 Sonja Prpar Mihevc
Sonja Prpar Mihevc Gregor Majdič
Gregor Majdič- 1Veterinary Faculty, Institute for Preclinical Sciences, University of Ljubljana, Ljubljana, Slovenia
- 2Medical Faculty, Institute for Physiology, University of Maribor, Maribor, Slovenia
Neurodegenerative diseases present a major and increasing burden in the societies worldwide. With aging populations, the prevalence of neurodegenerative diseases is increasing, yet there are no effective cures and very few treatment options are available. Alzheimer’s disease is one of the most prevalent neurodegenerative conditions and although the pathology is well studied, the pathogenesis of this debilitating illness is still poorly understood. This is, among other reasons, also due to the lack of good animal models as laboratory rodents do not develop spontaneous neurodegenerative diseases and human Alzheimer’s disease is only partially mimicked by transgenic rodent models. On the other hand, older dogs commonly develop canine cognitive dysfunction, a disease that is similar to Alzheimer’s disease in many aspects. Dogs show cognitive deficits that could be paralleled to human symptoms such as disorientation, memory loss, changes in behavior, and in their brains, beta amyloid plaques are commonly detected both in extracellular space as senile plaques and around the blood vessels. Dogs could be therefore potentially a very good model for studying pathological process and novel treatment options for Alzheimer’s disease. In the present article, we will review the current knowledge about the pathogenesis of canine cognitive dysfunction, its similarities and dissimilarities with Alzheimer’s disease, and developments of novel treatments for these two diseases with a focus on canine cognitive dysfunction.
Introduction
Neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease, Huntington’s disease, frontotemporal dementia, amyotrophic lateral sclerosis, and others are a major growing public health problem associated with aging, as aging is the greatest risk factor for neurodegeneration. The global number of people living with dementia more than doubled from 1990 to 2016 (GBD 2016 Dementia Collaborators, 2019). These diseases now affect nearly 50 million individuals, and the incidence is projected to triple by 2050. AD and other dementias are associated with alterations in cell type-specific function, including gliosis, neuronal dysfunction and cell death. The pathognomic cause is the condensation of certain proteins into insoluble aggregates and this aggregates damage vulnerable neurons and glial cells. These inclusions of misfolded proteins build up in the brain during normal aging and during the progression of adult-onset dementias.
Neurodegenerative diseases do not occur spontaneously in laboratory mice and rats, but do occur in several other mammalian species. Studies of neurodegenerative diseases in animals have shown strong similarities between cognitive dysfunction in dogs and human AD and between AD in humans and most other primates (Cummings et al., 1993; Cummings and Cotman, 1995; Dodart et al., 2002; Chambers et al., 2011; Braidy et al., 2015; Schütt et al., 2016). Canine cognitive dysfunction (CCD), also known as canine cognitive dysfunction syndrome (CDS) or canine dementia affects up to 60% of older dogs, mostly dogs older than 11 years (Fast et al., 2013b). The prevalence of CCD does not differ between breeds (Salvin et al., 2010) and there are no breed specific differences in clinical signs or pathology of the disease. However, as larger dog breeds have shorter life span than the smaller ones (Greer et al., 2007), clinical signs of CCD are more often observed and reported in smaller dogs (Vite and Head, 2014; Schmidt et al., 2015). Due to its similarity with AD, CCD is extremely interesting as a model for human disease. CCD is also a major problem for health of older dogs, and is thus also interesting from the point of view of the development of new veterinary diagnostic procedures/markers and medicines for treating this disease in dogs.
In dogs and humans, dementia often affects cerebral gyri (cerebral atrophy) and shows as widening of sulci together with ventricular enlargement (Borràs et al., 1999; Toepper, 2017). Neuronal loss and cortical atrophy has been described in several brain regions including cortex, hippocampus and parts of the limbic system in cognitively impaired dogs (Tapp et al., 2004; Siwak-Tapp et al., 2008), similarly to human brain affected by AD (West et al., 2000). In human patients, AD has a preclinical stage with the clinical symptoms not yet evident but with pathological changes in the brain already present, which is followed by mild cognitive impairment stage which gradually culminates in dementia due to AD, with severe cognitive and functional decline (Bature et al., 2017). The onset of AD, detected clinically at the mild cognitive impairment stage, is characterized by a subtle decline in memory functions, followed by changes in personality, deterioration of language functions and eventually motor dysfunction (Braak and Braak, 1997). These deficits in cognition and behavior are mirrored in the AD-related regional brain destruction. Similarly to these signs in humans, the most obvious signs of CCD, deduced by standard cognition tests, are loss of memory, poor or completely lost sense of orientation, changes in behavior and confusion (Bain et al., 2001). Most common symptoms include disorientation, anxiousness, dogs get easily scared, do not recognize their owner, become aggressive or apathetic, have difficulty controlling the secretion of body fluids, change circadian rhythms and others.
Neuropathological Features of Neurodegenerative Diseases
Role of Protein Aggregates in Neurodegenerative Diseases
Protein aggregation is an established pathogenic mechanism in AD, although little is known about the initiation of this process in vivo. As human brain research largely depends on the results of postmortem studies, an insight into the early stages of the disease, when protein aggregates are most likely to occur, is difficult. Novel and better animal models would therefore be very helpful to study the progress of AD in humans. The clinical course of the disease in humans could be monitored by brain imaging (CT, MRI). The exact connection between protein aggregates, such as extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles (NFTs), the latter composed of hyperphosphorylated microtubule associated protein (TAU), in the brains of patients with AD, and neurodegeneration is therefore still unknown. It does seem that the AD is caused by the accumulation of amyloid plaques and neurofibrillary fibers, although this is not absolutely confirmed and generally accepted (Medeiros et al., 2011). Stronger evidence that aggregates do trigger neurodegeneration (the so-called amyloid hypothesis) and are not just a consequence of the neurodegeneration, is provided by mutations in the gene for the amyloid beta precursor protein (APP) present in some human patients with AD (Selkoe, 1991; Hardy and Higgins, 1992; Hardy and Selkoe, 2002; Lansbury and Lashuel, 2006). Similarly, amyloid plaques are present in different brain regions in dogs with CCD clinical symptoms, and they can be present even before clinical signs of this disease become obvious.
In general, age-related neurodegenerative disorders are complex and multifaceted pathologies, wherein the formation of large aggregates and/or high concentrations of toxic proteins prevent the proper function of neuronal cells, leading to ischemia and eventually tissue removal. The spread of AD pathology follows a characteristic topographic pattern, different for the two proteins involved in the pathology, Aβ and TAU (Brettschneider et al., 2015). TAU aggregates first develop in the locus coeruleus, then in entorhinal cortex followed by hippocampus and neocortex. Aβ plaques firstly appear in the neocortex, and later in allocortical, diencephalic and basal ganglia structures and in the brainstem (Brettschneider et al., 2015; Jucker and Walker, 2018). These aggregates spread in a prion-like manner, forming intracellular and extracellular deposits. TAU, Aβ, and also Parkinson disease associated α-synuclein, exist in different conformational variants (‘strains’) that show seeding properties and exert different levels of neurotoxicity, which could be the source of heterogeneity of neurodegenerative diseases (Aguzzi, 2014). The seeding potential of these aggregates was recently substantiated by demonstrating propagation of structural variants of Aβ in their distinct conformations through template-directed folding of naïve Aβ peptides (Condello and Stöehr, 2018). Prion-like transmission and seeding has been also observed for TAU (DeVos et al., 2018). Similar mechanism may be involved in young, pre-depositing APP transgenic mice which developed cerebral β-amyloidosis and associated pathology after being intracerebrally injected with Aβ-containing brain extracts from human patients with AD (Meyer-Luehmann et al., 2006; Langer et al., 2011). Interestingly, even intraperitoneal inoculation with Aβ extracts induced β-amyloidosis in the brains of APP transgenic mice after prolonged incubation times (Eisele et al., 2010). Likewise, soluble oligomers from blood and cerebrospinal fluid (CSF) of an aged dog with CCD were neurotoxic to human neuroblastoma cell line, and canine CSF derived Aβ induced the in vitro aggregation of synthetic human Aβ peptides (Rusbridge et al., 2018). Spreading of misfolded Aβ oligomers in a prion-like mechanism might also exploit exosomes, which can seed Aβ not only as vehicles for the neuron-to-neuron transfer (Zheng et al., 2017; Sardar Sinha et al., 2018) but also between distant brain regions (i.e., from the dentate gyrus to other regions of hippocampus and to the cortex) (Zheng et al., 2017).
The Neuropathology in the Brain Parenchyma
The most prominent neuropathological signs of AD are accumulation of Aβ in a form of extracellular plaques in the brain parenchyma and also in the walls of blood vessels (cerebral amyloid angiopathy, CAA), and abnormally phosphorylated protein TAU that accumulates in NFTs (Braak and Braak, 1991; Nelson et al., 2012). These pathological features are believed to cause cognitive and behavioral changes. The amino acid sequence of APP and of the enzymes involved in the processing of Aβ peptides from APP, are highly homologous between humans and dogs (Johnstone et al., 1991; Sarasa and Pesini, 2009; Sarasa et al., 2010). Major canine APP isoforms are APP-770, APP-751, APP-714, and APP-695 (Sarasa et al., 2010). The alignment of the longest canine amyloid-beta precursor protein isoform (APP-770) sequence with protein sequence of human amyloid-beta precursor protein show 96.9% amino acid identity and 98.3% similarity (Figure 1A), making them almost identical. The expression patterns of canine APP isoforms are almost similar to the patterns previously detected for human APP isoforms (Yasojima et al., 2001; Sarasa et al., 2010). APP is a single-pass transmembrane protein with a large extracellular domain and a short cytoplasmic tail. The domain structure of APP-770 is shown in Figure 1B, the neuronal isoform APP-695 lacks the KPI domain (Müller et al., 2017). In contrary to APP, there is a difference in the TAU protein sequence between dogs and humans. Figure 1C shows the longest TAU isoform alignment between dog and human protein sequences with 84% similarity. Interestingly, the four microtubule-binding regions (4R) and the C-terminal regions (Figures 1C,D) are identical.

Figure 1. Protein sequences alignments between dog and human amyloid-beta precursor protein (APP) and TAU. (A) The alignment of the longest canine APP isoform (APP-770) sequence with protein sequence of human APP. Aligned sequences share 96.9% amino acid identity and 98.3% similarity. Of note, the sequence identity of canine APP-695 isoform, the predominant APP isoform in the canine brain, and human APP is 87.5%. Highlighted is Aβ domain and underlined the membrane bound part of this domain. (B) Domain structure of APP. Isoform APP-770 is depicted. HBD1/GFLD, heparin binding domain 1/growth factor like domain; CuBD, copper binding domain; ZnBD, zinc binding domain; KPI, Kunitz-type protease inhibitor domain (not present in APP-695); Aβ, amyloid beta domain, the latter anchors APP in the cell membrane. The α-, β-, and γ-secretase cleavage sites are directly adjacent or inside the Aβ domain region, which is also most mutation prone, thus enabling alternative cleavage of APP and its divergent aggregation propensities. (C) Alignment of the longest TAU protein isoform, out of six existing, of dog and human TAU. Aligned TAU sequences share 81.6% amino acid identity and 84.4% similarity. Highlighted are four microtubule-binding regions (4R). (D) Domain structure of TAU. The basic organization is shown. NTR, N-terminal region; PRR, proline-rich region; 4R, four microtubule-binding regions (some isoforms have three); CTR, C-terminal region. The PRR and 4R domains are subject of most posttranslational modifications, while the CTR enables interactions with microtubules and plasma membrane. EMBOSS Needle pairwise sequence alignment (Madeira et al., 2019) was performed. A line (|) indicates positions which have a single, fully conserved residue. A colon (:) indicates conservation between groups of strongly similar properties and a dot (.) indicates conservation between groups of weakly similar properties. The domain organization was depicted by software illustrator of biological sequences (IBS) (Liu et al., 2015). Figure made by the authors.
Amyloid-β was found to be present in the form of insoluble plaques in the area of the cerebral cortex in humans and dogs, and cognitive impairment in elderly dogs was in some studies strongly associated with the accumulation of Aβ in the brain (Cummings et al., 1993; Cummings and Cotman, 1995; Pugliese et al., 2006; Rofina et al., 2006; Barnes et al., 2016). However, in two other studies there was no significant correlation between the amount of Aβ brain load and CCD symptoms (Chambers et al., 2011; Ozawa et al., 2016) so the clear connection between Aβ load in the brain and clinical signs of CCD has not been firmly established yet. Although Aβ plaques are generally detected as extracellular deposits of diffuse and neuritic plaques in humans and dogs, some studies also reported the presence of Aβ deposits inside individual neurons in the canine brain (Cummings et al., 1993; Yu et al., 2011). Similarly, intraneuronal Aβ accumulation in human AD brains has been reported by many groups (reviewed in detail by Gouras et al., 2010).
In dogs, formation and maturation of Aβ deposits was observed by immunostaining throughout the canine cortical gray matter layers in a four-stage distribution, which is also characteristic for human AD, and this, according to some studies, correlates with the severity of cognitive deficit in the dog (Bosch et al., 2012) and varies as a function of age and size (weight) in companion dogs (Rofina et al., 2006; Schmidt et al., 2015). The Aβ load was higher in small and medium size dogs, which can be explained by the longer life span of smaller dogs and thus longer time necessary to accumulate these deposits (Schmidt et al., 2015). Most of the studies, on CCD brains, were made on geriatric dogs of different breeds and sizes, hence in this review the pathology is mainly described without further specifications of individual dogs characteristics. The canine prefrontal cortex is normally the site of disease onset (Figure 2), which spreads progressively to the parietal, entorhinal and occipital cortices, and lesions in certain brain regions correlate with certain behavioral deficits, as shown in Table 1. In separate studies amyloid plaques of mainly diffuse type were detected in several regions of canine brain, in the frontal and temporal cortex (Ishihara et al., 1991; Colle et al., 2000; Pugliese et al., 2006; Schmidt et al., 2015; Ozawa et al., 2016; Schütt et al., 2016; Smolek et al., 2016), in entorhinal cortex and hippocampus (Cummings et al., 1993; Colle et al., 2000; Yu et al., 2011; Schmidt et al., 2015; Borghys et al., 2017) and parietal cortex (Yu et al., 2011). Hippocampal deposits of a subspecies of pyroglutamyl Aβ, the highly neurotoxic pE3AA, were also detected in demented dogs (Schmidt et al., 2015). These plaques were more abundant in small and medium dogs (Schmidt et al., 2015). Studies further suggest that earlier assembly states of Aβ, such as oligomers and protofibrils, may be neurotoxic in the dog’s brain (Head et al., 2010). The levels of Aβ soluble oligomers in the CSF correlated inversely with Aβ load in the CCD affected beagle brains (Head et al., 2010). Precipitates from the CSF of demented Samoyed dog with CCD acted highly neurotoxic in in vitro tests and were more neurotoxic than the synthetic oligomeric and fibrillary forms of Aβ (Rusbridge et al., 2018).
