 Margaret Fahnestock
Margaret Fahnestock Arman Shekari
Arman Shekari- 1Department of Psychiatry and Behavioural Neurosciences, McMaster University, Hamilton, ON, Canada
- 2Neuroscience Graduate Program, McMaster University, Hamilton, ON, Canada
Profound and early basal forebrain cholinergic neuron (BFCN) degeneration is a hallmark of Alzheimer’s disease (AD). Loss of synapses between basal forebrain and hippocampal and cortical target tissue correlates highly with the degree of dementia and is thought to be a major contributor to memory loss. BFCNs depend for their survival, connectivity and function on the neurotrophin nerve growth factor (NGF) which is retrogradely transported from its sites of synthesis in the cortex and hippocampus. The form of NGF found in human brain is proNGF. ProNGF binds to the NGF receptors TrkA and p75NTR, but it binds more strongly to p75NTR and more weakly to TrkA than does mature NGF. This renders proNGF more sensitive to receptor balance than mature NGF. In the healthy brain, where BFCNs express both TrkA and p75NTR, proNGF is neurotrophic, activating TrkA-dependent signaling pathways such as MAPK and Akt-mTOR and eliciting cell survival and neurite outgrowth. However, if TrkA is lost or if p75NTR is increased, proNGF activates p75NTR-dependent apoptotic pathways such as JNK. This receptor sensitivity serves as a neurotrophic/apoptotic switch that eliminates BFCNs that cannot maintain TrkA/p75NTR balance and therefore synaptic connections with their targets. TrkA is increasingly lost in mild cognitive impairment (MCI) and AD. In addition, proNGF accumulates at BFCN terminals in cortex and hippocampus, reducing the amount of trophic factor that reaches BFCN cell bodies. The loss of TrkA and accumulation of proNGF occur early in MCI and correlate with cognitive impairment. Increased levels of proNGF and reduced levels of TrkA lead to BFCN neurodegeneration and eventual p75NTR-dependent apoptosis. In addition, in AD BFCNs suffer from reduced TrkA-dependent retrograde transport which reduces neurotrophic support. Thus, BFCNs are particularly vulnerable to AD due to their dependence upon retrograde trophic support from proNGF signaling and transport.
Basal Forebrain Cholinergic Neurons in Alzheimer’s Disease
The Alzheimer’s disease (AD) brain undergoes progressive dysfunction, degeneration and loss of neurons and synapses in selective areas of the brain (Coleman and Flood, 1987; Scheff et al., 1990). Amyloid-β, a component of plaques, has long been targeted as a precipitating insult in AD, triggering a cascade that leads to tau aggregation, impaired synaptic communication and neuronal death (Hardy and Selkoe, 2002). Tau, the primary component of neurofibrillary tangles, promotes the assembly and stabilization of microtubules in the cytoskeleton. When tau is abnormally phosphorylated, it detaches from microtubules and polymerizes into soluble oligomers, straight and paired helical filaments that assemble into neurofibrillary tangles. This tau disrupts axonal transport and is thought to lead to neuronal atrophy by loss of normal tau function and/or by gain of pathological function through toxicity of phosphorylated, truncated or aggregated forms of tau (Iqbal and Grundke-Iqbal, 2008; Ittner and Götz, 2011). As the disease progresses, both tau and amyloid pathology impact significant areas of the cortex and hippocampus. Efforts aimed at determining where the pathology begins have implicated TrkA-expressing cholinergic neurons in the basal forebrain in the very early stages of the disorder (Schmitz and Nathan Spreng, 2016).
Basal forebrain cholinergic neurons (BFCNs) are extremely important for learning, memory, and attention (Baxter and Chiba, 1999). They are among the first to degenerate and the most severely affected in aging and AD (Coyle et al., 1983; Hyman et al., 1984; Whitehouse, 1998; Schmitz and Nathan Spreng, 2016), but the reasons for the selective vulnerability of these neurons is unknown. BFCN degeneration correlates strongly with the degree of dementia and with AD pathology (Wilcock et al., 1982; Coyle et al., 1983; Bierer et al., 1995; Whitehouse, 1998). The vulnerability of BFCNs in AD may be related to disrupted communication with target neurons in the hippocampus and cortex, regions that also display pathology and neurodegeneration in AD.
