
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurosci. , 17 May 2019
Sec. Perception Science
Volume 13 - 2019 | https://doi.org/10.3389/fnins.2019.00484
This article is part of the Research Topic Neuromodulatory Interventions for Pain View all 12 articles
 He Zhu1,2
He Zhu1,2 Hong-Chun Xiang1
Hong-Chun Xiang1 Hong-Ping Li1
Hong-Ping Li1 Li-Xue Lin1
Li-Xue Lin1 Xue-Fei Hu1
Xue-Fei Hu1 Hong Zhang1
Hong Zhang1 Wang-Yang Meng1
Wang-Yang Meng1 Lu Liu1
Lu Liu1 Chao Chen1
Chao Chen1 Yang Shu3
Yang Shu3 Ru-Yue Zhang1
Ru-Yue Zhang1 Pei Zhang1
Pei Zhang1 Jun-Qiang Si2*
Jun-Qiang Si2* Man Li1*
Man Li1*Although electroacupuncture (EA) has become a worldwide practice, little is understood about its precise target in the central nervous system (CNS) and the cell type-specific analgesia mechanism. In the present study, we found that EA has significant antinociceptive effects both in inflammatory and neuropathic pain models. Chemogenetic inhibition of GABAergic neurons in the ventrolateral periaqueductal gray (vlPAG) replicated the effects of EA, whereas the combination of chemogenetic activation of GABAergic neurons and chemogenetic inhibition of glutamatergic neurons in the vlPAG was needed to reverse the effects of EA. Specifically knocking out CB1 receptors on GABAergic neurons in the vlPAG abolished the EA effect on pain hypersensitivity, while specifically knocking out CB1 receptors on glutamatergic neurons attenuated only a small portion of the EA effect. EA synchronously inhibits GABAergic neurons and activates glutamatergic neurons in the vlPAG through CB1 receptors to produce EA-induced analgesia. The CB1 receptors on GABAergic neurons localized in the vlPAG was the basis of the EA effect on pain hypersensitivity. This study provides new experimental evidence that EA can bidirectionally regulate GABAergic neurons and glutamatergic neurons via the CB1 receptors of the vlPAG to produce analgesia effects.
A series of randomized controlled trials of acupuncture and analgesia have shown that acupuncture has an analgesic effect in the context of chronic pain, such as knee pain, low back pain, migraine, and fibromyalgia (Liu et al., 2018; Mist and Jones, 2018; Musil et al., 2018). However, the specific target sites in the central nervous system (CNS) for electroacupuncture (EA) analgesia are still unclear, which seriously hinders the clinical promotion and application of EA analgesia.
Studies have confirmed that EA can induce the specific expression of c-fos in the periaqueductal gray (PAG) region (Fusumada et al., 2007). It has been demonstrated that the ventrolateral periaqueductal gray (vlPAG) is an essential part of the neural pathway that mediates pain regulation (Ho et al., 2011). Microinjection of GABA agonists into the vlPAG promotes pain, and microinjection of GABA antagonists produces antinociceptive effects by reducing inhibitory neurotransmission (Bobeck et al., 2014; Takasu et al., 2015). According to the GABA disinhibition hypothesis, tonically active GABAergic interneurons are present within the PAG, which release the neurotransmitter GABA that acts via GABAA receptors to inhibit spinally projecting output neurons (Basbaum and Fields, 1984). It was proposed that opioids and cannabinoids activate the descending pathway by indirectly suppressing the inhibitory influence of local GABAergic interneurons, thereby disinhibiting the antinociceptive pathway of the neuronal output to the spinal cord (Meng et al., 1998).
The endocannabinoid system, which is a crucial neuromodulatory system involved in the control of pain transmission and acupuncture analgesia within the CNS (Chen et al., 2009). Cannabinoid-induced antinociception plays a role in the activation of a descending inhibitory pain pathway (Palazzo et al., 2010). Cannabinoid receptor 1(CB1) are expressed in both nerve endings of GABAergic neurons and glutamatergic neurons in PAG and activation of the CB1 receptor may regulate GABAergic and glutamatergic neurotransmission (Drew et al., 2008; Tjen-A-Looi et al., 2009). Our previous results have shown that EA reversed the reduced expression of CB1 receptors and the 2-arachidonoylglycerol (2-AG) level in the midbrain in chronic pain conditions (Yuan et al., 2018). These results indicated that EA may exert an analgesic effect by acting on the CB1 receptor. Microinjection of the CB1 receptor antagonist AM251 into the vlPAG can reverse the EA effect on pain hypersensitivity, which further confirms the mechanism of EA exerting an antinociceptive effect through CB1 receptors in the vlPAG (Yuan et al., 2018). Despite the abundant evidence supporting the hypothesis that the CB1 receptor is involved in EA analgesia, the exact cell type involved in EA analgesia through vlPAG descending pain modulation has not been directly investigated. In this study, with the help of cell type-specific chemogenetic manipulations in the vlPAG, we attempt to verify the hypothesis that GABAergic neurons and glutamatergic neurons are involved in CB1-mediated EA antinociception.
All animal experiments were ratified by the Animal Care and Use Committee of Huazhong University of Science and Technology, the procedures conform to the ethical guidelines of the International Association for the Study of Pain (Demers et al., 2006). Eight-weeks-old male C57BL/6 mice (20–25 g) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. The mCnr1flox/flox mice were bought from the Cyagen biosciences laboratory (Nanjing). GAD67-GFP mice were kindly provided by Dr. Xiangning Li (Huazhong University of Science and Technology, Whuhan, China). The mice were individually housed in cages with a 12-h light/dark cycle and had free access to food and water. The mice used for EA experiments in CCI and KOA mice were divided into four groups (control, CCI or KOA, EA, and sham EA). The mice used for chemogenetic manipulation and knockout experiments in CCI and KOA mice were divided into three groups (control, chemogenetic manipulation or knockout, EA).
Adeno-associated viruses (AAV/2-9) were designed to achieve CRE-DIO system-mediated chemogenetic manipulation strategy: rAAV-mDlx-CRE-WPRE-pA combined with rAAV-hSyn-DIO-hM3D(Gq)-mCherry-WPRE-pA or rAAV-hSyn-DIO-hM4D(Gi)-mCherry-WPRE-pA were designed to excite or inhibit GABAergic neurons of vlPAG (Dimidschstein et al., 2016). rAAV-hSyn-mCherry-WPRE-pA was selected as control virus. Furthermore, rAAV-CaMKIIa-HA-KORD-IRES-mCitrine-WPRE-pA, selecting salvinorin B as DREAD, could inhibit glutaminergic neurons of vlPAG individually on the basis of above combined operations and without interactions.
All viruses used in this study were acquired from the Wuhan BrainVTA scientific and technical corporation. Before surgery, mice were anesthetized with isoflurane and fixed in the stereotaxic apparatus (RWD Instruments,China). Make a 1.5 cm length longitudinal incision along the midline of the skull, gently remove the periosteum from the exposed surface of the surgical area. Viruses injections were performed using the coordinates of vlPAG as following: −4.8 mm from bregma, −0.4 mm lateral from midline, and 2.8 mm ventral to skull. Desired viruses vectors (150 nL) were injected into the vlPAG at a rate of 50 nl per 60 s (Samineni et al., 2017). If the virus infection area exceeds the vlPAG area, it is not included in the statistics (Figure S5).
Three weeks after viruses injections, mice were intraperitoneally injected with clozapine N-oxide (CNO, Sigma) 60 min before the behavioral assessment. All baselines for thermal and mechanical sensitivity were recorded after the viruses injections and before the CNO administration. We administered 1 mg/kg CNO for both hM3Dq activation and hM4Di inhibition (Samineni et al., 2017). In rAAV-CaMKIIa-HA-KORD-IRES-mCitrine-WPRE-pA operation mice, we individually administered 5 mg/kg salvinorin B for inhibiting glutaminergic neurons of vlPAG even on the basis of GABAergic neurons have been excited by hM3Dq activation with CNO (Vardy et al., 2015). In the chemogenetic reversing experiment of EA antinociception, CNO was applied just before EA treatment.
Neuropathy was induced by chronic constriction injury (CCI) of the sciatic nerve using a similar procedure for rats (Bennett and Xie, 1988) which was adapted for mice (Sommer et al., 1998). Two loosely constrictive ligatures were tied around the left sciatic nerve to slow the blood flow of the sciatic myelin blood vessels without causing acute crushing damage. The sham group animals were anesthetized, their sciatic nerves were only exposed without constriction. Sham-operated animals were used as neuropathy controls.
Intra-articular injection of monosodium iodoacetate (MIA) (Sigma, UK) into the left knee joint was applied to induce KOA model. Five microliters of 5 mg/ml MIA in sterile saline (0.9%) were injected into the joint space of the left knee through the infrapatellar ligament with a 30-gauge needle (La Porta et al., 2013). This method causes histopathology changes in the cartilage (van Osch et al., 1994) and produces obvious joint pain in mice (Harvey and Dickenson, 2009). The sham-operated mice received an intra-articular injection of 5 μl of 0.9% sterile saline.
For the CCI model EA treatment group, the mouse received EA on the left “Huantiao” (GB30) and “Yanglingquan” (GB34) once a day, starting from the eighth day after operation. GB30 and GB34 were chosen based on their effect in improving inflammatory pain and neuropathic pain in mouse (Kang et al., 2007; Park et al., 2014; Lee et al., 2018). Two acupuncture needles were inserted 2–3 mm deep into two acupoints corresponding to GB30 and GB34 in humans. EA (1 mA and 0.1 ms) was carried out at 2 Hz for 30 min (Wu et al., 2013). Current was delivered with a modified constant current Han's Acupoint Nerve Stimulator (LH202, Huawei Co. Ltd., Beijing, China) (Wu et al., 2013).