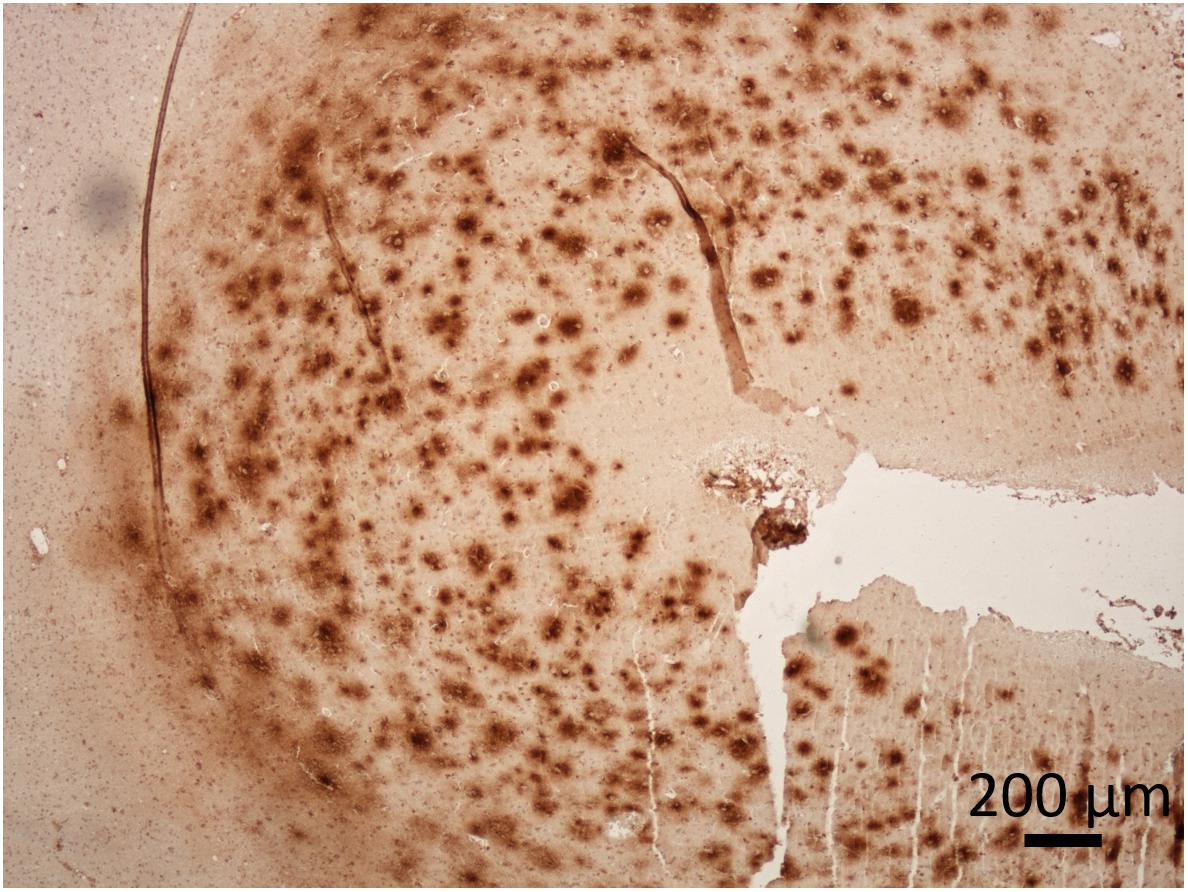
Figure 2. Presence of Aβ in the cerebral cortex of a dog with CCD. The dense plaques detected in superficial cortical layers and diffuse plaques in deeper layers of prefrontal cortex. The dog was 17-years-old of a mixed breed. Immunoperoxidase staining with antibodies against Aβ (purified anti-β-Amyloid, 17-24 Antibody, BioLegend, #800701) with diaminobenzidine (DAB) as chromogen (brown), counter stained with hematoxylin. Original microphotograph made by the authors.

Table 1. Affected brain regions and underlying cognitive deficits in dogs.
Besides Aβ plaques, recruitment and activation of astrocytes and microglial cells has been noticed in dogs with CCD (astrocytosis, microgliosis) (Smolek et al., 2016; Rusbridge et al., 2018) along with astrocyte hypertrophy (Borràs et al., 1999). Furthermore, the levels of pathological Aβ in the canine prefrontal cortex were positively correlated with age but neither with the severity of cognitive deficits (Gómez-Pinedo et al., 2016) nor the neuronal cell loss (Cummings et al., 1993). Neprilysin (NEP) mRNA, coding for a pivotal Aβ-degrading protein, was poorly expressed in the prefrontal cortex of aged dogs with CCD (Canudas et al., 2014), similar to human AD brain, where areas with higher Aβ aggregation express lower levels of NEP (Reilly, 2001).
Aged dogs with cognitive impairment exhibit degeneration of noradrenergic neurons, which correlates with higher levels of Aβ deposits in the prefrontal cortex (Insua et al., 2010). Likewise, in human AD, locus coeruleus degeneration and loss of cortical noradrenergic neurons occurs already at early stage (Kelly et al., 2017). The activity of the canine cholinergic system also declines with age (Araujo et al., 2005b). Number of basal forebrain cholinergic neurons was significantly reduced in aged cognitively impaired dogs in comparison to aged cognitive unimpaired and young dogs, but unlike for noradrenergic neurons, this reduction of cholinergic neurons did not correlate with the extent of Aβ cortical load (Insua et al., 2012). As in CCD, cholinergic neurons located in the basal forebrain, including the neurons that form the nucleus basalis of Meynert, are usually lost in AD (Whitehouse et al., 1981). In human patients, the cholinergic neurons appear most vulnerable to Aβ pathology, followed by glutamatergic and GABAergic neurons (Bell et al., 2006). The synaptic loss correlates with the disease progression and destruction of cholinergic neurons contributes to memory and attention deficits in AD (Terry et al., 1991). These alterations in neurotransmitter systems, with reduced neuronal and synaptic function have also been observed in dogs (Landsberg et al., 2012) and result in clinical symptoms of CCD. The employment of the canine model to examine the effect of the cholinesterase inhibitors in treatment of CCD is described in details under “Treatment and drug development”.
TAU protein, another important factor in neurodegenerative diseases, is encoded by MAPT (microtubule associated protein tau) gene. Mutations in this gene have not been linked to AD, but cause a familial form of frontotemporal dementia (Olszewska et al., 2016). However, in human AD, intraneuronal NFTs composed of hyperphosphorylated TAU and misfolded insoluble TAU protein aggregates and extracellular Aβ inclusions are both present and necessary for the diagnosis of the disease. In AD NFTs form initially in the locus coeruleus, then entorhinal cortex and further progress to the hippocampus, anterior cingulate cortex, visual association area and finally to the primary visual cortex in the occipital lobe (Ossenkoppele et al., 2016; Hoenig et al., 2018). Interestingly, TAU neurofibrillary inclusions were only rarely identified in canine brain, for instance only in one dog in one study (Smolek et al., 2016) and in three dogs in another study (Schmidt et al., 2015). Increased phosphorylation of TAU was observed at some amino acid sites in canine brain, although no study so far confirmed the presence of vast mature NFTs deposits as are typically observed in human AD. In one study, cytoplasmic deposits of phosphorylated TAU (pTAUThr181) were detected in the prefrontal cortex, but no NFTs were observed (Pugliese et al., 2006). Smolek et al. (2016) detected increased presence of phosphorylated TAU protein (pTAU) in synaptosomes of demented dogs. This could suggest that dementia in dogs might be partially caused by the weakening of the synaptic function, caused by pTAU, and not by the toxic effects of NFTs. This is further supported by the increase in TAU hyperphosphorylation in individual cortical neurons and by pTAU subcellular distribution shift from perinuclear to granular cytoplasmic and nuclear, which correlates with dog’s age (Pugliese et al., 2006). Expression of pTAUSer396 and accumulation of ubiquitin were also significantly increased in the parietal cortex and dorsal part of the hippocampus in old dogs when compared to expression in humans (Pugliese et al., 2006; Yu et al., 2011). Specifically, pTAUSer396 expressing astrocytes and neurons with co-localization of pTAUSer396 and ubiquitin were also observed in the parietal cortices and hippocampi of dogs with CCD (Yu et al., 2011). However, cytoplasmic aggregates of normally predominately nuclear proteins, TAR DNA-binding protein (TDP-43) and fused in sarcoma (FUS), which are strongly associated with frontotemporal dementia and amyotrophic lateral sclerosis in human patients, were not detected in the canine brain with CCD (Smolek et al., 2016).
Neurodegenerative diseases occur spontaneously in other domestic animals, especially in cats (Chambers et al., 2015), although brain pathology in these other species is much less studied. In the hippocampi of naturally aged domestic cats, Aβ accumulation, NFTs formation and neuronal loss were observed (Chambers et al., 2015) and this might contribute to cognitive decline in this species, although this has not yet been firmly established. Cognitive deficit may occur in the horse, as clinical signs similar to cognitive impairment have been observed, but no detailed studies focusing on age-related neurologic aberrances have been conducted so far. In one study, TAU protein was shown to be present in equine hippocampal neurons, but no NFTs were detected (Capucchio et al., 2010). Some horses develop equine motor neuron disease, but none have been associated with pathological accumulation of dementia related proteins (El-Assaad et al., 2012; Youssef et al., 2016). Interestingly, a recent study negated the belief that abnormally behaving stereotypic horses (Equus caballus) are cognitively impaired (Briefer Freymond et al., 2018).
Neurodegenerative diseases most likely occur in many other mammalian species, but there are very limited reports about these. The reports are focusing on the presence of Aβ deposits and phosphorylated TAU and/or NFTs, which are detected post mortem, not describing the cognitive deficit, this is of course more difficult to observe in wild life animals or in this aspect less characterized species. Diffuse deposits of Aβ were observed in the parietal cortex of dolphins and more compact senile plaques in their cerebellum (Di Guardo, 2018). Sheep and goats (Braak et al., 1994; Nelson et al., 1994; Nelson and Saper, 1995), cheetah (Serizawa et al., 2012), bison (Härtig et al., 2000, 2001), bears (Cork et al., 1988) and most species of nonhuman primates (Schultz et al., 2000a, b; Rosen et al., 2008; Oikawa et al., 2010; Perez et al., 2013, 2016; Rodriguez-Callejas et al., 2016; Reid et al., 2017) also develop amyloid plaques and/or harbor phosphorylated TAU laden neurons, but most of them do not have NFTs present in their central nervous systems (as reviewed in Youssef et al., 2016). However, AT8-immunostained pTAUSer202 and pTAUThr205 were found in the brains of brown lemur, rhesus monkey, baboons, rabbit, guanaco, and bison (Härtig et al., 2000). Furthermore, NFTs were revealed by Gallyas staining in aged bison (Härtig et al., 2001).
There have been speculations on the reasons for the lack of NFTs in the canine brain and in the brains of other animals (Kuroki et al., 1997; Youssef et al., 2016). First possibility is the dissimilarity between canine TAU protein sequence in comparison to human’s. Secondly, the lifespan of dogs might be too short to develop NFTs, as Aβ deposition precedes NTFs formation. Thirdly, although the amyloid protein sequence is highly conserved between species, its N-terminal part is not, which might influence TAU phosphorylation and its subsequent aggregation into NFTs. Therefore, it seems that only humans develop the full blown AD pathology with amyloid plaques, NFTs and dementia. There have been few reports of possible AD in aged great apes (Rosen et al., 2008; Perez et al., 2013, 2016), although also in these the TAU pathology was very limited. Several reasons for this exceptional vulnerability of humans to develop AD have been proposed (Arendt et al., 2017; Walker and Jucker, 2017). Predisposition to develop AD might arise as early as during embryonic development over the period of neurogenesis, this was inferred from the modular deposition of AD related cerebral deposits which mirrors the formation and migration of neurons and on a larger scale development of brain regions (Arendt et al., 2017). In humans, the relative brain size is much larger than in any other animal and this expansion is due to longer duration of brain development as well as higher number of neuronal progenitor cell division cycles, the latter giving rise to genomic aberrations, which in turn can be the origin of AD (Arendt et al., 2017). Furthermore, the neurons display genomic mosaicism, which was detected in neurons of approximately 10% of normally aged people. These neurons were shown to preferentially undergo apoptosis in AD (Arendt et al., 2010). The extent of neuronal genomic mosaicism is brain region specific and correlates with differential vulnerability to neurofibrillary pathology (Arendt et al., 2015). Moreover, the pathological TAU conversion might commence as early as in children (Braak and Del Tredici, 2012), although it has not been firmly established if these processes are transient in nature. The canine model would therefore serve as an accessible and suitable model to further delineate why, except for humans, are other species less susceptible to develop tauopathies.
The Neuropathology of Cerebral Blood Vessels
In patients with AD, accumulation of Aβ is often observed in the walls of blood vessels in the brain. This is called CAA and is caused by pathological deposits of Aβ and other proteins in the cerebral arterioles and capillaries of the leptomeninges and cortex. It is considered as an early and integral part of AD pathogenesis and the prevalence of CAA in AD is over 70% (Attems, 2005). CAA is a risk factor for hemorrhagic stroke and contributes to AD dementia. Atherosclerosis may also cause cerebrovascular dysfunction that may lead to cognitive decline as well as stroke (Shabir et al., 2018). The accumulation of amyloid in the walls of blood vessels is common in elderly people, patients with CAA and in almost all patients with AD (Kumar-Singh, 2008). The most CAA affected brain regions are the leptomeninges and frontal and temporal cortices (Borràs et al., 1999) with Aβ deposits in arteries, arterioles, capillaries, venules, and veins. The CAA disease progresses from mild, to moderate to severe stage, this is from almost intact blood vessels with minor Aβ deposits to extensive vascular Aβ load, which disrupts vessel architecture and exacerbates microangiopathies, microaneurysmal dilatation, and fibrinoid necrosis (Revesz et al., 2002).
In aged dogs Aβ deposits are detected both in the brain parenchyma and in the walls of cerebral blood vessels (Ishihara et al., 1991; Uchida et al., 1992; Borràs et al., 1999; Colle et al., 2000; Brettschneider et al., 2015; Schmidt et al., 2015; Ozawa et al., 2016; Nešić et al., 2017; Rusbridge et al., 2018), further suggesting similarities between AD in humans and CCD in dogs (Figures 2, 3). The amyloidotic blood vessels in the brains of dogs were observed by Congo red stainings as well as by antibodies directed against Aβ. Vascular Aβ deposition mainly consists of Aβ40 in humans, but both Aβ40 and Aβ42 were detected in dogs (Kumar-Singh, 2008; Chambers et al., 2012). In canine CAA, the most affected region is the frontal cortex, where Aβ is often present in microvascular parenchymal lesions in elderly dogs. In dogs with CCD an overlap between the density and anatomical distribution of CAA and parenchymal plaques was observed (Nešić et al., 2017). The CAA consistently increased with age, but did not correlate with the CCD score (Ozawa et al., 2016). Intracerebral hemorrhages have also been described in association with CAA in dogs (Uchida et al., 1990). A recent case reported multiple infarcts and brain hemorrhages in a dog with CAA and CCD, with cerebral and vascular Aβ deposits (Rodrigues et al., 2018). Pathological hallmarks of CCD and AD are summarized in Table 2.
