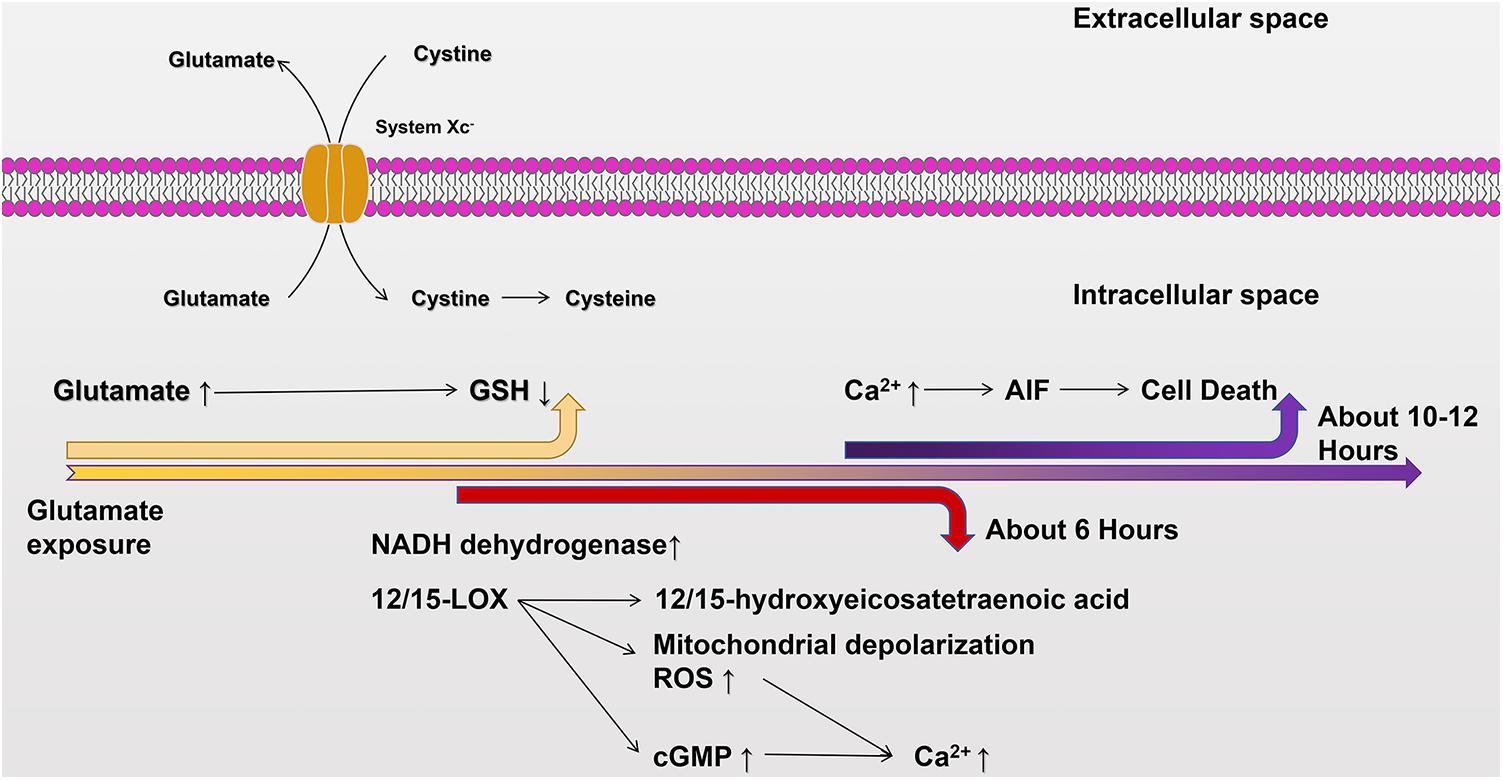
Figure 2. The cellular death pathway of oxidative glutamate toxicity. Excessive accumulation of glutamate in the extracellular environment inhibits cystine uptake through System Xc–, resulting in GSH deficiency in cells. Mitochondrial complex I increases about 6 h after glutamate exposure. At the same time, 12/15-LOX is activated and 12/15-hydroxyhexotetraenoic acid is produced, and 12/15-LOX directly damages mitochondria and increases production of ROS. Additionally, soluble functional acid cyclase is activated, and intracellular GMP content is increased. Subsequently, cGMP opens calcium channels and enables calcium influx. After glutamate exposure for about 10–12 h, ROS and intracellular calcium levels reach their peaks, and AIF is transferred from mitochondria to the nucleus, inducing nuclear coagulation and cellular death.
The excitotoxic cascade is triggered by excess extracellular glutamate, which may eventually lead to cell injury and death (Siesjö et al., 1989). Via excessive activation of NMDARs, increased Ca2+ influx further exacerbates ROS levels, leads to mitochondrial damage, and increases susceptibility to cellular death. Additionally, excessive activation of AMPARs and KARs induces Na+ overload, resulting in high intracellular permeability and acute cellular swelling. The increase of free radicals is particularly important for the pathogenesis of PD, because dopaminergic neurons in the SNpc are particularly susceptible to oxidative stress (Julie and Patrik, 2002; Yuan et al., 2008; Mosharov et al., 2009; Benjamin et al., 2010).
One of the findings of oxidative stress in the SNpc of PD patients is that GSH levels are significantly reduced only in the SNpc, which leads to changes in the GSH/GSSG ratio. In addition, the oxidation of lipids, proteins, and DNA, and the increase of total iron content in the SNpc of PD patients support the enhancement of the oxidation stress (Floor and Wetzel, 1998; Dexter et al., 2010). In this case, oxidative stress in the SNpc leads to excessive peroxide formation, and glutathione deficiency leads to a decreased ability of the brain to remove H2O2, and continuous accumulation of H2O2 induces cellular death (Wang et al., 2008; Saggu et al., 2010).
David et al. proposed that the excitotoxicity cascade can be divided into three parts. First, cellular death pathways start with the activation of NMDA receptors, after a brief exposure to a low concentration of glutamic acid. The second part involves 20–30% of the cell death pathway and has characteristics of oxidative glutamate toxicity, because it can be inhibited specifically by vitamin E, group I metabotropic receptor agonists, a caspase inhibitor, elevated extracellular cystine, and the removal of extracellular glutamate (Schubert and Piasecki, 2001). In addition, oxidative glutamate toxicity is higher than the glutamate concentration required for NMDA receptor activation (Murphy et al., 1989). This suggests that oxidative glutamate could be a part of an excited viral cascade and may provide a form of cell death that is independent of iGluRs. Therefore, neurons lacking postsynaptic glutamate receptors are also killed in this process, and oxidative glutamate toxicity may cause more damage than excitotoxicity.
