
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurosci. , 25 February 2020
Sec. Neurodegeneration
Volume 14 - 2020 | https://doi.org/10.3389/fnins.2020.00085
 Xiaomin Xu1,2
Xiaomin Xu1,2 Xiaokai Yang1Zhongliang Su1Hai Wang1,2Xiaoqing Li1,2
Xiaokai Yang1Zhongliang Su1Hai Wang1,2Xiaoqing Li1,2 Congcong Sun1,2Wenhuan Wang1,2Yao Chen3Chenhui Zhang1,2Hongping Zhang1
Congcong Sun1,2Wenhuan Wang1,2Yao Chen3Chenhui Zhang1,2Hongping Zhang1 Fan Jin4Jiayong Zheng1,2*
Fan Jin4Jiayong Zheng1,2*Giant axonal neuropathy (GAN) is a very rare autosomal recessive disorder characterized by abnormally large and dysfunctional neuronal axons. Mutations in the GAN gene have been identified as the cause of this disorder. In this report, we performed a detailed phenotypic assessment of a Chinese patient with GAN. An array-based exon capture test and targeted next-generation sequencing were used to detect the suspected mutation sites. Compound heterozygous mutations of p.S79L (c.236C > T) in the BTB domain and p.T489S (c.1466C > G) in the kelch domain were identified in the proband’s genome. S79L was a known mutation, and T489S was reported for the first time. The p.S79L and p.T489S were confirmed in the proband’s mother and father, respectively. Both mutations were located in highly conserved regions and affected the predicted protein crystal structures. The proband’s sural biopsy revealed the classical GAN phenotype of swollen axons filled with closely packed neurofilaments. The combined application of the next-generation sequencing platform and bioinformatics analyses was an effective method for diagnosing GAN. The novel compound mutations of S79L and T489S in the GAN gene were likely the cause of the patient’s GAN symptoms. Our findings enrich the spectrum of mutations associated with this rare type of axonopathy.
Giant axonal neuropathy (GAN; OMIM#256850) is a very rare autosomal recessive disorder caused by specific mutations in the GAN gene (Bomont et al., 2000). Since the first GAN report in 1972, more than 50 GAN gene mutations have been identified in 80 cases around the world (Normendez-Martinez et al., 2018). According to clinical symptoms, GAN can be divided into classical phenotype and mild phenotype; both shared the physiological features of dense nerve fiber accumulation. In addition, the classical clinical phenotype is characterized by severe peripheral neuropathy, kinky hair, early onset of cerebellar and pyramidal signs, and mental deterioration (Tazir et al., 2009; Vijaykumar et al., 2015). However, the mild clinical phenotype has a relatively slow onset and progression (Tazir et al., 2009; Koichihara et al., 2016).
Detecting GAN gene mutations is the current method used in diagnosing GAN (Normendez-Martinez et al., 2018). The GAN gene is located at 16q24.1 and encodes gigaxonin. Gigaxonin contains an N-terminus BTB domain, a C-terminus kelch domain, and a BACK domain between the N-terminal and C-terminal ends. The gigaxonin protein has been found to regulate microtubule-binding protein degradation (Bomont et al., 2000). The regulation of microtubule-binding protein degradation may be a common pathological target in neurodegenerative disorders that arise from alterations in the neurofilament network (Bomont et al., 2000). The GAN gene mutations, which result in changed gigaxonin structure and nerve signal transduction complications, are the cause of the pathophysiology observed in GAN (Israeli et al., 2016). In addition to the genetics studies, multiple functional studies expanded our knowledge of GAN. The regulation of intermediate filaments may be of importance in addition to the regulation of microtubule-associated proteins (Mahammad et al., 2013), and recent evidence shows that gigaxonin may regulate autophagy (Scrivo et al., 2019).
In this study, we report the clinical presentation and genetic profile of a Chinese patient with compound heterozygous mutations in the GAN gene.