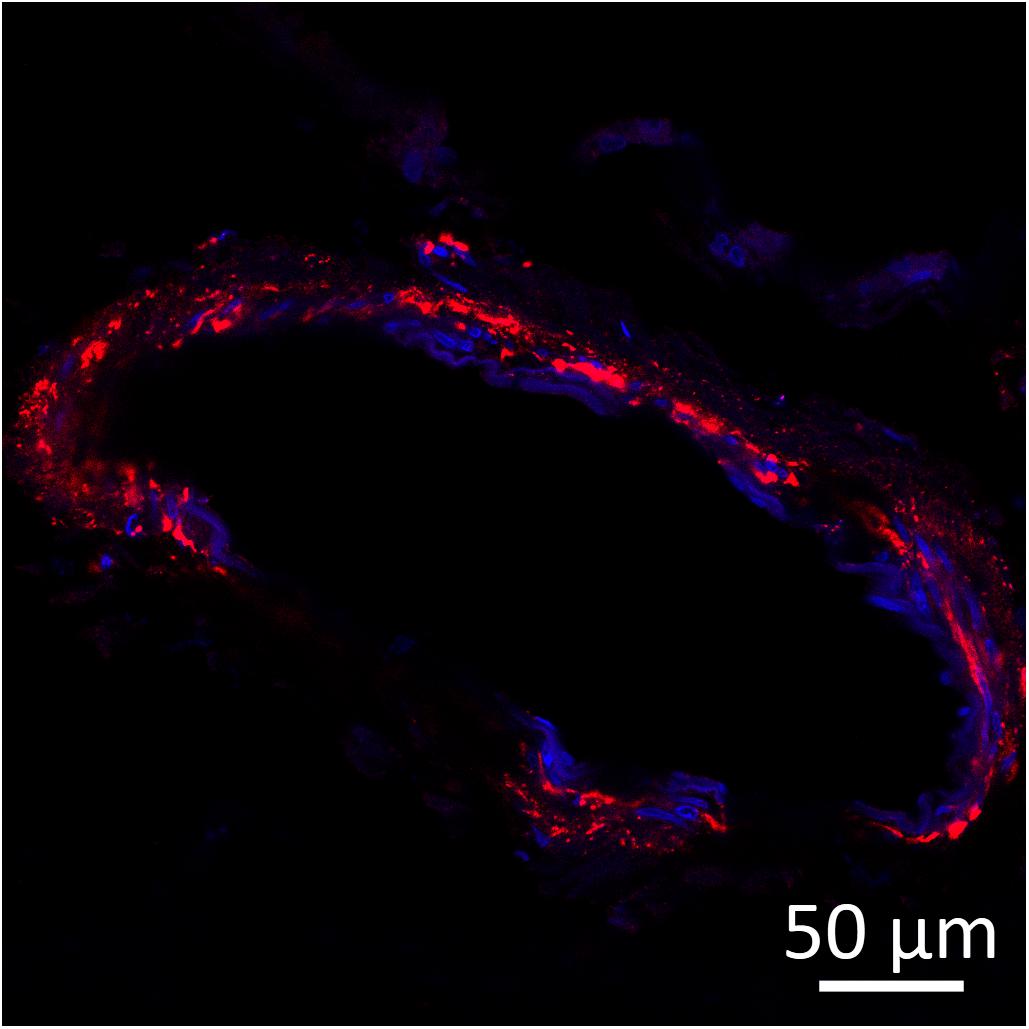
Figure 3. Amyloid beta (red) detected by immunofluorescence staining in the wall of a leptomeningeal blood vessel in the frontal cortex from a 15-years-old Pit Bull Terrier. Nuclei were counterstained with DAPI (blue). The antibody employed is the same as in Figure 2. Original microphotograph made by the authors.
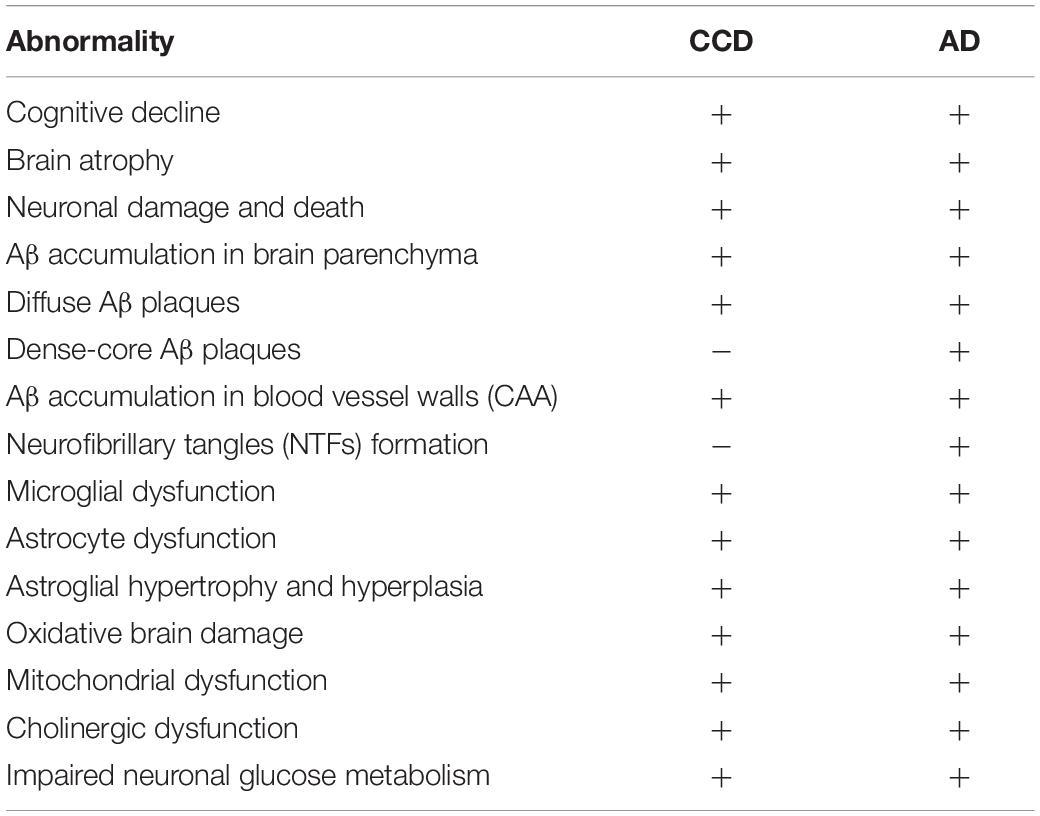
Table 2. Pathological hallmarks of CCD and/or AD.
Genetics
The incidence of human neurodegeneration and associated dementias is sporadic and is only rarely due to hereditary changes that are linked with genetic mutations. The majority of AD cases are sporadic late-onset (LOAD) with an unknown etiology (Cacace et al., 2016). The other rare AD form is genetically determined familial AD. Known specific gene mutations are the root cause for the development of familial form of AD (Selkoe, 1997). Most of the patients with early onset, familial form of AD have mutations in one of the two presenilin proteins (PSEN1 and PSEN2) (Bekris et al., 2010), which are proteins with a role in Aβ generation. Mutations also occur in APP gene (Selkoe, 1997). Polymorphism in APOEε4 allele of apolipoprotein E gene (APOE) is the major genetic risk factor for LOAD along with polymorphism in TREM2 gene, which has a role in maintaining normal immune functions in the brain (reviewed in detail in Wolfe et al., 2018). In population studies, several genetic loci have been identified as risk factors for AD (e.g., CLU, CR1, APP, PICALM, BIN1, ABCA7, MS4A, MEF2C CD33, EPHA1, CD2AP, APOE, TRIP4, TREM2, SORL1), although the connection between genes in these loci and pathogenesis of AD has not been established (Ashford, 2004; Karch et al., 2012; Mäkelä et al., 2018). Interestingly, the APOE4 polymorphism is unique to humans and its evolvement from the presumably ancestral primate form might have been instrumental for the AD pathogenesis (Mahley and Rall, 1999; Walker and Jucker, 2017).
Some presenilin mutations and mutations in APP have been connected to CAA (Hendriks et al., 1992; Grabowski et al., 2001; Rossi et al., 2004; Bugiani et al., 2010; Sellal et al., 2017) and also a polymorphism in the neprilysin gene was associated with CAA (Kumar-Singh, 2008), although the exact connections between these mutations and AD/CAA symptoms and progression are not yet understood. Furthermore, APOEε4 was shown to be a risk factor for CAA and both APOEε2 and ε4 alleles are associated with more severe forms of CAA (Nelson et al., 2013; Yu et al., 2015).
No mutations in specific genes have been reported in dogs with CCD so far. The only neurodegenerative disease in dogs that has been linked to specific mutation is degenerative myelopathy (DM), a condition caused by both demyelination and axonal loss in the canine spinal cord. In dogs with this disease, mutations have been found in a gene SOD1, coding for superoxide dismutase enzyme (Nakamae et al., 2015; Kobatake et al., 2016). Interestingly, this gene is in human patients associated with some forms of familial amyotrophic lateral sclerosis (Rosen et al., 1993). Beside mutations in SOD1 gene, risk for developing DM in dogs, as well as onset of disease, seem to be partially modulated by gene encoding SP110 nuclear body protein (SP110) at least in Pembroke Welsh Corgis (Ivansson et al., 2016). No other mutations or disease modifying loci related to neurodegeneration have been detected in dogs so far and thus, little is known about the potential genetic risks or genetic predisposition for CCD and CAA.
Biomarkers
Neurodegenerative diseases are particularly challenging diseases, as they are difficult to diagnose in the initial stages. Although many years of research have been devoted to the identification of suitable biomarkers, preferably in blood, that would allow early diagnosis or even prediction of AD in humans, such markers remain elusive. Numerous candidates have been identified in both blood and CSF, but none of the markers identified so far have been used routinely in the clinics. CSF levels of Aβ, total TAU and hyperphosphorylated TAU are the most often monitored variables, along with PET molecular imaging of amyloid and TAU deposition, in diagnosis of early stages of AD (McKhann et al., 2011).
In dogs there are no biological markers that would allow accurate and early diagnosis of CCD. In most cases assessment of cognitive functions through several neuropsychological tests and excluding other conditions with overlapping symptoms is sufficient to confirm diagnosis when the disease has progressed, but markers for detecting disease in early stages would be very useful in veterinary medicine.
In dogs with CCD, plasma Aβ42, a longer Aβ isoform which is more fibrillogenic and associated with disease, was monitored as a biomarker potentially linked to CCD (González-Martínez et al., 2011; Schütt et al., 2015). One study showed highest Aβ42 plasma levels in younger healthy dogs, and significantly higher Aβ42 plasma levels in mildly cognitively impaired dogs in comparison to severely impaired dogs (González-Martínez et al., 2011). However, another study reported the opposite with highest Aβ42 plasma levels in CCD dogs (Schütt et al., 2015). Plasma ratio of Aβ40 and Aβ42, was shown to be similar in human AD and CCD as in healthy individuals (Mayeux et al., 2003; Lopez et al., 2008; González-Martínez et al., 2011) although in some studies decreases in plasma Aβ42 were an indicator of faster progression of CCD and AD (Pesini et al., 2009; González-Martínez et al., 2011). This points to a possible disease mechanism wherein aggregation and accumulation of Aβ in the brain results in lower levels of Aβ in plasma and CSF. A more recent human study showed decrease in plasma Aβ during the dementia stage of AD and increased levels of Aβ in plasma during vascular disease (Janelidze et al., 2016).
In healthy aged dogs with age related Aβ deposits, a decrease in levels of Aβ42, but not Aβ40 was detected in CSF (Head et al., 2010). Similarly, in elderly people Aβ42 has been monitored in CSF and the decrease in Aβ42 is suggested as biomarker for Aβ deposition in the brain and has been observed alongside brain atrophy (Fagan et al., 2009; Racine et al., 2016). Although CSF Aβ content decreased in the aging dog (Head et al., 2010), high levels of Aβ in the CSF of young and middle-aged dogs also correlated with impaired learning (Borghys et al., 2017). This infers high CSF Aβ levels in younger dogs, which are not likely to harbor depositions of amyloid in their brains yet, as an early biomarker for the development of cognitive impairment. One study also reported an increase of lactate, pyruvate and potassium concentrations in CSF of dogs correlating with severe cognitive impairment (Pugliese et al., 2005), but this has not been firmly established or confirmed. Therefore, there are no biomarkers available to monitor and predict CCD progress in dogs. Future studies aiming to develop such biomarkers would be needed, hoping to provide biomarkers for early detection of this disease.
Diagnosis of CCD
Although CCD is highly prevalent the disease is severely under-diagnosed, affecting a growing population of aged dogs. The diagnosis of CCD is a diagnosis of elimination. The illness exacerbating symptoms, commonly also observed in CCD, must be excluded, such as brain tumors, hypertension, other neurological conditions, metabolic and hormonal imbalances, etc. Screening and diagnosis of CCD is primarily based on observation of clinical signs which are summarized by the acronym DISHAA [Disorientation, altered social Interactions, altered Sleep–wake cycles, House soiling and loss of other learned behaviors, altered Activity levels and increasing Anxiety (Ruehl et al., 1995; Neilson et al., 2001; Azkona et al., 2009; Rosado et al., 2012; Fast et al., 2013b; Madari et al., 2015)]. Sleeping during the day and restless at night, decreased interaction, disorientation at home and anxiety are common symptoms (Fast et al., 2013b).
The diagnosis depends largely on the owners and the veterinarians to observe and diagnose the disease, which is in most cases overlooked and symptoms attributed to the aging of dogs. To facilitate the detection of CCD, veterinarians can use a screening questionnaire that includes a list of possible signs. Several questionnaires are available and based on the scores the stage of dog’s cognitive decline can be identified (Colle et al., 2000; Neilson et al., 2001; Osella et al., 2007; Azkona et al., 2009; Golini et al., 2009; Salvin et al., 2011; Landsberg et al., 2012; Rosado et al., 2012; Fast et al., 2013b; Madari et al., 2015). Madari et al. (2015) have proposed criteria for discrimination of three stages of the disease, these are mild cognitive impairment, moderate cognitive impairment, and severe cognitive impairment. The severity and progression of CCD disease is identified by Canine Dementia Scale (CADES), which contains 17 items distributed into four domains related to changes in dogs’ behavior (spatial orientation, social interactions, sleep–wake cycles, and house soiling) (Madari et al., 2015). The Canine Cognitive Dysfunction Rating Scale (CCDR) is another questionnaire (Salvin et al., 2011), which is comprised of 13 behavioral items distributed into four domains, namely orientation, memory, apathy, impaired olfaction, and locomotion (Salvin et al., 2011). The cognitive deficits in dogs with CCD with regards to affected brain regions are summarized in Table 1. Cognitive impairment parallels the symptomology of AD. Performance on tasks involving complex learning and working memory are impaired first, along with executive function, visuospatial ability and complex learning deficits, with disease progression impairments in discrimination learning and behavioral changes are common (Neilson et al., 2001; Tapp et al., 2003; Studzinski et al., 2006; Madari et al., 2015).