Another major characteristic of PD is the formation of the Lewy bodies. Lewy bodies are formed by the combination of alpha-synuclein with ubiquitin (Simone, 2008), neurofilament protein, and alpha B crystalline. The Lewy body is thought to be an aggregating response in cells (Mikiei et al., 2004), which suggests that their function may be related to protein folding and aggregation defects (Mikiei et al., 2004; Kovacs, 2015) and may have protective effects on nerve endings (Sreeganga et al., 2005). The typical Lewy body is a postsynaptic cytoplasmic inclusion body consisting of a dense nucleus surrounded by a 10 nm wide ring of radiating fibers. As the main structural component of the Lewy body, alpha-synuclein mainly resides in neurons and is primarily expressed in cerebral cortex, hippocampus, substantia nigra, thalamus, and cerebellum, and is also found in glia. Alpha-synuclein is found in the cerebrospinal fluid of PD patients. In general, the secretion of synuclein in degenerating neurons is greatly increased compared with that of healthy neurons, and alpha-synuclein has enhanced immune characteristics when oxidized or nitrated. Previous studies have shown that alpha-synuclein is closely associated with glutamate excitotoxicity. Watson et al. reported that the relative release of glutamate was inhibited by the knockout of alpha-synuclein in a mouse model in SN terminals (Watson et al., 2009). In a transgenic mouse model of mutant alpha-synuclein (A53T), experiments showed that alpha-synuclein accumulation in astrocytes affected glutamate transport, triggering a rise of extracellular glutamatergic concentrations and excitotoxicity, which further exacerbated damage to dopaminergic neurons (Gu et al., 2010). These findings highlight that alpha-synuclein increases the release of glutamate. Furthermore, some studies have shown that the concentration of alpha-synuclein depends on the release of activity-dependent presynaptic glutamate from forebrain terminals (Sarafian et al., 2017). Additionally, overexpression of alpha-synuclein increases the phosphorylation of NMDARs, thereby increasing the expression of NR1 and NR2B subunits and increasing the sensitivity of NMDARs to glutamate excitotoxicity (Yang et al., 2016). In addition, some studies have found that alpha-synuclein can also enhance glutamate excitotoxicity by accelerating AMPAR signaling (Sandra et al., 2011). The above studies indicate that the abnormal aggregation of alpha-synuclein in PD is closely related to the excitatory toxicity of glutamic acid, which may be a potential neuropathological pathway.
Extensive microglial activation in and around the SNpc was first observed more than 30 years ago in autopsies of patients with PD (McGeer et al., 1988). It has been reported that macrophages may undergo pathological diffusion from blood vessels to the CNS during inflammation and transform into microglia cells, which promote the pathological development of intracranial nervous system diseases (Chen et al., 2016). Microglia mediating chronic inflammation is involved in the pathological processes of a variety of chronic neurodegenerative diseases. In these processes, microglia are activated over a time span of minutes and can last a long time, or even a lifetime (Ramlackhansingh et al., 2011), which then continuously release a series of inflammatory mediators, leading to oxidative stress. Microglia are activated by the increased production of pro-inflammatory and anti-inflammatory cytokines, chemokines, and other inflammatory molecules, and increased glutamate release, all of which remain in microglia (Hoeijmakers et al., 2016). When the destructive elements are fully activated in microglia, they will show strong inflammatory and excitatory toxic reactions, exceeding the response level of physiological activation of microglia (Norden et al., 2015). In this review, the two most studied neuronal excitatory factors, IL-1β and TNF-α, are discussed as examples. Studies have shown that TNF-α—when used alone in organotypic hippocampal–entorhinal cortex brain slices—causes minor neuronal damage; however, when high-frequency glutamate concentrations are increased, neurotoxicity is greatly accelerated (Zou and Crews, 2005). Additionally, for non-Ca2+-permeable AMPA receptors, studies have shown that low-dose TNF-α plays a neuroprotective role through tissue necrosis factor receptor-2 (TNFR-2), while high-dose TNF-α increases the synaptic transport of calcium-permeable AMPARs through the TNFR-1 and enhances excitotoxicity in a concentration-dependent manner (Blaylock, 2017). In addition, it has been demonstrated that the interaction between IL-1β receptors and NMDARs can also lead to increased excitotoxicity, and the expression of IL-1β receptors on the postsynaptic membrane is stimulated by NMDAR activation (Gardoni et al., 2011). As mentioned above, inflammation can change the effects of inflammatory factors, such as TNF-α and IL-1β, and can also enhance glutamine enzymes and inhibit glutamine dehydrogenase and glutamine synthase, thus enhancing glutamate excitotoxicity (Brown and Vilalta, 2015). These two enzymes play crucial roles in glutamate metabolism, and both are sensitive to oxidative stress in the body in an inflammatory environment (Levine, 1983; Aguirre et al., 1989). Glutamine synthase catalyzes the conversion of glutamic acid to glutamine to reduce the risk of excitatory intoxication. Glutamate dehydrogenase, which converts glutamate and ketoglutarate in both directions, also protects against elevated glutamate levels. Conversely, the abovementioned cysteine–glutamic acid reverse transporter (system Xc–) can exchange extracellular cystine with intracellular glutamic acid, thus providing the necessary raw materials for the synthesis of glutathione by glial cells; additionally, in the pathological state, it has also become a factor for the excitatory toxicity of glutamic acid. Overall, when inflammation is caused by the activation of microglia, there is a tight connection between inflammation and glutamate receptors. Inflammation can trigger the excessive release of glutamate. Additionally, the accumulation of extracellular glutamate can also strengthen inflammation, forming a vicious cycle (Blaylock, 2017).
Glutamate affects the CNS to a much greater extent than solely as a CNS courier. In PD, dopaminergic information transmitted by dopaminergic neurons decreases, while glutamatergic signaling in the basal ganglia increases and stimulates the release of dopamine through surviving dopaminergic neurons in the SNpc as a compensatory mechanism. However, by increasing glutamate and excessive activation of glutamate receptors, it could be a “critical strike” to dopaminergic neurons in PD patients. Excessive activation of NMDARs induces excessive influx of Ca2+ and further exacerbates ROS levels. Due to the high metabolism of the brain and the characteristics of relatively low regeneration, the brain is sensitive to oxidative damage produced by ROS levels and enhanced oxidative states will also lead to excessive oxidation of lipids, proteins, DNA, and increased total iron content in the SNpc. Overactivation of AMPARs and KARs induces Na+ overload, which leads to increased cellular permeability and results in cellular swelling and neuronal death. During abnormal rises in extracellular glutamate, the cystine/glutamate antiporter system Xc– has inhibited cystine transport into cells. Additionally, the ability of glutathione to be reduced by synthesis and the ability of the brain to remove peroxides are compromised; hence, peroxide is accumulated in cells and causes damage, which can also lead to cellular death.
