 María Teresa Flores-Dorantes
María Teresa Flores-Dorantes Yael Efren Díaz-López
Yael Efren Díaz-López Ruth Gutiérrez-Aguilar
Ruth Gutiérrez-Aguilar- 1Laboratorio de Biología Molecular y Farmacogenómica, Centro de Investigación de Ciencia y Tecnología Aplicada de Tabasco, División Académica de Ciencias Básicas, Universidad Juárez Autónoma de Tabasco, Villahermosa, Mexico
- 2Laboratorio de Enfermedades Metabólicas: Obesidad y Diabetes, Hospital Infantil de México “Federico Gómez,” Mexico City, Mexico
- 3División de Investigación, Facultad de Medicina, Universidad Nacional Autónoma de México (UNAM), Mexico City, Mexico
Obesity is a multifactorial disease in which environmental conditions and several genes play an important role in the development of this disease. Obesity is associated with neurodegenerative diseases (Alzheimer, Parkinson, and Huntington diseases) and with neurodevelopmental diseases (autism disorder, schizophrenia, and fragile X syndrome). Some of the environmental conditions that lead to obesity are physical activity, alcohol consumption, socioeconomic status, parent feeding behavior, and diet. Interestingly, some of these environmental conditions are shared with neurodegenerative and neurodevelopmental diseases. Obesity impairs neurodevelopment abilities as memory and fine-motor skills. Moreover, maternal obesity affects the cognitive function and mental health of the offspring. The common biological mechanisms involved in obesity and neurodegenerative/neurodevelopmental diseases are insulin resistance, pro-inflammatory cytokines, and oxidative damage, among others, leading to impaired brain development or cell death. Obesogenic environmental conditions are not the only factors that influence neurodegenerative and neurodevelopmental diseases. In fact, several genes implicated in the leptin–melanocortin pathway (LEP, LEPR, POMC, BDNF, MC4R, PCSK1, SIM1, BDNF, TrkB, etc.) are associated with obesity and neurodegenerative and neurodevelopmental diseases. Moreover, in the last decades, the discovery of new genes associated with obesity (FTO, NRXN3, NPC1, NEGR1, MTCH2, GNPDA2, among others) and with neurodegenerative or neurodevelopmental diseases (APOE, CD38, SIRT1, TNFα, PAI-1, TREM2, SYT4, FMR1, TET3, among others) had opened new pathways to comprehend the common mechanisms involved in these diseases. In conclusion, the obesogenic environmental conditions, the genes, and the interaction gene–environment would lead to a better understanding of the etiology of these diseases.
Introduction
Obesity is an excess of fat body mass that may decrement health. Obesity is caused by an energy imbalance due to an excess of food intake and less physical activity. The World Health Organization (WHO) has reported that worldwide obesity prevalence has tripled since 1975. In 2016, the prevalence of overweight and obesity was over 1.9 billion adults, 39% overweight, and 13% obese (World Health Organization [WHO], 2020). Body mass index (BMI), given by dividing the weight by height (kg/m2), is used for classifying people as obese (BMI above 30 kg/m2).
The obesity epidemic has dramatically increased in the last decades, and it goes in parallel with the change in our environment by unhealthy diet (sugar-sweetened beverages, fried foods, etc.), physical inactivity, sedentary lifestyle, and poor sleeping (Hruby et al., 2016).
In addition, genetics is also implicated in the development of obesity, explaining over 40% of the heritability of this disease. Several studies have demonstrated that genes in the melanocortin system control the energy balance [reviewed in Xu et al. (2011) and Hill and Faulkner (2017)]. Mutations in these genes, such as melanocortin 4 receptor (MC4R), have been associated with monogenic obesity (Farooqi et al., 2000). However, studies about common or polygenic obesity have revealed over 900 genetic variants associated with obesity by the approach of genome-wide association studies (GWAS) (Locke et al., 2015; Yengo et al., 2018).
Moreover, obesity is a risk factor to develop several neurological consequences (O’Brien et al., 2017), such as dementia and Alzheimer disease [AD, reviewed in Arnoldussen et al. (2014) and Anjum et al. (2018)]. Obesity is a comorbidity for psychiatric disorders that may influence behavior, cognition, and mood, suggesting that common biological pathways in the central nervous system (CNS) are implicated in obesity and psychiatric disorders (Proulx and Seeley, 2005).
In this journal’s issue, a compendium of papers focused on obesogenic environmental conditions that affect neurodevelopment and neurodegeneration was collected. In this review, we will address aspects, such as obesogenic environment leading to develop obesity. In addition, the principal genes that have been associated with obesity and that are implicated in neurodegenerative diseases (NDgDs) or neurodevelopmental diseases (NDvDs) as well as the new genes associated with NDgD or NDvD are described.
Obesogenic Environment
To prove that an obesogenic environment could play an important role in developing obesity, dogs exposed to an obesogenic environment (owned by obese people) presented a higher prevalence of obesity compared to dogs with lean owners (Mason, 1970). Then, obesogenic environmental conditions have been part of the increment on this disease.
Two important components, the environment (nurture) and the genes (nature), are risk factors to develop common obesity. These two components are frequently studied independently; however, the interaction between gene and environment can increase the susceptibility to develop the disease. The genes are part of nature and have been defined as the nucleotides that form our DNA (deoxyribonucleic acid). DNA differs in some base pairs among individuals, giving genetic variation and individual differences in a trait (Wood, 2019). However, environment is part of nurture; that is, a non-genetic thing that could modify a trait (Wood, 2019).
The United Kingdom Biobank (UK Biobank) has collected data of close to half a million United Kingdom citizens of middle to old age about lifestyle factors and genetics (Sudlow et al., 2015). More than 130 lifestyle factors, such as socioeconomic status, general health, mental health, sleep, physical activity, alcohol consumption, smoking, diet, the genetic risk score (GRS), among others, were analyzed to study the interaction between gene and environment leading to obesity. Gene–environment interaction is defined as the response of an individual to environmental stimuli based on his genotype. In other words, the genes confer susceptibility, but the environment could influence and modify the genotype (Reddon et al., 2016).
Analysis of the UK Biobank database showed that the most significant environmental factors that could influence the development of obesity, in the presence of genetic risk variants, were physical activity, alcohol consumption, and socioeconomic status (Sudlow et al., 2015; Rask-Andersen et al., 2017). Moreover, parent feeding behavior and diet had also been implicated in the development of obesity (Dalle Molle et al., 2017).
Physical Activity
When it comes to obesogenic environment, we need to think about the main arms of the energy balance, which are energy expenditure and energy intake. With respect to lower energy expenditure, a sedentary lifestyle, or physical inactivity, due to prolonged watching TV hours, interacts with the genetic predisposition causing the development of obesity (Qi et al., 2012b). A study analyzing 20,000 men demonstrated that having a physically active lifestyle reduces 40% of the genetic predisposition for obesity by analyzing 12 variants associated with obesity (Li et al., 2010). A meta-analysis of more than 110,000 individuals supported the previous results, indicating that the physical activity counterbalances the genetic predisposition to develop obesity (Ahmad et al., 2013).
Alcohol Consumption
Alcohol consumption was another gene–environment associated with BMI (Rask-Andersen et al., 2017). In fact, alcohol consumption might reduce the effect of the obesity genetic variants by reducing the BMI. This is consistent with data showing that alcoholic patients present lower physical activity (Liangpunsakul et al., 2010) and a lower BMI (Addolorato, 2000) as a consequence of an increase in lipolysis and disorders of lipid metabolism (Steiner and Lang, 2017). In fact, it has been demonstrated that chronic alcohol consumption could provoke lipodystrophy in rats, triggered by a disturbance on lipogenesis (Zhang et al., 2015). A higher lipolysis and fatty acid release will provoke the transportation of the fatty acids to the liver, leading to their accumulation and provoking hepatic steatosis over time (Kema et al., 2015).
Socioeconomic Status
Several studies have analyzed the relationship between socioeconomic status and obesity. It has been reported that obesity prevalence increases with higher deprivation levels (National Obesity Observatory, 2012) due to a worse diet (Darmon and Drewnowski, 2008) and less physical activity (Giles-Corti, 2002). Interestingly, in rich countries, the obesity prevalence was higher among poor people and did not change among the wealthiest. However, in poor countries, obesity and overweight is higher in the upper class (Templin et al., 2019). In developing countries, such as Mexico, a new middle class (poor people who became wealthier) has the most risk to become obese (Levasseur, 2015).
To address how education could influence BMI, a study with siblings sharing the same environment was performed. This study demonstrated that higher education levels correlate with a lower BMI (Kim, 2016). Educational level could affect the selection and purchase of healthy food due to the knowledge of healthy food (Bhurosy and Jeewon, 2014). Another study performed in children demonstrated that severe obesity increased with a lower household head education and lower urbanization level (Ogden et al., 2018).
Moreover, many “environmental layers” could affect obesity predisposition, such as intrauterine environment, mother–child interaction, food/community environment, parent feeding behavior, among others, representing the biological or social influence for any person to develop the disease (Dalle Molle et al., 2017). Of these layers, we will briefly describe parent feeding behavior and food (diet) as part of the energy intake, an important arm of the energy balance.
Parent Feeding Behavior
One of the environmental layers influencing child appetite is parent feeding behavior. For example, giving food as a reward of good behavior is associated with a higher intake of unhealthy foods and beverages (Dalle Molle et al., 2017).
A poor eating self-regulation in children has been associated with higher body weight (Hughes and Frazier-Wood, 2016). Picky eating is common in preschool children, which leads to parental anxiety and family conflicts (Kumar et al., 2018). Authoritative parenting has a positive correlation with non-picky eating in their toddler, suggesting that this parent feeding behavior could overcome the feeding difficulties (Podlesak et al., 2017). Lower weight has been predicted with mothers who promote positive child body image; however, feeding practices of pushing to eat have been associated with weight gain (Damiano et al., 2016). Then, nurture plays an important role in child eating behaviors and gene–environment interaction to shape child appetitive traits (Wood, 2019).
Diet
Diet is one of the main environmental factors that influence obesity development. An accelerated lifestyle has changed dietary habits, inciting people to look for ready-to-go food, instead of preparing their food at home. In fact, a United States National Nutritional Survey (from 1965 to 2008) reported that home-consumed diet has decreased 23% and most of the Americans have their calorie intake from processed foods purchased in restaurants or grocery stores (Smith et al., 2013). A study in the United Kingdom demonstrated that consumption of out-of-home (restaurants, cafes, and takeaways) meals has a higher energy intake (75–104 kcal per day) compared to those of people who rarely consume them (Goffe et al., 2017). On the other hand, frequent cooking at home has been associated with better healthy eating index, regardless low or high income (Wolfson et al., 2020).
Epidemiological studies have shown that in the period of 2005–2010, 14% of American young adults obtained their caloric needs from sugar-sweetened beverages (Ervin and Ogden, 2013). In Mexico, a country with one of the world’s highest obesity prevalence, 17.5% of children and 19% of the adults obtained their daily caloric intake from sweetened beverages (Stern et al., 2014).
A systematic review of 32 studies reported a positive association between sugar-sweetened beverage consumption and the obesity risk (Bucher Della Torre et al., 2016). Moreover, a meta-analysis showed that consumption of soft drinks has increased in the last decades, and 75% of beverages and food contained added sugar. Then, drinking sugar-sweetened beverages increases the risk of obesity, diabetes, and metabolic syndrome (Bray and Popkin, 2014). In addition, obesity-genetic predisposition is associated with the positive correlation between soft drink and fried food consumption and adiposity traits (Qi et al., 2014; Olsen et al., 2016). A review suggested that eating fried foods at least four times per week gives a higher risk on developing obesity and other chronic diseases, such as type 2 diabetes (T2D) and hypertension, leading to coronary artery disease (Gadiraju et al., 2015).
Another study showed that eating more fruits and vegetables and avoiding red and processed meats could improve cardiometabolic profiles (Schwedhelm et al., 2019).
For this reason, WHO has published dietary recommendations, suggesting a higher intake of fruits, vegetables, legumes, nuts, and whole grains, limiting to less than 10% of free sugars and 30% from fats of the total intake (World Health Organization [WHO]., 2018). Even though these recommendations have been published, it has been shown that there are several barriers to eat healthy food, such as household income and cooking and eating behaviors (Wolfson et al., 2019).
A recent study demonstrated that diets in different populations are “unhealthy, unsustainable, and unequitable” (Fanzo and Davis, 2019). As abovementioned, people are eating “unhealthy,” with high intake of sugar and fats. The diet is a link between humans and the environment. The production of plants and animals for human consumption is changing the environment, provoking biodiversity loss and becoming “unsustainable.” Finally, socioeconomic status marginalizes some people to access healthy food, the diet becoming “inequitable.”
Therefore, interventions to control obesity have to consider several environmental factors that had been associated with obesity. For example, in Latin America, some of the intervention programs have implemented elevated taxes for sugar-based foods, marketing control, and consumer education to select healthier food (Popkin and Reardon, 2018). The global network, International Network for Food and Obesity Monitoring and Action Support (INFORMAS), implemented in Mexico nine different actions to reduce obesity: provision and promotion of healthy food, increase the offer of healthy food, a comprehensive package, combat obesity using the money obtained from the taxes over the sugar-sweetened beverages, among others (Nieto et al., 2019).
As addressed above, environment has many different edges, so it cannot be simply studied. For example, using the new technologies such as sequencing, it could take decades, trying to sequence the whole environment. Therefore, it has been an easier option to sequence the human genome than the environment (Bogardus and Swinburn, 2017). Sequencing the human genome will help to understand how genetic mutations could provoke a disease (monogenetic) or how different genetic variants give susceptibility to develop the disease (polygenetic).
Genetics of Obesity
When we talk about genetics, we have to define heritability as “the proportion of observed variation in a particular trait that can be attributed to inherited genetic factors” (Merriam-Webster Dictionary, 2020). A systematic review of obesity heritability, based on the BMI of twins studies (140,525 twins) and family studies (42,968 family members), revealed that BMI heritability ranged from 47 to 90%. In addition, they demonstrated that the genetic contribution to BMI was higher during childhood than adulthood (Elks et al., 2012).
Obesity is classified in three different ways: monogenetic, polygenetic, or syndromic obesity. In this review, we will only focus on explaining monogenetic and polygenetic obesity.
Monogenic Obesity
Monogenetic obesity is caused by mutations (not common changes in the DNA) of one gene, typically causing severe early-onset obesity. This type of obesity is rare, and approximately 7.3% of the severe early-onset obesity is affected in one gene (Kleinendorst et al., 2018). Some of the genes associated with monogenetic severe obesity are: leptin (LEP), leptin receptor (LEPR), proopiomelanocortin (POMC), MC4R, preproconvertase 1 (PCSK1), single minded 1 (SIM1), brain-derived neurotrophic factor (BDNF), and tyrosine kinase receptor tropomycin-related kinase B (TrkB) (Ramachandrappa and Farooqi, 2011).
Most of the proteins involved in the monogenic obesity are involved in the leptin–melanocortin signaling pathway. This pathway is important in controlling the energy balance in the hypothalamus by coordinating the energy intake and the energy expenditure (Morton et al., 2014; van der Klaauw and Farooqi, 2015). Leptin stimulates neurons expressing POMC, producing melanocortin peptides that will bind the MC4R controlling the energy balance. Mutations in POMC and MC4R genes lead to severe obesity due to hyperphagia (Krude et al., 1998; Farooqi et al., 2003). Mutations in MC4R are the most common in monogenetic obesity, present in more than 5% of childhood obesity (Farooqi et al., 2000; Vaisse et al., 2000). Mutations on PCSK1 lead to a misprocessing of melanocortin peptides, then leading to obesity and abnormalities of glucose homeostasis and adrenal function (O’Rahilly et al., 1995; Jackson et al., 1997). The transcription factor SIM1 causes severe obesity due to hyperphagia and reduces the paraventricular nucleus (PVN) of the hypothalamus (Michaud, 2001; Ramachandrappa et al., 2013). BDNF is also expressed in the hypothalamus, and it is downstream MC4R signaling promoting anorectic signal and locomotor activity (Kernie, 2000; Lebrun et al., 2006). Mutations in BDNF and its receptor, TrkB, are associated with obesity and hyperphagia either in humans or mice (Xu et al., 2003; Yeo et al., 2004).
Details of the function of these genes will be presented in the section Monogenetic Obesity Genes.
Polygenetic Obesity
Common obesity is characterized to be associated with several variants or polymorphism of different genes, polygenetic. These polymorphisms or single nucleotide variants (SNVs) are changes of one nucleotide of the DNA sequence and are common variants in the population.
Fifteen years ago, technology had a great impact on genetics by creating new molecular biology methods able to analyze several variants at the same time. In addition, the formation of big consortia, such as the UK Biobank or Genetic Investigation of ANthropometric Traits (GIANT) consortium, helped increase the sample size for epidemiological or genetic studies.
The GWAS were created based on analyzing thousands or millions of variants (covering more than 75% of the human genome) at the same time in thousands of individuals (case-control cohorts). The GWAS are hypothesis-free studies, meaning that we ignore the biological effects of the variants analyzed.
In 2007, FTO was the first gene associated with obesity discovered by the GWAS approach (Frayling, 2007). Then in 2009, the first three obesity GWAS were published, reporting 16 loci associated with obesity (Meyre et al., 2009; Thorleifsson et al., 2009; Willer et al., 2009). Since then, several meta-analyses had identified different variants associated with obesity in different populations (Locke et al., 2015; Yengo et al., 2018) or with obesity-related traits, such as body fat distribution (Pulit et al., 2019) in more than 300,000 or 700,000 individuals. Now, 941 near-independent variants have been associated with BMI, and 346 loci were associated with body fat distribution (Yengo et al., 2018; Pulit et al., 2019).
Interestingly, many of the new genes associated with obesity were found to be involved in neurogenesis, in the development of the CNS, in pathways such as appetite and food intake regulation (Locke et al., 2015; Yengo et al., 2018). In addition, a human compendium of 578 genes associated with body weight and food intake and expressed in the brain was published by collecting data from several GWAS and some candidate genes (Ignatieva et al., 2016).
Some of the genes associated with obesity discovered by GWAS approach are MC4R, POMC, FTO, NRXN3, NPC1, NEGR1, GNPDA2, MTCH2, ETV5, among others. Some of them are related to synaptic function and neurotransmitter signaling (NEGR1, NRXN3, CADM2, GRID1, ELAVL4, and SCG3) and energy homeostasis (POMC, MC4R, BDNF, ETV5, HNF4G, and TLR4) (Locke et al., 2015).
The GWAS opened a new gate to understand the biological pathways involved in obesity. This hypothesis-free approach reveals new variants that are associated with obesity, without knowing their target gene and the effect that could have over it. Then, the challenge now is to go from genomics to physiology, describing the function of the new genes associated with obesity (Gutierrez-Aguilar et al., 2012, 2014).
Polygenic variants have a modest effect. Then, GRS was created to represent “the number of risk variants across all the genome,” meaning that the higher the GRS, the higher susceptibility to develop obesity (Loos and Janssens, 2017). Several efforts have been performed to obtain an algorithm that could predict the risk for developing obesity (Loos and Janssens, 2017; Khera et al., 2019). Finally, Khera et al. (2019) published a genome-wide polygenic score that integrates all available common variants for obesity into a quantitative measure of inherited susceptibility. This score is able to predict the risk of developing obesity, as well as differences in weight during childhood (Khera et al., 2019).
In the last 15 years of the genomic era, hundreds of variants have been associated with obesity. Ultimately, obesity genetic risk prediction could be used to know the individual’s genetic susceptibility to develop the disease. Therefore, as a principle of precision medicine, strategies and treatments could be personalized for each individual in the future, known as pharmacogenomics. However, biology is now the missing piece for solving the obesity puzzle. Many years may take to be able to understand the effect of each of the variants associated with obesity and to understand their function and their influence in the biology of obesity.
More details of the function of these genes will be presented in section Polygenetic Obesity Genes.
Gene–Environment Interaction Leading to Obesity
Many studies have reported that the predisposition to develop obesity is by the influence of either the environment or gene variants. In this section, we will discuss individual variants or the GRS (adding the effect of several gene variants associated with obesity) and their association with some obesity traits, as well as some environmental factors.
Individual variants are studied to simplify the understanding of the interaction between gene–environment. An example of this is the interaction of two well-known obesity variants (FTO rs9939609 and MC4R rs17782313) with lifestyle measured. The study demonstrated that changes in lifestyle by implementing physical activity and adherence to a Mediterranean diet can modulate the obesity risk conferred by FTO and MC4R variants (Corella et al., 2012). In addition, gene variants could have an effect on weight loss depending on the diet. FTO variant is associated with change in appetite or abdominal fat distribution when exposed to high or low protein diet (Zhang X. et al., 2012; Huang et al., 2014).
The BMI heritability in twin and family studies is 47–90% (Elks et al., 2012). However, an obesity GWAS meta-analysis, analyzing 941 gene variants, explained that those variants represented only [∼6% of the variance of BMI (Yengo et al., 2018)], suggesting that all those variants associated with obesity have modest effects. Then, where is the rest of the heritability? The answer to this question has been called the “missing heritability” that could partly be explained by the influence of the environment over the genes. Several reviews have summarized the state of the art of the interaction between genes and environment in obesity, eating behavior, and diet, among others (Dalle Molle et al., 2017; Heianza and Qi, 2017; Heianza and Qi, 2019; Li and Qi, 2019).
