 Audrey M. G. Ragagnin
Audrey M. G. Ragagnin Sina Shadfar1
Sina Shadfar1 Marta Vidal
Marta Vidal Julie D. Atkin
Julie D. Atkin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci., 27 June 2019
Sec. Neurodegeneration
Volume 13 - 2019 | https://doi.org/10.3389/fnins.2019.00532
This article is part of the Research TopicFrontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis: Genetics, Clinical and Pathological Features, and Disease MechanismsView all 13 articles
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the death of both upper and lower motor neurons (MNs) in the brain, brainstem and spinal cord. The neurodegenerative mechanisms leading to MN loss in ALS are not fully understood. Importantly, the reasons why MNs are specifically targeted in this disorder are unclear, when the proteins associated genetically or pathologically with ALS are expressed ubiquitously. Furthermore, MNs themselves are not affected equally; specific MNs subpopulations are more susceptible than others in both animal models and human patients. Corticospinal MNs and lower somatic MNs, which innervate voluntary muscles, degenerate more readily than specific subgroups of lower MNs, which remain resistant to degeneration, reflecting the clinical manifestations of ALS. In this review, we discuss the possible factors intrinsic to MNs that render them uniquely susceptible to neurodegeneration in ALS. We also speculate why some MN subpopulations are more vulnerable than others, focusing on both their molecular and physiological properties. Finally, we review the anatomical network and neuronal microenvironment as determinants of MN subtype vulnerability and hence the progression of ALS.
Amyotrophic lateral sclerosis (ALS) is a late-onset, progressive and fatal neurodegenerative disease which primarily affects motor neurons (MNs) of the motor cortex of the brain, brainstem motor nuclei and anterior horn of the spinal cord (Kiernan et al., 2011; Renton et al., 2014; Al Sultan et al., 2016; Taylor et al., 2016). ALS commonly begins in late-adulthood, when patients first experience focal symptoms, such as weakness in the limb or bulbar muscles, as well as widespread fasciculations. The disease then usually progresses in an organized way to adjacent areas of the central nervous system (CNS), and consequently symptoms appear in other regions of the body. Several clinical subsets of ALS can be distinguished by the anatomical location first affected (Renton et al., 2014; Taylor et al., 2016). This includes bulbar onset, where symptoms first appear in the muscles controlling speech, mastication and swallowing; and limb onset, where symptoms present initially in the upper (arm or hand) or lower limbs (leg or foot). Bulbar onset patients face a much worse prognosis than those with spinal onset ALS, where the average survival time following diagnosis is less than 2 years. However, in patients with the much rarer respiratory onset form (3–5%), the prognosis is even worse as the survival time following diagnosis is only 1.4 years (Swinnen and Robberecht, 2014). At disease end stage, only support and palliation are available, and patients usually die from respiratory failure, typically 3–5 years after diagnosis (Taylor et al., 2016). There are currently few effective treatments. Hence there is an urgent need to understand the underlying causes and risk factors for ALS to discover new therapeutic targets.
Neurons have complex and extended morphologies compared to other cell types, and within the CNS, neurons can vary greatly in their properties. MNs are unique cells amongst neurons because they are large, even by neuronal standards, with very long axons, up to 1 m in length in an adult human. MNs can be distinguished into two main categories according to their location in the CNS: upper MNs (UMNs) located in the cortex, and lower MNs (LMNs) located in the brainstem and spinal cord. The spinal MNs comprise both visceral MNs of the thoracic and sacral regions, which control autonomic functions, and somatic MNs, which regulate the contraction of skeletal muscles and thus control movement. The diversity of MNs reflects the variety of targets they innervate, including a wide range of muscle fiber types. UMNs and LMNs differ in the location of their cell bodies, the neurotransmitters released, their targeting and symptoms resulting from their injury.
It is unknown why MNs are specifically targeted in ALS and remarkably, MNs are not equally affected (Rochat et al., 2016; Nijssen et al., 2017). Whilst both UMNs and LMNs are involved, some LMN subtypes are relatively resistant to neurodegeneration. Spinal cord and hypoglossal MNs are amongst the first to degenerate, hence the ability to speak, breath and move is lost early in disease course. As ALS progresses, specific MN subtypes then preferentially deteriorate. However, some MNs are spared until disease end stage, such as oculomotor neurons and Onuf’s nuclei MNs, and as a result, patients retain normal visual, sexual and bladder function throughout the disease course. The resistant MNs differ significantly from the vulnerable MNs anatomically and functionally, and they possess distinct transcriptomes, metabolic and developmental profiles. Surprisingly, there are also differences in vulnerability amongst spinal MNs, because those that are part of the faster motor units degenerate before those in the slower motor units (Frey et al., 2000; Pun et al., 2006; Hegedus et al., 2007; Hadzipasic et al., 2014; Sharma et al., 2016; Spiller et al., 2016a), thus adding further complexity to the question of MN vulnerability.
ALS shares clinical and pathological features with frontotemporal dementia (FTD), a type of dementia that involves impaired judgment and executive skills. In FTD, the loss of cortical MNs is accompanied by loss of neurons in the frontal and temporal cortices, which correlates clinically with the symptoms of FTD (Neumann et al., 2006; Burrell et al., 2016). The relationship between ALS and FTD has been confirmed through genetic studies, and these two conditions are now considered to be at opposite ends of the same disease continuum (Taylor et al., 2016; Shahheydari et al., 2017). Hence, while ALS was historically judged as a disorder affecting the motor system only, it is now recognized that non-motor features are present (Fang et al., 2017). A wealth of evidence also demonstrates that ALS is a heterogeneous disorder. The clinical symptoms, including the proportion of UMN and LMN signs, age of onset, disease duration, and association with other conditions, are major features contributing to its highly variable phenotypes. As well as the development of FTD (Strong and Yang, 2011), ALS can also involve cognitive impairment in up to 50% of patients (Tsermentseli et al., 2012), the autonomic nervous system (Piccione et al., 2015), supranuclear gaze systems (van der Graaff et al., 2009; Donaghy et al., 2011), and extrapyramidal motor signs (Pradat et al., 2002). Sensory, olfactory and visual dysfunction have also been described in some patients (Bede et al., 2016). In addition, there are also other conditions affecting MNs that share similarities, but also striking differences, to ALS. In particular, primary lateral sclerosis (PLS) affects UMNs but it progresses much slowly than ALS. It also has a significantly lower mortality rate (Tartaglia et al., 2007), consistent with the relative resistant of LMNs in ALS.
One of the main pathological characteristics of ALS is the presence of insoluble protein inclusions in the soma of MNs. TAR DNA binding protein-43 (TDP-43) is the major component of these inclusions (Arai et al., 2006; Neumann et al., 2006) in almost all (∼97%) ALS patients and ∼50% FTD patients (Arai et al., 2006; Neumann et al., 2006; Mackenzie et al., 2007; Scotter et al., 2015; Le et al., 2016). Loss of TDP-43 from the nucleus is evident in MNs from ALS/FTD patient tissues, concomitant with the formation of TDP-43 inclusions in the cytoplasm of both MNs and glia. Neuropathological studies have also revealed that the clinical course of ALS reflects the presence of TDP-43 pathology, from its deposition at an initial site of onset, to its spread to contiguous regions of the CNS (Brettschneider et al., 2013). Mutations in TDP-43 are also present in 5% of familial forms of ALS (Sreedharan et al., 2008). In the genetic types of ALS, it remains unclear why MNs are specifically affected when the mutant proteins are ubiquitously expressed. Males are affected more by ALS than females, and ethnic populations show differences in the incidence rates of ALS, further highlighting the contribution of genetics to ALS.
Whilst our understanding of the etiology of ALS has increased significantly in recent years, major gaps in our knowledge remain. In this review, we address several unanswered questions regarding the unique susceptibility of specific types of MNs in ALS: Why does neurodegeneration spread throughout specific neural networks? How can ubiquitously expressed genes be selectively toxic to MNs? Why are some MN subtypes more vulnerable to degeneration than others? We also discuss the role of the neuronal network and the specific cellular microenvironment in driving cell-to-cell disease progression, plus the importance of genetics in influencing susceptibility of specific neuronal subpopulations. Finally, we discuss the role of aging as a potential risk factor for the susceptibility of specific MN subtypes. A thorough comprehension of why specific cell types degenerate is imperative to our understanding of ALS because it provides important clues as to what initiates neurodegeneration, and how this knowledge may be harnessed therapeutically.
In the CNS, the motor cortex, basal ganglia, cerebellum, and parts of the brainstem, are directly involved in the planning and initiation of movement. In contrast, the precise timing and pattern of movement is generated by MNs located in the spinal cord (Figure 1; Kiehn, 2016). The corticospinal (anterior and lateral) tract is the largest descending tract in humans. The lateral corticospinal tract originates in the primary motor cortex, which lies in the precentral gyrus and sends fibers to muscles in the extremity. This is via contralateral cortical innervation, so that the left motor cortex controls the right extremities and vice versa, to control the voluntary movement of contralateral limbs (Javed and Lui, 2018). MNs outputs are not confined to the peripheral muscles however, but also include excitatory terminals to a group of interneurons, Renshaw cells, and also to other MNs.
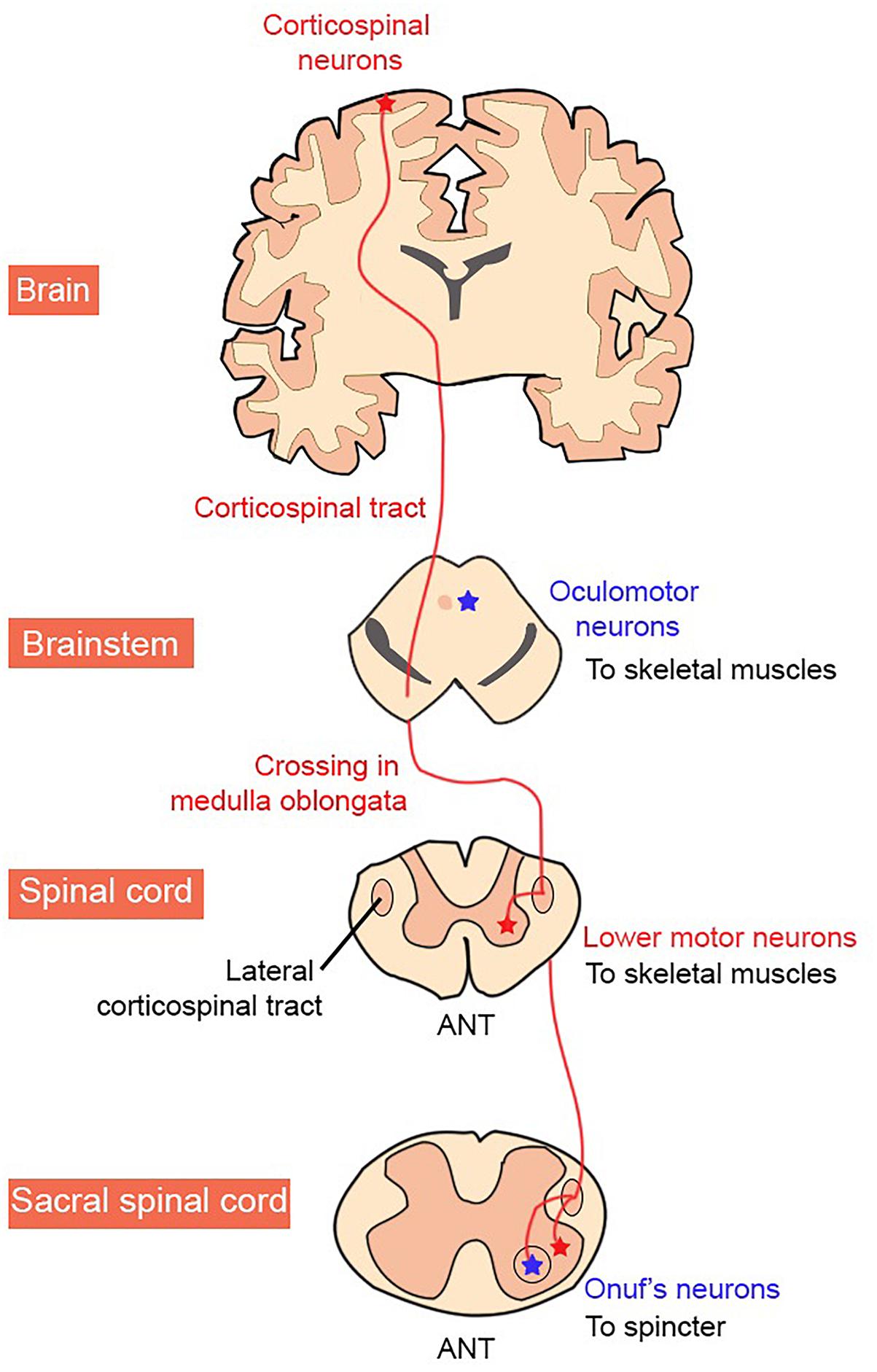
Figure 1. Organization of the human corticospinal tract. MN groups vulnerable and resistant to degeneration in ALS are shown in red and blue, respectively.
Glutamate (cortex, spinal cord) and acetylcholine (spinal cord) modulate excitatory input within neurons, whereas GABA and glycine facilitate inhibitory neurotransmission (Ramírez-Jarquín et al., 2014). At the neuromuscular junction (NMJ), only acetylcholine acts at the synapse but interestingly, synaptic transmission between MNs in the spinal cord involves both acetylcholine and glutamate (Bhumbra and Beato, 2018). Renshaw cells are excited through both acetylcholine and glutamate receptors and spinal MNs co-release glutamate to excite Renshaw cells and other MNs, but not to excite muscles (Nishimaru et al., 2005; Bories et al., 2007; Bhumbra and Beato, 2018). Hence, different synaptic transmission systems are present at different postsynaptic targets of MNs (Bhumbra and Beato, 2018).
However, MNs are not homogeneous throughout the CNS because they exhibit distinct morphologies and patterns of connectivity, which underlie their different physiological functions. Hence, within a single region, MNs that perform closely related functions can be further subdivided, both anatomically and physiologically. The identities of specific MN subtypes and their target projections are controlled by selective cell-type expression of transcription factors, notably members of the Hox, LIM, Nkx6, and ETS families (Stifani, 2014). This provides the fundamental mechanism for spinal MN diversification and connectivity to specific peripheral muscle targets. Thus, to generate movement, MNs integrate information from sensory structures and transform it into precise temporal and magnitudal activation of muscles.
A MN located in the spinal cord innervates up to several hundred fibers within one muscle, which together form the motor unit. Trains of action potentials within the axon cause the release of acetylcholine at the NMJ, which activates nicotinic receptors on the muscle fibers the MN innervates. This initiates a cascade of signaling events in the muscle fiber that leads to its contraction. A motor pool consists of all the individual MNs that innervate a single muscle. A muscle unit (one muscle and its motor pool) is composed of three different types of functional motor units consisting of alpha (α), beta (β), and gamma (γ) MNs, which are classified according to the contractile activity of the muscle fiber innervated. We will now discuss in more detail the anatomy of those structures involved in movement.
In the spinal cord, MNs are organized into columns (Table 1) based on the location of their target muscle [reviewed in Matise and Sharma (2013) and Stifani (2014)]. Within each column, the MNs innervating each muscle are clustered into motor pools, each containing of 20–300 cells depending on the muscle (Bryan et al., 1972; McHanwell and Biscoe, 1981). α-MNs located in the spinal cord are archetypal MNs that innervate extrafusal muscle fibers, thus creating force to move the skeleton (Table 2). In contrast, γ-MNs innervate intrafusal fibers, which modulate the sensitivity of muscle spindles to stretch (Table 2) (Hunt and Kuffler, 1951; Kuffler et al., 1951; Kanning et al., 2010). β-MNs are not as well characterized as α-MNs but they innervate both intrafusal and extrafusal muscle fibers (Bessou et al., 1965). Both α and γ-MNs have large dendritic trees but γ-MNs have fewer large dendrites than α-MNs (7–11) and they also branch less (Westbury, 1982). The somas of γ-MNs are smaller than those of α-MNs and they also possess thinner axons, which reflects their slower conduction velocity (<55 m/s in γ-MN vs. ∼70–90 m/s in α-MNs in cats) (Table 2) (Westbury, 1982). γ-MNs receive only indirect sensory inputs. Therefore, γ-MNs do not directly participate in spinal reflexes (Eccles et al., 1960; Stifani, 2014), but they contribute to the modulation of muscle contraction instead.
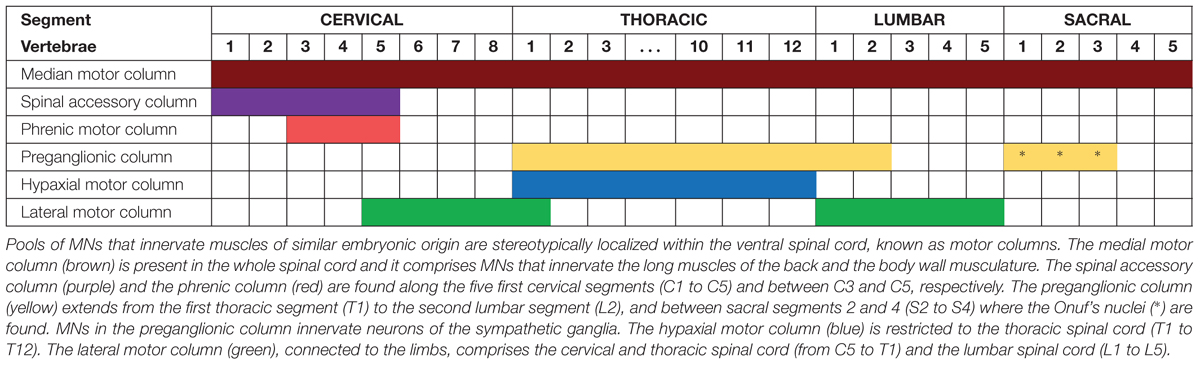
Table 1. Segmental organization of spinal cord columns.
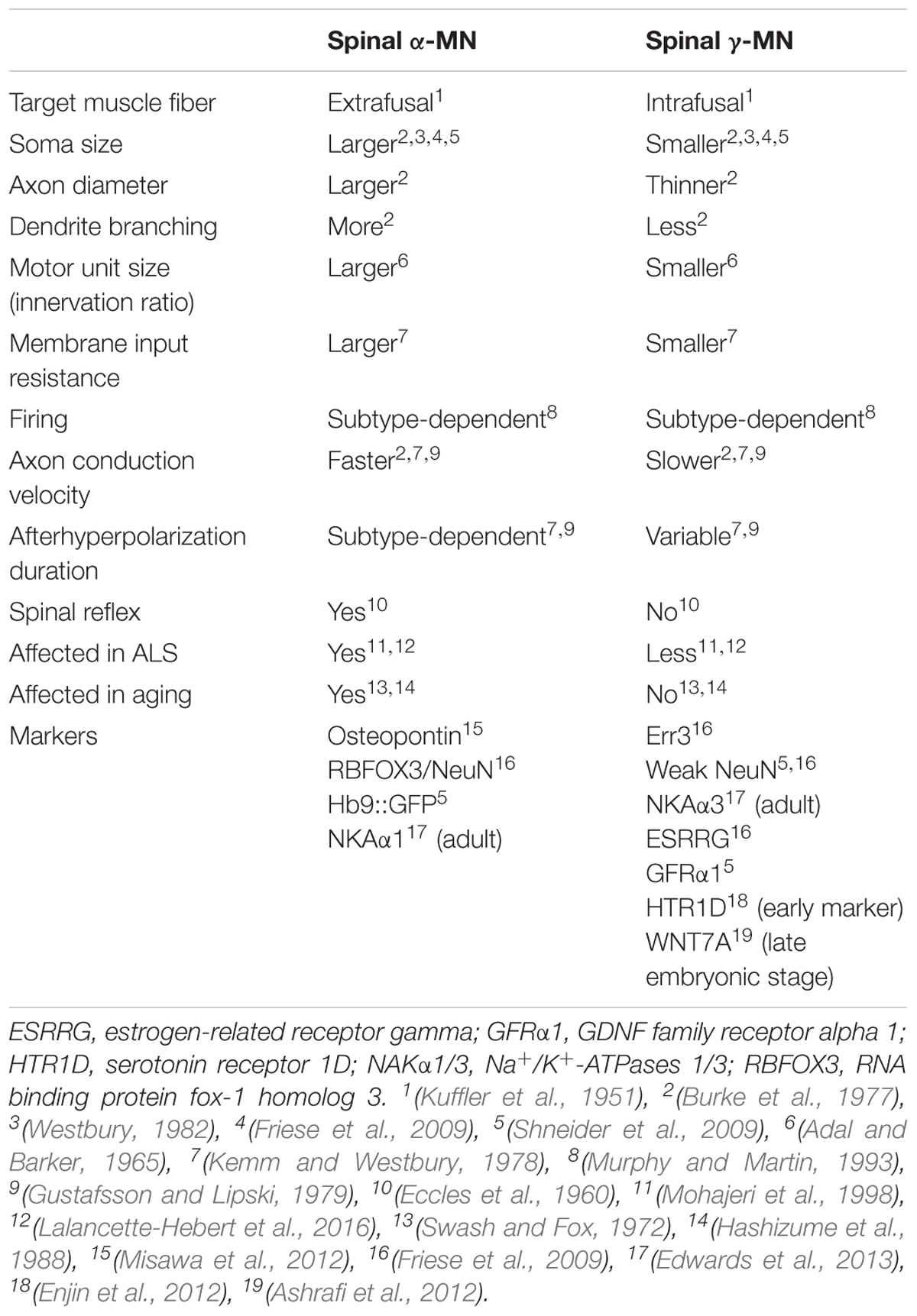
Table 2. Comparison of α- and γ-spinal motor neurons.
A distinct group of MNs in the sacral spinal cord termed ‘Onuf’s’ neurons, innervate the striated muscles of the external urethra, external anal sphincter via the pudental nerve, and the ischiocavernosus and bulbocavernosus muscles in males (Sato et al., 1978; Nagashima et al., 1979; Kuzuhara et al., 1980; Roppolo et al., 1985). These MNs are histologically similar to limb α-MNs (Mannen et al., 1977) and they are located anteromedial to the anterolateral nucleus and extend between the distal part of the S1 segment and the proximal part of S3.
α-motor units can be subdivided according to their contractile properties, into fast-twitch (F) and slow-twitch (S) fatigue-resistant types (Table 3) (Burke et al., 1973). In addition, fast-twitch α-motor units can be further categorized into fast-twitch fatigable [FF] and fast-twitch fatigue-resistant [FR] types, based on the length of time they sustain contraction. The basis of this classification is the duration of the twitch contraction time (Burke et al., 1973). F- and S-MNs also exhibit different afterhyperpolarization duration (AHP) properties. AHP is the phenomenon by which the membrane potential undershoots the resting potential following an action potential. S-MNs have a longer AHP than F-MNs, indicating that S-MNs have a longer “waiting period” before they can be stimulated by an action potential. Thus, they cannot fire at the same frequency as F-MNs (Eccles et al., 1957), so the larger FF-MNs take longer to reach an activation threshold. Similarly, other electrical properties differ between S- and F-MNs (Table 3), including their input resistance (a measure of resistance over the plasma membrane) and rheobase (a measure of the current needed to generate an action potential). S-MNs have a higher input resistance than F-MNs, underlying Hennenman’s size principle which postulates that S-motor units are the first to be recruited during movement, followed by FR and then FF units (Henneman, 1957; Mendell, 2005). Hence, a slow movement generating a small force will only recruit S-MNs, whereas a quick and strong movement will also recruit F-MNs, as well as S-MNs.
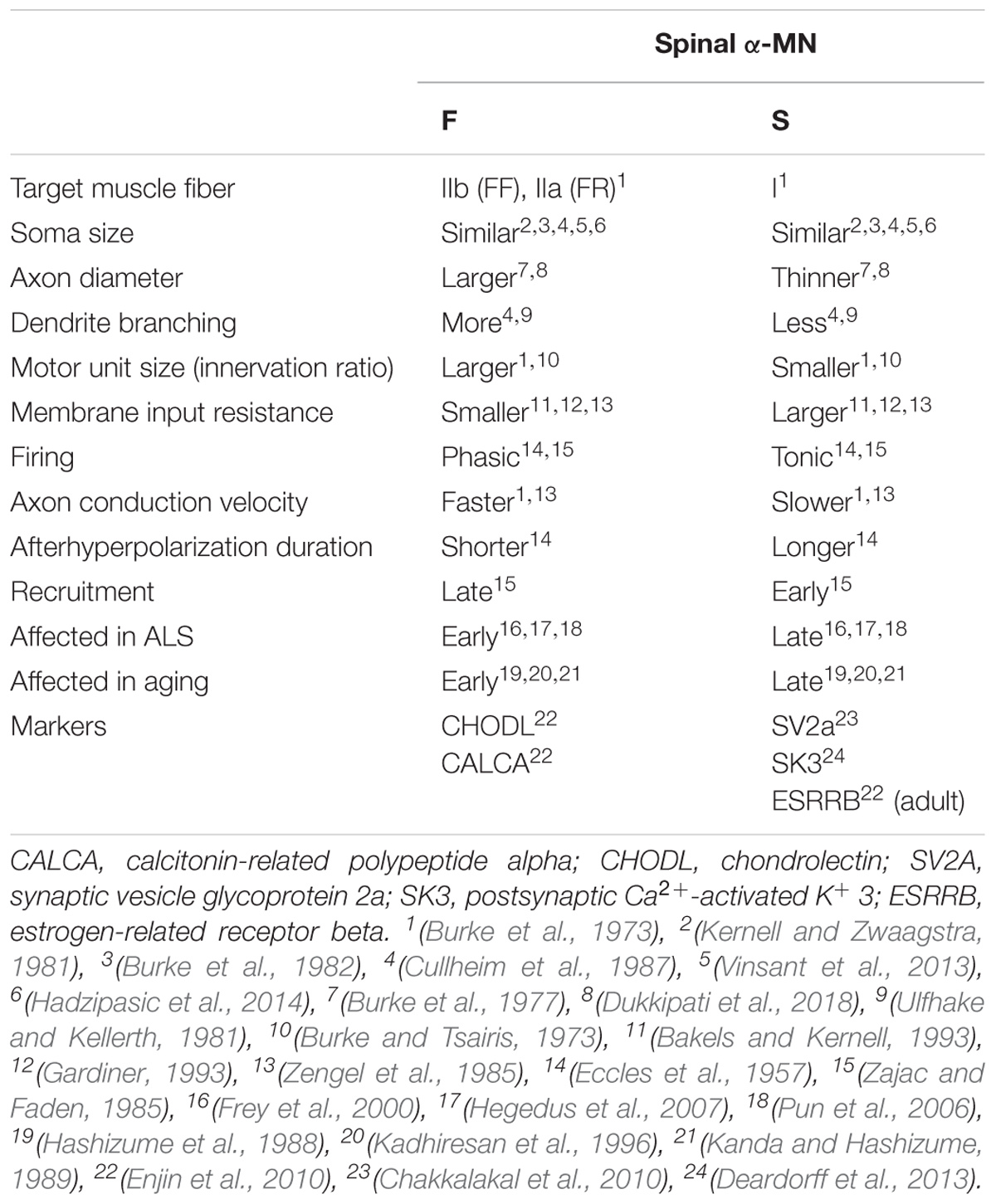
Table 3. Comparison of fast (FF, fast-fatigable; FR, fast-resistant) and slow (S) spinal α-motor neurons.
In addition, at least eleven types of interneurons are involved in the control of movement, as part of central pattern generators in the spinal cord. Interneurons arise from five progenitor cells and, according to the expression of distinct transcription factors, they mature into different lineages. This includes excitatory V2a, V3, MN and Hb9 neurons and inhibitory V0C/G,V0D, V0V, V1, V2b, Ia and Renshaw cells (belonging to the V1 interneuron subclass), which display specific locations and projections within the spinal cord (Ramírez-Jarquín et al., 2014).
Cranial nerve nuclei are populations of neurons in the brainstem that are associated with one or more cranial nerves. They provide afferent and efferent (sensory, motor, and autonomic) innervation to the structures of the head and neck (Sonne and Lopez-Ojeda, 2018). The more posterior and lateral nuclei tend to be sensory, and the more anterior nuclei are usually motor nuclei. Trigeminal MNs innervate the muscles of mastication, whereas facial MNs supply the superficial muscles of the face, and ambiguous MNs supply the muscles of the soft palate, pharynx, and larynx. The oculomotor (III), trochlear (IV) and abducens (VI) nuclei are somatic efferents innervating the extraocular muscles within the orbit. The oculomotor nucleus contains MNs that innervate four of the six extraocular muscles (superior, medial and inferior recti, inferior oblique), plus the levator palpebrae superioris muscle. These muscles display a unique composition of six fiber types, distinct from other skeletal muscles that possess marked fatigue resistance (Table 4). Oculomotor units are amongst the smallest of the motor units, in contrast to skeletal muscle motor units that have higher maximum MN discharge rates. Furthermore, α-MNs in oculomotor units have higher resting membrane potentials (∼61 mV) than spinal cord α-MNs (∼70 mV), and they also discharge at higher frequencies (∼100 Hz during steady state and ∼600 Hz during saccadic eye movements, compared to ∼100 Hz for spinal cord α-MNs) (Table 4) (Robinson, 1970; Fuchs et al., 1988; Torres-Torrelo et al., 2012). Oculomotor neurons are almost continually active at high frequencies when maintaining eye position (Fuchs et al., 1988; De La Cruz et al., 1989), and this level of activity places high metabolic demand on these cells (Robinson, 1970; Porter and Baker, 1996; Brockington et al., 2013).
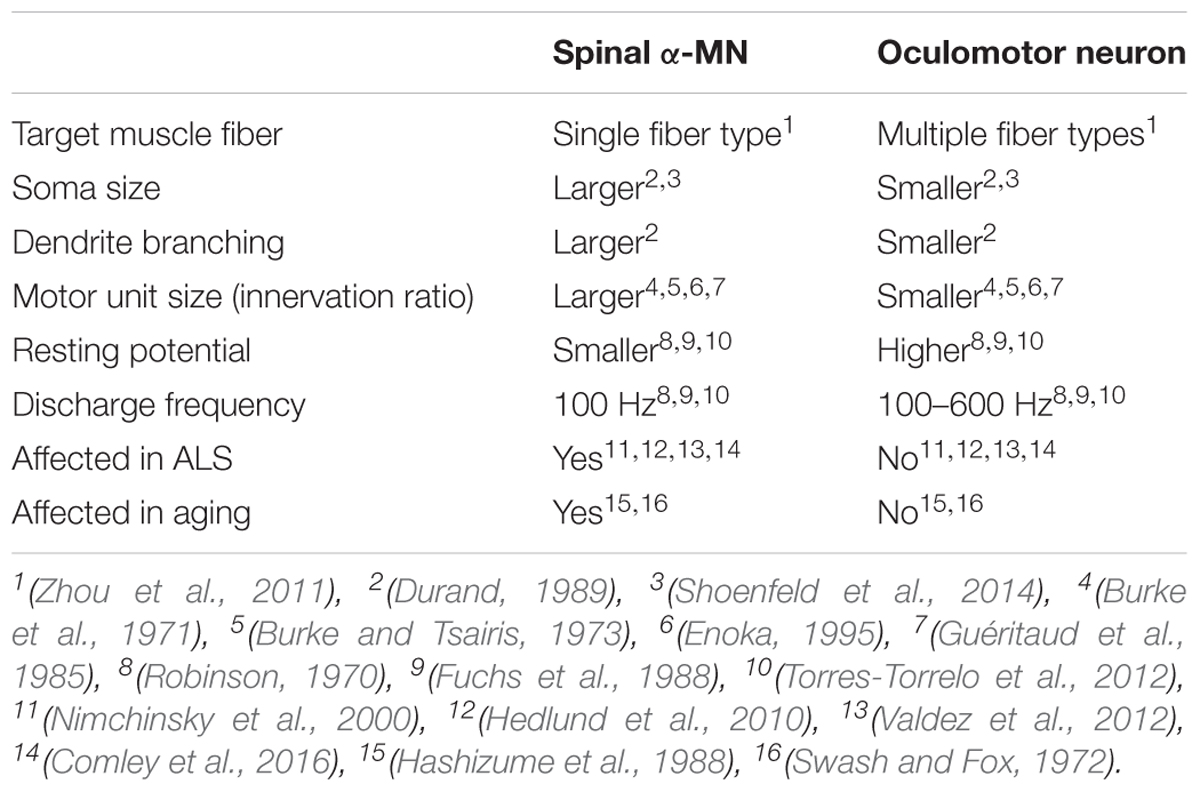
Table 4. Comparison of α-spinal motor neurons and oculomotor neurons.
The motor cortex is the region of the cerebral cortex responsible for mediating voluntary movements. In rodents, the primary cortex (M1) is large and comprises almost all of the frontal cortex (Gioanni and Lamarche, 1985; Neafsey et al., 1986; Brecht et al., 2004; Yu et al., 2008; Hira et al., 2013; Paxinos, 2014), whereas in primates, the frontal cortex is compartmentalized into specialized premotor subfields and M1 is relatively small in comparison (Ferrier, 1875; Leyton and Sherrington, 1917; Asanuma and Rosén, 1972; Dickey et al., 2013; Riehle et al., 2013; Young et al., 2013; Ebbesen and Brecht, 2017). M1 plays a central role in controlling movement. This involves specialized UMNs located in layer V of this region (Broadman area 4), the giant Betz cells or corticospinal MNs. These MNs are the cortical components of the MN circuit that initiates and modulates precise voluntary movement, through long-range projections to the spinal cord. Approximately ∼30–50% of corticospinal projections originate from M1 MNs and they begin modulating their firing rate several hundred ms before movement of the limb is initiated (Georgopoulos et al., 1982; Porter and Lemon, 1993). In most mammals, the axons of cortical MNs terminate at spinal interneurons, but they also make direct connections to MNs (Lemon, 2008; Rathelot and Strick, 2009). This constitutes the final efferent pathway to the muscle to generate or suppress movement (Ramírez-Jarquín and Tapia, 2018).
