 Zemfira N. Karamysheva
Zemfira N. Karamysheva Elena B. Tikhonova2
Elena B. Tikhonova2 Andrey L. Karamyshev
Andrey L. Karamyshev- 1Department of Biological Sciences, Texas Tech University, Lubbock, TX, United States
- 2Department of Cell Biology and Biochemistry, Texas Tech University Health Sciences Center, Lubbock, TX, United States
Frontotemporal lobar degeneration (FTLD) is a pathological process characterized by severe atrophy in the frontal and temporal lobes of the brain (Mackenzie et al., 2011). There are three major clinical syndromes in FTLD: behavioral variant of frontotemporal dementia (bvFTD), nonfluent variant of primary progressive aphasia (nfvPPA), and semantic variant of PPA (svPPA) (Gorno-Tempini et al., 2011; Rascovsky et al., 2011). bvFTD is the most common among three (Hernandez et al., 2018). It is associated with changes in personality and behavior accompanied with language deficits at later stages. In rare cases, FTLD subtypes may be associated with motor neuron disease worsening the patient survival time (Olney et al., 2005). FTLD also includes the clinical presentations of progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD), that are associated with parkinsonism, and other clinical features. PSP and CBD account for about 20–30% of patients in FTLD (Park and Chung, 2013). Unfortunately, there is no significant progress achieved in development of effective treatments for FTLD and current treatment options are purely symptomatic (Hodges and Piguet, 2018).
The pathological changes found in FTLD are very heterogenous in their nature. FTLD can be divided in three main histological subtypes according to the accumulation of neuronal protein inclusions (Mackenzie et al., 2011). The most common disease is characterized by the presence of inclusions containing the trans-activation response DNA-binding protein-43 (TDP-43) which is found to be abnormally phosphorylated and ubiquitinated in patients (Neumann et al., 2006). This subtype of pathology is classified as FTLD-TDP (Mackenzie et al., 2011). The second pathological subtype, FTLD-tau, includes cases with inclusions consisting of abnormally phosphorylated microtubule associated protein tau (Cairns et al., 2007). The third subtype, FTLD-FET, contains fused in sarcoma (FUS) RNA-binding protein, Ewing's sarcoma protein (EWS), and TATA-binding protein associated factor 15 (TAF15) in the pathological inclusions (Mackenzie and Neumann, 2012). About 40% of FTLD cases are familial and about 10% of cases exhibit autosomal dominant inheritance (Bang et al., 2015). Mutations in GRN (Baker et al., 2006; Cruts et al., 2006), MAPT (Hutton et al., 1998), CHMP2B (Skibinski et al., 2005), VCP (Watts et al., 2004), and C9orf72 (Renton et al., 2011) have been found associated with the disease. The most common known genetic causes of FTLD are connected with mutations in GRN, MAPT, and C9orf72 genes (Cruts et al., 2006; Gass et al., 2006, 2012; Mori et al., 2013; Hodges and Piguet, 2018). In this article we focus on progranulin (PGRN protein encoded by GRN gene) role in FTLD. Patients with progranulin mutations have ubiquitin and TDP-43 positive pathological inclusions (Baker et al., 2006; Cruts et al., 2006). In addition to its role in neurodegenerative diseases PGRN is also implicated in epithelial ovarian cancer and its level is highly elevated in various tumors (He and Bateman, 2003). It also has a role in metabolic diseases and its excess is associated with obesity and insulin resistance (Matsubara et al., 2012). PRGN is a multifunctional protein involved in regulation of many cellular processes including angiogenesis, cell proliferation, inflammation, tissue remodeling, and wound repair (Nguyen et al., 2013).