Second, in PD, the abnormal aggregation of alpha-synuclein may increase excitotoxicity by affecting the transport efficiency of glutamate transporters and increasing the phosphorylation of NMDARs. In addition, it is noteworthy that PD microglial cells, after being activated for a short time, will release a series of inflammatory mediators to generate oxidative stress. Neurodegeneration associated with this inflammation is an activated cellular mechanism secondary to signal transduction in inflammatory cells, including lipid peroxidation, reactive oxygen, reactive nitrogen, prostaglandins, proteases, and nitric oxide (Wei et al., 2005). Studies have shown that excitotoxicity may play a major role in the death of dopaminergic neurons during this process.
At present, dopamine replacement therapy is still the gold standard for the treatment of PD, although the efficacy of this therapy is not ideal. Research reports have indicated that Nedd4-2 knockdown attenuates astrogliosis and reactive microgliosis in PD model mice. This may be associated with glutamate excitotoxicity (Zhang et al., 2017). In addition, an antagonist of NMDAR attenuated motor symptoms in an MPTP-induced PD animal model (Pajarillo et al., 2019). Hence, further in-depth research into the different mechanisms of glutamate toxicity or the complexity related to other central neurotransmitters may provide more information for understanding the pathogenesis of PD. Therapeutic intervention to reduce glutamate toxicity will be a possible way to treat PD.
SQ: conceptualization and methodology. DM: visualization. SQ, JW, and FW: writing. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (grant nos. U1603281 and 81870991 to SQ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdulghani, M. A., Valiante, T. A., Carlen, P. L., and Pennefather, P. S. (1996). Metabotropic glutamate receptors coupled to IP3 production mediate inhibition of IAHP in rat dentate granule neurons. J. Neurophysiol. 76, 2691–2700. doi: 10.1152/jn.1996.76.4.2691
Aguirre, J., Rodríguez, R., and Hansberg, W. (1989). Oxidation of Neurospora crassa NADP-specific glutamate dehydrogenase by activated oxygen species. J. Bacteriol. 171, 6243–6250. doi: 10.1128/jb.171.11.6243-6250.1989
Arriza, J. L., Eliasof, S., Kavanaugh, M. P., and Amara, S. G. (1997). Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc. Natl. Acad. Sci. U.S.A. 94, 4155–4160. doi: 10.1073/pnas.94.8.4155
Arriza, J. L., Fairman, W. A., Wadiche, J. I., Murdoch, G. H., Kavanaugh, M. P., and Amara, S. G. (1994). Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J. Neurosci. 14, 5559–5569. doi: 10.1523/jneurosci.14-09-05559.1994
Arundine, M., and Tymianski, M. (2003). Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calc. 34, 325–337. doi: 10.1016/s0143-4160(03)00141-6
Bano, D., Young, K. W., Guerin, C. J., Lefeuvre, R., Rothwell, N. J., Naldini, L., et al. (2005). Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 120, 275–285. doi: 10.1016/j.cell.2004.11.049
Benazzouz, A., Breit, S., Koudsie, A., Pollak, P., Krack, P., and Benabid, A. L. (2002). Intraoperative microrecordings of the subthalamic nucleus in Parkinson’s disease. Mov. Disord. 17, S145–S149.
Benjamin, D., Jordi, B., Natalia, R. M., Celine, P., Ariadna, R., Patricia, B., et al. (2010). Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 30, 12535–12544. doi: 10.1523/jneurosci.1920-10.2010
Berger, U. V., DeSilva, T. M., Chen, W., and Rosenberg, P. A. (2005). Cellular and subcellular mRNA localization of glutamate transporter isoforms GLT1a and GLT1b in rat brain by in situ hybridization. J. Comp. Neurol. 492, 78–89. doi: 10.1002/cne.20737
Bergman, H., Wichmann, T., Karmon, B., and Delong, M. R. (1994). The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J. Neurophysiol. 72, 507–520. doi: 10.1152/jn.1994.72.2.507
Bianchi, M. G., Franchi-Gazzola, R., Reia, L., Allegri, M., Uggeri, J., Chiu, M., et al. (2012). Valproic acid induces the glutamate transporter excitatory amino acid transporter-3 in human oligodendroglioma cells. Neuroscience 227, 260–270. doi: 10.1016/j.neuroscience.2012.09.055
Blakely, R. D., and Bauman, A. L. (2000). Biogenic amine transporters: regulation in flux. Curr. Opin. Neurobiol. 10, 328–336. doi: 10.1016/s0959-4388(00)00088-x
Blaylock, R. L. (2017). Parkinson’s disease: microglial/macrophage-induced immunoexcitotoxicity as a central mechanism of neurodegeneration. Surg. Neurol. Int. 8:65. doi: 10.4103/sni.sni_441_16
Bonsi, P., Cuomo, D., Persis, C. D., Centonze, D., Bernardi, G., Calabresi, P., et al. (2005). Modulatory action of metabotropic glutamate receptor (mGluR) 5 on mGluR1 function in striatal cholinergic interneurons. Neuropharmacology 49, 104–113. doi: 10.1016/j.neuropharm.2005.05.012
Brown, G. C., and Vilalta, A. (2015). How microglia kill neurons. Brain Res. 1628, 288–297. doi: 10.1016/j.brainres.2015.08.031
Butcher, S. P., Sandberg, M., Hagberg, H., and Hamberger, A. (2010). Cellular origins of endogenous amino acids released into the extracellular fluid of the rat striatum during severe insulin-induced hypoglycemia. J. Neurochem. 48, 722–728. doi: 10.1111/j.1471-4159.1987.tb05576.x
Chai, J., Wu, Q., Shiozaki, E., Srinivasula, S. M., Alnemri, E. S., and Shi, Y. (2001). Crystal structure of a procaspase-7 zymogen : mechanisms of activation and substrate binding. Cell 107, 399–407. doi: 10.1016/s0092-8674(01)00544-x
Chaudhry, F. A., Lehre, K. P., Vanlookerencampagne, M., Ottersen, O. P., Danbolt, N. C., and Stormmathisen, J. (1995). Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 15, 711–720. doi: 10.1016/0896-6273(95)90158-2
Chen, L., Mo, M., Li, G., Cen, L., Wei, L., Xiao, Y., et al. (2016). The biomarkers of immune dysregulation and inflammation response in Parkinson disease. Transl. Neurodegen. 5:16.
