 Virginie Dinet
Virginie Dinet Klaus G. Petry
Klaus G. Petry Jerome Badaut
Jerome Badaut
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 12 November 2019
Sec. Neurodegeneration
Volume 13 - 2019 | https://doi.org/10.3389/fnins.2019.01178
This article is part of the Research Topic Cutting-Edge Approaches for CNS Protection and Repair: Focus on Vascular and Degenerative Disorders View all 83 articles
Traumatic brain injury (TBI) is the principal cause of death and disability in children and young adults. Clinical and preclinical research efforts have been carried out to understand the acute, life-threatening pathophysiological events happening after TBI. In the past few years, however, it was recognized that TBI causes significant morbidity weeks, months, or years after the initial injury, thereby contributing substantially to the overall burden of TBI and the decrease of life expectancy in these patients. Long-lasting sequels of TBI include cognitive decline/dementia, sensory-motor dysfunction, and psychiatric disorders, and most important for patients is the need for socio-economic rehabilitation affecting their quality of life. Cerebrovascular alterations have been described during the first week after TBI for direct consequence development of neuroinflammatory process in relation to brain edema. Within the brain–immune interactions, the complement system, which is a family of blood and cell surface proteins, participates in the pathophysiology process. In fact, the complement system is part of the primary defense and clearance component of innate and adaptive immune response. In this review, the complement activation after TBI will be described in relation to the activation of the microglia and astrocytes as well as the blood–brain barrier dysfunction during the first week after the injury. Considering the neuroinflammatory activity as a causal element of neurological handicaps, some major parallel lines of complement activity in multiple sclerosis and Alzheimer pathologies with regard to cognitive impairment will be discussed for chronic TBI. A better understanding of the role of complement activation could facilitate the development of new therapeutic approaches for TBI.
Traumatic brain injury (TBI) is a consequence of a direct or indirect external mechanical impact on the brain inducing the disruption of the normal structure and function of the brain. Despite its simple mechanical definition, TBI is a very complex and heterogeneous injury with various severities graded in clinic. The severity of TBI is predicated on alteration of consciousness, organized according to the Glasgow Coma Scale (GCS) for assessment of neurological function and neuroimaging as mild, moderate, or severe (Maas et al., 2008). TBI is a major burden worldwide, and in the United States, it represents more than 30% of all injury-related deaths (Faul et al., 2010). In Europe, it is about 75,000 deaths each year (Maas et al., 2015), and it affects all ages. It is important to underline that pediatric and elderly populations are more vulnerable than adults (Faul et al., 2010). Therefore, TBI has an economic toll on the society that includes medical expenses and losses of productivity (Corso et al., 2006; Gustavsson et al., 2011). A widespread research on TBI pathophysiology is still sparse compared to other brain diseases such as Alzheimer's disease (AD) and the progressive chronicity of multiple sclerosis (MS).
Pathophysiology following TBI is grouped into the following: (1) The primary acute event is the mechanical injury that can give extracranial and intracranial complications like epidural hematoma, subarachnoid hemorrhage, or hypoxia. Thus, TBI is particularly heterogeneous depending on the primary injury severity, location, and the age and sex of the patient. Important in the TBI pathogenesis is that the functional outcomes may deteriorate over time after initial admission of the patient (Baethmann et al., 1988). For example, the patient can communicate early after TBI and then worsen with loss of consciousness, with an early small contusion that becomes a much larger injury the day after. This evolution is due to the secondary injury events (Plesnila, 2016). (2) The secondary injury cascade includes the following landmarks: glutamatergic excitotoxicity, calcium overload, vascular dysfunction, and inflammation/neuroinflammation. Inflammation/neuroinflammation can last for several months and could contribute to chronic TBI (Plesnila, 2016). The pattern of “chronic brain disease” has been leading the concept of accelerating of brain aging post-TBI (Johnson et al., 2010; Pop and Badaut, 2011; Smith et al., 2013). Patients can present long-lasting behavioral dysfunctions including some psychiatric disorders after TBI (Smith et al., 2013). Very recently, a nationwide population cohort study was done in Denmark to monitor individuals who had TBI from 1977 to 2013. Over the study period, 4.7% of recruited people had at least one TBI, and about 112,218 (85.0%) of 132,093 individuals had a mild TBI for their first TBI diagnosis; those people were 24% more likely to develop dementia than those without a history of TBI. Additionally, the younger the person was at the time of the injury, the higher the risk of dementia development over time (Fann et al., 2018).
Several animal models have been developed in order to extend our understanding of the cellular and molecular mechanisms in post-TBI secondary injuries (Prins and Hovda, 2003; Petraglia et al., 2014). Having a wide variety of preclinical models is a good sketch to mimic the heterogeneity in human TBI. However, it is important to highlight that the definition of the degree of TBI severity differs between animal models, and it is difficult to compare findings between models (Badaut et al., 2019). Sometimes, it is challenging to make the translation with clinical severities. The molecular and cellular mechanisms of secondary injuries are dependent on injury severity and brain location. The post-TBI outcome is more severe for younger patients; therefore, it is important to consider the age of the animal in the preclinical models (McMillan and Teasdale, 2007; Himanen et al., 2011; Pop and Badaut, 2011). In fact, neuroinflammation/inflammation response will vary in function of the location and severity of the impact as well as the age of the animal. The review focuses on some of the cerebrovascular alterations and the importance of the inflammation through the exploration of the complement system as a new potential mechanism. Actually, the role of the complement pathway in TBI pathogenesis is not yet well-understood.
The neurovascular unit (NVU) is composed of neurons, a vascular tree with endothelial cells, smooth muscle cells, pericytes, astrocytes, and microglia (Zhang et al., 2012). Immune cells have been included as part of this physiological unit (Zhang et al., 2012). Cognitive impairments in AD as well as in healthy aging have been paralleled with NVU alterations (Iadecola, 2004; Raz et al., 2016). Similarly, NVU alteration is a typical landmark of the secondary injuries after TBI with, for example, a decrease of blood–brain perfusion with consequences on the good operation of the neurons (Pop and Badaut, 2011; Jullienne et al., 2016). In fact, preclinical TBI models unveil blood–brain barrier (BBB) disruption, hemorrhage, and cerebral blood flow alterations with hypo-perfusion, which are illustrations of the NVU alterations (Golding, 2002; DeWitt and Prough, 2003; Pop and Badaut, 2011). Cerebrovascular dysfunction has been described for almost two decades as a poor prognostic outcome. In fact, early decrease of the cerebral blood flow (CBF) post-injury induces hypoxic events in the brain tissue that directly contribute to the loss of neurons. In addition, the changes in BBB permeability play a role in edema formation with disruption of brain homeostasis. It exacerbates the cascade of secondary injury events including excitotoxicity (glutamate release and resulting higher metabolic demand) and inflammation (with the complement pathway, see below). TBI is a chronic brain disorder with molecular changes at the BBB several months after the initial injury (Jullienne et al., 2016). As it has been suggested in AD, the long-term alterations at the blood–brain interface could therefore be associated with premature aging of the brain after TBI (Pop and Badaut, 2011; Keightley et al., 2014; Jullienne et al., 2016).
Early decrease of CBF is a landmark of the pathophysiology post-TBI and its recovery represents a prognostic factor for outcome post-TBI (Bouma et al., 1991, 1992; DeWitt and Prough, 2003). Preclinical models appropriately reproduce CBF changes observed in clinic. Decreased CBF is observed after TBI using controlled cortical impact (CCI) and lateral fluid percussion (LFP) injury models and it is described up to 8 months after injury in adult rats (Bouma et al., 1991; Bryan et al., 1995; Plesnila et al., 2003; Hayward et al., 2010), which probably feeds the inflammatory process. Even for mild TBI, a decrease of CBF has also been described (Villapol et al., 2014; Long et al., 2015). In a mild CCI model, the reduction of CBF was described with restoration at 30 days post-TBI in young adult mice (Villapol et al., 2015). Importantly, hypo-perfusion adds to the development of the secondary injuries after TBI by decreasing glucose and oxygen delivery. Moreover, an ischemic event could be aggravated by the presence of cell hyper-metabolism during the first 6 h after concussion in rats (Yoshino et al., 1991). Consequently, decreased CBF and hyper-metabolism occurring early after TBI result in a mismatch between demand and supply, known as uncoupling of CBF and glucose metabolism, with direct consequence and aggravation of the secondary injuries and directly feeding the inflammation and neuroinflammation process. At the time, dysfunction in vascular perfusion is paralleled with BBB hyper-permeability and edema process.
The presence of hemorrhage is a marker of acute vascular dysfunction, and it is frequently observed in moderate and severe TBI. BBB and cerebral blood vessel alterations are frequently shadowed by edema formation with dramatic consequences on morbidity and mortality because it induces intracranial hypertension and contributes to the vicious cycle of the secondary injury cascade (Unterberg et al., 2004). Even for mild TBI characterized by the absence of major bleeding, the BBB structure can be damaged or altered during the acute period after the injury. Very importantly, the BBB dysfunction is not only an acute event; it could also be observed several months and years after the traumatic event in human post-mortem tissue (Hay et al., 2015) and in preclinical models (Pop et al., 2013; Jullienne et al., 2014).