PRGN is encoded by GRN gene that is located on chromosome 17q21 and consists of 13 exons with the Kozak sequence present in the second exon (Bhandari et al., 1992; Cruts and Van Broeckhoven, 2008) (Figure 1A). It encodes 593 amino acid long precursor protein with a predicted molecular mass of 63.5 kDa. PRGN contains a signal peptide (also known as a signal sequence) at the N-terminus to mediate its secretion, followed by 7.5 highly conserved cysteine-rich tandem repeats called granulins. Granulins are separated by divergent linker sequences. Cleavage of the signal peptide generates mature protein that is heavily glycosylated and migrates as 88 kDa protein. This protein is further processed by the cleavage at the linker regions to produce 6 kDa granulins or linked combinations of granulins (Cenik et al., 2012; Gass et al., 2012) (Figure 1B). PGRN does not have clear consensus sequence for protease cleavage and is cleaved by multiple intracellular and extracellular proteases such as elastase, proteinase 3, matrix metallopeptidase 12, or by cathepsins in the lysosomes (Gass et al., 2012; Nguyen et al., 2013; Zhou et al., 2017a). Both progranulin and 6 kDa granulins are shown to exist in vivo, however, their biological functions in the cell are not very clear. Recent data suggest that progranulin may be involved in anti-inflammatory activities through modulation of the TNF signaling while granulins are proinflammatory (Tang et al., 2011; Hu et al., 2014). The C-terminus of PRGN is necessary to bind sortilin, a receptor protein regulating intracellular protein trafficking in the Golgi (Hu et al., 2010). Lysosomal targeting of PGRN is carried out by two independent and complementary pathways. The first utilizes sortilin protein, protein trafficking receptor, located in Golgi and cell surface (Hu et al., 2010). The second, sortilin-independent pathway, is mediated by prosaposin (PSAP) through its interaction with mannose 6-phosphate receptor (M6PR) and low-density lipoprotein receptor-related protein 1 (LRP1) (Zhou et al., 2015). PSAP is the precursor of saposin protein essential for lysosomal degradation of glycosphingolipids. The role of PRGN and granulins in lysosome function is poorly understood, however, it has been recently revealed that deficiencies in granulins caused by mutations may play a role in lysosome dysfunction (Holler et al., 2017). Complete loss of PGRN due to homozygous GRN mutations was reported as a cause for neuronal ceroid lipofuscinosis (NCL) linking rare lysosomal impairment to neurodegeneration in FTLD (Smith et al., 2012; Gotzl et al., 2016). This disease leads to progressive degeneration of brain and loss of vision due to accumulation of ceroid lipofuscin, a lipid-containing pigment, associated with lysosome dysfunction (Kohlschutter and Schulz, 2009). It was shown that a lack of PGRN leads to decreased level of PSAP in neurons causing NCL (Zhou et al., 2017b). These discoveries suggested that PRGN and PSAP facilitate each other's lysosomal trafficking. Furthermore, studies of lysosome storage diseases from different groups suggested that PRGN might acts as a chaperone of lysosomal enzymes (Jian et al., 2016; Beel et al., 2017). Chaperone functions required direct association of PRGN with lysosomal proteins through granulin E domain and also involved recruitment of HSP70.
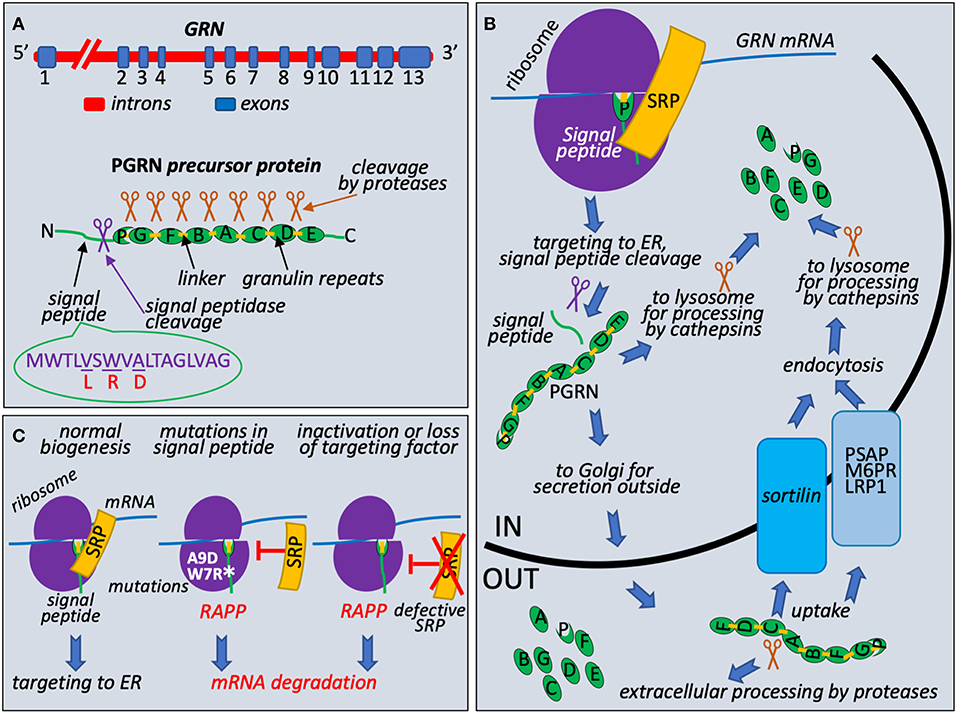