Basal forebrain cholinergic neurons are dependent upon the neurotrophin nerve growth factor (NGF) for survival and proper function. They do not make NGF themselves and must obtain it through retrograde transport from their targets. Older literature focused on the mature form of NGF, demonstrating that it is highly expressed in developing and adult hippocampus and cerebral cortex and is retrogradely transported from these target areas by BFCNs (Seiler and Schwab, 1984; DiStefano et al., 1992; Lapchak et al., 1993). NGF supports differentiation and survival of BFCNs in vitro (Hartikka and Hefti, 1988; Hatanaka et al., 1988; Friedman et al., 1993) and in vivo (Hefti, 1986; Williams et al., 1986; Lapchak and Hefti, 1991; Koliatsos et al., 1994). NGF increases acetylcholine (Ach) synthesis and release (Hatanaka et al., 1988; Takei et al., 1989; Lapchak and Hefti, 1991; Rylett et al., 1993; Pongrac and Rylett, 1996; Oosawa et al., 1999; Auld et al., 2001a,b) as well as activity and expression of cholinergic markers including choline acetyltransferase (ChAT; Williams and Rylett, 1990; Lorenzi et al., 1992; Koliatsos et al., 1994; Pongrac and Rylett, 1996) and vesicular Ach transporter (VAChT; Takei et al., 1997; Oosawa et al., 1999), which are decreased in AD (Bartus et al., 1982). NGF also increases expression of its own receptor, TrkA, in BFCN (Holtzman et al., 1992; Kojima et al., 1994, 1995; Li et al., 1995). Because BFCN rely on neurotrophins for their survival and function, it has been proposed that BFCN loss in aging and AD arises from lack of neurotrophic support (Appel, 1981; Hefti and Weiner, 1986; Price, 1986; Hefti et al., 1989). In fact, significant literature supports deficits in BDNF expression in AD (Fahnestock et al., 2002; Peng et al., 2005; Fahnestock, 2011) and also disruptions of NGF and its receptor, TrkA, with concomitant effects on attention, learning, and memory (Mufson et al., 1996, 2005, 2007; Counts et al., 2004; Peng et al., 2004; Perez et al., 2011; Parikh et al., 2013). However, contrary to initial hypotheses (Appel, 1981), loss of NGF expression does not occur in AD (Jetté et al., 1994; Fahnestock et al., 1996, 2001; Peng et al., 2004). We and others showed some years ago that despite normal levels of NGF mRNA expression in human brain tissue from AD subjects (Jetté et al., 1994), NGF-immunoreactive protein detected by ELISA or bioassay is increased in cortex and hippocampus and decreased in basal forebrain, suggesting that NGF-immunoreactive material accumulates in AD due to failed BFCN retrograde transport (Crutcher et al., 1993; Scott et al., 1995; Fahnestock et al., 1996; Narisawa-Saito et al., 1996). This immunoreactive material is entirely present as proNGF (Fahnestock et al., 2001). ProNGF protein is increased in BFCN target tissues both in AD (Fahnestock et al., 2001; Peng et al., 2004) and in the human tauopathy, Pick’s disease (Belrose et al., 2014). In AD, the accumulation of proNGF in cortex and hippocampus and its reduction in basal forebrain suggest a deficit in retrograde transport of proNGF leading to a lack of survival signaling and eventual neurodegeneration.
Animal models of AD further support the role of dysfunctional proNGF trafficking in AD, as the Ts65Dn mouse exhibits cholinergic degeneration and deficits in retrograde transport of proNGF (Salehi et al., 2006). However, this mouse also exhibits deficits in the NGF metabolic pathway responsible for processing proNGF to mature NGF (Iulita et al., 2014). This is consistent with an alternative hypothesis of proNGF accumulation in AD that suggests the accumulation of proNGF in AD is due to defective processing of proNGF into its mature form (Bruno and Cuello, 2006; Cuello and Bruno, 2007). This hypothesis is supported by decreases in tissue plasminogen activator and plasmin, which can process proNGF, and increases in the NGF degradative enzyme MMP-9 in MCI and in Down’s syndrome (Bruno et al., 2009a,b; Iulita et al., 2014). Both transport and processing mechanisms may be at work. However, if proNGF were processed to NGF in the normal brain, mature NGF should be detectable in normal human brain tissue, but it is not (Figure 1A; Fahnestock et al., 2001). Further, it is not clear that plasmin and MMP-9 are the endogenous proNGF/NGF processing enzymes in brain. Thus, although proNGF is acknowledged to be the form of NGF in the brain, the mechanism behind its accumulation in AD remains controversial.