MRI diagnosis is only rarely performed in dogs, due to possible complications during anesthesia and cost restraints. In dogs with CCD, as in humans with AD, MRI shows brain atrophy and include ventricular enlargement as well as widened and well-demarcated cerebral sulci. Measuring the thickness of the interthalamic adhesion in CCD was employed as a parameter for quantifying canine brain atrophy (Hasegawa et al., 2005; Noh et al., 2017). In demented canine brain MRI can be a useful tool to detect other abnormalities of the brain, possibly causing dementia such as leukoaraiosis (periventricular white matter hyperintensities) and brain microhemorrhages (Dewey et al., 2019). In human brain imaging a variant of amyloid-binding histological dye Thioflavin T, [11C]PiB, has been used as a tracer to detect Aβ using PET (positron-emission tomography) scans. In dogs this compound failed to detect the full amyloid load in the brain, established by ex vivo histopathological investigation (Fast et al., 2013a).
In general, the diagnosis of human AD is set similarly. First by ruling out other possible causes for symptoms and then by detection of CSF and plasma biomarker levels, tests of memory, problem solving, attention, counting, and language, which can be followed by MRI and PET scans (Hane et al., 2017). As in dogs, the cause of dementia can only be confirmed with certainty postmortem.
Treatment and Drug Development
Current treatment approaches for AD in humans are focused on helping people maintain mental function, manage behavioral symptoms, and slow or delay the symptoms of the disease. Unfortunately, there is no effective treatment for AD. Drugs that are used today in the management of AD can only alleviate the symptoms, and even that only temporarily. Cholinesterase inhibitors (donepezil, rivastigmine, and galantamine) have been approved for the management of AD in humans. Drugs acting as acetylcholinesterase inhibitors reduce the activity of the acetylcholine esterase which degrades acetylcholine, thus increasing the amount of acetylcholine available in the brain and therefore stimulate brain cells which receive more synaptic inputs (reviewed in Winblad et al., 2016). This very unspecific treatment can slow the disease progression for 6–12 months, and even achieve temporary improvement as shown for donepezil (Birks and Harvey, 2018), but does not cure the disease which will eventually progress. Another drug used for the treatment of AD is memantine that acts on the glutamatergic system by blocking NMDA (N-methyl-D-aspartate) receptors. A recent random-effects network meta-analysis of 41 randomized controlled trials reveled the most suitable dosages of cholinesterase inhibitors and memantine to treat patients with mild, moderate and severe AD (Dou et al., 2018). Although these pharmacological interventions have beneficial effects on cognition, function and global clinical impression, these treatments do not alleviate the neuropsychiatric symptoms (Dou et al., 2018). Unfortunately, there have been numerous clinical trials with many candidate drugs, but most of these had negative outcomes (Cummings et al., 2018).
Treatment of Canine Dementia
Current treatment options for CCD target prevention, slowing and/or improving the cognitive decline in dogs. Some drugs or food supplements are available for senior dogs and might act neuroprotective. Some enhance the blood flow into the brain, others work as antioxidants and more effort is now directed to slowing the progression of the disease instead of providing only symptomatic treatment. One commonly prescribed drug for cognitive impaired dogs is selegiline, which acts as an inhibitor of monoamine oxidase B (MAOB), thus reducing degradation of several neurotransmitters in the brain, and may have neuroprotective effects on dopaminergic, noradrenergic and cholinergic neurons (Landsberg, 2005; Magyar, 2011). Another drug that is occasionally used is nicergoline, which increases the blood flow through the brain. It may enhance neuronal transmission and act neuroprotective, increase dopamine and noradrenaline turnover and inhibit platelet aggregation (Landsberg, 2005). Propentofylline also has a neuroprotective role as it inhibits the production of free radicals and reduces the activation of microglial cell, thus acting anti-inflammatory (Frampton et al., 2003). Antidepressants such as selective serotonin reuptake inhibitors fluoxetine and sertraline, amitriptyline, paroxetine and anxiolytics benzodiazepines, gabapentin, valproic acid and buspirone can also be used to treat the anxiety and aggression which may accompany CCD. Clomipramin is an antidepressant commonly prescribed for dogs with anxiety (Landsberg, 2005), but these are all symptomatic treatments and do not treat the disease itself. S-adenosylmethionine tosylate supplementation was reported to be safe and effective in improving signs of age-related mental decline in dogs (Rème et al., 2008).
There are also some nutraceutical preparations available for dogs, which are based on natural products and/or supplement formulations. Behavioral enrichment alongside with antioxidant-rich diet and exercise is an approach for maintaining cognitive function and slowing the progression of CCD in senior pets. As means of preventative intervention, aging beagles were fed a diet rich in antioxidant, which improved cognition, maintained cognition and reduced oxidative damage and Aβ pathology in treated dogs (Milgram et al., 2004; Dowling and Head, 2012). Another longitudinal survey in beagles looked at the proteomic changes following administration of antioxidant-rich diet in combination with behavioral enrichment (Opii et al., 2008). Following treatment, the levels of oxidative stress biomarkers decreased and the increased expression levels of Cu/Zn superoxide dismutase, fructose-bisphosphate aldolase C, creatine kinase, glutamate dehydrogenase and glyceraldehyde-3-phosphate dehydrogenase correlated with improved cognition (Opii et al., 2008). In addition, some other studies implicated the nutrition as cognition modifying factor in dogs (Araujo et al., 2005a; Siwak et al., 2005; Osella et al., 2007; Christie et al., 2009; Head et al., 2009; Snigdha et al., 2011, 2012; Katina et al., 2016; Chapagain et al., 2018), highlighting combination of nutraceutical supplements directed at several mechanisms of pathological aging, in combination with behavioral enrichment, as more effective (Araujo et al., 2005a, 2008). Dogs receiving both an antioxidant-rich diet and environmental enrichment showed increased levels of brain-derived neurotrophic factor (BDNF) mRNA when compared to untreated aged dogs. As a result of increase in BDNF mRNA, the cognitive performance improved and the amount of cortical Aβ deposits decreased (Fahnestock et al., 2012). Interesting is the finding that neuronal loss in the hippocampus, occurring in the aged dogs, could be partially reversed by more engagement with the dog, i.e., with stimulation of brain function (Siwak et al., 2000).
Animal Models for Developing Novel Treatments
In comparison to transgenic mouse models, natural animal models better represent the pathophysiology of AD. Models of “physiologically” aged rats, degus and dogs are useful for studying mechanistic aspects of AD, which are also very valuable in the development of therapeutics that would alleviate age-related declines in cognitive function. Mouse models for AD research carry mutations, found in familial AD, and are artificially accumulating Aβ plaques and NFTs. Whether mice are good models have been thoroughly discussed elsewhere (Götz et al., 2018). Several drugs have cleared the amyloid load in mice but failed to do so in people with AD. Recently the genetic background and environmental factors have been demonstrated as the variability in AD development, which was partially recognized by incorporating genetic diversity into mouse models of AD (Neuner et al., 2018). Transgenic minipigs expressing APP695 or PSEN1 have also been developed but have not shown the histopathological nor the cognitive impairment signs (Holm et al., 2016). Therefore, natural animal models of species with spontaneously occurring neurodegeneration are potentially more useful in developing and testing novel treatments for such diseases.
To date, most of the drugs in development for AD treatment have been directed toward the removal of amyloid plaques or NFTs, not taking into the account the multifactorial causation of the disease. Several experimental drugs that have successfully removed plaques from mouse brains have not lessened the symptoms of AD in people. For instance, drugs acting as BACE1 (beta-site amyloid precursor protein cleaving enzyme-1) inhibitors had failed in Phase II/III clinical trials (Hawkes, 2017; Dobrowolska Zakaria and Vassar, 2018; Egan et al., 2018). A BACE1 inhibitor verubecestat successfully blocked the accumulation of amyloid protein in mice (Villarreal et al., 2017), rats and monkeys (Kennedy et al., 2016), but did not reduce cognitive or functional decline in patients with mild to moderate AD (Egan et al., 2018). Although decrease in Aβ biomarkers in CSF and brain has been noticed after treatment with BACE1 inhibitors, the failure to prevent cognitive decline might have been due to irreversible neurotoxic accumulation of Aβ that occurred prior to the start of the treatment. For this reason, focus is on the development of therapies that commence at presymptomatic stage (preclinical stage and the stage of mild cognitive impairment) although for this, development of novel, useful biomarkers, is also crucial.
As canine cognitive decline and human Alzheimer’s disease show neuropathological, cognitive and behavioral parallels, the testing of products for the treatment of AD in canine model could be useful to determine the efficacy of these compounds in humans, and also to develop novel therapeutic agents for the treatment of senior dogs. Several drugs have been tested in elderly dogs and their suitability and effectiveness correlated with results obtained in human trials, when available. Drugs tested in dogs are listed in Table 3.
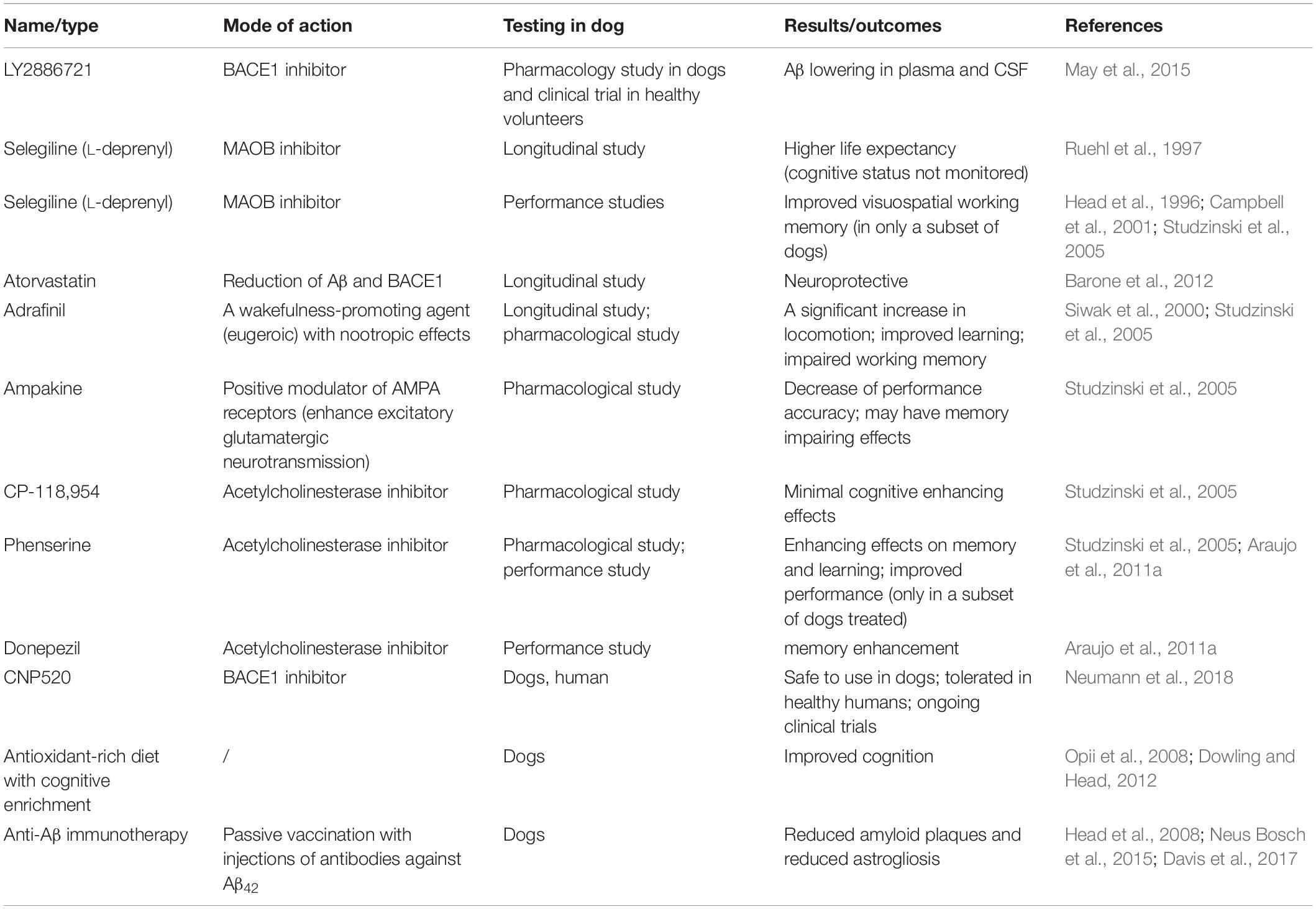
Table 3. Pharmacological interventions tested in dogs with cognitive decline.
BACE1 is a protease that controls the formation of Aβ and most likely plays an important role in the development of pathogenesis in AD. The usefulness of BACE1 small-molecule inhibitor LY2886721 has been tested in a dog model and in humans in the first clinical phase (May et al., 2015). It significantly reduced plasma and CSF Aβ levels both in dogs and healthy volunteers (May et al., 2015). After administration of two BACE1 inhibitors (cyclic sulfoxide hydroxyethylamine NB-B4 and oxazine derivative NB-C8) a unique pattern of secreted Aβ peptides was observed in canine CSF (Mattsson et al., 2012). Besides the expected reduced levels of Aβ40 and Aβ42, reduced levels of Aβ1–34 and increased levels of Aβ5–40 were detected, which were proposed as prognostic markers of BACE1 inhibition therapies (Mattsson et al., 2012). A recent survey, using BACE1 inhibitor CNP520, demonstrated reduced brain and CSF Aβ in rats, dogs, and reduced CSF Aβ in humans and was assessed to be well tolerated in healthy adults and further clinical trials are ongoing (Neumann et al., 2018). A cardiovascular disease drug, atorvastatin, has been investigated in a canine model of dementia whether it could reduce Aβ plaques, BACE1 protein levels and oxidative stress (Barone et al., 2012). It does offer some neuroprotective role through the up-regulation of enzyme biliverdin reductase-A (Barone et al., 2012). Selegiline (L-deprenyl), a type B monoamine oxidase inhibitor, has been reported to prolong the survival of aged dogs (Ruehl et al., 1997) and in some cases improve visuospatial working memory (Head et al., 1996). However, in subsequent studies only a small subset of dogs seemed to show improvement, pointing to a minor clinical relevance of selegiline administration (Campbell et al., 2001). Furthermore, several human AD trials provided no evidence of clinically meaningful benefits of selegiline administration for people with AD (Birks and Flicker, 2003). Interestingly, although selegiline was shown to be fairly ineffective in the treatment of CCD, it is still the only FDA-approved treatment for CCD.