For the EA treatment group of KOA, mice received EA administration on the left “Neixiyan” (Ex-LE4) and “Dubi” (ST35) for 7 days, starting from the fifteenth day after MIA injection. EA (1 mA and 0.1 ms) was carried out at 2 Hz for 30 min. Ex-LE4 and ST35 were chosen based on the fact that their using frequency is the highest in KOA (Wu et al., 2010).
For sham treatment control, acupuncture needles were inserted into above acupuncture points without electrical stimulation or manual manipulation. Previous studies showed that needles inserted into active acupoints, but no electrical or manual stimulation, do not produce analgesia (Lao et al., 2004). During EA treatment, each mouse was placed in a homemade bag. Control group also received the same method to exclude the stress response. The animals remained still during EA treatment and showed no evident signs of distress.
Mechanical allodynia and heat hyperalgesia were checked with von Frey filaments and the hot plate, respectively (Fernihough et al., 2004). The animals were habituated to the testing environment for 30 min. The behavioral tests were performed 3 times before model induction and every other day after model induction until the formal experimental operation. The behavioral tests were performed once a day during kinds of experimental operations (Figure S1).
The surface temperature of the hot-plate was maintained at 53°C. The withdrawal latency started from the mouse was put on the plate and terminated when a quick/prolonged withdrawal or flick of the paw was observed. Twenty seconds was set as a cut-off time for mice to prevent tissue damage (Chen et al., 2017). Thermal stimuli were delivered three times to hindpaw at 10 min intervals, and the mean value was calculated.
The tactile withdrawal threshold of mice was measured by using the “up-down” method (Chaplan et al., 1994). After an acclimation period of 30 min, we stimulated the plantar surface of the hindpaw vertically with a series of von Frey (Stoelting, Wood Dale, IL) hairs with logarithmically increasing stiffness, and bent the filament for 5 s to the central plantar surface with sufficient force. Brisk withdrawal or paw flinching was considered as a positive response. The test of tactile withdrawal threshold was repeated two times in each mouse, and the mean value was calculated.
The test trials consisted of one habituation (1st day), four conditionings (2nd−5th days), and one test (6th day) (Hnasko et al., 2005). In the case of EA treatment, the mice were confined in white box, thereby achieving EA treatment and white box condition matching. On the test day, animals were free to go any parts of the apparatus, and the time spent in bright or dark box was measured. Data were analyzed by the SuperMaze software (Xinsoft SuperMaze Animal Behavior Analysis System, Shanghai).
Mice under deep anesthesia with 10% chloralic hydras were transcardially perfused with 37°C normal saline followed by 4% paraformaldehyde in 0.1 M PBS (pH, 7.4; 4°C). The brain were removed immediately and post-fixed in the same fixative. Then, the tissues were cryoprotected in 20 and 30% sucrose in 0.1 M PBS for 24 h, respectively, at 4°C. The OCT embedded blocks were sectioned for 25-μm thickness. Sections from each group were rinsed in 0.01 M PBS and blocked for 2 h with blocking liquid (5% donkey serum and 0.2% tween-20 in 0.01 M PBS) at room temperature. The sections were probed with the following antibodies: rabbit anti-CB1 (1:200), mouse anti–GAD67 (1:200), guinea pig anti-VGLUT2 (1:100). Subsequently, the free-floating sections were washed with 0.01M PBS 3 times, and incubated with following secondary antibodies (Jackson ImmunoResearch) for 2 h: donkey anti-rabbit IgG conjugated with Dylight 594 (1:600), donkey anti-mouse IgG conjugated with Dylight 488 (1:600), donkey anti-guinea pig IgG conjugated with Dylight 488 (1:400). The sections were incubated with DAPI for the nucleus staining for 8 min, and washed 3 times in 0.01M PBS and then cover-slipped. The samples were studied under a fluorescence microscope (Olympus) for the immunefluorescence staining. Images were analyzed by using NIH Image J software (Bethesda, MD, USA). The layouts of images were based on the Photoshop (ADOBE company, USA).
Results of data are expressed as mean ± SEM. The thermal latency and withdrawal thresholds between different groups over time were tested with two-way analysis of variance (ANOVA) followed by Bonferroni post hoc tests. Other data were analyzed by One-way ANOVA and Newman-Keuls post hoc test (SPSS, Version 11.0). Results represented as significance based on a value of P < 0.05.
CCI or KOA induction significantly reduced mechanical withdrawal thresholds and thermal withdrawal latencies (Figures S2A–D).
The tactile threshold and thermal withdrawal latency were effectively increased by EA in CCI and KOA mice (Figures 1A–D).
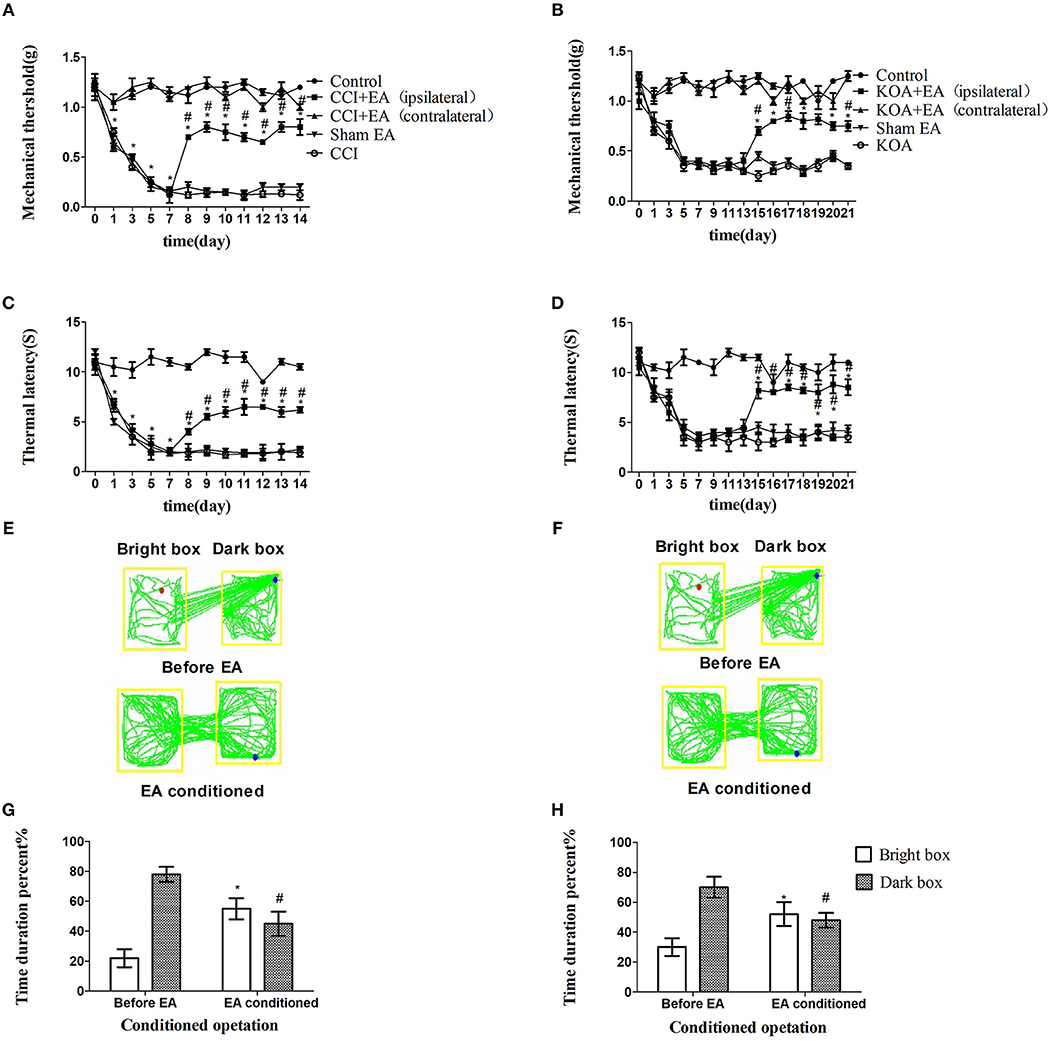
Figure 1. EA effectively reduces pain hypersensitivity in CCI and KOA mice. (A,B) Time course of tactile threshold in response to von Frey filaments. (C,D) The noxious heat stimulus (53°C) caused a change in the thermal thresholds. The effect of tactile (A) and thermal (C) withdrawal thresholds changes in CCI mice. The change of tactile (B) and thermal (D) withdrawal thresholds of KOA mice caused by EA. A motion map of the EA-conditioned CPP test in CCI (E) and KOA mice (F). G,H are summary values of motion trajectories. EA (1 mA and 0.1 ms) at 2 Hz was administered for 30 min, once a day starting from the 8th to the 14th day in the CCI model and starting from the 15th to 21st day in the KOA model. Control group consisted of sham-operated mice. Data are expressed as the means ± SEM (n = 12 in each group). In A–D, *p < 0.05, compared with the control group; #p < 0.05, compared with the model group; In panels G,H,*p < 0.05, compared with the bright box time duration percent before EA; #p < 0.05, compared with the dark box time duration percent before EA.
The repeated EA treatment also affected the behavior in the CPP test, as the mouse spent more time in the bright box after being paired with EA treatment (Figures 1E–H).
Since the PAG is a vital brain region related to the descending pain control system, both cannabinoids and opioids, released by EA, may inhibited GABAergic neurons to produce antinociceptive effect by GABAergic disinhibition mechanism (Osborne et al., 1996; Finn et al., 2003), we applied a chemogenetic strategy to assess this hypothesis.
The viruses combination cocktail of rAAV-mDlx-CRE-WPRE-pA and rAAV-hSyn-DIO-hM3D(Gi)-mCherry-WPRE-pA, designed to inhibit GABAergic neurons, was injected into the right side of the vlPAG (Figure 2A). The viruses combination was injected into the right side of the vlPAG of GAD67 GFP mouse to confirm the viruses combination efficiency (Figures 2B,C). A total of 80.5% of the viruses combination-tagged neurons in the vlPAG were marked together with GAD67 GFP-positive cells (Figure 2D).