Lesions to motor structures in humans and experimental animals lead to impairments in normal movement. In ALS, as MNs degenerate, the ability to control movement of the muscles is progressively lost. Specific MNs in the brain, brainstem and spinal cord are selectively targeted, and pathology appears first in these restricted MN populations. In fact, the name “Amyotrophic Lateral Sclerosis” reflects the strikingly selective degeneration of MNs in ALS. It is derived from a combination of three words; “Lateral” refers to the lateral spinal cord, given that corticospinal MNs are particularly vulnerable to degeneration; “Amyotrophic” is from the Greek “amyotrophia,” meaning lacking muscle nourishment; and “Sclerosis” (fibrosis) refers to gliosis of the crossed corticospinal tract in the dorsolateral quadrant of the spinal cord (Charcot, 1874; Frey et al., 2000; Pun et al., 2006). In the brain, UMNs in the primary cortex are also amongst the first to degenerate in ALS, and similarly, in the brainstem, the hypoglossal MNs that innervate the muscles of the tongue involved in swallowing and breathing, are also targeted early in disease course. In the brainstem, ALS can also affect trigeminal MNs, the facial MNs and ambiguous MNs. However, other MN subgroups within this region are relatively resistant to degeneration, including MNs of the oculomotor (III), trochlear (IV) and abducens (VI) nuclei, innervating the extraocular muscles (Mannen et al., 1977; Schrøder and Reske-Nielsen, 1984). Hence, eye movements remain relatively preserved throughout disease course (Kanning et al., 2010) and as a consequence, eye tracking devices are often used to aid communication in the later stages of ALS (Caligari et al., 2013). Whilst it has been reported that oculomotor neurons may be affected at disease end stage, this was recently attributed to dysfunction of the dorsolateral prefrontal cortex, the frontal eye field and the supplementary eye field, confirming the relative resistance of pure oculomotor functions in ALS (Shaunak et al., 1995; Proudfoot et al., 2015). Widespread loss of GABAergic interneurons has also been described in ALS, in both the cortex (Stephens et al., 2001; Maekawa et al., 2004) and the spinal cord (Stephens et al., 2006; Hossaini et al., 2011).
MRI studies of ALS patients has revealed that very specific neuronal networks are vulnerable to degeneration in ALS (Bede et al., 2016). However, whilst TDP-43 pathology is the signature pathological hallmark of almost all ALS cases, it can arise in areas of the CNS that are not particularly vulnerable to degeneration (Geser et al., 2008). Significant TDP-43 pathology is present in the substantia nigra and basal ganglia, which are not affected in ALS, as well as in the motor gyrus, midbrain and spinal cord. Curiously, pathological forms of TDP-43 are also detectable in the occipital lobe, amygdala, orbital gyrus and hippocampus (Geser et al., 2008). Hence, whilst major degeneration of corticobulbar, LMN, pyramidal and frontotemporal networks underlie the widespread clinical symptoms of ALS, it remains unclear how other circuits, such as the visual, sensory, autonomic and auditory systems, remain relatively protected in ALS. These unaffected networks, however, have not been well studied in ALS patients.
Most ALS cases occur without a clearly identified cause and are therefore referred to as sporadic ALS (SALS). In contrast, a positive family history is present in ∼10% of all patients (familial ALS; FALS) (van Blitterswijk et al., 2012; Nguyen et al., 2018) and these genetic mutations cause ALS in a mostly autosomal-dominant manner (Supplementary Table 1 and Figure 2). However, several recently discovered mutations have been described in patients diagnosed with SALS (Renton et al., 2014; Al Sultan et al., 2016; Taylor et al., 2016). The patterns of selective MN degeneration and vulnerability are similar between FALS and SALS (Comley et al., 2015), implying that shared molecular mechanisms exist between the two conditions.
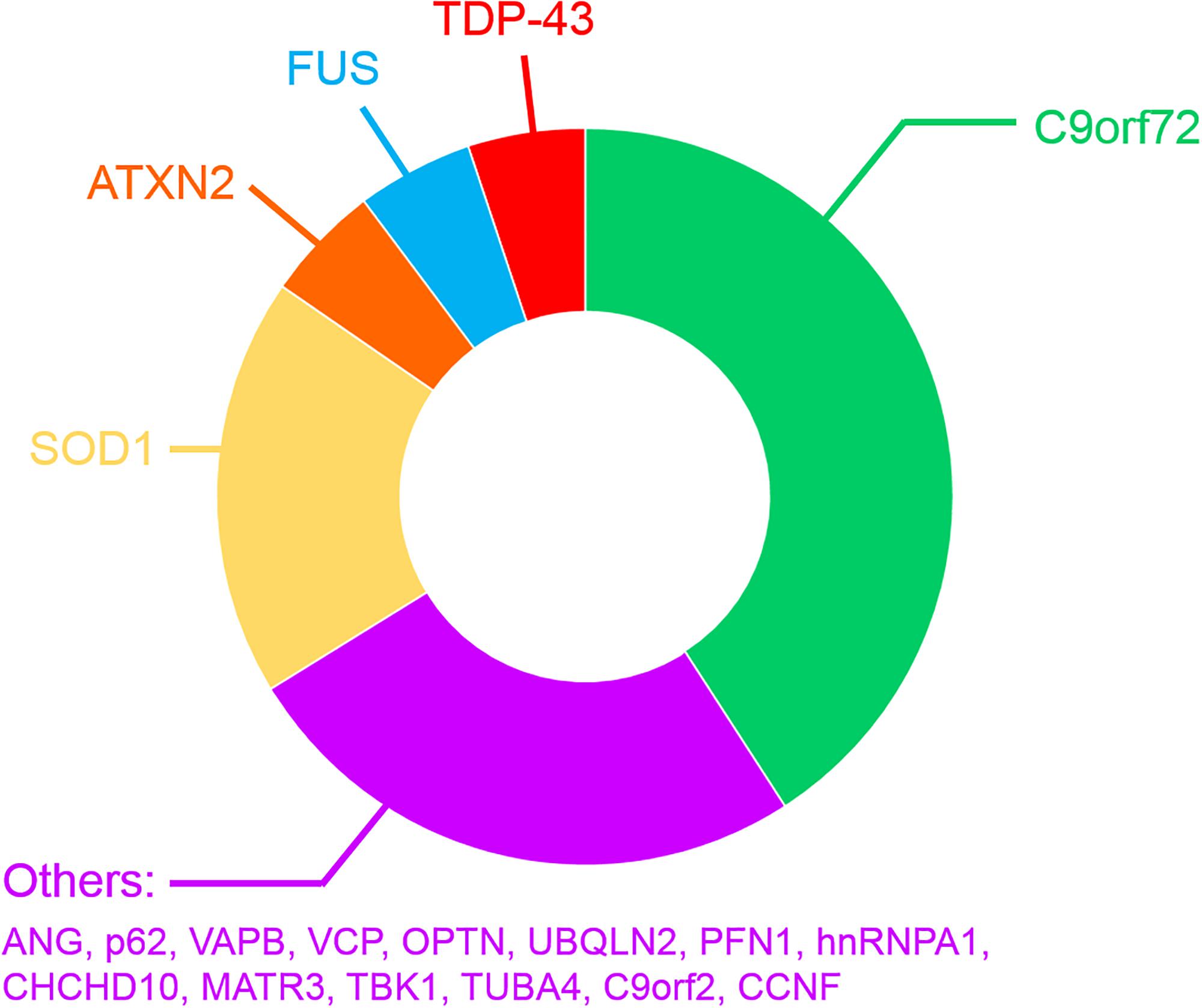
Figure 2. Frequency of mutated genes in FALS patients.
The first gene found to harbor mutations causing FALS encodes Cu/Zn superoxide dismutase (SOD1), an enzyme that detoxifies superoxide radicals (Rosen et al., 1993). Mutations in SOD1 account for 12–23.5% of FALS cases, representing 1–2.5% of all ALS, and 186 ALS mutations have now been described1. Since then, mutations in approximatively 26 genes have been identified (Supplementary Table 1 and Figure 2) using genome-wide or exome-wide association studies combined with segregation analysis. Hexanucleotide repeat expansions (GGGGCC) within the first intron of the chromosome 9 open reading frame 72 (C9orf72) gene are the most common cause of FALS and FTD (∼30–50% of FALS, ∼10% of SALS 25% of familial FTD and ∼5% of apparently sporadic ALS and FTD) (DeJesus-Hernandez et al., 2011b; Renton et al., 2011; Majounie et al., 2012; Devenney et al., 2014) (Supplementary Table 1 and Figure 2), in both Europe and North America (DeJesus-Hernandez et al., 2011b; Renton et al., 2011). However, this mutation is much rarer in Asian and Middle Eastern populations (Majounie et al., 2012; Woollacott and Mead, 2014). Healthy individuals possess ≤ 11 GGGGCC repeats in C9orf72 (Rutherford et al., 2012; Harms et al., 2013; van der Zee et al., 2013), whereas hundreds to thousands of repeats are present in ALS/FTD patients (Beck et al., 2013; Harms et al., 2013; van Blitterswijk et al., 2013; Suh et al., 2015). After C9orf72, mutations in SOD1 (20% of FALS), TARDPB encoding TDP-43 (5% of FALS, >50% of FTD) (Rutherford et al., 2008; Sreedharan et al., 2008; Borroni et al., 2010; Kirby et al., 2010), Fused in sarcoma encoding FUS (FUS, 5% of FALS) (Belzil et al., 2009; Blair et al., 2009; Chiò et al., 2009; Kwiatkowski et al., 2009; Neumann et al., 2009; Vance et al., 2009), and CCNF encoding cyclin F (0.6–3.3% of FALS-FTD) are more frequent than the remaining 20 genes mutated in the much rarer forms of FALS (Supplementary Table 1). The physiological functions and properties of the proteins encoded by these genes can be grouped according to their involvement in protein quality control, cytoskeletal dynamics, RNA homeostasis and the DNA damage response. However, it is possible that genetic inheritance could sometimes be missed, due to incomplete penetrance or an oligogenic mode of inheritance, whereby more than one mutated gene is necessary to fully present disease (Nguyen et al., 2018). Consistent with this notion, the frequency of ALS patients carrying two or more mutations in ALS-associated genes is in excess of what would be expected by chance (van Blitterswijk et al., 2012; Veldink, 2017; Zou et al., 2017; Nguyen et al., 2018).
TDP-43 is an ubiquitously expressed RNA-binding protein belonging to the heterogeneous nuclear ribonucleoprotein (hnRNP) family. Fifty three mutations in TARDBP have now been associated with FALS, located within all but one reside of the C-terminal domain of TDP-43 [Gitcho et al., 2008; Kabashi et al., 2008; Van Deerlin et al., 2008; http://alsod.iop.kcl.ac.uk/]. Pathological forms of TDP-43 – phosphorylated, fragmented, aggregated, ubiquitinated TDP-43 – were identified as the major component of MN inclusions (Neumann et al., 2006) in almost all ALS cases, including SALS (97%) (Arai et al., 2006; Neumann et al., 2006; Mackenzie et al., 2007; Scotter et al., 2015; Le et al., 2016). TDP-43 pathology is also observed in C9orf72 mutation cases in several brain regions, including the frontal, temporal and primary motor cortices, hippocampus, basal ganglia, amygdala, thalamus and midbrain (Murray et al., 2011; Hsiung et al., 2012; Mahoney et al., 2012; Irwin et al., 2013; Mackenzie et al., 2013; Balendra and Isaacs, 2018), highlighting an important role for TDP-43 in neurodegeneration in both SALS and FALS. Moreover, ALS and FTD cases bearing TDP-43 pathology are often referred to “TDP-43 proteinopathies” (Mackenzie et al., 2009). TDP-43 shares similar functional roles in RNA-binding, splicing and nucleocytosolic RNA transport as FUS. Fifty nine mutations in FUS have been identified in both SALS and FALS patients (Lattante et al., 2013; http://alsod.iop.kcl.ac.uk/) and FUS colocalises with TDP-43 in protein aggregates in MNs of a proportion of SALS and FALS patients (Kwiatkowski et al., 2009; Deng et al., 2010).
A wide range of cellular pathways have been implicated in ALS pathogenesis, as reviewed recently (Shin and Lee, 2013; Taylor et al., 2016; Balendra and Isaacs, 2018). These include altered RNA processing/metabolism, nucleolar dysfunction, RNA splicing transcriptional defects (Barmada, 2015; Fratta and Isaacs, 2018) and DNA damage (Konopka and Atkin, 2018; Penndorf et al., 2018). Proteostasis pathways have also been implicated, with impairments in autophagy and lysosomal function, the endoplasmic reticulum (ER), mitochondrial and the ubiquitin–proteasome systems described (Maharjan and Saxena, 2016; Ruegsegger and Saxena, 2016). Furthermore, several modes of vesicular trafficking are impaired in ALS, including nucleocytoplasmic (Kim and Taylor, 2017), ER-Golgi (Soo et al., 2015), and axonal forms of transport (De Vos and Hafezparast, 2017). In addition, defects in neuronal-specific processes, including hyper-excitability and hypo-excitability, glutamate excitotoxicity, and neuronal branching defects, have also been described in ALS (Fogarty, 2018).
Over the last 20 years, several transgenic mouse strains expressing human mutant SOD1 have been generated. These mice have been used to either examine disease mechanisms or trial potential therapeutic strategies for ALS, although the latter has led to questionable success (Perrin, 2014) (Tables 5, 6). The transgenic line harboring the Gly93 → Ala substitution (SOD1G93A) has been used most extensively (Gurney et al., 1994), followed by the SOD1G37R (Wong et al., 1995), SOD1G85R (Bruijn et al., 1997), SOD1G86R (Ripps et al., 1995) and SOD1D90A (Jonsson et al., 2006) models.
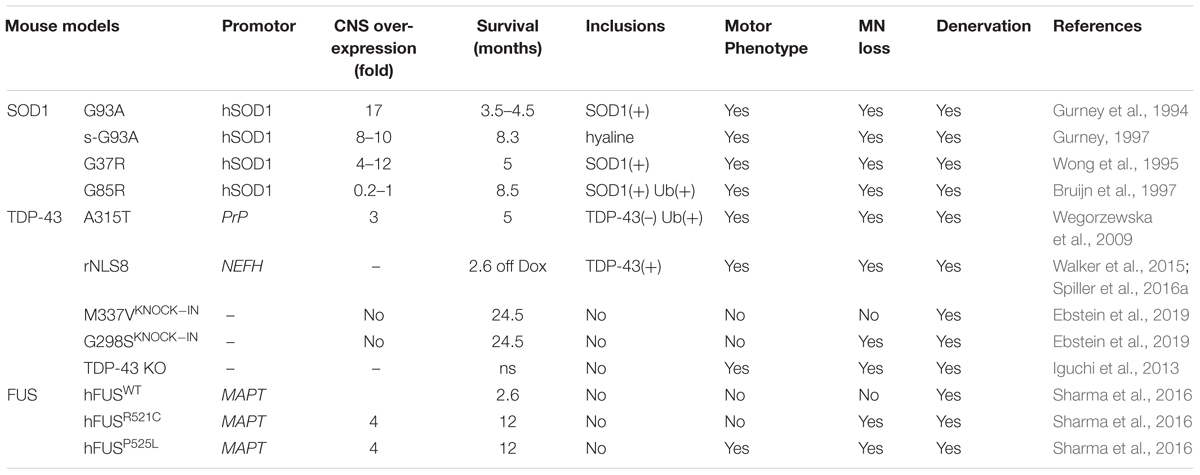
Table 5. SOD1, TDP-43 and FUS mouse models of ALS.
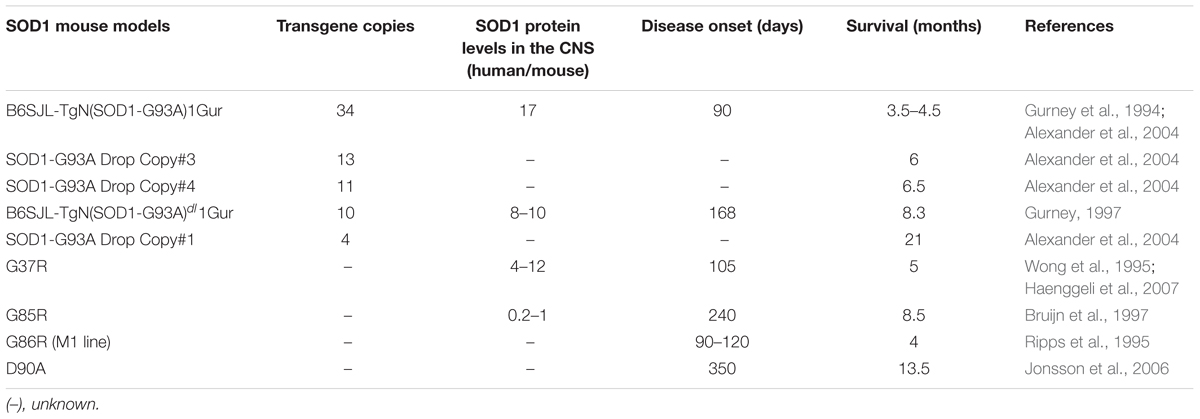
Table 6. Commonly used SOD1-transgenic mouse models of ALS and their phenotypes in relation to transgenic expression.
The B6SJL-TgN(SOD1-G93A)1Gur mouse (Gurney et al., 1994) carries 25 ± 1.5 copies of the transgene within chromosome 12 and as a result, it expresses very high levels of human mutant SOD1G93A (Alexander et al., 2004). Whilst these significant levels of overexpression are criticized as a major limitation (Alexander et al., 2004), these animals remain the most widely used mouse model for therapeutic studies in ALS (Gurney et al., 1994). These SOD1G93A mice become paralyzed in the hindlimbs as a result of MN loss from the spinal cord, resulting in death by 5 months of age. Another variant of this model, B6SJL-TgN(SOD1-G93A)dl1Gur, possesses fewer copies of the transgene; 8 ± 1.5 (Gurney, 1997; Alexander et al., 2004)2. This “low-copy” mouse, hereafter referred to as “G93A-slow” (s-SOD1G93A), develops a slower disease course in comparison, where paralysis begins at 6–8.5 months of age (Alexander et al., 2004; Muller et al., 2008; Acevedo-Arozena et al., 2011). In addition, several other “low-copy” mouse lines have subsequently been generated, with even fewer copies of the human SOD1G93A transgene. These models also exhibit greater life spans compared to the higher copy lines (Alexander et al., 2004) (Table 6). Similarly, four lines of mice expressing another SOD1 mutant, SOD1G37R, at different levels (5–14 times) have been produced, with variable phenotypes (Wong et al., 1995). Multiple mouse models based on transgenic expression of wild type or mutant TDP-43 have also been generated (Philips and Rothstein, 2015) (Table 5). Overexpressing human TDP-43 with a defective nuclear localization signal (NLS) in mice – in the absence of an ALS mutation – results in cytoplasmic expression of hTDP-43 and nuclear TDP-43 clearance. This results in a severe motor phenotype and reduced survival in the resulting ‘rNLS8’ mice compared to littermate controls (Walker et al., 2015). Several mouse models also exist based on transgenic expression of mutant FUS (Table 5). These mice display progressive, age- and mutation-dependent degeneration that also model aspects of ALS (Sharma et al., 2016). Furthermore, several newer models based on the C9orf72 repeat expansion have also been produced, although the phenotypes are more reminiscent of FTD rather than ALS (Batra and Lee, 2017).
The expression of specific proteins can vary between MN subpopulations and this may be linked to their vulnerability to degenerate. Evidence for this hypothesis comes from the existing mouse models of ALS. Whilst mutant SOD1G93A is expressed in all MNs in these mice (Jaarsma et al., 2008), its propensity to induce neurodegeneration and disease is proportional to its expression level (Table 6) (Gurney et al., 1994; Bruijn et al., 1997; Alexander et al., 2004). At lower levels of expression, pathology is restricted to MNs in the spinal cord and brainstem only, whereas higher expression levels also induce severe abnormalities in the brain. Fewer copies of the SOD1G37R transgene correlate with delayed disease progression and a significant increase in lifespan compared to animals with higher copy numbers (Table 6) (Zwiegers et al., 2014). Similarly, in TDP-43 models, higher levels of overexpression are associated with a worse phenotype (Philips and Rothstein, 2015). Moreover, disease is evident in both wildtype and mutant TDP-43 models, indicating that the expression levels of TDP-43, rather than the presence of a mutation per se, induces neurodegeneration. Hence, the effect of the TDP-43 mutation can be difficult to segregate from the effects of overexpression in these models (Philips and Rothstein, 2015). Both retaining the physiological expression levels and normal nuclear localization of TDP-43 have been linked to maintaining cellular homeostasis (Swarup et al., 2011; Philips and Rothstein, 2015). These studies together highlight the role of differing protein expression levels in the development and progression of ALS. However, further work is required to determine whether the expression levels of mutant ALS-associated proteins are different among MN subtypes, and whether this can differentially sensitize specific MNs to neurodegeneration and stress in ALS.
Rodent disease models are also useful in studies examining the selective vulnerability of specific MNs within an individual motor pool in ALS. Similar to human ALS, in mouse models based on mutant SOD1G93A, TDP-43A315T and FUSP525L, α-MNs selectively degenerate, while γ-MNs and MNs in the Onuf’s nucleus are spared (Mannen et al., 1977; Lalancette-Hebert et al., 2016). Also, as in ALS patients, the oculomotor MNs are spared in SOD1G93A (Niessen et al., 2006) and SOD1G86R (Nimchinsky et al., 2000) mice, whereas spinal cord MNs, trigeminal, facial and hypoglossal MNs are targeted (Niessen et al., 2006). In rNLS8 mice, MNs in the hypoglossal nucleus and the spinal cord are also involved, whereas those in the oculomotor, trigeminal, and facial nuclei are spared, despite widespread neuronal expression of cytoplasmic hTDP-43 (Spiller et al., 2016a). Atrophy of MNs in the trigeminal motor, facial and hypoglossal nuclei are also significantly smaller in TDP-43 knock-out mice, whereas MNs in the oculomotor nuclei are preserved (Iguchi et al., 2013). In addition, in another TDP-43 model, Prp-TDP43A315T mice, degeneration of specific neuronal populations occurs (Wegorzewska et al., 2009). Cytoplasmic ubiquitinated proteins accumulate in neurons of cortical layer V and in large neurons of the ventral horn and scattered interneurons, despite expression of the Prp-TDP-43A315T transgene in all neurons and glia (Wegorzewska et al., 2009). In a knock-in TDP-43 mouse model bearing a G298S mutation, MN loss was restricted to large-diameter α-MNs (Ebstein et al., 2019). Furthermore, in FUSP525L and FUSR521C mouse models, no significant MN loss was detected in oculomotor neurons, whereas spinal cord MNs were progressively lost during disease course (Sharma et al., 2016).
In mutant SOD1G93A mice, FF α-MNs are more susceptible to degenerate than FR α-MNs, resulting in the FF muscles becoming paralyzed before FR muscles (Hegedus et al., 2007). Furthermore, tonic S-units only disconnect from the muscle at disease end stage, meaning that S α-MNs are the least vulnerable within motor pools in SOD1G93A, SOD1G85R (Frey et al., 2000; Pun et al., 2006; Hegedus et al., 2007; Hadzipasic et al., 2014), TDP-43 rNLS8 (Spiller et al., 2016a), FUSR521C and FUSP525L transgenic models (Sharma et al., 2016). These findings together therefore provide strong evidence that there is a gradient of vulnerability amongst spinal MNs, whereby the faster, less excitable motor units are affected before the slower, more excitable types, at least in mouse models. Interestingly, selective denervation of MN subtypes occurs at the NMJ. Less denervation of the relatively resistant slow-twitch soleus muscle (Frey et al., 2000), compared to the vulnerable fast-twitch tibialis anterior muscle, occurs in TDP-43M337V, TDP-43G298S, FUSP525L, FUSR521C and TDP-43 rNLS8 mouse models (Sharma et al., 2016; Spiller et al., 2016a; Ebstein et al., 2019). In both the low- and high-copy s-SOD1G93A and SOD1G93A mice, the onset of interneuron degeneration also precedes the onset of behavioral motor manifestations and most MN degeneration (Chang and Martin, 2009; Jiang et al., 2009; Pullen and Athanasiou, 2009). Subtle changes to inhibitory synaptic inputs to MNs may therefore modulate MN excitability, leading to degeneration and motor symptoms in ALS/FTD.
Genetic mutations are present throughout life in ALS patients (summarized in Supplementary Table 1), but as only specific cellular populations are affected, this implies that the vulnerability of MN subtypes in ALS is not caused wholly by genetic factors. Hence, environmental or extrinsic factors, such as the neuronal circuitry or the microenvironment surrounding MNs, may explain the selective vulnerability of MNs in ALS/FTD.
The pattern of neurodegeneration in ALS/FTD is not random; it targets specific large-scale distributed networks in the brain and spinal cord. Motor manifestations begin in one region of the body in ∼98% of patients (Ravits et al., 2007) accompanied by unilateral, focal damage to MNs in the motor cortex or spinal cord, that innervate the corresponding peripheral body regions. It has been previously suggested that ALS targets specific evolutionarily linked, interdependent functions, and as the disease progresses these deficits combine into failure of specific networks (Eisen et al., 2014). More recently, several clinical studies have revealed that neurodegeneration and TDP-43 pathology spread to continuous anatomical regions during disease course (Ravits et al., 2007; Brettschneider et al., 2013; Walhout et al., 2018), and symptoms arise in the contralateral regions following a unilateral limb onset (Walhout et al., 2018). This also implies that neuronal circuitry might drive disease progression to specific MN populations in ALS/FTD. The spread of misfolded proteins from cell-to-cell, particularly TDP-43, provides a molecular explanation for the specific network and anatomical vulnerability observed in ALS. However, it must be noted that whilst contiguous spread is observed for most patients, this is not the case for all (Ravits and La Spada, 2009).
Increasing evidence suggests that ALS begins in the cortical regions of the brain, which is referred to as the “dying-forward hypothesis.” Features of cortical hyperexcitability – heralded by reduction in short interval intracortical inhibition – have been detected during the early phases of ALS in transcranial magnetic stimulation studies (Thomsen et al., 2014; Menon et al., 2015). This can precede the clinical onset of bulbar/spinal motor dysfunction by ∼3–6 months (Vucic et al., 2008; Bakulin et al., 2016). The dying forward hypothesis is consistent with Charcot, who first postulated that ALS begins in the cortex (Charcot, 1874). Clinical observations that MNs without monosynaptic connections to cortical MNs, such as the oculomotor, abducens, and Onuf’s nuclei, are spared in ALS, and that pure LMN forms of ALS are rare, also support this hypothesis. Further evidence is provided by the observation that MNs receiving direct, monosynaptic cortical input also predominantly develop TDP-43 pathology, while subcortical MNs do not (Eisen et al., 2017). Similarly, TDP-43 pathology develops in patients only in structures under the control of corticofugal projections (Brettschneider et al., 2013; Menon et al., 2015; Eisen et al., 2017)
TDP-43 pathology may then propagate through corticofugal axons to the spinal cord and regions of the brain (Braak et al., 2013; Eisen et al., 2017) in a time-dependant and region-specific manner (Brettschneider et al., 2013), consistent with the dying forward hypothesis (Figure 3). This sequential pattern of TDP-43 dissemination is consistent with the hypothesis that TDP-43 pathology is propagated synaptically from cell to cell (Brundin et al., 2010; Maniecka and Polymenidou, 2015), in a similar way to the pathogenic prion protein, a concept known as the “prion-like mechanism” (Lee and Kim, 2015; Ayers and Cashman, 2018). In this model, misfolded proteins act as template seeds to trigger aggregation of their natively folded counterparts. This results in the propagation of protein misfolding, leading to its orderly spread through the CNS (Soto, 2012; Maniecka and Polymenidou, 2015). However, the question of where disease begins remains controversial because many researchers still favor the “dying-back” hypothesis, in which ALS begins within the muscle cells or at the NMJ. This hypothesis proposes that there is a spread of pathology from LMNs to UMNs (Chou and Norris, 1993; Fischer et al., 2004; Pun et al., 2006; Turner et al., 2018), or else, a simultaneous involvement of both UMNS and LMNs (Turner et al., 2018). Whilst most of the evidence for the dying-back mechanism comes from animal models, studies of muscle biopsies from early stage ALS patients and long-term survivors have demonstrated significant morphological abnormalities and major denervation/re-innervation at the NMJ, implying that this region is targeted early in disease (Millecamps et al., 2010; reviewed in Arbour et al., 2017).
Figure 3. Schematic diagram representing the typical spread of neurodegeneration following an initial onset in motor neurons in ALS patients (n = 76 patients) (Brettschneider et al., 2013). Shading represents TDP-43 pathology.
There is evidence to support the prion-like model in ALS. The spread of neurodegeneration through adjacent anatomical regions of the CNS resembles the orderly spread of protein misfolding in prion disease. The in vitro cell-to-cell transmission of misfolded SOD1, TDP-43 and C9orf72 di-peptide repeat proteins has been demonstrated (Grad et al., 2011, 2014; Münch et al., 2011; Nonaka et al., 2013; Feiler et al., 2015; Porta et al., 2018). Similarly, the addition of cerebrospinal fluid from ALS/FTD patients (Ding et al., 2015), detergent-insoluble fractions of ALS-disease brains (Nonaka et al., 2013) or insoluble phosphorylated TDP-43 from post-mortem brain and spinal cord tissue (Smethurst et al., 2016), results in misfolding of TDP-43 when added to human cell lines. However, so far, only misfolded SOD1 and TDP-43 transmissibility has been demonstrated in vivo (Ayers et al., 2014, 2016; Porta et al., 2018). A recent study demonstrated that injection of brain-derived extracts from FTD patients into mice promoted the spatio-temporal transmission of TDP-43 pathology via the neuroanatomical connectome, suggesting that TDP-43 travels via axonal transport through connected regions of the CNS (Porta et al., 2018). Similarly, axonal transport is implicated in the spread of mutant SOD1 in mice (Ayers et al., 2016). Overexpression of misfolded TDP-43 or SOD1 facilitated the seeding ability of each inoculum, consistent with results obtained in vitro (Nonaka et al., 2013; Feiler et al., 2015; Smethurst et al., 2016).
Whilst these animal studies demonstrate that ALS spreads within MNs that are connected synaptically, a small portion of patients do not display this contiguous spreading of pathology, however. This implies the existence of alternative mechanisms of disease progression (Fujimura-Kiyono et al., 2011; Gargiulo-Monachelli et al., 2012), such as the transfer of misfolded proteins in nanotubules or exosomes (Nonaka et al., 2013; Sundaramoorthy et al., 2013; Grad et al., 2014; Ding et al., 2015; Feiler et al., 2015; Westergard et al., 2016). Interestingly, it has been suggested that the vulnerability of specific MN populations is associated with the spread of neurodegeneration in ALS (Fu et al., 2018).
There is increasing evidence for a role of the neighboring non-neuronal cells in ALS. Under normal conditions, glial cells provide nutritional and trophic support to MNs, but in ALS, they appear to exacerbate neurodegeneration in a non-cell autonomous fashion. These cells include microglia, astrocytes, oligodendrocytes and Schwann cells. Limiting the expression of mutant SOD1 to MNs only does not lead to neurodegeneration in mice (Pramatarova et al., 2001; Lino et al., 2002), and chimeric mouse studies have established that the presence of mutant SOD1G93A in glial cells induces neurodegeneration and MN loss (Papadeas et al., 2011). Both microglia and astrocytes appear to enhance disease progression by inducing neuroinflammation, whereas oligodendrocytes drive disease initiation. Non-neuronal cells may also be involved in the spread of pathological proteins in ALS (Thomas et al., 2017; Porta et al., 2018). However, whilst misfolded proteins released by MNs can be taken up by glial cells, they may be less toxic to these cells than to MNs (Benkler et al., 2018).
Microglia are the main immune cells of the CNS (Fujita and Kitamura, 1975; Hickey and Kimura, 1988; Lawson et al., 1990). In ALS patients, activated microglia increase in CNS regions that are susceptible to neurodegeneration (Kawamata et al., 1992) and in SOD1G93A mice, enhanced microglial reactivity precedes nerve denervation at the NMJ (Alexianu et al., 2001; Saxena et al., 2009). Microglia exist in both resting and activated states [reviewed in Perry and Holmes (2014)] and in ALS, activated microglia display two distinct phenotypes. The neuroprotective M2 phenotype promotes tissue repair and supports MN survival by releasing neuroprotective factors, and the toxic M1 phenotype produces cytokines, enhances inflammation, and induces cell death (Liao et al., 2012). Studies in mutant SOD1 mice reveal that the numbers of microglia increase during disease progression, but they vary between the neuroprotective M2 and toxic M1 phenotypes (Liao et al., 2012; Chiu et al., 2013). In lumbar spinal cords of pre-symptomatic SOD1G93A mice, the anti-inflammatory M2 microglia predominate (Gravel et al., 2016), whereas at disease onset and during progression, the proinflammatory M1 type is more common (Beers et al., 2011). Microglial-specific ablation of mutant SOD1G37R in mice does not affect disease initiation, but it significantly slows disease progression (Boillée et al., 2006b), indicating that microglia enhance the progression, but not the onset, of disease in transgenic mutant SOD1 mice. However, contradictory findings were obtained in the TDP-43 rNLS8 model, where microglia were neuroprotective and not neurotoxic (Spiller et al., 2018). Interestingly, knockdown of C9orf72 in mice alters microglial function and induces age-related neuroinflammation, but not neurodegeneration (Lall and Baloh, 2017). Further investigations are required to examine the role of microglia in other ALS disease models, and to determine whether MN subtypes display different vulnerabilities to microglia-mediated protective and/or toxicity in ALS.