The consequences of primary injury on the BBB properties significantly differ according to the age, the type, and the severity of the lesion. The “opening” of the BBB can be observed as early as 3 min after injury in a model of concussion (Povlishock et al., 1978). The “opening” of BBB can be characterized with the presence of two types of lesions on the luminal surface of pial arterioles: crater-shaped indentations and dome-shaped projections of the endothelial cell surface, suggesting necrotic cells (Wei et al., 1980). The tight junction protein complex is potentially altered after TBI. In fact, pial, and intracerebral blood vessels demonstrate decreased claudin-5 and occludin expressions early after injury (Nag et al., 2007). Similar changes in tight junction protein complex have been described in rodent models of mild TBI induced by blast shock waves, with specifically decreased expression in occludin, claudin-5, and zonula occludens protein-1 at 6 h and 24 h post-TBI (Abdul-Muneer et al., 2013). The changes in tight junction proteins have been paralleled with an increase of immunoglobulin G extravasation from 5 min to 48 h post-injury (Yeoh et al., 2013; Shetty et al., 2014). However, it is important to underline that tight junction complexes appear intact under electron microscopy during the first hours after a mild TBI; then, the expression of the tight junction protein is altered (Rafols et al., 2007). Therefore, the changes in expression of the tight junction proteins and increased IgG extravasation are not directly linked with physical rupture of the tight junctions in electron microscopy (Knowland et al., 2014; Haley and Lawrence, 2017). Similarly, IgG extravasation is increased close to the site of impact and surrounding tissue in the absence of a direct relation with changes in claudin-5 expression in a model of juvenile CCI (Pop and Badaut, 2011; Badaut et al., 2015). Similarly, IgG extravasation has been observed in the corpus callosum at 24 h after mild closed head injury, suggesting BBB hyper-permeability even for mild injury (Rodriguez-Grande et al., 2018).
Changes of expression of the tight junction proteins are accompanied by up-regulation of various matrix metalloproteases (MMPs), which have been involved in BBB alteration. In fact, MMP-9 and MMP-2 increase acutely after TBI in rodents (Wang et al., 2000; Zhang et al., 2010). MMP-3 activity is increased chronically after TBI and possibly plays a role in synaptic remodeling (Zhang et al., 2010). Up-regulation of MMPs alters proteins of the extracellular matrix and participates in BBB alteration and neurovascular unit dysfunction.
In summary, it is important to highlight that neurovascular unit disturbance, encompassing BBB and blood flow changes, fuels the inflammation and neuroinflammation process.
Astrocyte endfeet wrap the blood vessels and they play a key role in BBB properties in collaboration with pericytes (Zhang et al., 2012). Astrocytes become “reactive” and proliferate to form a glial scar in various severe brain injury models (Burda and Sofroniew, 2014). Depending on the timeline of the pathological process after injury, astrogliosis can be both beneficial and detrimental to the brain tissue adjacent to the lesion (Sofroniew, 2009). Astrocytes have various physiological functions such as providing energy substrates for neurons, regulating ion and neurotransmitter homeostasis, participating in synapse development and transmission, and regulation of CBF. Then, transformation of astrocyte in “reactive astrocyte” can have a direct and weighty impact on the brain functions on the post-injury outcomes (Sofroniew and Vinters, 2010). Astrocytes are among the first responder brain cells to TBI, and the mechanical forces of the primary injury trigger reactive astrocyte or astrogliosis (Burda et al., 2016). In fact, stretch injury or shear stress involves a rapid calcium influx in the astrocytes (Rzigalinski et al., 1998; Maneshi et al., 2015), by potential activation of astrocytic mechano-receptive channels (Burda et al., 2016). In parallel, activation of the astrocyte potentially contributes to the release of vasoactive substances (Howarth, 2014). In fact, release of isoprostanes (Hoffman et al., 1997, 2000) and endothelin 1 (Ostrow et al., 2000), powerful vasoconstrictors, have been described to increase after TBI in preclinical models (Petrov and Rafols, 2001; Armstead and Kreipke, 2011; Armstead and Raghupathi, 2011). Then, any changes in the calcium concentration within astrocytes could have a direct or indirect consequence on the regulation of CBF and perfusion post-injury. Then, calcium changes in astrocytes could contribute to the NVU dysfunction post-TBI with the increase of vasoconstrictor molecules.
Astrocytes play an important role in the integrity of the BBB (Abbott et al., 2006). Cytokines and inflammatory mediators are released by the astrocytes after injury with consequences on BBB properties (Chodobski et al., 2011; Sofroniew, 2014). For example, chemokines (Chodobski et al., 2011) and three isoforms of transforming growth factor–β (TGF- β) (Constam et al., 1992), potentially released by the astrocytes, have been implicated the BBB alterations after TBI (Shen et al., 2011). Very interestingly, increased TGF-β expression has been observed in patients with severe TBI where it parallels BBB dysfunction (Morganti-Kossmann et al., 1999). As already discussed, MMPs disturb the structure of the BBB after TBI and they are extensively produced by reactive astrocytes (Chen and Swanson, 2003). In the vicinity of the perivascular astrocyte endfoot, the water channel aquaporin 4 expression is altered after TBI, which is related to the formation of cerebral edema (Badaut et al., 2014; Gatto et al., 2015; Zhang et al., 2015). Increased aquaporin expression associated to edema brain formation event is also described in earlier stages of several neurodegenerative diseases (Sun et al., 2016; Yang et al., 2016; Gatto et al., 2018). There is a correlation between the level of aquaporin 4 expression and disruption of BBB (Fukuda and Badaut, 2012). The astrocyte endfeet play a critical role in the BBB integrity, and very interestingly, the physical interaction between astrocyte endfeet and the brain vasculature can also be altered after TBI (Villapol et al., 2014), with a direct contribution to the vascular dysfunction after injury.
Altogether, astrocyte phenotypic transformation can feed forward the brain-vascular pathology observed after TBI in relation to neuroinflammation and the complement pathway. In addition, the neurodegeneration proposed after TBI could be a result of astrocyte activation and chronic inflammation (Faden and Loane, 2015). Interestingly, astrocyte plays a potential role in immune response in the brain with expression of the specific receptors to complement proteins (see part 3). Therefore, it may be important to consider the contribution of reactive astrocytes into the long-term consequences of vascular dysfunction and a target of the activation of the complement pathway. This pathway could represent a new avenue for drug development targeting the NVU.
The complement system is part of an innate and adaptive immune response after infection acting as a primary defense and mechanism of clearance in order to preserve the organ against pathogens. It includes a wide collection of blood and cell surface proteins (C3, C5, CFB, CFD, CFH, C1q, etc.), whose activation results in a self-amplifying cascade of proteolytic reactions (Walport, 2001). Most of the plasmatic complement proteins (90%) are produced by the hepatocytes in the liver (C3, C5, CFB, CFH), released in the bloodstream to circulate as inactive forms. However, a local expression of complement proteins is also observed in cells (microglia/macrophages/astrocytes and neuronal cells) in the brain and the eye, mainly for C3 and its cleavage regulators (CFH and CFB).
The complement system is a key molecular pathway in various cellular and molecular activities, ranging from cell lysis to increase in the local inflammatory response. The complement activation is composed of three main pathways: classical, lectin, and alternate. They converge to the common effector pathway by forming the membrane attack complex (MAC) as illustrated in Figure 1.
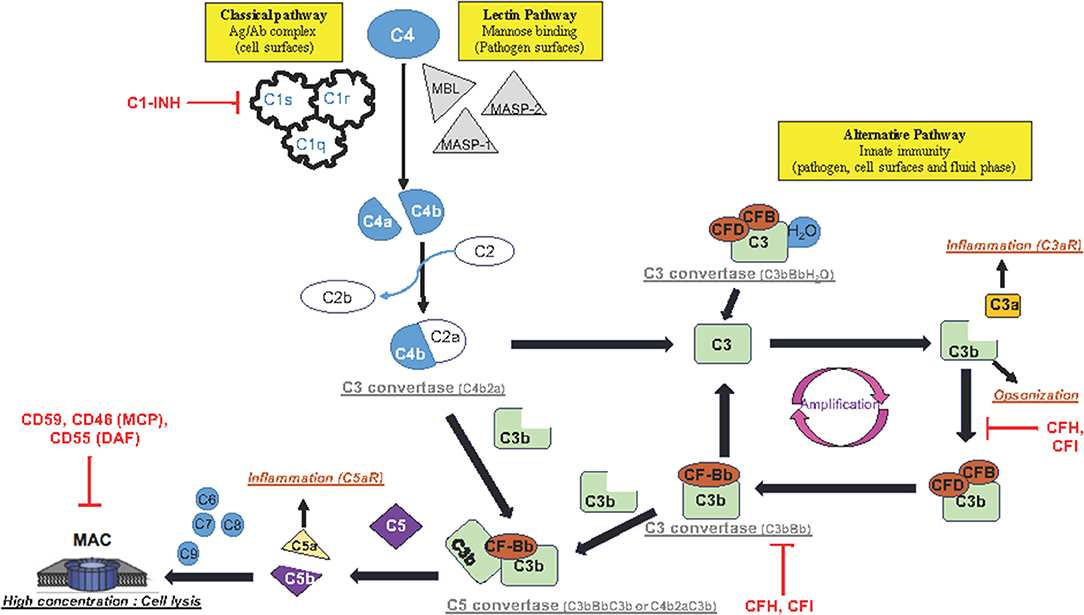
Figure 1. Schematic representation of complement activation pathways: The complement system is composed of three pathways (classical, lectin, and alternative pathway) that involve a collection of blood and cell surface proteins to eliminate pathogens from organism or damaged cells. This system consists of a series of proteins that interact with one another in a highly regulated manner. The classical pathway helps antibodies and phagocytic cells to clear pathogens or damaged cells. The lectin pathway is activated when MBL binds to mannose residues on the pathogen surface. This pathway is very similar to the classical pathway. The alternative pathway is an important part of innate immune system in which proteins cleave one another to form enzymatic complex able to initiate amplification of further cleavages and finally promotes inflammation by production of anaphylactic fragments. The final product of each activated complement pathway is the formation of the MAC in the target cell membrane. Once activated, the complement system has several major functions: lysis of pathogens or damaged cells, activation of inflammation, opsonization, and immune clearance.