Genetic risk score takes into account several genetic variants that could explain a trait. For example, some of the lifestyle factors that may modify the obesity GRS are sugar-sweetened beverages, where over 30 obesity gene variants were analyzed, and demonstrated interaction between genes and environment (Qi et al., 2012a; Brunkwall et al., 2016). As mentioned above, the UK Biobank data analysis demonstrated the relationship between genetic risk and environment (Sudlow et al., 2015; Rask-Andersen et al., 2017; Tyrrell et al., 2017; Nagpal et al., 2018). For example, the analysis of the GRS of 94 genetic variants were associated with physical activity, sedentary lifestyle, and socioeconomic status (Rask-Andersen et al., 2017).
Therefore, our genes confer susceptibility to develop obesity; however, we can modify the environment to reduce the risk. Then, improving adherence to healthy dietary patterns had a benefit, even in the presence of an obesity genetic risk (Wang et al., 2018c).
Then, gene–environment interactions are explaining part of the missing heritability; however, more research is needed to better understand how the genes give susceptibility to develop a disease in the presence of an environment that enhances the trait. Interestingly, some of the environmental factors associated with obesity are also risk factors to develop NDgD or NDvD, as described below.
Neurodegenerative and Neurodevelopmental Diseases
Neurodegenerative diseases and NDvDs are multifactorial disorders where environment and genetics play an important role for their development (Cardoso et al., 2019; Dunn et al., 2019).
Neuronal loss leads to neurodegeneration due to accumulation of abnormal proteins in the brain. Dementia could be described as the impairment of cognitive function, and the most common NDgDs are AD (60–70% of the cases) and Parkinson’s disease (PD).
Alzheimer disease is characterized by an abnormal accumulation and deposition of β-amyloid peptide on the amyloid plaque (Haass and Selkoe, 2007). In addition, TAU protein is hyperphosphorylated, producing paired helical filaments integrated in neurofibrillary tangles (Zenaro et al., 2017). On the other hand, PD is characterized by postural instability, resting tremor, stiffness, bradykinesia, caused by a progressive loss of dopaminergic neurons, and aggregates of α-synuclein (Poewe et al., 2017). Another NDgD is Huntington’s disease (HD) characterized by progressive brain disorder leading to movement, loss of cognitive conditions, emotional problems, and psychiatric symptoms (Snowden, 2017).
It has been demonstrated that obesity is a risk factor for mild cognitive impairment, independent of age (Elias et al., 2005; Hassing et al., 2010). Moreover, a meta-analysis reported the association between obesity and neurological disorders, demonstrating that obesity doubles the risk for AD (Anstey et al., 2011), and diabetes also confers risk (Profenno et al., 2010). Metabolic syndrome (obesity, T2D, hypertension, hypercholesterolemia, and hypertriglyceridemia) constitutes a risk factor for developing PD (Nam et al., 2018).
On the other hand, NDvDs are a group of conditions that affect the development of the nervous system, leading to learning disability, affecting self-control, emotions, and memory, as well as impairment in personal, occupational, and social functioning (Maher et al., 2017). Obesity has also been associated with neurodevelopmental disorders, such as autism (Criado et al., 2018) and schizophrenia (SCZ) (An et al., 2018).
Several authors have already reviewed the implications of obesity, and its metabolic dysfunctions could contribute to neurological consequences (O’Brien et al., 2017), as well as the impact on NDgD (Mazon et al., 2017). Briefly, overweight and obesity provoke metabolic changes that damage the CNS by altering synaptic plasticity and leading to neural death by either cell necrosis or apoptosis (Mazon et al., 2017). More details will be explained in Section “Common Biological Mechanisms Between Obesity and Neurodegenerative and Neurodevelopmental Diseases.” It is evident that obesity provokes metabolic and physiological changes that lead to the development of NDgD or NDvD.
Environmental Factors Influencing Neurodegenerative or Neurodevelopmental Diseases
Some of the environmental factors that have been implicated in a cognitive decline, dementia, or NDgD are physical activity, diet, and stress (lack of sleep) (Zhao et al., 2018; Gubert et al., 2020). In particular, for PD, lifestyle factors such as heavy alcohol consumption and cigarette were associated with the risk of developing the disease (Paul et al., 2019). Moreover, other illicit substances, heavy metals (iron, copper, manganese, lead, and mercury), pesticides, are other environmental factors that could provoke PD (Ball et al., 2019).
It has been described that obesity could lead to poorer neurodevelopment abilities, such as attention, inhibitory control, working memory, problem-solving, and fine motor skills (Mina et al., 2017). These abilities are impaired in NDvDs such as autism spectrum disorders (ASD), attention-deficit hyperactivity disorder (ADHD), and diseases associated with obesity (Manu et al., 2015; Criado et al., 2018; Hanć and Cortese, 2018).
A lower intelligence quotient score in childhood is associated with weight gain and obesity in later adulthood (Chandola et al., 2006; Yu et al., 2010). Moreover, midlife obesity is associated with lower cognitive ability, memory, verbal ability, and spatial abilities in late life (Dahl et al., 2010; Hassing et al., 2010). During early-life years, exposure to different factors such as education level, food deficiency, learning ability, family-related factors could increase the risk to develop cognitive impairment and dementia in advance age (Wang X. J. et al., 2019).
Maternal obesity, an environmental factor for the offspring, affects the cognitive function and mental health of the offspring. Maternal obesity could influence memory, learning, and some NDvDs, such as ADHD and ASD, as well as NDgD (Contu and Hawkes, 2017; Edlow, 2017). In addition, offspring of obese mothers present behavioral abnormalities, decreased sociability, anxiety, and feeding disorders (Edlow, 2017). In fact, maternal obesity increases the risk for intellectual disability in 1.3–3.6-fold, ADHD in 1.6–2.8-fold, and 2-fold in the difficulty of regulating emotions (Edlow, 2017).
Another NDvD is SCZ; its risk factors to develop it include obesity, poor diet, physical activity, genetic vulnerability, stress, environmental toxins, among others (Debnath et al., 2015).
Thus, obesity and NDgD/NDvD share the same environmental risk factors (physical activity, diet, socioeconomic status, stress, among others), suggesting that all these factors could modulate common biological mechanisms, regulating the CNS. In the next section, we address the common biological mechanism shared by obesity and NDgD/NDvD.
Neurodegenerative disease or NDvD development is influenced by exposure to environmental factors and by the genetic background of each person. Some of the genes associated with NDgD or NDvD are described in Section “Genes Associated With Neurodegenerative and Neurodevelopmental Diseases.”
Common Biological Mechanisms Between Obesity and Neurodegenerative and Neurodevelopmental Diseases
Several authors have reviewed the plausible common biological mechanisms between obesity and neurodegeneration/neurodevelopment, which are briefly explained below (Mazon et al., 2017; O’Brien et al., 2017; Pugazhenthi et al., 2017).
Obesity is a consequence of an excessive energy intake, provoking a hypertrophy of the adipose tissue. Consequently, dysfunction and inflammation of the adipose tissue trigger impaired insulin signaling, altered secretion of adipokines and cytokines, compromised triglyceride storage, and liberation of free fatty acids. The high levels of free fatty acids contribute to insulin resistance (IR) (Schneeberger et al., 2014), which has been linked to neurocognitive dysfunction (Stoeckel et al., 2016). IR and chronic hyperglycemia induce oxidative stress and inflammatory responses, provoking neuronal death and impairing cognitive processes (Treviño et al., 2015). Pro-inflammatory adipokines, including interleukin (IL)-1, IL-6, IL-1β, Tumor Necrosis Factor-Alpha (TNFα), plasminogen activator inhibitors (PAI-1), C-reactive protein, and leptin, have been proposed to be part of the neuroinflammation triggering neurodegeneration [reviewed in Arnoldussen et al. (2014)].
Leptin, a hormone secreted by the adipose tissue, reaches the hypothalamus (to control energy balance) and the hippocampus, regions involved in processes such as synaptic plasticity, memory, and cognition (Irving and Harvey, 2014). Besides the pro-inflammatory responses, other common biological features between obesity and neurodegeneration are oxidative damage, energy metabolism failures, and mitochondrial and neurotransmission systems dysfunction, ultimately leading to cell death [reviewed in Mazon et al. (2017)].
As mentioned above, maternal obesity exposes the offspring to inflammatory cytokines, which has been associated with low birth weight and premature birth, but interestingly is also associated with neural development, leading to disorders like SCZ, ADHD, and ASD (Buka et al., 2001; Donev and Thome, 2010; Blackmore et al., 2011; Angelidou et al., 2012). In fact, inflammatory mediators cross the blood–placenta barrier influencing fetal development. Then, maternal obesity produces a raise on inflammatory markers in the hippocampus of the offspring (Sullivan et al., 2015).
In addition, inflammation regulates serotonin function. Interferon alpha-treated rats showed decreased serotonergic axons in the amygdala and the ventral media prefrontal cortex (Ishikawa et al., 2007). Moreover, maternal high-fat diet consumption diminished the serotonin synthesis, leading to anxiety behaviors in the offspring (Sullivan et al., 2010).
Another reason how obesity affects the brain development is the fact that glucose can cross the blood–placenta barrier. However, the maternal insulin does not cross it, leading to insulin production by the baby. This hormone acts as a growth factor, then hyperinsulinemia in the prenatal period might alter brain development (Stachowiak et al., 2013). On the other hand, in ASD children, higher leptin levels were detected compared to healthy children (Ashwood et al., 2008).
Genes Associated With Obesity and Neurodegenerative and Neurodevelopmental Diseases
As we previously mentioned, many genes are involved in the susceptibility to develop obesity and NDgD/NDvD. However, in the next section, we will describe some of the most important genes associated with these diseases, their function, and the plausible mechanism overlapping among these complex diseases.
Genes Associated With Obesity and Its Impact on Neurodegenerative or Neurodevelopmental Diseases
Some of the genes that code for the leptin–melanocortin pathway proteins are associated with monogenetic or polygenic obesity.
In this section, we will briefly describe the genes involved in this pathway and that have been associated with monogenic obesity, as well as their effect in developing NDgD and NDvD (Table 1). These genes are LEP, LEPR, POMC, CART, NPY, MC4R, PCSK1, SIM1, BDNF, and TrKB.
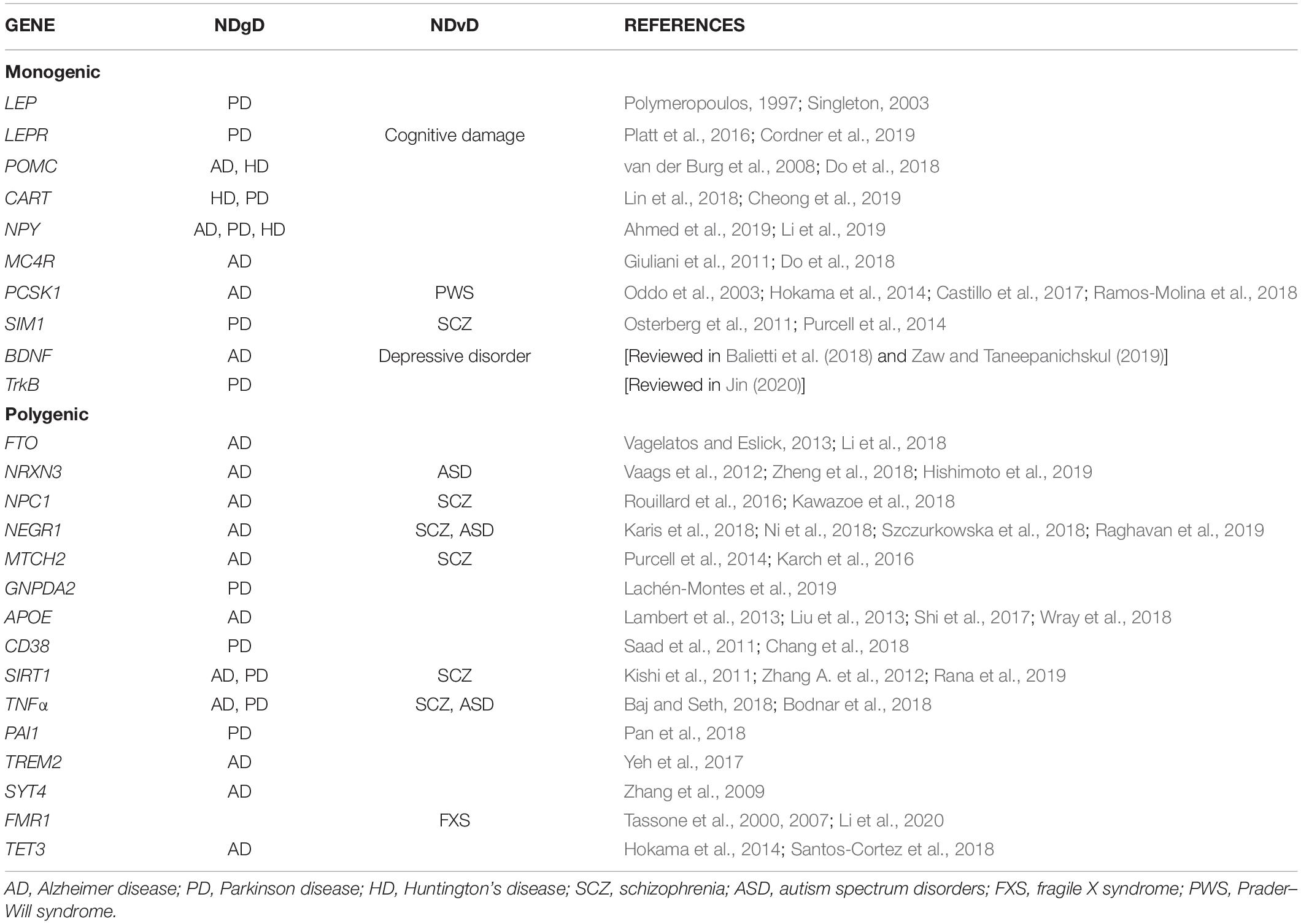
Table 1. Genes associated with obesity and NDgD or NDvD.
Monogenetic Obesity Genes
Leptin and leptin receptor (LEP and LEPR)
Leptin is a hormone expressed and secreted by adipose tissue (Masuzaki et al., 1995). It is a cytokine/adipokine essential for the regulation of energy balance through feeding behavior and energy expenditure (Zhang et al., 1994). Leptin is an anorexigenic hormone, stimulating the expression of anorexigenic neuropeptides (POMC and α-MSH) and inhibiting the expression of orexigenic neuropeptides (NPY and AGRP) (Jéquier, 2006). The desire to eat is reduced by leptin signals by binding to the leptin receptor in the arcuate nucleus of the hypothalamus and stimulating thermogenesis and satiety.
Leptin receptor β (LEPRβ) is expressed in the neocortex, hypothalamus, medulla, and cerebellum (Burguera et al., 2000). LEPRβ activates the Janus kinase 2 and signal transducer and activator of transcription 3 (JAK-2/STAT3) pathway (Baumann et al., 1996; Bjørbaek and Kahn, 2004; Arnoldussen et al., 2014). This signaling pathway is associated with multiple physiological and pathological regulation processes. It can induce the expression of high-mobility group box 1 (HMGB1), which promotes the release of cytokines as Tumor Necrosis Factor-Alpha (TNFα), inducing the inflammatory reaction (Liu et al., 2007).
Many mutations in the LEP and LEPR genes have a major influence on metabolism, leading to obesity (Ramachandrappa and Farooqi, 2011; Wasim et al., 2016). Both LEPR- and LEP-deficient individuals exhibit rapid weight gain in the first few months of life, with endocrine abnormalities and severe hyperphagia (Saeed et al., 2014).
Leptin plasma levels are highly correlated to IR, adipocyte number, and fat mass (Friedman and Halaas, 1998). However, in spite of high leptin levels during obesity, a failure in the essential leptin mechanisms (reduction in feeding behavior and increased energy expenditure) is present (Frederich et al., 1995) due to leptin resistance (Morris and Rui, 2009).
In the last few years, many studies have shown the role of leptin and leptin receptor in neurological and NDgD. In vitro studies showed that inhibition of the JAK/STAT3 pathway protects against α-synuclein (α-SYN), induced neuroinflammation and dopaminergic neurodegeneration (Qin et al., 2016). α-SYN is a presynaptic protein that plays a central role in the pathophysiology of PD through neuroinflammatory response (Roodveldt et al., 2008). Genetic missense mutations in α-SYN gene have been associated with familial forms of PD (Polymeropoulos, 1997; Singleton, 2003).
Moreover, leptin can reduce TAU phosphorylation triggering neuroprotective effects. A leptin-resistant mouse model (Lepr db/db) developed obesity and diabetic phenotypes showing high levels of TAU protein phosphorylation. TAU aggregation and hyperphosphorylation trigger cytotoxicity and development of NDgD. This phosphorylation increases the risk for developing dementia (Platt et al., 2016).
Recently, a study demonstrated that high-fat diet rat offspring showed cognitive damage and lower Insr, Lepr expression levels in hippocampus, persisting up to postnatal day 150. Thus, maternal exposure to high-fat diet during pregnancy and lactation can modulate cognition and behavior on adult offspring through Lepr (Cordner et al., 2019).
Proopiomelanocortin (POMC)
Proopiomelanocortin is a prohormone that suffers posttrans lational proteolysis, generating the active hormones α-, β-, and γ-, melanocyte-stimulating hormones (MSHs) and adrenocorticotropic hormone (ACTH), which all have a wide range of physiological actions (Cone, 2005; Toda et al., 2017). POMC is a key component of the melanocortin–leptin system, which regulates food intake and energy balance (Mountjoy, 2015). Then, mutations in POMC gene have been associated with morbid obesity (Ramachandrappa and Farooqi, 2011; Nordang et al., 2017).
In the brain, POMC is expressed in the arcuate nucleus (ARC) of the hypothalamus, the pituitary gland, and the brain stem (Toda et al., 2017). POMC neurons in the ARC integrate peripheral signals such as leptin, insulin, and glucose, which regulate energy balance by inducing satiety and higher energy expenditure. Satiety is mediated by the action of POMC peptides (α and β-MSH) on MC4R in the PVN of the hypothalamus (Andermann and Lowell, 2017; Toda et al., 2017; Candler et al., 2019).
The relationship of POMC with NDgD has been demonstrated with a specific AD animal model. This AD animal model (3xTg-AD) is a transgenic mouse expressing three dementia-relates genes: presenilin-1 (PS1m146V), amyloid precursor protein (APPSwe), and microtubule-associated protein tau (tauP301L). This model exhibited plaque and tangle pathology, synaptic dysfunction, and showed amyloid (β and TAU pathology (Oddo et al., 2003; Sterniczuk et al., 2010). Hypothalamic gene expression showed higher mRNA expression of genes related to inflammation and apoptosis, as well as lower levels of POMC and NPY-expressing neurons. However, voluntary exercise training reduced apoptosis and increased POMC and NPY-expressing neuronal populations (Do et al., 2018). Early exercise intervention can normalize hypothalamic inflammation, neurodegeneration, and the glucose metabolism in 3xTg-AD model, suggesting that exercise can reduce the progression of dementia and AD (Do et al., 2018).
On the other hand, an HD mouse model (R6/2), which has early dysregulations in the corticostriatal pathway (Cepeda et al., 2007) and hyperactive striatal neurons (Walker et al., 2008), exhibits a reduction of the feeding-related neuropeptides (POMC, NPY, and CART) (van der Burg et al., 2008). In addition, high-fat diet induced a reduction of synaptic inputs in hypothalamic nuclei [including lateral hypothalamus (LH) and ARC] and apoptosis of NPY/AGRP and POMC neurons. This dysfunction is associated with hypothalamic neurodegeneration (Tabrizi et al., 2011; Sousa-Ferreira et al., 2014). Expression of feeding-related neuropeptides in hypothalamic progenitor cells showed that POMC, CART, and NPY could be involved in the development of HD (Sousa-Ferreira et al., 2011).
Besides POMC, another anorexic neuropeptide is the cocaine and amphetamine regulated transcript (CART). CART is a neuropeptide expressed in the brain that is involved in reducing food intake and regulating energy homeostasis (Douglass et al., 1995; Banke et al., 2013). CART was associated with the development of HD (Gabery et al., 2010). Increased CART levels in the cerebrospinal fluid were associated with an increased number of CART immunopositivity neurons in the hypothalamus of HD patients (Cheong et al., 2019).
In patients with PD and ischemic stroke, levels of dopamine (DA) are reduced (Calne and Sandler, 1970; Bhakta et al., 2014). Administration of exogenous DA in ex vivo neurons induced CART expression and showed protection against brain damage by reducing inflammation activation (Lin et al., 2018). All of these confirm that CART could be involved in the development of NDgD as HD and PD.
On the other hand, NPY is an orexigenic neuropeptide associated with obesity, which is related to appetite regulation and development obesity (Wu et al., 2019). The NPY system is expressed in the peripheral nervous system and in the CNS (hippocampus, basal ganglia, and brain stem (Allen et al., 1986). Some monogenetic studies have reported the association of NPY with neurodevelopment, AD, PD, and HD (Ahmed et al., 2019; Li et al., 2019).
Melanocortin 4 receptor (MC4R)
Melanocortin is produced from the cleavage of the POMC precursor. This protein will then bind to one of its receptors. There are five known MCRs designated as MC1R through MC5R (Gantz et al., 1993, 1994; Roselli-Rehfuss et al., 1993; Yang et al., 2000). MC4R is predominantly expressed in the CNS including the hypothalamus, thalamus, hippocampus, brain stem, and cortex, although it is also detected in peripheral tissues. In addition, it could be expressed by neurons, microglia, and astrocytes (Chen et al., 2018). In particular, MC4R is activated by the POMC-derivate neuropeptides (α- and β-MSH) and blocked by agouti-related protein (AgRP) expressed in AgRP/NPY neurons in the ARC. The function of these neurons is modulated by signals from adipose tissue or the gut, such as leptin, ghrelin, and NPY (Clément et al., 2018).