Figure 1. Granulin biogenesis, quality control at the ribosome during its synthesis, and molecular mechanism of FTLD associated with mutations in the signal peptide of the granulin precursor. (A) Schematic presentation of GRN, precursor protein structure, and known missense mutations in the signal peptide. Progranulin pre-mRNA transcript is synthesized in the nucleus from GRN gene in the chromosome 17. It has 13 exons with exons 2–13 containing protein coding region. After splicing, mRNA is exported to cytoplasm for translation. Progranulin precursor (63.5 kDa protein) consists of N-terminal cleavable signal peptide (green line with indicated position of the cleavage by the signal peptidase shown as purple scissors) and 7.5 repeats (green ovals): P (half-repeat, paragranulin), G (granulin 1), F (granulin 2), B (granulin 3), A (granulin 4), C (granulin 5), D (granulin 6), and E (granulin 7). Repeats are connected by linker sequences (light orange boxes). Proteases are shown as brown scissors. Signal peptide sequence presented with known missense mutations L (V5L), R (W7R), D (A9D), corresponding amino acid residues in the wild-type signal peptide are underlined. (B) PGRN protein trafficking and processing. During early translation step on the ribosome (purple hemispheres), N-terminal signal peptide of progranulin (green line) is recognized by Signal Recognition Particle (SRP) (shown in orange) and ribosome-nascent chain complex is targeted to ER for signal peptide cleavage (purple scissors), posttranslational modifications, and further processing and transport. Full length protein could be processed to 6 kDa granulins in lysosomes by cathepsins (brown scissors are symbols for all proteases involved in posttranslational processing) or secreted outside and undergo extracellular processing. Uptake of full-length protein is governed by endocytosis with the help of sortilin receptor (blue box) or through alternative PSAP (prosaposin)-dependent pathway with involvement of mannose 6-phosphate receptor (M6PR) and low density lipoprotein receptor-related protein 1 (LRP1) (gray blue box). (C) Loss of interaction with targeting factor, SRP, activates RAPP pathway. During normal translational event, PGRN with N-terminal signal sequence is targeted to ER through interaction with SRP. Amino acid sequence of signal peptide and location of reported clinical mutations are shown on (A). When A9D or W7R mutations in signal peptide is detected or SRP is defective or lost, nascent chain is no longer targeted to ER by SRP. It leads to the RAPP pathway activation and degradation of the GRN mRNA.
Loss of the PGRN function can occur on the genomic, transcriptional, and posttranscriptional levels (Kleinberger et al., 2013). Mutations in GRN are one of the major causes of FTLD and found in 11.2% of patients, therefore progranulin is an important emerging target to develop better treatments (Abella et al., 2017). More than 100 different mutations were identified in the GRN gene, and at least 79 pathogenic mutations in 259 families have been associated with FTLD (Cruts et al., 2012) (http://www.molgen.ua.ac.be/FTDmutations/). Most common mutations include nonsense, frameshift and splice site mutations leading to generation of a premature stop codon that activate nonsense-mediated decay (NMD) (Baker et al., 2006; Cruts et al., 2006). Therefore, majority of the mutations are believed to act through a haploinsufficiency mechanism due to mutant mRNA degradation of the one allele and as a result reduced progranulin protein level (Cruts and Van Broeckhoven, 2008). Other mutations include genomic deletions or elimination of the initiation codon for protein synthesis. Loss of the PGRN function can also be mediated by mutations affecting the protein sorting, secretion, proteolytic processing, association with sortilin and cyclin T1, neurite outgrowth, and proinflammatory response (Kleinberger et al., 2013). Some missense and intronic mutations in GRN also contribute to the pathogenicity connected to FTLD due to the loss of functional protein (Abella et al., 2017).