Chen, W., Mahadomrongkul, V., Berger, U. V., Bassan, M., DeSilva, T., Tanaka, K., et al. (2004). The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J. Neurosci. 24, 1136–1148. doi: 10.1523/jneurosci.1586-03.2004
Choi, D. W. (1987). Ionic dependence of glutamate neurotoxicity. Neuroscience 7, 369–379. doi: 10.1523/jneurosci.07-02-00369.1987
Choi, D. W. (1988). Glutamate neurotoxicity and diseases of the nervous system. Neuron 1, 623–634. doi: 10.1016/0896-6273(88)90162-6
Chu, Z., and Hablitz, J. J. (2000). Quisqualate induces an inward current via mGluR activation in neocortical pyramidal neurons. Brain Res. 879, 88–92. doi: 10.1016/s0006-8993(00)02752-9
Chung, E. K. Y., Chen, L. W., Chan, Y. S., and Yung, K. K. L. (2010). Downregulation of glial glutamate transporters after dopamine denervation in the striatum of 6-hydroxydopamine-lesioned rats. J. Comp. Neurol. 511, 421–437. doi: 10.1002/cne.21852
Conti, F., Debiasi, S., Minelli, A., Rothstein, J. D., and Melone, M. (1998). EAAC1, a high-affinity glutamate tranporter, is localized to astrocytes and gabaergic neurons besides pyramidal cells in the rat cerebral cortex. Cereb. Cortex 8:108. doi: 10.1093/cercor/8.2.108
Crisman, T. J., Shaogang, Q., Kanner, B. I., and Forrest, L. R. (2009). Inward-facing conformation of glutamate transporters as revealed by their inverted-topology structural repeats. Proc. Natl. Acad. Sci. U.S.A. 106, 20752–20757. doi: 10.1073/pnas.0908570106
Dexter, D. T., Carter, C. J., Wells, F. R., Javoy-Agid, F., Agid, Y., Lees, A., et al. (2010). Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 52, 381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x
Dinesh, Y., Olga, B., Yan, J., and Eric, G. (2004). Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 431, 811–818. doi: 10.1038/nature03018
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Domercq, M., Sánchez-Gómez, M. V., Areso, P., and Matute, C. (2010). Expression of glutamate transporters in rat optic nerve oligodendrocytes. Eur. J. Neurosci. 11, 2226–2236. doi: 10.1046/j.1460-9568.1999.00639.x
Dutta, R., and Trapp, B. D. (2011). Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Prog. Neurobiol. 93, 1–12. doi: 10.1016/j.pneurobio.2010.09.005
Egger, G., Liang, G., Aparicio, A., and Jones, P. A. (2004). Epigenetics in human disease and prospects for epigenetic therapy. Nature 429, 457–463. doi: 10.1038/nature02625
Endoh, T. (2004). Characterization of modulatory effects of postsynaptic metabotropic glutamate receptors on calcium currents in rat nucleus tractus solitarius. Brain Res. 1024, 212–224. doi: 10.1016/j.brainres.2004.07.074
Erecinska, M., and Silver, I. A. (1990). Metabolism and role of glutamate in mammalian brain. Prog. Neurobiol. 35, 245–296. doi: 10.1016/0301-0082(90)90013-7
Fernández, E., Jiménezvidal, M., Calvo, M., Zorzano, A., Tebar, F., Palacín, M., et al. (2006). The structural and functional units of heteromeric amino acid transporters. J. Biol. Chem. 281, 26552–26561. doi: 10.1074/jbc.m604049200
Floor, E., and Wetzel, M. G. (1998). Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. Neurochemistry 70, 268–275. doi: 10.1046/j.1471-4159.1998.70010268.x
François, V., Closs, E. I., Wagner, C. A., Manuel, P., Hitoshi, E., and Yoshikatsu, K. (2004). CATs and HATs: the SLC7 family of amino acid transporters. Pflügers Archiv. Eur. J. Physiol. 447, 532–542. doi: 10.1007/s00424-003-1086-z
Furuta, A., Martin, L. J., Lin, C. L., Dykes-Hoberg, M., and Rothstein, J. D. (1997a). Cellular and synaptic localization of the neuronal glutamate transporters excitatory amino acid transporter 3 and 4. Neuroscience 81, 1031–1042. doi: 10.1016/s0306-4522(97)00252-2
Furuta, A., Rothstein, J. D., and Martin, L. J. (1997b). Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J. Neurosci. 17, 8363–8375.
Gardoni, F., Boraso, M., Zianni, E., Corsini, E., Galli, C. L., Cattabeni, F., et al. (2011). Distribution of interleukin-1 receptor complex at the synaptic membrane driven by interleukin-1β and NMDA stimulation. J. Neuroinflamm. 8:14. doi: 10.1186/1742-2094-8-14
Gras, G., Chrétien, F., Vallat-Decouvelaere, A. V., Le Porcheray, P. G. F., Bossuet, C., Léone, C., et al. (2010). Regulated expression of sodium-dependent glutamate transporters and synthetase: a neuroprotective role for activated microglia and macrophages in HIV infection? Brain Pathol. 13, 211–222. doi: 10.1111/j.1750-3639.2003.tb00020.x
Gu, X. L., Long, C. X., Sun, L., Xie, C., Lin, X., and Cai, H. (2010). Astrocytic expression of Parkinson’s disease-related A53T α-synuclein causes neurodegeneration in mice. Mol. Brain 3:12. doi: 10.1186/1756-6606-3-12
Hardingham, G. E., and Bading, H. (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696. doi: 10.1038/nrn2911
Hassani, O. K., Mouroux, M., and Féger, J. (1996). Increased subthalamic neuronal activity after nigral dopaminergic lesion independent of disinhibition via the globus pallidus. Neuroscience 72, 105–115. doi: 10.1016/0306-4522(95)00535-8
Hassel, B., Iversen, E. G., Gjerstad, L., and Taubøll, E. (2001). Up-regulation of hippocampal glutamate transport during chronic treatment with sodium valproate. J. Neurochem. 77, 1285–1292. doi: 10.1046/j.1471-4159.2001.00349.x
Hideyuki, T., Shijie, J., Jinyan, W., Guiqin, Z., Jun, K., Reiko, K., et al. (2006). Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 281, 21362–21368. doi: 10.1074/jbc.m600504200
Hoeijmakers, L., Heinen, Y., van Dam, A. M., Lucassen, P. J., and Korosi, A. (2016). Microglial priming and Alzheimer’s disease: a possible role for (Early) immune challenges and epigenetics? Front. Hum. Neurosci. 10:398. doi: 10.3389/fnhum.2016.00398
Holmer, H. K., Keyghobadi, M., Moore, C., and Meshul, C. K. (2005). l -dopa-induced reversal in striatal glutamate following partial depletion of nigrostriatal dopamine with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neuroscience 136, 333–341. doi: 10.1016/j.neuroscience.2005.08.003
Hutchison, W. D., Allan, R. J., Opitz, H., Levy, R., Dostrovsky, J. O., Lang, A. E., et al. (2010). Neurophysiological identification of the subthalamic nucleus in surgery for Parkinson’s disease. Ann. Neurol. 44, 622–628.