The MC4R signaling pathway is necessary to control the energy balance, thermogenesis, and peripheral glucose metabolism, which involves G protein-mediated activation of adenylate cyclase and augmented cAMP production (Vollbach et al., 2017). As mentioned above, mutations in the MC4R gene have been associated with early-onset obesity and severe hyperphagia, causing about 5% of severe obesity in children and adults (Farooqi et al., 2000; Vaisse et al., 2000; Ramachandrappa and Farooqi, 2011).
However, in the brain, MC4R is involved in anorexigenic, ant-inflammatory, and antiapoptotic effects (Caruso et al., 2013). In addition, AD transgenic mouse model 3xTg-AD showed lower levels of MC4R and AgRP mRNA compared to the control (Do et al., 2018), confirming the important role of MC4R in the development of AD.
Another study showed that MC4R activation can inhibit the overexpression of inflammatory cytokines (IL-6, IL-1, IL-1β, and TNFα) in cerebral ischemia and AD (Giuliani et al., 2011; Spaccapelo et al., 2013).
Preproconvertase-1 (PCSK1)
PCSK1 is the gene that encodes the proprotein convertase 1/3 (PC1/3), which is a principal processing enzyme of precursor proteins in the secretory pathway. It is expressed in the brain, neuroendocrine system, and enteroendocrine cells (Creemers, 2008; Choquet et al., 2011). PC1/3 is synthesized as proPC1/3, which is inactive and is quickly converted into PC1/3 by autocatalytic excision of the NH2-terminal propeptide in the endoplasmic reticulum (ER). For fully PC1/3 activation, a second internal rupture of the propeptide is required in the post-ER compartment (Muller and Lindberg, 1999). An example of PC1/3 substrate is POMC, which is expressed in different neural cell populations of the ARC. In addition, PC1/3 acts in concert with PC2 to process POMC and obtain different neuropeptides as α-MSH [reviewed in Stijnen et al. (2016)].
Deficiency of PSCK1 was associated with recessive monogenic obesity (Ramachandrappa and Farooqi, 2011). Mutations in PSCK1 were associated with early-onset obesity, hyperphagia, sensitive hypoglycemia, and endocrine disorders (Jackson et al., 1997; Farooqi et al., 2007). However, PCSK1-null mice are not obese but showed growth retardation and multiple neuroendocrine abnormalities (Zhu et al., 2002; Choquet et al., 2011; Creemers et al., 2012). Moreover, PCSK1 deficiency has been associated with a major neuroendocrine disease, the Prader–Willi Syndrome (PWS). This disease is a complex genetic disorder characterized by hypogonadism, obesity, hyperphagia, growth impairment, and cognitive impairments (Ramos-Molina et al., 2018).
On the other hand, PCSK1 expression in the hypothalamus is high in POMC and AgrP/NPY neurons, both leptin-responsive neuronal populations with ARC. PC1/3 activity is essential for pre-AGRP and POMC processing in the ARC (Ramos-Molina et al., 2016). The expression profiles in postmortem human brains showed downregulation in PCSK1 and other 11 metabolic genes (Hokama et al., 2014). In the cortex of 3xTg-AD mouse model, Pcsk1 expression was downregulated, which could correlate with cognitive impairment (Oddo et al., 2003; Hokama et al., 2014; Castillo et al., 2017).
Single-minded 1 (SIM1)
SIM1 is a member of the basic helix-loop-helix Per-Arnt-Sim (β-HLH-PAS) family of transcription factors. SIM1 is critical for the formation of the PVN in the hypothalamus in mice (Michaud et al., 1998). Homozygous Sim1-knockout mice (Sim1–/–) lack PVN and die perinatally. However, heterozygous Sim1+/– mice are viable, presenting an early-onset obesity, hyperphagia, and increased linear growth, similar to Mc4r-mutant mice (Michaud, 2001). A few mutations in SIM1 gene have been found in obese individuals (Ramachandrappa and Farooqi, 2011), affecting the SIM1 transcriptional activity (Zegers et al., 2014).
SIM1 gene, like other metabolic genes, participates in the development of NDgD and NDvD. Defects in the serotonergic systems are associated with depression, obsessive–compulsive disorder, and SCZ. In addition, degeneration of mesencephalic dopaminergic (mDA) neurons is associated with PD. Sim1–/– newborn mice were used to evaluate Sim1 impact in mDA neuron differentiation and rostral 5-hydroxytryptamin (5-HT) neurons. They found a reduction in the number of dorsal raphe nucleus (DRN) 5-HT neurons, suggesting that Sim1 may modulate serotonin release via regulator of G protein signaling 4 (RGS4) (Osterberg et al., 2011). The role of RGS4 in not well understood, but it can modulate 5-HT 1A-mediated neurotransmitter release in vitro and in vivo (Beyer et al., 2004; Ghavami et al., 2004). All of these suggest that SIM1 could have an important role in the development of NDgD as PD (Osterberg et al., 2011) and NDvD as SCZ (Purcell et al., 2014).
Brain-derived neurotrophic factor (BDNF)
BDNF is a neurotrophic factor that plays a fundamental role in the development and plasticity of the CNS. BDNF binds to the tropomyosin-related kinase receptor (TrkB) (Di Carlo et al., 2019). BDNF is a key factor in brain signaling and synaptic plasticity (Hofer et al., 1990; Kowiañski et al., 2018). BDNF/TrkB neurotrophic signaling regulates the migration, development, differentiation, and survival of fetal neurons (Chaldakov et al., 2007), and is a major participant in the regulation of food intake (Rosas-Vargas et al., 2011). Mutations in BDNF gene have been associated with monogenetic obesity (Ramachandrappa and Farooqi, 2011).
Recently, numerous studies have shown the important role of BDNF in the development of NDgD and neurodevelopment. BDNF was associated with HD and AD (Couly et al., 2018; Smith-Dijak et al., 2019). Deficient BDNF/TrkB activity triggers neurodegeneration in AD via the activation of JAK2/STAT3 pathway and increasing inflammatory cytokines in human AD brains (Wang Z. H. et al., 2019). BDNF levels and its signaling have been modulated in the etiopathogenesis of AD, which suggested that BDNF levels could be a biomarker for AD [reviewed in Balietti et al. (2018)]. Moreover, a recent study showed that the P42 peptide treatment alleviates HD deficits in motor performance by changing BDNF level and activity (Couly et al., 2018).
As previously reported, the exposition to heavy metal can cause cognitive impairment and depressive disorders through BDNF. In early pregnancy, higher arsenic levels in blood were associated with lower levels of BDNF. The heavy metal exposure could trigger maternal depressive disorder and newborn neurodevelopment by lower levels of BDNF (Zaw and Taneepanichskul, 2019).
Tropomyosin-related kinase B (TrkB)
TrkB or neurotrophic receptor tyrosine kinase 2 (NTRK2) is a receptor for neurotrophin (NT) 4, BDNF, and NT-3 (Klein et al., 1990; Squinto et al., 1991). Some neurological diseases, obesity, and eating disorders have been associated with dysregulation of TrkB (Desmet and Peeper, 2006; O’Rahilly and Farooqi, 2006; Luberg et al., 2010). Mutations in the gene encoding TrkB, NTRK2, have been associated with monogenetic severe obesity with developmental delay (Ramachandrappa and Farooqi, 2011). These mutations modify TrkB ability to stimulate neurite outgrowth in response to BDNF. Thus, reduced hypothalamic neurogenesis could play a role in obesity and severe hyperphagia (Gray et al., 2007). The dorsomedial hypothalamus (DMH) has a role in the regulation of energy expenditure. A recent study revealed that the activation of TrkB-expressing DMH neurons suppresses appetite and maintain physiological satiety. It indicates that BDNF can modulate in part on the DMH to control bodyweight, suggesting that activation of DMH by the TrkB neurons could be a powerful way to treat obesity (Liao et al., 2019).
Recently, studies showed that TrkB can be involved in the development of NDgD. TrkB is widely distributed in different regions of the human brain, specifically in the dopaminergic neurons of the substantia nigra. In PD patients, the TrkB expression in the substantia nigra is significantly lower [reviewed in Jin (2020)], suggesting a role of this gene in the development of NDgD.
Polygenetic Obesity Genes
Obesity is a multifactorial and polygenic disease in which variants of different genes are associated with this disease and with the development of NDgD and NDvD (Arnoldussen et al., 2014; Lee and Mattson, 2014). As mentioned above, over 900 genetic variants have been associated with BMI and 346 loci were associated with body fat distribution (Yengo et al., 2018; Pulit et al., 2019). In this review, we will briefly described a few of the genes associated with obesity that have been discovered by a GWAS approach and that had been replicated in different populations. The genes reviewed here are FTO, NRXN3, NPC1, NEGR1, MTCH2, and GNPDA2, which were chosen for their association with obesity and their influence on NDgD or NDvD.
Fat mass and obesity-associated gene (FTO)
FTO gene is one of the most studied genes due to its association with obesity (Locke et al., 2015). A common variant of the FTO gene was identified through a T2D GWAS in 2007, and it also showed a strong association with obesity (Frayling et al., 2007; Scuteri et al., 2007). FTO association with obesity is the most replicated in different populations worldwide (Dina et al., 2007; Andreasen et al., 2008; Villalobos-Comparán et al., 2008).
FTO protein function was first described as an N6-methyladenosine (m6A) demethylase dependent of iron and 2-oxoglutarate (Gerken et al., 2007; Jia et al., 2011). Then, FTO-deficient mouse model was studied to understand its physiological function. These mice display postnatal growth retardation and reduced food intake, with a reduction of adipose tissue (Fischer et al., 2009; Gao et al., 2010). On the contrary, the overexpression of FTO showed a reduction of adipose tissue (Church et al., 2010). However, it took 8 years to understand that FTO intronic variant associated with obesity does not regulate FTO expression. In fact, this variant disrupts the binding site of the ARID5B repressor, which regulates IRX3 and IRX5 expression, genes involved in adipogenesis, thermogenesis, and lipid accumulation (Claussnitzer et al., 2015). This is an example that the discovery of new genetic variants could unveil an unexpected impact on physiology.
Over the last few years, FTO was associated with NDgD, especially AD. Obesity and T2D in human and mice can activate FTO in the brain tissues by defective insulin signaling (Li et al., 2018). It is known that obesity and T2D are commonly associated with the development of AD (Vagelatos and Eslick, 2013). A recent study demonstrated that in the 3xTg-AD mouse model, Fto neuronal conditional silencing reduced the cognitive deficits, suggesting its implication on the insulin signaling defect present in AD (Li et al., 2018).
Neurexin 3 (NRXN3)
NRXN3 is a type of neurexins, which are neuron-specific cell surface proteins. Their structure suggests a role in the cell adhesion and cell recognition (Ushkaryov et al., 1992).
NRXN3 gene has been associated with waist circumference as an obesity trait (Heard-Costa et al., 2009). Moreover, this gene was also associated with increased BMI and visceral fat and decreased sleep duration (Prats-Puig et al., 2013).
NRXN3 is involved in synaptic function in the cognitive decline associated with aging and AD. Moreover, mutations in NRXN3 have been identified in AD patients (Zheng et al., 2018; Hishimoto et al., 2019), and rare deletions have been associated with ASD (Vaags et al., 2012).
Niemann–pick type C1 (NPC1)
NPC1 is a protein that regulates the transport of cholesterol and fatty acids from late endosomes/lysosomes to cellular structures as mitochondria and plasma membrane for maintaining lipid homeostasis [reviewed in King and Sharom (2012)]. In humans, GWAS showed common NPC1 variants associated with obesity (Meyre et al., 2009).
A rare autosomal recessive mutation in human NPC1 causes a disorder in lipid storage leading to progressive and lethal neurodegeneration and lung and liver failure [reviewed in Lamri et al. (2018)]. NPC disease is a lysosomal storage disorder, present in childhood with accumulation of visceral lipid and progressive neurodegenration, with characteristic dysphagia, cerebellar ataxia, and dementia resulting in a lower life expectancy (Newton et al., 2018). NPC1 gene has been associated with NDv and NDgD as AD (Rouillard et al., 2016) and SCZ (Kawazoe et al., 2018).
Neuronal growth regulator 1 (NEGR1)
NEGR1 is expressed in the rat brain. It has been proposed that NEGR1 could modulate the intracellular cholesterol trafficking, suggesting its implication in human obesity (Kim et al., 2017). NEGR1 was associated with obesity (Thorleifsson et al., 2009; Willer et al., 2009). Moreover, Negr1-deficient mice showed increased adiposity (Joo et al., 2019). The genetic variability in NEGR1 could be associated with psychological traits of patients with eating disorders, like bulimia (Gamero-Villarroel et al., 2015).
Moreover, a GWAS reported a NEGR1 variant associated with major depression (Wray et al., 2018). Recent studies on humans and animals support the idea that NEGR1 is involved in psychiatric disorders such as SCZ (Karis et al., 2018), ASD (Szczurkowska et al., 2018), and AD (Ni et al., 2018; Raghavan et al., 2019).
Negr1 deficiency in animals leads to impaired cortical development and impaired behavior, a conduct similarly observed in ASD patients (Szczurkowska et al., 2018). On the other hand, SCZ patients showed higher levels of Negr1 in the dorsolateral prefrontal cortex (Karis et al., 2018). Therefore, these data demonstrate the implication of NEGR1 in NDvD.
Mitochondrial carrier homolog 2 (MTCH2)
MTCH2 is a relative novel protein located in the inner membrane of mitochondria. It is expressed in white adipose tissue. MTCH2 has an important regulatory role in the differentiation and biology of the adipocyte (Bernhard et al., 2013). MTCH2 variants have been associated with increased BMI, obesity, and diabetes (Willer et al., 2009; Heid et al., 2010). A study on Mtch2-knockout mouse model reported that the animals die at an embryonic 7.5 day, suggesting that Mtch2 could play a specific role in embryonic development (Ruggiero et al., 2017).
It is relevant mentioning that the lower expression of MTCH2 was associated with late onset AD status in a GWAS (Karch et al., 2016). A case control study in Swedish population showed several variants associated with SCZ by exome-sequencing (Purcell et al., 2014). These data suggest the possible role that MTCH2 could have in the development of NDgD and NDvD.
Glucosamine 6 phosphate isomerase 2 (GNPDA2)
GNPDA2 gene encodes the enzyme glucosamine-6-phosphate deaminase (GlcN6P). This enzyme catalyzes the reversible reaction of D-glucosamine-6-phosphate into D-fructose-6-phosphate and ammonium (Arreola et al., 2003). This enzyme has a hydrolase activity and is involved in metabolic pathways, glucose and nucleotide metabolism. GNPDA2 is highly expressed by the brain (cortex and hypothalamus) (Willer et al., 2009).
GNPDA2 gene variant was associated with obesity in populations with European ancestry (Willer et al., 2009), and this association was replicated in other populations (Renström et al., 2009; Locke et al., 2015). In a diet-induced obesity model, high-fat diet led to a lower GNPDA2 hypothalamic expression compared to rats fed with chow diet (Gutierrez-Aguilar et al., 2012).
GNPDA2 was overexpressed in PD and AD cases and showed lower serum levels in PD patients. Interestingly, in PD patients, GNPDA2 showed an inverse correlation with α-synulein protein levels in the cerebrospinal fluid (Lachén-Montes et al., 2019), suggesting its implication in developing PD.
Genes Associated With Neurodegenerative and Neurodevelopmental Diseases
Genetic studies have shown genes implicated in developing NDgD and NDvD (Saad et al., 2011; Arnoldussen et al., 2014; Henriksen et al., 2017; Wu and Pan, 2018).
The heritability for AD is between 60 and 80% (Wingo, 2012). AD genetics has been reviewed elsewhere (Reitz, 2015; Goldman and Van Deerlin, 2018; Jung et al., 2018). However, only 1% of all AD is autosomal dominant genes, such as presenilin 1 (PSEN1), presenilin 2 (PSEN2), and amyloid precursor protein (APP) (Hinz and Geschwind, 2017).
With the genomic era, several GWAS and meta-analysis have been performed, identifying up to 29 risk loci associated with AD, and APOE is one of them (Lambert et al., 2013; Jansen et al., 2019).
Interestingly, some genes associated with obesity were also associated with AD by different genetic approaches: candidate gene approach FTO (Li et al., 2018), by differentially expressed genes NRXN3 (Zheng et al., 2018; Hishimoto et al., 2019), NPC1 (Rouillard et al., 2016), NEGR1 (Wray et al., 2018), or by GWAS approach: LEPR, BDNF, TNFα, and MTCH2 (Gao et al., 2015; Karch et al., 2016; Lemche, 2018).
In respect to PD, this disease only affects 2% of the population over 60 years old. Family-based genetic studies had identified 23 genes associated with PD, having diverse functions as deficiency of synaptic transmission, vesicular recycling, lysosomal dysfunction, mitophagy. Some of those genes are: synuclein-Alpha (SNCA), inherited in an autosomal dominant; PARKIN, autosomal recessive juvenile parkinsonism; PTEN-induced kinase (PINK1), recessive early onset, among others (Karimi-Moghadam et al., 2018).
Meta-analysis from PD GWAS report over 30 variants close to genes like CTSB, TMEM175, LRRK2, among others (Nalls et al., 2014; Chang et al., 2018). Some of the common genes that have been associated with obesity and PD are: GNPDA2 (Lachén-Montes et al., 2019) CD38, TNFα, and PAI-1 (Pan et al., 2018) that will be discussed below.
Huntington’s disease is another NDgD characterized by progressive neurodegeneration with severe neuronal loss (90%) that occurs in the lateral hypothalamus (Sprengelmeyer et al., 2006), as well as neuronal degeneration and hypothalamic dysfunction (van Duijn et al., 2014). In this disease, the gene HTT, encoding for the huntingtin protein, has a CAG trinucleotide expansion mutation, causing behavioral abnormalities, dementia, and progressive movement disorder (Ross et al., 2014). A GWAS reported a few gene variants associated with HD that are close to the MLH1, FAN1, MTRM10, RRM2B, and UBR5 genes. Some of these genes are implicated in DNA mismatch repair, structure-specific DNA handling, mitochondrial energetics, and oxidative stress (Lee et al., 2015). However, genes associated with obesity have been also described to be associated with HD, such as POMC, NPY, CART, and BDNF (Sousa-Ferreira et al., 2011).
Neurodevelopmental diseases are a group of early-onset neurological disorders, which also have a genetic background. These disorders include ASD that often present intellectual disability (ID), motor abnormalities, and epilepsy. Fragile X syndrome (FXS) is part of the syndromic ASD classification (Tãrlungeanu and Novarino, 2018).
Fragile X syndrome is an inherited disease linked to the X chromosome and is one cause of intellectual disability. This syndrome is associated with a triplet repeats of cytosine-guanine-guanine (CGG) in the FMR1 gene (O’Donnell and Warren, 2002; Visootsak et al., 2014). Usually, FXS patients have social anxiety, severe communication deficits, and stereotyped behavior (Hall et al., 2009). Some cases with FMR1 premutation showed lack of satiation, severe hyperphagia, and severe obesity (Martínez-Cerdeño et al., 2017).
Schizophrenia is another NDvD that affects 1% of the worldwide population. It is a chronic and severe mental disorder, characterized by cognitive impairment. More than 1,000 candidate genes have been proposed to be associated with SCZ. However, their functional implication in developing SCZ remains unclear. Hu et al. (2013) analyzed the new genetic variants associated with SCZ and identified pathways that might explain the biological function causing this disease. They propose that pathways involved in fatty acid degradation, glycan degradation, PPAR signaling, among others, could be involved in the development of this disease. However, the genes that have been associated with SCZ are NRXN3, and NEGR1, NPC1, and TNFα (Hu et al., 2013; Park et al., 2019).
Frequently, the associations between genes and diseases are studied separately. Interestingly, a recent genetic study analyzed the association of millions of genetic variants with BMI and major psychiatric disorders (SCZ, bipolar disorder, and major depression), instead of analyzing separately each disease. This study identified 111 genetic loci overlapping between BMI and the psychiatric disorders. Some of these variants are associated with genes expressed in the brain with plausible functions in CNS development, intracellular processes, and GABAergic and glutamatergic signaling (Bahrami et al., 2020); however, their functions have to be confirmed.
In the next section, we will briefly describe genes associated with NDgD as AD, PD, and HD that were discovered by a GWAS approach and meta-analysis (Qi et al., 2012b; Moss et al., 2017; Chang et al., 2018; Jansen et al., 2019) and that had previously shown strong association with monogenic or polygenic obesity (Tong, 2011; Hatziri et al., 2018; Wang et al., 2018a, b; Liu et al., 2019). Some of the genes mentioned below are APOE and TREM2 associated with AD; CD38 variants with AD and PD; and SYT4 with PD (Chang et al., 2018; Jansen et al., 2019). In addition, genetic studies found the association of SIRT1 with PD and AD (Zhang A. et al., 2012; Rana et al., 2019) and with SCZ (Kishi et al., 2011). A similar strategy to select the genes associated with NDvD as SCZ and ASD. However, there are just a few genes highly associated with the development of NDvD and obesity. Some of them were discovered by exome-sequencing studies as MTCH2 and SIM1, which were associated with the development of SCZ (Purcell et al., 2014). GWAS showed that FMR1 was associated with ASD and FXS (Tassone et al., 2000; Tassone et al., 2007) and TET3 gene variants with the development of NDvD (Santos-Cortez et al., 2018). However, little is known about the function of these genes and their implications in NDgD and NDvD.