Testing the same compounds in dogs and humans provided similar findings also in other studies. CP-118,954, an acetylcholinesterase inhibitor, showed minimal cognitive enhancing effects in dogs and human clinical trials were discontinued (Studzinski et al., 2005). Donepezil administration enhanced memory in dogs (Araujo et al., 2011a). Phenserine, an acetylcholinesterase inhibitor which also inhibits synthesis of Aβ and acts as a cognitive enhancing therapeutic, improved learning and working memory in geriatric dogs (Studzinski et al., 2005; Araujo et al., 2011a). Its administration showed similar results in Phase II human clinical trials, suggesting its effectiveness in improving memory in AD patients, but in Phase III human clinical trial, with patients with mild to moderate AD, no significant differences between the phenserine treated and placebo groups were noticed and the trial was discontinued (Thatte, 2005). There has been further discussions if this trial, and several other AD trials, failed due to procedural errors rather than due to a lack of drug efficacy (Winblad et al., 2010). In more recent studies patients with mild AD showed improvement in cognition following phenserine treatments, this was mirrored also by an increase in CSF Aβ40 (Kadir et al., 2008; Nordberg et al., 2015). There is another mode of action of phenserine in AD, namely inhibition of neuronal self-induced preprogrammed cell death, which further clinical trials might pursue (Becker et al., 2018). Ampakine (drug that alters the glutaminergic system) also failed in human AD trials and in dogs with age-associated memory disorder and dementia showed insignificant memory-enhancing effects after treatment with ampakine (Studzinski et al., 2005). Dogs, treated with adrafinil, a mild central nervous system stimulant used to relieve excessive sleepiness and inattention in elderly patients, were more attentive (Siwak et al., 2000). Cholinergic hypofunction might play a role in age-dependent cognitive decline in dogs (Araujo et al., 2011b), this was determined by administration of muscarinic cholinergic receptor antagonist scopolamine, which induces transient memory impairment. Similar findings were presented for aged and demented humans (Schliebs and Arendt, 2011). All these suggest that dogs might be a very good preclinical model for developing and testing new drugs for AD in humans.
Besides BACE1 and cholinesterase inhibitors several attempts have been made to develop immunotherapy treatments directed against Aβ or TAU (Bittar et al., 2018). A range of TAU-targeted immunotherapies have entered clinical development (recently reviewed in Novak et al., 2018). Clinical trials in patients with AD, which were administered Aβ peptide with conjugate to stimulate the immune response (i.e., active immunization) or anti-Aβ antibodies (i.e., passive immunization) have shown modest results and also serious side effects (reviewed in detail in Wisniewski and Drummond, 2016).
In dogs, Aβ immunotherapy could reduce the presence of amyloid plaques and astroglial reaction in aged individuals (Neus Bosch et al., 2015). A vaccine directed against fibrillary Aβ (anti-Aβ42) reduced the presence of Aβ plaques in canine prefrontal cortex and improved the function of the frontal cortex, thus resulting in cognitive benefits (Head et al., 2008). Anti-Aβ42 vaccination of dogs with CCD, in combination with behavioral enrichment, resulted in reduced presence of Aβ plaques in the brain, a significant maintenance of learning abilities over time and cognitive maintenance with no improvement in cognition (Davis et al., 2017). This anti-Aβ vaccine, however, also increased the frequency of brain microhemorrhages (Davis et al., 2017) which is commonly observed in the development of these immunotherapies also in human trials and could be very serious side effect. Similarly to pharmacological compounds, immunotherapies in both dogs and humans have provided modest improvements at most, yet similarities between human and canine patients again highlight the parallels between diseases and usefulness of dogs as a preclinical model for AD.
Conclusion
Risks for age-related AD and CCD are a complicated interplay between aging, genetic risk factors and environmental influences. CCD in dogs is similar to human AD with respect to APP processing, amyloid plaque deposition and cognitive dysfunction. As dogs age, they have been shown to accumulate amyloid plaques, but, dissimilar to human AD patients, dog brain rarely contains NFTs. Aβ peptide accumulates in the canine brain extracellular space in the form of soluble oligomers, fibrils and Aβ plaques. Its toxic accumulation is believed to be responsible for the neuronal dysfunction and degeneration although the severity of the disease does not always correlate with amyloid burden, but might correlate with toxic fibril polymorphs.
Dogs over 7–8 years seem to be interesting, naturally occurring model organism for aging/dementia that fairly faithfully recapitulate human disease. They are potentially much more useful as rodent models as in laboratory rodents neurodegenerative diseases do not occur spontaneously. Furthermore, genetically modified mice used in research often show very different neurodegeneration courses, as seen in humans. Dogs also spontaneously develop vasculopathies, such as CAA, and perhaps vascular dementia although this has not yet been truly studied, and are therefore also valuable as models to decipher the age and/or dementia related cerebrovascular changes that often accompany or even precede neurodegeneration in human patients.
There are no effective treatments for neurodegenerative disorders, practically all currently available treatments are symptomatic. This is partially related to the poor understanding of the pathogenesis of these diseases, and partly due to the lack of good animal models. Cholinesterase or secretase inhibitor therapy or immunotherapy has been attempted in dogs with CDD, with overlapping results to human AD trials. The neuroprotective effects of cognitive enrichment, alongside with antioxidant-rich diet, show benefits in managing the disease progression and severity in both humans and dogs. Similar results with various drugs do suggest that dogs are useful model to study both pathogenesis and novel treatments for AD in the future.
Taken together, animal studies are important to dissect the molecular and cellular processes specific to AD pathology. Dogs with CCD not only develop isomorphic changes in human cognition and brain pathology, but also accurately predicted the efficacy of known AD treatments and would be therefore good models for testing new substances that affect the lowering of Aβ levels or the reduction or degradation of aggregates associated with AD. This would benefit human patients, but could also help dog patients as any successful treatments could be also introduced into clinical veterinary medicine with the aim to successfully treat this debilitating disease in canine patients.
Author Contributions
Both authors have made a substantial contribution to this work.
Funding
This work was supported by European Social Fund and Slovenian Research Agency (ARRS) Research Program P4-0053.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aguzzi, A. (2014). Neurodegeneration: Alzheimer’s disease under strain. Nature 512, 32–34. doi: 10.1038/512032a
Araujo, J. A., Greig, N. H., Ingram, D. K., Sandin, J., de Rivera, C., and Milgram, N. W. (2011a). Cholinesterase inhibitors improve both memory and complex learning in aged beagle dogs. J. Alzheimers Dis. 26, 143–155. doi: 10.3233/JAD-2011-110005
Araujo, J. A., Landsberg, G. M., Milgram, N. W., and Miolo, A. (2008). Improvement of short-term memory performance in aged beagles by a nutraceutical supplement containing phosphatidylserine, Ginkgo biloba, vitamin E, and pyridoxine. Can. Vet. J. 49, 379–385.
Araujo, J. A., Nobrega, J. N., Raymond, R., and Milgram, N. W. (2011b). Aged dogs demonstrate both increased sensitivity to scopolamine impairment and decreased muscarinic receptor density. Pharmacol. Biochem. Behav. 98, 203–209. doi: 10.1016/j.pbb.2011.01.005
Araujo, J. A., Studzinski, C. M., Head, E., Cotman, C. W., and Milgram, N. W. (2005a). Assessment of nutritional interventions for modification of age-associated cognitive decline using a canine model of human aging. Age 27, 27–37. doi: 10.1007/s11357-005-4001-z
Araujo, J. A., Studzinski, C. M., and Milgram, N. W. (2005b). Further evidence for the cholinergic hypothesis of aging and dementia from the canine model of aging. Prog. Neuropsychopharmacol. Biol. Psychiatry 29, 411–422. doi: 10.1016/j.pnpbp.2004.12.008
Arendt, T., Brückner, M. K., and Lösche, A. (2015). Regional mosaic genomic heterogeneity in the elderly and in Alzheimer’s disease as a correlate of neuronal vulnerability. Acta Neuropathol. 130, 501–510. doi: 10.1007/s00401-015-1465-5
Arendt, T., Brückner, M. K., Mosch, B., and Lösche, A. (2010). Selective cell death of hyperploid neurons in Alzheimer’s disease. Am. J. Pathol. 177, 15–20. doi: 10.2353/ajpath.2010.090955
Arendt, T., Stieler, J., and Ueberham, U. (2017). Is sporadic Alzheimer’s disease a developmental disorder? J. Neurochem. 143, 396–408. doi: 10.1111/jnc.14036
Ashford, J. W. (2004). APOE genotype effects on Alzheimer’s disease onset and epidemiology. J. Mol. Neurosci. 23, 157–165.
Attems, J. (2005). Sporadic cerebral amyloid angiopathy: pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol. 110, 345–359. doi: 10.1007/s00401-005-1074-9
Azkona, G., García-Belenguer, S., Chacón, G., Rosado, B., León, M., and Palacio, J. (2009). Prevalence and risk factors of behavioural changes associated with age-related cognitive impairment in geriatric dogs. J. Small Anim. Pract. 50, 87–91. doi: 10.1111/j.1748-5827.2008.00718.x
Bain, M. J., Hart, B. L., Cliff, K. D., and Ruehl, W. W. (2001). Predicting behavioral changes associated with age-related cognitive impairment in dogs. J. Am. Vet. Med. Assoc. 218, 1792–1795. doi: 10.2460/javma.2001.218.1792
Barnes, J., Cotton, P., Robinson, S., and Jacobsen, M. (2016). Spontaneous pathology and routine clinical pathology parameters in aging beagle dogs: a comparison with adolescent and young adults. Vet. Pathol. 53, 447–455. doi: 10.1177/0300985815610390
Barone, E., Mancuso, C., Di Domenico, F., Sultana, R., Murphy, M. P., Head, E., et al. (2012). Biliverdin reductase-A: a novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease. J. Neurochem. 120, 135–146. doi: 10.1111/j.1471-4159.2011.07538.x
Bature, F., Guinn, B.-A., Pang, D., and Pappas, Y. (2017). Signs and symptoms preceding the diagnosis of Alzheimer’s disease: a systematic scoping review of literature from 1937 to 2016. BMJ Open 7:e015746. doi: 10.1136/bmjopen-2016-015746
Becker, R. E., Greig, N. H., Schneider, L. S., Ballard, C., Aarsland, D., Lahiri, D. K., et al. (2018). (-)-Phenserine and inhibiting apoptosis: in pursuit of a novel intervention for Alzheimer’s disease. Curr. Alzheimer Res. 15, 883–891. doi: 10.2174/1567205015666180110120026
Bekris, L. M., Yu, C.-E., Bird, T. D., and Tsuang, D. W. (2010). Genetics of Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 23, 213–227. doi: 10.1177/0891988710383571
Bell, K. F. S., Ducatenzeiler, A., Ribeiro-da-Silva, A., Duff, K., Bennett, D. A., and Cuello, A. C. (2006). The amyloid pathology progresses in a neurotransmitter-specific manner. Neurobiol. Aging 27, 1644–1657. doi: 10.1016/j.neurobiolaging.2005.09.034
Birks, J., and Flicker, L. (2003). Selegiline for Alzheimer’s disease. Cochrane Database Syst. Rev. 1:CD000442. doi: 10.1002/14651858.CD000442
Birks, J. S., and Harvey, R. J. (2018). Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst. Rev. 6:CD001190. doi: 10.1002/14651858.CD001190.pub3
Bittar, A., Sengupta, U., and Kayed, R. (2018). Prospects for strain-specific immunotherapy in Alzheimer’s disease and tauopathies. NPJ Vaccines 3:9. doi: 10.1038/s41541-018-0046-8
Borghys, H., Van Broeck, B., Dhuyvetter, D., Jacobs, T., de Waepenaert, K., Erkens, T., et al. (2017). Young to middle-aged dogs with high amyloid-β levels in cerebrospinal fluid are impaired on learning in standard cognition tests. J. Alzheimers Dis. 56, 763–774. doi: 10.3233/JAD-160434
Borràs, D., Ferrer, I., and Pumarola, M. (1999). Age-related changes in the brain of the dog. Vet. Pathol. 36, 202–211. doi: 10.1354/vp.36-3-202
Bosch, M. N., Pugliese, M., Gimeno-Bayón, J., Rodríguez, M. J., and Mahy, N. (2012). Dogs with cognitive dysfunction syndrome: a natural model of Alzheimer’s disease. Curr. Alzheimer Res. 9, 298–314. doi: 10.2174/156720512800107546
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/bf00308809
Braak, H., and Braak, E. (1997). Frequency of stages of alzheimer-related lesions in different age categories. Neurobiol. Aging 18, 351–357. doi: 10.1016/S0197-4580(97)00056-0
Braak, H., Braak, E., and Strothjohann, M. (1994). Abnormally phosphorylated tau protein related to the formation of neurofibrillary tangles and neuropil threads in the cerebral cortex of sheep and goat. Neurosci. Lett. 171, 1–4. doi: 10.1016/0304-3940(94)90589-4
Braak, H., and Del Tredici, K. (2012). Where, when, and in what form does sporadic Alzheimer’s disease begin? Curr. Opin. Neurol. 25, 708–714. doi: 10.1097/WCO.0b013e32835a3432
Braidy, N., Poljak, A., Jayasena, T., Mansour, H., Inestrosa, N. C., and Sachdev, P. S. (2015). Accelerating Alzheimer’s research through “natural” animal models. Curr. Opin. Psychiatry 28, 155–164. doi: 10.1097/YCO.0000000000000137
Brettschneider, J., Del Tredici, K., Lee, V. M.-Y., and Trojanowski, J. Q. (2015). Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci. 16, 109–120. doi: 10.1038/nrn3887
Briefer Freymond, S., Ruet, A., Grivaz, M., Fuentes, C., Zuberbühler, K., Bachmann, I., et al. (2018). Stereotypic horses (Equus caballus) are not cognitively impaired. Anim. Cogn. 22, 17–33. doi: 10.1007/s10071-018-1217-8
Bugiani, O., Giaccone, G., Rossi, G., Mangieri, M., Capobianco, R., Morbin, M., et al. (2010). Hereditary cerebral hemorrhage with amyloidosis associated with the E693K mutation of APP. Arch. Neurol. 67, 987–995. doi: 10.1001/archneurol.2010.178
Cacace, R., Sleegers, K., and Van Broeckhoven, C. (2016). Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 12, 733–748. doi: 10.1016/j.jalz.2016.01.012
Campbell, S., Trettien, A., and Kozan, B. (2001). A noncomparative open-label study evaluating the effect of selegiline hydrochloride in a clinical setting. Vet. Ther. 2, 24–39.