The patient described in this report was a 7-year-old boy conceived by unrelated parents. His hair was thick and curly. At 3 years of age, he developed a gait abnormality, walked slowly, fell easily when he ran, and had outward facing toes on his right foot. When he was 5 years old, he developed a mild obstructive sleep apnea–hypopnea syndrome, and his right hip was dislocated. After he underwent surgery on his hip, he suffered from muscle weakness and could not walk until 1 year later. At 7 years of age, the patient had normal intelligence, and his cervical region muscle strength was observed at level IV. He experienced a slight difficulty when trying to turn his neck. His muscle strength was at level V in the upper limbs, level IV in the in the lower limbs, and level IV in the feet, which was determined by dorsal extension tests. He performed poorly in the finger-to-nose test, especially when he was using his left hand. During this test, he clumsily alternated between the right and left hand. His pallesthesia of the upper and lower limbs was lost at distal locations more than proximal locations. The tendon reflex was absent, and he was unable to stand on one leg. Upon examining the cranial and spinal magnetic resonance imaging (MRI), electroencephalogram, and electrocardiogram test results, no abnormal signs were detected. Notably, the electromyography exam revealed neuronal damage, mainly located at the axons of multiple peripheral nerves. A subsequent electrophysiological examination supported the same conclusion. In the right median nerve, the level of CMAP was 2.1 m/s (amplitude, 2.6 mV; level of DL, 6.8 ms). In the left ulnar nerve, the level of CMAP was 62.2 m/s (amplitude, 2.4 mV; level of DL, 6.6 ms). The level of SNAP was 59.9 m/s in the left peroneus nerve (amplitude, 4.2 mV; level of DL, 1.4 ms), and 58.0 m/s in the right peroneus nerve (amplitude, 3.1 mV; level of DL, 1.3 ms). We calculated the Charcot–Marie–Tooth neuropathy score (CMTNS), and the result is 15 (moderate). No distal levels of CMAP or amplitude decreases were distinct from motor neurons of the lower limbs, and no distal SNAP levels from the upper limb sensory and bilateral plantar nerves were recorded. There was no history of neuromuscular disorders in the patient’s family, but his mother did have slightly curly hair. To preliminarily determine the etiology of the patient’s diagnosis, we used the targeted next-generation sequencing (Kooshavar et al., 2018; Langabeer et al., 2018).
After step-by-step analyses, only two compound heterozygous mutations in GAN survived our strict filtering process. The proband had heterozygous c.236C > T mutations in exon 2, which resulted in an amino acid substitution of serine to leucine (p.S79L) in the BTB domain (Figure 1A). Additionally, the proband had a heterozygous c.1466C > G mutation in exon 9, which resulted in a threonine-to-serine amino acid substitution (p.T489S) in the kelch domain (Figure 1A). The patient’s mother carried the c.236C > T mutation, and his father carried the c.1466C > G mutation. The c.236C > T (p.S79L) mutation was a known mutation (HGMD ID CM002979), and the c.1466C > G (p.T489S) mutation was a novel mutation. Although S79L mutation was reported (HGMD ID CM002979) here, we, for the first time, confirmed this mutation in Han Chinese. We then performed in silico predictions for pathogenicity for c.1466C > G mutation, which showed strong pathogenicity (Mutation Taster: disease causing; Polyphen: damaging; CADD score: 18.50). Both mutations were located in highly conserved regions of the DNA and affected the folding of the predicted protein crystal structures (Figures 1B,C).