Astrocytes perform multiple homeostatic functions in the CNS; they regulate the plasticity of synapses and synthesis of neurotransmitters (Ullian et al., 2004; Volterra and Meldolesi, 2005; Sloan and Barres, 2014), they maintain the blood brain barrier, and they provide neurotrophic support to MNs by releasing glial-derived neurotrophic factor (GDNF) and transforming growth factor β1 (TGF-β1) amongst others. Like microglia, during the neurodegenerative process, astrocytes can exist in two states, either reactive or activated, and activated astrocytes lose their neuroprotective functions and become neurotoxic during disease (Yamanaka et al., 2008; Ilieva et al., 2009; Valori et al., 2014; Das and Svendsen, 2015). Also, like microglia, astrocytes are implicated in the progression rather than onset of ALS. Deletion of SOD1 from astrocytes slowed disease progression, but not disease onset, in SOD1G93A mice (Yamanaka et al., 2008; Wang L. et al., 2011), whereas deletion of mutant SOD1 from MNs did delay onset (Boillée et al., 2006a; Wang L. et al., 2009). Furthermore, gene expression changes in MNs, astrocytes and oligodendrocytes start just before disease onset in SOD1G37R mice, but these alterations are first observed in MNs (Sun et al., 2015). Recently, two different subsets of reactive astrocytes were described in the adult CNS, A1 and A2 (Liddelow et al., 2017; Clarke et al., 2018; Miller, 2018) and the A1 reactive astrocytes were associated with the death of both neurons and oligodendrocytes (Liddelow et al., 2017).
There is increasing evidence that astrocytes mediate MN degeneration via the release of neurotoxic factors. Soluble toxic compounds produced by astrocytes expressing mutant SOD1 trigger the selective loss of spinal MNs (Nagai et al., 2007), but not spinal GABAergic neurons, consistent with the specific vulnerability of these cells in ALS (Nagai et al., 2007). Astrocytes in the ventral spinal cord can be distinguished from astrocytes in the dorsal spinal cord by expression of semaphorin A3 (Sema3a), which is implicated in the specific vulnerability to FF-MNs in ALS (see section “Neuroprotective and Neurotoxic Factor Expression in MN Subpopulations” below). Furthermore, astrocytes are also implicated in MN loss and disease progression by mediating AMPA receptor-induced excitotoxicity via EAAT2/GLT-1, as discussed below (section “Neuronal Excitability”). Expression of mutant TDP-43M337V in rat astrocytes led to down-regulation of neurotrophic genes, up-regulation of neurotoxic genes and progressive MN degeneration (Tong et al., 2013; Huang et al., 2014). Conditioned medium from primary astrocyte cultures of SOD1G86R and TDP-43A315T mice also induces MN death through activation of sodium channels and nitro-oxidative stress (Rojas et al., 2014). Furthermore, astrocytes expressing mutant FUSR521G trigger MN death by secreting pro-inflammatory tumor necrosis factor (TNF)-α (Kia et al., 2018). SOD1G93A aggregates in astrocytes appear in late disease stages, selectively in regions with extensive neuronal degeneration and prominent astrogliosis (Jaarsma et al., 2008). This raises the possibility that astroglial aggregate formation is triggered by MN degeneration, implying that disease may spread from neurons to glia (Jaarsma et al., 2008; Sun et al., 2015).
Together these studies suggest the involvement of astrocytes in the selective degeneration of MNs in ALS. Under normal conditions, astrocytes may be able to cope with the expression of low levels of misfolded proteins, but, during cell stress or in the context of MN degeneration, they become more vulnerable, and release factors toxic to MNs, thus producing a vicious cycle. However, the relative resistance of neuronal populations surrounded by reactive astrocytes indicates that the vulnerability of MNs is also determined by cell-autonomous components, such as their genetic background and transcriptional/translational profiles (Boillée et al., 2006a; Sun et al., 2015).
The two glial cell types responsible for myelination of axons have also been investigated in the context of ALS. Oligodendrocytes myelinate axons in the CNS whereas Schwann cells are responsible for myelination in the peripheral nervous system (PNS). Whilst they perform similar functions, there are also important differences between these two cell types. Schwann cells form a single myelin sheath around one single axon, whereas oligodendrocytes myelinate many different axons. Furthermore, there are differences in the protein composition of CNS and PNS myelin.
In ALS, TDP-43 pathology has been detected in oligodendrocytes in the motor cortex and spinal cord of both SALS and FALS patients (Arai et al., 2006; Mackenzie et al., 2007; Tan et al., 2007; Zhang et al., 2008; Seilhean et al., 2009; Murray et al., 2011; Philips et al., 2013). In addition, FUS forms cytoplasmic aggregates in oligodendrocytes from ALS patients bearing FUSR521C or FUSP525L mutations (Mackenzie et al., 2011). Degeneration of oligodendrocytes and their precursors was also linked with axon demyelination in both SALS and FALS patients (Kang et al., 2013). In SOD1G93A mice, oligodendrocyte loss in the spinal cord occurs before symptoms appear and importantly, before MN loss, implying that oligodendrocytes are associated with disease onset. This MN loss increases with disease progression, resulting in MNs with only partially myelinated axons in SOD1G93A mice and SOD1G93A rats (Niebroj-Dobosz et al., 2007; Kang et al., 2013; Philips et al., 2013). Whilst the proliferation of oligodendrocyte precursors may compensate for this loss, newly synthetized oligodendrocytes failed to mature and remain dysfunctional in SOD1G93A mice (Magnus et al., 2008; Philips et al., 2013). Recently, SOD1G85R was able to transfer from MNs to nearby oligodendrocytes (Thomas et al., 2017). The selective removal of mutant SOD1 from NG2+ oligodendrocyte progenitors, but not mature oligodendrocytes in SOD1G37R mice, leads to delayed disease onset and prolonged survival (Kang et al., 2013), further suggesting that mutant SOD1-induced oligodendrocyte defects are detrimental to MNs in ALS.
Schwann cells are required for the long-term maintenance of synapses at the NMJ (Reynolds and Woolf, 1992; Son and Thompson, 1995; Reddy et al., 2003). Early studies demonstrated that myelin is altered along peripheral nerves in ALS patients, implying that Schwann cells are involved in disease (Perrie et al., 1993). However, unlike the other glial cell types, more recent studies on the role of Schwann cells in ALS have reached conflicting conclusions. Knockdown of SOD1G37R within Schwann cells significantly accelerates disease progression, concomitant with a specific reduction in insulin-like growth factor (IGF-I), which is protective to MNs (see section “Neuroprotective and Neurotoxic Factor Expression in MN Subpopulations” below) (Lobsiger et al., 2009). This surprising finding, implying that SOD1G37R is protective in Schwann cells, could be linked to the dismutase activity of SOD1. Whereas SOD1G37R retains its enzymatic activity, SOD1G85R does not, and similar experiments performed in SOD1G85R mice resulted in opposite findings; Schwann cell specific knock-down of SOD1G85R delayed disease onset and extended survival (Wang et al., 2012). Furthermore, TGF-β1 produced by Schwann cells promotes synaptogenesis by increasing nerve-muscle contacts (Feng and Ko, 2008), in contrast to TGF-β1 expression in astrocytes which accelerates disease progression in SOD1 mice (Endo et al., 2015). Hence, the role of Schwann cells in ALS remains unclear.
Multiple cellular pathways are now implicated in the etiology of ALS, but it remains unclear how dysfunction of these diverse processes can result in the same disease phenotype. Furthermore, the same genetic mutation can result in either ALS, FTD or both conditions, implying that specific disease modifiers exist. Studies using in vivo and in vitro models of FALS suggest that the intrinsic properties of MNs are crucial for degeneration and/or protection (Boillée et al., 2006a). Importantly, resistant MN subtypes appear to display diverse gene expression profiles from susceptible MNs. Microarray analysis and laser capture microdissection of MNs isolated from oculomotor/trochlear nuclei, the hypoglossal nucleus and the lateral column of the cervical spinal cord in SOD1G93A rats (Hedlund et al., 2010), or in human brain and spinal cords (Brockington et al., 2013), have revealed marked differences between these subpopulations. Importantly, many of the genes that were differentially expressed encode proteins that function in pathways implicated in ALS pathogenesis, such as ER function, calcium regulation, mitochondrial function, ubiquitination, apoptosis, nitrogen metabolism, transport and cellular growth. Interestingly, oculomotor neurons possess a specific and relatively conserved protein signature between humans and rodents, implying that this contributes to the relative resistance of these MNs in ALS/FTD (Hedlund et al., 2010; Comley et al., 2015). Several of these proteins are known to be protective against MN neurodegeneration, such as insulin-like growth factors (IGF) and their receptors (see section “Neuroprotective and Neurotoxic Factor Expression in MN Subpopulations” below). Similarly, other genes highly expressed in vulnerable MNs are implicated in their susceptibility to degeneration, such as semaphorin A3 (Sema A3) and matrix metalloproteinase 9 (MMP-9) (see section “Neuroprotective and Neurotoxic Factor Expression in MN Subpopulations” below). Recently, a comprehensive bioinformatics meta-analysis of ALS modifier genes was performed from 72 published studies (Yanagi et al., 2019). A total of 946 modifier genes were identified and of these, 43 genes were identified as modifiers in more than one ALS gene/model. These included TDP-43, SOD1, ATXN2 and MMP9. Intrinsic factors in MNs might therefore underlie their relative vulnerability or resistance to neurodegeneration in ALS. The two pioneering studies linking gene expression differences to MN vulnerability in ALS (Hedlund et al., 2010; Brockington et al., 2013) have led to several subsequent reports, where the role of specific genes were examined further (summarized in Table 7, and discussed further in the sections below). However, it is also possible that the differences in gene expression reflect the diverse embryological origins or milieu of resistant and susceptible MN groups, or simply the structural and functional differences between oculomotor units and motor units of other skeletal muscles. To date, no studies have extensively characterized the specific transcriptional profile of vulnerable vs. susceptible MNs in TDP-43, C9orf72 FUS or other models of ALS, similar to those performed in SOD1G93A mice and ALS patients (Hedlund et al., 2010; Brockington et al., 2013).
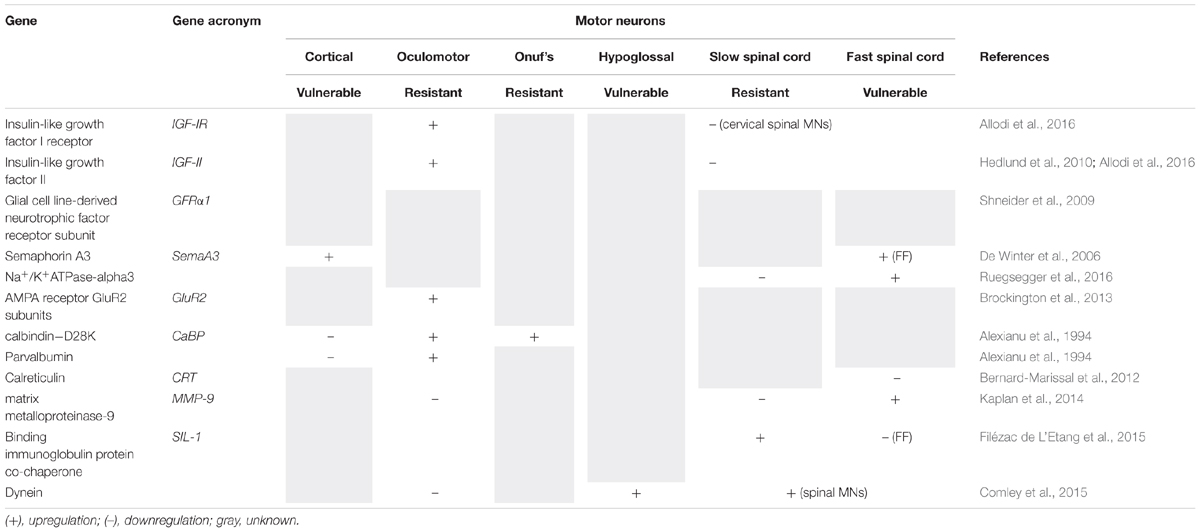
Table 7. Table with genes (described in this review) which are differently expressed among neuron subpopulations.
In addition to alterations in gene expression profiles, it is also possible that the resistant MNs in ALS display differing functional or morphological properties to those more susceptible to degeneration. A recent study demonstrated that cultures obtained from surviving MNs of SOD1G93A mice displayed more dendritic branching and axonal outgrowth, as well as increased actin based-growth cones, implying that they have more regenerative capacity (Osking et al., 2019).
Abnormal RNA homeostasis is increasingly implicated in the pathophysiology of ALS/FTD, consistent with the functions of TDP-43 and FUS in regulating RNA splicing and transport (Polymenidou et al., 2011; Tank et al., 2018). In the transgenic SOD1G93A rat, differences in the number of genes involved in transcription, RNA metabolism, RNA binding and splicing, and regulation of translation, were evident between neuronal populations located in the oculomotor/trochlear nucleus, the hypoglossal nucleus and the lateral column of the cervical spinal cord (Hedlund et al., 2010). These results therefore suggest that RNA homeostatic processes are involved in the differential vulnerability of specific subtypes of MNs in ALS. However, further studies in this area are required to investigate this possibility, particularly in relation to TDP-43 and FUS.
Differential expression of pro-survival or toxic factors is also implicated in the specific vulnerability of MN subtypes. The IGFs are proteins with high homology to insulin that form part of the IGF “axis” that promotes cell proliferation and inhibits apoptosis. In the normal rat, IGF-I is highly expressed in oculomotor neurons, where it is protective against glutamate-induced toxicity (Hedlund et al., 2010; Allodi et al., 2016). This may be due to activation of the PI3K/Akt and p44/42 MAPK pathways, which both inhibit apoptosis (Siddle et al., 2001; Sakowski et al., 2009). In addition, its associated receptor, IGF-I receptor (IGF-IR), is also highly expressed in oculomotor neurons and on the extraocular muscle endplate (Allodi et al., 2016). IGF-IR is important for the survival of neurons following hypoxic/ischemic injury (Vincent and Feldman, 2002; Liu et al., 2011) by upregulation of neuronal cellular inhibitor of apoptosis-1 (cIAP-1) and X-linked inhibitor of apoptosis (XIAP) (Liu et al., 2011). Delivery of IGF-II using AAV9 to the muscle of mutant SOD1G93A mice extended life-span by 10%, prevented the loss of MNs and induced motor axon regeneration (Allodi et al., 2016). These findings indicate that differential expression of IGF-II and IGF-IR in oculomotor neurons might contribute to their relative resistance to degeneration in ALS/FTD.
Conversely, aberrant expression of axon repulsion factors near the NMJ may contribute to neurodegeneration in ALS. Sema3A and its receptor neuropilin 1 (Nrp1) are involved in axon guidance during neural development (Huber et al., 2005; Moret et al., 2007). Sema3A is specifically upregulated in terminal Schwann cells near NMJs of vulnerable FF muscle fibers in mutant SOD1G93A mice (De Winter et al., 2006). Nrp1 is upregulated in axon terminals of the NMJ in this model and administration of an antibody against the Sema3A-binding domain of Nrp1 delayed the decline of motor functions while prolonging the lifespan of SOD1G93A mice (Venkova et al., 2014). Furthermore, Sema3A is upregulated in the motor cortex of ALS patients (Körner et al., 2016; Birger et al., 2018), but not in the spinal cord. Sema3A induces death of sensory, sympathetic, retinal and cortical neurons (Shirvan et al., 2002; Ben-Zvi et al., 2008; Jiang et al., 2010; Wehner et al., 2016), but not spinal neurons (Molofsky et al., 2014; Birger et al., 2018). Similarly, Sema3A induces apoptosis of human cortical neurons but promotes survival of spinal MNs (Birger et al., 2018). Furthermore, loss of Sema3A-expressing astrocytes in the ventral spinal cord leads to selective degeneration of α-MNs, but not γ-MNs (Hochstim et al., 2008; Molofsky et al., 2014). These data indicate that whilst Sema3A and Nrp1 contribute to the loss of MNs in ALS, some neuronal subpopulations are more susceptible than others. There is also evidence that other axon guidance proteins are associated with the susceptibility of MNs in ALS. Increased expression of ephrin A1 has been demonstrated in the vulnerable spinal MNs of ALS patients (Jiang et al., 2005). EPHA4, which is a disease modifier in zebrafish, rodent models and human ALS, encodes an Eph receptor tyrosine kinase, which is involved in axonal repulsion during development and in synapse formation, plasticity and memory in adults (Van Hoecke et al., 2012). The more vulnerable MNs express higher levels of EPHA4, and neuromuscular re-innervation is inhibited by Epha4. In ALS patients, EPHA4 expression also inversely correlates with disease onset and survival (Van Hoecke et al., 2012).
Matrix Metalloproteinase (MMP9) has been recently identified as another determinant of selective neuronal vulnerability in SOD1G93A mice (Kaplan et al., 2014). MMP-9 was strongly expressed by vulnerable FR spinal MNs, but not oculomotor, Onuf’s nuclei or S α-MNs, and it enhanced ER stress and mediated muscle denervation in this model (Kaplan et al., 2014). Delivery of MMP-9 into FF-MNs, but not in oculomotor neurons, accelerates denervation in SOD1G93A mice (Kaplan et al., 2014). Similarly, another study demonstrated that reduction of MMP-9 expression attenuated neuromuscular defects in rNLS8 mice expressing cytoplasmic hTDP43ΔNLS in neurons (Spiller et al., 2019). Edaravone, a free radical scavenger which inhibits MMP-9 expression, was recently approved for the treatment of ALS in Japan, South Korea, United States and Canada (Yoshino and Kimura, 2006; Ito et al., 2008; Yagi et al., 2009). Further molecular investigations into the differences and similarities between different motor units in ALS should yield additional insights into their vulnerability to neurodegeneration.
Polymorphisms in specific genes have also been linked to MN vulnerability. In SALS patients, variants in the gene encoding UNC13A are associated with greater susceptibility to disease and shorter survival (Diekstra et al., 2012). UNC13A functions in vesicle maturation during exocytosis and it regulates the release of neurotransmitters, including glutamate. Mutations in EPHA4 are also associated with longer survival (Van Hoecke et al., 2012), implying that Epha4 modulates the vulnerability of MNs in ALS. Furthermore, repeat expansions in the gene encoding ataxin 2 (ATXN2), which cause spinocerebellar ataxia type 2 (SCA2), are also increased in ALS patients compared to healthy controls (Ross et al., 2011). This implies that ATXN2 repeat expansions are also related to MN vulnerability to neurodegeneration in ALS.
The excitability properties of MNs are also implicated in the selective degeneration of specific MN subtypes in ALS. Alterations in MN excitability have been reported during the asymptomatic disease stage in the SOD1G93A (Saxena et al., 2013), s-SOD1G93A (Pambo-Pambo et al., 2009) and SOD1G85R (Bories et al., 2007) mouse models, in iPSC-derived MNs (Vucic et al., 2008; Wainger et al., 2014) and in SALS and FALS patients (Vucic and Kiernan, 2010; Devlin et al., 2015). Specific isoforms of the sodium–potassium pump (Na+/K+ATPase), which generates the Na+/K+ gradients that drive the action potential, are associated with the specific vulnerability of MN subtypes. Misfolded mutant SOD1 forms a complex with the α3 isoform of Na+/K+ATPase, and this leads to impairment in its ATPase activity. Altered levels of this isoform were also observed in spinal cords of SALS and non-SOD1 FALS patients (Ruegsegger et al., 2016). Importantly, α3 is the major isoform in vulnerable FF-MNs, whereas both α1 and α3 predominate in FR-MNs, and S-MNs express only α2. Furthermore, viral-mediated expression of a mutant Na+/K+ATPase-α3 that cannot bind to mutant SOD1 restored Na+/K+ATPase-α3 activity, delayed disease manifestations and increased lifespan in two different mutant SOD1 mouse models (SOD1G93A and SOD1G37R) (Ruegsegger et al., 2016). This indicates that modulating the activity of the α3 isoform of the Na+/K+ATPase, and therefore modulating the excitability status of MNs, is important in neurodegeneration in ALS.
However, increasing MN excitability is also neuroprotective to MNs in ALS. Enhancing MN excitability by delivering AMPA receptor agonists to mutant SOD1G93A mice reversed misfolded mutant protein accumulation, delayed pathology and extended survival, whereas reducing MN excitability by antagonist CNQX accelerated disease and induced early denervation, even in the more resistant S-MNs (Saxena et al., 2013). However, MN subpopulations can be differentially affected by changes in excitability. Disease resistant S-MNs exhibit hyper-excitability in ALS patients (de Carvalho and Swash, 2017) and early in disease in mutant SOD1G93A mice, whereas disease vulnerable FF-MNs are not hyper-excitable, again highlighting increased excitability as a protective property in ALS (Leroy et al., 2014). Also, the vulnerable masticatory trigeminal MNs from SOD1G93A mice exhibit a heterogeneous discharge pattern, unlike oculomotor neurons (Venugopal et al., 2015). However, MNs in FALS and SALS patients are hyperexcitable early in disease course, but then later become hypo-excitable (Vucic et al., 2008; Menon et al., 2015), indicating that modulation of neuronal excitability is a factor influencing the development of ALS.
Excitotoxicity is the process by which neurons degenerate from excessive stimulation by neurotransmitters such as glutamate, due to overactivation of NMDA or AMPA receptors. This can result from pathologically high levels of glutamate, or from excitotoxins like NMDA and kainic acid, which allow high levels of Ca2+ to enter the cell. One line of evidence supporting a role for excitotoxicity in ALS is that riluzole, one of the only two drugs available for ALS patients, has anti-excitotoxic properties (Bensimon et al., 1994; Lacomblez et al., 1996). Riluzole inhibits the release of glutamate due to inactivation of voltage-dependant Na+ channels on glutamatergic nerve terminals (Doble, 1996). Previous studies have suggested that MNs that are less susceptible to excitotoxicity are less prone to degenerate (Hedlund et al., 2010; Brockington et al., 2013).
Ca2+ enters neurons through ligand-gated channels or voltage-gated channels such as the voltage-gated-L-type Ca2+ channel (Cav1.3), which mediates the generation of persistent inward currents (Xu and Lipscombe, 2001). Cav1.3 is differentially expressed in MN subtypes, with more in the spinal cord compared to the oculomotor and hypoglossal nuclei (Shoenfeld et al., 2014). This Ca2+ inward current increases early in disease course in MNs of SOD1G93A mice, which is associated with an increase in Cav1.3 expression.
In addition, the presence of atypical AMPA receptors in MNs compared to other neurons might render them more permeable to Ca2+. Functional AMPA receptors normally form a tetrameric structure composed, in various combinations, of the four subunits, GluR1, GluR2, GluR3, and GluR4. The Ca2+ conductance of these receptors differs markedly depending on whether GluR2 is a component of the receptor. However, in MNs, AMPA receptors express proportionately fewer GluR2 subunits relative to other types (Kawahara et al., 2003; Sun et al., 2005), which may render them more permeable to Ca2+ and thus more vulnerable to excitotoxic injury than other cells. Consistent with this notion, more GluR1 and GluR2 subunits are present in oculomotor neurons compared to spinal MNs in humans (Brockington et al., 2013), and treatment with AMPA/kainate of slice preparations from the rat lumbar spinal cord and midbrain results in more Ca2+ influx in spinal cord MNs compared to oculomotor neurons (Brockington et al., 2013). MNs in culture or in vivo are selectively vulnerable to glutamate receptor agonists, particularly those that stimulate AMPA receptors and induce excitotoxicity (Carriedo et al., 1996; Urushitani et al., 1998; Fryer et al., 1999; Van and Robberecht, 2000), whereas NMDA does not damage spinal cord MNs (Curtis and Malik, 1985; Pisharodi and Nauta, 1985; Hugon et al., 1989; Urca and Urca, 1990; Nakamura et al., 1994; Ikonomidou et al., 1996; Kruman et al., 1999). Moreover, ALS-vulnerable α-spinal cord MNs display greater AMPA receptor current density than other spinal neurons (Vandenberghe et al., 2000). Furthermore, when this density is reduced pharmacologically to levels similar to spinal neurons, these MNs are no longer vulnerable to activation of AMPA receptors. Similarly, when mutant SOD1G93A mice are crossed with mice overexpressing the GluR2 subunit in cholinergic neurons, the resulting progeny possess AMPA receptors with reduced permeability to Ca2+ and prolonged survival compared to SOD1G93A mice (Tateno et al., 2004), highlighting the importance of AMPA receptors and GluR2 in ALS.
Editing of mRNA controls the ability of the GluA2 subunit to regulate Ca2+-permeability of AMPA receptors. RNA editing is a post-transcriptional modification (Gln; Q to Arg; R) in the GluA2 mRNA, and the AMPA receptor is Ca2+-impermeable if it contains the edited GluA2(R) subunit. Conversely, the receptor is Ca2+-permeable if it lacks GluA2 or if it contains the unedited GluA2(Q) subunit. Interestingly, spinal MNs in human ALS patients display less GluR2 Q/R site editing (Kawahara et al., 2004; Aizawa et al., 2010). GluR2 pre-mRNA is edited by the enzyme adenosine deaminase isoform 2 (ADAR2) (Kortenbruck et al., 2001) and reduced ADAR2 activity correlates with TDP-43 pathology in human MNs (Aizawa et al., 2010). Furthermore, when ADAR2 is conditionally knocked-down in MNs in mice, a decline in motor function and selective loss of MNs in the spinal cord and cranial motor nerve nuclei was observed (Hideyama et al., 2012). In contrast, MNs in the oculomotor nucleus were retained, despite a significant decrease in GluR2 Q/R site editing (Hideyama et al., 2010). Notably, cytoplasmic mislocalization of TDP-43 was present in the ADAR2-depleted MNs (Yamashita et al., 2012) and TDP-43 was also localized at the synapse, further highlighting a link between ADAR2, GluR2 and TDP-43 (Wang et al., 2008; Feiguin et al., 2009; Polymenidou et al., 2011; Gulino et al., 2015).
Motor neurons may be vulnerable to excitotoxicity because they possess a lower capacity than other neurons to buffer Ca2+ upon stimulation (Van Den Bosch et al., 2006). Several electrophysiological studies have demonstrated that susceptible MNs in ALS have a limited capacity to buffer Ca2+ compared to resistant MNs (Lips and Keller, 1998, 1999; Palecek et al., 1999; Vanselow and Keller, 2000). Ca2+-binding proteins, such as calbindin D28K and parvalbumin, protect neurons from Ca2+-mediated cell death by enhancing Ca2+ removal after stimulation (Chard et al., 1993). In human autopsy specimens, both proteins are absent in MN populations lost early in ALS (cortical, spinal and lower cranial MNs), whereas MNs targeted later in disease course (Onuf’s nucleus, oculomotor, trochlear, and abducens MNs) expressed markedly more of each (Alexianu et al., 1994). Similarly, in pre-symptomatic SOD1G93A mice, lower levels of the Ca2+ binding ER chaperone calreticulin (CRT) were detected in vulnerable FF-MNs of the tibialis anterior muscle, compared to resistant MNs of the soleus (Bernard-Marissal et al., 2012). Knock-down of CRT in vitro was sufficient to trigger MN death by the Fas/NO pathway (Bernard-Marissal et al., 2012). Furthermore, reduced CRT levels and activation of Fas both trigger ER stress and cell death specifically in vulnerable SOD1G93A-expressing MNs (Bernard-Marissal et al., 2012). These studies suggest that expression of Ca2+-binding proteins may confer resistance to excitotoxic stimuli (Alexianu et al., 1994; Obál et al., 2006). However, overexpression of parvalbumin in high-copy SOD1G93A mice was beneficial (Laslo et al., 2000), although these findings have been challenged (Beers et al., 2001). Also, the loss or reduction of parvalbumin and calbindin D-28k immunoreactivity in large MNs at early stages in SOD1-transgenic mice suggest that these Ca2+-binding proteins contribute to the selective vulnerability of MNs (Sasaki et al., 2006). Conversely, parvalbumin levels are significantly less in oculomotor neurons from SOD1G93A mice compared to spinal cord MNs (Comley et al., 2015). Hence, these conflicting data argue against the involvement of Ca2+-binding proteins in oculomotor neuron resistance to degeneration. However, together these studies suggest that neuronal excitability and excitotoxicity are determinants of the selective vulnerability of spinal cord neurons, and the relative resistance of oculomotor neurons, in ALS.
The ER is responsible for the folding and quality control of virtually all proteins that transit through the secretory pathway. Hence it is a fundamental aspect of proteostasis. Unfolded or misfolded proteins are retained in the ER, which activates the unfolded protein response (UPR). This aims to improve the cellular protein folding capacity by inhibiting translation, upregulating ER chaperones – such as immunoglobulin binding protein (BiP) and protein disulfide isomerase (PDI) – and stimulating protein degradation (Walter and Ron, 2011; Rozas et al., 2017; Shahheydari et al., 2017). Numerous ALS-related proteins chronically active the UPR, including ALS-associated mutant forms of SOD1 (Nishitoh et al., 2008), TDP-43 (Walker et al., 2013), C9orf72 (Dafinca et al., 2016), Vesicle-associated membrane protein-associated protein B (VAPB) (Suzuki et al., 2009) and FUS (Farg et al., 2012). ER stress has also been detected in sporadic ALS patients (Ilieva et al., 2007; Atkin et al., 2008). Furthermore, ER stress is linked to excitability in ALS. Mutant SOD1 induces a transcriptional signature characteristic of ER stress, which also disrupts MN excitability (Kiskinis et al., 2014). Similarly, modulating the excitability properties of human iPSC-derived MNs alters the UPR (Kiskinis et al., 2014). Conversely, treatment of MNs with salubrinal, an inhibitor of ER stress which inhibits eIF2α dephosphorylation (Boyce et al., 2005), reduced the excitability of MNs (Kiskinis et al., 2014). Similar results were obtained in MNs from patients carrying C9orf72 repeat expansions or VCP mutations (Kiskinis et al., 2014; Dafinca et al., 2016; Hall et al., 2017). Moreover, pharmacological reduction of neuronal excitability in SOD1G93A mice specifically reduced BiP accumulation in ipsilateral FALS α-MNs (Saxena et al., 2013). Hence, together these findings indicate that induction of the UPR and the electrical activity of MNs are both closely related in ALS.
An in vivo longitudinal analysis of MNs revealed that ER stress influences disease manifestations in SOD1G93A and SOD1G85R mouse models of FALS (Saxena et al., 2009). However, activation of the UPR is detrimental to mutant s-SOD1G93A mice, leading to failure to reinnervate NMJs. Conversely, treatment with salubrinal attenuated axon pathology and extended survival in mutant SOD1G93A mice (Saxena et al., 2009). Initiation of the UPR was detected specifically in FF-MNs in asymptomatic SOD1G93A mice, but not in S-MNs (Saxena et al., 2009). Hence these findings indicate that the more vulnerable MNs develop ER stress first, thus linking the UPR to MN susceptibility in ALS. FF-MNS may be more vulnerable to ER stress because they have much lower levels of BiP co-chaperone SIL1 compared to S-MNs (Filézac de L’Etang et al., 2015). SIL1 is protective against ER stress and reduces the formation of mutant SOD1 inclusions in vitro. Conversely SIL1 depletion leads to disturbed ER and nuclear envelope morphology, defective mitochondrial function, and ER stress, thus linking SIL1 to neurodegeneration (Roos et al., 2016). Furthermore, AAV-mediated overexpression of SIL1 in MNs of SOD1G93A mice preserves FF MN axons and prolongs survival by 25–30% compared to littermates (Filézac de L’Etang et al., 2015). In addition, SIL1 levels are reduced in MNs of mutant TDP-43A315T mice, and are increased in the surviving MNs of SALS patients, also implying that SIL1 is protective in ALS (Filézac de L’Etang et al., 2015).
Consistent with these studies, ER stress is present specifically in anterior horn MNs in knock-in mice expressing BiP artificially retained in the ER. Furthermore, this was accompanied by the accumulation of ubiquitinated proteins and wild type SOD1 (Mimura et al., 2008; Jin et al., 2014), reminiscent of SALS (Bosco et al., 2010). Significant changes in mRNAs of ER stress genes were also detected in the cerebellum by transcriptome analysis (Prudencio et al., 2015). These studies together link SIL1 and BiP to neurodegeneration in both neuronal subpopulations in ALS/FTD.
PDI is also upregulated in SOD1 mice and human SALS spinal cord tissues (Ilieva et al., 2007; Atkin et al., 2008; Sasaki, 2010; Walker et al., 2010; Chen et al., 2015; Sun et al., 2015). Wild type PDI overexpression and related family member Erp57 are protective in vitro in neuronal cells expressing mutant SOD1 (Walker et al., 2010; Jeon et al., 2014; Parakh et al., 2018a). Interestingly, mutations in PDI and Erp57 have been identified in ALS patients, and expression in zebrafish induces motor defects (Woehlbier et al., 2016). Furthermore, the levels of PDI in MNs are lower than in astrocytes and oligodendrocytes in SOD1G37R mice (Sun et al., 2015). This implies that MNs are intrinsically more vulnerable to unfolded protein accumulation than other cell types, which may also contribute to their susceptibility in ALS.
It should also be noted, however, that the ER in neurons (and therefore MNs) is not as well characterized as other cell types. In fact, most studies examining UPR mechanisms have involved non-neuronal cells. Neurons possess extensive ER which is distributed continuously throughout the axonal, dendritic and somatic compartments, implying that neurons make unique demands on the ER compared to other cell types (Ramírez and Couve, 2011). Hence, our current soma-centric view of the ER does not consider its role in neuronal processes and how this might relate to their specific functions. This is particularly true for large neurons, such as MNs with their extended axons. The findings that the most susceptible MNs develop ER stress first implies that the ER in MNs may confer unique susceptibility on these cells compared to other MNs and non-neuronal cells. However, this idea requires validation experimentally.