The classical pathway activation is antibody-dependent. It happens when the complement protein C1 complex, formed by C1q, C1r, and C1s proteins, interacts with antigen–IgM or aggregated antigen–IgG complex, or is antibody-independent. It also happens when poly-anions (e.g., heparin, protamine, DNA and RNA from apoptotic cells), gram-negative bacteria, or bound C-reactive protein reacts directly with the C1 complex. Once the C1 complex is activated, then C1s cleave C4 and C2 into C4a/C4b and C2a/C2b, respectively, and forms the C3 convertase of the classical pathway (C4b2a). C4b2a cleaves C3 into C3a and C3b fragments. The classical pathway is regulated by C1 inhibitor (C1-INH).
The lectin pathway is activated when mannose-binding lectin (MBL), a serum protein, binds to mannose or fructose groups on bacterial cell walls, yeast walls, or viruses, independently of the antibody complex. The lectin pathway shares similarities with the classical pathway.
The alternative pathway is activated after cleavage of a small amount of complement compound C3 caused by microbial cell surfaces or immunoglobulin. This pathway is functionally present in physiological situation, with a spontaneous (hydrolysis) cleavage of C3 to C3a/C3b. The C3b product is rapidly inactivated by its binding to proteins complement factor H (CFH) and associated with complement factor I (CFI) as a co-factor, and then its opsonization functions (tagging pathogens, immune complexes apoptotic cells for phagocytosis) are aborted. After injury, C3b product binds to complement factor B (CFB) and its co-factor complement factor D (CFD) to form a molecular complex “C3bBb,” called C3 convertase complex, which acts on additional C3 to form more C3b and C3a products (Figure 1). This function of C3b contributes to a positive feedback loop and amplifies a first C3 cleavage that is induced by either the classical pathway or both classical and alternative pathways, in order to produce high-level C3b and C3a proteins. It is important to highlight that each complement pathway (classical, lectin, and alternative pathways) activation ends up into the cleavage of C3.
Once activated, the complement pathway produces a high concentration of anaphylactic C3a/C5a and opsin C3b proteins (Figure 1). The receptors of these complement proteins (C3aR, C5aR, and CD11b, respectively) are expressed on cell membranes in various cerebral cell types such as endothelial cells, microglia/macrophage cells, and astrocytes. The wide distribution of the complement receptors underlines the variety of cellular functions to this system. Importantly, the complement system makes the link between the innate and acquired immunity by (a) enhancing antibody response and immunologic memory, (b) lysing the pathogens, and (c) clearing immune complexes and apoptotic cells.
The anaphylactic C3a molecule has a remote result because it diffuses away to play the role of an inflammation and chemotactic factor. In contrast, the produced C3b binds to the C3 convertase complex (C3bBb) to form a C5 convertase complex (C3bBbC3b). The C5 convertase complex is located in the cell membrane or in the bloodstream and cleaves complement 5 (C5) into C5b and C5a fragments. Similarly to C3a, the C5a product is described in the bloodstream to act remotely as an anaphylactic protein inducing a general inflammatory reaction. In contrast, C5b initiates the MAC in the cell membrane (Figure 1). Sequentially, C5b interacts successively with C6, C7, and C8 complement proteins, and finally with a number of C9 complement proteins to form MAC. Importantly, MAC is organized in a circle with, in its center, a pore that produces a hole in the cell membrane. High concentration of MAC induces lysis of the foreign cell (such as microbes or bacteria) or apoptosis (of host cell).
Very interestingly, MAC has been found to contribute to secondary damages in various preclinical models of human brain diseases (Leinhase et al., 2006; Stahel et al., 2009; Fluiter et al., 2014; Ruseva et al., 2015). However, depending on the level of expression of MAC, opposite physiological responses can be observed. In fact, a low level of the MAC (C5b9) complex on the cell surface promotes cell-cycle progression instead of cell lysis or apoptosis (Fosbrink et al., 2005). The role of MAC in cell-cycle progression is due to the transactivation of receptor tyrosine kinases, a regulator of cytosolic free Ca2+ concentration (Cybulsky et al., 1999), and activation of protein kinase C and cytosolic phospholipase A2-α (Cybulsky et al., 1995; Panesar et al., 1997).
It is important to have a fine-tuned regulation of MAC formation with a full panel of specific inhibitor proteins. Many of the complement regulator proteins are found in the blood plasma and on the cell membranes, and among them, CFH acts on the activity of the upstream C3 convertase complex (C3bBb) in the blood and on the cell surface. CFH removes “Bb fragment” from the C3 convertase enzymatic complex and inactivates C3 cleavage by competition with CFB for binding C3b; it stops the main amplification loop of the alternative complement pathways (Figure 1). Downstream, CD55 (or DAF: decay accelerating factor), CD46 (or MCP: membrane cofactor protein), and CD59 (known as protectin) have also been described as inhibitors of MAC formation in the cell membrane for both classical and alternative complement pathways (Figure 1).
The secondary injury events post-TBI include inflammatory mechanisms with early activation of the innate immune system, and then the chronic inflammation post-injury possibly contributes to the progressive neurodegenerative process (Francis et al., 2003; Schmidt et al., 2005; Stahel and Barnum, 2006).
The complement pathway represents a key element in the inflammatory response, and it contributes to neuronal cell death, edema, and infiltration of inflammatory cells after injury using rodent models of moderate and severe TBI (Kaczorowski et al., 1995; Ruseva et al., 2015; Rich et al., 2016). Involvement of the complement pathway observed in rodent TBI models has been confirmed with up-regulation of CFB (Kossmann et al., 1997), C3, C1q, and C4 (Stahel et al., 1998), and increase of soluble terminal complement complex C5b9 has been detected in ventricular cerebrospinal fluid (CSF) of severe TBI patients for up to 10 days after trauma, possibly due to the alteration of the BBB integrity (Stahel et al., 2001). CSF increase of complement proteins is paralleled by higher concentration of complement proteins in blood plasma of TBI patients (Manek et al., 2018). Expression of complement protein C3 and C9 has been described up to 6 months post-injury (Bao et al., 2018) in patients, suggesting a continuous complement pathway activation in the chronic TBI. Very interestingly, similar observations have been found in various preclinical rodent models, with higher levels of C9 and CFB in serum of rat during the first days after TBI (Thelin et al., 2018). Deposits of C3 have also been described in the peri-lesion tissue, while C9 protein has been reported in damaged neurons after cortical contusion in the adult rat (Bellander et al., 1996). Thus, a similar change in activation of the complement pathway has been described in clinical and in preclinical rodent models.
As presented above, C3 protein is common to the three complement pathways. In this line with the complement pathway inhibition in TBI, C3−/− mice exhibit less brain edema and microglia activation/infiltration compared to WT after TBI, suggesting a role of the complement pathways in the secondary injury cascade (Sewell et al., 2004; Yang et al., 2006) and inhibition of C3 could be a therapeutic strategy. Interestingly, severe TBI swine resuscitation with valproic acid (a histone deacetylase inhibitor) induces a down-regulation of the complement system activation, resulting in a significant decrease of lesion size after resuscitation (Dekker et al., 2014a,b; Bambakidis et al., 2016). Similarly, inhibition of MAC has been proposed to be protective. In fact, over-exuberant MAC formation contributes to neuronal cell death after severe TBI injury in mice lacking CD59, an inhibitor of MAC formation on the membrane cells (Figure 1) (Stahel et al., 2009). The inhibition of MAC formation using C6 antisense oligonucleotide or an inhibitor of C5 convertase complex reduces not only MAC deposition but also accumulation of microglia/macrophages, neuronal death, and axonal loss and enhances neurologic performances as compared to the placebo-treated TBI group (Fluiter et al., 2014).
The current paradigm is that MAC formation is predominantly implicated in development of post-TBI process neuropathology and neuroinflammation, but it is not excluded that C3 cleavage inhibition could prevent inflammation and/or cell death. Then, C3 and MAC would be good therapeutic targets to reduce TBI damage. Studies show that inhibition of C3 cleavage reduces post-injury of microglial and astrocyte activations, C3 deposits, and the extent of neuron cell death, and then triggers improvements in cognitive and functional recovery (Rich et al., 2016; Alawieh et al., 2018). It demonstrates a critical role of the complement system and mainly of the alternative pathway in the TBI neuropathology process.
Targeted deletion of cfb gene expression, the activator of alternate complement pathway by activation of C3 convertase, in transgenic mice clearly reveals a significant decrease of neuronal cell death post-TBI (Leinhase et al., 2006). In support to these results in transgenic mice, inhibition of the CFB by injection of anti-CFB monoclonal antibody attenuates both inflammation and cell death in TBI animals (Leinhase et al., 2007), confirming a key role of the alternative complement system in TBI.