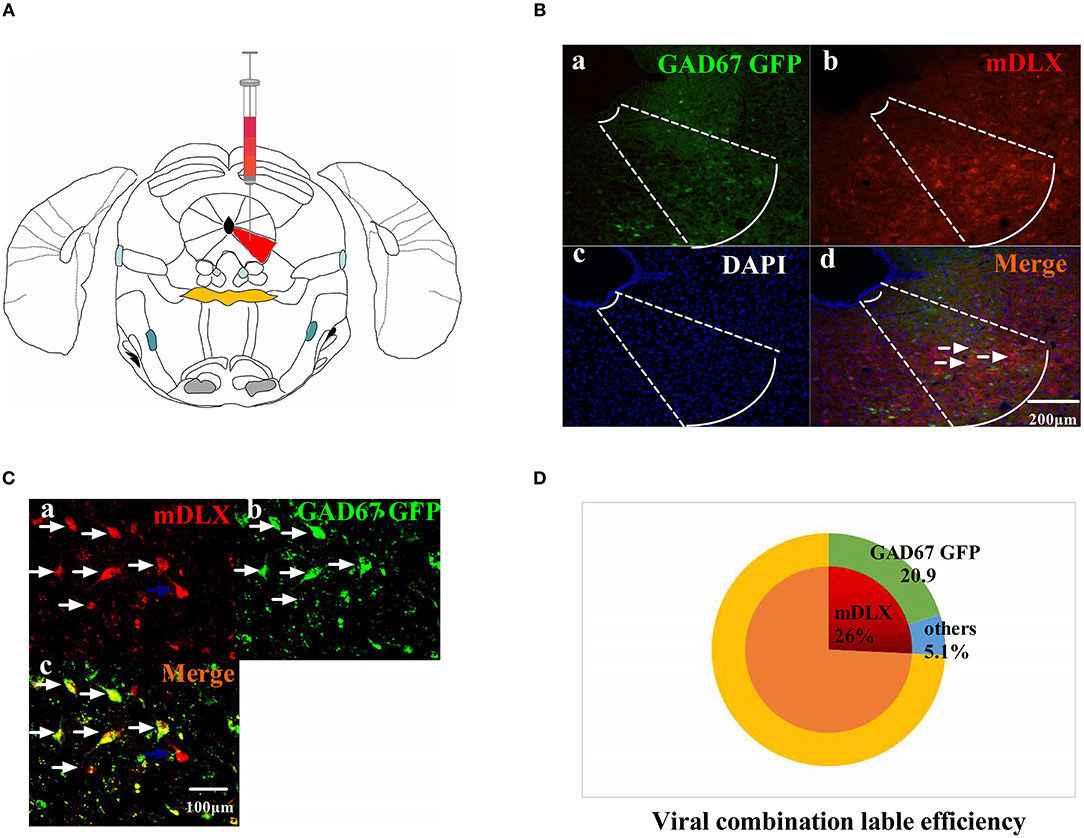
Figure 2. The viruses combination cocktail label efficiency. (A) Pattern map of the viruses injection area. The red color region is the vlPAG. (B) The viruses combination cocktail was injected into the right side of the vlPAG of GAD67 GFP mice. (a) GAD67 GFP cells in the vlPAG (green). (b) Viruses-labeled cells (red). (c) DAPI nuclear staining (blue). (d) Colocalization fluorescence (pink), arrows show typical fluorescent, colocalized cells. Scale bar, 200 μm. (C) High magnification images, scale bar, 100 μm. White arrows indicate cells that are co-labeled, and blue arrows indicate cells that are mDLX-labeled but not labeled with GAD67 GFP. (D) Summary data show the percentage of colocalized cells in the area of total viruses-labeled cells. Red sector, viruses-labeled cells; green sector, GAD67 GFP cells; blue sector, viruses-labeled other cells. Data are expressed as the means ± SEM (n = 3 mice in each group).
Chemogenetic inhibition of GABAergic neurons reliably replicated the effect of EA, including the antinociceptive effects (Figures 3A–H).
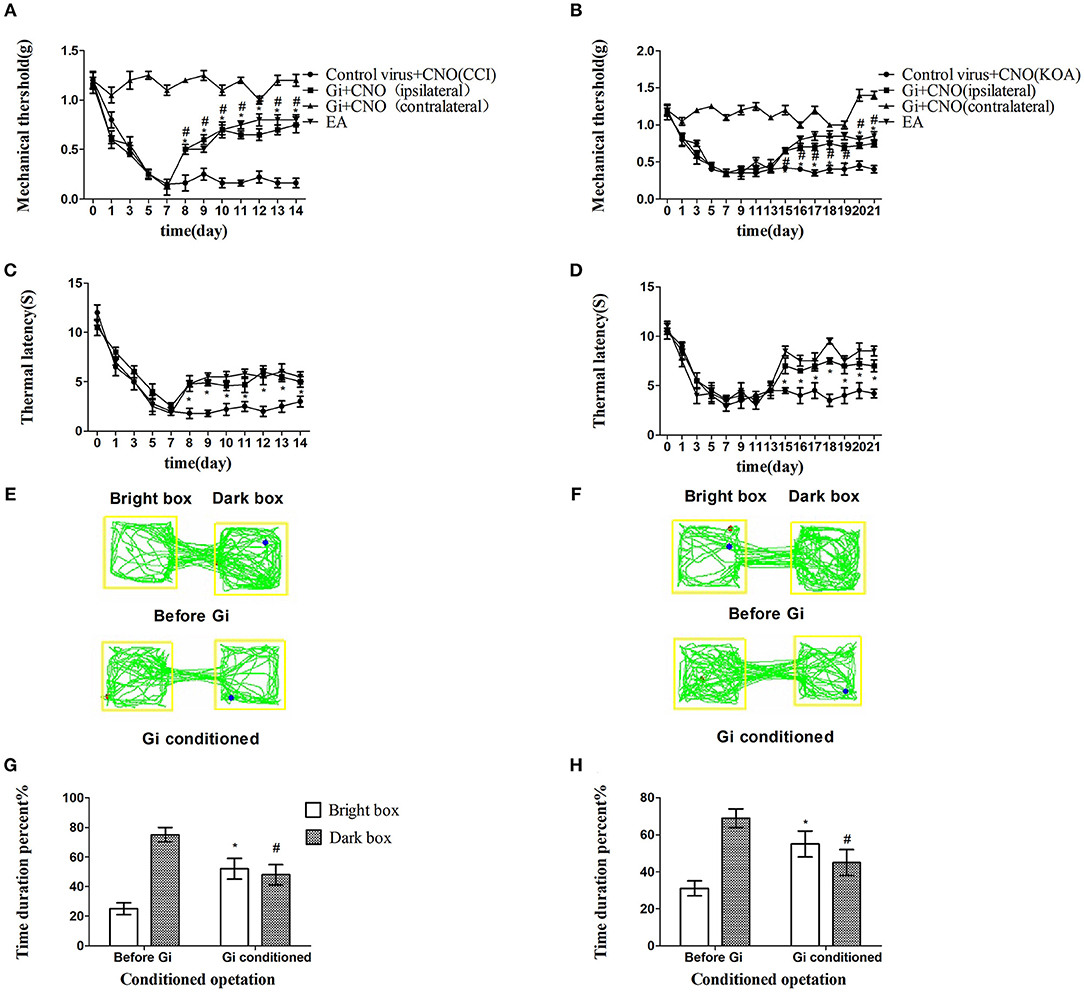
Figure 3. Chemogenetic inhibition of GABAergic neurons in the vlPAG replicated the effects of EA. (A,B) Time course of tactile threshold in response to von Frey filaments. (C,D) The noxious heat stimulus (53°C) caused a change in the thermal thresholds. Tactile (A) and thermal (C) withdrawal thresholds changes in CCI mice after chemogenetic inhibition. Changes in tactile (B) and thermal (D) withdrawal thresholds of KOA mice caused by chemogenetic inhibition. Motion map of chemogenetic inhibition of conditioning in the CPP test with CCI (E) and KOA mice (F). G,H are summary values of motion trajectories. Virus was injected 21 days before the behavior test. CNO (1 mg/kg) was administered once a day starting from the 8th to the 14th day in the CCI model and starting from the 15th to the 21st day in the KOA model. EA (1 mA and 0.1 ms) at 2 Hz was administered for 30 min. Once a day starting from the 8th to the 14th day in the CCI model and starting from the 15th to the 21st day in the KOA model, all behavior tests were completed 2 h after CNO administration. Control groups consisted of CCI or KOA mice injected with control virus and CNO but without EA treatment. Data are expressed as means ± SEM (n = 12 in each group). In A–D, *p < 0.05, compared with control group; #p < 0.05, compared with the model group; in G,H,*p < 0.05, compared with the bright box time duration percent before chemogenetic inhibition; #p < 0.05, compared with the dark box time duration percent before chemogenetic inhibition.
On the basis of chemogenetic inhibition of GABAergic neurons reliably replicating the antinociceptive effect of EA, we considered if chemogenetic activation could abolish the effect of EA. The viruses combination of rAAV-mDlx-CRE-WPRE-pA and rAAV-hSyn-DIO-hM3D(Gq)-mCherry-WPRE-pA, designed to activate GABAergic neurons, was injected into the right side of the vlPAG.
We unexpectedly found that chemogenetic activation of GABAergic neurons in the vlPAG only partly decreased the mechanical withdrawal threshold and thermal withdrawal latency in EA-treated mice (Figures 4A–D) while partly decreasing the bright box time in the CPP test (Figures 4E–H).