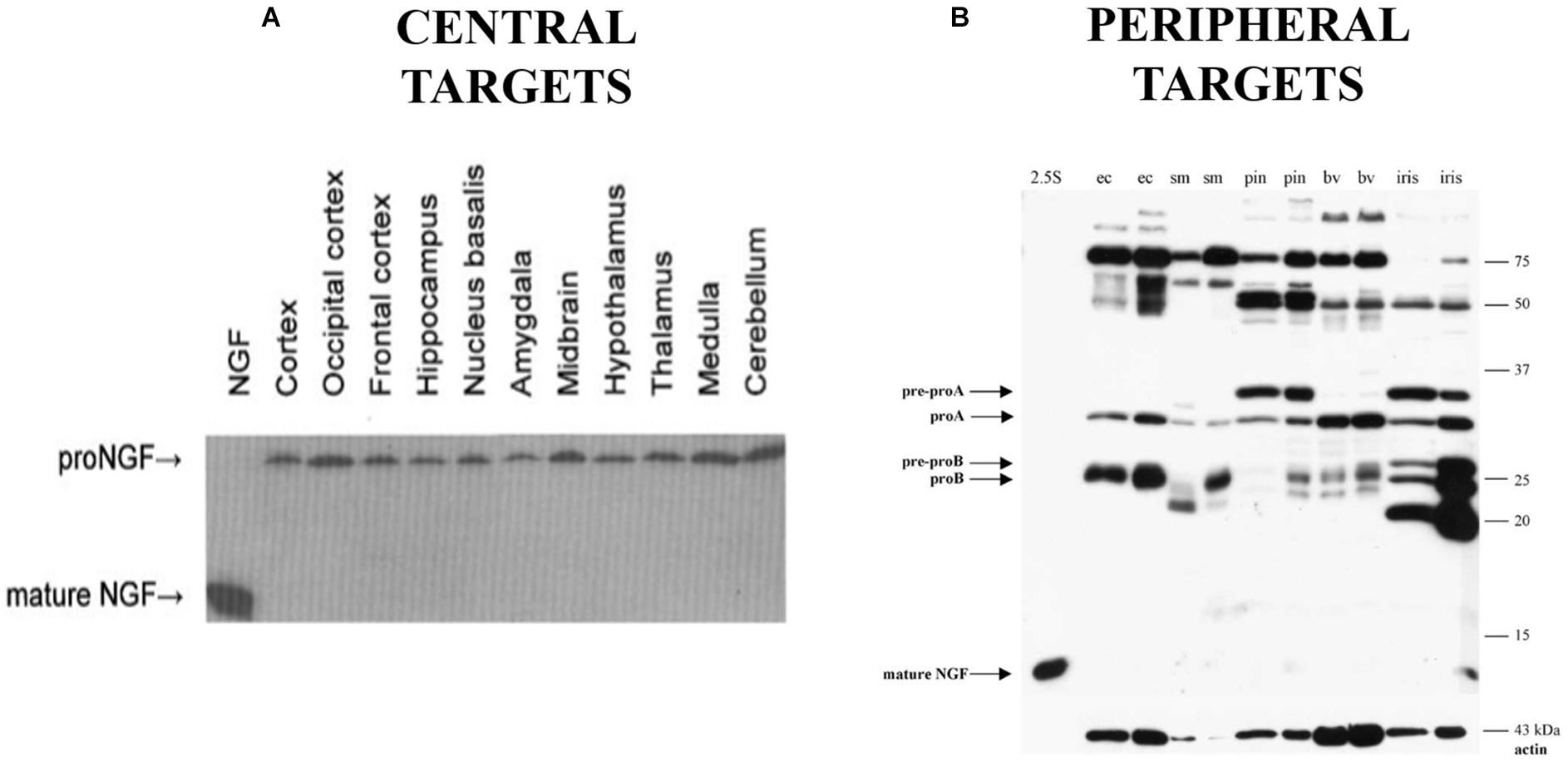
Figure 1. Western blot probing for NGF immunoreactivity in human brain tissue (A) and peripheral targets (B) demonstrating the presence of proNGF and the absence of mature NGF in the indicated areas (ec, external carotid; sm, submandibular gland; pin, pineal gland; bv, extracerebral blood vessels). (A) Reprinted from Fahnestock et al. (2001), with permission from Elsevier. (B) Reprinted from Bierl et al. (2005), with permission from Elsevier.