Both the classical and lectin pathways have also been assessed in preclinical TBI models and human studies: (1) C1-inhibitor injection after TBI mitigates motor deficits, cognitive dysfunction, and contusion volumes (Longhi et al., 2009), arguing for a role of the classical complement pathway in TBI secondary injuries. (2) Activation of the lectin complement pathway has been shown to be involved in secondary ischemic/inflammatory injury after severe TBI along with an increase of MBL protein in plasma of patients at 48 h post-injury (Osthoff et al., 2017). However, there is no correlation between lectin complement pathway activation and mortality/consciousness after severe TBI (Osthoff et al., 2017). In human and mice, MBL is expressed in the injured cortex from 30 min and persists up to 1 week post-TBI (Longhi et al., 2014). The inactivation of the lectin complement pathway induces a decrease of cortical cell death (Longhi et al., 2014), arguing that this deletion has a protective effect in TBI damages. Until now, no specific pharmacological compound has been found to limit TBI damages, but targeting complement pathway would be a novel therapeutic way.
Brain vasculature alterations and inflammatory processes implicating complement pathways are very likely hand in hand in the early secondary events post-injury. Several works clearly indicate that TBI is a chronic brain disorder with development of neurodegenerative disease landmarks such as accumulation of the amyloid-β (Aβ) (Johnson et al., 2010; Pop and Badaut, 2011; Pop et al., 2013). We and other groups have shown changes in the brain vasculature and their involvement in functional outcome after TBI, with BBB dysfunctions several months and years after the injury (Pop et al., 2013; Hay et al., 2015). We previously described increase of claudin-5 expression in large blood vessels, decrease of the efflux pump P-glycoprotein (P-gp), and increase of the perivascular matrix proteins perlecan and fibronectin up to 6 months post-injury in a juvenile TBI model (Pop et al., 2013; Jullienne et al., 2014). The matrix changes observed up to 6 months after juvenile TBI (Jullienne et al., 2014) have also been described in AD patients (Lepelletier et al., 2017). Changes in the matrix properties possibly participate in neurodegenerative processes by leading to the accumulation of Aβ, decreasing its clearance and its perivascular drainage (Pop et al., 2013; Jullienne et al., 2014). P-gp has been suggested to be a key player in Aβ clearance from the brain parenchyma. P-gp knock-out models have increased Aβ deposition after injection of Aβ in the brain compared to the wild type (Cirrito et al., 2005). Moreover, P-gp expression is decreased on endothelial cells in aged human and AD brains as well as in aged rodents (Silverberg et al., 2010). In addition to the vascular dysfunction, the presence of Aβ deposition could contribute to amplify the pathophysiology with activation of the complement pathways around the Aβ deposition, and the vicious cycle of chronic inflammation would accelerate the brain aging.
A number of chronic neurodegenerative diseases of the central nervous system (CNS) are characterized by prominent neuroinflammation marked by activated astrogliosis and microgliosis. In AD and MS affections of CNS, similar to what is seen in TBI, there is strong evidence of complement involvement in the pathogenesis that arises from a confluence of histochemical, genetic, and model data. The involvement of the complement system in the AD process comes from the identification of complement receptor 1, the C3b receptor expressed on macrophage/erythrocyte or monocyte cells, as a genetic risk to develop this neurodegenerative disease (Lambert et al., 2009; Crehan et al., 2012). In fact, erythrocyte cells capture complement-opsonized Aβ through CR1 receptor expressed on their membrane and facilitate clearance mechanism, which is relevant in AD pathophysiology (Brubaker et al., 2017). On the other hand, Aβ deposition promotes complement pathway activation by inhibition of CFI enzymatic activity, a cofactor of CFH (Lashkari et al., 2018). Thus, accumulation of Aβ contributes to increase C3 complement protein and ends up giving rise to the anaphylactic C3a/C5a proteins and MAC formation (Bradt et al., 1998; Lian et al., 2015). In fact, accumulation of these complement proteins generates damaged neurons, increases numbers of activated glial cells (Bradt et al., 1998), and, through neuronal C3a/C3aR signaling, disrupts dendritic morphology and network function (Lian et al., 2015). The Aβ-induced neuroinflammation response is also exaggerated by C5a/C5aR signaling (An et al., 2018), demonstrating another link between Aβ and complement activation. Similarly, the role of the complement pathway has been investigated in the context of the neuroinflammatory autoimmune neurodegenerative disease MS. Comparatively, these studies would help to understand the potential role of complement pathways in the long-term outcomes after TBI. With the aim to better understand the cause of MS, a demyelinating and neurodegenerative disease of the CNS, several investigations defined and quantified complement compounds in the blood circulation and CSF. In relation to the inflammatory activity of MS, to define potential biomarkers of the various MS disease forms, levels of C3, C4, and C9 were mostly investigated, providing convincing evidence of the role of complement compounds in MS disease development. The data obtained from various cohorts of patients, however, generated controversies. Indeed, early studies in patients with progressive MS revealed elevated levels of C4 in MS-serum and reduced C4 levels in MS-CSF (Jans et al., 1984). These findings were, however, not supported by others (Jongen et al., 2000). A more recent study defined elevated plasma C4a levels and elevated C3 levels in CSF of patients with active relapsing-remitting MS (RRMS) potentially providing furthermore follow-up markers for therapeutic efficacy (Tatomir et al., 2017). The levels of C9 compound and terminal complement complex (MAC) in CSF and/or blood also give heterogeneous quantitative data found to be reduced, unchanged, or increased (Ingram et al., 2009). Differences in the presence of the complement compounds in the CSF and blood circulation might be related to the modulation of the BBB by the inflammatory disease activity at the various disease MS forms. Histophathological post-mortem investigations of MS brain tissues revealed complement deposition and activation. Activated microglia is commonly observed in contact with axons suffering from demyelination. In brain tissue from patients with progressive MS, a combined immunohistopathology and in situ hybridization study showed the local expression of compounds of the complement activation pathways. Both transcripts for C1qA, activation fragment and opsonin C3b, and immunostained proteins C1q of the classical complement pathway and the activation CFB-fragment Bb of the alternative complement pathway were observed in neurons and/or glia within and in the vicinity of cortical and deep gray matter lesions. Furthermore, activated microglia expressing complement anaphylatoxin receptor were observed in these lesions in post-mortem brain tissue (Watkins et al., 2016). The deposits of activated C3 fragment on microglia in such chronic cortical lesions, but not in acute disease, indicate that C3 could function as a mediator for the removal of damaged nervous cells in the MS animal model of the relapsing experimental autoimmune encephalomyelitis (EAE) (Michailidou et al., 2017). The formation of MAC as a final complement product has been reported in acute and chronic white matter lesions of MS and would have a direct impact on nervous tissue destruction (Compston et al., 1989; Storch et al., 1998). Many experimental animal studies of MS support the complement-mediated role of nervous tissue degeneration. Further evidence for MAC in the inflammatory disease progression is documented in the EAE mouse model by the systemic curative administration of MAC inhibitors that prevents relapses after a first clinical attack (Michailidou et al., 2018). In EAE gray matter neurodegeneration, a significant synapse loss in the CA1 statum radiatum of the hippocampus reflecting cognitive impairment is caused by increased complement production and deposition of Cq1 and C3d that could make synapses vulnerable to phagocytosis by microglia. In particular, C3 mediates microglial activation and EAE motor impairments (Hammond et al., 2019). Altogether, there is convincing evidence that the complement activation is involved in MS disease development and the cerebral complement over-activation in inflammatory CNS lesions may be essential for the irreversible progression of MS. The knowledge gathered on the role of the complement pathway activation in MS patients and animal model of MS can be useful for understanding the chronic brain disorders post-TBI. The presence of the complement in serum can serve as a biomarker in TBI patients, and the potential inhibition of the complement can be a therapeutic strategy for the long-term consequences of TBI.
Complement pathway activation is present early after TBI and can contribute to the development of the secondary injuries by inducing neuronal cell loss and synapse pruning (Table 1). Very likely, the complement activation is still present in the long term after TBI (Table 1). The knowledge on complement activation in AD and MS strongly suggests that the activation of the complement pathways in the long term after TBI could be an interesting mechanism in order to develop new therapeutic approaches for chronic TBI. It is possible that the pruning of synapses by complement-mediated microglia activation causes long-term neurological handicaps including cognitive impairment in various models of neurodegenerative diseases such as Alzheimer's disease, viral encephalitis, TBI, and MS and potentially contributes to schizophrenia (Stevens et al., 2007; Paolicelli et al., 2011; Ingram et al., 2014; Hong et al., 2016; Sekar et al., 2016; Vasek et al., 2016; Alawieh et al., 2018; Hammond et al., 2019). Therefore, inhibition of the long-term complement activation could improve TBI outcomes.