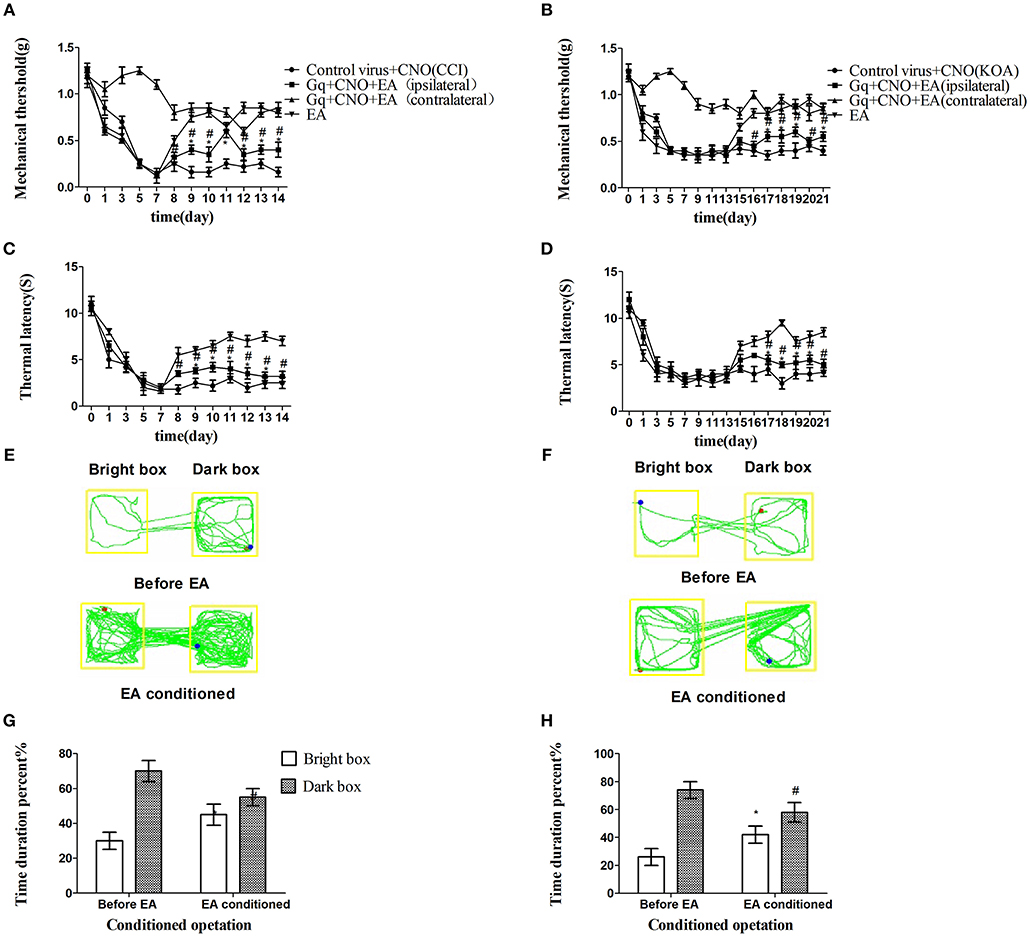
Figure 4. Chemogenetic activation of GABAergic neurons in the vlPAG only partly attenuated the effect of EA. (A,B) Time course of tactile threshold in response to von Frey filaments. (C,D) The noxious heat stimulus (53°C) caused a change in the thermal thresholds. Tactile (A) and thermal (C) withdrawal thresholds changes in CCI mice after chemogenetic activation. Changes in tactile (B) and thermal (D) withdrawal thresholds of KOA mice caused by chemogenetic activation. Motion map of chemogenetic activation of conditioning in the CPP test with CCI (E) and KOA mice (F). G,H are summary values of motion trajectories. Virus was injected 21 days before the behavior test. CNO (1 mg/kg) was administered once a day starting from the 8th to the 14th day in the CCI model and starting from the 15th to the 21st day in the KOA model. EA (1 mA and 0.1 ms) at 2 Hz was administered for 30 min. Once a day starting from the 8th to the 14th day in the CCI model and starting from the 15th to the 21st day in the KOA model, all behavior tests were completed 2 h after CNO administration. Control groups consisted of CCI or KOA mice injected with control virus and CNO but without EA treatment. Data are expressed as means ± SEM (n = 12 in each group). In A–D, *p < 0.05, compared with control group; #p < 0.05, compared with the model group; In G,H,*p < 0.05, compared with the bright box time duration percent before chemogenetic activation; #p < 0.05, compared with the dark box time duration percent before chemogenetic activation.
A large number of studies have shown that GABAergic neurons and glutamatergic neurons in the vlPAG play an important and complex role in nociceptive processes. Microinjection of glutamate receptor agonists or GABA antagonists into the vlPAG has a significant antinociceptive effect against noxious stimuli (Budai et al., 1998; Morgan et al., 2003). Since activation of GABAergic neurons alone in the vlPAG only partly attenuated the effect of EA, we speculated that glutamatergic and GABAergic neurons were both involved in these effects.
The rAAV-CaMKIIa-HA-KORD-IRES-mCitrine-WPRE-pA virus was applied to selectively inhibit the glutamatergic neurons of the vlPAG (Figure S4), on the basis that GABAergic neurons were excited by hM3Dq and CNO. These two systems were designed to work separately without interactions.
The combination of chemogenetic activation of GABAergic neurons and chemogenetic inhibition of glutamatergic neurons in the vlPAG powerfully decreased the mechanical withdrawal threshold and thermal withdrawal latency (Figures 5A–D) and the bright box time in the CPP test (Figures 5E–H).
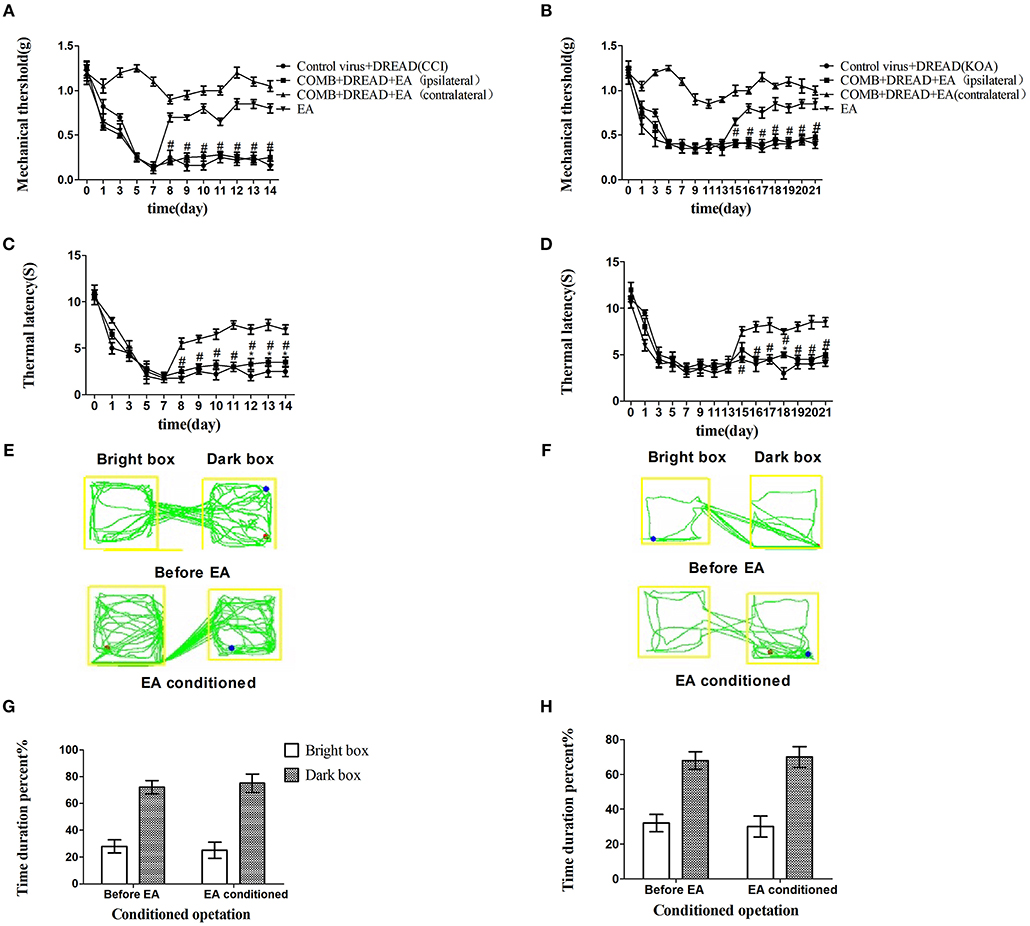
Figure 5. The combination of chemogenetic activation of GABAergic neurons and chemogenetic inhibition of glutamatergic neurons in the vlPAG effectively attenuate the effect of EA. COMB is abstract of combination operation. (A,B) Time course of tactile threshold in response to von Frey filaments. (C,D) The noxious heat stimulus (53°C) caused a change in the thermal thresholds. Tactile (A) and thermal (C) withdrawal thresholds changes in CCI mice after chemogenetic activation. Changes in tactile (B) and thermal (D) withdrawal thresholds of KOA mice caused by combination operation. Motion map of combination operation of conditioning in the CPP test with CCI (E) and KOA mice (F). G,H are summary values of motion trajectories. Virus was injected 21 days before the behavior test. CNO (1 mg/kg) and salvinorin B (5 mg/kg) were administered once a day starting from the 8th to the 14th day in the CCI model and starting from the 15th to the 21st day in the KOA model. EA (1 mA and 0.1 ms) at 2 Hz was administered for 30 min. Once a day starting from the 8th to the 14th day in the CCI model and starting from the 15th to the 21st day in the KOA model, all behavior tests were completed 2 h after CNO administration. Control groups consisted of CCI or KOA mice injected with control virus and DREAD but without EA treatment. Data are expressed as means ± SEM (n = 12 in each group). In A–D, *p < 0.05, compared with control group; #p < 0.05, compared with the model group; In G,H,*p < 0.05, compared with the bright box time duration percent before combination operation; #p < 0.05, compared with the dark box time duration percent before combination operation.