Apolipoprotein E (APOE)
Apolipoprotein E is a glycoprotein that is produced predominantly by astrocytes in the brain and peripherally in the liver (Huang and Mahley, 2014). There are three APOE alleles: E2, E3, and E4. Peripherally, APOE2 and APOE3 bind to high-density lipoproteins (HDLs), responsible for trafficking lipids from the periphery cells to the liver for elimination. APOE4 have greater affinity for very low-density lipoprotein, and it is less efficient at homeostatic maintenance (Huang and Mahley, 2014). However, APOE4 was associated with higher fasting glucose and insulin levels, as well as an increased metabolic syndrome risk with younger age onset (Torres-Perez et al., 2016). In obese men, APOE4 carriers have elevated levels of plasma cholesterol, triglycerides, glucose, and insulin and presents IR (Elosua et al., 2003; Jones and Rebeck, 2019).
The most common APOE3 allele has been associated with an average risk to develop AD. However, APOE4 homozygotes increase the risk 15 times to the development of AD. In addition, APOE4 is also associated with increased risk for CVD and metabolic syndrome (El-Lebedy et al., 2016).
Interestingly, APOE is one of the metabolic genes associated with the development of NDgD. APOE4 has shown a strongest association with the development of late-onset AD (Lambert et al., 2013; Jansen et al., 2019). APOE affects TAU pathogenesis, neuroinflammation, and TAU-mediated neurodegeneration (Yoshiyama et al., 2007). APOE4 carrier has increased Aβ accumulation and decreased clearance in AD brains (Liu et al., 2013; Shi et al., 2017).
Cluster of Differentiation 38 (CD38)
CD38 is a type II transmembrane glycoprotein (Jackson and Bell, 1990). It is a lymphocyte-specific antigen, has ectoenzymatic activity, and functions as a receptor and adhesion molecule (States et al., 1992). CD38 is highly expressed on plasma cells, red blood cells, platelets (Deaglio et al., 2001), and adipose tissue and in obese people (Nair et al., 2005; Mutch et al., 2009). CD38 is involved in different biological processes including cell proliferation, hormone secretion, muscle contraction, egg fertilization, and immune response. It is also involved in the catabolism of nicotinamide adenine dinucleotide (NAD+) and nicotinamide adenine dinucleotide phosphate (NADP) (Howard et al., 1993).
A CD38-deficient mouse model has higher metabolic rate and showed protection against diet-induced obesity through increasing NAD-dependent activation of sirtuin (SIRT) proliferator-activated receptor gamma coactivator 1-alpha (PGC1α), which is involved in the regulation of mitochondrial biogenesis and energy homeostasis (Barbosa et al., 2007). On the other hand, a recent study showed that CD38 participates in adipogenesis and lipogenesis of adipose tissues through regulating Sirt1-mediated signaling pathway (Wang et al., 2018b).
In addition to its role in obesity, CD38 is also expressed in neurons, microglial cells, and astrocytes [reviewed in Guerreiro et al. (2020)]. CD38 is highly expressed in the mouse brain during development (postnatal days 14 and 28). The CD38-knockout mice showed that the brain displayed a 10-fold NAD+ level more than wild type, suggesting that CD38 is one regulator of intracellular NAD+ levels in the brain.
Genomic studies demonstrated that CD38 was associated with PD (Saad et al., 2011) and was confirmed by a meta-analysis (Chang et al., 2018). It suggests that CD38 could play an important role in the development of NDgD as PD.
Sirtuin1 (SIRT1)
SIRT1 is a deacetylase NAD+-dependent that is expressed in the heart, adipose tissue, muscle, kidney, liver, and brain (basal ganglia, prefrontal cortex, and hippocampus, which are areas associated with NDgD) (Zakhary et al., 2010). SIRT1 is involved in important processes such as cell cycle regulation, energy metabolism modulation, mitochondrial biogenesis, glucose/cholesterol metabolism, etc. (Herranz and Serrano, 2010; Nogueiras et al., 2012). SIRT1 has been associated with obesity (Zillikens et al., 2009) and is involved in food intake regulation, life span, diabetes, and CVD (Pfluger et al., 2008; Nogueiras et al., 2012).
SIRT1 expression and activity were increased by resveratrol treatment, protecting neuronal cells (Seo et al., 2012; Herskovits and Guarente, 2014) and accelerating brain aging (Duan, 2013). SIRT1 has shown an important role in the regulation of other anti-aging genes as KL (Kloto), SHC1 or p66Shc (transforming protein SCH1), and FOXO1a/FOXO 3a (Forkhead box) by p53 transcription factor deacetylation (Seo et al., 2012; Herskovits and Guarente, 2014). Low expression of SIRT1 in adipose tissue was shown in obese subjects (Stefanowicz et al., 2018).
Moreover, low levels of SIRT1 are present in NDgD as AD and PD (Lutz et al., 2014; Singh et al., 2017). In addition, genetic studies found the association of SIRT1 with PD and AD (Zhang A. et al., 2012; Rana et al., 2019) and with SCZ (Kishi et al., 2011).
Tumor Necrosis Factor-Alpha (TNFα)
Tumor necrosis factor-alpha is a potent pleiotropic pro-inflammatory cytokine (Hajeer and Hutchinson, 2000, 2001). TNFα is produced by many cell types such as neutrophils, fibroblasts, keratinocytes, macrophages, natural killer cells, T and B cells, and tumor cells (Anderson et al., 2004). TNFα plays an essential role in chronic inflammation associated with different pathologies, such as obesity, T2D, AD, and PD (Wei et al., 2011).
Tumor necrosis factor-alpha has been associated with NDgD and NDvD in many reports (Arnoldussen et al., 2014). The neuroinflammation in the NDgD, such as AD, PD, amyotrophic lateral sclerosis, and multiple sclerosis is modulated by cytokines like TNFα (Baj and Seth, 2018). Higher levels of cytokines as TNFα, IL-10, TNFβ, and CRP were associated with NDvD, as SCZ and ASD (Bodnar et al., 2018).
Plasminogen Activator Inhibitor (PAI-1)
Plasminogen activator inhibitor is a single-chain glycoprotein belonging to the serine protease inhibitor (serpin) superfamily. There are four PAIs: PAIs-1 to 3 and protease nexins (Feinbloom and Bauer, 2005). PAI-1 inhibits the plasminogen activator and is produced by platelet, hepatocytes, adipocyte, vascular smooth muscle cell, fibroblast, and macrophages (Placencio and DeClerck, 2015). PAI-1 mRNA expression in visceral and subcutaneous adipose tissue was correlated with BMI and severe obesity (Alessi et al., 2000). Plasma PAI-1 activity and antigen were positively associated with BMI in hypertense men (Skurk et al., 2004). The expression of PAI-1 can be regulated by lipid/glucose metabolites, environmental factors, inflammation, age, BMI, and lifestyle. Also, chemical messengers like TNFα, hormones, inflammatory cytokines, growth factors, and endotoxins can modulate its expression (Oishi, 2009).
Recently, a new study showed the association of higher levels of PAI-1 with PD (Pan et al., 2018). PAI-1 polymorphisms can change focal and brain stem neurological signs in patients with traumatic brain injury (Pan et al., 2018). These results suggested that PAI-1 can participate in the development of NDgD (Arnoldussen et al., 2014).
Trigger Receptor Expressed on Myeloid Cells 2 (TREM2)
TREM2 is expressed on myeloid cells (macrophages, dendritic cells, and microglia) (Bouchon et al., 2001; Daws et al., 2001; Wang et al., 2015; Ulland et al., 2017) and in adipose tissue (Park et al., 2015). TREM2 regulates the behaviors of different cell biologicals: survival, proliferation, differentiation, phagocytosis, and inflammatory response (Zhong et al., 2015; Kober and Brett, 2017). It also acts as a lipid sensing receptor to recognize and bind lipids (Wang et al., 2015).
TREM2 gene expression was upregulated in adipose tissue in obesity animal models (Fujimoto et al., 2011; Grant et al., 2011; Park et al., 2015). Trem2–/– mice fed with a high-fat diet showed lower mass but higher hypertrophy in adipocytes and increased adipocyte death. In addition, these mice had deficient inflammatory response of adipose tissue macrophages and severe hepatic steatosis. They showed that the function of TREM2 is a feedback mechanism to control obesity-induced IR by regulating adipose tissue remodeling pathways (Liu et al., 2019).
Furthermore, TREM2 genetic variants have been associated with a higher risk to develop AD. The amyloid plaque compaction depends on TREM2 mechanism, forming a protective barrier that attenuates toxicity nearby neurons [reviewed in Yeh et al. (2017)].
Synaptotagmin-4 (SYT4)
SYT4 is insensitive to Ca2+. SYT4 is expressed in brain and neuroendocrine system and has been suggested to have a neuroendocrine role (Tong, 2011; Zhang et al., 2011). The upregulation of SYT4 inhibits the release of oxytocin, which is a characteristic in obese phenotype (Tong, 2011; Zhang et al., 2011). The negative regulation of SYT4 could potentially repair or reduce the degree of diabetes neuropathies (Rahimi et al., 2015); however, the physiological function of SYT4 remains unknown.
A specific mouse model showed that SYT4 upregulation, within dystrophic neurons, could reflect impaired protein degradation that happens in AD (Zhang et al., 2009), suggesting its implication in this disease.
Fragile X Mental Retardation 1 (FMR1)
FMR1 gene encodes the Fragile X mental retardation protein (FMRP). FMR1 is found in most adult and fetal tissues, especially in brain and testes (Berry-Kravis et al., 2002; Berry-Kravis et al., 2011). It has been reported that FXS patients have obesity (Martínez-Cerdeño et al., 2017).
Trinucleotide (CGG) repeat in FMR1 promotor region is associated with FXS, and a permutation is associated with fragile X-associated tremor/ataxia syndrome (FXSAS). Many individuals with permutation on the FMR1 gene have high levels of mRNA, but normal FMRP is synthesized (Tassone et al., 2000, 2007). Fxr1–/–-knockout mice die at 24 h of birth, and heterozygous mice exhibit abnormal limb musculature and learning and circadian rhythm deficits (Berry-Kravis et al., 2011; Francis et al., 2014). Analysis of graph diffusion and multitask clustering of FMR1 Clip-seq and transcriptional targets showed pathways regulated by FMR1 in human neural development (Li et al., 2020).
As we previously mentioned, the function or pathway of many of the genes discovered by GWAS is unknown. Such is the case of FMR1 with its implication in developing obesity or NDvD.
Ten Eleven Translocate 3 (TET3)
TET3 is a protein which belongs to the TET family, which catalyzes hydroxylation of 5-metylcytosine (5mC) to 5-hidroxymetylcytosine (5hmC) (Ito et al., 2010; Pastor et al., 2013). This is a key step in active DNA demethylation, which needs α-ketoglutarate (αKG) as a cofactor (Tahiliani et al., 2009). However, in maternal obesity, placental TET3 methylation is increased, without totally understanding its role in obesity (Mitsuya et al., 2017).
TET3 is the most highly expressed enzyme in the brain and is an essential enzyme in neuronal differentiation and in vivo early neocortical development (Lv et al., 2014; Li et al., 2015). In fact,Tet3-knockout mouse model showed defects in brain morphology, behavior, and motor development (Santos-Cortez et al., 2018).
Interestingly, some genomic studies have shown the association of TET3 gene variants with the development of NDvD (Santos-Cortez et al., 2018). Moreover, TET3 expression levels are downregulated in postmortem brains of AD individuals (Hokama et al., 2014). Therefore, TET3 is one of the new genes discovered to be associated with both NDgD/NDvD, and its mechanism leading to those diseases would have to be unveiled.
Discussion
In this review, we addressed the obesogenic environmental conditions that influence the development of NDgD or NDvD. Interestingly, some of the environmental factors that lead to the development of obesity, NDgD, or NDvD are common. Some of the environmental factors are physical activity, alcohol consumption, socioeconomic status, parent feeding behavior, and diet. All of these factors influence the development of obesity and impair abilities as memory, fine motor skills, and cognitive function.
Obesity is a comorbidity of NDgD and NDvD due to the common biological mechanisms that affect brain cell death or brain development. The adipose tissue hypertrophy provokes inflammation, impaired insulin signaling. The secretion of adipokines and pro-inflammatory cytokines is altered, as well as the lipid metabolism. High levels of free fatty acids contribute to IR and hyperglycemia and lead to impaired cognitive processes. Moreover, oxidative damage, energy metabolism failures, mitochondrial and neurotransmission systems dysfunction could lead to cell death and NDgD. On the other hand, maternal obesity produces high levels of inflammatory mediators that can cross the blood–placenta barrier, influencing fetal development. This fetal exposure could end in neurodevelopmental disorders like SCZ, ADHD, and ASD.
Genetics plays an important role in developing obesity, NDgD, and NDvD. We described the genes associated with obesity, either on monogenic or polygenic manner. For the monogenetic obesity, several of the genes are involved in the leptin–melanocortin pathway, mainly regulated in the hypothalamus. This pathway controls the energy homeostasis, given by the food intake and the energy expenditure. Surprisingly, all these genes were also associated with NDgD or NDvD by sharing regulation on specific brain regions (hypothalamus, hippocampus, thalamus, cortex, etc.) or by leading to synaptic dysfunction and amyloid β and TAU pathology.
Moreover, we described the new genes associated with obesity, NDgD, or NDvD, as discovered by a GWAS approach. This approach has helped to discover new genes without a previous established hypothesis, thus without knowledge limitations. These genomic studies had discovered gene variants with unknown functions that we might have never imagined to be implicated in a particular disease.
In this review, we mentioned just a few genes associated with obesity, NDgD, or NDvD, but a long list of genes are associated. However, the challenge now is to identify the function of all these genes and their plausible implication in these diseases, as well as the gene–environment interaction.
In fact, gene–environment interactions have explained part of the missing heritability, but more research is needed to better understand how the genes confer susceptibility to develop a disease in the presence of an environment that enhances the trait. Therefore, our genes confer susceptibility to develop obesity, NDgD, and NDvD; however, we can modify the environment to reduce the risk.
The prevalence of obesity has risen dramatically in the last decades. As obesity is a trigger for the development of many complex diseases, as NDgD and NDvD, it is plausible that in the next decades, the prevalence of these diseases will increase. Even though we cannot fully understand the etiology of obesity, many of the risk factors for the development of obesity are already known. Then, we can modify the environmental factors to prevent obesity and avoid the risk of developing NDgD and NDvD.
Conclusion
Obesity and NDgD/NDvD are diseases that share environmental and genetic factors, which lead to the development of all these diseases. Understanding the environment, the genes, and the gene–environment interaction involved in obesity and NDgD/NDvD, we will comprehend the etiology of these diseases. Then, by modifying our environmental factors and knowing the genetic susceptibility, we might be able to avoid the development of these diseases.
Author Contributions
MF-D analyzed, discussed, and wrote the genetic section. YD-L searched for the bibliography and analyzed the literature. RG-A conceived, analyzed, discussed, wrote, and edited all the manuscript. All authors revised and approved the last version of this manuscript.
Funding
We are very thankful to the Hospital Infantil de México “Federico Gómez” for the economic support to publish this paper. Also, we appreciate the discount Frontiers in Neuroscience gave us for this publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Addolorato, G. (2000). Body composition changes induced by chronic ethanol abuse: evaluation by dual energy x-ray absorptiometry. Am. J. Gastroenterol. 95, 2323–2327. doi: 10.1016/S0002-9270(00)01112-6
Ahmad, S., Rukh, G., Varga, T. V., Ali, A., Kurbasic, A., Shungin, D., et al. (2013). Gene × physical activity interactions in obesity: combined analysis of 111,421 individuals of european ancestry. PLoS Genet. 9:e1003607. doi: 10.1371/journal.pgen.1003607
Ahmed, R. M., Phan, K., Highton-Williamson, E., Strikwerda-Brown, C., Caga, J., Ramsey, E., et al. (2019). Eating peptides: biomarkers of neurodegeneration in amyotrophic lateral sclerosis and frontotemporal dementia. Ann. Clin. Transl. Neurol. 6, 486–495. doi: 10.1002/acn3.721
Alessi, M. C., Bastelica, D., Morange, P., Berthet, B., Leduc, I., Verdier, M., et al. (2000). Plasminogen activator inhibitor 1, transforming growth factor-beta1, and BMI are closely associated in human adipose tissue during morbid obesity. Diabetes Metab. Res. Rev. 49, 1374–1380. doi: 10.2337/diabetes.49.8.1374
Allen, Y. S., Bloom, S. R., and Polak, J. M. (1986). The neuropeptide Y-immunoreactive neuronal system: discovery, anatomy and involvement in neurodegenerative disease. Hum. Neurobiol. 5, 227–234.