Canudas, J., Insua, D., Sarasa, L., González-Martínez, A., Suárez, M. L., Santamarina, G., et al. (2014). Neprilysin is poorly expressed in the prefrontal cortex of aged dogs with cognitive dysfunction syndrome. Int. J. Alzheimers Dis. 2014:483281. doi: 10.1155/2014/483281
Capucchio, M. T., Márquez, M., Pregel, P., Foradada, L., Bravo, M., Mattutino, G., et al. (2010). Parenchymal and vascular lesions in ageing equine brains: histological and immunohistochemical studies. J. Comp. Pathol. 142, 61–73. doi: 10.1016/j.jcpa.2009.07.007
Chambers, J. K., Mutsuga, M., Uchida, K., and Nakayama, H. (2011). Characterization of AβpN3 deposition in the brains of dogs of various ages and other animal species. Amyloid 18, 63–71. doi: 10.3109/13506129.2011.570385
Chambers, J. K., Tokuda, T., Uchida, K., Ishii, R., Tatebe, H., Takahashi, E., et al. (2015). The domestic cat as a natural animal model of Alzheimer’s disease. Acta Neuropathol. Commun. 3:78. doi: 10.1186/s40478-015-0258-3
Chambers, J. K., Uchida, K., and Nakayama, H. (2012). White matter myelin loss in the brains of aged dogs. Exp. Gerontol. 47, 263–269. doi: 10.1016/j.exger.2011.12.003
Chapagain, D., Virányi, Z., Huber, L., Serra, J., Schoesswender, J., and Range, F. (2018). Effect of age and dietary intervention on discrimination learning in pet dogs. Front. Psychol. 9:2217. doi: 10.3389/fpsyg.2018.02217
Christie, L.-A., Opii, W. O., and Head, E. (2009). Strategies for improving cognition with aging: insights from a longitudinal study of antioxidant and behavioral enrichment in canines. Age 31, 211–220. doi: 10.1007/s11357-008-9063-2
Colle, M.-A., Hauw, J.-J., Crespeau, F., Uchihara, T., Akiyama, H., Checler, F., et al. (2000). Vascular and parenchymal Abeta deposition in the aging dog: correlation with behavior. Neurobiol. Aging 21, 695–704. doi: 10.1016/s0197-4580(00)00113-5
Condello, C., and Stöehr, J. (2018). Aβ propagation and strains: Implications for the phenotypic diversity in Alzheimer’s disease. Neurobiol. Dis. 109, 191–200. doi: 10.1016/j.nbd.2017.03.014
Cork, L. C., Powers, R. E., Selkoe, D. J., Davies, P., Geyer, J. J., and Price, D. L. (1988). Neurofibrillary Tangles and Senile Plaques in Aged Bears. J. Neuropathol. Exp. Neurol. 47, 629–641. doi: 10.1097/00005072-198811000-00006
Cummings, B. J., and Cotman, C. W. (1995). Image analysis of beta-amyloid load in Alzheimer’s disease and relation to dementia severity. Lancet 346, 1524–1528. doi: 10.1016/s0140-6736(95)92053-6
Cummings, B. J., Su, J. H., Cotman, C. W., White, R., and Russell, M. J. (1993). Beta-amyloid accumulation in aged canine brain: a model of early plaque formation in Alzheimer’s disease. Neurobiol. Aging 14, 547–560. doi: 10.1016/0197-4580(93)90038-d
Cummings, J., Ritter, A., and Zhong, K. (2018). Clinical trials for disease-modifying therapies in Alzheimer’s disease: a primer, lessons learned, and a blueprint for the future. J. Alzheimers Dis. 64, S3–S22. doi: 10.3233/JAD-179901
Davis, P. R., Giannini, G., Rudolph, K., Calloway, N., Royer, C. M., Beckett, T. L., et al. (2017). Aβ vaccination in combination with behavioral enrichment in aged beagles: effects on cognition, Aβ, and microhemorrhages. Neurobiol. Aging 49, 86–99. doi: 10.1016/j.neurobiolaging.2016.09.007
DeVos, S. L., Corjuc, B. T., Oakley, D. H., Nobuhara, C. K., Bannon, R. N., Chase, A., et al. (2018). Synaptic Tau seeding precedes Tau pathology in human Alzheimer’s disease brain. Front. Neurosci. 12:267. doi: 10.3389/fnins.2018.00267
Dewey, C. W., Davies, E. S., Xie, H., and Wakshlag, J. J. (2019). Canine cognitive dysfunction: pathophysiology, diagnosis, and treatment. Vet. Clin. North Am. Small Anim. Pract. 49, 477–499. doi: 10.1016/j.cvsm.2019.01.013
Di Guardo, G. (2018). Alzheimer’s disease, cellular prion protein, and dolphins. Alzheimers Dement. 14, 259–260. doi: 10.1016/j.jalz.2017.12.002
Dobrowolska Zakaria, J. A., and Vassar, R. J. (2018). A promising, novel, and unique BACE1 inhibitor emerges in the quest to prevent Alzheimer’s disease. EMBO Mol. Med. 10:e9717. doi: 10.15252/emmm.201809717
Dodart, J. C., Mathis, C., Bales, K. R., and Paul, S. M. (2002). Does my mouse have Alzheimer’s disease? Genes Brain Behav. 1, 142–155. doi: 10.1034/j.1601-183x.2002.10302.x
Dou, K.-X., Tan, M.-S., Tan, C.-C., Cao, X.-P., Hou, X.-H., Guo, Q.-H., et al. (2018). Comparative safety and effectiveness of cholinesterase inhibitors and memantine for Alzheimer’s disease: a network meta-analysis of 41 randomized controlled trials. Alzheimers Res. Ther. 10:126. doi: 10.1186/s13195-018-0457-9
Dowling, A. L. S., and Head, E. (2012). Antioxidants in the canine model of human aging. Biochim. Biophys. Acta 1822, 685–689. doi: 10.1016/j.bbadis.2011.09.020
Egan, M. F., Kost, J., Tariot, P. N., Aisen, P. S., Cummings, J. L., Vellas, B., et al. (2018). Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 378, 1691–1703. doi: 10.1056/NEJMoa1706441
Eisele, Y. S., Obermüller, U., Heilbronner, G., Baumann, F., Kaeser, S. A., Wolburg, H., et al. (2010). Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 330, 980–982. doi: 10.1126/science.1194516
El-Assaad, I., Di Bari, J. A., Yasuda, K., Divers, T. J., Summers, B. A., de Lahunta, A., et al. (2012). Differential expression of TAR DNA-binding protein (TDP-43) in the central nervous system of horses afflicted with equine motor neuron disease (EMND): a preliminary study of a potential pathologic marker. Vet. Res. Commun. 36, 221–226. doi: 10.1007/s11259-012-9533-y
Fagan, A. M., Head, D., Shah, A. R., Marcus, D., Mintun, M., Morris, J. C., et al. (2009). Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann. Neurol. 65, 176–183. doi: 10.1002/ana.21559
Fahnestock, M., Marchese, M., Head, E., Pop, V., Michalski, B., Milgram, W. N., et al. (2012). BDNF increases with behavioral enrichment and an antioxidant diet in the aged dog. Neurobiol. Aging 33, 546–554. doi: 10.1016/j.neurobiolaging.2010.03.019
Fast, R., Rodell, A., Gjedde, A., Mouridsen, K., Alstrup, A. K., Bjarkam, C. R., et al. (2013a). PiB fails to map amyloid deposits in cerebral cortex of aged dogs with canine cognitive dysfunction. Front. Aging Neurosci. 5:99. doi: 10.3389/fnagi.2013.00099
Fast, R., Schütt, T., Toft, N., Møller, A., and Berendt, M. (2013b). An observational study with long-term follow-up of canine cognitive dysfunction: clinical characteristics, survival, and risk factors. J. Vet. Intern. Med. 27, 822–829. doi: 10.1111/jvim.12109
Frampton, M., Harvey, R. J., and Kirchner, V. (2003). Propentofylline for dementia. Cochrane Database Syst. Rev. 2:CD002853. doi: 10.1002/14651858.CD002853
GBD 2016 Dementia Collaborators (2019). Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18, 88–106. doi: 10.1016/S1474-4422(18)30403-4
Golini, L., Colangeli, R., Tranquillo, V., and Mariscoli, M. (2009). Association between neurologic and cognitive dysfunction signs in a sample of aging dogs. J. Vet. Behav. 4, 25–30. doi: 10.1016/j.jveb.2008.09.033
Gómez-Pinedo, U., Villar-Quiles, R. N., Galán, L., Matías-Guiu, J. A., Benito-Martin, M. S., Guerrero-Sola, A., et al. (2016). Immununochemical markers of the amyloid cascade in the hippocampus in motor neuron diseases. Front. Neurol. 7:195. doi: 10.3389/fneur.2016.00195
González-Martínez, Á., Rosado, B., Pesini, P., Suárez, M.-L., Santamarina, G., García-Belenguer, S., et al. (2011). Plasma β-amyloid peptides in canine aging and cognitive dysfunction as a model of Alzheimer’s disease. Exp. Gerontol. 46, 590–596. doi: 10.1016/j.exger.2011.02.013
Götz, J., Bodea, L.-G., and Goedert, M. (2018). Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 19, 583–598. doi: 10.1038/s41583-018-0054-8
Gouras, G. K., Tampellini, D., Takahashi, R. H., and Capetillo-Zarate, E. (2010). Intraneuronal β-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol. 119, 523–541. doi: 10.1007/s00401-010-0679-9
Grabowski, T. J., Cho, H. S., Vonsattel, J. P., Rebeck, G. W., and Greenberg, S. M. (2001). Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol. 49, 697–705. doi: 10.1002/ana.1009
Greer, K. A., Canterberry, S. C., and Murphy, K. E. (2007). Statistical analysis regarding the effects of height and weight on life span of the domestic dog. Res. Vet. Sci. 82, 208–214. doi: 10.1016/j.rvsc.2006.06.005
Hane, F. T., Robinson, M., Lee, B. Y., Bai, O., Leonenko, Z., and Albert, M. S. (2017). Recent progress in Alzheimer’s disease research, part 3: diagnosis and treatment. J. Alzheimers Dis. 57, 645–665. doi: 10.3233/JAD-160907
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185.
Härtig, W., Klein, C., Brauer, K., Schüppel, K.-F., Arendt, T., Bigl, V., et al. (2001). Hyperphosphorylated protein tau is restricted to neurons devoid of perineuronal nets in the cortex of aged bison. Neurobiol. Aging 22, 25–33. doi: 10.1016/S0197-4580(00)00179-2
Härtig, W., Klein, C., Brauer, K., Schüppel, K. F., Arendt, T., Brückner, G., et al. (2000). Abnormally phosphorylated protein tau in the cortex of aged individuals of various mammalian orders. Acta Neuropathol. 100, 305–312. doi: 10.1007/s004010000183
Hasegawa, D., Yayoshi, N., Fujita, Y., Fujita, M., and Orima, H. (2005). Measurement of interthalamic adhesion thickness as a criteria for brain atrophy in dogs with and without cognitive dysfunction (dementia). Vet. Radiol. Ultrasound 46, 452–457. doi: 10.1111/j.1740-8261.2005.00083.x
Hawkes, N. (2017). Merck ends trial of potential Alzheimer’s drug verubecestat. BMJ 356:j845. doi: 10.1136/bmj.j845
Head, E., Hartley, J., Kameka, A. M., Mehta, R., Ivy, G. O., Ruehl, W. W., et al. (1996). The effects of L-deprenyl on spatial short term memory in young and aged dogs. Prog. Neuropsychopharmacol. Biol. Psychiatry 20, 515–530. doi: 10.1016/0278-5846(96)00014-0
Head, E., Nukala, V. N., Fenoglio, K. A., Muggenburg, B. A., Cotman, C. W., and Sullivan, P. G. (2009). Effects of age, dietary, and behavioral enrichment on brain mitochondria in a canine model of human aging. Exp. Neurol. 220, 171–176. doi: 10.1016/j.expneurol.2009.08.014
Head, E., Pop, V., Sarsoza, F., Kayed, R., Beckett, T. L., Studzinski, C. M., et al. (2010). Amyloid-beta peptide and oligomers in the brain and cerebrospinal fluid of aged canines. J. Alzheimers Dis. 20, 637–646. doi: 10.3233/JAD-2010-1397
Head, E., Pop, V., Vasilevko, V., Hill, M., Saing, T., Sarsoza, F., et al. (2008). A two-year study with fibrillar beta-amyloid (Abeta) immunization in aged canines: effects on cognitive function and brain Abeta. J. Neurosci. 28, 3555–3566. doi: 10.1523/JNEUROSCI.0208-08.2008
Hendriks, L., van Duijn, C. M., Cras, P., Cruts, M., Van Hul, W., van Harskamp, F., et al. (1992). Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the beta-amyloid precursor protein gene. Nat. Genet. 1, 218–221. doi: 10.1038/ng0692-218
Hoenig, M. C., Bischof, G. N., Seemiller, J., Hammes, J., Kukolja, J., Onur, Ö. A., et al. (2018). Networks of tau distribution in Alzheimer’s disease. Brain 141, 568–581. doi: 10.1093/brain/awx353
Holm, I. E., Alstrup, A. K. O., and Luo, Y. (2016). Genetically modified pig models for neurodegenerative disorders. J. Pathol. 238, 267–287. doi: 10.1002/path.4654
Insua, D., Corredoira, A., González-Martínez, A., Suárez, M.-L., Santamarina, G., Sarasa, M., et al. (2012). Expression of p75(NTR), a marker for basal forebrain cholinergic neurons, in young and aged dogs with or without cognitive dysfunction syndrome. J. Alzheimers Dis. 28, 291–296. doi: 10.3233/JAD-2011-110905
Insua, D., Suárez, M.-L., Santamarina, G., Sarasa, M., and Pesini, P. (2010). Dogs with canine counterpart of Alzheimer’s disease lose noradrenergic neurons. Neurobiol. Aging 31, 625–635. doi: 10.1016/j.neurobiolaging.2008.05.014
Ishihara, T., Gondo, T., Takahashi, M., Uchino, F., Ikeda, S., Allsop, D., et al. (1991). Immunohistochemical and immunoelectron microscopical characterization of cerebrovascular and senile plaque amyloid in aged dogs’ brains. Brain Res. 548, 196–205. doi: 10.1016/0006-8993(91)91122-h
Ivansson, E. L., Megquier, K., Kozyrev, S. V., Murén, E., Körberg, I. B., Swofford, R., et al. (2016). Variants within the SP110 nuclear body protein modify risk of canine degenerative myelopathy. Proc. Natl. Acad. Sci. U.S.A. 113, E3091–E3100. doi: 10.1073/pnas.1600084113
Janelidze, S., Stomrud, E., Palmqvist, S., Zetterberg, H., van Westen, D., Jeromin, A., et al. (2016). Plasma β-amyloid in Alzheimer’s disease and vascular disease. Sci. Rep. 6:26801. doi: 10.1038/srep26801
Johnstone, E. M., Chaney, M. O., Norris, F. H., Pascual, R., and Little, S. P. (1991). Conservation of the sequence of the Alzheimer’s disease amyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis. Brain Res. Mol. Brain Res. 10, 299–305. doi: 10.1016/0169-328x(91)90088-f
Jucker, M., and Walker, L. C. (2018). Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 21, 1341–1349. doi: 10.1038/s41593-018-0238-6
Kadir, A., Andreasen, N., Almkvist, O., Wall, A., Forsberg, A., Engler, H., et al. (2008). Effect of phenserine treatment on brain functional activity and amyloid in Alzheimer’s disease. Ann. Neurol. 63, 621–631. doi: 10.1002/ana.21345
Karch, C. M., Jeng, A. T., Nowotny, P., Cady, J., Cruchaga, C., and Goate, A. M. (2012). Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PLoS One 7:e50976. doi: 10.1371/journal.pone.0050976
Katina, S., Farbakova, J., Madari, A., Novak, M., and Zilka, N. (2016). Risk factors for canine cognitive dysfunction syndrome in Slovakia. Acta Vet. Scand. 58:17. doi: 10.1186/s13028-016-0196-5
Kelly, S. C., He, B., Perez, S. E., Ginsberg, S. D., Mufson, E. J., and Counts, S. E. (2017). Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease. Acta Neuropathol. Commun. 5:8. doi: 10.1186/s40478-017-0411-2
Kennedy, M. E., Stamford, A. W., Chen, X., Cox, K., Cumming, J. N., Dockendorf, M. F., et al. (2016). The BACE1 inhibitor verubecestat MK-8931 reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 8:363ra150. doi: 10.1126/scitranslmed.aad9704
Kobatake, Y., Sakai, H., Tsukui, T., Yamato, O., Kohyama, M., Sasaki, J., et al. (2016). Localization of a mutant SOD1 protein in E40K-heterozygous dogs: implications for non-cell-autonomous pathogenesis of degenerative myelopathy. J. Neurol. Sci. 372, 369–378. doi: 10.1016/j.jns.2016.10.034
Kumar-Singh, S. (2008). Cerebral amyloid angiopathy: pathogenetic mechanisms and link to dense amyloid plaques. Genes Brain Behav. 7,(Suppl. 1), 67–82. doi: 10.1111/j.1601-183X.2007.00380.x
Kuroki, K., Uchida, K., Kiatipattanasakul, W., Nakamura, S., Yamaguchi, R., Nakayama, H., et al. (1997). Immunohistochemical detection of tau protein in various non-human animal brains. Neuropathology 17, 174–180. doi: 10.1111/j.1440-1789.1997.tb00034.x
Landsberg, G. (2005). Therapeutic agents for the treatment of cognitive dysfunction syndrome in senior dogs. Prog. Neuropsychopharmacol. Biol. Psychiatry 29, 471–479. doi: 10.1016/j.pnpbp.2004.12.012
Landsberg, G. M., Nichol, J., and Araujo, J. A. (2012). Cognitive dysfunction syndrome: a disease of canine and feline brain aging. Vet. Clin. North Am. Small Anim. Pract. 42, 749–768, vii. doi: 10.1016/j.cvsm.2012.04.003
Langer, F., Eisele, Y. S., Fritschi, S. K., Staufenbiel, M., Walker, L. C., and Jucker, M. (2011). Soluble Aβ seeds are potent inducers of cerebral β-amyloid deposition. J. Neurosci. 31, 14488–14495. doi: 10.1523/JNEUROSCI.3088-11.2011
Lansbury, P. T., and Lashuel, H. A. (2006). A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature 443, 774–779. doi: 10.1038/nature05290
Liu, W., Xie, Y., Ma, J., Luo, X., Nie, P., Zuo, Z., et al. (2015). IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics 31, 3359–3361. doi: 10.1093/bioinformatics/btv362
Lopez, O. L., Kuller, L. H., Mehta, P. D., Becker, J. T., Gach, H. M., Sweet, R. A., et al. (2008). Plasma amyloid levels and the risk of AD in normal subjects in the Cardiovascular Health Study. Neurology 70, 1664–1671. doi: 10.1212/01.wnl.0000306696.82017.66
Madari, A., Farbakova, J., Katina, S., Smolek, T., Novak, P., Weissova, T., et al. (2015). Assessment of severity and progression of canine cognitive dysfunction syndrome using the CAnine DEmentia Scale (CADES). Appl Anim. Behav. Sci. 171, 138–145. doi: 10.1016/j.applanim.2015.08.034
Madeira, F., Park, Y. M., Lee, J., Buso, N., Gur, T., Madhusoodanan, N., et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. [Epub ahead of print].