Studies have found that CB1 receptors are distributed on the axon terminals of GABAergic and glutamatergic PAG neurons (Tsou et al., 1998; Vaughan et al., 2000). On this basis, it was important to explore whether the CB1 receptors on GABAergic neurons or the CB1 receptors on glutamatergic neurons participate in the analgesic action of EA.
We injected rAAV-mDlx-CRE-WPRE-pA into the right side of the vlPAG of mCnr1flox/flox mice to specifically knock out the CB1 receptor on GABAergic neurons localized in the vlPAG (Figures 6A,C). Specifically knocking out the CB1 receptor on GABAergic neurons abolished the EA effect on pain hypersensitivity, as it decreased the mechanical withdrawal threshold, thermal withdrawal latency (Figures 7A–D), and the bright box time in the CPP test (Figures 7E–H).
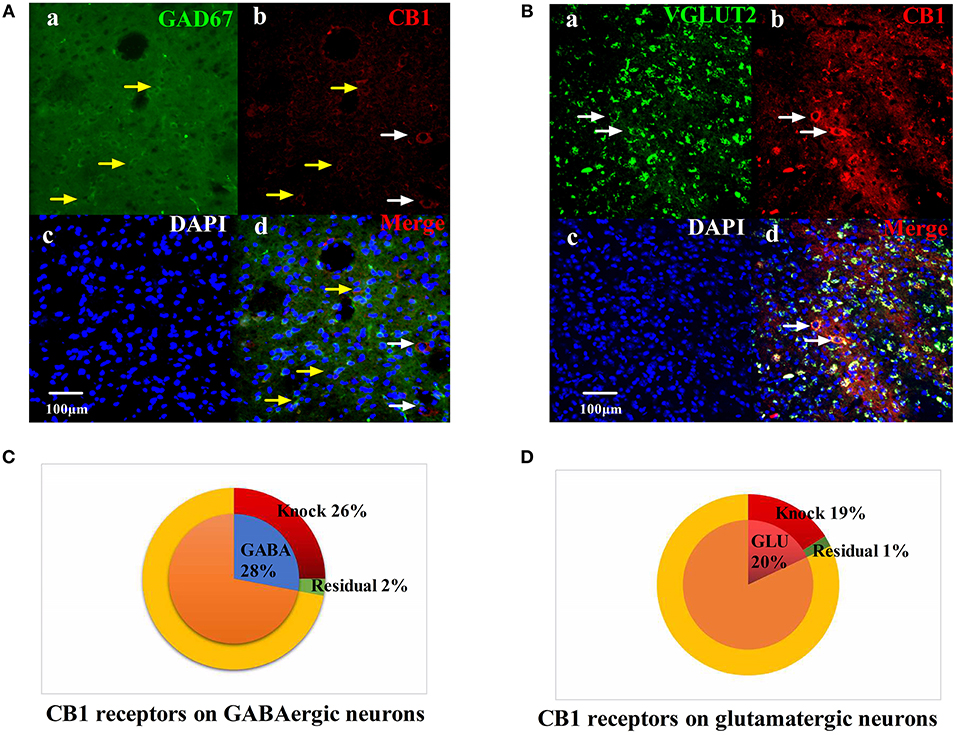
Figure 6. Specific knockout of the CB1 receptor on GABAergic neurons localized in the vlPAG. (A) rAAV-mDlx-CRE-WPRE-pA was injected into the right side of the vlPAG of mCnr1flox/flox mice. (a) GABA-immunoreactive cellsin the vlPAG (green). (b) CB1-immunoreactive cells (red). (c) DAPI nuclear staining (blue). (d) GABA-immunoreactive and DAPI nuclear staining colocalization fluorescence (purple); Yellow arrows show CB1 receptors not knocked out on GABAergic neurons. White arrows show CB1 receptors not knocked out on other large diameter neurons. Scale bar, 100 μm. (B) rAAV-CaMKII-CRE-WPRE-pA was injected into the right side of the vlPAG of mCnr1flox/flox mice. (a) GLU-immunoreactive cells (green). (b) CB1-immunoreactive cells in the vlPAG (red). (c) DAPI nuclear staining (blue). (d) GLU-immunoreactive and DAPI nuclear staining colocalization fluorescence (purple); White arrows show CB1 receptors not knocked out on glutamatergic neurons. Scale bar, 100 μm. (C) Summary data show that the percentage of CB1-immunoreactive cells was knocked out in the area of GABA-immunoreactive cells. Blue sector, GABA-immunoreactive cells; red sector, CB1-immunoreactive cells were knocked out; green sector, residual cells. (D) Summary data shows the percentage of CB1-immunoreactive cells knocked out in the area of glutamatergic-immunoreactive cells. Pink sector, glutamatergic-immunoreactive cells; red sector, CB1-immunoreactive cells been knocked out; green sector, residual cells. Data are expressed as the means ± SEM (n = 3 mice in each group).
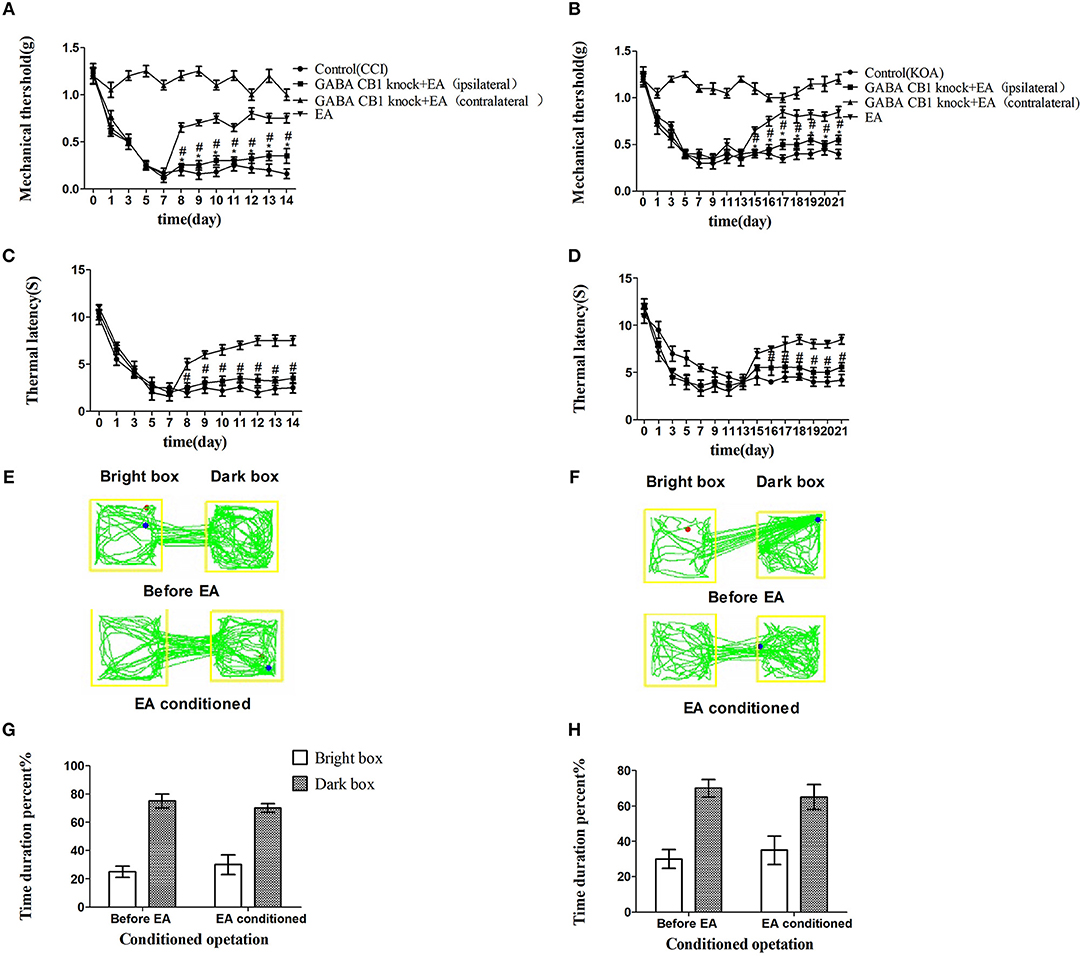
Figure 7. Specifically knocking out CB1 receptor of GABAergic neurons abolished the EA effect on pain hypersensitivity. (A,B) Time course of tactile threshold in response to von Frey filaments. (C,D) The noxious heat stimulus (53°C) caused a change in the thermal thresholds. Tactile (A) and thermal (C) withdrawal thresholds changes in CCI mice. Changes in tactile (B) and thermal (D) withdrawal thresholds of KOA mice. Motion map of EA conditioned CPP test of CCI (E) and KOA mice (F) on the basis of specifically knocking out CB1 receptors of GABAergic neurons. G,H are statistical charts of motion trajectories. The rAAV-mDlx-CRE-WPRE-pA was injected into the right side of vlPAG of mCnr1flox/flox 21 days before behavior test. Same volume of saline was injected into the right side of vlPAG of mCnr1flox/flox as control group. EA (1mA and 0.1ms) at 2 Hz was administered for 30 min, once a day starting from 8th day to 14th in CCI model and starting from 15th day to 21th in KOA model. Control groups are CCI or KOA mice without EA treatment. Data are expressed as means ± SEM (n = 5 in each group). In A–D, *p < 0.05, compared with the control group; #p < 0.05, compared with the model group; In G,H,*p < 0.05, compared with the bright box time duration percent before EA; #p < 0.05, compared with the dark box time duration percent before EA.
On the other hand, rAAV-CaMKII-CRE-WPRE-pA was injected into the right side of the vlPAG of mCnr1flox/flox mice for the purpose of specifically knocking out the CB1 receptor on glutamatergic neurons localized in the vlPAG (Figures 6B,D) and only slightly decreased the EA effect on pain hypersensitivity (Figures S3A–H). It seems that the CB1 receptor on GABAergic neurons localized in the vlPAG was the basis of the EA effect on pain hypersensitivity.