Neurons utilize most of their energy at the synapse, which consumes more than a third of the overall cellular ATP (Harris et al., 2012; Niven, 2016). The properties and types of ion channels expressed in a MN influence the energy required to generate an action potential, and the Na+/K+ pump is estimated to account for 20–40% of the brain’s energy consumption (Purves et al., 2001). The size and shape of a MN also affects its electrical properties, and the distance over which signals must spread. MNs have particularly high energetic demands, even compared to other neurons. They also have large numbers of NMJs as well as high intracellular Ca2+ flux as discussed above.
More than 90% of ATP generation in the CNS occurs via mitochondrial oxidative phosphorylation (Hyder et al., 2013; Vandoorne et al., 2018). Reductions in energy metabolism have been reported in ALS (Vandoorne et al., 2018) and mitochondrial abnormalities, such as swelling and morphological changes, are among the earliest signs of pathology in SOD1G93A and SOD1G37R mice (Wong et al., 1995; Kong and Xu, 1998), FUSR521C rats (Huang et al., 2012; So et al., 2018) and wild type TDP-43 mice (Shan et al., 2010; Xu et al., 2010). Moreover, mitochondrial abnormalities are also present in MNs of ALS patient tissues (Fujita et al., 1996; Sasaki and Iwata, 1996; Swerdlow et al., 1998; Dhaliwal and Grewal, 2000; Sasaki et al., 2007). Furthermore, mutant SOD1 specifically associates with mitochondria and interferes with their function (Liu et al., 2004; Pasinelli et al., 2004; Ferri et al., 2006; Sotelo-Silveira et al., 2009; Vande Velde et al., 2011). Decreased activity of mitochondrial respiratory chain complexes was also present in spinal cord sections (Borthwick et al., 1999) and homogenates (Wiedemann et al., 2002) from ALS patients. Consistent with these findings, genes involved in mitochondrial function were upregulated in rat oculomotor neurons compared to hypoglossal and cervical spinal cord MNs. However, it should be noted that the higher firing rate of the former might confer some resistance to energy imbalance (Hedlund et al., 2010; Brockington et al., 2013).
In vulnerable MNs lacking Ca2+-binding proteins calbindin and parvalbumin, Ca2+ is largely taken up by mitochondria (Lautenschläger et al., 2013). As a result, extensive mitochondrial transport to the dendritic space is required to maintain Ca2+ homeostasis. The normal distribution of mitochondria is also perturbed in ALS patient MNs. Whereas they are depleted in distal dendrites and axons, mitochondria also accumulate in the soma and proximal axon hillock (Sasaki et al., 2007). Disturbed mitochondrial dynamics were also described in MNs in mutant SOD1G93A (De Vos et al., 2007; Sotelo-Silveira et al., 2009; Bilsland et al., 2010; Magrané et al., 2014) and TDP-43A315T (Magrané et al., 2014) mice. In addition, iPSC-derived A4V MNs exhibit disturbances in mitochondrial morphology and motility within the axon (Kiskinis et al., 2014). Similarly, expression of mutant TDP-43 in spinal cord primary neurons leads to abnormal distribution of mitochondria (Wang et al., 2013). Dysfunctional Ca2+ uptake by mitochondria may therefore result in elevated intracellular Ca2+ levels, thus contributing to neurodegeneration.
Compared to FF-MNs, S-MNs have smaller soma and axons, less dendritic branching, and fewer neuromuscular terminals (Kanning et al., 2010). This results in higher input resistance and therefore less energy is required to initiate an action potential in comparison. Moreover, S-MNs contain more mitochondria compared to FF-MNs (Kanning et al., 2010). These two properties may therefore render FF-MNs more vulnerable to depletion of energy than S-MNs. Indeed, a computational analysis study estimated that the energy requirements of FF-MNs are considerably larger than S-MNs for a similar discharge (Le Masson et al., 2014), rendering the former more sensitive to ATP imbalance. Furthermore, the muscle fiber types associated with FF- and S-MNs differ in their major energy source. The slow twitch muscles use mainly oxidative metabolism, whereas the fast-twitch fibers use glycolysis. Hence, the heightened vulnerability of MN subpopulations may relate to their bioenergetic and morphological characteristics. Both the direct interaction of misfolded ALS mutant proteins with mitochondria and the secondary overload of ion uptake could account for mitochondrial metabolism failure, leading to reduced ATP availability (Israelson et al., 2010).
Motor neurons can vary widely in their size and this can impact on their physiological functions. There is also increasing evidence that vulnerability to degeneration is related to MN size. The disease-vulnerable FF-MNs somas are larger than the S-MN resistant types, and they possess larger motor units. Moreover, the size of a MN also correlates inversely with its excitability, discharge behavior, firing rate, recruitment during movement, and vulnerability to degeneration in ALS (Henneman, 1957; Le Masson et al., 2014). The soma of MNs from male SOD1G93A mice is larger than those of wild type male mice (Shoenfeld et al., 2014). Furthermore, a recent study demonstrated that not only are the larger MN subtypes more vulnerable to neurodegeneration in SOD1G93A mice, but MNs also increase in size during disease in multiple regions of the spinal cord. Interestingly, in silico modeling predicted that the excitability properties of these cells were also altered (Dukkipati et al., 2018). Hence, MN size may alter during disease progression, and this plasticity may impact on the vulnerability of MN subtypes.
Oxidative stress arises when reactive oxygen species (ROS) or nitrogen species (RNS) accumulate within cells. This can lead to oxidative modifications and altered functional states of proteins, nucleic acids and lipids. Oxidative stress is linked to neurodegeneration in ALS (Carrí et al., 2003) and oxidation products, such as malondialdehyde, hydroxynonenal, and oxidized proteins, DNA or membrane phospholipids, are elevated in SALS and FALS patients (Shaw et al., 1995; Beal et al., 1997; Ferrante et al., 1997; Bogdanov et al., 2000; Shibata et al., 2001) and mouse models of ALS (Gurney et al., 1994; Andrus et al., 1998; Bogdanov et al., 1998; Hall et al., 1998; Liu et al., 1998, 1999; Rizzardini et al., 2003). Mitochondria damage in ALS has also been attributed to intracellular oxidative stress (Fujita et al., 1996). The normal physiological function of SOD1 is the detoxification of superoxide radicals, although loss of SOD1 function is no longer favored as a disease mechanism in ALS (Saccon et al., 2013). However, mutations in SOD1 increase neuronal vulnerability to oxidative stress (Franco et al., 2013; Tsang et al., 2014). Moreover, in response to elevated ROS, SOD1 relocates from the cytoplasm to the nucleus, where it regulates the expression of oxidative resistance and repair genes (Tsang et al., 2014).
Some neurons exhibit differential vulnerability to oxidative damage. Cerebellar granule and hippocampal CA1 neurons are more sensitive to oxidative stress than cerebral cortical and hippocampal CA3 neurons (Wang X. et al., 2009; Wang and Michaelis, 2010). Hence, it is possible that similar differences in vulnerability to oxidative stress might exist between MN populations. However, this possibility needs to be confirmed experimentally.
Efficient intracellular trafficking is required to maintain the structure and function of MNs, particularly because MNs have very long axons that connect the soma with distant synaptic sites [reviewed in De Vos and Hafezparast (2017)]. Disorganization of the neuronal cytoskeleton and inhibition of axonal, ER-Golgi, endosomal and nucleocytoplasmic transport, are now widely reported features of ALS [reviewed in Parakh et al. (2018b) and Burk and Pasterkamp (2019)]. Importantly, defects in trafficking could reduce the supply of components necessary for synaptic and/or somal function, and prevent clearance of waste products from the synapse, together contributing to neurodegeneration in ALS.
The existence of mutations in genes encoding cytoskeletal proteins or the cellular transport machinery highlights the involvement of these processes in ALS/FTD. These include tubulin α4A (Smith et al., 2014a; Perrone et al., 2017), a major component of microtubules, neurofilament heavy chain (Figlewicz et al., 1994), a type of intermediate filament, and profilin-1 (Wu et al., 2012; Dillen et al., 2013; Smith et al., 2014b), which is involved in actin polymerization. Similarly, dynactin-1, involved in axonal transport (Puls et al., 2003; Münch et al., 2004; Münch et al., 2005; Liu et al., 2017) and SCFD1 (Sec1 family domain containing 1), involved in ER to Golgi transport (van Rheenen et al., 2016), are also mutated in a small proportion of patients, further implying that protein transport is impaired in ALS/FTD.
Axonal transport defects may be an important factor underlying the selective vulnerability of MNs or MN subtypes in ALS/FTD. Abnormal accumulation of phosphorylated neurofilaments, mitochondria and lysosomes in the proximal axon of large MNs and axonal spheroids, are present in SALS and FALS patients (Hirano et al., 1984; Corbo and Hays, 1992; Okada et al., 1995; Rouleau et al., 1996; Sasaki and Iwata, 1996). Mutant SOD1 slows both anterograde (Williamson and Cleveland, 1999) and retrograde (Chen et al., 2007; Perlson et al., 2009) axonal transport. Cytoskeletal and motor proteins are differentially expressed in spinal MNs compared to oculomotor neurons. This includes peripherin (Hedlund et al., 2010; Comley et al., 2015), which is also found in ubiquitinated inclusions in the spinal cord of FALS (Robertson et al., 2003) and SALS patients (He and Hays, 2004). Overexpression of peripherin leads to defective axonal transport (Millecamps et al., 2006) and late-onset MN degeneration (Beaulieu et al., 1999), implying that differential expression of peripherin contributes to neurodegeneration.
Axonal transport requires the efficient regulation of both dynein and kinesin molecular motors (Melkov et al., 2016), which mediate transport in the retrograde and anterograde directions respectively. Dynein is differentially expressed in vulnerable and susceptible MNs because higher levels are present in spinal and hypoglossal MNs compared to oculomotor neurons (Ilieva et al., 2008). However, dynein levels were significantly decreased in motor nuclei in SOD1G93A mice compared to wild type mice although its expression in MNs was equivalent (Comley et al., 2015). Similar patterns were observed in ALS patients (Comley et al., 2015). Disruption of dynein inhibits axonal transport and results in abnormal redistribution of mitochondria (Varadi et al., 2004) and late-onset degeneration in mice (LaMonte et al., 2002). Several FALS-linked SOD1 mutants co-localize with dynein/dynactin in vitro and SOD1G93A mice (Ligon et al., 2005; Zhang et al., 2007; Shi et al., 2010), which perturbs axonal transport and synaptic mitochondrial content (De Vos et al., 2007). The lower expression of dynein in oculomotor neurons might therefore confer resistance to axonal transport defects in ALS. However, it is also possible that this simply reflects less need for retrograde transport in oculomotor neurons due to their smaller cell bodies, shorter axons and lower requirements for energy, compared to spinal and hypoglossal MNs. Nevertheless, the inefficient axonal transport of mitochondria may confer loss of energy at the synapse in vulnerable MN subpopulations. These MNs require more energy to function than other cells, leading to disturbed synaptic activity.
Kinesin-dependant axonal transport is also disrupted in ALS. Oxidized forms of wild type SOD1 immunopurified from SALS tissues inhibited kinesin-based fast axonal transport (Bosco et al., 2010). However, no interaction between members of the kinesin family (KIF5A, 5B or 5C) and SOD1 was detected in SOD1G93A mice. High expression of KIF proteins is also associated with neurodegeneration. KIF5C was abundantly expressed in vulnerable spinal MNs in SOD1G93A mice (Kanai et al., 2000), but a marked reduction in KIF3Aβ levels was detected in the motor cortex of SALS patients (Pantelidou et al., 2007). Furthermore, reduced kinesin-associated protein 3 (KIFAP3) expression was linked to an increase in the survival of ALS patients (Landers et al., 2009) and changes in the transport of choline acetyltransferase transporter (ChAT) along axons. KIF5C is expressed more in rat spinal MNs than oculomotor and hypoglossal MNs (Hedlund et al., 2010), However, further work is necessary to determine if this is related to ALS, and to examine whether KIFs are differentially expressed in neuronal subtypes.
Defects in the secretory pathway are also linked to ALS. Depletion of TDP-43 inhibits endosomal trafficking and results in lack of neurotrophic signaling and neurodegeneration (Schwenk et al., 2016). Similarly, inhibition of the first part of the classical secretory pathway, ER-Golgi transport, is also induced by mutant SOD1, TDP-43 and FUS (Sundaramoorthy et al., 2013; Soo et al., 2015). This mechanism has been described as a possible trigger for ER stress (Soo et al., 2015), which, as detailed above, is linked to neuronal susceptibility. Both endosomal and ER-Golgi transport are also linked to transport within the axon. However, it remains to be determined if these other forms of trafficking are directly associated with selective neuronal susceptibility in ALS.
Defective nucleocytoplasmic transport is emerging as an important cellular mechanism in the initiation or progression of ALS. Nuclear pore pathology is present in the brain of SALS and C9orf72 patients (Zhang K. et al., 2015; Chou et al., 2018). C9orf72 repeat expansions impair protein trafficking from the cytoplasm to the nucleus, and reduce the proportion of nuclear TDP-43 in patient-derived MNs (Zhang K. et al., 2015), thereby mimicking the nuclear depletion of TDP-43 in ALS patients (Neumann et al., 2006). Proteins involved in nucleocytoplasmic transport are abnormally localized in aggregates in the cortex of C9orf72 ALS patients, patient-derived MNs and the brain of C9orf72 mouse models (Zhang K. et al., 2015, Zhang et al., 2016). Similarly, TDP-43 pathology disrupts nuclear pore complexes and lamina morphology in cell lines and patient-derived MNs. Furthermore, insoluble TDP-43 aggregates also contain components of the nucleocytoplasmic machinery (Chou et al., 2018). Both protein import and RNA export were impaired by mutant TDP-43 in the brain of SALS mouse primary neurons (Chou et al., 2018). A recent meta-analysis of ALS modifier genes identified several genes encoding proteins involved in nucleocytoplasmic shuttling (Yanagi et al., 2019). In fact, the most enriched gene ontology term in this study was “protein import into the nucleus,” and it included KPNB1, encoding importin subunit beta-1, which was identified as a genetic modifier in three separate ALS models. Interestingly, the gene encoding lamin B1 subunit 1, which is involved in nuclear stability, was upregulated in oculomotor neurons compared to hypoglossal MNs and spinal cord MNs (Hedlund et al., 2010). Furthermore, lamin B1 is also known to possess cellular protective functions such as controlling the cellular response to oxidative stress (Malhas et al., 2009), DNA repair (Butin-Israeli et al., 2015) and RNA synthesis (Tang et al., 2008). It is therefore tempting to speculate that lamin B1 confers resistance to specific MN populations when highly expressed. However, further work is necessary to examine this possibility.
Although genetic mutations are present throughout life, ALS most commonly develops in mid-adulthood (50–60 years), implying that the normal aging process renders MNs vulnerable to degeneration. However, there is considerable variability in disease progression amongst mutation carriers, even within the same families. Hence, this implies that there is no simple correlation between genetics and disease phenotypes, suggesting that environmental factors and the normal aging process are relevant to understand neuronal vulnerability in ALS/FTD.
Aging results in the accumulation of detrimental biological changes over time. The reduction of muscle mass and strength (sarcopenia) is one of the major causes of disability in older persons (Enoka et al., 2003; Lauretani et al., 2003; Delmonico et al., 2009; Clark and Manini, 2012), which affects gait speed, balance, and the command of fine motor skills (Fried et al., 2004; Sorond et al., 2015). The deterioration of motor functions with advancing age therefore increases the risk of injury and age-associated diseases such as ALS/FTD (Spiller et al., 2016b; Niccoli et al., 2017).
Aging-associated muscle weakness also results from impairment of the activity of MNs contacting skeletal muscles (Fiatarone and Evans, 1993; Manini et al., 2013). High resolution structural MRI imaging reveals prominent atrophy in the primary motor cortex (Salat et al., 2004), as early as middle life in humans. Age-related decreases in white matter mass and myelinated nerve fiber length also correlate with reductions in the size of the motor cortex (Marner et al., 2003). However, loss of neurons during normal human aging is restricted to specific regions of the CNS only, and the number of cells lost is only slight, contrary to previous convictions that significant loss of neurons occur in the human cortex (Pannese, 2011). Instead, age-related changes observed in aged rhesus monkeys and mice appear to involve loss of dendrites and axons, and demyelination, resulting in significant loss of synapses without loss of the neuronal soma (Pannese, 2011). Similarly, there are fewer cholinergic and glutamatergic synaptic inputs directly abutting α-MNs in aged animals, indicating that aging causes α-MNs to shed synaptic inputs. Thus, both impairment of axon function and substantial loss of synaptic inputs may contribute to age-related dysfunction of α-MNs, without loss of the soma (Maxwell et al., 2018). As a consequence, motor units are gradually lost over the first six decades of life, and this accelerates thereafter (Deschenes, 2011). These studies together indicate that neuronal atrophy and axonal impairment, with reduced neuromuscular activity in the absence of MN loss, occur with normal aging.
A major component of aging-related muscle weakness is breakdown in communication between the brain and NMJ. This is related to increased neural noise which reduces the accuracy of neural transmission (Manini et al., 2013). This can result in activation of the motor unit, so that it becomes erratic, and together with diminished glutamate uptake into MNs, leads to an inability to exert muscle force and motor control (Manini et al., 2013). Furthermore, susceptibility of neurons to cellular stress, due to impairment of proteostasis and/or increased oxidative or metabolic stress during normal aging, may render MNs vulnerable to degeneration. Hence, genetic and environmental factors may combine to determine whether a MN can withstand an age-related disease such as ALS or not (Mattson and Magnus, 2006).
During the aging process, a decline in the normal cellular ability to maintain proteostasis is observed and, as a result, damaged proteins accumulate (Kikis et al., 2010). Thus the normal aging process in MNs that are already weakened by ALS-associated insults, such as the presence of misfolded proteins or environmental factors, may combine to induce neurodegeneration. MN populations that are more susceptible in ALS may therefore be less able to tolerate disturbances in proteostasis than the more resistant populations (Neumann et al., 2006; Kikis et al., 2010).
Mitochondria play a crucial role in neuronal aging. Normal features observed in the aging brain include the accumulation of mutations in mitochondrial DNA, the production of ROS, mitochondrial metabolic abnormalities and altered Ca2+ storage (Sun et al., 2016). Remarkably, mitochondria in different regions of the CNS are not equally affected during aging. The sensitivity of the mitochondrial permeability transition pore to Ca2+ in the cortex and hippocampus is greater than that of the striatum and the cerebellum in aged rats (LaFrance et al., 2005; Brown et al., 2006). The cellular location of mitochondria is also relevant to the aging processes. Synaptic mitochondria are more prone to oxidative stress-induced damage than mitochondria located in the soma (Brown et al., 2006; Reddy and Beal, 2008). In addition, synaptic mitochondria display a limited capacity to accumulate Ca2+, unlike those located in the soma (Brown et al., 2006). Furthermore, marked differences have been described between mitochondria located in the spinal cord and those found in distal axons of MNs from aged rats. In the axon termini at the NMJ, mitochondria swelling, fusion and an abundance of megamitochondria (giant mitochondria) during aging have been reported (García et al., 2013). These studies therefore imply that mitochondria become dysfunctional in aged MNs, which might sensitize vulnerable MN populations to ALS/FTD. Mitochondria located at the synapse may also be particularly vulnerable to these age-related processes.
The mammalian genome is under constant attack from both endogenous and exogenous sources. This can result in DNA damage, mutations and impaired cellular viability if not repaired correctly (Madabhushi et al., 2014). There is a significant increase in DNA damage during aging due to reduced capacity of DNA repair. Moreover, erroneous repair of DNA lesions can result in further mutations in the aged brain (Vijg and Suh, 2013). DNA damage is increasingly implicated in neurodegenerative disorders, including ALS, where it is induced by the C9orf72 repeat expansion (Farg et al., 2017; Walker et al., 2017). Interestingly, there is also evidence that both FUS and TDP-43 function in the DNA damage response, in either prevention of damage or repair of R loop-associated DNA damage (Hill et al., 2016). In addition, impairment of the DNA damage response due to the presence of ALS/FTD-associated FUS mutations induces neurodegeneration (Higelin et al., 2016; Naumann et al., 2018). It is therefore possible that the normal aging process results in an impaired ability to repair DNA in MNs. This may be an important source of cellular stress that precipitates neurodegeneration in cells already exposed to pathological events throughout life. However, recent work suggests that mutant SOD1G93A does not impact on DNA strand integrity, implying that DNA damage is not present in all forms of ALS (Penndorf et al., 2017).
Motor neurons are unique cells compared to other neurons. They are large cells, with extraordinarily long axons, and very high energetic requirements, which may render them uniquely susceptible to degeneration in ALS. Remarkably, however, not all MNs are equally affected, and there are marked differences in vulnerabilities between MN subtypes, even within the same motor unit. The resistant MNs possess distinct morphological and functional characteristics, as well as different gene expression profiles, compared to the more vulnerable groups (Figure 4). Importantly, the oculomotor neurons continue to function, even in the late stages of ALS when the vulnerable spinal and other MNs are significantly depleted. These oculomotor neurons are anatomically and functionally very different from all other motor units: they are much smaller, and their function involves sensing rather than movement, hence different circuits are involved. In contrast, spinal MNs are more prone to hyperexcitation and they express high levels of AMPA receptors, they are more prone to develop ER stress, and they do not buffer Ca2+ as well as the more resistant MN types. These properties may confer unique sensitivity to neurodegeneration in ALS. Interestingly, even within spinal MNs, there are distinct differences in vulnerability, because FF-MNs degenerate first, followed by FR-MNs, and the more resistant S-MNs degenerate later. Similarly, these cells also display differences in excitability and ER stress.
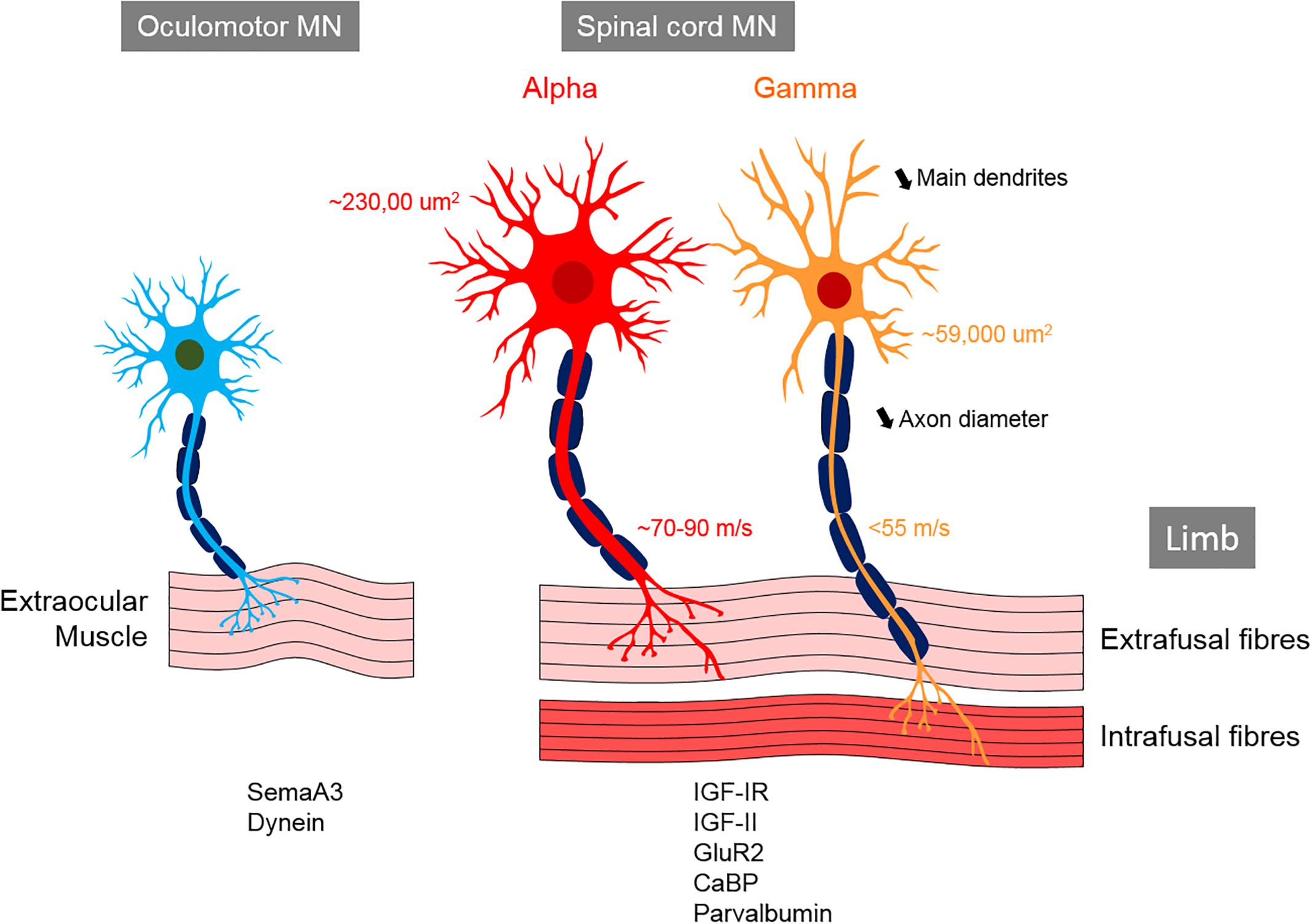
Figure 4. Reported differences between the vulnerable (ventral spinal cord MNs) and resistant (oculomotor) motor neurons in ALS. The surface area and axonal conduction velocities referred to here were obtained from studies in cats (Westbury, 1982). The α-MNs innervate highly contracting extrafusal fibers, whereas γ-MNs innervate intrafusal fibers that contract much less; oculomotor neurons innervate the extraocular muscles in the orbit. α-MNs are larger than γ-MNs and oculomotor neurons and possess more dendritic trees. α-MNs are further subdivided based on their size and function. The proteins listed at the bottom of the figure are those enriched in each MN population.
A hypothetical model is presented in Figure 5, summarizing the possible molecular mechanisms involved in MN vulnerability in ALS. The regulation of synaptic plasticity and neuronal excitability may underlie susceptibility in ALS involving nuclear-cytoplasmic defects, ER stress, transport dysfunction and mitochondrial alterations. From an initial site of onset, neurodegeneration begins in susceptible MN groups, and then spreads contiguously throughout the neuroanatomy, in a defined pattern, to the surrounding cells. This therefore highlights the role of impaired neurotransmission in triggering and propagating neurodegeneration in ALS. Glial cells are involved in both the onset and progression of ALS.
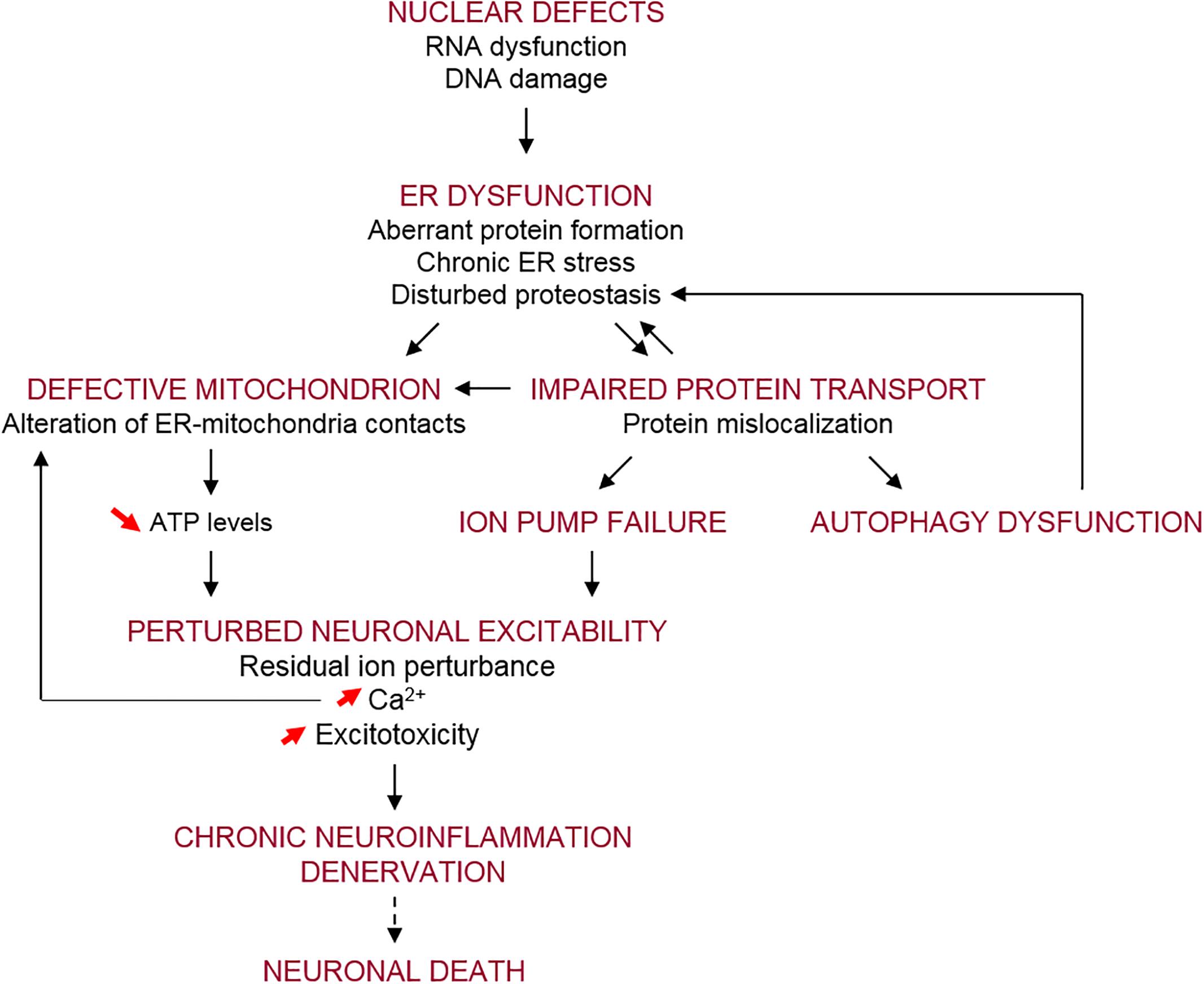
Figure 5. Diagram showing a hypothetic cascade of cellular events leading to neurodegeneration and neuronal death in motor neurons in ALS/FTD. This schematic diagram summarizes the key features occurring in vulnerable MNs. Resistant MNs are protected by the expression of a genes controlling cellular mechanisms that are defective in ALS/FTD (RNA dysfunction, ER stress, mitochondrial defects, protein transport dysfunction, dysregulation of neuronal excitability and excitotoxicity). These processes can be exacerbated by age, environmental and genetic mutations.
The susceptibility of specific MN groups, however, is further complicated by the heterogeneous nature of ALS, even within the same families, and the different patterns of motor involvement. Stratification of ALS patients into distinct subtypes and investigations into MNs susceptibilities may reveal more insights why specific groups of MNs degenerate first in ALS in the future. However, the blurring of some neurodegenerative disorders, including ALS and FTD, and the presence of C9orf72 mutations in several other neurodegenerative conditions as well as ALS, is another confounding factor. Understanding the fundamental mechanisms dictating MN vulnerability in ALS is central to our understanding of this devastating disorder. Hence, studies in this area may lead to novel therapeutic insights in the future.
MV wrote the “Site-Specific Onset and Spread of Neurodegeneration in ALS” section. MJ wrote the “Role of Glial Cells in Driving Disease Progression” section. SS wrote the “Aging” section. AR conceived and prepared the figures, and wrote the “Introduction,” and “Anatomy of the Motor System,” “Genetic Mutations and Risk Factors in ALS,” and “Intrinsic Factors Specific to MN Subpopulations” sections. JA conceived the article, wrote the “Conclusion” section, contributed text in numerous sections, and edited the manuscript throughout for content and style consistency.
This work was supported by the Australian National Health and Medical Research Council, FightMND Foundation, and Motor Neuron Disease Research Institute of Australia. SS was supported by an International Research Training Program Scholarship (iRTP). MJ was supported by the International Macquarie University Research Excellence Scholarship (iMQRES).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2019.00532/full#supplementary-material
Acevedo-Arozena, A., Kalmar, B., Essa, S., Ricketts, T., Joyce, P., Kent, R., et al. (2011). A comprehensive assessment of the SOD1G93A low-copy transgenic mouse, which models human amyotrophic lateral sclerosis. Dis. Model Mech. 4, 686–700. doi: 10.1242/dmm.00723
Adal, M. N., and Barker, D. (1965). Intramuscular branching of fusimotor fibres. J. Physiol. 177, 288–299. doi: 10.1113/jphysiol.1965.sp007592
Aizawa, H., Sawada, J., Hideyama, T., Yamashita, T., Katayama, T., Hasebe, N., et al. (2010). TDP-43 pathology in sporadic ALS occurs in motor neurons lacking the RNA editing enzyme ADAR2. Acta Neuropathol. 120, 75–84. doi: 10.1007/s00401-010-0678-x
Al Sultan, A., Waller, R., Heath, P., and Kirby, J. (2016). The genetics of amyotrophic lateral sclerosis: current insights. Degen. Neurol. Neuromusc. Dis. 2016, 49–64.