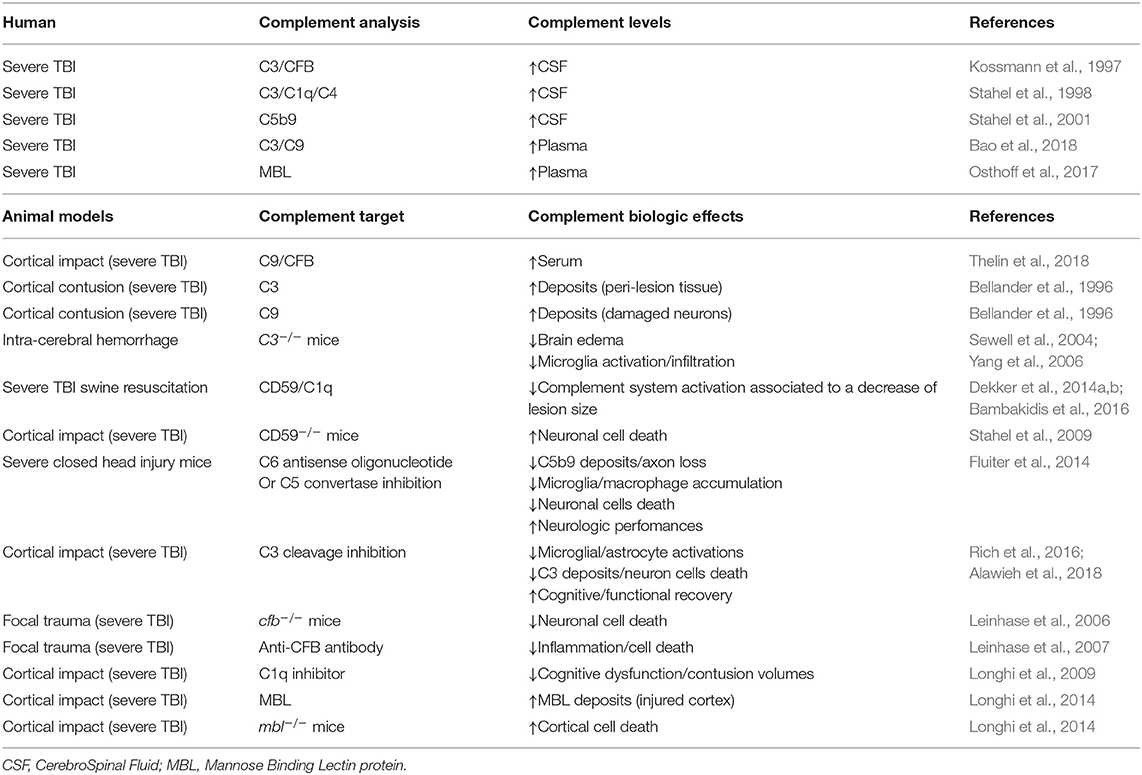
Table 1. Summary of complement effects on the TBI process in animals models and human studies.
VD and JB contributed to the design and concept of the review manuscript. VD, KP, and JB wrote and revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by grants from the Eranet neuron consortia TRAINS (JB), TRAIL-Labex ANR (JB), ANR-17-CE37-0011, Nanospace (JB), INSERM (VD and KP), and ANR (KP).
Abbott, N. J., Rönnbäck, L., and Hansson, E. (2006). Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7, 41–53. doi: 10.1038/nrn1824
Abdul-Muneer, P. M., Schuetz, H., Wang, F., Skotak, M., Jones, J., Gorantla, S., et al. (2013). Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic. Biol. Med. 60, 282–291. doi: 10.1016/j.freeradbiomed.2013.02.029
Alawieh, A., Langley, E. F., Weber, S., Adkins, D., and Tomlinson, S. (2018). Identifying the role of complement in triggering neuroinflammation after traumatic brain injury. J. Neurosci. 38, 2519–2532. doi: 10.1523/JNEUROSCI.2197-17.2018
An, X.-Q., Xi, W., Gu, C.-Y., and Huang, X. (2018). Complement protein C5a enhances the β-amyloid-induced neuro-inflammatory response in microglia in Alzheimer's disease. Med. Sci. 34, 116–120. doi: 10.1051/medsci/201834f120
Armstead, W. M., and Kreipke, C. W. (2011). Endothelin-1 is upregulated after traumatic brain injury: a cross-species, cross-model analysis. Neurol. Res. 33, 133–136. doi: 10.1179/016164111X12881719352174
Armstead, W. M., and Raghupathi, R. (2011). Endothelin and the neurovascular unit in pediatric traumatic brain injury. Neurol. Res. 33, 127–132. doi: 10.1179/016164111X12881719352138
Badaut, J., Adami, A., Huang, L., and Obenaus, A. (2019). Noninvasive magnetic resonance imaging stratifies injury severity in a rodent model of male juvenile traumatic brain injury. J. Neurosci. Res. doi: 10.1002/jnr.24415. [Epub ahead of print].
Badaut, J., Ajao, D. O., Sorensen, D. W., Fukuda, A. M., and Pellerin, L. (2015). Caveolin expression changes in the neurovascular unit after juvenile traumatic brain injury: signs of blood-brain barrier healing? Neuroscience 285, 215–226. doi: 10.1016/j.neuroscience.2014.10.035
Badaut, J., Fukuda, A. M., Jullienne, A., and Petry, K. G. (2014). Aquaporin and brain diseases. Biochim. Biophys. Acta 1840, 1554–1565. doi: 10.1016/j.bbagen.2013.10.032
Baethmann, A., Maier-Hauff, K., Kempski, O., Unterberg, A., Wahl, M., and Schürer, L. (1988). Mediators of brain edema and secondary brain damage. Crit. Care Med. 16, 972–978. doi: 10.1097/00003246-198810000-00008
Bambakidis, T., Dekker, S. E., Sillesen, M., Liu, B., Johnson, C. N., Jin, G., et al. (2016). Resuscitation with valproic acid alters inflammatory genes in a porcine model of combined traumatic brain injury and hemorrhagic shock. J. Neurotrauma 33, 1514–1521. doi: 10.1089/neu.2015.4163
Bao, W., He, F., Yu, L., Gao, J., Meng, F., Ding, Y., et al. (2018). Complement cascade on severe traumatic brain injury patients at the chronic unconscious stage: implication for pathogenesis. Expert Rev. Mol. Diagn. 18, 761–766. doi: 10.1080/14737159.2018.1471985
Bellander, B. M., von Holst, H., Fredman, P., and Svensson, M. (1996). Activation of the complement cascade and increase of clusterin in the brain following a cortical contusion in the adult rat. J. Neurosurg. 85, 468–475. doi: 10.3171/jns.1996.85.3.0468
Bouma, G. J., Muizelaar, J. P., Choi, S. C., Newlon, P. G., and Young, H. F. (1991). Cerebral circulation and metabolism after severe traumatic brain injury: the elusive role of ischemia. J. Neurosurg. 75, 685–693. doi: 10.3171/jns.1991.75.5.0685
Bouma, G. J., Muizelaar, J. P., Stringer, W. A., Choi, S. C., Fatouros, P., and Young, H. F. (1992). Ultra-early evaluation of regional cerebral blood flow in severely head-injured patients using xenon-enhanced computerized tomography. J. Neurosurg. 77, 360–368. doi: 10.3171/jns.1992.77.3.0360
Bradt, B. M., Kolb, W. P., and Cooper, N. R. (1998). Complement-dependent proinflammatory properties of the Alzheimer's disease beta-peptide. J. Exp. Med. 188, 431–438. doi: 10.1084/jem.188.3.431
Brubaker, W. D., Crane, A., Johansson, J. U., Yen, K., Garfinkel, K., Mastroeni, D., et al. (2017). Peripheral complement interactions with amyloid β peptide: erythrocyte clearance mechanisms. Alzheimers Dement. 13, 1397–1409. doi: 10.1016/j.jalz.2017.03.010
Bryan, R. M., Cherian, L., and Robertson, C. (1995). Regional cerebral blood flow after controlled cortical impact injury in rats. Anesth. Analg. 80, 687–695. doi: 10.1213/00000539-199504000-00007
Burda, J. E., Bernstein, A. M., and Sofroniew, M. V. (2016). Astrocyte roles in traumatic brain injury. Exp. Neurol. 275 Pt 3, 305–315. doi: 10.1016/j.expneurol.2015.03.020
Burda, J. E., and Sofroniew, M. V. (2014). Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248. doi: 10.1016/j.neuron.2013.12.034
Chen, Y., and Swanson, R. A. (2003). Astrocytes and brain injury. J. Cereb. Blood Flow Metab. 23, 137–149. doi: 10.1097/01.WCB.0000044631.80210.3C
Chodobski, A., Zink, B. J., and Szmydynger-Chodobska, J. (2011). Blood-brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res. 2, 492–516. doi: 10.1007/s12975-011-0125-x
Cirrito, J. R., Deane, R., Fagan, A. M., Spinner, M. L., Parsadanian, M., Finn, M. B., et al. (2005). P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Invest. 115, 3285–3290. doi: 10.1172/JCI25247
Compston, D. A., Morgan, B. P., Campbell, A. K., Wilkins, P., Cole, G., Thomas, N. D., et al. (1989). Immunocytochemical localization of the terminal complement complex in multiple sclerosis. Neuropathol. Appl. Neurobiol. 15, 307–316. doi: 10.1111/j.1365-2990.1989.tb01231.x
Constam, D. B., Philipp, J., Malipiero, U. V., ten Dijke, P., Schachner, M., and Fontana, A. (1992). Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J. Immunol. 148, 1404–1410.