An, H., Du, X., Huang, X., Qi, L., Jia, Q., Yin, G., et al. (2018). Obesity, altered oxidative stress, and clinical correlates in chronic schizophrenia patients. Transl. Psychiatry 8, 258. doi: 10.1038/s41398-018-0303-7
Andermann, M. L., and Lowell, B. B. (2017). Toward a wiring diagram understanding of appetite control. Neuron 95, 757–778. doi: 10.1016/j.neuron.2017.06.014
Anderson, G., DeWitte, M., and Nakada, M. T. (2004). Tumor necrosis factor-? in the pathogenesis and treatment of cancer. Curr. Opin. Pharmacol. 4, 314–320. doi: 10.1016/j.coph.2004.04.004
Andreasen, C. H., Stender-Petersen, K. L., Mogensen, M. S., Torekov, S. S., Wegner, L., Andersen, G., et al. (2008). Low physical activity acentuates the effect of rs9939609 polymorphism. Diabetes 57, 95–101. doi: 10.2337/db07-0910
Angelidou, A., Asadi, S., Alysandratos, K.-D., Karagkouni, A., Kourembanas, S., and Theoharides, T. C. (2012). Perinatal stress, brain inflammation and risk of autism-Review and proposal. BMC Pediatr. 12:89. doi: 10.1186/1471-2431-12-89
Anjum, I., Fayyaz, M., Wajid, A., Sohail, W., and Ali, A. (2018). Does obesity increase the risk of dementia: a literature review. Cureus 10:e2660. doi: 10.7759/cureus.2660
Anstey, K. J., Cherbuin, N., Budge, M., and Young, J. (2011). Body mass index in midlife and late-life as a risk factor for dementia: a meta-analysis of prospective studies. Obes. Rev. 12, e426–e437. doi: 10.1111/j.1467-789X.2010.00825.x
Arnoldussen, I. A. C., Kiliaan, A. J., and Gustafson, D. R. (2014). Obesity and dementia: adipokines interact with the brain. Eur. Neuropsychopharmacol. 24, 1982–1999. doi: 10.1016/j.euroneuro.2014.03.002
Arreola, R., Valderrama, B., Morante, M. L., and Horjales, E. (2003). Two mammalian glucosamine-6-phosphate deaminases: a structural and genetic study. FEBS Lett. 551, 63–70. doi: 10.1016/S0014-5793(03)00896-2
Ashwood, P., Kwong, C., Hansen, R., Hertz-Picciotto, I., Croen, L., Krakowiak, P., et al. (2008). Brief report: plasma leptin levels are elevated in autism: Association with early onset phenotype? J. Autism Dev. Disord. 38, 169–175. doi: 10.1007/s10803-006-0353-1
Bahrami, S., Steen, N. E., Shadrin, A., O’Connell, K., Frei, O., Bettella, F., et al. (2020). Shared genetic loci between body mass index and major psychiatric disorders. JAMA Psychiatry. 77, 1–11. doi: 10.1001/jamapsychiatry.2019.4188
Baj, T., and Seth, R. (2018). Role of curcumin in regulation of TNFα mediated brain inflammatory responses. Recent Pat. Inflamm. Allergy Drug Discov. 12, 69–77. doi: 10.2174/1872213x12666180703163824
Balietti, M., Giuli, C., and Conti, F. (2018). Peripheral blood brain-derived neurotrophic factor as a biomarker of Alzheimer’s disease: Are there methodological biases? Mol. Neurobiol. 55, 6661–6672. doi: 10.1007/s12035-017-0866-y
Ball, N., Teo, W.-P., Chandra, S., and Chapman, J. (2019). Parkinson’s Disease and the Environment. Front. Neurol. 10:218. doi: 10.3389/fneur.2019.00218
Banke, E., Riva, M., Shcherbina, L., Wierup, N., and Degerman, E. (2013). Cocaine- and amphetamine-regulated transcript is expressed in adipocytes and regulate lipid- and glucose homeostasis. Regul. Pept. 182, 35–40. doi: 10.1016/j.regpep.2012.12.011
Barbosa, M. T. P., Soares, S. M., Novak, C. M., Sinclair, D., Levine, J. A., Aksoy, P., et al. (2007). The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 21, 3629–3639. doi: 10.1096/fj.07-8290com
Baumann, H., Morella, K. K., White, D. W., Dembski, M., Bailon, P. S., Kim, H., et al. (1996). The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc. Natl. Acad. Sci. U.S.A. 93, 8374–8378. doi: 10.1073/pnas.93.16.8374
Bernhard, F., Landgraf, K., Klöting, N., Berthold, A., Büttner, P., Friebe, D., et al. (2013). Functional relevance of genes implicated by obesity genome-wide association study signals for human adipocyte biology. Diabetologia 56, 311–322. doi: 10.1007/s00125-012-2773-0
Berry-Kravis, E., Grossman, A. W., Crnic, L. S., and Greenough, W. T. (2002). Understanding fragile X syndrome. Curr. Paediatr. 12,, 316–324. doi: 10.1054/cupe.2002.0305
Berry-Kravis, E., Knox, A., and Hervey, C. (2011). Targeted treatments for fragile X syndrome. J. Neurodev. Disord. 3, 193–210. doi: 10.1007/s11689-011-9074-7
Beyer, C. E., Ghavami, A., Lin, Q., Sung, A., Rhodes, K. J., Dawson, L. A., et al. (2004). Regulators of G-protein signaling 4: modulation of 5-HT 1A- mediated neurotransmitter release in vivo. Brain Res. 1022, 214–220. doi: 10.1016/j.brainres.2004.06.073
Bhakta, B. B., Hartley, S., Holloway, I., Couzens, J. A., Ford, G. A., Meads, D., et al. (2014). The DARS (Dopamine Augmented Rehabilitation in Stroke) trial: protocol for a randomised controlled trial of Co-careldopa treatment in addition to routine NHS occupational and physical therapy after stroke. Trials 15:316. doi: 10.1186/1745-6215-15-316
Bhurosy, T., and Jeewon, R. (2014). Overweight and obesity epidemic in developing countries: A problem with diet, physical activity, or socioeconomic status? Sci. World J. 2014:964236. doi: 10.1155/2014/964236
Bjørbaek, C., and Kahn, B. B. (2004). Leptin signaling in the central nervous system and the periphery. Recent Prog. Horm. Res. 59, 305–331. doi: 10.1210/rp.59.1.305
Blackmore, E. R., Moynihan, J. A., Rubinow, D. R., Pressman, E. K., Gilchrist, M., and O’Connor, T. G. (2011). Psychiatric symptoms and proinflammatory cytokines in pregnancy. Psychosom. Med. 73, 656–673. doi: 10.1097/PSY.0b013e31822fc277
Bodnar, T. S., Raineki, C., Wertelecki, W., Yevtushok, L., Plotka, L., Zymak-Zakutnya, N., et al. (2018). Altered maternal immune networks are associated with adverse child neurodevelopment: Impact of alcohol consumption during pregnancy. Brain. Behav. Immun. 73, 205–215. doi: 10.1016/j.bbi.2018.05.004
Bogardus, C., and Swinburn, B. (2017). Obesity triggers: sequencing the genome versus sequencing the environment. Obesity 25, 1861–1863. doi: 10.1002/oby.21985
Bouchon, A., Hernández-Munain, C., Cella, M., and Colonna, M. (2001). A Dap12-mediated pathway regulates expression of Cc chemokine receptor 7 and maturation of human dendritic cells. J. Exp. Med. 194, 1111–1122. doi: 10.1084/jem.194.8.1111
Bray, G. A., and Popkin, B. M. (2014). Dietary sugar and body weight: Have we reached a crisis in the epidemic of obesity and diabetes? Diabetes Care 37, 950–956. doi: 10.2337/dc13-2085
Brunkwall, L., Chen, Y., Hindy, G., Rukh, G., Ericson, U., Barroso, I., et al. (2016). Sugar-sweetened beverage consumption and genetic predisposition to obesity in 2 Swedish cohorts. Am. J. Clin. Nutr. 104, 809–815. doi: 10.3945/ajcn.115.126052
Bucher Della Torre, S., Keller, A., Laure Depeyre, J., and Kruseman, M. (2016). Sugar-sweetened beverages and obesity risk in children and adolescents: a systematic analysis on how methodological quality may influence conclusions. J. Acad. Nutr. Diet. 116, 638–659. doi: 10.1016/j.jand.2015.05.020
Buka, S. L., Tsuang, M. T., Torrey, E. F., Klebanoff, M. A., Wagner, R. L., and Yolken, R. H. (2001). Maternal cytokine levels during pregnancy and adult psychosis. Brain Behav. Immun. 15, 411–420. doi: 10.1006/brbi.2001.0644
Burguera, B., Couce, M. E., Long, J., Lamsam, J., Laakso, K., Jensen, M. D., et al. (2000). The long form of the leptin receptor (OB-Rb) is widely expressed in the human brain. Neuroendocrinology 71, 187–195. doi: 10.1159/000054536
Calne, D. B., and Sandler, M. (1970). L-Dopa and Parkinsonism. Nature 227, 21–24. doi: 10.1038/226021a0
Candler, T., Kühnen, P., Prentice, A. M., and Silver, M. (2019). Epigenetic regulation of POMC; implications for nutritional programming, obesity and metabolic disease. Front. Neuroendocrinol. 54:100773. doi: 10.1016/j.yfrne.2019.100773
Cardoso, A. R., Lopes-Marques, M., Silva, R. M., Serrano, C., Amorim, A., Prata, M. J., et al. (2019). Essential genetic findings in neurodevelopmental disorders. Hum. Genomics 13:31. doi: 10.1186/s40246-019-0216-4
Caruso, C., Carniglia, L., Durand, D., Scimonelli, T. N., and Lasaga, M. (2013). Astrocytes: new targets of melanocortin 4 receptor actions. J. Mol. Endocrinol. 51, R33–R50. doi: 10.1530/JME-13-0064
Castillo, E., Leon, J., Mazzei, G., Abolhassani, N., Haruyama, N., Saito, T., et al. (2017). Comparative profiling of cortical gene expression in Alzheimer’s disease patients and mouse models demonstrates a link between amyloidosis and neuroinflammation. Sci. Rep. 7:17762. doi: 10.1038/s41598-017-17999-3
Cepeda, C., Wu, N., Andre, V., Cummings, D., and Levine, M. (2007). The corticostriatal pathway in Huntington’s disease. Prog. Neurobiol. 81, 253–271. doi: 10.1016/j.pneurobio.2006.11.001
Chaldakov, G. N., Tonchev, A. B., Manni, L., Hristova, M. G., Nikolova, V., Fiore, M., et al. (2007). Comment on: Krabbe KS, Nielsen AR, Krogh-Madsen R et al (2007) Brain-derived neurotrophic factor (BDNF) and type 2 diabetes. Diabetologia 50, 431–438. doi: 10.1007/s00125-007-0706-0
Chandola, T., Deary, I. J., Blane, D., and Batty, G. D. (2006). Childhood IQ in relation to obesity and weight gain in adult life: the National Child Development (1958) Study. Int. J. Obes. 30, 1422–1432. doi: 10.1038/sj.ijo.0803279
Chang, D., Nalls, M. A., Hunkapiller, J., Brug, D., Cai, F., Kerchner, G. A., et al. (2018). A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 49, 1511–1516. doi: 10.1038/ng.3955.A
Chen, S., Zhao, L., Sherchan, P., Ding, Y., Yu, J., Nowrangi, D., et al. (2018). Activation of melanocortin receptor 4 with RO27-3225 attenuates neuroinflammation through AMPK/JNK/p38 MAPK pathway after intracerebral hemorrhage in mice. J. Neuroinflammation 15, 106. doi: 10.1186/s12974-018-1140-6
Cheong, R. Y., Gabery, S., and Petersén, Å. (2019). The role of hypothalamic pathology for non-motor features of Huntington’s disease. J. Huntingtons Dis. 8, 375–391. doi: 10.3233/JHD-190372
Choquet, H., Stijnen, P., and Creemers, J. W. M. (2011). Genetic and functional characterization of PCSK1. Methods Mol. Biol. 768, 247–253. doi: 10.1007/978-1-61779-204-5_13
Church, C., Moir, L., McMurray, F., Girard, C., Banks, G. T., Teboul, L., et al. (2010). Overexpression of Fto leads to increased food intake and results in obesity. Nat. Genet. 42, 1086–1092. doi: 10.1038/ng.713
Claussnitzer, M., Dankel, S. N., Kim, K.-H., Quon, G., Meuleman, W., Haugen, C., et al. (2015). FTO obesity variant circuitry and adipocyte browning in humans. N. Engl. J. Med. 373, 895–907. doi: 10.1056/NEJMoa1502214
Clément, K., Biebermann, H., Farooqi, I. S., Van der Ploeg, L., Wolters, B., Poitou, C., et al. (2018). MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat. Med. 24, 551–555. doi: 10.1038/s41591-018-0015-9
Cone, R. D. (2005). Anatomy and regulation of the central melanocortin system. Nat. Neurosci. 8, 571–578. doi: 10.1038/nn1455
Contu, L., and Hawkes, C. A. (2017). A review of the impact of maternal obesity on the cognitive function and mental health of the offspring. Int. J. Mol. Sci. 18:1093. doi: 10.3390/ijms18051093
Cordner, Z. A., Khambadkone, S. G., Boersma, G. J., Song, L., Summers, T. N., Moran, T. H., et al. (2019). Maternal high-fat diet results in cognitive impairment and hippocampal gene expression changes in rat offspring. Exp. Neurol. 318, 92–100. doi: 10.1016/j.expneurol.2019.04.018
Corella, D., Ortega-Azorín, C., Sorlí, J. V., Covas, M. I., Carrasco, P., Salas-Salvadó, J., et al. (2012). Statistical and biological gene-lifestyle interactions of MC4R and FTO with diet and physical activity on obesity: new effects on alcohol consumption. PLoS One 7:e52344. doi: 10.1371/journal.pone.0052344
Couly, S., Paucard, A., Bonneaud, N., Maurice, T., Benigno, L., Jourdan, C., et al. (2018). Improvement of BDNF signalling by P42 peptide in Huntington’s disease. Hum. Mol. Genet. 27, 3012–3028. doi: 10.1093/hmg/ddy207
Creemers, J. W. M. (2008). Knock-out mouse models of proprotein convertases: unique functions or redundancy? Front. Biosci. 13, 4960–4971. doi: 10.2741/3055
Creemers, J. W. M., Choquet, H., Stijnen, P., Vatin, V., Pigeyre, M., Beckers, S., et al. (2012). Heterozygous mutations causing partial prohormone convertase 1 deficiency contribute to human obesity. Diabetes Metab. Res. Rev. 61, 383–390. doi: 10.2337/db11-0305
Criado, K. K., Sharp, W. G., McCracken, C. E., De Vinck-Baroody, O., Dong, L., Aman, M. G., et al. (2018). Overweight and obese status in children with autism spectrum disorder and disruptive behavior. Autism 22, 450–459. doi: 10.1177/1362361316683888
Dahl, A., Hassing, L. B., Fransson, E., Berg, S., Gatz, M., Reynolds, C. A., et al. (2010). Being overweight in midlife is associated with lower cognitive ability and steeper cognitive decline in late life. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 65A, 57–62. doi: 10.1093/gerona/glp035
Dalle Molle, R., Fatemi, H., Dagher, A., Levitan, R. D., Silveira, P. P., and Dubé, L. (2017). Gene and environment interaction: Is the differential susceptibility hypothesis relevant for obesity? Neurosci. Biobehav. Rev. 73, 326–339. doi: 10.1016/j.neubiorev.2016.12.028
Damiano, S. R., Hart, L. M., and Paxton, S. J. (2016). Correlates of parental feeding practices with pre-schoolers: parental body image and eating knowledge, attitudes, and behaviours. Appetite 101, 192–198. doi: 10.1016/j.appet.2016.03.008
Darmon, N., and Drewnowski, A. (2008). Does social class predict diet quality? Am. J. Clin. Nutr. 87, 1107–1117. doi: 10.1093/ajcn/87.5.1107
Daws, M. R., Lanier, L. L., Seaman, W. E., and Ryan, J. C. (2001). Cloning and characterization of a novel mouse myeloid DAP12-associated receptor family. Eur. J. Immunol. 31, 783–791. doi: 10.1002/1521-4141(200103)31:3<783::aid-immu783>3.0.co;2-u
Deaglio, S., Mehta, K., and Malavasi, F. (2001). Human CD38: a (r)evolutionary story of enzymes and receptors. Leuk. Res. 25, 1–12. doi: 10.1016/S0145-2126(00)00093-X
Debnath, M., Venkatasubramanian, G., and Berk, M. (2015). Fetal programming of schizophrenia: select mechanisms. Neurosci. Biobehav. Rev. 49, 90–104. doi: 10.1016/j.neubiorev.2014.12.003
Desmet, C. J., and Peeper, D. S. (2006). The neurotrophic receptor TrkB: A drug target in anti-cancer therapy? Cell. Mol. Life Sci. 63, 755–759. doi: 10.1007/s00018-005-5490-8
Di Carlo, P., Punzi, G., and Ursini, G. (2019). Brain-derived neurotrophic factor and schizophrenia. Psychiatr. Genet. 29, 200–210. doi: 10.1097/YPG.0000000000000237
Dina, C., Meyre, D., Gallina, S., Durand, E., Körner, A., Jacobson, P., et al. (2007). Variation in FTO contributes to childhood obesity and severe adult obesity. Nat. Genet. 39, 724–726. doi: 10.1038/ng2048
Do, K., Laing, B. T., Landry, T., Bunner, W., Mersaud, N., Matsubara, T., et al. (2018). The effects of exercise on hypothalamic neurodegeneration of Alzheimer’s disease mouse model. PLoS One 13:e0190205. doi: 10.1371/journal.pone.0190205
Donev, R., and Thome, J. (2010). Inflammation: good or bad for ADHD? ADHD Atten. Deficit Hyperact. Disord. 2, 257–266. doi: 10.1007/s12402-010-0038-7
Douglass, J., McKinzie, A., and Couceyro, P. (1995). PCR differential display identifies a rat brain mRNA that is transcriptionally regulated by cocaine and amphetamine. J. Neurosci. 15, 2471–2481. doi: 10.1523/JNEUROSCI.15-03-02471.1995
Duan, W. (2013). Sirtuins: from metabolic regulation to brain aging. Front. Aging Neurosci. 5:36. doi: 10.3389/fnagi.2013.00036
Dunn, A. R., O’Connell, K. M. S., and Kaczorowski, C. C. (2019). Gene-by-environment interactions in Alzheimer’s disease and Parkinson’s disease. Neurosci. Biobehav. Rev. 103, 73–80. doi: 10.1016/j.neubiorev.2019.06.018
Edlow, A. G. (2017). Maternal obesity and neurodevelopmental and psychiatric disorders in offspring. Prenat. Diagn. 37, 95–110. doi: 10.1002/pd.4932
Elias, M. F., Elias, P. K., Sullivan, L. M., Wolf, P. A., and D’Agostino, R. B. (2005). Obesity, diabetes and cognitive deficit: the Framingham Heart Study. Neurobiol. Aging 26, 11–16. doi: 10.1016/j.neurobiolaging.2005.08.019
Elks, C. E., den Hoed, M., Zhao, J. H., Sharp, S. J., Wareham, N. J., Loos, R. J. F., et al. (2012). Variability in the heritability of body mass index: a systematic review and meta-regression. Front. Endocrinol. 3:29. doi: 10.3389/fendo.2012.00029
El-Lebedy, D., Raslan, H. M., and Mohammed, A. M. (2016). Apolipoprotein E gene polymorphism and risk of type 2 diabetes and cardiovascular disease. Cardiovasc. Diabetol. 15:12. doi: 10.1186/s12933-016-0329-1
Elosua, R., Demissie, S., Cupples, L. A., Meigs, J. B., Wilson, P. W. F., Schaefer, E. J., et al. (2003). Obesity modulates the association among APOE genotype, insulin, and glucose in men. Obes. Res. 11, 1502–1508. doi: 10.1038/oby.2003.201
Ervin, R. B., and Ogden, C. L. (2013). Consumption of added sugars among U.S. adults, 2005-2010. NCHS Data Brief. 122, 1–8.
Fanzo, J., and Davis, C. (2019). Can Diets Be Healthy, Sustainable, and Equitable? Curr. Obes. Rep. 8, 495–503. doi: 10.1007/s13679-019-00362-0
Farooqi, I. S., Keogh, J. M., Yeo, G. S. H., Lank, E. J., Cheetham, T., and O’Rahilly, S. (2003). Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N. Engl. J. Med. 348, 1085–1095. doi: 10.1056/NEJMoa022050
Farooqi, I. S., Volders, K., Stanhope, R., Heuschkel, R., White, A., Lank, E., et al. (2007). Hyperphagia and early-onset obesity due to a novel homozygous missense mutation in prohormone convertase 1/3. J. Clin. Endocrinol. Metab. 92, 3369–3373. doi: 10.1210/jc.2007-0687
Farooqi, I. S., Yeo, G. S. H., Keogh, J. M., Aminian, S., Jebb, S. A., Butler, G., et al. (2000). Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J. Clin. Invest. 106, 271–279. doi: 10.1172/JCI9397
Feinbloom, D., and Bauer, K. A. (2005). Assessment of hemostatic risk factors in predicting arterial thrombotic events. Arterioscler. Thromb. Vasc. Biol. 25, 2043–2053. doi: 10.1161/01.ATV.0000181762.31694.da
Fischer, J., Koch, L., Emmerling, C., Vierkotten, J., Peters, T., Brüning, J. C., et al. (2009). Inactivation of the Fto gene protects from obesity. Nature 458, 894–898. doi: 10.1038/nature07848
Francis, S. M., Sagar, A., Levin-Decanini, T., Liu, W., Carter, C. S., and Jacob, S. (2014). Oxytocin and vasopressin systems in genetic syndromes and neurodevelopmental disorders. Brain Res. 1580, 199–218. doi: 10.1016/j.brainres.2014.01.021
Frayling, T. M. (2007). Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nat. Rev. Genet. 8, 657–662. doi: 10.1038/nrg2178
Frayling, T. M., Timpson, N. J., Weedon, M. N., Zeggini, E., Freathy, R. M., Lindgren, C. M., et al. (2007). A Common Variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889–894. doi: 10.1126/science.1141634
Frederich, R. C., Hamann, A., Anderson, S., Löllmann, B., Lowell, B. B., and Flier, J. S. (1995). Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat. Med. 1, 1311–1314. doi: 10.1038/nm1295-1311
Friedman, J. M., and Halaas, J. L. (1998). Leptin and the regulation of body weight in mammals. Nature 395, 763–770. doi: 10.1038/27376
Fujimoto, S., Goda, T., and Mochizuki, K. (2011). In vivo evidence of enhanced di-methylation of histone H3 K4 on upregulated genes in adipose tissue of diabetic db/db mice. Biochem. Biophys. Res. Commun. 404, 223–227. doi: 10.1016/j.bbrc.2010.11.097
Gabery, S., Murphy, K., Schultz, K., Loy, C. T., McCusker, E., Kirik, D., et al. (2010). Changes in key hypothalamic neuropeptide populations in Huntington disease revealed by neuropathological analyses. Acta Neuropathol. 120, 777–788. doi: 10.1007/s00401-010-0742-6
Gadiraju, T., Patel, Y., Gaziano, J., and Djoussé, L. (2015). Fried food consumption and cardiovascular health: a review of current evidence. Nutrients 7, 8424–8430. doi: 10.3390/nu7105404
Gamero-Villarroel, C., González, L. M., Gordillo, I., Carrillo, J. A., García-Herráiz, A., Flores, I., et al. (2015). Impact of NEGR1 genetic variability on psychological traits of patients with eating disorders. Pharmacogenomics J. 15, 278–283. doi: 10.1038/tpj.2014.53
Gantz, I., Miwa, H., Konda, Y., Shimoto, Y., Tashiro, T., Watson, S. J., et al. (1993). Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J. Biol. Chem. 268, 15174–15179.