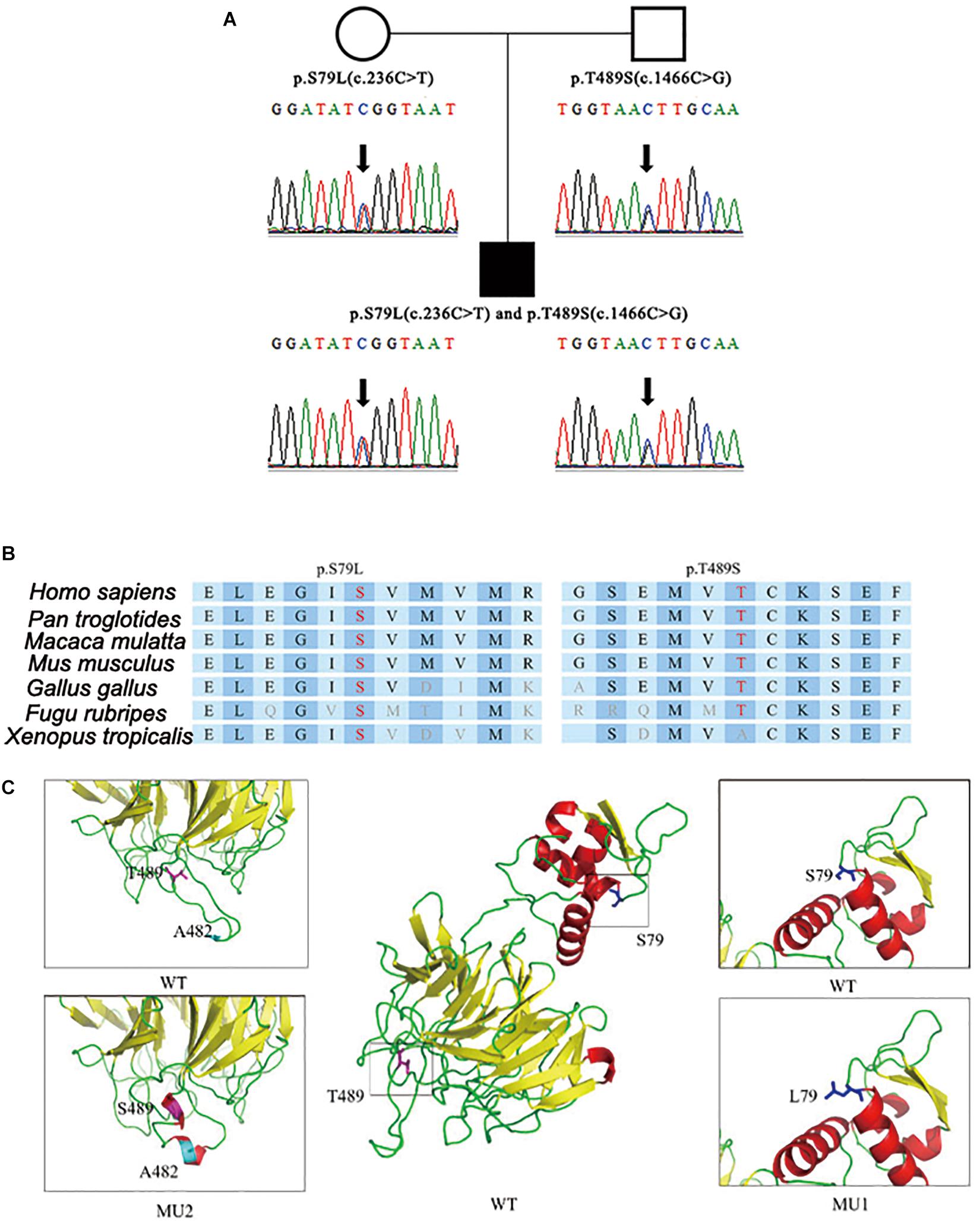
Figure 1. Identification of compound heterozygous mutations in the GAN gene. (A) Compound heterozygous mutations p.S79L (c.236C > T) and p.T489S (c.1466C > G) were identified in the patient’s copies of the GAN gene. (B) The location of the mutations with respect to the topological model of the GAN polypeptide. (C) Predicted crystal structures of wild-type and mutant GAN proteins. The proposed locations of the two mutations are shown in blue and pink. This model is based on template c4yy8B and covered 375 residues (32–575). The red coloring indicates helices, the yellow coloring indicates β-sheets, and the green coloring indicates loops.
On light microscopy, parts of the nerve fibers were observed to have tumid axis-cylinder spreads, in which the neurons were depleted of their myelin sheaths (Figure 2A). Medullated fibers were mildly reduced, and thin myelinated fibers were randomly spread (Figure 2B). Immunohistochemical staining corroborated the results (Figure 2C). Tissue cells, labeled by CD163, infiltrated the nerve fibers, while lymphocytes were not present, as shown by CD8 and CD20. The PAS staining showed normal glycogen content (Figure 2D). An ultrastructural examination exposed neuronal swelling and demyelinated areas in the neural axons, which were distributed among some of the myelinated fibers (Figure 3A). However, parts of these fibers were atrophic. Giant axons were filled with closely packed and irregularly oriented neurofilaments, and the microtubules are arranged in clusters (Figure 3B). All of the affected axons exhibited thin or absent myelin sheaths (Figures 3C,D).