Alexander, G. M., Erwin, K. L., Byers, N., Deitch, J. S., Augelli, B. J., Blankenhorn, E. P., et al. (2004). Effect of transgene copy number on survival in the G93A SOD1 transgenic mouse model of ALS. Brain Res. Mol. Brain Res. 130, 7–15. doi: 10.1016/j.molbrainres.2004.07.002
Alexianu, M. E., Ho, B. K., Mohamed, A. H., La Bella, V., Smith, R. G., and Appel, S. H. (1994). The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 36, 846–858. doi: 10.1002/ana.410360608
Alexianu, M. E., Kozovska, M., and Appel, S. H. (2001). Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology 57, 1282–1289. doi: 10.1212/wnl.57.7.1282
Allodi, I., Comley, L., Nichterwitz, S., Nizzardo, M., Simone, C., Benitez, J. A., et al. (2016). Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALS. Sci. Rep. 6:25960. doi: 10.1038/srep25960
Andersen, P. M., Sims, K. B., Xin, W. W., Kiely, R., O’Neill, G., Ravits, J., et al. (2003). Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 4, 62–73. doi: 10.1080/14660820310011700
Andrus, P. K., Fleck, T. J., Gurney, M. E., and Hall, E. D. (1998). Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 71, 2041–2048. doi: 10.1046/j.1471-4159.1998.71052041.x
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi: 10.1016/j.bbrc.2006.10.093
Arbour, D., Vande Velde, C., and Robitaille, R. (2017). New perspectives on amyotrophic lateral sclerosis: the role of glial cells at the neuromuscular junction. J. Physiol. 595, 647–661. doi: 10.1113/JP270213
Asanuma, H., and Rosén, I. (1972). Topographical organization of cortical efferent zones projecting to distal forelimb muscles in the monkey. Exp. Brain Res. 14, 243–256. doi: 10.1007/BF00816161
Ashrafi, S., Lalancette-Hébert, M., Friese, A., Sigrist, M., Arber, S., Shneider, N. A., et al. (2012). Wnt7A identifies embryonic γ-motor neurons and reveals early postnatal dependence of γ-motor neurons on a muscle spindle-derived signal. J. Neurosci. 32, 8725–8731. doi: 10.1523/JNEUROSCI.1160-12.2012
Atkin, J. D., Farg, M. A., Walker, A. K., McLean, C., Tomas, D., and Horne, M. K. (2008). Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 30, 400–407. doi: 10.1016/j.nbd.2008.02.009
Ayers, J. I., and Cashman, N. R. (2018). Prion-like mechanisms in amyotrophic lateral sclerosis. Handb. Clin. Neurol. 153, 337–354. doi: 10.1016/B978-0-444-63945-5.00018-0
Ayers, J. I., Fromholt, S., Koch, M., DeBosier, A., McMahon, B., Xu, G., et al. (2014). Experimental transmissibility of mutant SOD1 motor neuron disease. Acta Neuropathol. 128, 791–803. doi: 10.1007/s00401-014-1342-7
Ayers, J. I., Fromholt, S. E., O’Neal, V. M., Diamond, J. H., and Borchelt, D. R. (2016). Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol. 131, 103–114. doi: 10.1007/s00401-015-1514-0
Bakels, R., and Kernell, D. (1993). Matching between motoneurone and muscle unit properties in rat medial gastrocnemius. J. Physiol. 463, 307–324. doi: 10.1113/jphysiol.1993.sp019596
Bakulin, I. S., Chervyakov, A. V., Suponeva, N. A., Zakharova, M. N., and Piradov, M. A. (2016). Motor Cortex Hyperexcitability, Neuroplasticity, and Degeneration in Amyotrophic Lateral Sclerosis. Update on Amyotrophic Lateral Sclerosis. London: Intechopen.
Balendra, R., and Isaacs, A. M. (2018). C9orf72 -mediated ALS and FTD: multiple pathways to disease. Nat. Rev. Neurol. 14:544. doi: 10.1038/s41582-018-0047-2
Bannwarth, S., Ait-El-Mkadem, S., Chaussenot, A., Genin, E. C., Lacas-Gervais, S., Fragaki, K., et al. (2014). A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137, 2329–2345. doi: 10.1093/brain/awu138
Barmada, S. J. (2015). Linking RNA dysfunction and neurodegeneration in amyotrophic lateral sclerosis. Neurotherapeutics 12, 340–351. doi: 10.1007/s13311-015-0340-3
Batra, R., and Lee, C. W. (2017). Mouse models of C9orf72 hexanucleotide repeat expansion in amyotrophic lateral sclerosis/ frontotemporal dementia. Front. Cell Neurosci. 11:196. doi: 10.3389/fncel.2017.00196
Beal, M. F., Ferrante, R. J., Browne, S. E., Matthews, R. T., Kowall, N. W., and Brown, R. H. (1997). Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann. Neurol. 42, 644–654. doi: 10.1002/ana.410420416
Beaulieu, J. M., Nguyen, M. D., and Julien, J. P. (1999). Late onset of motor neurons in mice overexpressing wild-type peripherin. J. Cell Biol. 147, 531–544. doi: 10.1083/jcb.147.3.531
Beck, J., Poulter, M., Hensman, D., Rohrer, J. D., Mahoney, C. J., Adamson, G., et al. (2013). Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am. J. Hum. Genet. 92, 345–353. doi: 10.1016/j.ajhg.2013.01.011
Bede, P., Iyer, P. M., Schuster, C., Elamin, M., Mclaughlin, R. L., Kenna, K., et al. (2016). The selective anatomical vulnerability of ALS: “disease-defining” and “disease-defying” brain regions. Amyotroph. Lateral Scler. Frontotemporal Degener. 17, 561–570. doi: 10.3109/21678421.2016.1173702
Beers, D. R., Henkel, J. S., Zhao, W., Wang, J., Huang, A., Wen, S., et al. (2011). Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain J. Neurol. 134, 1293–1314. doi: 10.1093/brain/awr074
Beers, D. R., Ho, B.-K., Siklós, L., Alexianu, M. E., Mosier, D. R., Mohamed, A. H., et al. (2001). Parvalbumin overexpression alters immune-mediated increases in intracellular calcium, and delays disease onset in a transgenic model of familial amyotrophic lateral sclerosis. J. Neurochem. 79, 499–509. doi: 10.1046/j.1471-4159.2001.00582.x
Belzil, V. V., Valdmanis, P. N., Dion, P. A., Daoud, H., Kabashi, E., Noreau, A., et al. (2009). Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology 73, 1176–1179. doi: 10.1212/WNL.0b013e3181bbfeef
Benkler, C., O’Neil, A. L., Slepian, S., Qian, F., Weinreb, P. H., and Rubin, L. L. (2018). Aggregated SOD1 causes selective death of cultured human motor neurons. Sci. Rep. 8:16393. doi: 10.1038/s41598-018-34759-z
Bensimon, G., Lacomblez, L., and Meininger, V. (1994). A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole study group. N. Engl. J. Med. 330, 585–591. doi: 10.1056/NEJM199403033300901
Ben-Zvi, A., Manor, O., Schachner, M., Yaron, A., Tessier-Lavigne, M., and Behar, O. (2008). The Semaphorin receptor PlexinA3 mediates neuronal apoptosis during dorsal root ganglia development. J. Neurosci. 28, 12427–12432. doi: 10.1523/JNEUROSCI.3573-08.2008
Bernard-Marissal, N., Moumen, A., Sunyach, C., Pellegrino, C., Dudley, K., Henderson, C. E., et al. (2012). Reduced calreticulin levels link endoplasmic reticulum stress and Fas-triggered cell death in motoneurons vulnerable to ALS. J. Neurosci. 32, 4901–4912. doi: 10.1523/JNEUROSCI.5431-11.2012
Bessou, P., Emonet-Dénand, F., and Laporte, Y. (1965). Motor fibres innervating extrafusal and intrafusal muscle fibres in the cat. J. Physiol. 180, 649–672. doi: 10.1113/jphysiol.1965.sp007722
Bhumbra, G. S., and Beato, M. (2018). Recurrent excitation between motoneurones propagates across segments and is purely glutamatergic. PLoS Biol. 16:e2003586. doi: 10.1371/journal.pbio.2003586
Bilsland, L. G., Sahai, E., Kelly, G., Golding, M., Greensmith, L., and Schiavo, G. (2010). Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. U.S.A. 107, 20523–20528. doi: 10.1073/pnas.1006869107
Birger, A., Ottolenghi, M., Perez, L., Reubinoff, B., and Behar, O. (2018). ALS-related human cortical and motor neurons survival is differentially affected by Sema3A. Cell Death Dis. 9:256. doi: 10.1038/s41419-018-0294-6
Blair, I. P., Williams, K. L., Warraich, S. T., Durnall, J. C., Thoeng, A. D., Manavis, J., et al. (2009). FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J. Neurol. Neurosurg. Psychiatry 81, 639–645. doi: 10.1136/jnnp.2009.194399
Boehringer, A., Garcia-Mansfield, K., Singh, G., Bakkar, N., Pirrotte, P., and Bowser, R. (2017). ALS associated mutations in matrin 3 alter protein-protein interactions and impede mRNA nuclear export. Sci. Rep. 7:14529. doi: 10.1038/s41598-017-14924-6
Bogdanov, M., Brown, R. H., Matson, W., Smart, R., Hayden, D., O’Donnell, H., et al. (2000). Increased oxidative damage to DNA in ALS patients. Free Radic. Biol. Med. 29, 652–658.
Bogdanov, M. B., Ramos, L. E., Xu, Z., and Beal, M. F. (1998). Elevated “hydroxyl radical” generation in vivo in an animal model of amyotrophic lateral sclerosis. J. Neurochem. 71, 1321–1324. doi: 10.1046/j.1471-4159.1998.71031321.x
Boillée, S., Vande Velde, C., and Cleveland, D. W. (2006a). ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59. doi: 10.1016/j.neuron.2006.09.018
Boillée, S., Yamanaka, K., Lobsiger, C. S., Copeland, N. G., Jenkins, N. A., Kassiotis, G., et al. (2006b). Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392. doi: 10.1126/science.1123511
Bories, C., Amendola, J., Lamotte d’Incamps, B., and Durand, J. (2007). Early electrophysiological abnormalities in lumbar motoneurons in a transgenic mouse model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 25, 451–459. doi: 10.1111/j.1460-9568.2007.05306.x
Borroni, B., Archetti, S., Del Bo, R., Papetti, A., Buratti, E., Bonvicini, C., et al. (2010). TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features, and disease course. Rejuven. Res 13, 509–517. doi: 10.1089/rej.2010.1017
Borthwick, G. M., Johnson, M. A., Ince, P. G., Shaw, P. J., and Turnbull, D. M. (1999). Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann. Neurol. 46, 787–790. doi: 10.1002/1531-8249(199911)46:5<787::aid-ana17>3.0.co;2-8
Bosco, D. A., Morfini, G., Karabacak, N. M., Song, Y., Gros-Louis, F., Pasinelli, P., et al. (2010). Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403. doi: 10.1038/nn.2660
Boyce, M., Bryant, K. F., Jousse, C., Long, K., Harding, H. P., Scheuner, D., et al. (2005). A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307, 935–939. doi: 10.1126/science.1101902
Braak, H., Brettschneider, J., Ludolph, A. C., Lee, V. M., Trojanowski, J. Q., and Del Tredici, K. (2013). Amyotrophic lateral sclerosis–a model of corticofugal axonal spread. Nat. Rev. Neurol. 9, 708–714. doi: 10.1038/nrneurol.2013.221
Bradshaw, W. J., Rehman, S., Pham, T. T. K., Thiyagarajan, N., Lee, R. L., Subramanian, V., et al. (2017). Structural insights into human angiogenin variants implicated in Parkinson’s disease and amyotrophic lateral sclerosis. Sci. Rep. 7:41996. doi: 10.1038/srep41996
Brecht, M., Krauss, A., Muhammad, S., Sinai-Esfahani, L., Bellanca, S., and Margrie, T. W. (2004). Organization of rat vibrissa motor cortex and adjacent areas according to cytoarchitectonics, microstimulation, and intracellular stimulation of identified cells. J. Comp. Neurol. 479, 360–373. doi: 10.1002/cne.20306
Brenner, D., Müller, K., Wieland, T., Weydt, P., Böhm, S., Lulé, D., et al. (2016). NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 139:e28. doi: 10.1093/brain/aww033
Brettschneider, J., Del Tredici, K., Toledo, J. B., Robinson, J. L., Irwin, D. J., Grossman, M., et al. (2013). Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 74, 20–38. doi: 10.1002/ana.23937
Brockington, A., Ning, K., Heath, P. R., Wood, E., Kirby, J., Fusi, N., et al. (2013). Unravelling the enigma of selective vulnerability in neurodegeneration: motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 125, 95–109. doi: 10.1007/s00401-012-1058-5
Brown, M. R., Sullivan, P. G., and Geddes, J. W. (2006). Synaptic mitochondria are more susceptible to ca2+overload than nonsynaptic mitochondria. J. Biol. Chem. 281, 11658–11668. doi: 10.1074/jbc.M510303200
Bruijn, L. I., Becher, M. W., Lee, M. K., Anderson, K. L., Jenkins, N. A., Copeland, N. G., et al. (1997). ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18, 327–338. doi: 10.1016/s0896-6273(00)80272-x
Brundin, P., Melki, R., and Kopito, R. (2010). Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 11, 301–307. doi: 10.1038/nrm2873
Bryan, R. N., Trevino, D. L., and Willis, W. D. (1972). Evidence for a common location of alpha and gamma motoneurons. Brain Res. 38, 193–196. doi: 10.1016/0006-8993(72)90602-6
Burk, K., and Pasterkamp, R. J. (2019). Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. doi: 10.1007/s00401-019-01964-7 [Epub ahead of print].
Burke, R. E., Dum, R. P., Fleshman, J. W., Glenn, L. L., Lev-Tov, A., O’Donovan, M. J., et al. (1982). A HRP study of the relation between cell size and motor unit type in cat ankle extensor motoneurons. J. Comp. Neurol. 209, 17–28. doi: 10.1002/cne.902090103
Burke, R. E., Levine, D. N., Tsairis, P., and Zajac, F. E. (1973). Physiological types and histochemical profiles in motor units of the cat gastrocnemius. J. Physiol. 234, 723–748. doi: 10.1113/jphysiol.1973.sp010369
Burke, R. E., Levine, D. N., and Zajac, F. E. (1971). Mammalian motor units: physiological-histochemical correlation in three types in cat gastrocnemius. Science 174, 709–712. doi: 10.1126/science.174.4010.709
Burke, R. E., Strick, P. L., Kanda, K., Kim, C. C., and Walmsley, B. (1977). Anatomy of medial gastrocnemius and soleus motor nuclei in cat spinal cord. J. Neurophysiol. 40, 667–680. doi: 10.1152/jn.1977.40.3.667
Burke, R. E., and Tsairis, P. (1973). Anatomy and innervation ratios in motor units of cat gastrocnemius. J. Physiol. 234, 749–765. doi: 10.1113/jphysiol.1973.sp010370
Burrell, J. R., Halliday, G. M., Kril, J. J., Ittner, L. M., Götz, J., Kiernan, M. C., et al. (2016). The frontotemporal dementia-motor neuron disease continuum. Lancet 388, 919–931. doi: 10.1016/S0140-6736(16)00737-6
Butin-Israeli, V., Adam, S. A., Jain, N., Otte, G. L., Neems, D., Wiesmüller, L., et al. (2015). Role of lamin B1 in chromatin instability. Mol. Cell Biol. 35, 884–898. doi: 10.1128/MCB.01145-14
Caligari, M., Godi, M., Guglielmetti, S., Franchignoni, F., and Nardone, A. (2013). Eye tracking communication devices in amyotrophic lateral sclerosis: impact on disability and quality of life. Amyotroph. Lateral Scler. Frontotemporal Degen. 14, 546–552. doi: 10.3109/21678421.2013.803576
Carrí, M. T., Ferri, A., Cozzolino, M., Calabrese, L., and Rotilio, G. (2003). Neurodegeneration in amyotrophic lateral sclerosis: the role of oxidative stress and altered homeostasis of metals. Brain Res. Bull. 61, 365–374. doi: 10.1016/s0361-9230(03)00179-5
Carriedo, S. G., Yin, H. Z., and Weiss, J. H. (1996). Motor neurons are selectively vulnerable to AMPA/Kainate receptor-mediated injury in vitro. J. Neurosci. 16, 4069–4079. doi: 10.1523/JNEUROSCI.16-13-04069.1996
Chakkalakal, J. V., Nishimune, H., Ruas, J. L., Spiegelman, B. M., and Sanes, J. R. (2010). Retrograde influence of muscle fibers on their innervation revealed by a novel marker for slow motoneurons. Development 137, 3489–3499. doi: 10.1242/dev.053348
Chang, Q., and Martin, L. J. (2009). Glycinergic innervation of motoneurons is deficient in amyotrophic lateral sclerosis mice: a quantitative confocal analysis. Am. J. Pathol. 174, 574–585. doi: 10.2353/ajpath.2009.080557
Charcot, J.-M. (1874). De la sclérose latérale amyotrophique. Prog. Med. 2, 325–327, 341–342, 453–455.
Chard, P. S., Bleakman, D., Christakos, S., Fullmer, C. S., and Miller, R. J. (1993). Calcium buffering properties of calbindin D28k and parvalbumin in rat sensory neurones. J. Physiol. 472, 341–357. doi: 10.1113/jphysiol.1993.sp019950
Chen, D., Wang, Y., and Chin, E. R. (2015). Activation of the endoplasmic reticulum stress response in skeletal muscle of G93A∗SOD1 amyotrophic lateral sclerosis mice. Front. Cell Neurosci. 9:170. doi: 10.3389/fncel.2015.00170
Chen, X.-J., Levedakou, E. N., Millen, K. J., Wollmann, R. L., Soliven, B., and Popko, B. (2007). Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic dynein heavy chain 1 gene. J. Neurosci. 27, 14515–14524. doi: 10.1523/JNEUROSCI.4338-07.2007
Chiò, A., Restagno, G., Brunetti, M., Ossola, I., Calvo, A., Mora, G., et al. (2009). Two Italian kindreds with familial amyotrophic lateral sclerosis due to FUS mutation. Neurobiol. Aging 30, 1272–1275. doi: 10.1016/j.neurobiolaging.2009.05.001
Chiu, I. M., Morimoto, E. T. A., Goodarzi, H., Liao, J. T., O’Keeffe, S., Phatnani, H. P., et al. (2013). A neurodegeneration-specific gene expression signature and immune profile of acutely isolated microglia from an ALS mouse model. Cell Rep. 4, 385–401. doi: 10.1016/j.celrep.2013.06.018
Chou, C.-C., Zhang, Y., Umoh, M. E., Vaughan, S. W., Lorenzini, I., Liu, F., et al. (2018). TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 21, 228–239. doi: 10.1038/s41593-017-0047-3
Chou, S. M., and Norris, F. H. (1993). Amyotrophic lateral sclerosis: lower motor neuron disease spreading to upper motor neurons. Muscle Nerve 16, 864–869. doi: 10.1002/mus.880160810
Cirulli, E. T., Lasseigne, B. N., Petrovski, S., Sapp, P. C., Dion, P. A., Leblond, C. S., et al. (2015). Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441. doi: 10.1126/science.aaa3650
Clark, B. C., and Manini, T. M. (2012). What is dynapenia? Nutrition 28, 495–503. doi: 10.1016/j.nut.2011.12.002
Clarke, L. E., Liddelow, S. A., Chakraborty, C., Münch, A. E., Heiman, M., and Barres, B. A. (2018). Normal aging induces A1-like astrocyte reactivity. PNAS 115, E1896–E1905. doi: 10.1073/pnas.1800165115
Comley, L., Allodi, I., Nichterwitz, S., Nizzardo, M., Simone, C., Corti, S., et al. (2015). Motor neurons with differential vulnerability to degeneration show distinct protein signatures in health and ALS. Neuroscience 291, 216–229. doi: 10.1016/j.neuroscience.2015.02.013
Comley, L. H., Nijssen, J., Frost-Nylen, J., and Hedlund, E. (2016). Cross-disease comparison of amyotrophic lateral sclerosis and spinal muscular atrophy reveals conservation of selective vulnerability but differential neuromuscular junction pathology. J. Comp. Neurol. 524, 1424–1442. doi: 10.1002/cne.23917
Corbo, M., and Hays, A. P. (1992). Peripherin and neurofilament protein coexist in spinal spheroids of motor neuron disease. J. Neuropathol. Exp. Neurol. 51, 531–537. doi: 10.1097/00005072-199209000-00008
Cullheim, S., Fleshman, J. W., Glenn, L. L., and Burke, R. E. (1987). Membrane area and dendritic structure in type-identified triceps surae alpha motoneurons. J. Comp. Neurol. 255, 68–81. doi: 10.1002/cne.902550106
Curtis, D. R., and Malik, R. (1985). A neurophysiological analysis of the effect of kainic acid on nerve fibres and terminals in the cat spinal cord. J. Physiol. 368, 99–108. doi: 10.1113/jphysiol.1985.sp015848
Dafinca, R., Scaber, J., Ababneh, N., Lalic, T., Weir, G., Christian, H., et al. (2016). C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 34, 2063–2078. doi: 10.1002/stem.2388
Das, M. M., and Svendsen, C. N. (2015). Astrocytes show reduced support of motor neurons with aging that is accelerated in a rodent model of ALS. Neurobiol. Aging 36, 1130–1139. doi: 10.1016/j.neurobiolaging.2014.09.020
de Carvalho, M., and Swash, M. (2017). Physiology of the fasciculation potentials in amyotrophic lateral sclerosis: which motor units fasciculate? J. Physiol. Sci. 67, 569–576. doi: 10.1007/s12576-016-0484-x
De La Cruz, R. R., Escudero, M., and Delgado-García, J. M. (1989). Behaviour of medial rectus motoneurons in the alert cat. Eur. J. Neurosci. 1, 288–295. doi: 10.1111/j.1460-9568.1989.tb00796.x
De Vos, K. J., Chapman, A. L., Tennant, M. E., Manser, C., Tudor, E. L., Lau, K.-F., et al. (2007). Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 16, 2720–2728. doi: 10.1093/hmg/ddm226
De Vos, K. J., and Hafezparast, M. (2017). Neurobiology of axonal transport defects in motor neuron diseases: opportunities for translational research? Neurobiol. Dis. 105, 283–299. doi: 10.1016/j.nbd.2017.02.004
De Winter, F., Vo, T., Stam, F. J., Wisman, L. A. B., Bär, P. R., Niclou, S. P., et al. (2006). The expression of the chemorepellent Semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol. Cell. Neurosci. 32, 102–117. doi: 10.1016/j.mcn.2006.03.002
Deardorff, A. S., Romer, S. H., Deng, Z., Bullinger, K. L., Nardelli, P., Cope, T. C., et al. (2013). Expression of postsynaptic Ca2+-activated K+ (SK) channels at C-bouton synapses in mammalian lumbar α-motoneurons. J. Physiol. 591, 875–897. doi: 10.1113/jphysiol.2012.240879
DeJesus-Hernandez, M., Desaro, P., Johnston, A., Ross, O. A., Wszolek, Z. K., Ertekin-Taner, N., et al. (2011a). Novel p.Ile151Val mutation in VCP in a patient of African American descent with sporadic ALS. Neurology 77, 1102–1103. doi: 10.1212/WNL.0b013e31822e563c
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011b). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-Linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Delmonico, M. J., Harris, T. B., Visser, M., Park, S. W., Conroy, M. B., Velasquez-Mieyer, P., et al. (2009). Longitudinal study of muscle strength, quality, and adipose tissue infiltration. Am. J. Clin. Nutr. 90, 1579–1585. doi: 10.3945/ajcn.2009.28047
Deng, H.-X., Chen, W., Hong, S.-T., Boycott, K. M., Gorrie, G. H., Siddique, N., et al. (2011). Mutations in UBQLN2 cause dominant X-linked juvenile and adult onset ALS and ALS/dementia. Nature 477, 211–215. doi: 10.1038/nature10353
Deng, H.-X., Zhai, H., Bigio, E. H., Yan, J., Fecto, F., Ajroud, K., et al. (2010). FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 67, 739–748. doi: 10.1002/ana.22051
Deschenes, M. R. (2011). Motor unit and neuromuscular junction remodeling with aging. Curr. Aging Sci. 4, 209–220. doi: 10.2174/1874609811104030209
Deshaies, J.-E., Shkreta, L., Moszczynski, A. J., Sidibé, H., Semmler, S., Fouillen, A., et al. (2018). TDP-43 regulates the alternative splicing of hnRNP A1 to yield an aggregation-prone variant in amyotrophic lateral sclerosis. Brain 141, 1320–1333. doi: 10.1093/brain/awy062
Devenney, E., Hornberger, M., Irish, M., Mioshi, E., Burrell, J., Tan, R., et al. (2014). Frontotemporal dementia associated with the C9ORF72 mutation: a unique clinical profile. JAMA Neurol. 71, 331–339. doi: 10.1001/jamaneurol.2013.6002
Devlin, A.-C., Burr, K., Borooah, S., Foster, J. D., Cleary, E. M., Geti, I., et al. (2015). Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat. Commun. 6:5999. doi: 10.1038/ncomms6999
Dhaliwal, G. K., and Grewal, R. P. (2000). Mitochondrial DNA deletion mutation levels are elevated in ALS brains. Neuroreport 11, 2507–2509. doi: 10.1097/00001756-200008030-00032
Dickey, A. S., Amit, Y., and Hatsopoulos, N. G. (2013). Heterogeneous neural coding of corrective movements in motor cortex. Front. Neural Circ. 7:51. doi: 10.3389/fncir.2013.00051
Diekstra, F. P., van Vught, P. W. J., van Rheenen, W., Koppers, M., Pasterkamp, R. J., van Es, M. A., et al. (2012). UNC13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol. Aging 33, 630.e3–630.e8. doi: 10.1016/j.neurobiolaging.2011.10.029
Dillen, L., Van Langenhove, T., Engelborghs, S., Vandenbulcke, M., Sarafov, S., Tournev, I., et al. (2013). Explorative genetic study of UBQLN2 and PFN1 in an extended flanders-belgian cohort of frontotemporal lobar degeneration patients. Neurobiol. Aging 34, 1711.e1–1711.e5. doi: 10.1016/j.neurobiolaging.2012.12.007
Ding, X., Ma, M., Teng, J., Teng, R. K. F., Zhou, S., Yin, J., et al. (2015). Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes and TNTs-like structure. Oncotarget 6, 24178–24191. doi: 10.18632/oncotarget.4680
Dols-Icardo, O., Nebot, I., Gorostidi, A., Ortega-Cubero, S., Hernández, I., Rojas-García, R., et al. (2015). Analysis of the CHCHD10 gene in patients with frontotemporal dementia and amyotrophic lateral sclerosis from Spain. Brain 138:e400. doi: 10.1093/brain/awv175
Donaghy, C., Thurtell, M. J., Pioro, E. P., Gibson, J. M., and Leigh, R. J. (2011). Eye movements in amyotrophic lateral sclerosis and its mimics: a review with illustrative cases. J. Neurol. Neurosurg. Psychiatry 82, 110–116. doi: 10.1136/jnnp.2010.212407
Dukkipati, S. S., Garrett, T. L., and Elbasiouny, S. M. (2018). The vulnerability of spinal motoneurons and soma size plasticity in a mouse model of amyotrophic lateral sclerosis. J. Physiol. 596, 1723–1745. doi: 10.1113/JP275498
Durand, J. (1989). Intracellular study of oculomotor neurons in the rat. Neuroscience 30, 639–649. doi: 10.1016/0306-4522(89)90157-7
Ebbesen, C. L., and Brecht, M. (2017). Motor cortex—to act or not to act? Nat. Rev. Neurosci. 18, 694–705. doi: 10.1038/nrn.2017.119
Ebstein, S. Y., Yagudayeva, I., and Shneider, N. A. (2019). Mutant TDP-43 causes early-stage dose-dependent motor neuron degeneration in a TARDBP knockin mouse model of ALS. Cell Reports 26, 364.e4–373.e4. doi: 10.1016/j.celrep.2018.12.045
Eccles, J. C., Eccles, R. M., Iggo, A., and Lundberg, A. (1960). Electrophysiological studies on gamma motoneurones. Acta Physiol. Scand. 50, 32–40. doi: 10.1111/j.1748-1716.1960.tb02070.x
Eccles, J. C., Eccles, R. M., and Lundberg, A. (1957). Durations of after-hyperpolarization of motoneurones supplying fast and slow muscles. Nature 179, 866–868. doi: 10.1038/179866b0
Edwards, I. J., Bruce, G., Lawrenson, C., Howe, L., Clapcote, S. J., Deuchars, S. A., et al. (2013). Na+/K+ ATPase α1 and α3 isoforms are differentially expressed in α- and γ-motoneurons. J. Neurosci. 33, 9913–9919. doi: 10.1523/JNEUROSCI.5584-12.2013
Eisen, A., Braak, H., Tredici, K. D., Lemon, R., Ludolph, A. C., and Kiernan, M. C. (2017). Cortical influences drive amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 88, 917–924. doi: 10.1136/jnnp-2017-315573
Eisen, A., Turner, M. R., and Lemon, R. (2014). Tools and talk: an evolutionary perspective on the functional deficits associated with amyotrophic lateral sclerosis. Muscle Nerve 49, 469–477. doi: 10.1002/mus.24132
Elden, A. C., Kim, H.-J., Hart, M. P., Chen-Plotkin, A. S., Johnson, B. S., Fang, X., et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466:1069. doi: 10.1038/nature09320
Endo, F., Komine, O., Fujimori-Tonou, N., Katsuno, M., Jin, S., Watanabe, S., et al. (2015). Astrocyte-derived TGF-β1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep. 11, 592–604. doi: 10.1016/j.celrep.2015.03.053
Enjin, A., Leão, K. E., Mikulovic, S., Le Merre, P., Tourtellotte, W. G., and Kullander, K. (2012). Sensorimotor function is modulated by the serotonin receptor 1d, a novel marker for gamma motor neurons. Mol. Cell. Neurosci. 49, 322–332. doi: 10.1016/j.mcn.2012.01.003
Enjin, A., Rabe, N., Nakanishi, S. T., Vallstedt, A., Gezelius, H., Memic, F., et al. (2010). Identification of novel spinal cholinergic genetic subtypes disclose Chodl and Pitx2 as markers for fast motor neurons and partition cells. J. Comp. Neurol. 518, 2284–2304. doi: 10.1002/cne.22332
Enoka, R. M. (1995). Morphological features and activation patterns of motor units. J. Clin. Neurophysiol. 12, 538–559. doi: 10.1097/00004691-199511000-00002
Enoka, R. M., Christou, E. A., Hunter, S. K., Kornatz, K. W., Semmler, J. G., Taylor, A. M., et al. (2003). Mechanisms that contribute to differences in motor performance between young and old adults. J. Electromyogr. Kinesiol. 13, 1–12. doi: 10.1016/s1050-6411(02)00084-6
Fang, T., Jozsa, F., and Al-Chalabi, A. (2017). Nonmotor symptoms in amyotrophic lateral sclerosis: a systematic review. Int. Rev. Neurobiol. 134, 1409–1441. doi: 10.1016/bs.irn.2017.04.009
Fang, X., Lin, H., Wang, X., Zuo, Q., Qin, J., and Zhang, P. (2015). The NEK1 interactor, C21ORF2, is required for efficient DNA damage repair. Acta Biochim. Biophys. Sin. 47, 834–841. doi: 10.1093/abbs/gmv076
Farg, M. A., Konopka, A., Soo, K. Y., Ito, D., and Atkin, J. D. (2017). The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum. Mol. Genet. 26, 2882–2896. doi: 10.1093/hmg/ddx170
Farg, M. A., Soo, K. Y., Walker, A. K., Pham, H., Orian, J., Horne, M. K., et al. (2012). Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol. Aging 33, 2855–2868. doi: 10.1016/j.neurobiolaging.2012.02.009
Farg, M. A., Sundaramoorthy, V., Sultana, J. M., Yang, S., Atkinson, R. A., Levina, V., et al. (2014). C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 23, 3579–3595. doi: 10.1093/hmg/ddu068
Fecto, F., Yan, J., Vemula, S. P., Liu, E., Yang, Y., Chen, W., et al. (2011). SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 68, 1440–1446. doi: 10.1001/archneurol.2011.250
Feiguin, F., Godena, V. K., Romano, G., D’Ambrogio, A., Klima, R., and Baralle, F. E. (2009). Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 583, 1586–1592. doi: 10.1016/j.febslet.2009.04.019
Feiler, M. S., Strobel, B., Freischmidt, A., Helferich, A. M., Kappel, J., Brewer, B. M., et al. (2015). TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 211, 897–911. doi: 10.1083/jcb.201504057
Feit, H., Silbergleit, A., Schneider, L. B., Gutierrez, J. A., Fitoussi, R.-P., Réyes, C., et al. (1998). Vocal cord and pharyngeal weakness with autosomal dominant distal myopathy: clinical description and gene localization to 5q31. Am. J. Hum. Genet. 63, 1732–1742. doi: 10.1086/302166
Feng, Z., and Ko, C.-P. (2008). Schwann cells promote synaptogenesis at the neuromuscular junction via transforming growth factor-β1. J. Neurosci. 28, 9599–9609. doi: 10.1523/JNEUROSCI.2589-08.2008
Ferrante, R. J., Browne, S. E., Shinobu, L. A., Bowling, A. C., Baik, M. J., MacGarvey, U., et al. (1997). Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 69, 2064–2074. doi: 10.1046/j.1471-4159.1997.69052064.x
Ferri, A., Cozzolino, M., Crosio, C., Nencini, M., Casciati, A., Gralla, E. B., et al. (2006). Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. U.S.A. 103, 13860–13865. doi: 10.1073/pnas.0605814103
Ferrier, D. (1875). Experiments on the brain of monkeys.—No. I. Proc. R. Soc. Lond. 23, 409–430. doi: 10.1098/rspl.1874.0058
Fiatarone, M. A., and Evans, W. J. (1993). The etiology and reversibility of muscle dysfunction in the aged. J. Gerontol. 48, 77–83. doi: 10.1093/geronj/48.special_issue.77
Figlewicz, D. A., Krizus, A., Martinoli, M. G., Meininger, V., Dib, M., Rouleau, G. A., et al. (1994). Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum. Mol. Genet. 3, 1757–1761. doi: 10.1093/hmg/3.10.1757
Filézac de L’Etang, A., Maharjan, N., Cordeiro Braña, M., Ruegsegger, C., Rehmann, R., Goswami, A., et al. (2015). Marinesco-Sjögren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress in ALS. Nat. Neurosci. 18, 227–238. doi: 10.1038/nn.3903
Fischer, L. R., Culver, D. G., Tennant, P., Davis, A. A., Wang, M., Castellano-Sanchez, A., et al. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240. doi: 10.1016/j.expneurol.2003.10.004
Fogarty, M. J. (2018). Driven to decay: Excitability and synaptic abnormalities in amyotrophic lateral sclerosis. Brain Res. Bull. 140, 318–333. doi: 10.1016/j.brainresbull.2018.05.023
Franco, M. C., Dennys, C. N., Estévez, F. H. R., and Estévez, A. G. (2013). “Superoxide Dismutase and Oxidative Stress in Amyotrophic Lateral Sclerosis,” in Current Advances in Amyotrophic Lateral Sclerosis, ed. A. Estévez (London: IntechOpen).