Corso, P., Finkelstein, E., Miller, T., Fiebelkorn, I., and Zaloshnja, E. (2006). Incidence and lifetime costs of injuries in the United States. Inj. Prev. 12, 212–218. doi: 10.1136/ip.2005.010983
Crehan, H., Holton, P., Wray, S., Pocock, J., Guerreiro, R., and Hardy, J. (2012). Complement receptor 1 (CR1) and Alzheimer's disease. Immunobiology 217, 244–250. doi: 10.1016/j.imbio.2011.07.017
Cybulsky, A. V., Monge, J. C., Papillon, J., and McTavish, A. J. (1995). Complement C5b-9 activates cytosolic phospholipase A2 in glomerular epithelial cells. Am. J. Physiol. 269, F739–749. doi: 10.1152/ajprenal.1995.269.5.F739
Cybulsky, A. V., Takano, T., Papillon, J., and McTavish, A. J. (1999). Complement C5b-9 induces receptor tyrosine kinase transactivation in glomerular epithelial cells. Am. J. Pathol. 155, 1701–1711. doi: 10.1016/S0002-9440(10)65485-5
Dekker, S. E., Bambakidis, T., Sillesen, M., Liu, B., Johnson, C. N., Jin, G., et al. (2014a). Effect of pharmacologic resuscitation on the brain gene expression profiles in a swine model of traumatic brain injury and hemorrhage. J Trauma Acute Care Surg. 77, 906–912. doi: 10.1097/TA.0000000000000345
Dekker, S. E., Sillesen, M., Bambakidis, T., Andjelkovic, A. V., Jin, G., Liu, B., et al. (2014b). Treatment with a histone deacetylase inhibitor, valproic acid, is associated with increased platelet activation in a large animal model of traumatic brain injury and hemorrhagic shock. J. Surg. Res. 190, 312–318. doi: 10.1016/j.jss.2014.02.049
DeWitt, D. S., and Prough, D. S. (2003). Traumatic cerebral vascular injury: the effects of concussive brain injury on the cerebral vasculature. J. Neurotrauma 20, 795–825. doi: 10.1089/089771503322385755
Faden, A. I., and Loane, D. J. (2015). Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics 12, 143–150. doi: 10.1007/s13311-014-0319-5
Fann, J. R., Ribe, A. R., Pedersen, H. S., Fenger-Grøn, M., Christensen, J., Benros, M. E., et al. (2018). Long-term risk of dementia among people with traumatic brain injury in Denmark: a population-based observational cohort study. Lancet Psychiatry 5, 424–431. doi: 10.1016/S2215-0366(18)30065-8
Faul, M., Xu, L., Wald, M. M., and Coronado, V. G. (2010). Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths, 2002–2006. Atlanta, GA: CDC.
Fluiter, K., Opperhuizen, A. L., Morgan, B. P., Baas, F., and Ramaglia, V. (2014). Inhibition of the membrane attack complex of the complement system reduces secondary neuroaxonal loss and promotes neurologic recovery after traumatic brain injury in mice. J. Immunol. 192, 2339–2348. doi: 10.4049/jimmunol.1302793
Fosbrink, M., Niculescu, F., and Rus, H. (2005). The role of c5b-9 terminal complement complex in activation of the cell cycle and transcription. Immunol. Res. 31, 37–46. doi: 10.1385/IR:31:1:37
Francis, K., van Beek, J., Canova, C., Neal, J. W., and Gasque, P. (2003). Innate immunity and brain inflammation: the key role of complement. Expert Rev. Mol. Med. 5, 1–19. doi: 10.1017/S1462399403006252
Fukuda, A. M., and Badaut, J. (2012). Aquaporin 4: a player in cerebral edema and neuroinflammation. J Neuroinflammation 9:279. doi: 10.1186/1742-2094-9-279
Gatto, R., Chauhan, M., and Chauhan, N. (2015). Anti-edema effects of rhEpo in experimental traumatic brain injury. Restor. Neurol. Neurosci. 33, 927–941. doi: 10.3233/RNN-150577
Gatto, R. G., Amin, M. Y., Deyoung, D., Hey, M., Mareci, T. H., and Magin, R. L. (2018). Ultra-high field diffusion MRI reveals early axonal pathology in spinal cord of ALS mice. Transl. Neurodegener 7:20. doi: 10.1186/s40035-018-0122-z
Golding, E. M. (2002). Sequelae following traumatic brain injury. The cerebrovascular perspective. Brain Res. Brain Res. Rev. 38, 377–388. doi: 10.1016/S0165-0173(02)00141-8
Gustavsson, A., Svensson, M., Jacobi, F., Allgulander, C., Alonso, J., Beghi, E., et al. (2011). Cost of disorders of the brain in Europe 2010. Eur. Neuropsychopharmacol. 21, 718–779. doi: 10.1016/j.euroneuro.2011.08.008
Haley, M. J., and Lawrence, C. B. (2017). The blood-brain barrier after stroke: structural studies and the role of transcytotic vesicles. J. Cereb. Blood Flow Metab. 37, 456–470. doi: 10.1177/0271678X16629976
Hammond, J. W., Bellizzi, M. J., Ware, C., Qiu, W. Q., Saminathan, P., Li, H., et al. (2019). Complement-dependent synapse loss and microgliosis in a mouse model of multiple sclerosis. bioRxiv [preprint] 720649. doi: 10.1101/720649
Hay, J. R., Johnson, V. E., Young, A. M. H., Smith, D. H., and Stewart, W. (2015). Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J. Neuropathol. Exp. Neurol. 74, 1147–1157. doi: 10.1093/jnen/74.12.1147
Hayward, N. M. E. A., Immonen, R., Tuunanen, P. I., Ndode-Ekane, X. E., Gröhn, O., and Pitkänen, A. (2010). Association of chronic vascular changes with functional outcome after traumatic brain injury in rats. J. Neurotrauma 27, 2203–2219. doi: 10.1089/neu.2010.1448
Himanen, L., Portin, R., Hämäläinen, P., Hurme, S., Hiekkanen, H., and Tenovuo, O. (2011). Risk factors for reduced survival after traumatic brain injury: a 30-year follow-up study. Brain Inj. 25, 443–452. doi: 10.3109/02699052.2011.556580
Hoffman, S. W., Moore, S., and Ellis, E. F. (1997). Isoprostanes: free radical-generated prostaglandins with constrictor effects on cerebral arterioles. Stroke 28, 844–849. doi: 10.1161/01.STR.28.4.844
Hoffman, S. W., Rzigalinski, B. A., Willoughby, K. A., and Ellis, E. F. (2000). Astrocytes generate isoprostanes in response to trauma or oxygen radicals. J. Neurotrauma 17, 415–420. doi: 10.1089/neu.2000.17.415
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Howarth, C. (2014). The contribution of astrocytes to the regulation of cerebral blood flow. Front. Neurosci. 8:103. doi: 10.3389/fnins.2014.00103
Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat. Rev. Neurosci. 5, 347–360. doi: 10.1038/nrn1387
Ingram, G., Hakobyan, S., Robertson, N. P., and Morgan, B. P. (2009). Complement in multiple sclerosis: its role in disease and potential as a biomarker. Clin. Exp. Immunol. 155, 128–139. doi: 10.1111/j.1365-2249.2008.03830.x
Ingram, G., Loveless, S., Howell, O. W., Hakobyan, S., Dancey, B., Harris, C. L., et al. (2014). Complement activation in multiple sclerosis plaques: an immunohistochemical analysis. Acta Neuropathol. Commun. 2:53. doi: 10.1186/2051-5960-2-53
Jans, H., Heltberg, A., Zeeberg, I., Kristensen, J. H., Fog, T., and Raun, N. E. (1984). Immune complexes and the complement factors C4 and C3 in cerebrospinal fluid and serum from patients with chronic progressive multiple sclerosis. Acta Neurol. Scand. 69, 34–38. doi: 10.1111/j.1600-0404.1984.tb07777.x
Johnson, V. E., Stewart, W., and Smith, D. H. (2010). Traumatic brain injury and amyloid-β pathology: a link to Alzheimer's disease? Nat. Rev. Neurosci. 11, 361–370. doi: 10.1038/nrn2808
Jongen, P. J., Doesburg, W. H., Ibrahim-Stappers, J. L., Lemmens, W. A., Hommes, O. R., and Lamers, K. J. (2000). Cerebrospinal fluid C3 and C4 indexes in immunological disorders of the central nervous system. Acta Neurol. Scand. 101, 116–121. doi: 10.1034/j.1600-0404.2000.101002116.x
Jullienne, A., Obenaus, A., Ichkova, A., Savona-Baron, C., Pearce, W. J., and Badaut, J. (2016). Chronic cerebrovascular dysfunction after traumatic brain injury. J. Neurosci. Res. 94, 609–622. doi: 10.1002/jnr.23732
Jullienne, A., Roberts, J. M., Pop, V., Paul Murphy, M., Head, E., Bix, G. J., et al. (2014). Juvenile traumatic brain injury induces long-term perivascular matrix changes alongside amyloid-beta accumulation. J. Cereb. Blood Flow Metab. 34, 1637–1645. doi: 10.1038/jcbfm.2014.124
Kaczorowski, S. L., Schiding, J. K., Toth, C. A., and Kochanek, P. M. (1995). Effect of soluble complement receptor-1 on neutrophil accumulation after traumatic brain injury in rats. J. Cereb. Blood Flow Metab. 15, 860–864. doi: 10.1038/jcbfm.1995.107
Keightley, M. L., Sinopoli, K. J., Davis, K. D., Mikulis, D. J., Wennberg, R., Tartaglia, M. C., et al. (2014). Is there evidence for neurodegenerative change following traumatic brain injury in children and youth? A scoping review. Front. Hum. Neurosci. 8:139. doi: 10.3389/fnhum.2014.00139
Knowland, D., Arac, A., Sekiguchi, K. J., Hsu, M., Lutz, S. E., Perrino, J., et al. (2014). Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 82, 603–617. doi: 10.1016/j.neuron.2014.03.003