Magyar, K. (2011). The pharmacology of selegiline. Int. Rev. Neurobiol. 100, 65–84. doi: 10.1016/B978-0-12-386467-3.00004-2
Mahley, R. W., and Rall, S. C. (1999). Is epsilon4 the ancestral human apoE allele? Neurobiol. Aging 20, 429–430. doi: 10.1016/s0197-4580(99)00081-0
Mäkelä, M., Kaivola, K., Valori, M., Paetau, A., Polvikoski, T., Singleton, A. B., et al. (2018). Alzheimer risk loci and associated neuropathology in a population-based study (Vantaa 85+). Neurol. Genet. 4:e211. doi: 10.1212/NXG.0000000000000211
Martin, S. B., Dowling, A. L. S., and Head, E. (2011). Therapeutic interventions targeting Beta amyloid pathogenesis in an aging dog model. Curr. Neuropharmacol. 9, 651–661. doi: 10.2174/157015911798376217
Mattsson, N., Rajendran, L., Zetterberg, H., Gustavsson, M., Andreasson, U., Olsson, M., et al. (2012). BACE1 inhibition induces a specific cerebrospinal fluid β-amyloid pattern that identifies drug effects in the central nervous system. PLoS One 7:e31084. doi: 10.1371/journal.pone.0031084
May, P. C., Willis, B. A., Lowe, S. L., Dean, R. A., Monk, S. A., Cocke, P. J., et al. (2015). The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. J. Neurosci. 35, 1199–1210. doi: 10.1523/JNEUROSCI.4129-14.2015
Mayeux, R., Honig, L. S., Tang, M.-X., Manly, J., Stern, Y., Schupf, N., et al. (2003). Plasma A[beta]40 and A[beta]42 and Alzheimer’s disease: relation to age, mortality, and risk. Neurology 61, 1185–1190.
McKhann, G. M., Knopman, D. S., Chertkow, H., Hyman, B. T., Jack, C. R., Kawas, C. H., et al. (2011). The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269. doi: 10.1016/j.jalz.2011.03.005
Medeiros, R., Baglietto-Vargas, D., and LaFerla, F. M. (2011). The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci. Ther. 17, 514–524. doi: 10.1111/j.1755-5949.2010.00177.x
Meyer-Luehmann, M., Coomaraswamy, J., Bolmont, T., Kaeser, S., Schaefer, C., Kilger, E., et al. (2006). Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 313, 1781–1784. doi: 10.1126/science.1131864
Milgram, N. W., Head, E., Zicker, S. C., Ikeda-Douglas, C., Murphey, H., Muggenberg, B. A., et al. (2004). Long-term treatment with antioxidants and a program of behavioral enrichment reduces age-dependent impairment in discrimination and reversal learning in beagle dogs. Exp. Gerontol. 39, 753–765. doi: 10.1016/j.exger.2004.01.007
Müller, U. C., Deller, T., and Korte, M. (2017). Not just amyloid: physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 18, 281–298. doi: 10.1038/nrn.2017.29
Nakamae, S., Kobatake, Y., Suzuki, R., Tsukui, T., Kato, S., Yamato, O., et al. (2015). Accumulation and aggregate formation of mutant superoxide dismutase 1 in canine degenerative myelopathy. Neuroscience 303, 229–240. doi: 10.1016/j.neuroscience.2015.06.066
Neilson, J. C., Hart, B. L., Cliff, K. D., and Ruehl, W. W. (2001). Prevalence of behavioral changes associated with age-related cognitive impairment in dogs. J. Am. Vet. Med. Assoc. 218, 1787–1791. doi: 10.2460/javma.2001.218.1787
Nelson, P. T., Alafuzoff, I., Bigio, E. H., Bouras, C., Braak, H., Cairns, N. J., et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. doi: 10.1097/NEN.0b013e31825018f7
Nelson, P. T., Greenberg, S. G., and Saper, C. B. (1994). Neurofibrillary tangles in the cerebral cortex of sheep. Neurosci. Lett. 170, 187–190. doi: 10.1016/0304-3940(94)90270-4
Nelson, P. T., Pious, N. M., Jicha, G. A., Wilcock, D. M., Fardo, D. A., Estus, S., et al. (2013). APOE-ε2 and APOE-ε4 Correlate with Increased Amyloid Accumulation in Cerebral Vasculature. J. Neuropathol. Exp. Neurol. 72, 708–715. doi: 10.1097/NEN.0b013e31829a25b9
Nelson, P. T., and Saper, C. B. (1995). Ultrastructure of neurofibrillary tangles in the cerebral cortex of sheep. Neurobiol. Aging 16, 315–323. doi: 10.1016/0197-4580(94)00175-z
Nešić, S., Kukolj, V., Marinkoviæ, D., Vuèiæeviæ, I., and Jovanoviæ, M. (2017). Histological and immunohistochemical characteristics of cerebral amyloid angiopathy in elderly dogs. Vet. Q. 37, 1–7. doi: 10.1080/01652176.2016.1235301
Neumann, U., Ufer, M., Jacobson, L. H., Rouzade-Dominguez, M.-L., Huledal, G., Kolly, C., et al. (2018). The BACE-1 inhibitor CNP520 for prevention trials in Alzheimer’s disease. EMBO Mol. Med. 10:e9316. doi: 10.15252/emmm.201809316
Neuner, S. M., Heuer, S. E., Huentelman, M. J., O’Connell, K. M. S., and Kaczorowski, C. C. (2018). Harnessing genetic complexity to enhance translatability of Alzheimer’s disease mouse models: a path toward precision medicine. Neuron 101, 399–411.e5. doi: 10.1016/j.neuron.2018.11.040
Neus Bosch, M., Pugliese, M., Andrade, C., Gimeno-Bayón, J., Mahy, N., and Rodriguez, M. J. (2015). Amyloid-β immunotherapy reduces amyloid plaques and astroglial reaction in aged domestic dogs. Neurodegener. Dis. 15, 24–37. doi: 10.1159/000368672
Noh, D., Choi, S., Choi, H., Lee, Y., and Lee, K. (2017). Evaluation of interthalamic adhesion size as an indicator of brain atrophy in dogs with and without cognitive dysfunction. Vet. Radiol. Ultrasound 58, 581–587. doi: 10.1111/vru.12528
Nordberg, A., Kadir, A., Andreasen, N., Almkvist, O., Wall, A., Långström, B., et al. (2015). Correlations between Alzheimer’s Disease Cerebrospinal Fluid Biomarkers and Cerebral Glucose Metabolism after 12 Months of Phenserine Treatment. J. Alzheimers Dis. 47, 691–704. doi: 10.3233/JAD-132474
Novak, P., Kontsekova, E., Zilka, N., and Novak, M. (2018). Ten Years of Tau-Targeted Immunotherapy: The Path Walked and the Roads Ahead. Front. Neurosci. 12:798. doi: 10.3389/fnins.2018.00798
Oikawa, N., Kimura, N., and Yanagisawa, K. (2010). Alzheimer-type tau pathology in advanced aged nonhuman primate brains harboring substantial amyloid deposition. Brain Res. 1315, 137–149. doi: 10.1016/j.brainres.2009.12.005
Olszewska, D. A., Lonergan, R., Fallon, E. M., and Lynch, T. (2016). Genetics of Frontotemporal Dementia. Curr. Neurol. Neurosci. Rep. 16:107. doi: 10.1007/s11910-016-0707-9
Opii, W. O., Joshi, G., Head, E., Milgram, N. W., Muggenburg, B. A., Klein, J. B., et al. (2008). Proteomic identification of brain proteins in the canine model of human aging following a long-term treatment with antioxidants and a program of behavioral enrichment: relevance to Alzheimer’s disease. Neurobiol. Aging 29, 51–70. doi: 10.1016/j.neurobiolaging.2006.09.012
Osella, M. C., Re, G., Odore, R., Girardi, C., Badino, P., Barbero, R., et al. (2007). Canine cognitive dysfunction syndrome: prevalence, clinical signs and treatment with a neuroprotective nutraceutical. Appl. Anim. Behav. Sci. 105, 297–310. doi: 10.1016/j.applanim.2006.11.007
Ossenkoppele, R., Schonhaut, D. R., Schöll, M., Lockhart, S. N., Ayakta, N., Baker, S. L., et al. (2016). Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 139, 1551–1567. doi: 10.1093/brain/aww027
Ozawa, M., Chambers, J. K., Uchida, K., and Nakayama, H. (2016). The Relation between canine cognitive dysfunction and age-related brain lesions. J. Vet. Med. Sci. 78, 997–1006. doi: 10.1292/jvms.15-0624
Perez, S. E., Raghanti, M. A., Hof, P. R., Kramer, L., Ikonomovic, M. D., Lacor, P. N., et al. (2013). Alzheimer’s disease pathology in the neocortex and hippocampus of the western lowland gorilla (Gorilla gorilla gorilla). J. Comp. Neurol. 521, 4318–4338. doi: 10.1002/cne.23428
Perez, S. E., Sherwood, C. C., Cranfield, M. R., Erwin, J. M., Mudakikwa, A., Hof, P. R., et al. (2016). Early Alzheimer’s disease-type pathology in the frontal cortex of wild mountain gorillas (Gorilla beringei beringei). Neurobiol. Aging 39, 195–201. doi: 10.1016/j.neurobiolaging.2015.12.017
Pesini, P., Boada, M., Martínez-Lage, P., Tárraga, L., Monleón, I., Pérez-Grijalva, V., et al. (2009). Differences in beta amyloid pool in blood discriminate between amnesic mild cognitive impairment patients and healthy controls. J. Nutr. Health Aging 13:850.
Pugliese, M., Carrasco, J. L., Andrade, C., Mas, E., Mascort, J., and Mahy, N. (2005). Severe cognitive impairment correlates with higher cerebrospinal fluid levels of lactate and pyruvate in a canine model of senile dementia. Prog. Neuropsychopharmacol. Biol. Psychiatry 29, 603–610. doi: 10.1016/j.pnpbp.2005.01.017
Pugliese, M., Mascort, J., Mahy, N., and Ferrer, I. (2006). Diffuse beta-amyloid plaques and hyperphosphorylated tau are unrelated processes in aged dogs with behavioral deficits. Acta Neuropathol. 112, 175–183. doi: 10.1007/s00401-006-0087-3
Racine, A. M., Koscik, R. L., Nicholas, C. R., Clark, L. R., Okonkwo, O. C., Oh, J. M., et al. (2016). Cerebrospinal fluid ratios with Aβ42 predict preclinical brain β-amyloid accumulation. Alzheimers Dement. 2, 27–38. doi: 10.1016/j.dadm.2015.11.006
Reid, S. J., Mckean, N. E., Henty, K., Portelius, E., Blennow, K., Rudiger, S. R., et al. (2017). Alzheimer’s disease markers in the aged sheep (Ovis aries). Neurobiol. Aging 58, 112–119. doi: 10.1016/j.neurobiolaging.2017.06.020
Reilly, C. E. (2001). Neprilysin content is reduced in Alzheimer brain areas. J. Neurol. 248, 159–160. doi: 10.1007/s004150170259
Rème, C. A., Dramard, V., Kern, L., Hofmans, J., Halsberghe, C., and Mombiela, D. V. (2008). Effect of S-adenosylmethionine tablets on the reduction of age-related mental decline in dogs: a double-blinded, placebo-controlled trial. Vet. Ther. 9, 69–82.