Gantz, I., Shimoto, Y., Konda, Y., Miwa, H., Dickinson, C. J., and Yamada, T. (1994). Molecular cloning, expression, and characterization of a fifth melanocortin receptor. Biochem. Biophys. Res. Commun. 200, 1214–1220. doi: 10.1006/bbrc.1994.1580
Gao, H., Tao, Y., He, Q., Song, F., and Saffen, D. (2015). Functional enrichment analysis of three Alzheimer’s disease genome-wide association studies identities DAB1 as a novel candidate liability/protective gene. Biochem. Biophys. Res. Commun. 463, 490–495. doi: 10.1016/j.bbrc.2015.05.044
Gao, X., Shin, Y. H., Li, M., Wang, F., Tong, Q., and Zhang, P. (2010). The fat mass and obesity associated gene FTO functions in the brain to regulate postnatal growth in mice. PLoS One 5:e14005. doi: 10.1371/journal.pone.0014005
Gerken, T., Girard, C. A., Tung, Y.-C. L., Webby, C. J., Saudek, V., Hewitson, K. S., et al. (2007). The Obesity-Associated FTO Gene Encodes a 2-Oxoglutarate-Dependent Nucleic Acid Demethylase. Science 318, 1469–1472. doi: 10.1126/science.1151710
Ghavami, A., Hunt, R. A., Olsen, M. A., Zhang, J., Smith, D. L., Kalgaonkar, S., et al. (2004). Differential effects of regulator of G protein signaling (RGS) proteins on serotonin 5-HT1A, 5-HT2A, and dopamine D2 receptor-mediated signaling and adenylyl cyclase activity. Cell. Signal. 16, 711–721. doi: 10.1016/j.cellsig.2003.11.006
Giles-Corti, B. (2002). Socioeconomic status differences in recreational physical activity levels and real and perceived access to a supportive physical environment. Prev. Med. 35, 601–611. doi: 10.1006/pmed.2002.1115
Giuliani, D., Zaffe, D., Ottani, A., Spaccapelo, L., Galantucci, M., Minutoli, L., et al. (2011). Treatment of cerebral ischemia with melanocortins acting at MC4 receptors induces marked neurogenesis and long-lasting functional recovery. Acta Neuropathol. 122, 443–453. doi: 10.1007/s00401-011-0873-4
Goffe, L., Rushton, S., White, M., Adamson, A., and Adams, J. (2017). Relationship between mean daily energy intake and frequency of consumption of out-of-home meals in the UK National Diet and Nutrition Survey. Int. J. Behav. Nutr. Phys. Act. 14:131. doi: 10.1186/s12966-017-0589-5
Goldman, J. S., and Van Deerlin, V. M. (2018). Alzheimer’s disease and frontotemporal dementia: the current state of genetics and genetic testing since the advent of next-generation sequencing. Mol. Diagn. Ther. 22, 505–513. doi: 10.1007/s40291-018-0347-7
Grant, R. W., Vester Boler, B. M., Ridge, T. K., Graves, T. K., and Swanson, K. S. (2011). Adipose tissue transcriptome changes during obesity development in female dogs. Physiol. Genomics 43, 295–307. doi: 10.1152/physiolgenomics.00190.2010
Gray, J., Yeo, G., Hung, C., Keogh, J., Clayton, P., Banerjee, K., et al. (2007). Functional characterization of human NTRK2 mutations identified in patients with severe early-onset obesity. Int. J. Obes. 31, 359–364. doi: 10.1038/sj.ijo.0803390
Gubert, C., Kong, G., Renoir, T., and Hannan, A. J. (2020). Exercise, diet and stress as modulators of gut microbiota: implications for neurodegenerative diseases. Neurobiol. Dis. 134:104621. doi: 10.1016/j.nbd.2019.104621
Guerreiro, S., Privat, A.-L., Bressac, L., and Toulorge, D. (2020). CD38 in neurodegeneration and neuroinflammation. Cells 9:471. doi: 10.3390/cells9020471
Gutierrez-Aguilar, R., Kim, D.-H., Casimir, M., Dai, X.-Q., Pfluger, P. T., Park, J., et al. (2014). The role of the transcription factor ETV5 in insulin exocytosis. Diabetologia 57, 383–391. doi: 10.1007/s00125-013-3096-5
Gutierrez-Aguilar, R., Kim, D.-H., Woods, S. C., and Seeley, R. J. (2012). Expression of new loci associated with obesity in diet-induced obese rats: from genetics to physiology. Obesity 20, 306–312. doi: 10.1038/oby.2011.236
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Hajeer, A. H., and Hutchinson, I. V. (2000). TNFα gene polymorphism: clinical and biological implications. Microsc. Res. Tech. 50, 216–228. doi: 10.1002/1097-0029(20000801)50:3<216::aid-jemt5>3.0.co;2-q
Hajeer, A. H., and Hutchinson, I. V. (2001). Influence of TNFα gene polymorphisms on TNFα production and disease. Hum. Immunol. 62, 1191–1199. doi: 10.1016/S0198-8859(01)00322-6
Hall, S. S., Lightbody, A. A., Huffman, L. C., Lazzeroni, L. C., and Reiss, A. L. (2009). Physiological correlates of social avoidance behavior in children and adolescents with fragile X syndrome. J. Am. Acad. Child Adolesc. Psychiatry 48, 320–329. doi: 10.1097/CHI.0b013e318195bd15
Hanć, T., and Cortese, S. (2018). Attention deficit/hyperactivity-disorder and obesity: a review and model of current hypotheses explaining their comorbidity. Neurosci. Biobehav. Rev. 92, 16–28. doi: 10.1016/j.neubiorev.2018.05.017
Hassing, L. B., Dahl, A. K., Pedersen, N. L., and Johansson, B. (2010). Overweight in midlife is related to lower cognitive function 30 years later: a prospective study with longitudinal assessments. Dement. Geriatr. Cogn. Disord. 29, 543–552. doi: 10.1159/000314874
Hatziri, A., Kalogeropoulou, C., Xepapadaki, E., Birli, E., Karavia, E. A., Papakosta, E., et al. (2018). Site-specific effects of apolipoprotein E expression on diet-induced obesity and white adipose tissue metabolic activation. Biochim. Biophys. Acta 1864, 471–480. doi: 10.1016/j.bbadis.2017.11.007
Heard-Costa, N. L., Zillikens, M. C., Monda, K. L., Johansson, Å., Harris, T. B., Fu, M., et al. (2009). NRXN3 is a novel locus for waist circumference: a genome-wide association study from the CHARGE consortium. PLoS Genet. 5:e1000539. doi: 10.1371/journal.pgen.1000539
Heianza, Y., and Qi, L. (2017). Gene-diet interaction and precision nutrition in obesity. Int. J. Mol. Sci. 18:787. doi: 10.3390/ijms18040787
Heianza, Y., and Qi, L. (2019). Impact of genes and environment on obesity and cardiovascular disease. Endocrinology 160, 81–100. doi: 10.1210/en.2018-00591
Heid, I. M., Jackson, A. U., Randall, J. C., Winkler, T. W., Qi, L., Ssteinthorsdottir, V., et al. (2010). Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat. Genet. 42, 949–960. doi: 10.1038/ng.685
Henriksen, M. G., Nordgaard, J., and Jansson, L. B. (2017). Genetics of schizophrenia: overview of methods, findings and limitations. Front. Hum. Neurosci. 11:322. doi: 10.3389/fnhum.2017.00322
Herranz, D., and Serrano, M. (2010). SIRT1: recent lessons from mouse models. Nat. Rev. Cancer 10, 819–823. doi: 10.1038/nrc2962
Herskovits, A. Z., and Guarente, L. (2014). SIRT1 in neurodevelopment and brain senescence. Neuron 81, 471–483. doi: 10.1016/j.neuron.2014.01.028
Hill, J. W., and Faulkner, L. D. (2017). The role of the melanocortin system in metabolic disease: new developments and advances. Neuroendocrinology 104, 330–346. doi: 10.1159/000450649
Hinz, F. I., and Geschwind, D. H. (2017). Molecular genetics of neurodegenerative dementias. Cold Spring Harb. Perspect. Biol. 9:a023705. doi: 10.1101/cshperspect.a023705
Hishimoto, A., Pletnikova, O., Lang, D. L., Troncoso, J. C., Egan, J. M., and Liu, Q.-R. (2019). Neurexin 3 transmembrane and soluble isoform expression and splicing haplotype are associated with neuron inflammasome and Alzheimer’s disease. Alzheimers Res. Ther. 11:28. doi: 10.1186/s13195-019-0475-2
Hofer, M., Pagliusi, S. R., Hohn, A., Leibrock, J., and Barde, Y. A. (1990). Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J. 9, 2459–2464. doi: 10.1002/j.1460-2075.1990.tb07423.x
Hokama, M., Oka, S., Leon, J., Ninomiya, T., Honda, H., Sasaki, K., et al. (2014). Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb. Cortex 24, 2476–2488. doi: 10.1093/cercor/bht101
Howard, M., Grimaldi, J., Bazan, J., Lund, F., Santos-Argumedo, L., Parkhouse, R., et al. (1993). Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 262, 1056–1059. doi: 10.1126/science.8235624
Hruby, A., Manson, J. E., Qi, L., Malik, V. S., Rimm, E. B., Sun, Q., et al. (2016). Determinants and consequences of obesity. Am. J. Public Health 106, 1656–1662. doi: 10.2105/AJPH.2016.303326
Hu, X., Zhang, J., Jin, C., Mi, W., Wang, F., Ma, W., et al. (2013). Association study of NRXN3 polymorphisms with schizophrenia and risperidone-induced bodyweight gain in Chinese Han population. Prog. Neuropsychopharmacol. Biol. Psychiatry 43, 197–202. doi: 10.1016/j.pnpbp.2012.12.007
Huang, T., Qi, Q., Li, Y., Hu, F. B., Bray, G. A., Sacks, F. M., et al. (2014). FTO genotype, dietary protein, and change in appetite: the Preventing Overweight Using Novel Dietary Strategies trial. Am. J. Clin. Nutr. 99, 1126–1130. doi: 10.3945/ajcn.113.082164
Huang, Y., and Mahley, R. W. (2014). Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 72, 3–12. doi: 10.1016/j.nbd.2014.08.025
Hughes, S. O., and Frazier-Wood, A. C. (2016). Satiety and the self-regulation of food take in children: a potential role for gene-environment interplay. Curr. Obes. Rep. 5, 81–87. doi: 10.1007/s13679-016-0194-y
Ignatieva, E. V., Afonnikov, D. A., Saik, O. V., Rogaev, E. I., and Kolchanov, N. A. (2016). A compendium of human genes regulating feeding behavior and body weight, its functional characterization and identification of GWAS genes involved in brain-specific PPI network. BMC Genet. 17:158. doi: 10.1186/s12863-016-0466-2
Irving, A. J., and Harvey, J. (2014). Leptin regulation of hippocampal synaptic function in health and disease. Philos. Trans. R. Soc. B Biol. Sci. 369:20130155. doi: 10.1098/rstb.2013.0155
Ishikawa, J., Ishikawa, A., and Nakamura, S. (2007). Interferon-?? reduces the density of monoaminergic axons in the rat brain. Neuroreport 18, 137–140. doi: 10.1097/WNR.0b013e328010231a
Ito, S., D’Alessio, A. C., Taranova, O. V., Hong, K., Sowers, L. C., and Zhang, Y. (2010). Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133. doi: 10.1038/nature09303
Jackson, D. G., and Bell, J. I. (1990). Isolation of a cDNA encoding the human CD38 (T10) molecule, a cell surface glycoprotein with an unusual discontinuous pattern of expression during lymphocyte differentiation. J. Immunol. 144, 2811–2815.
Jackson, R. S., Creemers, J. W. M., Ohagi, S., Raffin-Sanson, M.-L., Sanders, L., Montague, C. T., et al. (1997). Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat. Genet. 16, 303–306. doi: 10.1038/ng0797-303
Jansen, I. E., Savage, J. E., Watanabe, K., Bryois, J., Williams, D. M., Steinberg, S., et al. (2019). Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413.
Jéquier, E. (2006). Leptin Signaling, Adiposity, and Energy Balance. Ann. N. Y. Acad. Sci. 967, 379–388. doi: 10.1111/j.1749-6632.2002.tb04293.x
Jia, G., Fu, Y., Zhao, X., Dai, Q., Zheng, G., Yang, Y., et al. (2011). N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887. doi: 10.1038/nchembio.687
Jin, W. (2020). Regulation of BDNF-TrkB Signaling and Potential Therapeutic Strategies for Parkinson’s Disease. J. Clin. Med. 9:257. doi: 10.3390/jcm9010257
Jones, N. S., and Rebeck, G. W. (2019). The synergistic effects of APOE genotype and obesity on Alzheimer’s Disease risk. Int. J. Mol. Sci. 20:63. doi: 10.3390/ijms20010063
Joo, Y., Kim, H., Lee, S., and Lee, S. (2019). Neuronal growth regulator 1-deficient mice show increased adiposity and decreased muscle mass. Int. J. Obes. 43, 1769–1782. doi: 10.1038/s41366-019-0376-2
Jung, Y., Kim, Y., Bhalla, M., Lee, S., and Seo, J. (2018). Genomics: new light on Alzheimer’s disease research. Int. J. Mol. Sci. 19:3771. doi: 10.3390/ijms19123771
Karch, C. M., Ezerskiy, L. A., Bertelsen, S., Goate, A. M., Albert, M. S., Albin, R. L., et al. (2016). Alzheimer’s disease risk polymorphisms regulate gene expression in the ZCWPW1 and the CELF1 loci. PLoS One 11:148717. doi: 10.1371/journal.pone.0148717
Karimi-Moghadam, A., Charsouei, S., Bell, B., and Jabalameli, M. R. (2018). Parkinson disease from mendelian forms to genetic susceptibility: new molecular insights into the neurodegeneration process. Cell. Mol. Neurobiol. 38, 1153–1178. doi: 10.1007/s10571-018-0587-4
Karis, K., Eskla, K.-L., Kaare, M., Täht, K., Tuusov, J., Visnapuu, T., et al. (2018). Altered expression profile of IgLON family of neural cell adhesion molecules in the dorsolateral prefrontal cortex of schizophrenic patients. Front. Mol. Neurosci. 11:8. doi: 10.3389/fnmol.2018.00008
Kawazoe, T., Yamamoto, T., Narita, A., Ohno, K., Adachi, K., Nanba, E., et al. (2018). Phenotypic variability of Niemann-Pick disease type C including a case with clinically pure schizophrenia: a case report. BMC Neurol. 18:117. doi: 10.1186/s12883-018-1124-2
Kema, V. H., Mojerla, N. R., Khan, I., and Mandal, P. (2015). Effect of alcohol on adipose tissue: a review on ethanol mediated adipose tissue injury. Adipocyte 4, 225–231. doi: 10.1080/21623945.2015.1017170
Kernie, S. G. (2000). BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 19, 1290–1300. doi: 10.1093/emboj/19.6.1290
Khera, A. V., Chaffin, M., Wade, K. H., Zahid, S., Brancale, J., Xia, R., et al. (2019). Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell 177, 587–596.e9. doi: 10.1016/j.cell.2019.03.028
Kim, H., Chun, Y., Che, L., Kim, J., Lee, S., and Lee, S. (2017). The new obesity-associated protein, neuronal growth regulator 1 (NEGR1), is implicated in Niemann-Pick disease Type C (NPC2)-mediated cholesterol trafficking. Biochem. Biophys. Res. Commun. 482, 1367–1374. doi: 10.1016/j.bbrc.2016.12.043
Kim, Y.-J. (2016). The long-run effect of education on obesity in the US. Econ. Hum. Biol. 21, 100–109. doi: 10.1016/j.ehb.2015.12.003
King, G., and Sharom, F. J. (2012). Proteins that bind and move lipids: MsbA and NPC1. Crit. Rev. Biochem. Mol. Biol. 47, 75–95. doi: 10.3109/10409238.2011.636505
Kishi, T., Fukuo, Y., Kitajima, T., Okochi, T., Yamanouchi, Y., Kinoshita, Y., et al. (2011). SIRT1 gene, schizophrenia and bipolar disorder in the Japanese population: an association study. Genes. Brain. Behav. 10, 257–263. doi: 10.1111/j.1601-183X.2010.00661.x
Klein, R., Conway, D., Parada, L. F., and Barbacid, M. (1990). The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell 61, 647–656. doi: 10.1016/0092-8674(90)90476-U
Kleinendorst, L., Massink, M. P. G., Cooiman, M. I., Savas, M., van der Baan-Slootweg, O. H., Roelants, R. J., et al. (2018). Genetic obesity: next-generation sequencing results of 1230 patients with obesity. J. Med. Genet. 55, 578–586. doi: 10.1136/jmedgenet-2018-105315
Kober, D. L., and Brett, T. J. (2017). TREM2-Ligand Interactions in Health and Disease. J. Mol. Biol. 429, 1607–1629. doi: 10.1016/j.jmb.2017.04.004
Kowiañski, P., Lietzau, G., Czuba, E., Waśkow, M., Steliga, A., and Moryś, J. (2018). BDNF: a key factor with multipotent impact on brain signaling and synaptic plasticity. Cell. Mol. Neurobiol. 38, 579–593. doi: 10.1007/s10571-017-0510-4
Krude, H., Biebermann, H., Luck, W., Horn, R., Brabant, G., and Grüters, A. (1998). Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 19, 155–157. doi: 10.1038/509
Kumar, K. P., Srikrishna, S., Pavan, I., and Chary, E. (2018). Prevalence of picky eating behavior and its impact on growth in preschool children. Int. J. Contemp. Pediatr. 5:714. doi: 10.18203/2349-3291.ijcp20181036
Lachén-Montes, M., González-Morales, A., Iloro, I., Elortza, F., Ferrer, I., Gveric, D., et al. (2019). Unveiling the olfactory proteostatic disarrangement in Parkinson’s disease by proteome-wide profiling. Neurobiol. Aging 73, 123-134. doi: 10.1016/j.neurobiolaging.2018.09.018
Lambert, J.-C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Lamri, A., Pigeyre, M., Garver, W. S., and Meyre, D. (2018). The extending spectrum of NPC1-Related human disorders: from Niemann-Pick C1 disease to obesity. Endocr. Rev. 39, 192–220. doi: 10.1210/er.2017-00176
Lebrun, B., Bariohay, B., Moyse, E., and Jean, A. (2006). Brain-derived neurotrophic factor (BDNF) and food intake regulation: a minireview. Auton. Neurosci. 126-127, 30–38. doi: 10.1016/j.autneu.2006.02.027
Lee, E. B., and Mattson, M. P. (2014). The neuropathology of obesity: insights from human disease. Acta Neuropathol. 127, 3–28. doi: 10.1007/s00401-013-1190-x
Lee, J.-M., Wheeler, V. C., Chao, M. J., Vonsattel, J. P. G., Pinto, R. M., Lucente, D., et al. (2015). Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 162, 516–526. doi: 10.1016/j.cell.2015.07.003
Lemche, E. (2018). Early life stress and epigenetics in late-onset Alzheimer’s dementia: a systematic review. Curr. Genomics 19, 522–602. doi: 10.2174/1389202919666171229145156
Levasseur, P. (2015). Causal effects of socioeconomic status on central adiposity risks: evidence using panel data from urban Mexico. Soc. Sci. Med. 136–137, 165–174. doi: 10.1016/j.socscimed.2015.05.018
Li, C., Wu, X., Liu, S., Zhao, Y., Zhu, J., and Liu, K. (2019). Roles of neuropeptide Y in neurodegenerative and neuroimmune diseases. Front. Neurosci. 13:869. doi: 10.3389/fnins.2019.00869
Li, H., Ren, Y., Mao, K., Hua, F., Yang, Y., Wei, N., et al. (2018). FTO is involved in Alzheimer’s disease by targeting TSC1-mTOR-Tau signaling. Biochem. Biophys. Res. Commun. 498, 234–239. doi: 10.1016/j.bbrc.2018.02.201
Li, M., Shin, J., Risgaard, R. D., Parries, M. J., Wang, J., Chasman, D., et al. (2020). Identification of FMR1-regulated molecular networks in human neurodevelopment. Genome Res. 30, 361–374. doi: 10.1101/gr.251405.119
Li, S., Zhao, J. H., Luan, J., Ekelund, U., Luben, R. N., Khaw, K.-T., et al. (2010). Physical activity attenuates the genetic predisposition to obesity in 20,000 men and women from EPIC-norfolk prospective population study. PLoS Med. 7:e1000332. doi: 10.1371/journal.pmed.1000332
Li, T., Yang, D., Li, J., Tang, Y., Yang, J., and Le, W. (2015). Critical Role of Tet3 in neural progenitor cell maintenance and terminal differentiation. Mol. Neurobiol. 51, 142–154. doi: 10.1007/s12035-014-8734-5
Li, X., and Qi, L. (2019). Gene–environment interactions on body fat distribution. Int. J. Mol. Sci. 20:3690. doi: 10.3390/ijms20153690
Liangpunsakul, S., Crabb, D. W., and Qi, R. (2010). Relationship among alcohol intake, body fat, and physical activity: a population-based study. Ann. Epidemiol. 20, 670–675. doi: 10.1016/j.annepidem.2010.05.014
Liao, G.-Y., Kinney, C. E., An, J. J., and Xu, B. (2019). TrkB-expressing neurons in the dorsomedial hypothalamus are necessary and sufficient to suppress homeostatic feeding. Proc. Natl. Acad. Sci. U.S.A. 116, 3256–3261. doi: 10.1073/pnas.1815744116
Lin, L., Sun, D., Chang, J., Ma, M., Zhou, X., Zhao, M., et al. (2018). Cocaine- and amphetamine-regulated transcript (CART) is associated with dopamine and is protective against ischemic stroke. Mol. Med. Rep. 18, 3298–3304. doi: 10.3892/mmr.2018.9296
Liu, C., Li, P., Li, H., Wang, S., Ding, L., Wang, H., et al. (2019). TREM2 regulates obesity-induced insulin resistance via adipose tissue remodeling in mice of high-fat feeding. J. Transl. Med. 17:300. doi: 10.1186/s12967-019-2050-9
Liu, C. C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein e and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
Liu, H., Yao, Y.-M., Yu, Y., Dong, N., Yin, H.-N., and Sheng, Z.-Y. (2007). Role of Janus kinase/signal transducer and activator of transcription pathway in regulation of expression and inflammation-promoting activity of high mobility group box protein 1 in rat peritoneal macrophages. Shock 27, 55–60. doi: 10.1097/01.shk.0000233197.40989.31
Locke, A. E., Kahali, B., Berndt, S. I., Justice, A. E., Pers, T. H., Day, F. R., et al. (2015). Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206. doi: 10.1038/nature14177
Loos, R. J. F., and Janssens, A. C. J. W. (2017). Predicting polygenic obesity using genetic information. Cell Metab. 25, 535–543. doi: 10.1016/j.cmet.2017.02.013
Luberg, K., Wong, J., Weickert, C. S., and Timmusk, T. (2010). Human TrkB gene: novel alternative transcripts, protein isoforms and expression pattern in the prefrontal cerebral cortex during postnatal development. J. Neurochem. 113, 952–964. doi: 10.1111/j.1471-4159.2010.06662.x
Lutz, M. I., Milenkovic, I., Regelsberger, G., and Kovacs, G. G. (2014). Distinct patterns of sirtuin expression during progression of Alzheimer’s disease. Neuromol. Med. 16, 405–414. doi: 10.1007/s12017-014-8288-8
Lv, X., Jiang, H., Liu, Y., Lei, X., and Jiao, J. (2014). Micro RNA -15b promotes neurogenesis and inhibits neural progenitor proliferation by directly repressing TET 3 during early neocortical development. EMBO Rep. 15, 1305–1314. doi: 10.15252/embr.201438923
Maher, G. M., O’Keeffe, G. W., Kenny, L. C., Kearney, P. M., Dinan, T. G., and Khashan, A. S. (2017). Hypertensive disorders of pregnancy and risk of neurodevelopmental disorders in the offspring: a systematic review and meta-analysis protocol. BMJ Open 7:e018313. doi: 10.1136/bmjopen-2017-018313
Manu, P., Dima, L., Shulman, M., Vancampfort, D., De Hert, M., and Correll, C. U. (2015). Weight gain and obesity in schizophrenia: epidemiology, pathobiology, and management. Acta Psychiatr. Scand. 132, 97–108. doi: 10.1111/a.12445
Martínez-Cerdeño, V., Lechpammer, M., Noctor, S., Ariza, J., Hagerman, P., and Hagerman, R. (2017). FMR1 premutation with Prader-Willi phenotype and fragile X-associated tremor/ataxia syndrome. Clin. Case Rep. 5, 625–629. doi: 10.1002/ccr3.834
Masuzaki, H., Ogawa, Y., Isse, N., Satoh, N., Okazaki, T., Shigemoto, M., et al. (1995). Human obese gene expression: adipocyte-specific expression and regional differences in the adipose tissue. Diabetes Metab. Res. Rev. 44, 855–858. doi: 10.2337/diab.44.7.855
Mazon, J. N., de Mello, A. H., Ferreira, G. K., and Rezin, G. T. (2017). The impact of obesity on neurodegenerative diseases. Life Sci. 182, 22–28. doi: 10.1016/j.lfs.2017.06.002
Merriam-Webster Dictionary (2020). Merriam-Webster.com Dictionary, s.v. “heritability,”. Available online at: https://www.merriam-webster.com/dictionary/heritability (accessed August 17, 2020).