Kossmann, T., Stahel, P. F., Morganti-Kossmann, M. C., Jones, J. L., and Barnum, S. R. (1997). Elevated levels of the complement components C3 and factor B in ventricular cerebrospinal fluid of patients with traumatic brain injury. J. Neuroimmunol. 73, 63–69. doi: 10.1016/S0165-5728(96)00164-6
Lambert, J.-C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat. Genet. 41, 1094–1099. doi: 10.1038/ng.439
Lashkari, K., Teague, G., Chen, H., Lin, Y.-Q., Kumar, S., McLaughlin, M. M., et al. (2018). A monoclonal antibody targeting amyloid β (Aβ) restores complement factor I bioactivity: potential implications in age-related macular degeneration and Alzheimer's disease. PLoS ONE 13:e0195751. doi: 10.1371/journal.pone.0195751
Leinhase, I., Holers, V. M., Thurman, J. M., Harhausen, D., Schmidt, O. I., Pietzcker, M., et al. (2006). Reduced neuronal cell death after experimental brain injury in mice lacking a functional alternative pathway of complement activation. BMC Neurosci. 7:55. doi: 10.1186/1471-2202-7-55
Leinhase, I., Rozanski, M., Harhausen, D., Thurman, J. M., Schmidt, O. I., Hossini, A. M., et al. (2007). Inhibition of the alternative complement activation pathway in traumatic brain injury by a monoclonal anti-factor B antibody: a randomized placebo-controlled study in mice. J Neuroinflammation 4:13. doi: 10.1186/1742-2094-4-13
Lepelletier, F.-X., Mann, D. M. A., Robinson, A. C., Pinteaux, E., and Boutin, H. (2017). Early changes in extracellular matrix in Alzheimer's disease. Neuropathol. Appl. Neurobiol. 43, 167–182. doi: 10.1111/nan.12295
Lian, H., Yang, L., Cole, A., Sun, L., Chiang, A. C.-A., Fowler, S. W., et al. (2015). NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer's disease. Neuron 85, 101–115. doi: 10.1016/j.neuron.2014.11.018
Long, J. A., Watts, L. T., Li, W., Shen, Q., Muir, E. R., Huang, S., et al. (2015). The effects of perturbed cerebral blood flow and cerebrovascular reactivity on structural MRI and behavioral readouts in mild traumatic brain injury. J. Cereb. Blood Flow Metab. 35, 1852–1861. doi: 10.1038/jcbfm.2015.143
Longhi, L., Orsini, F., De Blasio, D., Fumagalli, S., Ortolano, F., Locatelli, M., et al. (2014). Mannose-binding lectin is expressed after clinical and experimental traumatic brain injury and its deletion is protective. Crit. Care Med. 42, 1910–1918. doi: 10.1097/CCM.0000000000000399
Longhi, L., Perego, C., Ortolano, F., Zanier, E. R., Bianchi, P., Stocchetti, N., et al. (2009). C1-inhibitor attenuates neurobehavioral deficits and reduces contusion volume after controlled cortical impact brain injury in mice. Crit. Care Med. 37, 659–665. doi: 10.1097/CCM.0b013e318195998a
Maas, A. I. R., Menon, D. K., Steyerberg, E. W., Citerio, G., Lecky, F., Manley, G. T., et al. (2015). Collaborative European NeuroTrauma Effectiveness Research in Traumatic Brain Injury (CENTER-TBI): a prospective longitudinal observational study. Neurosurgery 76, 67–80. doi: 10.1227/NEU.0000000000000575
Maas, A. I. R., Stocchetti, N., and Bullock, R. (2008). Moderate and severe traumatic brain injury in adults. Lancet Neurol. 7, 728–741. doi: 10.1016/S1474-4422(08)70164-9
Manek, R., Moghieb, A., Yang, Z., Kumar, D., Kobessiy, F., Sarkis, G. A., et al. (2018). Protein biomarkers and neuroproteomics characterization of microvesicles/exosomes from human cerebrospinal fluid following traumatic brain injury. Mol. Neurobiol. 55, 6112–6128. doi: 10.1007/s12035-017-0821-y
Maneshi, M. M., Sachs, F., and Hua, S. Z. (2015). A threshold shear force for calcium influx in an astrocyte model of traumatic brain injury. J. Neurotrauma 32, 1020–1029. doi: 10.1089/neu.2014.3677
McMillan, T. M., and Teasdale, G. M. (2007). Death rate is increased for at least 7 years after head injury: a prospective study. Brain 130, 2520–2527. doi: 10.1093/brain/awm185
Michailidou, I., Jongejan, A., Vreijling, J. P., Georgakopoulou, T., de Wissel, M. B., Wolterman, R. A., et al. (2018). Systemic inhibition of the membrane attack complex impedes neuroinflammation in chronic relapsing experimental autoimmune encephalomyelitis. Acta Neuropathol. Commun. 6:36. doi: 10.1186/s40478-018-0536-y
Michailidou, I., Naessens, D. M. P., Hametner, S., Guldenaar, W., Kooi, E.-J., Geurts, J. J. G., et al. (2017). Complement C3 on microglial clusters in multiple sclerosis occur in chronic but not acute disease: implication for disease pathogenesis. Glia 65, 264–277. doi: 10.1002/glia.23090
Morganti-Kossmann, M. C., Hans, V. H., Lenzlinger, P. M., Dubs, R., Ludwig, E., Trentz, O., et al. (1999). TGF-beta is elevated in the CSF of patients with severe traumatic brain injuries and parallels blood-brain barrier function. J. Neurotrauma 16, 617–628. doi: 10.1089/neu.1999.16.617
Nag, S., Venugopalan, R., and Stewart, D. J. (2007). Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta Neuropathol. 114, 459–469. doi: 10.1007/s00401-007-0274-x
Osthoff, M., Walder, B., Delhumeau, C., Trendelenburg, M., and Turck, N. (2017). Association of lectin pathway protein levels and genetic variants early after injury with outcomes after severe traumatic brain injury: a prospective cohort study. J. Neurotrauma 34, 2560–2566. doi: 10.1089/neu.2016.4941
Ostrow, L. W., Langan, T. J., and Sachs, F. (2000). Stretch-induced endothelin-1 production by astrocytes. J. Cardiovasc. Pharmacol. 36, S274–277. doi: 10.1097/00005344-200036001-00081
Panesar, M., Papillon, J., McTavish, A. J., and Cybulsky, A. V. (1997). Activation of phospholipase A2 by complement C5b-9 in glomerular epithelial cells. J. Immunol. 159, 3584–3594.
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Petraglia, A. L., Dashnaw, M. L., Turner, R. C., and Bailes, J. E. (2014). Models of mild traumatic brain injury: translation of physiological and anatomic injury. Neurosurgery 75 (Suppl. 4), S34–S49. doi: 10.1227/NEU.0000000000000472
Petrov, T., and Rafols, J. A. (2001). Acute alterations of endothelin-1 and iNOS expression and control of the brain microcirculation after head trauma. Neurol. Res. 23, 139–143. doi: 10.1179/016164101101198479
Plesnila, N. (2016). The immune system in traumatic brain injury. Curr. Opin. Pharmacol. 26, 110–117. doi: 10.1016/j.coph.2015.10.008
Plesnila, N., Friedrich, D., Eriskat, J., Baethmann, A., and Stoffel, M. (2003). Relative cerebral blood flow during the secondary expansion of a cortical lesion in rats. Neurosci. Lett. 345, 85–88. doi: 10.1016/S0304-3940(03)00396-3
Pop, V., and Badaut, J. (2011). A neurovascular perspective for long-term changes after brain trauma. Transl Stroke Res. 2, 533–545. doi: 10.1007/s12975-011-0126-9
Pop, V., Sorensen, D. W., Kamper, J. E., Ajao, D. O., Murphy, M. P., Head, E., et al. (2013). Early brain injury alters the blood-brain barrier phenotype in parallel with β-amyloid and cognitive changes in adulthood. J. Cereb. Blood Flow Metab. 33, 205–214. doi: 10.1038/jcbfm.2012.154
Povlishock, J. T., Becker, D. P., Sullivan, H. G., and Miller, J. D. (1978). Vascular permeability alterations to horseradish peroxidase in experimental brain injury. Brain Res. 153, 223–239. doi: 10.1016/0006-8993(78)90404-3
Prins, M. L., and Hovda, D. A. (2003). Developing experimental models to address traumatic brain injury in children. J. Neurotrauma 20, 123–137. doi: 10.1089/08977150360547053
Rafols, J. A., Kreipke, C. W., and Petrov, T. (2007). Alterations in cerebral cortex microvessels and the microcirculation in a rat model of traumatic brain injury: a correlative EM and laser Doppler flowmetry study. Neurol. Res. 29, 339–347. doi: 10.1179/016164107X204648
Raz, L., Knoefel, J., and Bhaskar, K. (2016). The neuropathology and cerebrovascular mechanisms of dementia. J. Cereb. Blood Flow Metab. 36, 172–186. doi: 10.1038/jcbfm.2015.164
Rich, M. C., Keene, C. N., Neher, M. D., Johnson, K., Yu, Z.-X., Ganivet, A., et al. (2016). Site-targeted complement inhibition by a complement receptor 2-conjugated inhibitor (mTT30) ameliorates post-injury neuropathology in mouse brains. Neurosci. Lett. 617, 188–194. doi: 10.1016/j.neulet.2016.02.025
Rodriguez-Grande, B., Obenaus, A., Ichkova, A., Aussudre, J., Bessy, T., Barse, E., et al. (2018). Gliovascular changes precede white matter damage and long-term disorders in juvenile mild closed head injury. Glia 66, 1663–1677. doi: 10.1002/glia.23336