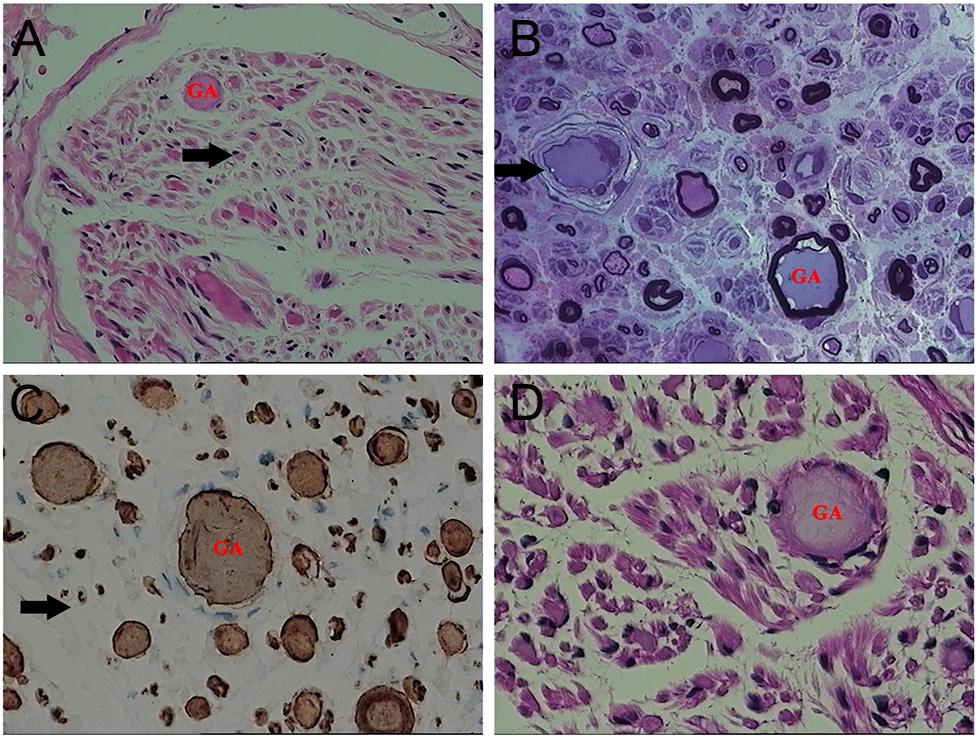
Figure 2. Microscopic examination of the patient’s sural nerve revealed a giant axon (GA). (A) HE staining revealed tumefaction of the neural axon and a few regenerating clusters (arrow). (B) Part of the axon atrophied mildly, and the vascular wall (arrow) was thickened and layered. (C) A myelinated nerve fiber (arrow) is slightly reduced. (D) No obvious increase in glycogen was observed.
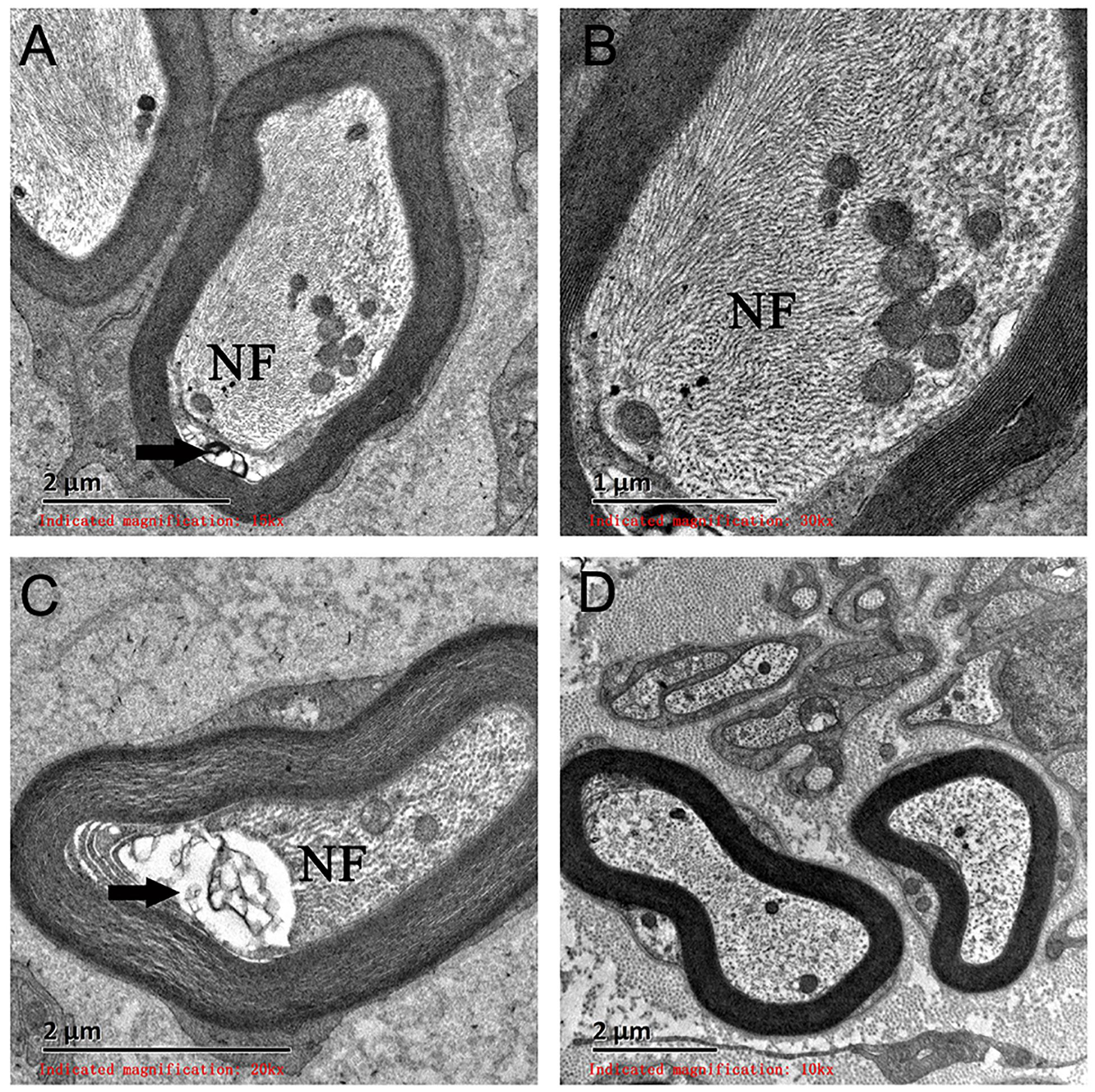
Figure 3. Electron microscopy examination of a giant axon (GA). (A,B) Electron microscopy showed giant axons filled with closely packed neurofilaments. (C) Neural axons were mildly atrophied (arrow) and separated from the myelin sheath. (D) A normal neural axon. NF, neurofilaments.
DNA samples were extracted from peripheral blood samples using the QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany). The patient’s mutation was detected by targeted next-generation sequencing, which was designed by BGI (China) and performed with the Human Sequence Capture 2.1 M Array (Roche NimbleGen, Madison, WI, United States). All of the mutations were verified by PCR and Sanger sequencing. Variant analyses were performed using an established bioinformatics pipeline as previously described (Huang et al., 2015a, b, 2017). Briefly, variants with a minor allele frequency of >0.005 in any of the variant databases were excluded. The effects of the candidate variants were assessed using in silico prediction programs, including Polyphen-2, MutationTaster, and CADD. We predicted the topological model using web resources, including SMART, to explore protein domain architectures and RaptorX structure prediction web server to generate both the wild-type and mutant protein models, and visualized any changes in protein folding and structure using the PyMol software. GeneMANIA was used to study the protein–protein interaction (PPI).
Sural nerve biopsy samples were obtained from the patient. First, HE staining was performed to fix one piece of the nerve in 10% formaldehyde. This piece was subsequently paraffin embedded, cut into 8-μm sections, and stained with hematoxylin and eosin. Electron microscopy was then performed on the rest of the sample, which was fixed in 2.5% glutaraldehyde and embedded in Epon 812. Semithin sections, prepared for light microscopy, were stained with toluidine blue. Ultrathin sections were contrasted with uranyl acetate and lead citrate, and then examined under an electron microscope.