The vlPAG, as an essential part of the neural pathway that mediates pain sensation, has been extensively studied (Vaughan et al., 1997; Ho et al., 2013; Tovote et al., 2016). Consistent with these studies, we found that inhibition of vlPAG neurons by chemogenetics can produce significant antinociceptive effects. We also found that chemogenetic activation of vlPAG neurons resulted in noxious hypersensitivity, and the results of this two-way manipulation were consistent with the bidirectional regulation of the vlPAG in nociceptive regulation (Koutsikou et al., 2015; Hernandez-Leon et al., 2016; Samineni et al., 2017). The vlPAG is critical for the mechanisms of EA-induced analgesia, and we have demonstrated in previous work that during KOA chronic pain, EA exerted an analgesic effect by increasing the levels of CB1 receptors and 2-AG in the vlPAG that had been significantly reduced (Yuan et al., 2018). In this study, we provided new experimental evidence that chemogenetic inhibition of GABAergic neurons in the vlPAG was able to replicate the antinociceptive effect of EA and accordingly further verify that the vlPAG is essential for EA analgesia.
The cellular mechanisms of analgesia and hyperalgesia, which involve in inhibitory and excitatory neurotransmission in the vlPAG have not been directly evaluated. It is not known how distinct neuron subpopulations in the vlPAG are engaged in the descending pain modulation pathway. For the first time, we showed that chemogenetic inhibition of GABAergic vlPAG neurons produced antinociceptive effects, while the combination of chemogenetic activation of GABAergic neurons and chemogenetic inhibition of glutamatergic neurons in vlPAG effectively attenuated the effect of EA. It is hypothesized that GABAergic neurons have a tonic inhibitory effect on vlPAG glutamatergic neurons, while glutamate neurons are output neurons that project to the rostral ventromedial medulla (RVM) (Vaughan et al., 1997; Morgan et al., 2008; Ho et al., 2013). As we can see from Figure 8, chemogenetic inhibition of GABAergic neurons in the vlPAG also produces activation of glutamatergic neurons according to the GABA disinhibition hypothesis, whereas selective activation of GABAergic neurons is not sufficient to attenuate the effect of EA on the condition that some glutamatergic neurons are still directly activated by EA (Figure 8C). This is the potential reason why specific chemogenetic inhibition of GABAergic neurons is sufficient to simulate the EA effect, while the reverse of the EA effect requires the combination of chemogenetic activation of GABAergic neurons and chemogenetic inhibition of glutamatergic neurons in the vlPAG. More studies are necessary to determine the neural circuitry of the vlPAG and connectivity with other brain regions. It is also possible that that GABAergic and glutamatergic neurons in the vlPAG have local circuits, such as synaptic connections between GABAergic neurons and glutamatergic neurons in the vlPAG. Future studies should examine the exact physiological function of GABAergic and glutamatergic neurons in the vlPAG local circuits and through synaptic connections between brain regions.
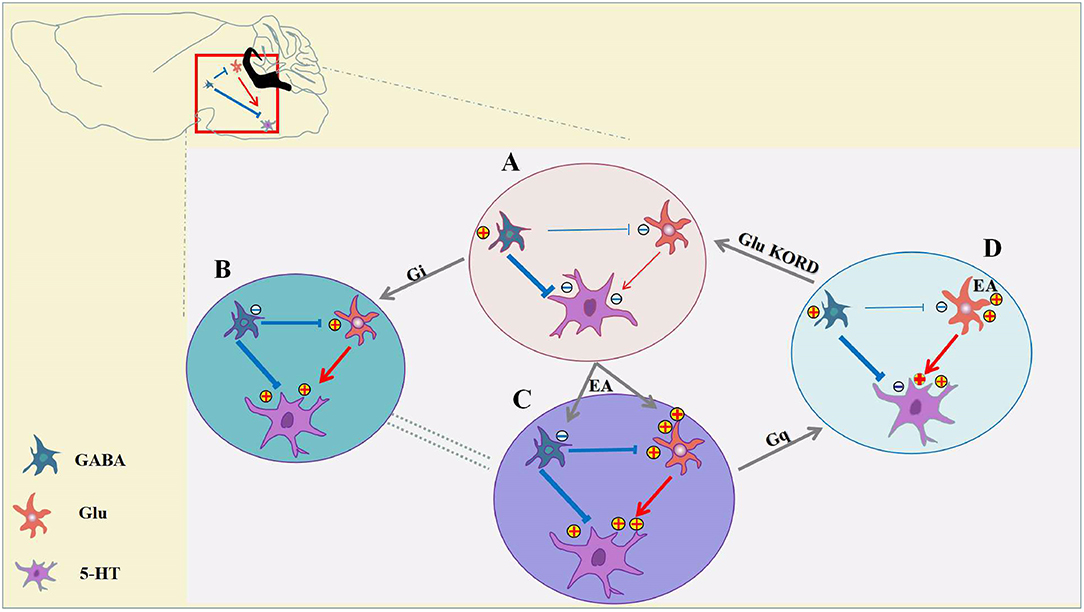
Figure 8. Inhibition of GABAergic neurons and excitation of glutamatergic neurons in the vlPAG participate in EA analgesia. (A) Under pain conditions, GABAergic neurons are excited, resulting in glutamatergic, and serotoninergic neuron inhibition. (B) Chemogenetic inhibition of GABAergic neurons results in activation of glutamatergic neurons and serotoninergic neurons, and the descending inhibitory pain pathway would be activated to relieve the pain. (C) EA synchronously inhibits GABAergic neurons and activates glutamatergic neurons, thereby allowing serotoninergic neurons to be excited sufficiently. (D) With chemogenetic inhibition of GABAergic neurons, glutamatergic neurons are still activated by EA, and serotoninergic neurons in the descending inhibitory pain pathway remained partially activated. Chemogenetic activation of GABAergic neurons and inhibition of glutamatergic neurons is sufficient to attenuate the effects of EA.
Endogenous cannabinoid ligands and CB1 receptors are widely present in the nociceptive and descending inhibitory pathways (Mitrirattanakul et al., 2006). CB1 receptors are expressed in both nerve endings of GABAergic neurons and glutamatergic neurons in the PAG, which provides histological evidence that the activation of the CB1 receptor may regulate GABAergic and glutamatergic neurotransmission (Drew et al., 2009; Hu et al., 2014). WIN55212-2 (WIN), a CB1R agonist, modulates various K+ and Ca2+currents that could activate GABAergic and glutamatergic neurons (Straiker et al., 1999; Yazulla et al., 2000; Fan and Yazulla, 2003; Straiker and Sullivan, 2003; Yazulla, 2008; Schwitzer et al., 2016).
Consistent with previous electrophysiology results, we experimentally confirmed that the CB1 receptors on GABAergic and glutamatergic neurons are involved in the analgesia of EA. Using the Cre/loxP system, we are able to specifically knock out the CB1 receptor on GABAergic neurons or glutamatergic neurons localized in the vlPAG. Knocking out the CB1 receptor on GABAergic neurons abolished most of the EA effect on pain hypersensitivity, while knocking out the CB1 receptor on glutamatergic neurons localized in the vlPAG, only lessened some of the EA effect on pain hypersensitivity. According to our results, EA can simultaneously inhibit GABAergic neurons and excite glutamatergic neurons in the vlPAG via CB1 receptors. Chemogenic CB1 knock-out on GABAergic neurons abolished the effect of EA while chemogenic activation of GABAergic neurons only partly attenuated the effect of EA. It suggested that the CB1 receptors on GABAergic neurons may also be required for activation of the CB1 receptor on glutamatergic neurons localized in the vlPAG, which conforms to the theory of ON- and OFF-cells in the vlPAG (Heinricher et al., 1987). According to Wang's research (Wang et al., 2016), a CB1R agonist reduced the mIPSC frequency regardless of whether AMPA receptors were blocked, while it affected the mEPSC frequency when inhibitory inputs were present. It indicated that the activation of GABAergic neurons may be necessary for glutamatergic neuron excitation via CB1 receptors.
Through chemical genetic manipulations, we have observed for the first time that EA can exert an analgesic effect by simultaneously inhibiting GABAergic neurons and stimulating glutamatergic neurons. We know that when pain occurs, it is accompanied by a functional imbalance between GABAergic neurons and glutamatergic neurons. The relative balance of GABAergic neurons and glutamatergic neurons is critical for the maintenance of homeostasis (Siegfried and de Souza, 1989; Schmidtko et al., 2008). The microinjection of GABAA receptor antagonists or glutamate agonists into the vlPAG produces antinociceptive effects (Bobeck et al., 2009; Liao et al., 2011). According to recent research by Vijay K, activation of glutamatergic neurons or inhibition of GABAergic neurons by chemical genetics achieves an effective inhibition of nociceptive sensation, in spite of that experiment being performed in normal mice rather than in a mouse pain model, as in the present experiments (Samineni et al., 2017). Despite the support of those experiments, the effect of clinical application of GABAA receptor antagonists or glutamate agonists is not optimistic (de Meij et al., 2014; Bruhn et al., 2017). In clinical treatment, it is indeed difficult to find a drug that can simultaneously act on both GABAergic and glutamatergic neurons, as they belong to different neuroreceptor systems with two distinct functions. According to our results, EA may simultaneously inhibit GABAergic neurons and activate glutamatergic neurons in the vlPAG via CB1 receptors to exert antinociceptive effects. The results of this study provide experimental evidence contributing to our understanding of the dual target adjustment mechanism of EA analgesia and homeostasis recovery under pain conditions.