Unusual and intriguing molecular mechanism of FTLD that is associated with mutations in progranulin signal sequences was recently discovered (Pinarbasi et al., 2018). Progranulin is a secreted protein and it is synthesized as a precursor with signal peptide (Figure 1A). Signal Recognition Particle (SRP) recognizes signal peptides co-translationally during protein synthesis at the ribosome and targets ribosome nascent complexes to endoplasmic reticulum (ER) membrane for the protein translocation to the ER lumen and further processing and transport outside of the cells (Figure 1B). It is well-established that integrity of the signal peptides is important for protein targeting and transport (Karamyshev et al., 1998; Kalinin et al., 1999; Karamyshev and Johnson, 2005; Nilsson et al., 2015). Despite the absence of the strong amino acid homology between signal peptides of different proteins they have similar organization and contain n-terminal, hydrophobic core or h-region, and c-terminal parts (von Heijne, 1985). Amino acid substitutions that decrease hydrophobicity of the h-region inhibit interaction with SRP (Nilsson et al., 2015). As we recently discovered, the loss of SRP interaction activates the protein quality control pathway named RAPP (regulation of aberrant protein production) leading to mRNA degradation of the defective proteins (Karamyshev et al., 2014; Karamyshev and Karamysheva, 2018). Among more than 100 of different mutations in the progranulin three missense mutations lead to amino acid alterations in the signal peptide hydrophobic core; they are V5L, W7R, and A9D (Gass et al., 2006; Mukherjee et al., 2006; Lopez de Munain et al., 2008; Cruts et al., 2012) (Figure 1A). While V5L and W7R mutations are not well-studied in patients, it was demonstrated that the A9D mutation resulted in decreased GRN mRNA and protein levels (Mukherjee et al., 2006, 2008). However, the mechanism of the reduced mRNA level was not clear at that time. Further detailed experimental examination of the PGRN signal peptide mutations showed that W7R and A9D inhibited signal peptide interaction with SRP and pathologically activated the RAPP pathway leading to degradation of the defective GRN mRNAs establishing the molecular mechanism of the familial FTLD through mRNA degradation (Pinarbasi et al., 2018) (Figure 1C). Remarkably, the mechanism of GRN mRNA degradation was specific to the mutated mRNAs only and did not affect the wild-type GRN mRNA when they were co-expressed. The mRNA degradation was initiated by the loss of SRP interaction with the signal peptide due to W7R or A9D mutation. RAPP activation is a unique feature of the pathway—it recognizes defective proteins and degrades their mRNA templates. Interestingly, V5L mutation did not interfere with SRP interactions and did not induce the RAPP pathway, and the mutated mRNA did not degrade, suggesting that the V5L is a benign polymorphism and most likely does not lead to a disease. Analysis of the signal peptide hydrophobicity profiles revealed that W7R or A9D mutations decreased hydrophobicity while V5L did not. This observation may be used for theoretical prediction of the impact of the uncharacterized mutations for RAPP activation and mRNA degradation. Noteworthy, the depletion of SRP54 (one of the six SRP subunits) led to mRNA degradation of the wild-type protein (Figure 1C). This fact suggests that defects in SRP subunits may be a molecular basis of sporadic human diseases. Indeed, it was found recently that several mutations in SRP54 are associated with inherited neutropenia and Shwachman-Diamond-like syndrome (Carapito et al., 2017).
Polypeptide nascent chain interactions at the ribosome are important for proper protein folding, transport, and modification. As it is discussed above, the loss of the SRP signal peptide interaction leads to dramatic consequences: elimination of the defective protein mRNA in the RAPP pathway and as a result to decrease of PGRN protein level and finally to FTLD. Most likely, the induction of the RAPP pathway is not limited to the mutant PGRNs, and may be associated with signal peptide mutations in other secretory proteins leading to the diverse group of the human diseases caused by the pathological RAPP activation.
In conclusion, it seems that the decrease or loss of GRN expression in many different familial FTLDs is associated with mRNA degradation, although the nature of the mutations is different. The nonsense, frameshift, and splice site mutations generate premature stop codons that induce NMD, while the mutations in the signal peptide activate RAPP. Regardless of the pathway engaged, the GRN mRNA is degraded that may lead to PGRN haploinsufficiency and the disease. These observations open the necessity of deep exploration of the molecular mechanisms of mRNA degradation pathways in neurodegenerative diseases that may eventually lead to development better pharmacological treatments in the future.
Author Contributions
AK and ZK wrote the manuscript. ET designed and prepared the figure, all authors discussed and edited the manuscript.