Giant axonal neuropathy is a rare autosomal recessive neurodegenerative disease caused by mutations in the GAN gene. In 1972, the GAN clinical phenotype and neuropathology were first described (Asbury et al., 1972; Berg et al., 1972). The classical clinical presentation characteristics of GAN include disorder in the lower limb gait, myasthenia, ataxia, and gradual upper limb dysfunction occurring around 3 years of age. As the disease progresses, the patient becomes dysarthric and dysphagic, and ultimately passes away due to respiratory failure. In 2000, Bomont et al. reported that mutations in the GAN gene were the underlying cause of the GAN, and Bruno et al. subsequently confirmed this conclusion (Bomont et al., 2000; Bruno et al., 2004). The GAN gene encodes gigaxonin, which belongs to the BTB/kelch cytoskeleton protein family, contains an N-terminus BTB domain, a C-terminus kelch domain, and a BACK domain between the N and C terminuses. Gigaxonin has been proposed to be an E3 ligase substrate adaptor, which may be important for the turnover or regulation of intermediate filaments. Recently, Scrivo et al. demonstrated that GAN, in its capacity as an E3 ligase, regulates the ATG16L1 turnover and, in this way, controls autophagosome production and fine tunes the autophagy machinery (Scrivo et al., 2019).
Since the discovery of gigaxonin, at least 50 mutations, including missense, nonsense, insert, and splice site mutations in 11 exons of the GAN gene, have been reported (Johnson-Kerner et al., 2014). These mutations are scattered in the three domains and cause either the classical clinical phenotype or the mild clinical phenotype of GAN. GAN gene mutation detection is the accepted way to definitively diagnose GAN beyond observing the clinical presentations. We added functional analysis using protein–protein interaction (PPI) (Supplementary Figure S1) using GeneMANIA. From Supplementary Figure S1, we can see the different types of networks between related genes, suggesting that the MAP1B, TBCB, and PRPH genes are possibly involved in GAN. Interestingly, the role of MAP1B in GAN has been reported in a few studies (Ding et al., 2002; Allen et al., 2005; Ding et al., 2006).
In this report, we described a Chinese patient with GAN who had tight curly hair. The differential diagnosis of GAN was also considered. For instance, Charcot–Marie–Tooth type 2E and 1F caused by mutations in NEFL has been excluded. Electromyography showed multiple peripheral nerves that were damaged, which primarily consisted of axonal lesions and demyelination. Though the boy had the typical GAN phenotype, we could not confirm the pathogenesis without consent for the sural nerve biopsy. Instead, we employed the targeted next-generation sequencing method to test all of the known skeletal, muscle, and nervous system monogenic diseases, and used PCR to verify the mutation sites of the family. The next-generation sequencing method has proven to be a very powerful approach to identify mutations for rare diseases (Huang et al., 2018, 2019). The sequencing results showed that the patient had compound heterozygous mutations of p.S79L (c.236C > T) and p.T489S (c.1466C > G). The p.S79L mutation, reported by Bomont et al., was located in the N-terminus BTB domain of gigaxonin, which synthesizes a functional ubiquitin ligase complex with Cul3 and Rbx1, and then mediates downstream protein degradation (Bomont et al., 2000). Of note, p.S79L was previously only reported in Caucasian population. In this study, we, for the first time, confirmed this mutation in Han Chinese, indicating S79L as a common mutation in Caucasians and East Asians. However, the c.1466C > G was a novel missense mutation. This mutation resulted in amino acid substitutions of threonine to serine (p.T489S) in the C-terminus kelch domain of gigaxonin, which interacted with microtubule-associated protein 1B (MAP1B), microtubule-associated protein 8, and tubulin folding cofactor B. Similar to the other members of the BTB/kelch proteins, gigaxonin seems to interact with cytoskeletal proteins involved in various cellular processes, such as actin cytoskeleton interaction (mayven, kelch, and ENC-1 proteins), cytoplasmic sequestration of transcription factors (Keap1 protein), and cell morphology (calicin) (Ding et al., 2002, 2006). Allen et al. concluded that overexpression of gigaxonin could aggravate the degradation of MAP1B, and the ablation of gigaxonin resulted in substantial accumulation of MAP1B, both of which affect transport processes indirectly by altering microtubule dynamics (Allen et al., 2005). After we obtained consent for the sural nerve biopsy, the results from this test showed that the nerve tracts had giant axonal fibers that had been demyelinated. As a result, we were able to confirm that the GAN was the likely cause of pathogenesis.