In conclusion, we have demonstrated a novel role of GABAergic neurons and glutamatergic neurons in the vlPAG in EA antinociceptive effects. EA can simultaneously inhibit GABAergic neurons and excite glutamatergic neurons in the vlPAG via CB1 receptors, thereby exerting antinociceptive effects. Knocking out the CB1 receptor on GABAergic neurons is sufficient to abolish the EA effect on pain hypersensitivity, while the combination of activation of GABAergic neurons and inhibition of glutamatergic neurons in vlPAG is sufficient to attenuate the effect of EA. EA synchronously regulates the GABAergic system and the glutamatergic system with distinct functions through the CB1 receptor. This finding may provide new ideas for the clinical treatment of pain and the development of analgesic drugs.
J-QS and ML designed this experiment. HeZ mainly completed this experiment. H-CX, H-PL, L-XL, and X-FH provided help in brain stereotactic injection work. HoZ, W-YM, LL, CC, YS, and R-YZ assisted in the completion of animal behavior experiments. PZ, J-QS, and ML provided help in writing and modifying this paper.
This work was supported by a grant from the National Natural Science Foundation of China (81473768) and the Natural Science Foundation of Jiangsu Province (No. BK 20160548 to YS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2019.00484/full#supplementary-material
Basbaum, A. I., and Fields, H. L. (1984). Endogenous pain control systems: brainstem spinal pathways and endorphin circuitry. Annu. Rev. Neurosci. 7, 309–338. doi: 10.1146/annurev.ne.07.030184.001521
Bennett, G. J., and Xie, Y. K. (1988). A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33, 87–107. doi: 10.1016/0304-3959(88)90209-6
Bobeck, E. N., Chen, Q., Morgan, M. M., and Ingram, S. L. (2014). Contribution of adenylyl cyclase modulation of pre- and postsynaptic GABA neurotransmission to morphine antinociception and tolerance. Neuropsychopharmacology 39, 2142–2152. doi: 10.1038/npp.2014.62
Bobeck, E. N., McNeal, A. L., and Morgan, M. M. (2009). Drug dependent sex-differences in periaqueducatal gray mediated antinociception in the rat. Pain 147, 210–216. doi: 10.1016/j.pain.2009.09.008
Bruhn, J., Scheffer, G. J., and van Geffen, G. J. (2017). Clinical application of perioperative multimodal analgesia. Curr. Opin. Support. Palliat. Care 11, 106–111. doi: 10.1097/SPC.0000000000000267
Budai, D., Harasawa, I., and Fields, H. L. (1998). Midbrain periaqueductal gray (PAG) inhibits nociceptive inputs to sacral dorsal horn nociceptive neurons through alpha2-adrenergic receptors. J. Neurophysiol. 80, 2244–2254. doi: 10.1152/jn.1998.80.5.2244
Chaplan, S. R., Bach, F. W., Pogrel, J. W., Chung, J. M., and Yaksh, T. L. (1994). Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63. doi: 10.1016/0165-0270(94)90144-9
Chen, G., Kim, Y. H., Li, H., Luo, H., Liu, D. L., Zhang, Z. J., et al. (2017). PD-L1 inhibits acute and chronic pain by suppressing nociceptive neuron activity via PD-1. Nat. Neurosci. 20, 917–926. doi: 10.1038/nn.4571
Chen, L., Zhang, J., Li, F., Qiu, Y., Wang, L., Li, Y. H., et al. (2009). Endogenous anandamide and cannabinoid receptor-2 contribute to electroacupuncture analgesia in rats. J. Pain 10, 732–739. doi: 10.1016/j.jpain.2008.12.012
de Meij, N., Köke, A., van der Weijden, T., van Kleef, M., and Patijn, J. (2014). Pain treatment facilities: do we need quantity or quality? J. Eval. Clin. Pract. 20, 578–581. doi: 10.1111/jep.12148
Demers, G., Griffin, G., De Vroey, G., Haywood, J. R., Zurlo, J., and Bédard, M. (2006). Animal research. Harmonization of animal care and use guidance. Science 312, 700–701. doi: 10.1126/science.1124036
Dimidschstein, J., Chen, Q., Tremblay, R., Rogers, S. L., Saldi, G. A., Guo, L., et al. (2016). A viral strategy for targeting and manipulating interneurons across vertebrate species. Nat. Neurosci. 19, 1743–1749. doi: 10.1038/nn.4430
Drew, G. M., Lau, B. K., and Vaughan, C. W. (2009). Substance P drives endocannabinoid-mediated disinhibition in a midbrain descending analgesic pathway. J. Neurosci. 29, 7220–7229. doi: 10.1523/JNEUROSCI.4362-08.2009
Drew, G. M., Mitchell, V. A., and Vaughan, C. W. (2008). Glutamate spillover modulates GABAergic synaptic transmission in the rat midbrain periaqueductal grey via metabotropic glutamate receptors and endocannabinoid signaling. J. Neurosci. 28, 808–815. doi: 10.1523/JNEUROSCI.4876-07.2008
Fan, S. F., and Yazulla, S. (2003). Biphasic modulation of voltage-dependent currents of retinal cones by cannabinoid CB1 receptor agonist WIN 55212-2. Vis. Neurosci. 20, 177–188. doi: 10.1017/S095252380320208X
Fernihough, J., Gentry, C., Malcangio, M., Fox, A., Rediske, J., Pellas, T., et al. (2004). Pain related behaviour in two models of osteoarthritis in the rat knee. Pain 112, 83–93. doi: 10.1016/j.pain.2004.08.004
Finn, D. P., Jhaveri, M. D., Beckett, S. R., Roe, C. H., Kendall, D. A., Marsden, C. A., et al. (2003). Effects of direct periaqueductal grey administration of a cannabinoid receptor agonist on nociceptive and aversive responses in rats. Neuropharmacology 45, 594–604. doi: 10.1016/S0028-3908(03)00235-1
Fusumada, K., Yokoyama, T., Miki, T., Wang, Z. Y., Yang, W., Lee, N. S., et al. (2007). c-Fos expression in the periaqueductal gray is induced by electroacupuncture in the rat, with possible reference to GABAergic neurons. Okajimas Folia Anat. Jpn. 84, 1–9. doi: 10.2535/ofaj.84.1
Harvey, V. L., and Dickenson, A. H. (2009). Behavioural and electrophysiological characterisation of experimentally induced osteoarthritis and neuropathy in C57Bl/6 mice. Mol. Pain 5, 18–18. doi: 10.1186/1744-8069-5-18
Heinricher, M. M., Cheng, Z. F., and Fields, H. L. (1987). Evidence for two classes of nociceptive modulating neurons in the periaqueductal gray. J. Neurosci. 7, 271–278. doi: 10.1523/JNEUROSCI.07-01-00271.1987
Hernandez-Leon, A., Fernández-Guasti, A., and González-Trujano, M. E. (2016). Rutin antinociception involves opioidergic mechanism and descending modulation of ventrolateral periaqueductal grey matter in rats. Eur. J. Pain 20, 274–283. doi: 10.1002/ejp.720
Hnasko, T. S., Sotak, B. N., and Palmiter, R. D. (2005). Morphine reward in dopamine-deficient mice. Nature 438, 854–857. doi: 10.1038/nature04172
Ho, Y. C., Cheng, J. K., and Chiou, L. C. (2013). Hypofunction of Glutamatergic neurotransmission in the periaqueductal gray contributes to nerve-injury-induced neuropathic pain. J. Neurosci. 33, 7825–7836. doi: 10.1523/JNEUROSCI.5583-12.2013
Ho, Y. C., Lee, H. J., Tung, L. W., Liao, Y. Y., Fu, S. Y., Teng, S. F., et al. (2011). Activation of orexin 1 receptors in the periaqueductal gray of male rats leads to antinociception via retrograde endocannabinoid (2-Arachidonoylglycerol)-induced disinhibition. J. Neurosci. 31, 14600–14610. doi: 10.1523/JNEUROSCI.2671-11.2011
Hu, S. S., Ho, Y. C., and Chiou, L. C. (2014). No more pain upon Gq-protein-coupled receptor activation: role of endocannabinoids. Eur. J. Neurosci. 39, 467–484. doi: 10.1111/ejn.12475
Kang, J. M., Park, H. J., Choi, Y. G., Choe, I. H., Park, J. H., Kim, Y. S., et al. (2007). Acupuncture inhibits microglial activation and inflammatory events in the MPTP-induced mouse model. Brain Res. 1131, 211–219. doi: 10.1016/j.brainres.2006.10.089
Koutsikou, S., Watson, T. C., Crook, J. J., Leith, J. L., Lawrenson, C. L., Apps, R., et al. (2015). The periaqueductal gray orchestrates sensory and motor circuits at multiple levels of the neuraxis. J. Neurosci. 35, 14132–14147. doi: 10.1523/JNEUROSCI.0261-15.2015
La Porta, C., Bura, S. A., Aracil-Fernández, A., Manzanares, J., and Maldonado, R. (2013). Role of CB1 and CB2 cannabinoid receptors in the development of joint pain induced by monosodium iodoacetate. Pain 154, 160–174. doi: 10.1016/j.pain.2012.10.009
Lao, L., Zhang, R. X., Zhang, G., Wang, X., Berman, B. M., and Ren, K. (2004). A parametric study of electroacupuncture on persistent hyperalgesia and Fos protein expression in rats. Brain Res. 1020, 18–29. doi: 10.1016/j.brainres.2004.01.092
Lee, Y., Choi, G., Jeon, H., Kim, D., Ryu, S., Koo, S., et al. (2018). Acupuncture stimulation at GB34 suppresses 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced oxidative stress in the striatum of mice. J. Physiol. Sci. 68, 455–462. doi: 10.1007/s12576-017-0547-7