Funding
This work was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number R03NS102645 to AK. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abella, V., Pino, J., Scotece, M., Conde, J., Lago, F., Gonzalez-Gay, M. A., et al. (2017). Progranulin as a biomarker and potential therapeutic agent. Drug Discov. Today 22, 1557–1564. doi: 10.1016/j.drudis.2017.06.006
Baker, M., Mackenzie, I. R., Pickering-Brown, S. M., Gass, J., Rademakers, R., Lindholm, C., et al. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. doi: 10.1038/nature05016
Bang, J., Spina, S., and Miller, B. L. (2015). Frontotemporal dementia. Lancet 386, 1672–1682. doi: 10.1016/S0140-6736(15)00461-4
Beel, S., Moisse, M., Damme, M., De Muynck, L., Robberecht, W., Van Den Bosch, L., et al. (2017). Progranulin functions as a cathepsin D chaperone to stimulate axonal outgrowth in vivo. Hum. Mol. Genet. 26, 2850–2863. doi: 10.1093/hmg/ddx162
Bhandari, V., Palfree, R. G., and Bateman, A. (1992). Isolation and sequence of the granulin precursor cDNA from human bone marrow reveals tandem cysteine-rich granulin domains. Proc Natl Acad Sci U.S.A. 89, 1715–1719.
Cairns, N. J., Bigio, E. H., Mackenzie, I. R., Neumann, M., Lee, V. M., Hatanpaa, K. J., et al. (2007). Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 114, 5–22. doi: 10.1007/s00401-007-0237-2
Carapito, R., Konantz, M., Paillard, C., Miao, Z., Pichot, A., Leduc, M. S., et al. (2017). Mutations in signal recognition particle SRP54 cause syndromic neutropenia with Shwachman-Diamond-like features. J. Clin. Invest. 127, 4090–4103. doi: 10.1172/JCI92876
Cenik, B., Sephton, C. F., Kutluk Cenik, B., Herz, J., and Yu, G. (2012). Progranulin: a proteolytically processed protein at the crossroads of inflammation and neurodegeneration. J. Biol. Chem. 287, 32298–32306. doi: 10.1074/jbc.R112.399170
Cruts, M., Gijselinck, I., van der Zee, J., Engelborghs, S., Wils, H., Pirici, D., et al. (2006). Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924. doi: 10.1038/nature05017
Cruts, M., Theuns, J., and Van Broeckhoven, C. (2012). Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 33, 1340–1344. doi: 10.1002/humu.22117
Cruts, M., and Van Broeckhoven, C. (2008). Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet. 24, 186–194. doi: 10.1016/j.tig.2008.01.004
Gass, J., Cannon, A., Mackenzie, I. R., Boeve, B., Baker, M., Adamson, J., et al. (2006). Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet. 15, 2988–3001. doi: 10.1093/hmg/ddl241
Gass, J., Prudencio, M., Stetler, C., and Petrucelli, L. (2012). Progranulin: an emerging target for FTLD therapies. Brain Res. 1462, 118–128. doi: 10.1016/j.brainres.2012.01.047
Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Kertesz, A., Mendez, M., Cappa, S. F., et al. (2011). Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014. doi: 10.1212/WNL.0b013e31821103e6
Gotzl, J. K., Lang, C. M., Haass, C., and Capell, A. (2016). Impaired protein degradation in FTLD and related disorders. Ageing Res. Rev. 32, 122–139. doi: 10.1016/j.arr.2016.04.008
He, Z., and Bateman, A. (2003). Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J. Mol. Med. 81, 600–612. doi: 10.1007/s00109-003-0474-3
Hernandez, I., Fernandez, M. V., Tarraga, L., Boada, M., and Ruiz, A. (2018). Frontotemporal lobar degeneration (FTLD): review and update for clinical neurologists. Curr. Alzheimer Res. 15, 511–530. doi: 10.2174/1567205014666170725130819.