Ruseva, M. M., Ramaglia, V., Morgan, B. P., and Harris, C. L. (2015). An anticomplement agent that homes to the damaged brain and promotes recovery after traumatic brain injury in mice. Proc. Natl. Acad. Sci. U.S.A. 112, 14319–14324. doi: 10.1073/pnas.1513698112
Rzigalinski, B. A., Weber, J. T., Willoughby, K. A., and Ellis, E. F. (1998). Intracellular free calcium dynamics in stretch-injured astrocytes. J. Neurochem. 70, 2377–2385. doi: 10.1046/j.1471-4159.1998.70062377.x
Schmidt, O. I., Heyde, C. E., Ertel, W., and Stahel, P. F. (2005). Closed head injury–an inflammatory disease? Brain Res. Brain Res. Rev. 48, 388–399. doi: 10.1016/j.brainresrev.2004.12.028
Sekar, A., Bialas, A. R., de Rivera, H., Davis, A., Hammond, T. R., Kamitaki, N., et al. (2016). Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183. doi: 10.1038/nature16549
Sewell, D. L., Nacewicz, B., Liu, F., Macvilay, S., Erdei, A., Lambris, J. D., et al. (2004). Complement C3 and C5 play critical roles in traumatic brain cryoinjury: blocking effects on neutrophil extravasation by C5a receptor antagonist. J. Neuroimmunol. 155, 55–63. doi: 10.1016/j.jneuroim.2004.06.003
Shen, W., Li, S., Chung, S. H., Zhu, L., Stayt, J., Su, T., et al. (2011). Tyrosine phosphorylation of VE-cadherin and claudin-5 is associated with TGF-β1-induced permeability of centrally derived vascular endothelium. Eur. J. Cell Biol. 90, 323–332. doi: 10.1016/j.ejcb.2010.10.013
Shetty, A. K., Mishra, V., Kodali, M., and Hattiangady, B. (2014). Blood brain barrier dysfunction and delayed neurological deficits in mild traumatic brain injury induced by blast shock waves. Front. Cell Neurosci. 8:232. doi: 10.3389/fncel.2014.00404
Silverberg, G. D., Messier, A. A., Miller, M. C., Machan, J. T., Majmudar, S. S., Stopa, E. G., et al. (2010). Amyloid efflux transporter expression at the blood-brain barrier declines in normal aging. J. Neuropathol. Exp. Neurol. 69, 1034–1043. doi: 10.1097/NEN.0b013e3181f46e25
Smith, D. H., Johnson, V. E., and Stewart, W. (2013). Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat. Rev. Neurol. 9, 211–221. doi: 10.1038/nrneurol.2013.29
Sofroniew, M. V. (2009). Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647. doi: 10.1016/j.tins.2009.08.002
Sofroniew, M. V. (2014). Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist 20, 160–172. doi: 10.1177/1073858413504466
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. doi: 10.1007/s00401-009-0619-8
Stahel, P. F., and Barnum, S. R. (2006). The role of the complement system in CNS inflammatory diseases. Expert Rev. Clin. Immunol. 2, 445–456. doi: 10.1586/1744666X.2.3.445
Stahel, P. F., Flierl, M. A., Morgan, B. P., Persigehl, I., Stoll, C., Conrad, C., et al. (2009). Absence of the complement regulatory molecule CD59a leads to exacerbated neuropathology after traumatic brain injury in mice. J. Neuroinflammation 6:2. doi: 10.1186/1742-2094-6-2
Stahel, P. F., Morganti-Kossmann, M. C., and Kossmann, T. (1998). The role of the complement system in traumatic brain injury. Brain Res. Brain Res. Rev. 27, 243–256. doi: 10.1016/S0165-0173(98)00015-0
Stahel, P. F., Morganti-Kossmann, M. C., Perez, D., Redaelli, C., Gloor, B., Trentz, O., et al. (2001). Intrathecal levels of complement-derived soluble membrane attack complex (sC5b-9) correlate with blood-brain barrier dysfunction in patients with traumatic brain injury. J. Neurotrauma 18, 773–781. doi: 10.1089/089771501316919139
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi: 10.1016/j.cell.2007.10.036
Storch, M. K., Piddlesden, S., Haltia, M., Iivanainen, M., Morgan, P., and Lassmann, H. (1998). Multiple sclerosis: in situ evidence for antibody- and complement-mediated demyelination. Ann. Neurol. 43, 465–471. doi: 10.1002/ana.410430409
Sun, H., Liang, R., Yang, B., Zhou, Y., Liu, M., Fang, F., et al. (2016). Aquaporin-4 mediates communication between astrocyte and microglia: implications of neuroinflammation in experimental Parkinson's disease. Neuroscience 317, 65–75. doi: 10.1016/j.neuroscience.2016.01.003
Tatomir, A., Talpos-Caia, A., Anselmo, F., Kruszewski, A. M., Boodhoo, D., Rus, V., et al. (2017). The complement system as a biomarker of disease activity and response to treatment in multiple sclerosis. Immunol. Res. 65, 1103–1109. doi: 10.1007/s12026-017-8961-8
Thelin, E. P., Just, D., Frostell, A., Häggmark-Månberg, A., Risling, M., Svensson, M., et al. (2018). Protein profiling in serum after traumatic brain injury in rats reveals potential injury markers. Behav. Brain Res. 340, 71–80. doi: 10.1016/j.bbr.2016.08.058
Unterberg, A. W., Stover, J., Kress, B., and Kiening, K. L. (2004). Edema and brain trauma. Neuroscience 129, 1021–1029. doi: 10.1016/j.neuroscience.2004.06.046
Vasek, M. J., Garber, C., Dorsey, D., Durrant, D. M., Bollman, B., Soung, A., et al. (2016). A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534, 538–543. doi: 10.1038/nature18283
Villapol, S., Balarezo, M. G., Affram, K., Saavedra, J. M., and Symes, A. J. (2015). Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain 138, 3299–3315. doi: 10.1093/brain/awv172
Villapol, S., Byrnes, K. R., and Symes, A. J. (2014). Temporal dynamics of cerebral blood flow, cortical damage, apoptosis, astrocyte-vasculature interaction and astrogliosis in the pericontusional region after traumatic brain injury. Front. Neurol. 5:82. doi: 10.3389/fneur.2014.00082
Walport, M. J. (2001). Complement. First of two parts. N. Engl. J. Med. 344, 1058–1066. doi: 10.1056/NEJM200104053441406
Wang, X., Jung, J., Asahi, M., Chwang, W., Russo, L., Moskowitz, M. A., et al. (2000). Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J. Neurosci. 20, 7037–7042. doi: 10.1523/JNEUROSCI.20-18-07037.2000
Watkins, L. M., Neal, J. W., Loveless, S., Michailidou, I., Ramaglia, V., Rees, M. I., et al. (2016). Complement is activated in progressive multiple sclerosis cortical grey matter lesions. J. Neuroinflammation 13:161. doi: 10.1186/s12974-016-0611-x
Wei, E. P., Dietrich, W. D., Povlishock, J. T., Navari, R. M., and Kontos, H. A. (1980). Functional, morphological, and metabolic abnormalities of the cerebral microcirculation after concussive brain injury in cats. Circ. Res. 46, 37–47. doi: 10.1161/01.RES.46.1.37
Yang, C., Huang, X., Huang, X., Mai, H., Li, J., Jiang, T., et al. (2016). Aquaporin-4 and Alzheimer's Disease. J. Alzheimers Dis. 52, 391–402. doi: 10.3233/JAD-150949
Yang, S., Nakamura, T., Hua, Y., Keep, R. F., Younger, J. G., He, Y., et al. (2006). The role of complement C3 in intracerebral hemorrhage-induced brain injury. J. Cereb. Blood Flow Metab. 26, 1490–1495. doi: 10.1038/sj.jcbfm.9600305
Yeoh, S., Bell, E. D., and Monson, K. L. (2013). Distribution of blood-brain barrier disruption in primary blast injury. Ann. Biomed. Eng. 41, 2206–2214. doi: 10.1007/s10439-013-0805-7
Yoshino, A., Hovda, D. A., Kawamata, T., Katayama, Y., and Becker, D. P. (1991). Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: evidence of a hyper- and subsequent hypometabolic state. Brain Res. 561, 106–119. doi: 10.1016/0006-8993(91)90755-K
Zhang, C., Chen, J., and Lu, H. (2015). Expression of aquaporin-4 and pathological characteristics of brain injury in a rat model of traumatic brain injury. Mol. Med. Rep. 12, 7351–7357. doi: 10.3892/mmr.2015.4372
Zhang, H., Adwanikar, H., Werb, Z., and Noble-Haeusslein, L. J. (2010). Matrix metalloproteinases and neurotrauma: evolving roles in injury and reparative processes. Neuroscientist 16, 156–170. doi: 10.1177/1073858409355830
Keywords: traumatic brain injury, neuroinflammation, complement, blood-brain barrier, astrocyte
Citation: Dinet V, Petry KG and Badaut J (2019) Brain–Immune Interactions and Neuroinflammation After Traumatic Brain Injury. Front. Neurosci. 13:1178. doi: 10.3389/fnins.2019.01178
Received: 27 July 2019; Accepted: 18 October 2019;
Published: 12 November 2019.
Edited by:
Emmanuel Pinteaux, University of Manchester, United KingdomReviewed by:
Rodolfo Gabriel Gatto, University of Illinois at Chicago, United StatesCopyright © 2019 Dinet, Petry and Badaut. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Virginie Dinet, dmlyZ2luaWUuZGluZXRAaW5zZXJtLmZy; Jerome Badaut, amVyb21lLmJhZGF1dEB1LWJvcmRlYXV4LmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.