Meyre, D., Delplanque, J., Chèvre, J.-C., Lecoeur, C., Lobbens, S., Gallina, S., et al. (2009). Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat. Genet. 41, 157–159. doi: 10.1038/ng.301
Michaud, J. L. (2001). Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum. Mol. Genet. 10, 1465–1473. doi: 10.1093/hmg/10.14.1465
Michaud, J. L., Rosenquist, T., May, N. R., and Fan, C.-M. (1998). Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes Dev. 12, 3264–3275. doi: 10.1101/gad.12.20.3264
Mina, T. H., Lahti, M., Drake, A. J., Denison, F. C., Räikkönen, K., Norman, J. E., et al. (2017). Prenatal exposure to maternal very severe obesity is associated with impaired neurodevelopment and executive functioning in children. Pediatr. Res. 82, 47–54. doi: 10.1038/pr.2017.43
Mitsuya, K., Parker, A. N., Liu, L., Ruan, J., Vissers, M. C. M., and Myatt, L. (2017). Alterations in the placental methylome with maternal obesity and evidence for metabolic regulation. PLoS One 12:e0186115. doi: 10.1371/journal.pone.0186115
Morris, D. L., and Rui, L. (2009). Recent advances in understanding leptin signaling and leptin resistance. Am. J. Physiol. Metab. 297, E1247–E1259. doi: 10.1152/ajpendo.00274.2009
Morton, G. J., Meek, T. H., and Schwartz, M. W. (2014). Neurobiology of food intake in health and disease. Nat. Rev. Neurosci. 15, 367–378. doi: 10.1038/nrn3745
Moss, D. J. H., Pardiñas, A. F., Langbehn, D., Lo, K., Leavitt, B. R., Roos, R., et al. (2017). Identification of genetic variants associated with Huntington’s disease progression: a genome-wide association study. Lancet Neurol. 16, 701–711. doi: 10.1016/S1474-4422(17)30161-8
Mountjoy, K. G. (2015). Pro-Opiomelanocortin (POMC) Neurones, POMC-derived peptides, melanocortin receptors and obesity: how understanding of this system has changed over the last decade. J. Neuroendocrinol. 27, 406–418. doi: 10.1111/jne.12285
Muller, L., and Lindberg, I. (1999). The cell biology of the prohormone convertases PCI and PC2. Prog. Nucleic Acid Res. Mol. Biol. 63, 69–108. doi: 10.1016/S0079-6603(08)60720-5
Mutch, D. M., Tordjman, J., Pelloux, V., Hanczar, B., Henegar, C., Poitou, C., et al. (2009). Needle and surgical biopsy techniques differentially affect adipose tissue gene expression profiles. Am. J. Clin. Nutr. 89, 51–57. doi: 10.3945/ajcn.2008.26802
Nagpal, S., Gibson, G., and Marigorta, U. (2018). Pervasive modulation of obesity risk by the environment and genomic background. Genes 9:411. doi: 10.3390/genes9080411
Nair, S., Lee, Y. H., Rousseau, E., Cam, M., Tataranni, P. A., Baier, L. J., et al. (2005). Increased expression of inflammation-related genes in cultured preadipocytes/stromal vascular cells from obese compared with non-obese Pima Indians. Diabetologia 48, 1784–1788. doi: 10.1007/s00125-005-1868-2
Nalls, M. A., Pankratz, N., Lill, C. M., Do, C. B., Hernandez, D. G., Saad, M., et al. (2014). Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989–993. doi: 10.1530/ERC-14-0411.Persistent
Nam, G. E., Kim, S. M., Han, K., Kim, N. H., Chung, H. S., Kim, J. W., et al. (2018). Metabolic syndrome and risk of Parkinson disease: a nationwide cohort study. PLoS Med. 15:e1002640. doi: 10.1371/journal.pmed.1002640
National Obesity Observatory (2012). Adult Obesity and Socioeconomic Status. Oxford: National Obesity Observatory.
Newton, J., Milstien, S., and Spiegel, S. (2018). Niemann-Pick type C disease: the atypical sphingolipidosis. Adv. Biol. Regul. 70, 82–88. doi: 10.1016/j.jbior.2018.08.001
Ni, H., Xu, M., Zhan, G. L., Fan, Y., Zhou, H., Jiang, H. Y., et al. (2018). The GWAS risk genes for depression may be actively involved in Alzheimer’s disease. J. Alzheimers Dis. 64, 1–13. doi: 10.3233/JAD-180276
Nieto, C., Rodríguez, E., Sánchez-Bazán, K., Tolentino-Mayo, L., Carriedo-Lutzenkirchen, A., Vandevijvere, S., et al. (2019). The INFORMAS healthy food environment policy index (Food-EPI) in Mexico: an assessment of implementation gaps and priority recommendations. Obes. Rev. 20, 67–77. doi: 10.1111/obr.12814
Nogueiras, R., Habegger, K. M., Chaudhary, N., Finan, B., Banks, A. S., Dietrich, M. O., et al. (2012). Sirtuin 1 and Sirtuin 3: physiological modulators of metabolism. Physiol. Rev. 92, 1479–1514. doi: 10.1152/physrev.00022.2011
Nordang, G. B. N., Busk, Ø. L., Tveten, K., Hanevik, H. I., Fell, A. K. M., Hjelmesćth, J., et al. (2017). Next-generation sequencing of the monogenic obesity genes LEP, LEPR, MC4R, PCSK1 and POMC in a Norwegian cohort of patients with morbid obesity and normal weight controls. Mol. Genet. Metab. 121, 51–56. doi: 10.1016/j.ymgme.2017.03.007
O’Brien, P. D., Hinder, L. M., Callaghan, B. C., and Feldman, E. L. (2017). Neurological consequences of obesity. Lancet Neurol. 16, 465–477. doi: 10.1016/S1474-4422(17)30084-4
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., et al. (2003). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. doi: 10.1016/s0896-6273(03)00434-3
O’Donnell, W. T., and Warren, S. T. (2002). A decade of molecular studies of fragile X syndrome. Annu. Rev. Neurosci. 25, 315–338. doi: 10.1146/annurev.neuro.25.112701.142909
Ogden, C. L., Fryar, C. D., Hales, C. M., Carroll, M. D., Aoki, Y., and Freedman, D. S. (2018). Differences in obesity prevalence by demographics and urbanization in US children and adolescents, 2013-2016. JAMA 319, 2410–2418. doi: 10.1001/jama.2018.5158
Oishi, K. (2009). Plasminogen activator inhibitor-1 and the circadian clock in metabolic disorders. Clin. Exp. Hypertens. 31, 208–219. doi: 10.1080/10641960902822468
Olsen, N. J., Ängquist, L., Larsen, S. C., Linneberg, A., Skaaby, T., Husemoen, L. L. N., et al. (2016). Interactions between genetic variants associated with adiposity traits and soft drinks in relation to longitudinal changes in body weight and waist circumference. Am. J. Clin. Nutr. 104, 816–826. doi: 10.3945/ajcn.115.122820
O’Rahilly, S., and Farooqi, I. S. (2006). Genetics of obesity. Philos. Trans. R. Soc. B Biol. Sci. 361, 1095–1105. doi: 10.1098/rstb.2006.1850
O’Rahilly, S., Gray, H., Humphreys, P. J., Krook, A., Polonsky, K. S., White, A., et al. (1995). Impaired processing of prohormones associated with abnormalities of glucose homeostasis and adrenal function. N. Engl. J. Med. 333, 1386–1391. doi: 10.1056/NEJM199511233332104
Osterberg, N., Wiehle, M., Oehlke, O., Heidrich, S., Xu, C., Fan, C.-M., et al. (2011). Sim1 is a novel regulator in the differentiation of mouse dorsal raphe serotonergic neurons. PLoS One 6:e19239. doi: 10.1371/journal.pone.0019239
Pan, H., Zhao, Y., Zhai, Z., Zheng, J., Zhou, Y., Zhai, Q., et al. (2018). Role of plasminogen activator inhibitor-1 in the diagnosis and prognosis of patients with Parkinson’s disease. Exp. Ther. Med. 15, 5517–5522. doi: 10.3892/etm.2018.6076
Park, K., Noh, K., and Lee, S. J. (2019). Negr1 KO mice show socially submissive phenotype when co-housed with wild-type mice. IBRO Rep. 6:S442. doi: 10.1016/j.ibror.2019.07.1400
Park, M., Yi, J. W., Kim, E. M., Yoon, I. J., Lee, E. H., Lee, H. Y., et al. (2015). Triggering receptor expressed on myeloid cells 2 (TREM2) promotes adipogenesis and diet-induced obesity. Diabetes 64, 117–127. doi: 10.2337/db13-1869
Pastor, W. A., Aravind, L., and Rao, A. (2013). TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 14, 341–356. doi: 10.1038/nrm3589
Paul, K. C., Chuang, Y.-H., Shih, I.-F., Keener, A., Bordelon, Y., Bronstein, J. M., et al. (2019). The association between lifestyle factors and Parkinson’s disease progression and mortality. Mov. Disord. 34, 58–66. doi: 10.1002/mds.27577
Pfluger, P. T., Herranz, D., Velasco-Miguel, S., Serrano, M., and Tschop, M. H. (2008). Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. U.S.A. 105, 9793–9798. doi: 10.1073/pnas.0802917105
Placencio, V. R., and DeClerck, Y. A. (2015). Plasminogen activator inhibitor-1 in cancer: rationale and insight for future therapeutic testing. Cancer Res. 75, 2969–2974. doi: 10.1158/0008-5472.CAN-15-0876
Platt, T. L., Beckett, T. L., Kohler, K., Niedowicz, D. M., and Murphy, M. P. (2016). Obesity, diabetes, and leptin resistance promote tau pathology in a mouse model of disease. Neuroscience 315, 162–174. doi: 10.1016/j.neuroscience.2015.12.011
Podlesak, A. K., Mozer, M. E., Smith-Simpson, S., Lee, S.-Y., and Donovan, S. M. (2017). Associations between parenting style and parent and toddler mealtime behaviors. Curr. Dev. Nutr. 1:e000570. doi: 10.3945/cdn.117.000570
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Prim. 3:17013. doi: 10.1038/nrdp.2017.13
Polymeropoulos, M. H. (1997). Mutation in the -synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Popkin, B. M., and Reardon, T. (2018). Obesity and the food system transformation in Latin America. Obes. Rev. 19, 1028–1064. doi: 10.1111/obr.12694
Prats-Puig, A., Grau-Cabrera, P., Riera-Pérez, E., Cortés-Marina, R., Fortea, E., Soriano-Rodríguez, P., et al. (2013). Variations in the obesity genes FTO, TMEM18 and NRXN3 influence the vulnerability of children to weight gain induced by short sleep duration. Int. J. Obes. 37, 182–187. doi: 10.1038/ijo.2012.27
Profenno, L. A., Porsteinsson, A. P., and Faraone, S. V. (2010). Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol. Psychiatry 67, 505–512. doi: 10.1016/j.biopsych.2009.02.013
Proulx, K., and Seeley, R. J. (2005). The regulation of energy balance by the central nervous system. Psychiatr. Clin. North Am. 28, 25–38. doi: 10.1016/j.psc.2004.09.005
Pugazhenthi, S., Qin, L., and Reddy, P. H. (2017). Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta 1863, 1037–1045. doi: 10.1016/j.bbadis.2016.04.017
Pulit, S. L., Stoneman, C., Morris, A. P., Wood, A. R., Glastonbury, C. A., Tyrrell, J., et al. (2019). Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet. 28, 166–174. doi: 10.1093/hmg/ddy327
Purcell, S. M., Moran, J. L., Fromer, M., Ruderfer, D., Solovieff, N., Roussos, P., et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. doi: 10.1038/nature12975
Qi, Q., Chu, A. Y., Kang, J. H., Huang, J., Rose, L. M., Jensen, M. K., et al. (2014). Fried food consumption, genetic risk, and body mass index: gene-diet interaction analysis in three US cohort studies. BMJ 348:g1610. doi: 10.1136/bmj.g1610
Qi, Q., Chu, A. Y., Kang, J. H., Jensen, M. K., Curhan, G. C., Pasquale, L. R., et al. (2012a). Sugar-sweetened beverages and genetic risk of obesity. N. Engl. J. Med. 367, 1387–1396. doi: 10.1056/NEJMoa1203039
Qi, Q., Li, Y., Chomistek, A. K., Kang, J. H., Curhan, G. C., Pasquale, L. R., et al. (2012b). Television watching, leisure time physical activity, and the genetic predisposition in relation to body mass index in women and men. Circulation 126, 1821–1827. doi: 10.1161/CIRCULATIONAHA.112.098061
Qin, H., Buckley, J. A., Li, X., Liu, Y., Fox, T. H., Meares, G. P., et al. (2016). Inhibition of the JAK/STAT pathway protects against α-synuclein-induced neuroinflammation and dopaminergic neurodegeneration. J. Neurosci. 36, 5144–5159. doi: 10.1523/JNEUROSCI.4658-15.2016
Raghavan, N. S., Vardarajan, B., and Mayeux, R. (2019). Genomic variation in educational attainment modifies Alzheimer disease risk. Neurol. Genet. 5:e310. doi: 10.1212/NXG.0000000000000310
Rahimi, M., Vinciguerra, M., Daghighi, M., Özcan, B., Akbarkhanzadeh, V., Sheedfar, F., et al. (2015). Age-related obesity and type 2 diabetes dysregulate neuronal associated genes and proteins in humans. Oncotarget 33, 1112–1118. doi: 10.18632/oncotarget.4904
Ramachandrappa, S., and Farooqi, I. S. (2011). Genetic approaches to understanding human obesity. J. Clin. Invest. 121, 2080–2086. doi: 10.1172/JCI46044
Ramachandrappa, S., Raimondo, A., Cali, A. M. G., Keogh, J. M., Henning, E., Saeed, S., et al. (2013). Rare variants in single-minded 1 (SIM1) are associated with severe obesity. J. Clin. Invest. 123, 3042–3050. doi: 10.1172/JCI68016
Ramos-Molina, B., Molina-Vega, M., Fernández-García, J., and Creemers, J. (2018). Hyperphagia and Obesity in Prader–Willi Syndrome: PCSK1 Deficiency and Beyond? Genes 9:288. doi: 10.3390/genes9060288
Ramos-Molina, B., Martin, M. G., and Lindberg, I. (2016). PCSK1 variants and human obesity. Prog. Mol. Biol. Transl. Sci. 140, 47–74. doi: 10.1016/bs.pmbts.2015.12.001
Rana, P., Franco, E. F., Rao, Y., Syed, K., Barh, D., Azevedo, V., et al. (2019). Evaluation of the Common Molecular Basis in Alzheimer’s and Parkinson’s Diseases. Int. J. Mol. Sci. 20:3730. doi: 10.3390/ijms20153730
Rask-Andersen, M., Karlsson, T., Ek, W. E., and Johansson, Å. (2017). Gene-environment interaction study for BMI reveals interactions between genetic factors and physical activity, alcohol consumption and socioeconomic status. PLoS Genet. 13:e1006977. doi: 10.1371/journal.pgen.1006977
Reddon, H., Guéant, J.-L., and Meyre, D. (2016). The importance of gene–environment interactions in human obesity. Clin. Sci. 130, 1571–1597. doi: 10.1042/CS20160221
Reitz, C. (2015). Genetic diagnosis and prognosis of Alzheimer’s disease: challenges and opportunities. Expert Rev. Mol. Diagn. 15, 339–348. doi: 10.1586/14737159.2015.1002469
Renström, F., Payne, F., Nordström, A., Brito, E. C., Rolandsson, O., Hallmans, G., et al. (2009). Replication and extension of genome-wide association study results for obesity in 4923 adults from northern Sweden. Hum. Mol. Genet. 18, 1489–1496. doi: 10.1093/hmg/ddp041
Roodveldt, C., Christodoulou, J., and Dobson, C. M. (2008). Immunological features of α-synuclein in Parkinson’s disease. J. Cell. Mol. Med. 12, 1820–1829. doi: 10.1111/j.1582-4934.2008.00450.x
Rosas-Vargas, H., Martínez-Ezquerro, J. D., and Bienvenu, T. (2011). Brain-derived neurotrophic factor, food intake regulation, and obesity. Arch. Med. Res. 42, 482–494. doi: 10.1016/j.arcmed.2011.09.005
Roselli-Rehfuss, L., Mountjoy, K. G., Robbins, L. S., Mortrud, M. T., Low, M. J., Tatro, J. B., et al. (1993). Identification of a receptor for gamma melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proc. Natl. Acad. Sci. U.S.A. 90, 8856–8860. doi: 10.1073/pnas.90.19.8856
Ross, C. A., Aylward, E. H., Wild, E. J., Langbehn, D. R., Long, J. D., Warner, J. H., et al. (2014). Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 10, 204–216. doi: 10.1038/nrneurol.2014.24
Rouillard, A. D., Gundersen, G. W., Fernandez, N. F., Wang, Z., Monteiro, C. D., McDermott, M. G., et al. (2016). The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016:baw100. doi: 10.1093/database/baw100
Ruggiero, A., Aloni, E., Korkotian, E., Zaltsman, Y., Oni-Biton, E., Kuperman, Y., et al. (2017). Loss of forebrain MTCH2 decreases mitochondria motility and calcium handling and impairs hippocampal-dependent cognitive functions. Sci. Rep. 7:44401. doi: 10.1038/srep44401
Saad, M., Lesage, S., Saint-Pierre, A., Corvol, J. C., Zelenika, D., Lambert, J. C., et al. (2011). Genome-wide association study confirms BST1 and suggests a locus on 12q24 as the risk loci for Parkinson’s disease in the European population. Hum. Mol. Genet. 20, 615–627. doi: 10.1093/hmg/ddq497
Saeed, S., Bech, P. R., Hafeez, T., Alam, R., Falchi, M., Ghatei, M. A., et al. (2014). Changes in levels of peripheral hormones controlling appetite are inconsistent with hyperphagia in leptin-deficient subjects. Endocrine 45, 401–408.
Santos-Cortez, R. L. P., Khan, V., Khan, F. S., Mughal, Z.-N., Chakchouk, I., Lee, K., et al. (2018). Novel candidate genes and variants underlying autosomal recessive neurodevelopmental disorders with intellectual disability. Hum. Genet. 137, 735–752. doi: 10.1007/s00439-018-1928-6
Schneeberger, M., Gomis, R., and Claret, M. (2014). Hypothalamic and brainstem neuronal circuits controlling homeostatic energy balance. J. Endocrinol. 220, T25–T46. doi: 10.1530/JOE-13-0398
Schwedhelm, C., Schwingshackl, L., Agogo, G. O., Sonestedt, E., Boeing, H., and Knüppel, S. (2019). Associations of food groups and cardiometabolic and inflammatory biomarkers: Does the meal matter? Br. J. Nutr. 122, 707–716. doi: 10.1017/S000711451900151X
Scuteri, A., Sanna, S., Chen, W. M., Uda, M., Albai, G., Strait, J., et al. (2007). Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 3:e115. doi: 10.1371/journal.pgen.0030115
Seo, J.-S., Moon, M.-H., Jeong, J.-K., Seol, J.-W., Lee, Y.-J., Park, B.-H., et al. (2012). SIRT1, a histone deacetylase, regulates prion protein-induced neuronal cell death. Neurobiol. Aging 33, 1110–1120. doi: 10.1016/j.neurobiolaging.2010.09.019
Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. doi: 10.1038/nature24016
Singh, P., Hanson, P. S., and Morris, C. M. (2017). SIRT1 ameliorates oxidative stress induced neural cell death and is down-regulated in Parkinson’s disease. BMC Neurosci. 18:46. doi: 10.1186/s12868-017-0364-1
Singleton, A. B. (2003). -Synuclein locus triplication causes Parkinson’s disease. Science 302, 841–841. doi: 10.1126/science.1090278
Skurk, T., Lee, Y.-M., Nicuta-Rolfs, T.-O., Haastert, B., Wirth, A., and Hauner, H. (2004). Effect of the angiotensin II receptor blocker candesartan on fibrinolysis in patients with mild hypertension. Diabetes Obes. Metab. 6, 56–62. doi: 10.1111/j.1463-1326.2004.00316.x
Smith, L. P., Ng, S. W., and Popkin, B. M. (2013). Trends in US home food preparation and consumption: analysis of national nutrition surveys and time use studies from 1965–1966 to 2007–2008. Nutr. J. 12:45.