Hodges, J. R., and Piguet, O. (2018). Progress and challenges in frontotemporal dementia research: a 20-year review. J. Alzheimers Dis. 62, 1467–1480. doi: 10.3233/JAD-171087
Holler, C. J., Taylor, G., Deng, Q., and Kukar, T. (2017). Intracellular proteolysis of progranulin generates stable, lysosomal granulins that are haploinsufficient in patients with frontotemporal dementia caused by GRN mutations. eNeuro 4:ENEURO.0100-17.2017. doi: 10.1523/ENEURO.0100-17.2017
Hu, F., Padukkavidana, T., Vaegter, C. B., Brady, O. A., Zheng, Y., Mackenzie, I. R., et al. (2010). Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68, 654–667. doi: 10.1016/j.neuron.2010.09.034
Hu, Y., Xiao, H., Shi, T., Oppenheim, J. J., and Chen, X. (2014). Progranulin promotes tumour necrosis factor-induced proliferation of suppressive mouse CD4(+) Foxp3(+) regulatory T cells. Immunology 142, 193–201. doi: 10.1111/imm.12241
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., et al. (1998). Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. doi: 10.1038/31508
Jian, J., Tian, Q. Y., Hettinghouse, A., Zhao, S., Liu, H., Wei, J., et al. (2016). Progranulin recruits HSP70 to beta-glucocerebrosidase and is therapeutic against gaucher disease. EBioMedicine 13, 212–224. doi: 10.1016/j.ebiom.2016.10.010
Kalinin, A. E., Mikhaleva, N. I., Karamyshev, A. L., Karamysheva, Z. N., and Nesmeyanova, M. A. (1999). Interaction of mutant alkaline phosphatase precursors with membrane phospholipids in vivo and in vitro. Biochemistry 64, 1021–1029.
Karamyshev, A. L., and Johnson, A. E. (2005). Selective SecA association with signal sequences in ribosome-bound nascent chains: a potential role for SecA in ribosome targeting to the bacterial membrane. J. Biol. Chem. 280, 37930–37940. doi: 10.1074/jbc.M509100200
Karamyshev, A. L., and Karamysheva, Z. N. (2018). Lost in translation: ribosome-associated mRNA and protein quality controls. Front. Genet. 9:431. doi: 10.3389/fgene.2018.00431
Karamyshev, A. L., Karamysheva, Z. N., Kajava, A. V., Ksenzenko, V. N., and Nesmeyanova, M. A. (1998). Processing of Escherichia coli alkaline phosphatase: role of the primary structure of the signal peptide cleavage region. J. Mol. Biol. 277, 859–870. doi: 10.1006/jmbi.1997.1617
Karamyshev, A. L., Patrick, A. E., Karamysheva, Z. N., Griesemer, D. S., Hudson, H., Tjon-Kon-Sang, S., et al. (2014). Inefficient SRP interaction with a nascent chain triggers a mRNA quality control pathway. Cell 156, 146–157. doi: 10.1016/j.cell.2013.12.017
Kleinberger, G., Capell, A., Haass, C., and Van Broeckhoven, C. (2013). Mechanisms of granulin deficiency: lessons from cellular and animal models. Mol. Neurobiol. 47, 337–360. doi: 10.1007/s12035-012-8380-8
Kohlschutter, A., and Schulz, A. (2009). Towards understanding the neuronal ceroid lipofuscinoses. Brain Dev. 31, 499–502. doi: 10.1016/j.braindev.2008.12.008
Lopez de Munain, A., Alzualde, A., Gorostidi, A., Otaegui, D., Ruiz-Martinez, J., Indakoetxea, B., et al. (2008). Mutations in progranulin gene: clinical, pathological, and ribonucleic acid expression findings. Biol. Psychiatry 63, 946–952. doi: 10.1016/j.biopsych.2007.08.015
Mackenzie, I. R., and Neumann, M. (2012). FET proteins in frontotemporal dementia and amyotrophic lateral sclerosis. Brain Res. 1462, 40–43. doi: 10.1016/j.brainres.2011.12.010
Mackenzie, I. R., Neumann, M., Baborie, A., Sampathu, D. M., Du Plessis, D., Jaros, E., et al. (2011). A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 122, 111–113. doi: 10.1007/s00401-011-0845-8
Matsubara, T., Mita, A., Minami, K., Hosooka, T., Kitazawa, S., Takahashi, K., et al. (2012). PGRN is a key adipokine mediating high fat diet-induced insulin resistance and obesity through IL-6 in adipose tissue. Cell Metab. 15, 38–50. doi: 10.1016/j.cmet.2011.12.002
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Mukherjee, O., Pastor, P., Cairns, N. J., Chakraverty, S., Kauwe, J. S., Shears, S., et al. (2006). HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann. Neurol. 60, 314–322. doi: 10.1002/ana.20963
Mukherjee, O., Wang, J., Gitcho, M., Chakraverty, S., Taylor-Reinwald, L., Shears, S., et al. (2008). Molecular characterization of novel progranulin (GRN) mutations in frontotemporal dementia. Hum. Mutat. 29, 512–521. doi: 10.1002/humu.20681
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nguyen, A. D., Nguyen, T. A., Martens, L. H., Mitic, L. L., and Farese, R. V. Jr. (2013). Progranulin: at the interface of neurodegenerative and metabolic diseases. Trends Endocrinol. Metab. 24, 597–606. doi: 10.1016/j.tem.2013.08.003
Nilsson, I., Lara, P., Hessa, T., Johnson, A. E., von Heijne, G., and Karamyshev, A. L. (2015). The code for directing proteins for translocation across ER membrane: SRP cotranslationally recognizes specific features of a signal sequence. J. Mol. Biol. 427 (6 Pt A), 1191–1201. doi: 10.1016/j.jmb.2014.06.014.