Fratta, P., and Isaacs, A. M. (2018). The snowball effect of RNA binding protein dysfunction in amyotrophic lateral sclerosis. Brain 141, 1236–1238. doi: 10.1093/brain/awy091
Freischmidt, A., Wieland, T., Richter, B., Ruf, W., Schaeffer, V., Müller, K., et al. (2015). Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636. doi: 10.1038/nn.4000
Frey, D., Schneider, C., Xu, L., Borg, J., Spooren, W., and Caroni, P. (2000). Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J. Neurosci. 20, 2534–2542. doi: 10.1523/jneurosci.20-07-02534.2000
Fried, L. P., Ferrucci, L., Darer, J., Williamson, J. D., and Anderson, G. (2004). Untangling the concepts of disability, frailty, and comorbidity: implications for improved targeting and care. J. Gerontol. A Biol. Sci. Med. Sci. 59, 255–263.
Friese, A., Kaltschmidt, J. A., Ladle, D. R., Sigrist, M., Jessell, T. M., and Arber, S. (2009). Gamma and alpha motor neurons distinguished by expression of transcription factor Err3. Proc. Natl. Acad. Sci. U.S.A. 106, 13588–13593. doi: 10.1073/pnas.0906809106
Fryer, H. J., Knox, R. J., Strittmatter, S. M., and Kalb, R. G. (1999). Excitotoxic death of a subset of embryonic rat motor neurons in vitro. J. Neurochem. 72, 500–513. doi: 10.1046/j.1471-4159.1999.0720500.x
Fu, H., Hardy, J., and Duff, K. E. (2018). Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 21:1350. doi: 10.1038/s41593-018-0221-2
Fuchs, A. F., Scudder, C. A., and Kaneko, C. R. (1988). Discharge patterns and recruitment order of identified motoneurons and internuclear neurons in the monkey abducens nucleus. J. Neurophysiol. 60, 1874–1895. doi: 10.1152/jn.1988.60.6.1874
Fujimura-Kiyono, C., Kimura, F., Ishida, S., Nakajima, H., Hosokawa, T., Sugino, M., et al. (2011). Onset and spreading patterns of lower motor neuron involvements predict survival in sporadic amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 82, 1244–1249. doi: 10.1136/jnnp-2011-300141
Fujita, K., Yamauchi, M., Shibayama, K., Ando, M., Honda, M., and Nagata, Y. (1996). Decreased cytochrome c oxidase activity but unchanged superoxide dismutase and glutathione peroxidase activities in the spinal cords of patients with amyotrophic lateral sclerosis. J. Neurosci. Res. 45, 276–281. doi: 10.1002/(sici)1097-4547(19960801)45:3<276::aid-jnr9>3.0.co;2-a
Fujita, S., and Kitamura, T. (1975). Origin of brain macrophages and the nature of the so-called microglia. Acta Neuropathol. Suppl. 6, 291–296. doi: 10.1007/978-3-662-08456-4_51
García, M. L., Fernández, A., and Solas, M. T. (2013). Mitochondria, motor neurons and aging. J. Neurol. Sci. 330, 18–26. doi: 10.1016/j.jns.2013.03.019
Gardiner, P. F. (1993). Physiological properties of motoneurons innervating different muscle unit types in rat gastrocnemius. J. Neurophysiol. 69, 1160–1170. doi: 10.1152/jn.1993.69.4.1160
Gargiulo-Monachelli, G. M., Janota, F., Bettini, M., Shoesmith, C. L., Strong, M. J., and Sica, R. E. P. (2012). Regional spread pattern predicts survival in patients with sporadic amyotrophic lateral sclerosis. Eur. J. Neurol. 19, 834–841. doi: 10.1111/j.1468-1331.2011.03616.x
Georgopoulos, A. P., Kalaska, J. F., Caminiti, R., and Massey, J. T. (1982). On the relations between the direction of two-dimensional arm movements and cell discharge in primate motor cortex. J. Neurosci. 2, 1527–1537. doi: 10.1523/jneurosci.02-11-01527.1982
Geser, F., Brandmeir, N. J., Kwong, L. K., Martinez-Lage, M., Elman, L., McCluskey, L., et al. (2008). Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch. Neurol. 65, 636–641. doi: 10.1001/archneur.65.5.636
Gioanni, Y., and Lamarche, M. (1985). A reappraisal of rat motor cortex organization by intracortical microstimulation. Brain Res. 344, 49–61. doi: 10.1016/0006-8993(85)91188-6
Gitcho, M. A., Baloh, R. H., Chakraverty, S., Mayo, K., Norton, J. B., Levitch, D., et al. (2008). TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 63, 535–538. doi: 10.1002/ana.21344
Grad, L. I., Guest, W. C., Yanai, A., Pokrishevsky, E., O’Neill, M. A., Gibbs, E., et al. (2011). Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. PNAS 108, 16398–16403. doi: 10.1073/pnas.1102645108
Grad, L. I., Yerbury, J. J., Turner, B. J., Guest, W. C., Pokrishevsky, E., O’Neill, M. A., et al. (2014). Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. U.S.A. 111, 3620–3625. doi: 10.1073/pnas.1312245111
Gravel, M., Béland, L.-C., Soucy, G., Abdelhamid, E., Rahimian, R., Gravel, C., et al. (2016). IL-10 controls early microglial phenotypes and disease onset in als caused by misfolded superoxide dismutase 1. J. Neurosci. 36, 1031–1048. doi: 10.1523/JNEUROSCI.0854-15.2016
Greenway, M. J., Andersen, P. M., Russ, C., Ennis, S., Cashman, S., Donaghy, C., et al. (2006). ANG mutations segregate with familial and “sporadic” amyotrophic lateral sclerosis. Nat. Genet. 38, 411–413. doi: 10.1038/ng1742
Guéritaud, J. P., Horcholle-Bossavit, G., Jami, L., Thiesson, D., and Tyc-Dumont, S. (1985). Resistance to glycogen depletion of motor units in the cat rectus lateralis muscle. Exp. Brain Res. 60, 542–550.
Gulino, R., Forte, S., Parenti, R., and Gulisano, M. (2015). TDP-43 as a modulator of synaptic plasticity in a mouse model of spinal motoneuron degeneration. CNS Neurol. Disord. Drug Targets 14, 55–60. doi: 10.2174/1871527314666150116115414
Gurney, M. E. (1997). The use of transgenic mouse models of amyotrophic lateral sclerosis in preclinical drug studies. J. Neurol. Sci. 152(Suppl. 1),S67–S73.
Gurney, M. E., Pu, H., Chiu, A. Y., Canto, M. D., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775. doi: 10.1126/science.8209258
Gustafsson, B., and Lipski, J. (1979). Do γ-motoneurones lack a long-lasting afterhyperpolarization? Brain Res. 172, 349–353. doi: 10.1016/0006-8993(79)90545-6
Hadzipasic, M., Tahvildari, B., Nagy, M., Bian, M., Horwich, A. L., and McCormick, D. A. (2014). Selective degeneration of a physiological subtype of spinal motor neuron in mice with SOD1-linked ALS. Proc. Natl. Acad. Sci. U.S.A. 111, 16883–16888. doi: 10.1073/pnas.1419497111
Haenggeli, C., Julien, J.-P., Mosley, R. L., Perez, N., Dhar, A., Gendelman, H. E., et al. (2007). Therapeutic immunization with a glatiramer acetate derivative does not alter survival in G93A and G37R SOD1 mouse models of familial ALS. Neurobiol. Dis. 26, 146–152. doi: 10.1016/j.nbd.2006.12.013
Hall, C. E., Yao, Z., Choi, M., Tyzack, G. E., Serio, A., Luisier, R., et al. (2017). Progressive motor neuron pathology and the role of astrocytes in a human stem cell model of vcp-related ALS. Cell Rep. 19, 1739–1749. doi: 10.1016/j.celrep.2017.05.024
Hall, E. D., Andrus, P. K., Oostveen, J. A., Fleck, T. J., and Gurney, M. E. (1998). Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS. J. Neurosci. Res. 53, 66–77. doi: 10.1002/(sici)1097-4547(19980701)53:1<66::aid-jnr7>3.3.co;2-k
Harms, M. B., Cady, J., Zaidman, C., Cooper, P., Bali, T., Allred, P., et al. (2013). Lack of C9ORF72 coding mutations supports a gain of function for repeat expansions in amyotrophic lateral sclerosis. Neurobiol. Aging 34, 2234.e13–2234.e19. doi: 10.1016/j.neurobiolaging.2013.03.006
Harris, J. J., Jolivet, R., and Attwell, D. (2012). Synaptic energy use and supply. Neuron 75, 762–777. doi: 10.1016/j.neuron.2012.08.019
Hashizume, K., Kanda, K., and Burke, R. E. (1988). Medial gastrocnemius motor nucleus in the rat: age-related changes in the number and size of motoneurons. J. Comp. Neurol. 269, 425–430. doi: 10.1002/cne.902690309
He, C. Z., and Hays, A. P. (2004). Expression of peripherin in ubiquinated inclusions of amyotrophic lateral sclerosis. J. Neurol. Sci. 217, 47–54. doi: 10.1016/j.jns.2003.08.016
Hedlund, E., Karlsson, M., Osborn, T., Ludwig, W., and Isacson, O. (2010). Global gene expression profiling of somatic motor neuron populations with different vulnerability identify molecules and pathways of degeneration and protection. Brain 133, 2313–2330. doi: 10.1093/brain/awq167
Hegedus, J., Putman, C. T., and Gordon, T. (2007). Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 28, 154–164. doi: 10.1016/j.nbd.2007.07.003
Henneman, E. (1957). Relation between size of neurons and their susceptibility to discharge. Science 126, 1345–1347. doi: 10.1126/science.126.3287.1345
Hickey, W. F., and Kimura, H. (1988). Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science 239, 290–292. doi: 10.1126/science.3276004
Hideyama, T., Yamashita, T., Aizawa, H., Tsuji, S., Kakita, A., Takahashi, H., et al. (2012). Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol. Dis. 45, 1121–1128. doi: 10.1016/j.nbd.2011.12.033
Hideyama, T., Yamashita, T., Suzuki, T., Tsuji, S., Higuchi, M., Seeburg, P. H., et al. (2010). Induced loss of ADAR2 engenders slow death of motor neurons from Q/R site-unedited GluR2. J. Neurosci. 30, 11917–11925. doi: 10.1523/JNEUROSCI.2021-10.2010
Higelin, J., Demestre, M., Putz, S., Delling, J. P., Jacob, C., Lutz, A.-K., et al. (2016). FUS mislocalization and vulnerability to DNA damage in ALS patients derived hiPSCs and aging motoneurons. Front. Cell. Neurosci. 10:290. doi: 10.3389/fncel.2016.00290
Hill, S. J., Mordes, D. A., Cameron, L. A., Neuberg, D. S., Landini, S., Eggan, K., et al. (2016). Two familial ALS proteins function in prevention/repair of transcription-associated DNA damage. PNAS 113, E7701–E7709. doi: 10.1073/pnas.1611673113
Hira, R., Ohkubo, F., Tanaka, Y. R., Masamizu, Y., Augustine, G. J., Kasai, H., et al. (2013). In vivo optogenetic tracing of functional corticocortical connections between motor forelimb areas. Front. Neural Circ. 7:55. doi: 10.3389/fncir.2013.00055
Hirano, A., Nakano, I., Kurland, L. T., Mulder, D. W., Holley, P. W., and Saccomanno, G. (1984). Fine structural study of neurofibrillary changes in a family with amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 43, 471–480. doi: 10.1097/00005072-198409000-00002
Hochstim, C., Deneen, B., Lukaszewicz, A., Zhou, Q., and Anderson, D. J. (2008). Identification of positionally distinct astrocyte subtypes whose identities are specified by a homeodomain code. Cell 133, 510–522. doi: 10.1016/j.cell.2008.02.046
Hossaini, M., Cano, S. C., van Dis, V., Haasdijk, E. D., Hoogenraad, C. C., Holstege, J. C., et al. (2011). Spinal inhibitory interneuron pathology follows motor neuron degeneration independent of glial mutant superoxide dismutase 1 expression in SOD1-ALS Mice. J. Neuropathol. Exp. Neurol. 70, 662–677. doi: 10.1097/NEN.0b013e31822581ac
Hsiung, G.-Y. R., DeJesus-Hernandez, M., Feldman, H. H., Sengdy, P., Bouchard-Kerr, P., Dwosh, E., et al. (2012). Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 135, 709–722. doi: 10.1093/brain/awr354
Huang, C., Huang, B., Bi, F., Yan, L. H., Tong, J., Huang, J., et al. (2014). Profiling the genes affected by pathogenic TDP-43 in astrocytes. J. Neurochem. 129, 932–939. doi: 10.1111/jnc.12660
Huang, C., Tong, J., Bi, F., Wu, Q., Huang, B., Zhou, H., et al. (2012). Entorhinal cortical neurons are the primary targets of FUS mislocalization and ubiquitin aggregation in FUS transgenic rats. Hum. Mol. Genet. 21, 4602–4614. doi: 10.1093/hmg/dds299
Huber, A. B., Kania, A., Tran, T. S., Gu, C., De Marco Garcia, N., Lieberam, I., et al. (2005). Distinct roles for secreted semaphorin signaling in spinal motor axon guidance. Neuron 48, 949–964. doi: 10.1016/j.neuron.2005.12.003
Hugon, J., Vallat, J. M., Spencer, P. S., Leboutet, M. J., and Barthe, D. (1989). Kainic acid induces early and delayed degenerative neuronal changes in rat spinal cord. Neurosci. Lett. 104, 258–262. doi: 10.1016/0304-3940(89)90585-5
Hunt, C. C., and Kuffler, S. W. (1951). Further study of efferent small-nerve fibres to mammalian muscle spindles. multiple spindle innervation and activity during contraction. J. Physiol. 113, 283–297. doi: 10.1113/jphysiol.1951.sp004572
Hyder, F., Rothman, D. L., and Bennett, M. R. (2013). Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. U.S.A. 110, 3549–3554. doi: 10.1073/pnas.1214912110
Iguchi, Y., Katsuno, M., Niwa, J., Takagi, S., Ishigaki, S., Ikenaka, K., et al. (2013). Loss of TDP-43 causes age-dependent progressive motor neuron degeneration. Brain 136, 1371–1382. doi: 10.1093/brain/awt029
Ikonomidou, C., Qin Qin, Y., Labruyere, J., and Olney, J. W. (1996). Motor neuron degeneration induced by excitotoxin agonists has features in common with those seen in the SOD-1 transgenic mouse model of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 55, 211–224. doi: 10.1097/00005072-199602000-00010
Ilieva, E. V., Ayala, V., Jové, M., Dalfó, E., Cacabelos, D., Povedano, M., et al. (2007). Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 130, 3111–3123. doi: 10.1093/brain/awm190
Ilieva, H., Polymenidou, M., and Cleveland, D. W. (2009). Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 187, 761–772. doi: 10.1083/jcb.200908164
Ilieva, H. S., Yamanaka, K., Malkmus, S., Kakinohana, O., Yaksh, T., Marsala, M., et al. (2008). Mutant dynein (Loa) triggers proprioceptive axon loss that extends survival only in the SOD1 ALS model with highest motor neuron death. Proc. Natl. Acad. Sci. U.S.A. 105, 12599–12604. doi: 10.1073/pnas.0805422105
Irwin, D. J., McMillan, C. T., Brettschneider, J., Libon, D. J., Powers, J., Rascovsky, K., et al. (2013). Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 84, 163–169. doi: 10.1136/jnnp-2012-303507
Israelson, A., Arbel, N., Da Cruz, S., Ilieva, H., Yamanaka, K., Shoshan-Barmatz, V., et al. (2010). Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 67, 575–587. doi: 10.1016/j.neuron.2010.07.019
Ito, H., Wate, R., Zhang, J., Ohnishi, S., Kaneko, S., Ito, H., et al. (2008). Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Exp. Neurol. 213, 448–455. doi: 10.1016/j.expneurol.2008.07.017
Jaarsma, D., Teuling, E., Haasdijk, E. D., De Zeeuw, C. I., and Hoogenraad, C. C. (2008). Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J. Neurosci. 28, 2075–2088. doi: 10.1523/JNEUROSCI.5258-07.2008
Javed, K., and Lui, F. (2018). “Neuroanatomy, Lateral Corticospinal Tract,” in StatPearls, ed. M. Varacallo (Treasure Island FL: StatPearls Publishing).
Jeon, G. S., Nakamura, T., Lee, J.-S., Choi, W.-J., Ahn, S.-W., Lee, K.-W., et al. (2014). Potential effect of s-nitrosylated protein disulfide isomerase on mutant SOD1 aggregation and neuronal cell death in amyotrophic lateral sclerosis. Mol. Neurobiol. 49, 796–807. doi: 10.1007/s12035-013-8562-z
Jiang, M., Schuster, J. E., Fu, R., Siddique, T., and Heckman, C. J. (2009). Progressive changes in synaptic inputs to motoneurons in adult sacral spinal cord of a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 29, 15031–15038. doi: 10.1523/JNEUROSCI.0574-09.2009
Jiang, S. X., Whitehead, S., Aylsworth, A., Slinn, J., Zurakowski, B., Chan, K., et al. (2010). Neuropilin 1 directly interacts with Fer kinase to mediate semaphorin 3A-induced death of cortical neurons. J. Biol. Chem. 285, 9908–9918. doi: 10.1074/jbc.M109.080689
Jiang, Y.-M., Yamamoto, M., Kobayashi, Y., Yoshihara, T., Liang, Y., Terao, S., et al. (2005). Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 57, 236–251. doi: 10.1002/ana.20379
Jin, H., Mimura, N., Kashio, M., Koseki, H., and Aoe, T. (2014). Late-onset of spinal neurodegeneration in knock-in mice expressing a mutant biP. PLoS One 9:e112837. doi: 10.1371/journal.pone.0112837
Johnson, J. O., Glynn, S. M., Gibbs, J. R., Nalls, M. A., Sabatelli, M., Restagno, G., et al. (2014a). Mutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosis. Brain 137:e311. doi: 10.1093/brain/awu265
Johnson, J. O., Pioro, E. P., Boehringer, A., Chia, R., Feit, H., Renton, A. E., et al. (2014b). Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 17:664.
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., Van Deerlin, V. M., Trojanowski, J. Q., et al. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864. doi: 10.1016/j.neuron.2010.11.036
Jonsson, P. A., Graffmo, K. S., Brännström, T., Nilsson, P., Andersen, P. M., and Marklund, S. L. (2006). Motor neuron disease in mice expressing the wild type-like D90A mutant superoxide dismutase-1. J. Neuropathol. Exp. Neurol. 65, 1126–1136. doi: 10.1097/01.jnen.0000248545.36046.3c
Kabashi, E., El Oussini, H., Bercier, V., Gros-Louis, F., Valdmanis, P. N., McDearmid, J., et al. (2013). Investigating the contribution of VAPB/ALS8 loss of function in amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 2350–2360. doi: 10.1093/hmg/ddt080
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., McConkey, B. J., Vande Velde, C., et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. doi: 10.1038/ng.132
Kadhiresan, V. A., Hassett, C. A., and Faulkner, J. A. (1996). Properties of single motor units in medial gastrocnemius muscles of adult and old rats. J. Physiol. 493, 543–552. doi: 10.1113/jphysiol.1996.sp021402
Kanai, Y., Okada, Y., Tanaka, Y., Harada, A., Terada, S., and Hirokawa, N. (2000). KIF5C, a novel neuronal kinesin enriched in motor neurons. J. Neurosci. 20, 6374–6384. doi: 10.1523/JNEUROSCI.20-17-06374.2000
Kanda, K., and Hashizume, K. (1989). Changes in properties of the medial gastrocnemius motor units in aging rats. J. Neurophysiol. 61, 737–746. doi: 10.1152/jn.1989.61.4.737
Kang, S. H., Li, Y., Fukaya, M., Lorenzini, I., Cleveland, D. W., Ostrow, L. W., et al. (2013). Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 16, 571–579. doi: 10.1038/nn.3357
Kanning, K. C., Kaplan, A., and Henderson, C. E. (2010). Motor neuron diversity in development and disease. Ann. Rev. Neurosci. 33, 409–440. doi: 10.1146/annurev.neuro.051508.135722
Kaplan, A., Spiller, K. J., Towne, C., Kanning, K. C., Choe, G. T., Geber, A., et al. (2014). Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron 81, 333–348. doi: 10.1016/j.neuron.2013.12.009
Kawahara, Y., Ito, K., Sun, H., Aizawa, H., Kanazawa, I., and Kwak, S. (2004). Glutamate receptors: RNA editing and death of motor neurons. Nature 427:801. doi: 10.1038/427801a
Kawahara, Y., Kwak, S., Sun, H., Ito, K., Hashida, H., Aizawa, H., et al. (2003). Human spinal motoneurons express low relative abundance of GluR2 mRNA: an implication for excitotoxicity in ALS. J. Neurochem. 85, 680–689. doi: 10.1046/j.1471-4159.2003.01703.x
Kawamata, T., Akiyama, H., Yamada, T., and McGeer, P. L. (1992). Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am. J. Pathol. 140, 691–707.
Kemm, R. E., and Westbury, D. R. (1978). Some properties of spinal gamma-motoneurones in the cat, determined by micro-electrode recording. J. Physiol. 282, 59–71. doi: 10.1113/jphysiol.1978.sp012448
Kernell, D., and Zwaagstra, B. (1981). Input conductance axonal conduction velocity and cell size among hindlimb motoneurones of the cat. Brain Res. 204, 311–326. doi: 10.1016/0006-8993(81)90591-6
Kia, A., McAvoy, K., Krishnamurthy, K., Trotti, D., and Pasinelli, P. (2018). Astrocytes expressing ALS-linked mutant FUS induce motor neuron death through release of tumor necrosis factor-alpha. Glia 66, 1016–1033. doi: 10.1002/glia.23298
Kiehn, O. (2016). Decoding the organization of spinal circuits that control locomotion. Nat. Rev. Neurosci. 17, 224–238. doi: 10.1038/nrn.2016.9
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., et al. (2011). Amyotrophic lateral sclerosis. Lancet 377, 942–955. doi: 10.1016/S0140-6736(10)61156-7
Kikis, E. A., Gidalevitz, T., and Morimoto, R. I. (2010). Protein homeostasis in models of aging and age-related conformational disease. Adv. Exp. Med. Biol. 694, 138–159. doi: 10.1007/978-1-4419-7002-2_11
Kim, H. J., Kim, N. C., Wang, Y.-D., Scarborough, E. A., Moore, J., Diaz, Z., et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467. doi: 10.1038/nature11922
Kim, H.-J., Kwon, M.-J., Choi, W.-J., Oh, K.-W., Oh, S.-I., Ki, C.-S., et al. (2014). Mutations in UBQLN2 and SIGMAR1 genes are rare in Korean patients with amyotrophic lateral sclerosis. Neurobiol. Aging 35, 1957.e7–1957.e8. doi: 10.1016/j.neurobiolaging.2014.03.001
Kim, H. J., and Taylor, J. P. (2017). Lost in transportation: nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron 96, 285–297. doi: 10.1016/j.neuron.2017.07.029
Kirby, J., Goodall, E. F., Smith, W., Highley, J. R., Masanzu, R., Hartley, J. A., et al. (2010). Broad clinical phenotypes associated with TAR-DNA binding protein (TARDBP) mutations in amyotrophic lateral sclerosis. Neurogenetics 11, 217–225. doi: 10.1007/s10048-009-0218-9
Kiskinis, E., Sandoe, J., Williams, L. A., Boulting, G. L., Moccia, R., Wainger, B. J., et al. (2014). Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14, 781–795. doi: 10.1016/j.stem.2014.03.004
Kong, J., and Xu, Z. (1998). Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 18, 3241–3250. doi: 10.1523/jneurosci.18-09-03241.1998
Konopka, A., and Atkin, J. D. (2018). The emerging role of DNA damage in the pathogenesis of the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 19:E3137. doi: 10.3390/ijms19103137
Körner, S., Böselt, S., Wichmann, K., Thau-Habermann, N., Zapf, A., Knippenberg, S., et al. (2016). The axon guidance protein semaphorin 3A is increased in the motor cortex of patients with amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. doi: 10.1093/jnen/nlw003 [Epub ahead of print].