Iacovelli, L., Bruno, V., Salvatore, L., Melchiorri, D., Gradini, R., Caricasole, A., et al. (2010). Native group-III metabotropic glutamate receptors are coupled to the mitogen-activated protein kinase/phosphatidylinositol-3-kinase pathways. J. Neurochem. 82, 216–223. doi: 10.1046/j.1471-4159.2002.00929.x
Jankovic, J. (2008). Parkinson’s disease: clinical features and diagnosis. Neurol. Neurosurg. Psychiatry 79, 368–376.
Johnson, J. Jr., Pajarillo, E., Karki, P., Kim, J., Son, D. S., Aschner, M., et al. (2018). Valproic acid attenuates manganese-induced reduction in expression of GLT-1 and GLAST with concomitant changes in murine dopaminergic neurotoxicity. Neurotoxicology 67, 112–120. doi: 10.1016/j.neuro.2018.05.001
Joly, C., Gomeza, J., Brabet, I., Curry, K., Bockaert, J., and Pin, J. P. (1995). Molecular, functional, and pharmacological characterization of the metabotropic glutamate receptor type 5 splice variants: comparison with mGluR1. J. Neurosci. 15, 3970–3981. doi: 10.1523/jneurosci.15-05-03970.1995
Julie, L., and Patrik, B. (2002). Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat. Rev. Neurosci. 3, 932–942. doi: 10.1038/nrn983
Kanai, Y., Segawa, H., Miyamoto, K., Uchino, H., Takeda, E., and Endou, H. (1998). Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). Biol. Chem. 273, 23629–23632. doi: 10.1074/jbc.273.37.23629
Koch, H. P., and Peter, L. H. (2005). Small-scale molecular motions accomplish glutamate uptake in human glutamate transporters. J. Neurosci. 25, 1730–1736. doi: 10.1523/jneurosci.4138-04.2005
Kovacs, G. G. (2015). Neuropathology of tauopathies: principles and practice. Neuropathol. Appl. Neurobiol. 41, 3–23. doi: 10.1111/nan.12208
Krzyżanowska, W., Pomierny, B., Filip, M., and Pera, J. (2014). Glutamate transporters in brain ischemia: to modulate or not? Acta Pharmacol. Sin. 35, 444–462. doi: 10.1038/aps.2014.1
Landshamer, S., Hoehn, M., Barth, N., Duvezin-Caubet, S., Schwake, G., Tobaben, S., et al. (2008). Bid-induced release of AIF from mitochondria causes immediate neuronal cell death. Cell Death Differ. 15, 1553–1563.
Lehre, K. P., Levy, L. M., Ottersen, O. P., Storm-Mathisen, J., and Danbolt, N. C. (1995). Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J. Neurosci. 15, 1835–1853. doi: 10.1523/jneurosci.15-03-01835.1995
Levine, R. L. (1983). Oxidative modification of glutamine synthetase. I. Inactivation is due to loss of one histidine residue. J. Biol. Chem. 258, 11823–11827.
Li, Y., Maher, P., and Schubert, D. (1997a). A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron 19, 453–463. doi: 10.1016/s0896-6273(00)80953-8
Li, Y., Maher, P., and Schubert, D. (1997b). Requirement for cGMP in nerve cell death caused by glutathione depletion. J. Cell Biol. 139, 1317–1324. doi: 10.1083/jcb.139.5.1317
Lin, C. L., Kong, Q., Cuny, G. D., and Glicksman, M. A. (2012). Glutamate transporter EAAT2: a new target for the treatment of neurodegenerative diseases. Future Med. Chem. 4, 1689–1700. doi: 10.4155/fmc.12.122
Lipton, S. A. (2006). Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat. Rev. Drug Discov. 5, 160–170. doi: 10.1038/nrd1958
Löschmann, P. A., De Groote, C., Smith, L., Wüllner, U., Fischer, G., Kemp, J. A., et al. (2004). Antiparkinsonian activity of Ro 25-6981, a NR2B subunit specific NMDA receptor antagonist, in animal models of Parkinson’s disease. Exp. Neurol. 187, 86–93. doi: 10.1016/j.expneurol.2004.01.018
Luc, P., and Magistretti, P. J. (2004). Neuroenergetics: calling upon astrocytes to satisfy hungry neurons. Neuroscientist a review. J. Bring. Neurobiol. Neurol. Psychiatry 10, 53–62. doi: 10.1177/1073858403260159
Madji Hounoum, B., Blasco, H., Coque, E., Vourc’h, P., Emond, P., Corcia, P., et al. (2018). The metabolic disturbances of motoneurons exposed to glutamate. Mol. Neurobiol. 55, 7669–7676. doi: 10.1007/s12035-018-0945-8
Makowske, M., and Christensen, H. N. (2012). Contrasts in transport systems for anionic amino acids in hepatocytes and a hepatoma cell line HTC. J. Biol. Chem. 257, 5663–5670.