In conclusion, compound heterozygous mutations of p.S79L (c.236C > T) and p.T489S (c.1466C > G) in the GAN gene were etiology of the GAN disorder in this Chinese patient. These findings assist in adding to the body of knowledge about genetic mechanisms that can result in GAN.
All datasets generated for this study are included in the article/Supplementary Material.
We obtained written informed consent for genomic analysis and sural nerve biopsy of the patient and her parents in accordance with the Declaration of Helsinki. The study was approved by the Wenzhou People’s Hospital (China) Ethics Committee. The patient and his parents gave written informed consent to the publication of the information and images related to this case report.
JZ and FJ conceived the idea. XY, ZS, HZ, and HW collected the samples. XL, CZ, and CS performed the experiments. YC and WW performed the data analyses. XX wrote the manuscript. All authors have read and approved the final version of the manuscript.
This study was supported by the Zhejiang Provincial Natural Science Foundation (LY19H040008), the Key Plan of Medical and Health Care of Zhejiang Province (2019KY666), and a funding from the Department of Science and Technology of Wenzhou (Y20170602).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the participants for their participation in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2020.00085/full#supplementary-material
Allen, E., Ding, J., Wang, W., Pramanik, S., Chou, J., Yau, V., et al. (2005). Gigaxonin-controlled degradation of MAP1B light chain is critical to neuronal survival. Nature 438, 224–228. doi: 10.1038/nature04256
Asbury, A. K., Gale, M. K., Cox, S. C., Baringer, J. R., and Berg, B. O. (1972). Giant axonal neuropathy–a unique case with segmental neurofilamentous masses. Acta Neuropathol. 20, 237–247. doi: 10.1007/bf00686905
Berg, B. O., Rosenberg, S. H., and Asbury, A. K. (1972). Giant axonal neuropathy. Pediatrics 49, 894–899.
Bomont, P., Cavalier, L., Blondeau, F., Ben Hamida, C., Belal, S., Tazir, M., et al. (2000). The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat. Genet. 26, 370–374. doi: 10.1038/81701
Bruno, C., Bertini, E., Federico, A., Tonoli, E., Lispi, M. L., Cassandrini, D., et al. (2004). Clinical and molecular findings in patients with giant axonal neuropathy (GAN). Neurology 62, 13–16. doi: 10.1212/01.wnl.0000101676.41505.a7
Ding, J., Allen, E., Wang, W., Valle, A., Wu, C., Nardine, T., et al. (2006). Gene targeting of GAN in mouse causes a toxic accumulation of microtubule-associated protein 8 and impaired retrograde axonal transport. Hum. Mol. Genet. 15, 1451–1463. doi: 10.1093/hmg/ddl069
Ding, J., Liu, J. J., Kowal, A. S., Nardine, T., Bhattacharya, P., Lee, A., et al. (2002). Microtubule-associated protein 1B: a neuronal binding partner for gigaxonin. J. Cell Biol. 158, 427–433. doi: 10.1083/jcb.200202055
Huang, X. F., Huang, F., Wu, K. C., Wu, J., Chen, J., Pang, C. P., et al. (2015a). Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 17, 271–278. doi: 10.1038/gim.2014.138
Huang, X. F., Mao, J. Y., Huang, Z. Q., Rao, F. Q., Cheng, F. F., Li, F. F., et al. (2017). Genome-wide detection of copy number variations in unsolved inherited retinal disease. Invest. Ophthalmol. Vis. Sci. 58, 424–429. doi: 10.1167/iovs.16-20705
Huang, X. F., Wu, J., Lv, J. N., Zhang, X., and Jin, Z. B. (2015b). Identification of false-negative mutations missed by next-generation sequencing in retinitis pigmentosa patients: a complementary approach to clinical genetic diagnostic testing. Genet. Med. 17, 307–311. doi: 10.1038/gim.2014.193