Liao, H. T., Lee, H. J., Ho, Y. C., and Chiou, L. C. (2011). Capsaicin in the periaqueductal gray induces analgesia via metabotropic glutamate receptor-mediated endocannabinoid retrograde disinhibition. Br. J. Pharmacol. 163, 330–345. doi: 10.1111/j.1476-5381.2011.01214.x
Liu, L., Zhao, L. P., Zhang, C. S., Zeng, L., Wang, K., Zhao, J., et al. (2018). Acupuncture as prophylaxis for chronic migraine: a protocol for a single-blinded, double-dummy randomised controlled trial. BMJ Open 8:e020653. doi: 10.1136/bmjopen-2017-020653
Meng, I. D., Manning, B. H., Martin, W. J., and Fields, H. L. (1998). An analgesia circuit activated by cannabinoids. Nature 395, 381–383. doi: 10.1038/26481
Mist, S. D., and Jones, K. D. (2018). Randomized controlled trial of acupuncture for women with fibromyalgia: group acupuncture with traditional chinese medicine diagnosis-based point selection. Pain Med. 19, 1862–1871. doi: 10.1093/pm/pnx322
Mitrirattanakul, S., Ramakul, N., Guerrero, A. V., Matsuka, Y., Ono, T., Iwase, H., et al. (2006). Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain 126, 102–114. doi: 10.1016/j.pain.2006.06.016
Morgan, M., Nanzer, A., Camacho-Hubner, C., Kaltsas, G. A., and Monson, J. P. (2003). Hypoglycaemia secondary to the secretion of pro-insulin like growth factor II by a metastatic neuroendocrine tumour with sarcomatous differentiation. Hormones 2, 125–129. doi: 10.14310/horm.2002.1192
Morgan, M. M., Whittier, K. L., Hegarty, D. M., and Aicher, S. A. (2008). Periaqueductal gray neurons project to spinally projecting GABAergic neurons in the rostral ventromedial medulla. Pain 140, 376–386. doi: 10.1016/j.pain.2008.09.009
Musil, F., Pokladnikova, J., Pavelek, Z., Wang, B., Guan, X., and Valis, M. (2018). Acupuncture in migraine prophylaxis in Czech patients: an open-label randomized controlled trial. Neuropsychiatr. Dis. Treat. 14, 1221–1228. doi: 10.2147/NDT.S155119
Osborne, P. B., Vaughan, C. W., Wilson, H. I., and Christie, M. J. (1996). Opioid inhibition of rat periaqueductal grey neurones with identified projections to rostral ventromedial medulla in vitro. J. Physiol. 490, 383–389. doi: 10.1113/jphysiol.1996.sp021152
Palazzo, E., Luongo, L., Novellis, V., Rossi, F., and Maione, S. (2010). The role of cannabinoid receptors in the descending modulation of pain. Pharmaceuticals 3, 2661–2673. doi: 10.3390/ph3082661
Park, J. Y., Park, J. J., Jeon, S., Doo, A. R., Kim, S. N., Lee, H., et al. (2014). From peripheral to central: the role of ERK signaling pathway in acupuncture analgesia. J. Pain 15, 535–549. doi: 10.1016/j.jpain.2014.01.498
Samineni, V. K., Grajales-Reyes, J. G., Copits, B. A., O'Brien, D. E., Trigg, S. L., Gomez, A. M., et al. (2017). Divergent modulation of nociception by glutamatergic and GABAergic neuronal subpopulations in the periaqueductal gray. eNeuro 4:ENEURO.0129-16.2017. doi: 10.1523/ENEURO.0129-16.2017
Schmidtko, A., Luo, C., Gao, W., Geisslinger, G., Kuner, R., and Tegeder, I. (2008). Genetic deletion of synapsin II reduces neuropathic pain due to reduced glutamate but increased GABA in the spinal cord dorsal horn. Pain 139, 632–643. doi: 10.1016/j.pain.2008.06.018
Schwitzer, T., Schwan, R., Angioi-Duprez, K., Giersch, A., and Laprevote, V. (2016). The endocannabinoid system in the retina: from physiology to practical and therapeutic applications. Neural Plast. 2016:2916732. doi: 10.1155/2016/2916732
Siegfried, B., and de Souza, R. L. (1989). NMDA receptor blockade in the periaqueductal grey prevents stress-induced analgesia in attacked mice. Eur. J. Pharmacol. 168, 239–242. doi: 10.1016/0014-2999(89)90570-0
Sommer, C., Schmidt, C., and George, A. (1998). Hyperalgesia in experimental neuropathy is dependent on the TNF receptor 1. Exp. Neurol. 151, 138–142. doi: 10.1006/exnr.1998.6797
Straiker, A., Stella, N., Piomelli, D., Mackie, K., Karten, H. J., and Maguire, G. (1999). Cannabinoid CB1 receptors and ligands in vertebrate retina: Localization and function of an endogenous signaling system. Proc. Natl. Acad. Sci. U.S.A. 96, 14565–14570. doi: 10.1073/pnas.96.25.14565
Straiker, A., and Sullivan, J. M. (2003). Cannabinoid receptor activation differentially modulates ion channels in photoreceptors of the tiger salamander. J. Neurophysiol. 89, 2647–2654. doi: 10.1152/jn.00268.2002
Takasu, K., Ogawa, K., Nakamura, A., Kanbara, T., Ono, H., Tomii, T., et al. (2015). Enhanced GABAergic synaptic transmission at VLPAG neurons and potent modulation by oxycodone in a bone cancer pain model. Br. J. Pharmacol. 172, 2148–2164. doi: 10.1111/bph.13039
Tjen-A-Looi, S. C., Li, P., and Longhurst, J. C. (2009). Processing cardiovascular information in the vlPAG during electroacupuncture in rats: roles of endocannabinoids and GABA. J. App. Physiol. 106, 1793–1799. doi: 10.1152/japplphysiol.00142.2009
Tovote, P., Esposito, M. S., Botta, P., Chaudun, F., Fadok, J. P., Markovic, M., et al. (2016). Midbrain circuits for defensive behaviour. Nature 534, 206–212. doi: 10.1038/nature17996
Tsou, K., Nogueron, M. I., Muthian, S., Sañudo-Pena, M. C., Hillard, C. J., Deutsch, D. G., et al. (1998). Fatty acid amide hydrolase is located preferentially in large neurons in the rat central nervous system as revealed by immunohistochemistry. Neurosci. Lett. 254, 137–140. doi: 10.1016/S0304-3940(98)00700-9
van Osch, G. J., van der Kraan, P. M., and van den Berg, W. B. (1994). Site-specific cartilage changes in murine degenerative knee joint disease induced by iodoacetate and collagenase. J. Orthop. Res. 12, 168–175. doi: 10.1002/jor.1100120204
Vardy, E., Robinson, J. E., Li, C., Olsen, R. H. J., DiBerto, J. F., Giguere, P. M., et al. (2015). A new DREADD facilitates the multiplexed chemogenetic interrogation of behavior. Neuron 86, 936–946. doi: 10.1016/j.neuron.2015.03.065
Vaughan, C. W., Connor, M., Bagley, E. E., and Christie, M. J. (2000). Actions of cannabinoids on membrane properties and synaptic transmission in rat periaqueductal gray neurons in vitro. Mol. Pharmacol. 57, 288–295. doi: 10.1002/bjs.5237
Vaughan, C. W., Ingram, S. L., Connor, M. A., and Christie, M. J. (1997). How opioids inhibit GABA-mediated neurotransmission. Nature 390, 611–614. doi: 10.1038/37610
Wang, X. H., Wu, Y., Yang, X. F., Miao, Y., Zhang, C. Q., Dong, L. D., et al. (2016). Cannabinoid CB1 receptor signaling dichotomously modulates inhibitory and excitatory synaptic transmission in rat inner retina. Brain Struct. Funct. 221, 301–316. doi: 10.1007/s00429-014-0908-4
Wu, C. H., Lv, Z. T., Zhao, Y., Gao, Y., Li, J. Q., Gao, F., et al. (2013). Electroacupuncture improves thermal and mechanical sensitivities in a rat model of postherpetic neuralgia. Mol. Pain 9, 18–18. doi: 10.1186/1744-8069-9-18
Wu, M. X., Li, X. H., Lin, M. N., Jia, X. R., Mu, R., Wan, W. R., et al. (2010). Clinical study on the treatment of knee osteoarthritis of Shen -Sui insufficiency syndrome type by electroacupuncture. Chin. J. Integr. Med. 16, 291–297. doi: 10.1007/s11655-010-0513-1
Yazulla, S. (2008). Endocannabinoids in the retina: from marijuana to neuroprotection. Prog. Retin. Eye Res. 27, 501–526. doi: 10.1016/j.preteyeres.2008.07.002
Yazulla, S., Studholme, K. M., McIntosh, H. H., and Fan, S. F. (2000). Cannabinoid receptors on goldfish retinal bipolar cells: electron-microscope immunocytochemistry and whole-cell recordings. Vis. Neurosci. 17, 391–401. doi: 10.1017/S0952523800173079
Keywords: pain, electroacupuncture, chemogenetics, GABAergic neuron, glutamatergic neuron, vlPAG
Citation: Zhu H, Xiang H-C, Li H-P, Lin L-X, Hu X-F, Zhang H, Meng W-Y, Liu L, Chen C, Shu Y, Zhang R-Y, Zhang P, Si J-Q and Li M (2019) Inhibition of GABAergic Neurons and Excitation of Glutamatergic Neurons in the Ventrolateral Periaqueductal Gray Participate in Electroacupuncture Analgesia Mediated by Cannabinoid Receptor. Front. Neurosci. 13:484. doi: 10.3389/fnins.2019.00484
Received: 29 January 2019; Accepted: 29 April 2019;
Published: 17 May 2019.
Edited by:
Lauren C. Heathcote, Stanford University, United StatesReviewed by:
Christoph Thoeringer, Technische Universität München, GermanyCopyright © 2019 Zhu, Xiang, Li, Lin, Hu, Zhang, Meng, Liu, Chen, Shu, Zhang, Zhang, Si and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun-Qiang Si, c2lqdW5xaWFuZzExQGhvdG1haWwuY29t
Man Li, bGltYW43M0BtYWlscy50am11LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.