Manev, H., Costa, E., Wroblewski, J. T., and Guidotti, A. (1990). Abusive stimulation of excitatory amino acid receptors: a strategy to limit neurotoxicity. Faseb 4:2789. doi: 10.1096/fasebj.4.10.2165013
Manev, H., Favaron, M., Siman, R., Guidotti, A., and Costa, E. (2010). Glutamate neurotoxicity is independent of calpain I inhibition in primary cultures of cerebellar granule cells. J. Neurochem. 57, 1288–1295. doi: 10.1111/j.1471-4159.1991.tb08292.x
McGeer, P. L., Itagaki, S., Boyes, B. E., and McGeer, E. G. (1988). Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291. doi: 10.1212/wnl.38.8.1285
Meldrum, B., and Garthwaite, J. (1990). Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol. Sci. 11, 379–387. doi: 10.1016/0165-6147(90)90184-a
Meredith, G. E., Totterdell, S., Beales, M., and Meshul, C. K. (2009). Impaired glutamate homeostasis and programmed cell death in a chronic MPTP mouse model of Parkinson’s disease. Exp. Neurol. 219, 334–340. doi: 10.1016/j.expneurol.2009.06.005
Mikiei, T., Man, K. Y., Gwang, L., Eunsung, J., Takeshi, I., and Maral, M. M. (2004). A ggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J. Biol. Chem. 279, 4625–4631. doi: 10.1074/jbc.m310994200
Mosharov, E. V., Larsen, K. E., Kanter, E., Phillips, K. A., Wilson, K., Schmitz, Y., et al. (2009). Interplay between cytosolic dopamine, calcium, and α-Synuclein causes selective death of substantia nigra neurons. Neuron 62, 218–229. doi: 10.1016/j.neuron.2009.01.033
Murphy, T. H., Miyamoto, M., Sastre, A., Schnaar, R. L., and Coyle, J. T. (1989). Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2, 1547–1558. doi: 10.1016/0896-6273(89)90043-3
Murrough, J. W., Abdallah, C. G., and Mathew, S. J. (2017). Targeting glutamate signalling in depression: progress and prospects. Nature reviews. Drug Discov. 16, 472–486. doi: 10.1038/nrd.2017.16
Nakanishi, S. (1994). Metabotropic glutamate receptors: Synaptic transmission, modulation, and plasticity. Neuron 13, 1031–1037. doi: 10.1016/0896-6273(94)90043-4
Nedergaard, M., Takano, T., and Hansen, A. J. (2002). Beyond the role of glutamate as a neurotransmitter. Nat. Rev. Neurosci. 3, 748–755. doi: 10.1038/nrn916
Niciu, M. J., Kelmendi, B., and Sanacora, G. (2012). Overview of glutamatergic neurotransmission in the nervous system. Pharmacol. Biochem. Behav. 100, 656–664. doi: 10.1016/j.pbb.2011.08.008
Nicoletti, F., Bockaert, J., Collingridge, G. L., Conn, P. J., Ferraguti, F., Schoepp, D. D., et al. (2011). Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology 60, 1017–1041. doi: 10.1016/j.neuropharm.2010.10.022
Niswender, C. M., and Conn, P. J. (2010). Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322. doi: 10.1146/annurev.pharmtox.011008.145533
Norden, D. M., Muccigrosso, M. M., and Godbout, J. P. (2015). Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 96, 29–41. doi: 10.1016/j.neuropharm.2014.10.028
Ouardouz, M., Coderre, E., Zamponi, G. W., Hameed, S., Yin, X., Trapp, B. D., et al. (2009). Glutamate receptors on myelinated spinal cord axons: II. AMPA and GluR5 receptors. Ann. Neurol. 65, 160–166. doi: 10.1002/ana.21539
Pajarillo, E., Rizor, A., Lee, J., Aschner, M., and Lee, E. (2019). The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: potential targets for neurotherapeutics. Neuropharmacology 161:107559. doi: 10.1016/j.neuropharm.2019.03.002
Palencia, G., Garcia, E., Osorio-Rico, L., Trejo-Solís, C., Escamilla-Ramírez, A., and Sotelo, J. (2015). Neuroprotective effect of thalidomide on MPTP-induced toxicity. Neurotoxicology 47, 82–87. doi: 10.1016/j.neuro.2015.02.004
Paoletti, P., and Neyton, J. (2007). NMDA receptor subunits: function and pharmacology. Curr. Opin. Pharmacol. 7, 39–47. doi: 10.1016/j.coph.2006.08.011
Perisic, T., Holsboer, F., Rein, T., and Zschocke, J. (2012). The CpG island shore of the GLT-1 gene acts as a methylation-sensitive enhancer. Glia 60, 1345–1355. doi: 10.1002/glia.22353
Pin, J. P., and Duvoisin, R. (1995). The metabotropic glutamate receptors: structure and functions. Neuropharmacology 34, 1–26. doi: 10.1016/0028-3908(94)00129-g
Pines, G., Danbolt, N. C., Bjørås, M., Zhang, Y., Bendahan, A., Eide, L., et al. (1992). Erratum: cloning and expression of a rat brain L-glutamate transporter. Nature 360, 768–768. doi: 10.1038/360768a0
Pitt, D., Werner, P., and Raine, C. S. (2000). Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 6, 67–70. doi: 10.1038/71555
Platt, S. R. (2007). The role of glutamate in central nervous system health and disease–a review. Vet. J. 173, 278–286. doi: 10.1016/j.tvjl.2005.11.007
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Prim. 3:17013.
Ramlackhansingh, A. F., Brooks, D. J., Greenwood, R. J., Bose, S. K., Turkheimer, F. E., Kinnunen, K. M., et al. (2011). Inflammation after trauma: microglial activation and traumatic brain injury. Ann. Neurol. 70, 374–383. doi: 10.1002/ana.22455
Rondard, P., and Pin, J. P. (2015). Dynamics and modulation of metabotropic glutamate receptors. Curr. Opin. Pharmacol. 20, 95–101. doi: 10.1016/j.coph.2014.12.001
Rong, X., Zhang, X., and Qu, S. (2015). A complex relative motion between hairpin loop 2 and transmembrane domain 5 in the glutamate transporter GLT-1. Int. J. Biochem. 60, 1–7. doi: 10.1016/j.biocel.2014.12.012
Rosenmund, C., Stern-Bach, Y., and Stevens, C. F. (1998). The tetrameric structure of a glutamate receptor channel. Science 280, 1596–1599. doi: 10.1126/science.280.5369.1596
Rothstein, J. D., Martin, L., Levey, A. I., Dykes-Hoberg, M., Jin, L., Wu, D., et al. (1994). Localization of neuronal and glial glutamate transporters. Neuron 13, 713–725. doi: 10.1016/0896-6273(94)90038-8
Saggu, H., Cooksey, J., Dexter, D., Wells, F. R., Lees, A., Jenner, P., et al. (2010). A selective increase in particulate superoxide dismutase activity in parkinsonian substantia nigra. J. Neurochem. 53, 692–697. doi: 10.1111/j.1471-4159.1989.tb11759.x
Sandra, H., Tobias, H. G., Neville, V., Danzer, K. M., Bastian, H., Armin, G., et al. (2011). AMPA-receptor-mediated excitatory synaptic transmission is enhanced by iron-induced α-synuclein oligomers. J. Neurochem. 117, 868–878. doi: 10.1111/j.1471-4159.2011.07254.x
Sarafian, T. A., Littlejohn, K., Yuan, S., Fernandez, C., Cilluffo, M., Koo, B. K., et al. (2017). Stimulation of synaptoneurosome glutamate release by monomeric and fibrillated α-synuclein. J. Neurosci. Res. 95, 1871–1887. doi: 10.1002/jnr.24024
Sato, H., Tamba, M., Ishii, T., and Bannai, S. (1999). Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 274, 11455–11458. doi: 10.1074/jbc.274.17.11455
Schmitt, A., Asan, E., Puschel, B., Jons, T., and Kugler, P. (1996). Expression of the glutamate transporter GLT1 in neural cells of the rat central nervous system: non-radioactive in situ hybridization and comparative immunocytochemistry. Neuroscience 71, 989–1004. doi: 10.1016/0306-4522(95)00477-7
Schoepp, D. D. (2001). Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J. Pharmacol. Exp. Ther. 299, 12–20.