Huang, X. F., Xiang, L., Cheng, W., Cheng, F. F., He, K. W., Zhang, B. W., et al. (2018). Mutation of IPO13 causes recessive ocular coloboma, microphthalmia, and cataract. Exp. Mol. Med. 50:53. doi: 10.1038/s12276-018-0079-0
Huang, X. F., Xiang, L., Fang, X. L., Liu, W. Q., Zhuang, Y. Y., Chen, Z. J., et al. (2019). Functional characterization of CEP250 variant identified in nonsyndromic retinitis pigmentosa. Hum. Mutat. 40, 1039–1045. doi: 10.1002/humu.23759
Israeli, E., Dryanovski, D. I., Schumacker, P. T., Chandel, N. S., Singer, J. D., Julien, J. P., et al. (2016). Intermediate filament aggregates cause mitochondrial dysmotility and increase energy demands in giant axonal neuropathy. Hum. Mol. Genet. 25, 2143–2157. doi: 10.1093/hmg/ddw081
Johnson-Kerner, B. L., Roth, L., Greene, J. P., Wichterle, H., and Sproule, D. M. (2014). Giant axonal neuropathy: an updated perspective on its pathology and pathogenesis. Muscle Nerve 50, 467–476. doi: 10.1002/mus.24321
Koichihara, R., Saito, T., Ishiyama, A., Komaki, H., Yuasa, S., Saito, Y., et al. (2016). A mild case of giant axonal neuropathy without central nervous system manifestation. Brain Dev. 38, 350–353. doi: 10.1016/j.braindev.2015.09.001
Kooshavar, D., Razipour, M., Movasat, M., and Keramatipour, M. (2018). Targeted next generation sequencing identified a novel mutation in MYO7A causing Usher syndrome type 1 in an Iranian consanguineous pedigree. Int. J. PediatrOtorhinolaryngol. 104, 10–13. doi: 10.1016/j.ijporl.2017.10.022
Langabeer, S. E., Haslam, K., Kelly, J., Quinn, J., Morrell, R., and Conneally, E. (2018). Targeted next-generation sequencing identifies clinically relevant mutations in patients with chronic neutrophilic leukemia at diagnosis and blast crisis. Clin. Transl. Oncol. 20, 420–423. doi: 10.1007/s12094-017-1722-1722
Mahammad, S., Murthy, S. N., Didonna, A., Grin, B., Israeli, E., Perrot, R., et al. (2013). Giant axonal neuropathy-associated gigaxonin mutations impair intermediate filament protein degradation. J. Clin. Invest. 123, 1964–1975. doi: 10.1172/JCI66387
Normendez-Martinez, M. I., Monterde-Cruz, L., Martinez, R., Marquez-Harper, M., Esquitin-Garduno, N., Valdes-Flores, M., et al. (2018). Two novel mutations in the GAN gene causing giant axonal neuropathy. World J. Pediatr. 14, 298–304. doi: 10.1007/s12519-018-0140-z
Scrivo, A., Codogno, P., and Bomont, P. (2019). Gigaxonin E3 ligase governs ATG16L1 turnover to control autophagosome production. Nat. Commun. 10:780. doi: 10.1038/s41467-019-08331-w
Tazir, M., Nouioua, S., Magy, L., Huehne, K., Assami, S., Urtizberea, A., et al. (2009). Phenotypic variability in giant axonal neuropathy. Neuromusc. Disord. 19, 270–274. doi: 10.1016/j.nmd.2009.01.011
Keywords: giant axonal neuropathy, point mutation, compound heterozygosity, targeted next-generation sequencing, disease attributes
Citation: Xu X, Yang X, Su Z, Wang H, Li X, Sun C, Wang W, Chen Y, Zhang C, Zhang H, Jin F and Zheng J (2020) Identification of Novel Compound Heterozygous Mutations in the GAN Gene of a Chinese Patient Diagnosed With Giant Axonal Neuropathy. Front. Neurosci. 14:85. doi: 10.3389/fnins.2020.00085
Received: 03 April 2019; Accepted: 21 January 2020;
Published: 25 February 2020.
Edited by:
Odete A. B. da Cruz e Silva, University of Aveiro, PortugalReviewed by:
Emil Ylikallio, University of Helsinki, FinlandCopyright © 2020 Xu, Yang, Su, Wang, Li, Sun, Wang, Chen, Zhang, Zhang, Jin and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiayong Zheng, d3J5emhlbmdAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.