Olney, R. K., Murphy, J., Forshew, D., Garwood, E., Miller, B. L., Langmore, S., et al. (2005). The effects of executive and behavioral dysfunction on the course of ALS. Neurology 65, 1774–1777. doi: 10.1212/01.wnl.0000188759.87240.8b
Park, H. K., and Chung, S. J. (2013). New perspective on parkinsonism in frontotemporal lobar degeneration. J Mov Disord 6, 1–8. doi: 10.14802/jmd.13001
Pinarbasi, E. S., Karamyshev, A. L., Tikhonova, E. B., Wu, I. H., Hudson, H., and Thomas, P. J. (2018). Pathogenic signal sequence mutations in progranulin disrupt SRP interactions required for mRNA stability. Cell Rep. 23, 2844–2851. doi: 10.1016/j.celrep.2018.05.003
Rascovsky, K., Hodges, J. R., Knopman, D., Mendez, M. F., Kramer, J. H., Neuhaus, J., et al. (2011). Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134 (Pt 9), 2456–2477. doi: 10.1093/brain/awr179
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Skibinski, G., Parkinson, N. J., Brown, J. M., Chakrabarti, L., Lloyd, S. L., Hummerich, H., et al. (2005). Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 37, 806–808. doi: 10.1038/ng1609
Smith, K. R., Damiano, J., Franceschetti, S., Carpenter, S., Canafoglia, L., Morbin, M., et al. (2012). Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am. J. Hum. Genet. 90, 1102–1107. doi: 10.1016/j.ajhg.2012.04.021
Tang, W., Lu, Y., Tian, Q. Y., Zhang, Y., Guo, F. J., Liu, G. Y., et al. (2011). The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science 332, 478–484. doi: 10.1126/science.1199214
Watts, G. D., Wymer, J., Kovach, M. J., Mehta, S. G., Mumm, S., Darvish, D., et al. (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381. doi: 10.1038/ng1332
Zhou, X., Paushter, D. H., Feng, T., Sun, L., Reinheckel, T., and Hu, F. (2017a). Lysosomal processing of progranulin. Mol. Neurodegener. 12:62. doi: 10.1186/s13024-017-0205-9.
Zhou, X., Sun, L., Bastos de Oliveira, F., Qi, X., Brown, W. J., Smolka, M. B., et al. (2015). Prosaposin facilitates sortilin-independent lysosomal trafficking of progranulin. J. Cell Biol. 210, 991–1002. doi: 10.1083/jcb.201502029.
Keywords: frontotemporal lobar degeneration (FTLD), granulin, disease-causing mutations, protein quality control, protein targeting and transport, signal peptide, signal recognition particle (SRP), RNA degradation
Citation: Karamysheva ZN, Tikhonova EB and Karamyshev AL (2019) Granulin in Frontotemporal Lobar Degeneration: Molecular Mechanisms of the Disease. Front. Neurosci. 13:395. doi: 10.3389/fnins.2019.00395
Received: 15 February 2019; Accepted: 08 April 2019;
Published: 25 April 2019.
Edited by:
Alberto Lleo, Hospital de la Santa Creu i Sant Pau, SpainReviewed by:
Marc Suárez-Calvet, BarcelonaBeta Brain Research Center, SpainCopyright © 2019 Karamysheva, Tikhonova and Karamyshev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zemfira N. Karamysheva, emVtZmlyYS5rYXJhbXlzaGV2YUB0dHUuZWR1
Andrey L. Karamyshev, YW5kcmV5LmthcmFteXNoZXZAdHR1aHNjLmVkdQ==