Revesz, T., Holton, J. L., Lashley, T., Plant, G., Rostagno, A., Ghiso, J., et al. (2002). Sporadic and familial cerebral amyloid angiopathies. Brain Pathol. 12, 343–357. doi: 10.1111/j.1750-3639.2002.tb00449.x
Rodrigues, L. L., Mesquita, L. P., Costa, R. C., Gomes, R. G., Biihrer, D. A., and Maiorka, P. C. (2018). Multiple infarcts and hemorrhages in the central nervous system of a dog with cerebral amyloid angiopathy: a case report. BMC Vet. Res. 14:370. doi: 10.1186/s12917-018-1700-0
Rodriguez-Callejas, J. D., Fuchs, E., and Perez-Cruz, C. (2016). Evidence of Tau Hyperphosphorylation and Dystrophic Microglia in the Common Marmoset. Front. Aging Neurosci. 8:315. doi: 10.3389/fnagi.2016.00315
Rofina, J. E., van Ederen, A. M., Toussaint, M. J. M., Secrève, M., van der Spek, A., van der Meer, I., et al. (2006). Cognitive disturbances in old dogs suffering from the canine counterpart of Alzheimer’s disease. Brain Res. 1069, 216–226. doi: 10.1016/j.brainres.2005.11.021
Rosado, B., González-Martínez, A., Pesini, P., García-Belenguer, S., Palacio, J., Villegas, A., et al. (2012). Effect of age and severity of cognitive dysfunction on spontaneous activity in pet dogs - part 1: locomotor and exploratory behaviour. Vet. J. 194, 189–195. doi: 10.1016/j.tvjl.2012.03.025
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Rosen, R. F., Farberg, A. S., Gearing, M., Dooyema, J., Long, P. M., Anderson, D. C., et al. (2008). Tauopathy with paired helical filaments in an aged chimpanzee. J. Comp. Neurol. 509, 259–270. doi: 10.1002/cne.21744
Rossi, G., Giaccone, G., Maletta, R., Morbin, M., Capobianco, R., Mangieri, M., et al. (2004). A family with Alzheimer disease and strokes associated with A713T mutation of the APP gene. Neurology 63, 910–912. doi: 10.1212/01.wnl.0000137048.80666.86
Ruehl, W. W., Bruyette, D. S., DePaoli, A., Cotman, C. W., Head, E., Milgram, N. W., et al. (1995). Canine cognitive dysfunction as a model for human age-related cognitive decline, dementia and Alzheimer’s disease: clinical presentation, cognitive testing, pathology and response to 1-deprenyl therapy. Prog. Brain Res. 106, 217–225. doi: 10.1016/s0079-6123(08)61218-2
Ruehl, W. W., Entriken, T. L., Muggenburg, B. A., Bruyette, D. S., Griffith, W. C., and Hahn, F. F. (1997). Treatment with L-deprenyl prolongs life in elderly dogs. Life Sci. 61, 1037–1044. doi: 10.1016/s0024-3205(97)00611-5
Rusbridge, C., Salguero, F. J., David, M. A., Faller, K. M. E., Bras, J. T., Guerreiro, R. J., et al. (2018). An aged Canid with behavioral deficits exhibits blood and cerebrospinal fluid amyloid beta oligomers. Front. Aging Neurosci. 10:7. doi: 10.3389/fnagi.2018.00007
Salvin, H. E., McGreevy, P. D., Sachdev, P. S., and Valenzuela, M. J. (2011). The canine cognitive dysfunction rating scale (CCDR): a data-driven and ecologically relevant assessment tool. Vet. J. 188, 331–336. doi: 10.1016/j.tvjl.2010.05.014
Salvin, H. E., McGreevy, P. D., Sachdev, P. S., and Valenzuela, M. J. (2010). Under diagnosis of canine cognitive dysfunction: a cross-sectional survey of older companion dogs. Vet. J. 184, 277–281. doi: 10.1016/j.tvjl.2009.11.007
Sarasa, L., Gallego, C., Monleón, I., Olvera, A., Canudas, J., Montañés, M., et al. (2010). Cloning, sequencing and expression in the dog of the main amyloid precursor protein isoforms and some of the enzymes related with their processing. Neuroscience 171, 1091–1101. doi: 10.1016/j.neuroscience.2010.09.042
Sarasa, M., and Pesini, P. (2009). Natural non-trasgenic animal models for research in Alzheimer’s disease. Curr. Alzheimer Res. 6, 171–178. doi: 10.2174/156720509787602834
Sardar Sinha, M., Ansell-Schultz, A., Civitelli, L., Hildesjö, C., Larsson, M., Lannfelt, L., et al. (2018). Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 136, 41–56. doi: 10.1007/s00401-018-1868-1
Schliebs, R., and Arendt, T. (2011). The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 221, 555–563. doi: 10.1016/j.bbr.2010.11.058
Schmidt, F., Boltze, J., Jäger, C., Hofmann, S., Willems, N., Seeger, J., et al. (2015). Detection and Quantification of β-Amyloid, Pyroglutamyl Aβ, and Tau in Aged Canines. J. Neuropathol. Exp. Neurol. 74, 912–923. doi: 10.1097/NEN.0000000000000230
Schultz, C., Dehghani, F., Hubbard, G. B., Thal, D. R., Struckhoff, G., Braak, E., et al. (2000a). Filamentous tau pathology in nerve cells, astrocytes, and oligodendrocytes of aged baboons. J. Neuropathol. Exp. Neurol. 59, 39–52. doi: 10.1093/jnen/59.1.39
Schultz, C., Hubbard, G. B., Rüb, U., Braak, E., and Braak, H. (2000b). Age-related progression of tau pathology in brains of baboons. Neurobiol. Aging 21, 905–912. doi: 10.1016/s0197-4580(00)00176-7
Schütt, T., Helboe, L., Pedersen, L. Ø., Waldemar, G., Berendt, M., and Pedersen, J. T. (2016). Dogs with cognitive dysfunction as a spontaneous model for early Alzheimer’s disease: a translational study of neuropathological and inflammatory markers. J. Alzheimers Dis. 52, 433–449. doi: 10.3233/JAD-151085
Schütt, T., Toft, N., and Berendt, M. (2015). Cognitive Function, Progression of Age-related Behavioral Changes, Biomarkers, and Survival in Dogs More Than 8 Years Old. J. Vet. Intern. Med. 29, 1569–1577. doi: 10.1111/jvim.13633
Selkoe, D. J. (1997). Alzheimer’s disease: genotypes, phenotypes, and treatments. Science 275, 630–631. doi: 10.1126/science.275.5300.630
Sellal, F., Wallon, D., Martinez-Almoyna, L., Marelli, C., Dhar, A., Oesterlé, H., et al. (2017). APP Mutations in Cerebral Amyloid Angiopathy with or without Cortical Calcifications: Report of Three Families and a Literature Review. J. Alzheimers Dis. 56, 37–46. doi: 10.3233/JAD-160709
Serizawa, S., Chambers, J. K., and Une, Y. (2012). Beta amyloid deposition and neurofibrillary tangles spontaneously occur in the brains of captive cheetahs (Acinonyx jubatus). Vet. Pathol. 49, 304–312. doi: 10.1177/0300985811410719
Shabir, O., Berwick, J., and Francis, S. E. (2018). Neurovascular dysfunction in vascular dementia, Alzheimer’s and atherosclerosis. BMC Neurosci. 19:62. doi: 10.1186/s12868-018-0465-5
Siwak, C. T., Gruet, P., Woehrlé, F., Muggenburg, B. A., Murphey, H. L., and Milgram, N. W. (2000). Comparison of the effects of adrafinil, propentofylline, and nicergoline on behavior in aged dogs. Am. J. Vet. Res. 61, 1410–1414. doi: 10.2460/ajvr.2000.61.1410
Siwak, C. T., Tapp, P. D., Head, E., Zicker, S. C., Murphey, H. L., Muggenburg, B. A., et al. (2005). Chronic antioxidant and mitochondrial cofactor administration improves discrimination learning in aged but not young dogs. Prog. Neuropsychopharmacol. Biol. Psychiatry 29, 461–469. doi: 10.1016/j.pnpbp.2004.12.011
Siwak-Tapp, C. T., Head, E., Muggenburg, B. A., Milgram, N. W., and Cotman, C. W. (2008). Region specific neuron loss in the aged canine hippocampus is reduced by enrichment. Neurobiol. Aging 29, 39–50. doi: 10.1016/j.neurobiolaging.2006.09.018
Smolek, T., Madari, A., Farbakova, J., Kandrac, O., Jadhav, S., Cente, M., et al. (2016). Tau hyperphosphorylation in synaptosomes and neuroinflammation are associated with canine cognitive impairment. J. Comp. Neurol. 524, 874–895. doi: 10.1002/cne.23877
Snigdha, S., Astarita, G., Piomelli, D., and Cotman, C. W. (2012). Effects of diet and behavioral enrichment on free fatty acids in the aged canine brain. Neuroscience 202, 326–333. doi: 10.1016/j.neuroscience.2011.12.002
Snigdha, S., Berchtold, N., Astarita, G., Saing, T., Piomelli, D., and Cotman, C. W. (2011). Dietary and behavioral interventions protect against age related activation of caspase cascades in the canine brain. PLoS One 6:e24652. doi: 10.1371/journal.pone.0024652
Studzinski, C. M., Araujo, J. A., and Milgram, N. W. (2005). The canine model of human cognitive aging and dementia: pharmacological validity of the model for assessment of human cognitive-enhancing drugs. Prog. Neuropsychopharmacol. Biol. Psychiatry 29, 489–498. doi: 10.1016/j.pnpbp.2004.12.014
Studzinski, C. M., Christie, L.-A., Araujo, J. A., Burnham, W. M., Head, E., Cotman, C. W., et al. (2006). Visuospatial function in the beagle dog: an early marker of cognitive decline in a model of human aging and dementia. Neurobiol. Learn. Mem. 86, 197–204. doi: 10.1016/j.nlm.2006.02.005
Tapp, P. D., Siwak, C. T., Estrada, J., Head, E., Muggenburg, B. A., Cotman, C. W., et al. (2003). Size and reversal learning in the beagle dog as a measure of executive function and inhibitory control in aging. Learn. Mem. 10, 64–73. doi: 10.1101/lm.54403
Tapp, P. D., Siwak, C. T., Gao, F. Q., Chiou, J.-Y., Black, S. E., Head, E., et al. (2004). Frontal lobe volume, function, and beta-amyloid pathology in a canine model of aging. J. Neurosci. 24, 8205–8213. doi: 10.1523/JNEUROSCI.1339-04.2004
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., et al. (1991). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. doi: 10.1002/ana.410300410
Toepper, M. (2017). Dissociating Normal Aging from Alzheimer’s Disease: A View from Cognitive Neuroscience. J. Alzheimers Dis. 57, 331–352. doi: 10.3233/JAD-161099
Uchida, K., Miyauchi, Y., Nakayama, H., and Goto, N. (1990). Amyloid angiopathy with cerebral hemorrhage and senile plaque in aged dogs. Nippon Juigaku Zasshi 52, 605–611. doi: 10.1292/jvms1939.52.605
Uchida, K., Nakayama, H., Tateyama, S., and Goto, N. (1992). Immunohistochemical analysis of constituents of senile plaques and cerebro-vascular amyloid in aged dogs. J. Vet. Med. Sci. 54, 1023–1029. doi: 10.1292/jvms.54.1023
Villarreal, S., Zhao, F., Hyde, L. A., Holder, D., Forest, T., Sondey, M., et al. (2017). Chronic Verubecestat Treatment Suppresses Amyloid Accumulation in Advanced Aged Tg2576-AβPPswe Mice Without Inducing Microhemorrhage. J. Alzheimers Dis. 59, 1393–1413. doi: 10.3233/JAD-170056
Vite, C. H., and Head, E. (2014). Aging in the canine and feline brain. Vet. Clin. North Am. Small Anim. Pract. 44, 1113–1129. doi: 10.1016/j.cvsm.2014.07.008
Walker, L. C., and Jucker, M. (2017). The Exceptional Vulnerability of Humans to Alzheimer’s Disease. Trends Mol. Med. 23, 534–545. doi: 10.1016/j.molmed.2017.04.001
West, M. J., Kawas, C. H., Martin, L. J., and Troncoso, J. C. (2000). The CA1 region of the human hippocampus is a hot spot in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 908, 255–259. doi: 10.1111/j.1749-6632.2000.tb06652.x
Whitehouse, P. J., Price, D. L., Clark, A. W., Coyle, J. T., and DeLong, M. R. (1981). Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. 10, 122–126. doi: 10.1002/ana.410100203
Winblad, B., Amouyel, P., Andrieu, S., Ballard, C., Brayne, C., Brodaty, H., et al. (2016). Defeating Alzheimer’s disease and other dementias: a priority for European science and society. Lancet Neurol. 15, 455–532. doi: 10.1016/S1474-4422(16)00062-4
Winblad, B., Giacobini, E., Frölich, L., Friedhoff, L. T., Bruinsma, G., Becker, R. E., et al. (2010). Phenserine Efficacy in Alzheimer’s disease. J. Alzheimers Dis. 22, 1201–1208. doi: 10.3233/JAD-2010-101311
Wisniewski, T., and Drummond, E. (2016). Developing therapeutic vaccines against Alzheimer’s disease. Expert Rev. Vaccines 15, 401–415. doi: 10.1586/14760584.2016.1121815
Wolfe, C. M., Fitz, N. F., Nam, K. N., Lefterov, I., and Koldamova, R. (2018). The Role of APOE and TREM2 in Alzheimer’s Disease-Current Understanding and Perspectives. Int. J. Mol. Sci. 20:E81. doi: 10.3390/ijms20010081
Yasojima, K., McGeer, E. G., and McGeer, P. L. (2001). Relationship between beta amyloid peptide generating molecules and neprilysin in Alzheimer disease and normal brain. Brain Res. 919, 115–121. doi: 10.1016/s0006-8993(01)03008-6
Youssef, S. A., Capucchio, M. T., Rofina, J. E., Chambers, J. K., Uchida, K., Nakayama, H., et al. (2016). Pathology of the aging brain in domestic and laboratory animals, and animal models of human neurodegenerative diseases. Vet. Pathol. 53, 327–348. doi: 10.1177/0300985815623997
Yu, C.-H., Song, G.-S., Yhee, J.-Y., Kim, J.-H., Im, K.-S., Nho, W.-G., et al. (2011). Histopathological and immunohistochemical comparison of the brain of human patients with Alzheimer’s disease and the brain of aged dogs with cognitive dysfunction. J. Comp. Pathol. 145, 45–58. doi: 10.1016/j.jcpa.2010.11.004
Yu, L., Boyle, P. A., Nag, S., Leurgans, S., Buchman, A. S., Wilson, R. S., et al. (2015). APOE and cerebral amyloid angiopathy in community dwelling older persons. Neurobiol. Aging 36, 2946–2953. doi: 10.1016/j.neurobiolaging.2015.08.008
Keywords: brain, neurodegeneration, canine cognitive dysfunction, Alzheimer’s disease, amyloid beta, TAU, animal model, treatment
Citation: Prpar Mihevc S and Majdič G (2019) Canine Cognitive Dysfunction and Alzheimer’s Disease – Two Facets of the Same Disease? Front. Neurosci. 13:604. doi: 10.3389/fnins.2019.00604
Received: 24 March 2019; Accepted: 27 May 2019;
Published: 12 June 2019.
Edited by:
Marietta Zille, Universität zu Lübeck, GermanyReviewed by:
Ling Li, University of Minnesota Twin Cities, United StatesWolfgang Härtig, Leipzig University, Germany
Björn Nitzsche, Leipzig University, Germany
Copyright © 2019 Prpar Mihevc and Majdič. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gregor Majdič, Z3JlZ29yLm1hamRpY0B2Zi51bmktbGouc2k=