Kortenbruck, G., Berger, E., Speckmann, E., and Musshoff, U. (2001). RNA editing at the Q/R site for the glutamate receptor subunits GLUR2, GLUR5, and GLUR6 in hippocampus and temporal cortex from epileptic patients - sciencedirect. Neurobiol. Dis. 8, 459–468. doi: 10.1006/nbdi.2001.0394
Kruman, I. I., Pedersen, W. A., Springer, J. E., and Mattson, M. P. (1999). ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis. Exp. Neurol. 160, 28–39. doi: 10.1006/exnr.1999.7190
Kuffler, S. W., Hunt, C. C., and Quilliam, J. P. (1951). Function of medullated small-nerve fibers in mammalian ventral roots; efferent muscle spindle innervation. J. Neurophysiol. 14, 29–54. doi: 10.1152/jn.1951.14.1.29
Kuijpers, M., van Dis, V., Haasdijk, E. D., Harterink, M., Vocking, K., Post, J. A., et al. (2013). Amyotrophic lateral sclerosis (ALS)-associated VAPB-P56S inclusions represent an ER quality control compartment. Acta Neuropathol. Commun. 1:24. doi: 10.1186/2051-5960-1-24
Kuzuhara, S., Kanazawa, I., and Nakanishi, T. (1980). Topographical localization of the Onuf’s nuclear neurons innervating the rectal and vesical striated sphincter muscles: a retrograde fluorescent double labeling in cat and dog. Neurosci. Lett. 16, 125–130. doi: 10.1016/0304-3940(80)90331-6
Kwiatkowski, T. J., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Lacomblez, L., Bensimon, G., Leigh, P. N., Guillet, P., and Meininger, V. (1996). Dose-ranging study of riluzole in amyotrophic lateral sclerosis. amyotrophic lateral sclerosis/riluzole study group II. Lancet 347, 1425–1431. doi: 10.1016/s0140-6736(96)91680-3
Laffita-Mesa, J. M., Pupo, J. M. R., Sera, R. M., Mojena, Y. V., Kourí, V., Laguna-Salvia, L., et al. (2013). De novo mutations in ataxin-2 gene and ALS risk. PLoS One 8:e70560. doi: 10.1371/journal.pone.0070560
LaFrance, R., Brustovetsky, N., Sherburne, C., Delong, D., and Dubinsky, J. M. (2005). Age-related changes in regional brain mitochondria from Fischer 344 rats. Aging Cell 4, 139–145. doi: 10.1111/j.1474-9726.2005.00156.x
Lalancette-Hebert, M., Sharma, A., Lyashchenko, A. K., and Shneider, N. A. (2016). Gamma motor neurons survive and exacerbate alpha motor neuron degeneration in ALS. PNAS 113, E8316–E8325. doi: 10.1073/pnas.1605210113
Lall, D., and Baloh, R. H. (2017). Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Invest. 127, 3250–3258. doi: 10.1172/JCI90607
LaMonte, B. H., Wallace, K. E., Holloway, B. A., Shelly, S. S., Ascaño, J., Tokito, M., et al. (2002). Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron 34, 715–727. doi: 10.1016/s0896-6273(02)00696-7
Landers, J. E., Melki, J., Meininger, V., Glass, J. D., van den Berg, L. H., van Es, M. A., et al. (2009). Reduced expression of the kinesin-associated protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 106, 9004–9009. doi: 10.1073/pnas.0812937106
Laslo, P., Lipski, J., Nicholson, L. F., Miles, G. B., and Funk, G. D. (2000). Calcium binding proteins in motoneurons at low and high risk for degeneration in ALS. Neuroreport 11, 3305–3308. doi: 10.1097/00001756-200010200-00009
Lattante, S., Rouleau, G. A., and Kabashi, E. (2013). TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum. Mutat. 34, 812–826. doi: 10.1002/humu.22319
Lauretani, F., Russo, C. R., Bandinelli, S., Bartali, B., Cavazzini, C., Di Iorio, A., et al. (2003). Age-associated changes in skeletal muscles and their effect on mobility: an operational diagnosis of sarcopenia. J. Appl. Physiol. 95, 1851–1860. doi: 10.1152/japplphysiol.00246.2003
Lautenschläger, J., Prell, T., Ruhmer, J., Weidemann, L., Witte, O. W., and Grosskreutz, J. (2013). Overexpression of human mutated G93A SOD1 changes dynamics of the ER mitochondria calcium cycle specifically in mouse embryonic motor neurons. Exp. Neurol. 247, 91–100. doi: 10.1016/j.expneurol.2013.03.027
Lawson, L. J., Perry, V. H., Dri, P., and Gordon, S. (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39, 151–170. doi: 10.1016/0306-4522(90)90229-w
Le, N. T. T., Chang, L., Kovlyagina, I., Georgiou, P., Safren, N., Braunstein, K. E., et al. (2016). Motor neuron disease, TDP-43 pathology, and memory deficits in mice expressing ALS-FTD-linked UBQLN2 mutations. Proc. Natl. Acad. Sci. U.S.A. 113, E7580–E7589. doi: 10.1073/pnas.1608432113
Le Ber, I., Camuzat, A., Guerreiro, R., Bouya-Ahmed, K., Bras, J., Nicolas, G., et al. (2013). SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol. 70, 1403–1410. doi: 10.1001/jamaneurol.2013.3849
Le Masson, G., Przedborski, S., and Abbott, L. F. (2014). A computational model of motor neuron degeneration. Neuron 83, 975–988. doi: 10.1016/j.neuron.2014.07.001
Lee, S., and Kim, H.-J. (2015). Prion-like mechanism in amyotrophic lateral sclerosis: are protein aggregates the key? Exp. Neurobiol. 24, 1–7. doi: 10.5607/en.2015.24.1.1
Lemon, R. N. (2008). Descending pathways in motor control. Annu. Rev. Neurosci. 31, 195–218. doi: 10.1146/annurev.neuro.31.060407.125547
Leroy, F., Lamotte d’Incamps, B., Imhoff-Manuel, R. D., and Zytnicki, D. (2014). Early intrinsic hyperexcitability does not contribute to motoneuron degeneration in amyotrophic lateral sclerosis. eLife 3:e04046. doi: 10.7554/eLife.04046
Leyton, A. S. F., and Sherrington, C. S. (1917). Observations on the excitable cortex of the chimpanzee, orang-utan, and gorilla. Q. J. Exp. Physiol. 11, 135–222. doi: 10.1113/expphysiol.1917.sp000240
Liao, B., Zhao, W., Beers, D. R., Henkel, J. S., and Appel, S. H. (2012). Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 237, 147–152. doi: 10.1016/j.expneurol.2012.06.011
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Ligon, L. A., LaMonte, B. H., Wallace, K. E., Weber, N., Kalb, R. G., and Holzbaur, E. L. F. (2005). Mutant superoxide dismutase disrupts cytoplasmic dynein in motor neurons. Neuroreport 16, 533–536. doi: 10.1097/00001756-200504250-00002
Lin, F. R. (2011). Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum. Mol. Genet. 20, 1697–1700. doi: 10.1093/hmg/ddr045
Lino, M. M., Schneider, C., and Caroni, P. (2002). Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J. Neurosci. 22, 4825–4832. doi: 10.1523/jneurosci.22-12-04825.2002
Lips, M. B., and Keller, B. U. (1998). Endogenous calcium buffering in motoneurones of the nucleus hypoglossus from mouse. J. Physiol. 511, 105–117. doi: 10.1111/j.1469-7793.1998.105bi.x
Lips, M. B., and Keller, B. U. (1999). Activity-related calcium dynamics in motoneurons of the nucleus hypoglossus from mouse. J. Neurophysiol. 82, 2936–2946. doi: 10.1152/jn.1999.82.6.2936
Liu, D., Wen, J., Liu, J., and Li, L. (1999). The roles of free radicals in amyotrophic lateral sclerosis: reactive oxygen species and elevated oxidation of protein, DNA, and membrane phospholipids. FASEB J. 13, 2318–2328. doi: 10.1096/fasebj.13.15.2318
Liu, J., Lillo, C., Jonsson, P. A., Vande Velde, C., Ward, C. M., Miller, T. M., et al. (2004). Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 43, 5–17. doi: 10.1016/j.neuron.2004.06.016
Liu, R., Althaus, J. S., Ellerbrock, B. R., Becker, D. A., and Gurney, M. E. (1998). Enhanced oxygen radical production in a transgenic mouse model of familial amyotrophic lateral sclerosis. Ann. Neurol. 44, 763–770. doi: 10.1002/ana.410440510
Liu, W., D’Ercole, J. A., and Ye, P. (2011). Blunting type 1 insulin-like growth factor receptor expression exacerbates neuronal apoptosis following hypoxic/ischemic injury. BMC Neurosci. 12:64. doi: 10.1186/1471-2202-12-64
Liu, X., Yang, L., Tang, L., Chen, L., Liu, X., and Fan, D. (2017). DCTN1 gene analysis in Chinese patients with sporadic amyotrophic lateral sclerosis. PLoS One 12:e0182572. doi: 10.1371/journal.pone.0182572
Lobsiger, C. S., Boillee, S., McAlonis-Downes, M., Khan, A. M., Feltri, M. L., Yamanaka, K., et al. (2009). Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc. Natl. Acad. Sci. U.S.A. 106, 4465–4470. doi: 10.1073/pnas.0813339106
Mackenzie, I. R., Arzberger, T., Kremmer, E., Troost, D., Lorenzl, S., Mori, K., et al. (2013). Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 126, 859–879. doi: 10.1007/s00401-013-1181-y
Mackenzie, I. R. A., Ansorge, O., Strong, M., Bilbao, J., Zinman, L., Ang, L.-C., et al. (2011). Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta Neuropathol. 122, 87–98. doi: 10.1007/s00401-011-0838-7
Mackenzie, I. R. A., Bigio, E. H., Ince, P. G., Geser, F., Neumann, M., Cairns, N. J., et al. (2007). Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 61, 427–434. doi: 10.1002/ana.21147
Mackenzie, I. R. A., Neumann, M., Bigio, E. H., Cairns, N. J., Alafuzoff, I., Kril, J., et al. (2009). Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 117, 15–18. doi: 10.1007/s00401-008-0460-5
Madabhushi, R., Pan, L., and Tsai, L.-H. (2014). DNA damage and its links to neurodegeneration. Neuron 83, 266–282. doi: 10.1016/j.neuron.2014.06.034
Maekawa, S., Al-Sarraj, S., Kibble, M., Landau, S., Parnavelas, J., Cotter, D., et al. (2004). Cortical selective vulnerability in motor neuron disease: a morphometric study. Brain 127, 1237–1251. doi: 10.1093/brain/awh132
Magnus, T., Carmen, J., Deleon, J., Xue, H., Pardo, A. C., Lepore, A. C., et al. (2008). Adult glial precursor proliferation in mutant SOD1G93A mice. Glia 56, 200–208. doi: 10.1002/glia.20604
Magrané, J., Cortez, C., Gan, W.-B., and Manfredi, G. (2014). Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 23, 1413–1424. doi: 10.1093/hmg/ddt528
Maharjan, N., and Saxena, S. (2016). ER strikes again: proteostasis dysfunction in ALS. EMBO J. 35, 798–800. doi: 10.15252/embj.201694117
Mahoney, C. J., Beck, J., Rohrer, J. D., Lashley, T., Mok, K., Shakespeare, T., et al. (2012). Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain 135, 736–750. doi: 10.1093/brain/awr361
Majounie, E., Renton, A. E., Mok, K., Dopper, E. G. P., Waite, A., Rollinson, S., et al. (2012). Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330. doi: 10.1016/S1474-4422(12)70043-1
Malhas, A. N., Lee, C. F., and Vaux, D. J. (2009). Lamin B1 controls oxidative stress responses via Oct-1. J. Cell Biol. 184, 45–55. doi: 10.1083/jcb.200804155
Maniecka, Z., and Polymenidou, M. (2015). From nucleation to widespread propagation: a prion-like concept for ALS. Virus Res. 207, 94–105. doi: 10.1016/j.virusres.2014.12.032
Manini, T. M., Hong, S. L., and Clark, B. C. (2013). Aging and muscle: a neuron’s perspective. Curr. Opin. Clin. Nutr. Metab. Care 16, 21–26. doi: 10.1097/MCO.0b013e32835b5880
Mannen, T., Iwata, M., Toyokura, Y., and Nagashima, K. (1977). Preservation of a certain motoneurone group of the sacral cord in amyotrophic lateral sclerosis: its clinical significance. J. Neurol. Neurosurg. Psychiatry 40, 464–469. doi: 10.1136/jnnp.40.5.464
Marner, L., Nyengaard, J. R., Tang, Y., and Pakkenberg, B. (2003). Marked loss of myelinated nerve fibers in the human brain with age. J. Comp. Neurol. 462, 144–152. doi: 10.1002/cne.10714
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., et al. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. doi: 10.1038/nature08971
Matise, M., and Sharma, K. (2013). “Chapter 21 - The Specification and Generation of Neurons in the Ventral Spinal Cord,” in Patterning and Cell Type Specification in the Developing CNS and PNS, eds J. L. R. Rubenstein and P. Rakic (Oxford: Academic Press), 401–415. doi: 10.1016/b978-0-12-397265-1.00101-5
Mattson, M. P., and Magnus, T. (2006). Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 7, 278–294. doi: 10.1038/nrn1886
Maxwell, N., Castro, R. W., Sutherland, N. M., Vaughan, K. L., Szarowicz, M. D., de Cabo, R., et al. (2018). α-Motor neurons are spared from aging while their synaptic inputs degenerate in monkeys and mice. Aging Cell 17:e12726. doi: 10.1111/acel.12726
McHanwell, S., and Biscoe, T. J. (1981). The localization of motoneurons supplying the hindlimb muscles of the mouse. Philos. Trans. R. Soc. Lond. B Biol. Sci. 293, 477–508. doi: 10.1098/rstb.1981.0082
Melkov, A., Baskar, R., Alcalay, Y., and Abdu, U. (2016). A new mode of mitochondrial transport and polarized sorting regulated by Dynein, Milton and Miro. Development 143, 4203–4213. doi: 10.1242/dev.138289
Mendell, L. M. (2005). The size principle: a rule describing the recruitment of motoneurons. J. Neurophysiol. 93, 3024–3026. doi: 10.1152/classicessays.00025.2005
Menon, P., Kiernan, M. C., and Vucic, S. (2015). Cortical hyperexcitability precedes lower motor neuron dysfunction in ALS. Clin. Neurophysiol. 126, 803–809. doi: 10.1016/j.clinph.2014.04.023
Millecamps, S., Da Barroca, S., Cazeneuve, C., Salachas, F., Pradat, P.-F., Danel-Brunaud, V., et al. (2010). Questioning on the role of D amino acid oxidase in familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 107:E107. doi: 10.1073/pnas.1006190107
Millecamps, S., Robertson, J., Lariviere, R., Mallet, J., and Julien, J.-P. (2006). Defective axonal transport of neurofilament proteins in neurons overexpressing peripherin. J. Neurochem. 98, 926–938. doi: 10.1111/j.1471-4159.2006.03932.x
Miller, S. J. (2018). Astrocyte heterogeneity in the adult central nervous system. Front. Cell Neurosci. 12:401. doi: 10.3389/fncel.2018.00401
Mimura, N., Yuasa, S., Soma, M., Jin, H., Kimura, K., Goto, S., et al. (2008). Altered quality control in the endoplasmic reticulum causes cortical dysplasia in knock-in mice expressing a mutant BiP. Mol. Cell. Biol. 28, 293–301. doi: 10.1128/MCB.00473-07
Misawa, H., Hara, M., Tanabe, S., Niikura, M., Moriwaki, Y., and Okuda, T. (2012). Osteopontin is an alpha motor neuron marker in the mouse spinal cord. J. Neurosci. Res. 90, 732–742. doi: 10.1002/jnr.22813
Mitchell, J., Paul, P., Chen, H.-J., Morris, A., Payling, M., Falchi, M., et al. (2010). Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc. Natl. Acad. Sci. U.S.A. 107, 7556–7561. doi: 10.1073/pnas.0914128107
Mohajeri, M. H., Figlewicz, D. A., and Bohn, M. C. (1998). Selective loss of alpha motoneurons innervating the medial gastrocnemius muscle in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 150, 329–336. doi: 10.1006/exnr.1998.6758
Molofsky, A. V., Kelley, K. W., Tsai, H.-H., Redmond, S. A., Chang, S. M., Madireddy, L., et al. (2014). Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature 509, 189–194. doi: 10.1038/nature13161
Moret, F., Renaudot, C., Bozon, M., and Castellani, V. (2007). Semaphorin and neuropilin co-expression in motoneurons sets axon sensitivity to environmental semaphorin sources during motor axon pathfinding. Development 134, 4491–4501. doi: 10.1242/dev.011452
Muller, F. L., Liu, Y., Jernigan, A., Borchelt, D., Richardson, A., and Van Remmen, H. (2008). MnSOD deficiency has a differential effect on disease progression in two different ALS mutant mouse models. Muscle Nerve 38, 1173–1183. doi: 10.1002/mus.21049
Müller, T. J., Kraya, T., Stoltenburg-Didinger, G., Hanisch, F., Kornhuber, M., Stoevesandt, D., et al. (2014). Phenotype of matrin-3–related distal myopathy in 16 G erman patients. Ann. Neurol. 76, 669–680. doi: 10.1002/ana.24255
Münch, C., O’Brien, J., and Bertolotti, A. (2011). Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. U.S.A. 108, 3548–3553. doi: 10.1073/pnas.1017275108
Münch, C., Rosenbohm, A., Sperfeld, A.-D., Uttner, I., Reske, S., Krause, B. J., et al. (2005). Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann. Neurol. 58, 777–780. doi: 10.1002/ana.20631
Münch, C., Sedlmeier, R., Meyer, T., Homberg, V., Sperfeld, A. D., Kurt, A., et al. (2004). Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63, 724–726. doi: 10.1212/01.wnl.0000134608.83927.b1
Murphy, P. R., and Martin, H. A. (1993). Fusimotor discharge patterns during rhythmic movements. Trends Neurosci. 16, 273–278. doi: 10.1016/0166-2236(93)90181-K
Murray, M. E., DeJesus-Hernandez, M., Rutherford, N. J., Baker, M., Duara, R., Graff-Radford, N. R., et al. (2011). Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 122, 673–690. doi: 10.1007/s00401-011-0907-y
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi: 10.1038/nn1876
Nagashima, T., Beppu, H., Uono, M., and Yamada, H. (1979). Demonstration of neuronal localization in Onufrowcz’s group-X in rabbit by double labeling method. Acta Histochem. Cytochem. 12, 409–415. doi: 10.1267/ahc.12.409
Nakamura, R., Kamakura, K., and Kwak, S. (1994). Late-onset selective neuronal damage in the rat spinal cord induced by continuous intrathecal administration of AMPA. Brain Res. 654, 279–285. doi: 10.1016/0006-8993(94)90490-1
Naumann, M., Pal, A., Goswami, A., Lojewski, X., Japtok, J., Vehlow, A., et al. (2018). Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 9:335. doi: 10.1038/s41467-017-02299-1
Neafsey, E. J., Bold, E. L., Haas, G., Hurley-Gius, K. M., Quirk, G., Sievert, C. F., et al. (1986). The organization of the rat motor cortex: a microstimulation mapping study. Brain Res. 396, 77–96. doi: 10.1016/s0006-8993(86)80191-3
Neumann, M., Rademakers, R., Roeber, S., Baker, M., Kretzschmar, H. A., and Mackenzie, I. R. (2009). A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132, 2922–2931. doi: 10.1093/brain/awp214
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nguyen, H. P., Broeckhoven, C. V., and van der Zee, J. (2018). ALS Genes in the genomic era and their implications for FTD. Trends Genet. 34, 404–423. doi: 10.1016/j.tig.2018.03.001
Niccoli, T., Partridge, L., and Isaacs, A. M. (2017). Ageing as a risk factor for ALS/FTD. Hum. Mol. Genet. 26, R105–R113. doi: 10.1093/hmg/ddx247
Niebroj-Dobosz, I., Rafałowska, J., Fidziańska, A., Gadamski, R., and Grieb, P. (2007). Myelin composition of spinal cord in a model of amyotrophic lateral sclerosis (ALS) in SOD1G93A transgenic rats. Folia Neuropathol. 45, 236–241.
Niessen, H. G., Angenstein, F., Sander, K., Kunz, W. S., Teuchert, M., Ludolph, A. C., et al. (2006). In vivo quantification of spinal and bulbar motor neuron degeneration in the G93A-SOD1 transgenic mouse model of ALS by T2 relaxation time and apparent diffusion coefficient. Exp. Neurol. 201, 293–300. doi: 10.1016/j.expneurol.2006.04.007
Nijssen, J., Comley, L. H., and Hedlund, E. (2017). Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 133, 863–885. doi: 10.1007/s00401-017-1708-8
Nimchinsky, E. A., Young, W. G., Yeung, G., Shah, R. A., Gordon, J. W., Bloom, F. E., et al. (2000). Differential vulnerability of oculomotor, facial, and hypoglossal nuclei in G86R superoxide dismutase transgenic mice. J. Comp. Neurol. 416, 112–125. doi: 10.1002/(sici)1096-9861(20000103)416:1<112::aid-cne9>3.0.co;2-k
Nishimaru, H., Restrepo, C. E., Ryge, J., Yanagawa, Y., and Kiehn, O. (2005). Mammalian motor neurons corelease glutamate and acetylcholine at central synapses. Proc. Natl. Acad. Sci. U.S.A. 102, 5245–5249. doi: 10.1073/pnas.0501331102
Nishimura, A. L., Mitne-Neto, M., Silva, H. C. A., Richieri-Costa, A., Middleton, S., Cascio, D., et al. (2004). A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75, 822–831. doi: 10.1086/425287
Nishitoh, H., Kadowaki, H., Nagai, A., Maruyama, T., Yokota, T., Fukutomi, H., et al. (2008). ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 22, 1451–1464. doi: 10.1101/gad.1640108
Niven, J. E. (2016). Neuronal energy consumption: biophysics, efficiency and evolution. Curr. Opin. Neurobiol. 41, 129–135. doi: 10.1016/j.conb.2016.09.004
Nonaka, T., Masuda-Suzukake, M., Arai, T., Hasegawa, Y., Akatsu, H., Obi, T., et al. (2013). Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 4, 124–134. doi: 10.1016/j.celrep.2013.06.007
Obál, I., Engelhardt, J. I., and Siklós, L. (2006). Axotomy induces contrasting changes in calcium and calcium-binding proteins in oculomotor and hypoglossal nuclei of Balb/c mice. J. Comp. Neurol. 499, 17–32. doi: 10.1002/cne.21041
Okada, Y., Yamazaki, H., Sekine-Aizawa, Y., and Hirokawa, N. (1995). The neuron-specific kinesin superfamily protein KIF1A is a uniqye monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell 81, 769–780. doi: 10.1016/0092-8674(95)90538-3
Osking, Z., Ayers, J. I., Hildebrandt, R., Skruber, K., Brown, H., Ryu, D., et al. (2019). ALS-linked SOD1 mutants enhance neurite outgrowth and branching in adult motor neurons. Science 11, 294–304. doi: 10.1016/j.isci.2018.12.026
Palecek, J., Lips, M. B., and Keller, B. U. (1999). Calcium dynamics and buffering in motoneurones of the mouse spinal cord. J. Physiol. 520, 485–502. doi: 10.1111/j.1469-7793.1999.00485.x
Pambo-Pambo, A., Durand, J., and Gueritaud, J.-P. (2009). Early excitability changes in lumbar motoneurons of transgenic SOD1G85R and SOD1G(93A-Low) mice. J. Neurophysiol. 102, 3627–3642. doi: 10.1152/jn.00482.2009
Pannese, E. (2011). Morphological changes in nerve cells during normal aging. Brain Struct. Funct. 216, 85–89. doi: 10.1007/s00429-011-0308-y
Pantelidou, M., Zographos, S. E., Lederer, C. W., Kyriakides, T., Pfaffl, M. W., and Santama, N. (2007). Differential expression of molecular motors in the motor cortex of sporadic ALS. Neurobiol. Dis. 26, 577–589. doi: 10.1016/j.nbd.2007.02.005
Papadeas, S. T., Kraig, S. E., O’Banion, C., Lepore, A. C., and Maragakis, N. J. (2011). Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc. Natl. Acad. Sci. U.S.A. 108, 17803–17808. doi: 10.1073/pnas.1103141108
Parakh, S., Jagaraj, C. J., Vidal, M., Ragagnin, A. M. G., Perri, E. R., Konopka, A., et al. (2018a). ERp57 is protective against mutant SOD1-induced cellular pathology in amyotrophic lateral sclerosis. Hum. Mol. Genet. 27, 1311–1331. doi: 10.1093/hmg/ddy041
Parakh, S., Perri, E. R., Jagaraj, C. J., Ragagnin, A. M. G., and Atkin, J. D. (2018b). Rab-dependent cellular trafficking and amyotrophic lateral sclerosis. Crit. Rev. Biochem. Mol. Biol. 53, 623–651. doi: 10.1080/10409238.2018.1553926
Pasinelli, P., Belford, M. E., Lennon, N., Bacskai, B. J., Hyman, B. T., Trotti, D., et al. (2004). Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 43, 19–30. doi: 10.1016/j.neuron.2004.06.021
Penndorf, D., Tadiæ, V., Witte, O. W., Grosskreutz, J., and Kretz, A. (2017). DNA strand breaks and TDP-43 mislocation are absent in the murine hSOD1G93A model of amyotrophic lateral sclerosis in vivo and in vitro. PLoS One 12:e0183684. doi: 10.1371/journal.pone.0183684
Penndorf, D., Witte, O. W., and Kretz, A. (2018). DNA plasticity and damage in amyotrophic lateral sclerosis. Neural Regen. Res. 13, 173–180. doi: 10.4103/1673-5374.226377
Perlson, E., Jeong, G.-B., Ross, J. L., Dixit, R., Wallace, K. E., Kalb, R. G., et al. (2009). A switch in retrograde signaling from survival to stress in rapid-onset neurodegeneration. J. Neurosci. 29, 9903–9917. doi: 10.1523/JNEUROSCI.0813-09.2009
Perrie, W. T., Lee, G. T., Curtis, E. M., Sparke, J., Buller, J. R., and Rossi, M. L. (1993). Changes in the myelinated axons of femoral nerve in amyotrophic lateral sclerosis. J. Neural Transm. Suppl. 39, 223–233.
Perrin, S. (2014). Preclinical research: make mouse studies work. Nature 507, 423–425. doi: 10.1038/507423a
Perrone, F., Nguyen, H. P., Van Mossevelde, S., Moisse, M., Sieben, A., Santens, P., et al. (2017). Investigating the role of ALS genes CHCHD10 and TUBA4A in Belgian FTD-ALS spectrum patients. Neurobiol. Aging 51, 177.e9–177.e16. doi: 10.1016/j.neurobiolaging.2016.12.008
Perry, V. H., and Holmes, C. (2014). Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 10, 217–224. doi: 10.1038/nrneurol.2014.38
Philips, T., Bento-Abreu, A., Nonneman, A., Haeck, W., Staats, K., Geelen, V., et al. (2013). Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 136, 471–482. doi: 10.1093/brain/aws339
Philips, T., and Rothstein, J. D. (2015). Rodent models of amyotrophic lateral sclerosis. Curr. Protoc. Pharmacol. 69, 5.67.1–5.67.21. doi: 10.1002/0471141755.ph0567s69
Piccione, E. A., Sletten, D. M., Staff, N. P., and Low, P. A. (2015). Autonomic system and ALS. Muscle Nerve 51, 676–679. doi: 10.1002/mus.24457
Pisharodi, M., and Nauta, H. J. (1985). An animal model for neuron-specific spinal cord lesions by the microinjection of N-methylaspartate, kainic acid, and quisqualic acid. Appl. Neurophysiol. 48, 226–233. doi: 10.1159/000101132
Polci, R., Peng, A., Chen, P.-L., Riley, D. J., and Chen, Y. (2004). NIMA-related protein kinase 1 is involved early in the ionizing radiation-induced DNA damage response. Cancer Res. 64, 8800–8803. doi: 10.1158/0008-5472.CAN-04-2243
Polymenidou, M., Lagier-Tourenne, C., Hutt, K. R., Huelga, S. C., Moran, J., Liang, T. Y., et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468. doi: 10.1038/nn.2779
Porta, S., Xu, Y., Restrepo, C. R., Kwong, L. K., Zhang, B., Brown, H. J., et al. (2018). Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun. 9:4220. doi: 10.1038/s41467-018-06548-9
Porter, J. D., and Baker, R. S. (1996). Muscles of a different “color”: the unusual properties of the extraocular muscles may predispose or protect them in neurogenic and myogenic disease. Neurology 46, 30–37. doi: 10.1212/wnl.46.1.30
Porter, R., and Lemon, R. (1993). Corticospinal Function and Voluntary Movement. Oxford: Clarendon Press.
Pottier, C., Bieniek, K. F., Finch, N., van de Vorst, M., Baker, M., Perkersen, R., et al. (2015). Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 130, 77–92. doi: 10.1007/s00401-015-1436-x
Pradat, P.-F., Salachas, F., Lacomblez, L., Patte, N., Leforestier, N., Gaura, V., et al. (2002). Association of chorea and motor neuron disease. Mov. Disord. 17, 419–420. doi: 10.1002/mds.10039
Pramatarova, A., Laganière, J., Roussel, J., Brisebois, K., and Rouleau, G. A. (2001). Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J. Neurosci. 21, 3369–3374. doi: 10.1523/jneurosci.21-10-03369.2001
Proudfoot, M., Menke, R. A. L., Sharma, R., Berna, C. M., Hicks, S. L., Kennard, C., et al. (2015). Eye-tracking in amyotrophic lateral sclerosis: a longitudinal study of saccadic and cognitive tasks. Amyotroph. Lateral Scler. Frontotemporal Degener. 17, 101–111. doi: 10.3109/21678421.2015.1054292
Prudencio, M., Belzil, V. V., Batra, R., Ross, C. A., Gendron, T. F., Pregent, L. J., et al. (2015). Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 18, 1175–1182. doi: 10.1038/nn.4065
Pullen, A. H., and Athanasiou, D. (2009). Increase in presynaptic territory of C-terminals on lumbar motoneurons of G93A SOD1 mice during disease progression. Eur. J. Neurosci. 29, 551–561. doi: 10.1111/j.1460-9568.2008.06602.x
Puls, I., Jonnakuty, C., LaMonte, B. H., Holzbaur, E. L. F., Tokito, M., Mann, E., et al. (2003). Mutant dynactin in motor neuron disease. Nat. Genet. 33, 455–456. doi: 10.1038/ng1123
Pun, S., Santos, A. F., Saxena, S., Xu, L., and Caroni, P. (2006). Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat. Neurosci. 9, 408–419. doi: 10.1038/nn1653
Purves, D., Augustine, G. J., Fitzpatrick, D., Katz, L. C., LaMantia, A.-S., McNamara, J. O., et al. (2001). Functional Properties of the Na+/K+ Pump. Available at: https://www.ncbi.nlm.nih.gov/books/NBK10857/ (accessed December 14, 2018).
Ramírez, O. A., and Couve, A. (2011). The endoplasmic reticulum and protein trafficking in dendrites and axons. Trends Cell Biol. 21, 219–227. doi: 10.1016/j.tcb.2010.12.003
Ramírez-Jarquín, U. N., Lazo-Gómez, R., Tovar-Y-Romo, L. B., and Tapia, R. (2014). Spinal inhibitory circuits and their role in motor neuron degeneration. Neuropharmacology 82, 101–107. doi: 10.1016/j.neuropharm.2013.10.003
Ramírez-Jarquín, U. N., and Tapia, R. (2018). Excitatory and inhibitory neuronal circuits in the spinal cord and their role in the control of motor neuron function and degeneration. ACS Chem. Neurosci. 9, 211–216. doi: 10.1021/acschemneuro.7b00503
Rathelot, J.-A., and Strick, P. L. (2009). Subdivisions of primary motor cortex based on cortico-motoneuronal cells. Proc. Natl. Acad. Sci. U.S.A. 106, 918–923. doi: 10.1073/pnas.0808362106
Ravits, J., Paul, P., and Jorg, C. (2007). Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 68:1571. doi: 10.1212/01.wnl.0000260965.20021.47
Ravits, J. M., and La Spada, A. R. (2009). ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology 73, 805–811. doi: 10.1212/WNL.0b013e3181b6bbbd
Reddy, L. V., Koirala, S., Sugiura, Y., Herrera, A. A., and Ko, C. P. (2003). Glial cells maintain synaptic structure and function and promote development of the neuromuscular junction in vivo. Neuron 40, 563–580. doi: 10.1016/s0896-6273(03)00682-2
Reddy, P. H., and Beal, M. F. (2008). Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 14, 45–53. doi: 10.1016/j.molmed.2007.12.002
Renton, A. E., Chiò, A., and Traynor, B. J. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23. doi: 10.1038/nn.3584
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-Linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Reynolds, M. L., and Woolf, C. J. (1992). Terminal Schwann cells elaborate extensive processes following denervation of the motor endplate. J. Neurocytol. 21, 50–66. doi: 10.1007/bf01206897
Riehle, A., Wirtssohn, S., Grün, S., and Brochier, T. (2013). Mapping the spatio-temporal structure of motor cortical LFP and spiking activities during reach-to-grasp movements. Front. Neural Circ. 7:48. doi: 10.3389/fncir.2013.00048
Ripps, M. E., Huntley, G. W., Hof, P. R., Morrison, J. H., and Gordon, J. W. (1995). Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 92, 689–693. doi: 10.1073/pnas.92.3.689
Rizzardini, M., Lupi, M., Bernasconi, S., Mangolini, A., and Cantoni, L. (2003). Mitochondrial dysfunction and death in motor neurons exposed to the glutathione-depleting agent ethacrynic acid. J. Neurol. Sci. 207, 51–58. doi: 10.1016/s0022-510x(02)00357-x
Robertson, J., Doroudchi, M. M., Nguyen, M. D., Durham, H. D., Strong, M. J., Shaw, G., et al. (2003). A neurotoxic peripherin splice variant in a mouse model of ALS. J. Cell Biol. 160, 939–949. doi: 10.1083/jcb.200205027
Robinson, D. A. (1970). Oculomotor unit behavior in the monkey. J. Neurophysiol. 33, 393–403. doi: 10.1152/jn.1970.33.3.393
Rochat, C., Bernard-Marissal, N., and Schneider, B. L. (2016). Selective Vulnerability of Neuronal Subtypes in ALS: A Fertile Ground for the Identification of Therapeutic Targets. London: IntechOpen.
Rojas, F., Cortes, N., Abarzua, S., Dyrda, A., and van Zundert, B. (2014). Astrocytes expressing mutant SOD1 and TDP43 trigger motoneuron death that is mediated via sodium channels and nitroxidative stress. Front. Cell Neurosci. 8:24. doi: 10.3389/fncel.2014.00024
Roos, A., Kollipara, L., Buchkremer, S., Labisch, T., Brauers, E., Gatz, C., et al. (2016). Cellular signature of SIL1 depletion: disease pathogenesis due to alterations in protein composition beyond the ER machinery. Mol. Neurobiol. 53, 5527–5541. doi: 10.1007/s12035-015-9456-z
Roppolo, J. R., Nadelhaft, I., and de Groat, W. C. (1985). The organization of pudendal motoneurons and primary afferent projections in the spinal cord of the rhesus monkey revealed by horseradish peroxidase. J. Comp. Neurol. 234, 475–488. doi: 10.1002/cne.902340406
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Ross, O. A., Rutherford, N. J., Baker, M., Soto-Ortolaza, A. I., Carrasquillo, M. M., DeJesus-Hernandez, M., et al. (2011). Ataxin-2 repeat-length variation and neurodegeneration. Hum. Mol. Genet. 20, 3207–3212. doi: 10.1093/hmg/ddr227
Rouleau, G. A., Clark, A. W., Rooke, K., Pramatarova, A., Krizus, A., Suchowersky, O., et al. (1996). SOD1 mutation is assosiated with accumulation of neurofilaments in amyotrophic lateral scelaries. Ann. Neurol. 39, 128–131. doi: 10.1002/ana.410390119
Rozas, P., Bargsted, L., Martínez, F., Hetz, C., and Medinas, D. B. (2017). The ER proteostasis network in ALS: determining the differential motoneuron vulnerability. Neurosci. Lett. 636, 9–15. doi: 10.1016/j.neulet.2016.04.066
Rubino, E., Rainero, I., Chiò, A., Rogaeva, E., Galimberti, D., Fenoglio, P., et al. (2012). SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 79, 1556–1562. doi: 10.1212/WNL.0b013e31826e25df
Ruegsegger, C., Maharjan, N., Goswami, A., Filézac de L’Etang, A., Weis, J., Troost, D., et al. (2016). Aberrant association of misfolded SOD1 with Na+/K+ATPase-α3 impairs its activity and contributes to motor neuron vulnerability in ALS. Acta Neuropathol. 131, 427–451. doi: 10.1007/s00401-015-1510-4
Ruegsegger, C., and Saxena, S. (2016). Proteostasis impairment in ALS. Brain Res. 1648, 571–579. doi: 10.1016/j.brainres.2016.03.032
Rutherford, N. J., Heckman, M. G., Dejesus-Hernandez, M., Baker, M. C., Soto-Ortolaza, A. I., Rayaprolu, S., et al. (2012). Length of normal alleles of C9ORF72 GGGGCC repeat do not influence disease phenotype. Neurobiol. Aging 33, 2950.e5–2950.e7. doi: 10.1016/j.neurobiolaging.2012.07.005
Rutherford, N. J., Zhang, Y.-J., Baker, M., Gass, J. M., Finch, N. A., Xu, Y.-F., et al. (2008). Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 4:e1000193. doi: 10.1371/journal.pgen.1000193
Saccon, R. A., Bunton-Stasyshyn, R. K. A., Fisher, E. M. C., and Fratta, P. (2013). Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 136, 2342–2358. doi: 10.1093/brain/awt097
Sakowski, S. A., Schuyler, A. D., and Feldman, E. L. (2009). Insulin-like growth factor-I for the treatment of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 10, 63–73. doi: 10.1080/17482960802160370
Salat, D. H., Buckner, R. L., Snyder, A. Z., Greve, D. N., Desikan, R. S. R., Busa, E., et al. (2004). Thinning of the cerebral cortex in aging. Cereb. Cortex 14, 721–730. doi: 10.1093/cercor/bhh032
Sasaki, S. (2010). Endoplasmic reticulum stress in motor neurons of the spinal cord in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 69, 346–355. doi: 10.1097/NEN.0b013e3181d44992
Sasaki, S., Horie, Y., and Iwata, M. (2007). Mitochondrial alterations in dorsal root ganglion cells in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 114, 633–639. doi: 10.1007/s00401-007-0299-1
Sasaki, S., and Iwata, M. (1996). Dendritic synapses of anterior horn neurons in amyotrophic lateral sclerosis: an ultrastructural study. Acta Neuropathol. 91, 278–283. doi: 10.1007/s004010050426
Sasaki, S., Warita, H., Komori, T., Murakami, T., Abe, K., and Iwata, M. (2006). Parvalbumin and calbindin D-28k immunoreactivity in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1083, 196–203. doi: 10.1016/j.brainres.2006.01.129
Sato, M., Mizuno, N., and Konishi, A. (1978). Localization of motoneurons innervating perineal muscles: a HRP study in cat. Brain Res. 140, 149–154. doi: 10.1016/0006-8993(78)90244-5
Saxena, S., Cabuy, E., and Caroni, P. (2009). A role for motoneuron subtype–selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636. doi: 10.1038/nn.2297
Saxena, S., Roselli, F., Singh, K., Leptien, K., Julien, J.-P., Gros-Louis, F., et al. (2013). Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80, 80–96. doi: 10.1016/j.neuron.2013.07.027
Schrøder, H. D., and Reske-Nielsen, E. (1984). Preservation of the nucleus X-pelvic floor motosystem in amyotrophic lateral sclerosis. Clin. Neuropathol. 3, 210–216.