Smith-Dijak, A. I., Sepers, M. D., and Raymond, L. A. (2019). Alterations in synaptic function and plasticity in Huntington disease. J. Neurochem. 150, 346–365. doi: 10.1111/jnc.14723
Snowden, J. S. (2017). The neuropsychology of Huntington’s disease. Arch. Clin. Neuropsychol. 32, 876–887. doi: 10.1093/arclin/acx086
Sousa-Ferreira, L., Álvaro, A. R., Aveleira, C., Santana, M., Brandão, I., Kügler, S., et al. (2011). Proliferative hypothalamic neurospheres express NPY, AGRP, POMC, CART and Orexin-A and differentiate to functional neurons. PLoS One 6:e19745. doi: 10.1371/journal.pone.0019745
Sousa-Ferreira, L., de Almeida, L. P., and Cavadas, C. (2014). Role of hypothalamic neurogenesis in feeding regulation. Trends Endocrinol. Metab. 25, 80–88. doi: 10.1016/j.tem.2013.10.005
Spaccapelo, L., Galantucci, M., Neri, L., Contri, M., Pizzala, R., D’Amico, R., et al. (2013). Up-regulation of the canonical Wnt-3A and Sonic hedgehog signaling underlies melanocortin-induced neurogenesis after cerebral ischemia. Eur. J. Pharmacol. 707, 78–86. doi: 10.1016/j.ejphar.2013.03.030
Sprengelmeyer, R., Schroeder, U., Young, A. W., and Epplen, J. T. (2006). Disgust in pre-clinical Huntington’s disease: a longitudinal study. Neuropsychologia 44, 518–533. doi: 10.1016/j.neuropsychologia.2005.07.003
Squinto, S. P., Stitt, T. N., Aldrich, T. H., Davis, S., Blanco, S. M., RadzieJewski, C., et al. (1991). trkB encodes a functional receptor for brain-derived neurotrophic factor and neurotrophin-3 but not nerve growth factor. Cell 65, 885–893. doi: 10.1016/0092-8674(91)90395-F
Stachowiak, E. K., Oommen, S., Vasu, V. T., Srinivasan, M., Stachowiak, M., Gohil, K., et al. (2013). Maternal obesity affects gene expression and cellular development in fetal brains. Nutr. Neurosci. 16, 96–103. doi: 10.1179/1476830512Y.0000000035
States, D. J., Walseth, T. F., and Lee, H. C. (1992). Similarities in amino acid sequences of Aplysia ADP-ribosyl cyclase and human lymphocyte antigen CD38. Trends Biochem. Sci. 17:495. doi: 10.1016/0968-0004(92)90337-9
Stefanowicz, M., Nikołajuk, A., Matulewicz, N., and Karczewska-Kupczewska, M. (2018). Adipose tissue, but not skeletal muscle, sirtuin 1 expression is decreased in obesity and related to insulin sensitivity. Endocrine 60, 263–271. doi: 10.1007/s12020-018-1544-1
Steiner, J., and Lang, C. (2017). Alcohol, adipose tissue and lipid dysregulation. Biomolecules 7:16. doi: 10.3390/biom7010016
Stern, D., Piernas, C., Barquera, S., Rivera, J. A., and Popkin, B. M. (2014). Caloric beverages were major sources of energy among children and adults in Mexico, 1999–2012. J. Nutr. 144, 949–956. doi: 10.3945/jn.114.190652
Sterniczuk, R., Antle, M. C., LaFerla, F. M., and Dyck, R. H. (2010). Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: Part 2. Behavioral and cognitive changes. Brain Res. 1348, 149–155. doi: 10.1016/j.brainres.2010.06.011
Stijnen, P., Ramos-Molina, B., O’Rahilly, S., and Creemers, J. W. M. (2016). PCSK1 mutations and human endocrinopathies: from obesity to gastrointestinal disorders. Endocr. Rev. 37, 347–371. doi: 10.1210/er.2015-1117
Stoeckel, L. E., Arvanitakis, Z., Gandy, S., Small, D., Kahn, C. R., Pascual-Leone, A., et al. (2016). Complex mechanisms linking neurocognitive dysfunction to insulin resistance and other metabolic dysfunction. F1000Res. 5:353. doi: 10.12688/f1000research.8300.2
Sudlow, C., Gallacher, J., Allen, N., Beral, V., Burton, P., Danesh, J., et al. (2015). UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12:e1001779. doi: 10.1371/journal.pmed.1001779
Sullivan, E. L., Grayson, B., Takahashi, D., Robertson, N., Maier, A., Bethea, C. L., et al. (2010). Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. J. Neurosci. 30, 3826–3830. doi: 10.1523/JNEUROSCI.5560-09.2010
Sullivan, E. L., Riper, K. M., Lockard, R., and Valleau, J. C. (2015). Maternal high-fat diet programming of the neuroendocrine system and behavior. Horm. Behav. 76, 153–161. doi: 10.1016/j.yhbeh.2015.04.008
Szczurkowska, J., Pischedda, F., Pinto, B., Managò, F., Haas, C. A., Summa, M., et al. (2018). NEGR1 and FGFR2 cooperatively regulate cortical development and core behaviours related to autism disorders in mice. Brain 141, 2772–2794. doi: 10.1093/brain/awy190
Tabrizi, S. J., Scahill, R. I., Durr, A., Roos, R. A. C., Leavitt, B. R., Jones, R., et al. (2011). Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol. 10, 31–42. doi: 10.1016/S1474-4422(10)70276-3
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., et al. (2009). Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 324, 930–935. doi: 10.1126/science.1170116
Tãrlungeanu, D. C., and Novarino, G. (2018). Genomics in neurodevelopmental disorders: an avenue to personalized medicine. Exp. Mol. Med. 50:100. doi: 10.1038/s12276-018-0129-7
Tassone, F., Beilina, A., Carosi, C., Albertosi, S., Bagni, C., Li, L., et al. (2007). Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA 13, 555–562. doi: 10.1261/rna.280807
Tassone, F., Hagerman, R. J., Chamberlain, W. D., and Hagerman, P. J. (2000). Transcription of the FMR1 gene in individuals with fragile X syndrome. Am. J. Med. Genet. 97, 195–203.
Templin, T., Cravo Oliveira Hashiguchi, T., Thomson, B., Dieleman, J., and Bendavid, E. (2019). The overweight and obesity transition from the wealthy to the poor in low- and middle-income countries: a survey of household data from 103 countries. PLoS Med. 16:e1002968. doi: 10.1371/journal.pmed.1002968
Thorleifsson, G., Walters, G. B., Gudbjartsson, D. F., Steinthorsdottir, V., Sulem, P. A., Helgadottir, A., et al. (2009). Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat. Genet. 41, 18–24.
Toda, C., Santoro, A., Kim, J. D., and Diano, S. (2017). POMC neurons: from birth to death. Annu. Rev. Physiol. 79, 209–236.
Tong, Q. (2011). Synaptotagmin 4: A new antiobesity target? Neuron 69, 401–403. doi: 10.1016/j.neuron.2011.01.018
Torres-Perez, E., Ledesma, M., Garcia-Sobreviela, M. P., Leon-Latre, M., and Arbones-Mainar, J. M. (2016). Apolipoprotein E4 association with metabolic syndrome depends on body fatness. Atherosclerosis 245, 35–42. doi: 10.1016/j.atherosclerosis.2015.11.029
Treviño, S., Aguilar-Alonso, P., Flores Hernandez, J. A., Brambila, E., Guevara, J., Flores, G., et al. (2015). A high calorie diet causes memory loss, metabolic syndrome and oxidative stress into hippocampus and temporal cortex of rats. Synapse 69, 421–433. doi: 10.1002/syn.21832
Tyrrell, J., Wood, A. R., Ames, R. M., Yaghootkar, H., Beaumont, R. N., Jones, S. E., et al. (2017). Gene–obesogenic environment interactions in the UK Biobank study. Int. J. Epidemiol. 46, 559–575. doi: 10.1093/ije/dyw337
Ulland, T. K., Song, W. M., Huang, S. C. C., Ulrich, J. D., Sergushichev, A., Beatty, W. L., et al. (2017). TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170, 649–663.e13. doi: 10.1016/j.cell.2017.07.023
Ushkaryov, Y. A., Petrenko, A. G., Geppert, M., and Südhof, T. C. (1992). Neurexins: synaptic cell surface proteins related to the α-latrotoxin receptor and laminin. Science 257, 50–56. doi: 10.1126/science.1621094
Vaags, A. K., Lionel, A. C., Sato, D., Goodenberger, M., Stein, Q. P., Curran, S., et al. (2012). Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am. J. Hum. Genet. 90, 133–141. doi: 10.1016/j.ajhg.2011.11.025
Vagelatos, N. T., and Eslick, G. D. (2013). Type 2 diabetes as a risk factor for Alzheimer’s disease: the confounders, interactions, and neuropathology associated with this relationship. Epidemiol. Rev. 35, 152–160. doi: 10.1093/epirev/mxs012
Vaisse, C., Clement, K., Durand, E., Hercberg, S., Guy-Grand, B., and Froguel, P. (2000). Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J. Clin. Invest. 106, 253–262. doi: 10.1172/JCI9238
van der Burg, J. M. M., Bacos, K., Wood, N. I., Lindqvist, A., Wierup, N., Woodman, B., et al. (2008). Increased metabolism in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 29, 41–51. doi: 10.1016/j.nbd.2007.07.029
van der Klaauw, A. A., and Farooqi, I. S. (2015). The hunger genes: pathways to obesity. Cell 161, 119–132. doi: 10.1016/j.cell.2015.03.008
van Duijn, E., Craufurd, D., Hubers, A. A. M., Giltay, E. J., Bonelli, R., Rickards, H., et al. (2014). Neuropsychiatric symptoms in a European Huntington’s disease cohort (REGISTRY). J. Neurol. Neurosurg. Psychiatry 85, 1411–1418.
Villalobos-Comparán, M., Teresa Flores-Dorantes, M., Teresa Villarreal-Molina, M., Rodríguez-Cruz, M., García-Ulloa, A. C., Robles, L., et al. (2008). The FTO gene is associated with adulthood obesity in the Mexican population. Obesity 16, 2296–2301. doi: 10.1038/oby.2008.367
Visootsak, J., Hipp, H., Clark, H., Berry-Kravis, E., Anderson, T., and Laney, D. (2014). Climbing the branches of a family tree: diagnosis of fragile X syndrome. J. Pediatr. 164, 1292–1295. doi: 10.1016/j.jpeds.2014.01.051
Vollbach, H., Brandt, S., Lahr, G., Denzer, C., Von Schnurbein, J., Debatin, K. M., et al. (2017). Prevalence and phenotypic characterization of MC4R variants in a large pediatric cohort. Int. J. Obes. 41, 13–22. doi: 10.1038/ijo.2016.161
Walker, A. G., Miller, B. R., Fritsch, J. N., Barton, S. J., and Rebec, G. V. (2008). Altered information processing in the prefrontal cortex of Huntington’s disease mouse models. J. Neurosci. 28, 8973–8982. doi: 10.1523/JNEUROSCI.2804-08.2008
Wang, L.-F., Huang, C.-C., Xiao, Y.-F., Guan, X.-H., Wang, X.-N., Cao, Q., et al. (2018a). CD38 deficiency protects heart from high fat diet-induced oxidative stress via activating Sirt3/FOXO3 pathway. Cell. Physiol. Biochem. 48, 2350–2363. doi: 10.1159/000492651
Wang, L. F., Miao, L. J., Wang, X. N., Huang, C. C., Qian, Y. S., Huang, X., et al. (2018b). CD38 deficiency suppresses adipogenesis and lipogenesis in adipose tissues through activating Sirt1/PPARγ signaling pathway. J. Cell. Mol. Med. 22, 101–110. doi: 10.1111/jcmm.13297
Wang, T., Heianza, Y., Sun, D., Huang, T., Ma, W., Rimm, E. B., et al. (2018c). Improving adherence to healthy dietary patterns, genetic risk, and long term weight gain: gene-diet interaction analysis in two prospective cohort studies. BMJ 360:j5644. doi: 10.1136/bmj.j5644
Wang, X. J., Xu, W., Li, J. Q., Cao, X. P., Tan, L., and Yu, J. T. (2019). Early-life risk factors for dementia and cognitive impairment in later life: a systematic review and meta-analysis. J. Alzheimers Dis. 67, 221–229. doi: 10.3233/JAD-180856
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Wang, Z. H., Xiang, J., Liu, X., Yu, S. P., Manfredsson, F. P., Sandoval, I. M., et al. (2019). Deficiency in BDNF/TrkB Neurotrophic Activity Stimulates δ-Secretase by Upregulating C/EBPβ in Alzheimer’s Disease. Cell Rep. 28, 655. doi: 10.1016/j.celrep.2019.06.054
Wasim, M., Awan, F. R., Najam, S. S., Khan, A. R., and Khan, H. N. (2016). Role of leptin deficiency, inefficiency, and leptin receptors in obesity. Biochem. Genet. 54, 565–572. doi: 10.1007/s10528-016-9751-z
Wei, Y., Liu, F., Li, B., Chen, X., Ma, Y., Yan, L., et al. (2011). Polymorphisms of tumor necrosis factor-alpha and hepatocellular carcinoma risk: a HuGE systematic review and meta-analysis. Dig. Dis. Sci. 56, 2227–2236. doi: 10.1007/s10620-011-1617-y
Willer, C. J., Speliotes, E. K., Loos, R. J. F., Li, S., Lindgren, C. M., Heid, I. M., et al. (2009). Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet. 41, 25–34. doi: 10.1038/ng.287
Wingo, T. S. (2012). Autosomal recessive causes likely in early-onset Alzheimer disease. Arch. Neurol. 69, 59–64. doi: 10.1001/archneurol.2011.221
Wolfson, J. A., Leung, C. W., and Richardson, C. R. (2020). More frequent cooking at home is associated with higher Healthy Eating Index-2015 score. Public Health Nutr. 23, 2384–2394. doi: 10.1017/S1368980019003549
Wolfson, J. A., Ramsing, R., Richardson, C. R., and Palmer, A. (2019). Barriers to healthy food access: associations with household income and cooking behavior. Prev. Med. Rep. 13, 298–305. doi: 10.1016/j.pmedr.2019.01.023
Wood, A. C. (2019). Gene-environment interplay in child eating behaviors: What the role of “Nature” means for the effects of “Nurture”. Curr. Nutr. Rep. 7, 294–302. doi: 10.1007/s13668-018-0254-x.Gene-Environment
Wray, N. R., Ripke, S., Mattheisen, M., Trzaskowski, M., Byrne, E. M., Abdellaoui, A., et al. (2018). Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681. doi: 10.1038/s41588-018-0090-3
Wu, C., and Pan, W. (2018). Integration of enhancer-promoter interactions with GWAS summary results identifies novel schizophrenia-associated genes and pathways. Genetics 209, 699–709. doi: 10.1534/genetics.118.300805
Wu, Y., He, H., Cheng, Z., Bai, Y., and Ma, X. (2019). The Role of Neuropeptide Y and Peptide YY in the Development of Obesity via Gut-brain Axis. Curr. Protein Pept. Sci. 20, 750–758. doi: 10.2174/1389203720666190125105401
Xu, B., Goulding, E. H., Zang, K., Cepoi, D., Cone, R. D., Jones, K. R., et al. (2003). Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat. Neurosci. 6, 736–742. doi: 10.1038/nn1073
Xu, Y., Elmquist, J. K., and Fukuda, M. (2011). Central nervous control of energy and glucose balance: focus on the central melanocortin system. Ann. N. Y. Acad. Sci. 1243, 1–14. doi: 10.1111/j.1749-6632.2011.06248.x
Yang, Y., Fong, T. M., Dickinson, C. J., Mao, C., Li, J.-Y., Tota, M. R., et al. (2000). Molecular Determinants of Ligand Binding to the Human Melanocortin-4 Receptor †. Biochemistry 39, 14900–14911. doi: 10.1021/bi001684q
Yeh, F. L., Hansen, D. V., and Sheng, M. (2017). TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol. Med. 23, 512–533. doi: 10.1016/j.molmed.2017.03.008
Yengo, L., Sidorenko, J., Kemper, K. E., Zheng, Z., Wood, A. R., Weedon, M. N., et al. (2018). Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum. Mol. Genet. 27, 3641–3649. doi: 10.1093/hmg/ddy271
Yeo, G. S. H., Connie Hung, C.-C., Rochford, J., Keogh, J., Gray, J., Sivaramakrishnan, S., et al. (2004). A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat. Neurosci. 7, 1187–1189. doi: 10.1038/nn1336
Yoshiyama, Y., Higuchi, M., Zhang, B., Huang, S. M., Iwata, N., Saido, T. C. C., et al. (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. doi: 10.1016/j.neuron.2007.01.010
Yu, Z. B., Han, S. P., Cao, X. G., and Guo, X. R. (2010). Intelligence in relation to obesity: a systematic review and meta-analysis. Obes. Rev. 11, 656–670. doi: 10.1111/j.1467-789X.2009.00656.x
Zakhary, S. M., Ayubcha, D., Dileo, J. N., Jose, R., Leheste, J. R., Horowitz, J. M., et al. (2010). Distribution analysis of deacetylase SIRT1 in rodent and human nervous systems. Anat. Rec. 293, 1024–1032. doi: 10.1002/ar.21116
Zaw, Y. H., and Taneepanichskul, N. (2019). Blood heavy metals and brain-derived neurotrophic factor in the first trimester of pregnancy among migrant workers. PLoS One 14:e0218409. doi: 10.1371/journal.pone.0218409
Zegers, D., Beckers, S., Hendrickx, R., Van Camp, J. K., de Craemer, V., Verrijken, A., et al. (2014). Mutation screen of the SIM1 gene in pediatric patients with early-onset obesity. Int. J. Obes. 38, 1000–1004. doi: 10.1038/ijo.2013.188
Zenaro, E., Piacentino, G., and Constantin, G. (2017). The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 107, 41–56. doi: 10.1016/j.nbd.2016.07.007
Zhang, A., Wang, H., Qin, X., Pang, S., and Yan, B. (2012). Genetic analysis of SIRT1 gene promoter in sporadic Parkinson’s disease. Biochem. Biophys. Res. Commun. 422, 693–696. doi: 10.1016/j.bbrc.2012.05.059
Zhang, G., Bai, H., Zhang, H., Dean, C., Wu, Q., Li, J., et al. (2011). Neuropeptide exocytosis involving synaptotagmin-4 and oxytocin in hypothalamic programming of body weight and energy balance. Neuron 69, 523–535. doi: 10.1016/j.neuron.2010.12.036
Zhang, W., Zhong, W., Sun, X., Sun, Q., Tan, X., Li, Q., et al. (2015). Visceral white adipose tissue is susceptible to alcohol-induced Lipodystrophy in rats: role of acetaldehyde. Alcohol. Clin. Exp. Res. 39, 416–423. doi: 10.1111/acer.12646
Zhang, X., Qi, Q., Zhang, C., Smith, S. R., Hu, F. B., Sacks, F. M., et al. (2012). FTO genotype and 2-year change in body composition and fat distribution in response to weight-loss diets: the POUNDS LOST Trial. Diabetes Metab. Res. Rev. 61, 3005–3011. doi: 10.2337/db11-1799
Zhang, Y., Proenca, R., Maffei, M., Barone, M., Leopold, L., and Friedman, J. M. (1994). Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425–432. doi: 10.1038/372425a0
Zhang, Z., Bhalla, A., Dean, C., Chapman, E. R., and Jackson, M. B. (2009). Synaptotagmin IV: a multifunctional regulator of peptidergic nerve terminals. Nat. Neurosci. 12, 163–171. doi: 10.1038/nn.2252
Zhao, C., Noble, J. M., Marder, K., Hartman, J. S., Gu, Y., and Scarmeas, N. (2018). Dietary patterns, physical activity, sleep, and risk for dementia and cognitive decline. Curr. Nutr. Rep. 7, 335–345. doi: 10.1007/s13668-018-0247-9
Zheng, J.-J., Li, W.-X., Liu, J.-Q., Guo, Y.-C., Wang, Q., Li, G.-H., et al. (2018). Low expression of aging-related NRXN3 is associated with Alzheimer disease. Medicine 97:e11343. doi: 10.1097/MD.0000000000011343
Zhong, L., Chen, X. F., Zhang, Z. L., Wang, Z., Shi, X. Z., Xu, K., et al. (2015). DAP12 stabilizes the C-terminal fragment of the triggering receptor expressed on myeloid cells-2 (TREM2) and protects against LPS-induced pro-inflammatory response. J. Biol. Chem. doi: 10.1074/jbc.M115.645986
Zhu, X., Zhou, A., Dey, A., Norrbom, C., Carroll, R., Zhang, C., et al. (2002). Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc. Natl. Acad. Sci. U.S.A. 99, 10293–10298. doi: 10.1073/pnas.162352599
Keywords: obesity, neurodegenerative diseases, neurodevelopmental diseases, environment, genes
Citation: Flores-Dorantes MT, Díaz-López YE and Gutiérrez-Aguilar R (2020) Environment and Gene Association With Obesity and Their Impact on Neurodegenerative and Neurodevelopmental Diseases. Front. Neurosci. 14:863. doi: 10.3389/fnins.2020.00863
Received: 24 May 2020; Accepted: 24 July 2020;
Published: 28 August 2020.
Edited by:
Kioko Rubi Guzman-Ramos, Metropolitan Autonomous University, MexicoReviewed by:
Ian James Martins, The University of Western Australia, AustraliaKeiko Iwata, University of Fukui, Japan
Copyright © 2020 Flores-Dorantes, Díaz-López and Gutiérrez-Aguilar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruth Gutiérrez-Aguilar, cnV0aGd1dHpAdW5hbS5teA==; cnV0aGd1dGllcnJlemhpbWZnQGdtYWlsLmNvbQ==