Schousboe, A., Scafidi, S., Bak, L. K., Waagepetersen, H. S., and McKenna, M. C. (2014). Glutamate metabolism in the brain focusing on astrocytes. Adv. Neurobiol. 11, 13–30. doi: 10.1007/978-3-319-08894-5_2
Schubert, D., and Piasecki, D. (2001). Oxidative glutamate toxicity can be a component of the excitotoxicity cascade. J. Neurosci. 21:7455. doi: 10.1523/jneurosci.21-19-07455.2001
Shaogang, Q., and Kanner, B. I. (2008). Substrates and non-transportable analogues induce structural rearrangements at the extracellular entrance of the glial glutamate transporter GLT-1/EAAT2. J. Biol. Chem. 283, 26391–26400. doi: 10.1074/jbc.m802401200
Sheldon, A. L., and Robinson, M. B. (2007). The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem. Int. 51, 333–355. doi: 10.1016/j.neuint.2007.03.012
Shen, J., Petersen, K. F., Behar, K. L., Brown, P., Nixon, T. W., Mason, G. F., et al. (1999). Determination of the Rate of the Glutamate/Glutamine Cycle in the Human Brain by in vivo 13C NMR. Proc. Natl. Acad. Sci. U.S.A. 96, 8235–8240. doi: 10.1073/pnas.96.14.8235
Shigemoto, R., Kinoshita, A., Wada, E., Nomura, S., Ohishi, H., Takada, M., et al. (1997). Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J. Neurosci. 17, 7503–7522. doi: 10.1523/jneurosci.17-19-07503.1997
Shigemoto, R., Nomura, S., Ohishi, H., Sugihara, H., Nakanishi, S., and Mizuno, N. (1993). Immunohistochemical localization of a metabotropic glutamate receptor, mGluR5, in the rat brain. Neurosci. Lett. 163, 53–57. doi: 10.1016/0304-3940(93)90227-c
Sibson, N. R., Dhankhar, A., Mason, G. F., Rothman, D. L., Behar, K. L., and Shulman, R. G. (1998). Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc. Natl. Acad. Sci. U.S.A. 95, 316–321. doi: 10.1073/pnas.95.1.316
Siesjö, B. K., Bengtsson, F., Grampp, W., and Theander, S. (1989). Calcium, excitotoxins, and neuronal death in the brain. Ann. N. Y. Acad. Sci. 568, 234–251. doi: 10.1111/j.1749-6632.1989.tb12513.x
Simone, E. (2008). Ubiquitination of alpha-synuclein and autophagy in Parkinson’s disease. Autophagy 4, 372–374. doi: 10.4161/auto.5604
Sladeczek, F., Momiyama, A., and Takahashi, T. (1993). Presynaptic inhibitory action of a metabotropic glutamate receptor agonist on excitatory transmission in visual cortical neurons. Proc. Biol. Sci. 253, 297–303. doi: 10.1098/rspb.1993.0117
Sonsalla, P. K., Albers, D. S., and Zeevalk, G. D. (1998). Role of glutamate in neurodegeneration of dopamine neurons in several animal models of parkinsonism. Amino Acids 14, 69–74. doi: 10.1007/bf01345245
Sreeganga, C., Gilbert, G., Rafael, F. C., Schlüter, O. M., and Südhof, T. C. (2005). Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396. doi: 10.1016/j.cell.2005.09.028
Stefanie, P., Ken, A., Xiaoying, W., Lo, E. H., and Klaus, V. L. (2010). 12/15-Lipoxygenase targets neuronal mitochondria under oxidative stress. J. Neurochem. 111, 882–889. doi: 10.1111/j.1471-4159.2009.06379.x
Storck, T., Schulte, S., Hofmann, K., and Stoffel, W. (1992). Structure, expression, and functional analysis of a Na(+)-dependent glutamate/aspartate transporter from rat brain. Proc. Natl. Acad. Sci. U.S.A. 89, 10955–10959. doi: 10.1073/pnas.89.22.10955
Takeuchi, H., Jin, S., Wang, J., Zhang, G., Kawanokuchi, J., Kuno, R., et al. (2006). Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 281, 21362–21368.