Schwenk, B. M., Hartmann, H., Serdaroglu, A., Schludi, M. H., Hornburg, D., Meissner, F., et al. (2016). TDP-43 loss of function inhibits endosomal trafficking and alters trophic signaling in neurons. EMBO J. 35, 2350–2370. doi: 10.15252/embj.201694221
Scotter, E. L., Chen, H.-J., and Shaw, C. E. (2015). TDP-43 Proteinopathy and ALS: insights into disease mechanisms and therapeutic targets. Neurotherapeutics 12, 352–363. doi: 10.1007/s13311-015-0338-x
Seilhean, D., Cazeneuve, C., Thuriès, V., Russaouen, O., Millecamps, S., Salachas, F., et al. (2009). Accumulation of TDP-43 and alpha-actin in an amyotrophic lateral sclerosis patient with the K17I ANG mutation. Acta Neuropathol. 118, 561–573. doi: 10.1007/s00401-009-0545-9
Shahheydari, H., Ragagnin, A., Walker, A. K., Toth, R. P., Vidal, M., Jagaraj, C. J., et al. (2017). Protein quality control and the amyotrophic lateral sclerosis/frontotemporal dementia continuum. Front. Mol. Neurosci. 10:119. doi: 10.3389/fnmol.2017.00119
Shan, X., Chiang, P.-M., Price, D. L., and Wong, P. C. (2010). Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 107, 16325–16330. doi: 10.1073/pnas.1003459107
Sharma, A., Lyashchenko, A. K., Lu, L., Nasrabady, S. E., Elmaleh, M., Mendelsohn, M., et al. (2016). ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 7:10465. doi: 10.1038/ncomms10465
Shaunak, S., Orrell, R. W., O’Sullivan, E., Hawken, M. B., Lane, R. J., Henderson, L., et al. (1995). Oculomotor function in amyotrophic lateral sclerosis: evidence for frontal impairment. Ann. Neurol. 38, 38–44. doi: 10.1002/ana.410380109
Shaw, P. J., Ince, P. G., Falkous, G., and Mantle, D. (1995). Oxidative damage to protein in sporadic motor neuron disease spinal cord. Ann. Neurol. 38, 691–695. doi: 10.1002/ana.410380424
Shi, P., Ström, A.-L., Gal, J., and Zhu, H. (2010). Effects of ALS-related SOD1 mutants on dynein- and KIF5-mediated retrograde and anterograde axonal transport. Biochim. Biophys. Acta 1802, 707–716. doi: 10.1016/j.bbadis.2010.05.008
Shibata, N., Nagai, R., Uchida, K., Horiuchi, S., Yamada, S., Hirano, A., et al. (2001). Morphological evidence for lipid peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic lateral sclerosis patients. Brain Res. 917, 97–104. doi: 10.1016/s0006-8993(01)02926-2
Shin, J. H., and Lee, J. K. (2013). “Multiple Routes of Motor Neuron Degeneration in ALS,” in Current Advances in Amyotrophic Lateral Sclerosis, ed. A. Estévez (London: Intechopen). doi: 10.1080/14660820050515395
Shirvan, A., Kimron, M., Holdengreber, V., Ziv, I., Ben-Shaul, Y., Melamed, S., et al. (2002). Anti-semaphorin 3A antibodies rescue retinal ganglion cells from cell death following optic nerve axotomy. J. Biol. Chem. 277, 49799–49807. doi: 10.1074/jbc.M204793200
Shneider, N. A., Brown, M. N., Smith, C. A., Pickel, J., and Alvarez, F. J. (2009). Gamma motor neurons express distinct genetic markers at birth and require muscle spindle-derived GDNF for postnatal survival. Neural Dev. 4:42. doi: 10.1186/1749-8104-4-42
Shoenfeld, L., Westenbroek, R. E., Fisher, E., Quinlan, K. A., Tysseling, V. M., Powers, R. K., et al. (2014). Soma size and Cav1.3 channel expression in vulnerable and resistant motoneuron populations of the SOD1G93A mouse model of ALS. Physiol. Rep. 2:e12113. doi: 10.14814/phy2.12113
Siddle, K., Ursø, B., Niesler, C. A., Cope, D. L., Molina, L., Surinya, K. H., et al. (2001). Specificity in ligand binding and intracellular signalling by insulin and insulin-like growth factor receptors. Biochem. Soc. Trans. 29, 513–525. doi: 10.1042/bst0290513
Sloan, S. A., and Barres, B. A. (2014). Mechanisms of astrocyte development and their contributions to neurodevelopmental disorders. Curr. Opin. Neurobiol. 27, 75–81. doi: 10.1016/j.conb.2014.03.005
Smethurst, P., Newcombe, J., Troakes, C., Simone, R., Chen, Y.-R., Patani, R., et al. (2016). In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol. Dis. 96, 236–247. doi: 10.1016/j.nbd.2016.08.007
Smith, B. N., Ticozzi, N., Fallini, C., Gkazi, A. S., Topp, S., Kenna, K. P., et al. (2014a). Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84, 324–331. doi: 10.1016/j.neuron.2014.09.027
Smith, B. N., Vance, C., Scotter, E. L., Troakes, C., Wong, C. H., Topp, S., et al. (2014b). Novel mutations support a role for Profilin1 in the pathogenesis of ALS. Neurobiol. Aging 36, 1602.e17–1602.e27. doi: 10.1016/j.neurobiolaging.2014.10.032
So, E., Mitchell, J. C., Memmi, C., Chennell, G., Vizcay-Barrena, G., Allison, L., et al. (2018). Mitochondrial abnormalities and disruption of the neuromuscular junction precede the clinical phenotype and motor neuron loss in hFUSWT transgenic mice. Hum. Mol. Genet. 27, 463–474. doi: 10.1093/hmg/ddx415
Son, Y. J., and Thompson, W. J. (1995). Schwann cell processes guide regeneration of peripheral axons. Neuron 14, 125–132. doi: 10.1016/0896-6273(95)90246-5
Sonne, J., and Lopez-Ojeda, W. (2018). “Neuroanatomy, Cranial Nerve,” in StatPearls, ed. M. Varacallo (Treasure Island FL: StatPearls Publishing).
Soo, K. Y., Halloran, M., Sundaramoorthy, V., Parakh, S., Toth, R. P., Southam, K. A., et al. (2015). Rab1-dependent ER-Golgi transport dysfunction is a common pathogenic mechanism in SOD1, TDP-43 and FUS-associated ALS. Acta Neuropathol. 130, 679–697. doi: 10.1007/s00401-015-1468-2
Sorond, F. A., Cruz-Almeida, Y., Clark, D. J., Viswanathan, A., Scherzer, C. R., De Jager, P., et al. (2015). Aging, the central nervous system, and mobility in older adults: neural mechanisms of mobility impairment. J. Gerontol. A Biol. Sci. Med. Sci. 70, 1526–1532. doi: 10.1093/gerona/glv130
Sotelo-Silveira, J. R., Lepanto, P., Elizondo, V., Horjales, S., Palacios, F., Martinez-Palma, L., et al. (2009). Axonal mitochondrial clusters containing mutant SOD1 in transgenic models of ALS. Antioxid. Redox Signal. 11, 1535–1545. doi: 10.1089/ars.2009.2614
Soto, C. (2012). Transmissible proteins: expanding the prion heresy. Cell 149, 968–977. doi: 10.1016/j.cell.2012.05.007
Spiller, K. J., Cheung, C. J., Restrepo, C. R., Kwong, L. K., Stieber, A. M., Trojanowski, J. Q., et al. (2016a). Selective motor neuron resistance and recovery in a new inducible mouse model of TDP-43 proteinopathy. J. Neurosci. 36, 7707–7717. doi: 10.1523/JNEUROSCI.1457-16.2016
Spiller, K. J., Restrepo, C. R., Khan, T., Stieber, A. M., Kwong, L. K., Trojanowski, J. Q., et al. (2016b). Progression of motor neuron disease is accelerated and the ability to recover is compromised with advanced age in rNLS8 mice. Acta Neuropathol. Commun. 4:105. doi: 10.1186/s40478-016-0377-5
Spiller, K. J., Khan, T., Dominique, M. A., Restrepo, C. R., Cotton-Samuel, D., Levitan, M., et al. (2019). Reduction of matrix metalloproteinase 9 (MMP-9) protects motor neurons from TDP-43-triggered death in rNLS8 mice. Neurobiol. Dis. 124, 133–140. doi: 10.1016/j.nbd.2018.11.013
Spiller, K. J., Restrepo, C. R., Khan, T., Dominique, M. A., Fang, T. C., Canter, R. G., et al. (2018). Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 21, 329–340. doi: 10.1038/s41593-018-0083-7
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Stephens, B., Guiloff, R. J., Navarrete, R., Newman, P., Nikhar, N., and Lewis, P. (2006). Widespread loss of neuronal populations in the spinal ventral horn in sporadic motor neuron disease. A morphometric study. J. Neurol. Sci. 244, 41–58. doi: 10.1016/j.jns.2005.12.003
Stephens, B., Navarrete, R., and Guiloff, R. J. (2001). Ubiquitin immunoreactivity in presumed spinal interneurones in motor neurone disease. Neuropathol. Appl. Neurobiol. 27, 352–361. doi: 10.1046/j.1365-2990.2001.00354.x
Stifani, N. (2014). Motor neurons and the generation of spinal motor neuron diversity. Front. Cell Neurosci. 8:293. doi: 10.3389/fncel.2014.00293
Strong, M. J., and Yang, W. (2011). The frontotemporal syndromes of ALS. clinicopathological correlates. J. Mol. Neurosci. 45, 648–655. doi: 10.1007/s12031-011-9609-0
Suga, A., Mizota, A., Kato, M., Kuniyoshi, K., Yoshitake, K., Sultan, W., et al. (2016). Identification of novel mutations in the LRR-Cap domain of C21orf2 in japanese patients with retinitis pigmentosa and cone-rod dystrophy. Invest. Ophthalmol. Vis. Sci. 57, 4255–4263. doi: 10.1167/iovs.16-19450
Suh, E., Lee, E. B., Neal, D., Wood, E. M., Toledo, J. B., Rennert, L., et al. (2015). Semi-automated quantification of C9orf72 expansion size reveals inverse correlation between hexanucleotide repeat number and disease duration in frontotemporal degeneration. Acta Neuropathol. 130, 363–372. doi: 10.1007/s00401-015-1445-9
Sun, H., Kawahara, Y., Ito, K., Kanazawa, I., and Kwak, S. (2005). Expression profile of AMPA receptor subunit mRNA in single adult rat brain and spinal cord neurons in situ. Neurosci. Res. 52, 228–234. doi: 10.1016/j.neures.2005.03.008
Sun, N., Youle, R. J., and Finkel, T. (2016). The mitochondrial basis of aging. Mol. Cell 61, 654–666. doi: 10.1016/j.molcel.2016.01.028
Sun, S., Sun, Y., Ling, S.-C., Ferraiuolo, L., McAlonis-Downes, M., Zou, Y., et al. (2015). Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc. Natl. Acad. Sci. U.S.A. 112, E6993–E7002. doi: 10.1073/pnas.1520639112
Sundaramoorthy, V., Walker, A. K., Yerbury, J., Soo, K. Y., Farg, M. A., Hoang, V., et al. (2013). Extracellular wildtype and mutant SOD1 induces ER-Golgi pathology characteristic of amyotrophic lateral sclerosis in neuronal cells. Cell. Mol. Life Sci. 70, 4181–4195. doi: 10.1007/s00018-013-1385-2
Suzuki, H., Kanekura, K., Levine, T. P., Kohno, K., Olkkonen, V. M., Aiso, S., et al. (2009). ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. J. Neurochem. 108, 973–985. doi: 10.1111/j.0022-3042.2008.05857.x
Swarup, V., Phaneuf, D., Dupré, N., Petri, S., Strong, M., Kriz, J., et al. (2011). Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB-mediated pathogenic pathways. J. Exp. Med. 208, 2429–2447. doi: 10.1084/jem.20111313
Swash, M., and Fox, K. P. (1972). The effect of age on human skeletal muscle. studies of the morphology and innervation of muscle spindles. J. Neurol. Sci. 16, 417–432. doi: 10.1016/0022-510x(72)90048-2
Swerdlow, R. H., Parks, J. K., Cassarino, D. S., Trimmer, P. A., Miller, S. W., Maguire, D. J., et al. (1998). Mitochondria in sporadic amyotrophic lateral sclerosis. Exp. Neurol. 153, 135–142. doi: 10.1006/exnr.1998.6866
Swinnen, B., and Robberecht, W. (2014). The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 10, 661–670. doi: 10.1038/nrneurol.2014.184
Synofzik, M., Maetzler, W., Grehl, T., Prudlo, J., vom Hagen, J. M., Haack, T., et al. (2012). Screening in ALS and FTD patients reveals 3 novel UBQLN2 mutations outside the PXX domain and a pure FTD phenotype. Neurobiol. Aging 33:2949.e13–2949.e17. doi: 10.1016/j.neurobiolaging.2012.07.002
Tan, C.-F., Eguchi, H., Tagawa, A., Onodera, O., Iwasaki, T., Tsujino, A., et al. (2007). TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol. 113, 535–542. doi: 10.1007/s00401-007-0206-9
Tang, C. W., Maya-Mendoza, A., Martin, C., Zeng, K., Chen, S., Feret, D., et al. (2008). The integrity of a lamin-B1-dependent nucleoskeleton is a fundamental determinant of RNA synthesis in human cells. J. Cell Sci. 121, 1014–1024. doi: 10.1242/jcs.020982
Tank, E. M., Figueroa-Romero, C., Hinder, L. M., Bedi, K., Archbold, H. C., Li, X., et al. (2018). Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 9:2845. doi: 10.1038/s41467-018-05049-z
Tartaglia, M. C., Rowe, A., Findlater, K., Orange, J. B., Grace, G., and Strong, M. J. (2007). Differentiation between primary lateral sclerosis and amyotrophic lateral sclerosis: examination of symptoms and signs at disease onset and during follow-up. Arch. Neurol. 64, 232–236. doi: 10.1001/archneur.64.2.232
Tateno, M., Sadakata, H., Tanaka, M., Itohara, S., Shin, R.-M., Miura, M., et al. (2004). Calcium-permeable AMPA receptors promote misfolding of mutant SOD1 protein and development of amyotrophic lateral sclerosis in a transgenic mouse model. Hum. Mol. Genet. 13, 2183–2196. doi: 10.1093/hmg/ddh246
Taylor, J. P., Brown, R. H., and Cleveland, D. W. (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. doi: 10.1038/nature20413
Teyssou, E., Chartier, L., Amador, M.-D.-M., Lam, R., Lautrette, G., Nicol, M., et al. (2017). Novel UBQLN2 mutations linked to amyotrophic lateral sclerosis and atypical hereditary spastic paraplegia phenotype through defective HSP70-mediated proteolysis. Neurobiol. Aging 58, 239.e11–239.e20. doi: 10.1016/j.neurobiolaging.2017.06.018
Thomas, E. V., Fenton, W. A., McGrath, J., and Horwich, A. L. (2017). Transfer of pathogenic and nonpathogenic cytosolic proteins between spinal cord motor neurons in vivo in chimeric mice. Proc. Natl. Acad. Sci. U.S.A. 114, E3139–E3148. doi: 10.1073/pnas.1701465114
Thomsen, G. M., Gowing, G., Latter, J., Chen, M., Vit, J.-P., Staggenborg, K., et al. (2014). Delayed disease onset and extended survival in the SOD1G93A rat model of amyotrophic lateral sclerosis after suppression of mutant SOD1 in the motor cortex. J. Neurosci. 34, 15587–15600. doi: 10.1523/JNEUROSCI.2037-14.2014
Ticozzi, N., Vance, C., LeClerc, A. L., Keagle, P., Glass, J. D., McKenna-Yasek, D., et al. (2011). Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am. J. Med. Genet. 156, 285–290. doi: 10.1002/ajmg.b.31158
Tong, J., Huang, C., Bi, F., Wu, Q., Huang, B., Liu, X., et al. (2013). Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J. 32, 1917–1926. doi: 10.1038/emboj.2013.122
Torres-Torrelo, J., Rodríguez-Rosell, D., Nunez-Abades, P., Carrascal, L., and Torres, B. (2012). Glutamate modulates the firing rate in oculomotor nucleus motoneurons as a function of the recruitment threshold current. J. Physiol. 590, 3113–3127. doi: 10.1113/jphysiol.2011.226985
Tsang, C. K., Liu, Y., Thomas, J., Zhang, Y., and Zheng, X. F. S. (2014). Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat. Comm. 5:3446. doi: 10.1038/ncomms4446
Tsermentseli, S., Leigh, P. N., and Goldstein, L. H. (2012). The anatomy of cognitive impairment in amyotrophic lateral sclerosis: more than frontal lobe dysfunction. Cortex 48, 166–182. doi: 10.1016/j.cortex.2011.02.004
Turner, M. R., Eisen, A., Kiernan, M. C., Ravits, J., and Swash, M. (2018). Kinnier Wilson’s puzzling features of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 89, 657–666. doi: 10.1136/jnnp-2017-317217
Ulfhake, B., and Kellerth, J.-O. (1981). A quantitative light microscopic study of the dendrites of cat spinal α-motoneurons after intracellular staining with horseradish peroxidase. J. Comp. Neurol. 202, 571–583. doi: 10.1002/cne.902020409
Ullian, E. M., Christopherson, K. S., and Barres, B. A. (2004). Role for glia in synaptogenesis. Glia 47, 209–216. doi: 10.1002/glia.20082
Urca, G., and Urca, R. (1990). Neurotoxic effects of excitatory amino acids in the mouse spinal cord: quisqualate and kainate but not N-methyl-l-aspartate induce permanent neural damage. Brain Res. 529, 7–15. doi: 10.1016/0006-8993(90)90805-L
Urushitani, M., Shimohama, S., Kihara, T., Sawada, H., Akaike, A., Ibi, M., et al. (1998). Mechanism of selective motor neuronal death after exposure of spinal cord to glutamate: involvement of glutamate-induced nitric oxide in motor neuron toxicity and nonmotor neuron protection. Ann. Neurol. 44, 796–807. doi: 10.1002/ana.410440514
Valdez, G., Tapia, J. C., Lichtman, J. W., Fox, M. A., and Sanes, J. R. (2012). Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLoS One 7:e34640. doi: 10.1371/journal.pone.0034640
Valori, C. F., Brambilla, L., Martorana, F., and Rossi, D. (2014). The multifaceted role of glial cells in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 71, 287–297. doi: 10.1007/s00018-013-1429-7
Van, L. D. B., and Robberecht, W. (2000). Different receptors mediate motor neuron death induced by short and long exposures to excitotoxicity. Brain Res. Bull. 53, 383–388. doi: 10.1016/S0361-9230(00)00371-3
van Blitterswijk, M., DeJesus-Hernandez, M., Niemantsverdriet, E., Murray, M. E., Heckman, M. G., Diehl, N. N., et al. (2013). Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. 12, 978–988. doi: 10.1016/S1474-4422(13)70210-2
van Blitterswijk, M., van Es, M. A., Hennekam, E. A. M., Dooijes, D., van Rheenen, W., Medic, J., et al. (2012). Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum. Mol. Genet. 21, 3776–3784. doi: 10.1093/hmg/dds199
Van Deerlin, V. M., Leverenz, J. B., Bekris, L. M., Bird, T. D., Yuan, W., Elman, L. B., et al. (2008). TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 7, 409–416. doi: 10.1016/S1474-4422(08)70071-1
Van Den Bosch, L., Van Damme, P., Bogaert, E., and Robberecht, W. (2006). The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 1068–1082. doi: 10.1016/j.bbadis.2006.05.002
van der Graaff, M. M., de Jong, J. M. B. V., Baas, F., and de Visser, M. (2009). Upper motor neuron and extra-motor neuron involvement in amyotrophic lateral sclerosis: a clinical and brain imaging review. Neuromuscul. Disord. 19, 53–58. doi: 10.1016/j.nmd.2008.10.002
van der Zee, J., Gijselinck, I., Dillen, L., Van Langenhove, T., Theuns, J., Engelborghs, S., et al. (2013). A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum. Mutat. 34, 363–373. doi: 10.1002/humu.22244
Van Hoecke, A., Schoonaert, L., Lemmens, R., Timmers, M., Staats, K. A., Laird, A. S., et al. (2012). EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat. Med. 18, 1418–1422. doi: 10.1038/nm.2901
van Rheenen, W., Shatunov, A., Dekker, A. M., McLaughlin, R. L., Diekstra, F. P., Pulit, S. L., et al. (2016). Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 48, 1043–1048. doi: 10.1038/ng.3622
Vance, C., Rogelj, B., Hortobágyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Vande Velde, C., McDonald, K. K., Boukhedimi, Y., McAlonis-Downes, M., Lobsiger, C. S., Bel Hadj, S., et al. (2011). Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS One 6:e22031. doi: 10.1371/journal.pone.0022031
Vandenberghe, W., Ihle, E. C., Patneau, D. K., Robberecht, W., and Brorson, J. R. (2000). AMPA receptor current density, not desensitization, predicts selective motoneuron vulnerability. J. Neurosci. 20, 7158–7166. doi: 10.1523/jneurosci.20-19-07158.2000
Vandoorne, T., De Bock, K., and Van Den Bosch, L. (2018). Energy metabolism in ALS: an underappreciated opportunity? Acta Neuropathol. 135, 489–509. doi: 10.1007/s00401-018-1835-x
Vanselow, B. K., and Keller, B. U. (2000). Calcium dynamics and buffering in oculomotor neurones from mouse that are particularly resistant during amyotrophic lateral sclerosis (ALS)-related motoneurone disease. J. Physiol. 525, 433–445. doi: 10.1111/j.1469-7793.2000.t01-1-00433.x
Varadi, A., Johnson-Cadwell, L. I., Cirulli, V., Yoon, Y., Allan, V. J., and Rutter, G. A. (2004). Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynamin-related protein-1. J. Cell. Sci. 117, 4389–4400. doi: 10.1242/jcs.01299
Veldink, J. H. (2017). ALS genetic epidemiology “How simplex is the genetic epidemiology of ALS?”. J. Neurol. Neurosurg. Psychiatry 88:537. doi: 10.1136/jnnp-2016-315469
Venkova, K., Christov, A., Kamaluddin, Z., Kobalka, P., Siddiqui, S., and Hensley, K. (2014). Semaphorin 3A signaling through neuropilin-1 is an early trigger for distal axonopathy in the SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 73, 702–713. doi: 10.1097/NEN.0000000000000086
Venugopal, S., Hsiao, C.-F., Sonoda, T., Wiedau-Pazos, M., and Chandler, S. H. (2015). Homeostatic dysregulation in membrane properties of masticatory motoneurons compared with oculomotor neurons in a mouse model for amyotrophic lateral sclerosis. J. Neurosci. 35, 707–720. doi: 10.1523/JNEUROSCI.1682-14.2015
Vijg, J., and Suh, Y. (2013). Genome instability and aging. Annu. Rev. Physiol. 75, 645–668. doi: 10.1146/annurev-physiol-030212-183715
Vincent, A. M., and Feldman, E. L. (2002). Control of cell survival by IGF signaling pathways. Growth Horm. IGF Res. 12, 193–197. doi: 10.1016/s1096-6374(02)00017-5
Vinsant, S., Mansfield, C., Jimenez-Moreno, R., Del Gaizo Moore, V., Yoshikawa, M., Hampton, T. G., et al. (2013). Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part II, results and discussion. Brain Behav. 3, 431–457. doi: 10.1002/brb3.142
Volterra, A., and Meldolesi, J. (2005). Astrocytes, from brain glue to communication elements: the revolution continues. Nat. Rev. Neurosci. 6, 626–640. doi: 10.1038/nrn1722
Vucic, S., and Kiernan, M. C. (2010). Upregulation of persistent sodium conductances in familial ALS. J. Neurol. Neurosurg. Psychiatry 81, 222–227. doi: 10.1136/jnnp.2009.183079
Vucic, S., Nicholson, G. A., and Kiernan, M. C. (2008). Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 131, 1540–1550. doi: 10.1093/brain/awn071
Wainger, B. J., Kiskinis, E., Mellin, C., Wiskow, O., Han, S. S. W., Sandoe, J., et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7, 1–11. doi: 10.1016/j.celrep.2014.03.019
Walhout, R., Verstraete, E., van den Heuvel, M. P., Veldink, J. H., and van den Berg, L. H. (2018). Patterns of symptom development in patients with motor neuron disease. Amyotroph. Lateral. Scler. Frontotemporal Degener. 19, 21–28. doi: 10.1080/21678421.2017.1386688
Walker, A. K., Farg, M. A., Bye, C. R., McLean, C. A., Horne, M. K., and Atkin, J. D. (2010). Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain 133, 105–116. doi: 10.1093/brain/awp267
Walker, A. K., Soo, K. Y., Sundaramoorthy, V., Parakh, S., Ma, Y., Farg, M. A., et al. (2013). ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS One 8:e81170. doi: 10.1371/journal.pone.0081170
Walker, A. K., Spiller, K. J., Ge, G., Zheng, A., Xu, Y., Zhou, M., et al. (2015). Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 130, 643–660. doi: 10.1007/s00401-015-1460-x
Walker, C., Herranz-Martin, S., Karyka, E., Liao, C., Lewis, K., Elsayed, W., et al. (2017). C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat. Neurosci. 20, 1225–1235. doi: 10.1038/nn.4604
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. doi: 10.1126/science.1209038
Wang, I.-F., Wu, L.-S., Chang, H.-Y., and Shen, C.-K. J. (2008). TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J. Neurochem. 105, 797–806. doi: 10.1111/j.1471-4159.2007.05190.x
Wang, L., Gutmann, D. H., and Roos, R. P. (2011). Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum. Mol. Genet. 20, 286–293. doi: 10.1093/hmg/ddq463
Wang, L., Pytel, P., Feltri, M. L., Wrabetz, L., and Roos, R. P. (2012). Selective knockdown of mutant SOD1 in Schwann cells ameliorates disease in G85R mutant SOD1 transgenic mice. Neurobiol. Dis. 48, 52–57. doi: 10.1016/j.nbd.2012.05.014
Wang, L., Sharma, K., Grisotti, G., and Roos, R. P. (2009). The effect of mutant SOD1 dismutase activity on non-cell autonomous degeneration in familial amyotrophic lateral sclerosis. Neurobiol. Dis. 35, 234–240. doi: 10.1016/j.nbd.2009.05.002
Wang, X., Zaidi, A., Pal, R., Garrett, A. S., Braceras, R., Chen, X., et al. (2009). Genomic and biochemical approaches in the discovery of mechanisms for selective neuronal vulnerability to oxidative stress. BMC Neurosci. 10:12. doi: 10.1186/1471-2202-10-12
Wang, W., Li, L., Lin, W.-L., Dickson, D. W., Petrucelli, L., Zhang, T., et al. (2013). The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 22, 4706–4719. doi: 10.1093/hmg/ddt319
Wang, X., and Michaelis, E. K. (2010). Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2:12. doi: 10.3389/fnagi.2010.00012
Wegorzewska, I., Bell, S., Cairns, N. J., Miller, T. M., and Baloh, R. H. (2009). TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. U.S.A. 106, 18809–18814. doi: 10.1073/pnas.0908767106
Wehner, A. B., Abdesselem, H., Dickendesher, T. L., Imai, F., Yoshida, Y., Giger, R. J., et al. (2016). Semaphorin 3A is a retrograde cell death signal in developing sympathetic neurons. Development 143, 1560–1570. doi: 10.1242/dev.134627
Westbury, D. R. (1982). A comparison of the structures of α- and γ-spinal motoneurones of the cat. J. Physiol. 325, 79–91. doi: 10.1113/jphysiol.1982.sp014137
Westergard, T., Jensen, B. K., Wen, X., Cai, J., Kropf, E., Iacovitti, L., et al. (2016). Cell-to-cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Rep. 17, 645–652. doi: 10.1016/j.celrep.2016.09.032
Wheway, G., Schmidts, M., Mans, D. A., Szymanska, K., Nguyen, T.-M. T., Racher, H., et al. (2015). An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell Biol. 17, 1074–1087. doi: 10.1038/ncb3201
Wiedemann, F. R., Manfredi, G., Mawrin, C., Beal, M. F., and Schon, E. A. (2002). Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 80, 616–625. doi: 10.1046/j.0022-3042.2001.00731.x
Williams, K. L., Topp, S., Yang, S., Smith, B., Fifita, J. A., Warraich, S. T., et al. (2016). CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 7:11253. doi: 10.1038/ncomms11253
Williams, K. L., Warraich, S. T., Yang, S., Solski, J. A., Fernando, R., Rouleau, G. A., et al. (2012). UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol. Aging 33, 2527.e3–2527.e10. doi: 10.1016/j.neurobiolaging.2012.05.008
Williamson, T. L., and Cleveland, D. W. (1999). Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat. Neurosci. 2, 50–56. doi: 10.1038/4553
Woehlbier, U., Colombo, A., Saaranen, M. J., Pérez, V., Ojeda, J., Bustos, F. J., et al. (2016). ALS-linked protein disulfide isomerase variants cause motor dysfunction. EMBO J. 35, 845–865. doi: 10.15252/embj.201592224
Wong, P. C., Pardo, C. A., Borchelt, D. R., Lee, M. K., Copeland, N. G., Jenkins, N. A., et al. (1995). An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14, 1105–1116. doi: 10.1016/0896-6273(95)90259-7
Woollacott, I. O. C., and Mead, S. (2014). The C9ORF72 expansion mutation: gene structure, phenotypic and diagnostic issues. Acta Neuropathol. 127, 319–332. doi: 10.1007/s00401-014-1253-7
Wu, C.-H., Fallini, C., Ticozzi, N., Keagle, P. J., Sapp, P. C., Piotrowska, K., et al. (2012). Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503. doi: 10.1038/nature11280
Wu, D., Yu, W., Kishikawa, H., Folkerth, R. D., Iafrate, A. J., Shen, Y., et al. (2007). Angiogenin loss-of-function mutations in amyotrophic lateral sclerosis. Ann. Neurol. 62, 609–617. doi: 10.1002/ana.21221
Xu, W., and Lipscombe, D. (2001). Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J. Neurosci. 21, 5944–5951. doi: 10.1523/jneurosci.21-16-05944.2001
Xu, Y.-F., Gendron, T. F., Zhang, Y.-J., Lin, W.-L., D’Alton, S., Sheng, H., et al. (2010). Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 30, 10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010
Yagi, K., Kitazato, K. T., Uno, M., Tada, Y., Kinouchi, T., Shimada, K., et al. (2009). Edaravone, a free radical scavenger, inhibits MMP-9-related brain hemorrhage in rats treated with tissue plasminogen activator. Stroke 40, 626–631. doi: 10.1161/STROKEAHA.108.520262
Yamamoto, Y., Mizuno, R., Nishimura, T., Ogawa, Y., Yoshikawa, H., Fujimura, H., et al. (1994). Cloning and expression of myelin-associated oligodendrocytic basic protein. A novel basic protein constituting the central nervous system myelin. J. Biol. Chem. 269, 31725–31730.
Yamanaka, K., Chun, S. J., Boillee, S., Fujimori-Tonou, N., Yamashita, H., Gutmann, D. H., et al. (2008). Astrocytes as determinants of disease progression in inherited ALS. Nat. Neurosci. 11, 251–253. doi: 10.1038/nn2047
Yamashita, T., Hideyama, T., Hachiga, K., Teramoto, S., Takano, J., Iwata, N., et al. (2012). A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat. Commun. 3:1307. doi: 10.1038/ncomms2303
Yanagi, K. S., Wu, Z., Amaya, J., Chapkis, N., Duffy, A. M., Hajdarovic, K. H., et al. (2019). Meta-analysis of genetic modifiers reveals candidate dysregulated pathways in amyotrophic lateral sclerosis. Neuroscience 396, A3–A20. doi: 10.1016/j.neuroscience.2018.10.033
Yoshino, H., and Kimura, A. (2006). Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph. Lateral. Scler. 7, 241–245. doi: 10.1080/17482960600881870
Young, N. A., Collins, C. E., and Kaas, J. H. (2013). Cell and neuron densities in the primary motor cortex of primates. Front. Neural Circ. 7:30. doi: 10.3389/fncir.2013.00030
Yu, J., Anderson, C. T., Kiritani, T., Sheets, P. L., Wokosin, D. L., Wood, L., et al. (2008). Local-circuit phenotypes of layer 5 neurons in motor-frontal cortex of YFP-H mice. Front. Neural Circ. 2:6. doi: 10.3389/neuro.04.006.2008
Zajac, F. E., and Faden, J. S. (1985). Relationship among recruitment order, axonal conduction velocity, and muscle-unit properties of type-identified motor units in cat plantaris muscle. J. Neurophysiol. 53, 1303–1322. doi: 10.1152/jn.1985.53.5.1303
Zengel, J. E., Reid, S. A., Sypert, G. W., and Munson, J. B. (1985). Membrane electrical properties and prediction of motor-unit type of medial gastrocnemius motoneurons in the cat. J. Neurophysiol. 53, 1323–1344. doi: 10.1152/jn.1985.53.5.1323
Zhang, F., Ström, A.-L., Fukada, K., Lee, S., Hayward, L. J., and Zhu, H. (2007). Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex. J. Biol. Chem. 282, 16691–16699. doi: 10.1074/jbc.M609743200
Zhang, H., Tan, C.-F., Mori, F., Tanji, K., Kakita, A., Takahashi, H., et al. (2008). TDP-43-immunoreactive neuronal and glial inclusions in the neostriatum in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol. 115, 115–122. doi: 10.1007/s00401-007-0285-7
Zhang, K., Donnelly, C. J., Haeusler, A. R., Grima, J. C., Machamer, J. B., Steinwald, P., et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61. doi: 10.1038/nature14973
Zhang, M., Xi, Z., Zinman, L., Bruni, A. C., Maletta, R. G., Curcio, S. A. M., et al. (2015). Mutation analysis of CHCHD10 in different neurodegenerative diseases. Brain 138:e380. doi: 10.1093/brain/awv082
Zhang, Y.-J., Gendron, T. F., Grima, J. C., Sasaguri, H., Jansen-West, K., Xu, Y.-F., et al. (2016). C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci. 19, 668–677. doi: 10.1038/nn.4272
Zhou, Y., Liu, D., and Kaminski, H. J. (2011). Pitx2 regulates myosin heavy chain isoform expression and multi-innervation in extraocular muscle. J. Physiol. 589, 4601–4614. doi: 10.1113/jphysiol.2011.207076
Zou, Z.-Y., Zhou, Z.-R., Che, C.-H., Liu, C.-Y., He, R.-L., and Huang, H.-P. (2017). Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 88, 540–549. doi: 10.1136/jnnp-2016-315018
Keywords: amyotrophic lateral sclerosis, neurodegeneration, selective vulnerability, fast and slow motor units, frontotemporal dementia
Citation: Ragagnin AMG, Shadfar S, Vidal M, Jamali MS and Atkin JD (2019) Motor Neuron Susceptibility in ALS/FTD. Front. Neurosci. 13:532. doi: 10.3389/fnins.2019.00532
Received: 06 March 2019; Accepted: 08 May 2019;
Published: 27 June 2019.
Edited by:
Alberto Lleo, Hospital de la Santa Creu i Sant Pau, SpainReviewed by:
Mamede De Carvalho, Universidade de Lisboa, PortugalCopyright © 2019 Ragagnin, Shadfar, Vidal, Jamali and Atkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie D. Atkin, anVsaWUuYXRraW5AbXEuZWR1LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.