Tan, S., Sagara, Y., Liu, Y., Maher, P., and Schubert, D. (1998). The regulation of reactive oxygen species production during programmed cell death. J. Cell Biol. 141, 1423–1432. doi: 10.1083/jcb.141.6.1423
Tan, S., Schubert, D., and Maher, P. (2001). Oxytosis: a novel form of programmed cell death. Curr. Top. Med. Chem. 1, 497–506. doi: 10.2174/1568026013394741
Tanaka, K., Watase, K., Manabe, T., Yamada, K., Watanabe, M., Takahashi, K., et al. (1997). Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276, 1699–1702. doi: 10.1126/science.276.5319.1699
Teichman, S., Qu, S., and Kanner, B. I. (2009). The equivalent of a thallium binding residue from an archeal homolog controls cation interactions in brain glutamate transporters. Proc. Natl. Acad. Sci. U.S.A. 106, 14297–14302. doi: 10.1073/pnas.0904625106
Torres, G. E., and Caron, G. M. G. (2003). Plasma membrane monoamine transporters: structure, regulation and function. Nat. Rev. Neurosci. 4, 13–25. doi: 10.1038/nrn1008
Vargas-Caballero, M., and Robinson, H. P. (2004). Fast and slow voltage-dependent dynamics of magnesium block in the NMDA receptor: the asymmetric trapping block model. J. Neurosci. 24, 6171–6180. doi: 10.1523/jneurosci.1380-04.2004
Wang, H., Tamba, M., Kimata, M., Sakamoto, K., Bannai, S., and Sato, H. (2003). Expression of the activity of cystine/glutamate exchange transporter, system x(c)(-), by xCT and rBAT. Biochem. Biophys. Res. Commun. 305, 611–618. doi: 10.1016/s0006-291x(03)00808-8
Wang, T., Cai, Q., Yang, W. J., Fan, H. H., Yi, J. F., and Xu, F. (2018). MicroRNA-219 alleviates glutamate-induced neurotoxicity in cultured hippocampal neurons by targeting calmodulin-dependent protein kinase II gamma. Neural Regen. Res. 13, 1216–1224. doi: 10.4103/1673-5374.235059
Wang, X., Wang, J., Lin, S., Geng, Y., Wang, J., and Jiang, B. J. J. O. E. (2008). Sp1 is involved in H 2 O 2 -induced PUMA gene expression and apoptosis in colorectal cancer cells. J. Exp. Clin. Cancer Res. 27, 44–44. doi: 10.1186/1756-9966-27-44
Watson, J. B., Hatami, A., David, H., Masliah, E., Roberts, K., Evans, C. E., et al. (2009). Alterations in corticostriatal synaptic plasticity in mice overexpressing human α-synuclein. Neuroscience 159, 501–513. doi: 10.1016/j.neuroscience.2009.01.021
Wei, Z., Tongguang, W., Zhong, P., Miller, D. S., Xuefei, W., Block, M. L., et al. (2005). Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 19, 533–542. doi: 10.1096/fj.04-2751com
Wen-Hui, H., Walters, W. M., Xiao-Mei, X., Karmally, S. A., and Bethea, J. R. (2003). Neuronal glutamate transporter EAAT4 is expressed in astrocytes. Glia 44, 13–25. doi: 10.1002/glia.10268
Wetherington, J., Serrano, G., and Dingledine, R. (2008). Astrocytes in the epileptic brain. Neuron 58, 168–178. doi: 10.1016/j.neuron.2008.04.002
Wu, D. M., Wang, S., Wen, X., Han, X. R., Wang, Y. J., Shen, M., et al. (2019). Suppression of microRNA-342-3p increases glutamate transporters and prevents dopaminergic neuron loss through activating the Wnt signaling pathway via p21-activated kinase 1 in mice with Parkinson’s disease. J. Cell. Physiol. 234, 9033–9044. doi: 10.1002/jcp.27577
Wu, X., Meng, X., Tan, F., Jiao, Z., Zhang, X., Tong, H., et al. (2019). Regulatory mechanism of miR-543-3p on GLT-1 in a mouse model of Parkinson’s disease. ACS Chem. Neurosci. 10, 1791–1800. doi: 10.1021/acschemneuro.8b00683
Xie, Y., Hou, W., Song, X., Yu, Y., Huang, J., Sun, X., et al. (2016). Ferroptosis: process and function. Cell Death Differ. 23, 369–379. doi: 10.1038/cdd.2015.158
Yang, J., Hertz, E., Zhang, X., Leinartaite, L., Lundius, E. G., Li, J., et al. (2016). Overexpression of alpha-synuclein simultaneously increases glutamate NMDA receptor phosphorylation and reduces glucocerebrosidase activity. Neurosci. Lett. 611, 51–58. doi: 10.1016/j.neulet.2015.11.023
Yang, Y., Gozen, O., Vidensky, S., Robinson, M. B., and Rothstein, J. D. (2010). Epigenetic regulation of neuron-dependent induction of astroglial synaptic protein GLT1. Glia 58, 277–286.
Yernool, D., Boudker, O., Folta-Stogniew, E., and Gouaux, E. (2003). Trimeric subunit stoichiometry of the glutamate transporters from Bacillus caldotenax and Bacillus stearothermophilus. Biochemistry 42, 12981–12988. doi: 10.1021/bi030161q
Yuan, Y., Jie, L., Chaoliang, W., Kaitao, L., Wenjun, X., Yanru, W., et al. (2008). Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc. Res. 77, 432–441. doi: 10.1093/cvr/cvm047
Yunlong, Z., Xiuping, Z., and Shaogang, Q. (2014). Cysteine mutagenesis reveals alternate proximity between transmembrane domain 2 and hairpin loop 1 of the glutamate transporter EAAT1. Amino Acids 46, 1697–1705. doi: 10.1007/s00726-014-1731-1
Zerangue, N., and Kavanaugh, M. P. (1996). Flux coupling in a neuronal glutamate transporter. Nature 383, 634–637. doi: 10.1038/383634a0
Zhang, X., and Qu, S. (2012). The accessibility in the external part of the TM5 of the glutamate transporter EAAT1 is conformationally sensitive during the transport cycle. PLoS One 7:e30961. doi: 10.1371/journal.pone.0030961
Zhang, Y., He, X., Meng, X., Wu, X., Tong, H., Zhang, X., et al. (2017). Regulation of glutamate transporter trafficking by Nedd4-2 in a Parkinson’s disease model. Cell Death Dis. 8:e2574. doi: 10.1038/cddis.2016.454
Zhang, Y., Tan, F., Xu, P., and Qu, S. (2016). Recent advance in the relationship between excitatory amino acid transporters and Parkinson’s disease. Neural Plast. 2016:8941327.
Zou, J. Y., and Crews, F. T. (2005). TNFα potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NFκB inhibition. Brain Res. 1034, 11–24. doi: 10.1016/j.brainres.2004.11.014
Keywords: glutamate, neurotransmitter, excitotoxicity, oxidative glutamate toxicity, immunoexcitotoxicity
Citation: Wang J, Wang F, Mai D and Qu S (2020) Molecular Mechanisms of Glutamate Toxicity in Parkinson’s Disease. Front. Neurosci. 14:585584. doi: 10.3389/fnins.2020.585584
Received: 31 July 2020; Accepted: 28 October 2020;
Published: 26 November 2020.
Edited by:
Rodrigo Franco, University of Nebraska-Lincoln, United StatesReviewed by:
Luciene Bruno Vieira, Federal University of Minas Gerais, BrazilCopyright © 2020 Wang, Wang, Mai and Qu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shaogang Qu, c2dxOTUyOEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.