Biological Relevance of Neurotrophins
All neuronal cells require neurotrophic factors for their proper function and survival. NGF belongs to the neurotrophin family of neurotrophic factors, secreted proteins that are required for maintenance of connectivity, synaptic plasticity and survival (Maisonpierre et al., 1990; Fahnestock, 1991, 2011; Ip et al., 1992). It has been amply demonstrated both in vitro and in vivo that a deficit of neurotrophins can lead to cell dysfunction and death, and that neurotrophins can be used to rescue compromised neuronal populations from atrophy or death (Koliatsos et al., 1991; Mount et al., 1994; Rice et al., 1998; Schober et al., 1998; Klein et al., 2000). The overlapping but distinct specificities of the neurotrophins for various subsets of neurons has been proposed to explain selective neuronal impairments in particular diseases, raising the possibility that individual neurotrophins may be used therapeutically to target specific types of neurons (Hefti, 1994; Lindsay et al., 1994; Apfel, 2002; Lambiase et al., 2003; Duman and Monteggia, 2006). However, neurotrophins are not simple survival factors and can trigger cellular dysfunction and cell death under certain conditions related to their processing and receptor availability. This duality in neurotrophin function makes them critical for maintaining the balance of survival and death in the nervous system, a balance that is disrupted in AD.
Post-Translational Processing of Neurotrophin Precursors to Produce Mature Neurotrophins
The first member of the neurotrophin family to be discovered was NGF (Levi-Montalcini and Angeletti, 1968; Levi-Montalcini, 1987). NGF supports the development, survival and function of PNS (sympathetic and sensory) and CNS (cholinergic) neurons (Levi-Montalcini and Angeletti, 1968; Thoenen and Barde, 1980; Ruit et al., 1992). Murine NGF is translated from two major alternatively spliced transcripts to produce 34 and 27 kDa prepro species (Selby et al., 1987; Fahnestock, 1991). Removal of the signal sequence reduces these translation products to proNGF species of 32 and 25 kDa (Edwards et al., 1986, 1988b; Selby et al., 1987), which occur widely (Berger and Shooter, 1977; Edwards et al., 1988b; Dicou, 1992; Seidah et al., 1996b; Chen et al., 1997; Reinshagen et al., 2000; Fahnestock et al., 2001; Bierl et al., 2005) and were initially thought to be biologically inactive (Edwards et al., 1988a; Suter et al., 1991). ProNGF can undergo further post-translational processing at both amino- and carboxyl-terminal ends to generate a mature, biologically active product of 13.2 kDa (Fahnestock, 1991). Processing of proneurotrophins may occur either intra- or extracellularly. The kallikrein γNGF, which in mouse submandibular gland is found intracellularly in a complex with NGF, processes proNGF to produce intermediate and mature forms of NGF (Greene et al., 1968; Edwards et al., 1988a; Jongstra-Bilen et al., 1989). Furin and other prohormone convertases are able to process proNGF, proBDNF, and proNT-3 to their mature forms (Bresnahan et al., 1990; Seidah et al., 1996a,b). ProNGF can also be processed extracellularly by plasmin and by matrix metalloproteases (Lee et al., 2001; Bruno and Cuello, 2006). In the CNS, proNGF is largely unprocessed (Fahnestock et al., 2001).
We now know that proNGF possesses biological activity independent from its mature isoform (Lee et al., 2001; Fahnestock et al., 2004a,b; Masoudi et al., 2009). Based on the mouse submandibular gland as a model, it was previously thought that proNGF is mainly processed into the mature form which accounts for the biological activity in most tissues (Edwards et al., 1988a; Suter et al., 1991). ELISA assays and immunohistochemistry for NGF are widely used and were thought to measure the mature 13.2 kDa NGF protein as the major form of NGF (Randolph et al., 2007). Little had been done to study NGF biosynthesis in other species or tissues, particularly CNS tissue. In 2001, we were the first to demonstrate (Fahnestock et al., 2001) that in rodent and human brain, NGF exists as 32 kDa proNGF (Figure 1A). In rodent brain there is some but very little mature NGF, while mature NGF is completely absent from human brain. The proNGF found in these tissues is not simply an intracellular precursor; it has since been accepted that secretion of unprocessed proNGF occurs from many cells and tissues, including neurons and astrocytes (Chen et al., 1997; Delsite and Djakiew, 1999; Mowla et al., 1999; Yardley et al., 2000; Lee et al., 2001; Fahnestock et al., 2004a; Bruno and Cuello, 2006), and that proNGF is the major species in both PNS (Figure 1B) and CNS tissues (Figure 1A; Fahnestock et al., 2001; Bierl et al., 2005). This distinction between the pro and mature isoforms of NGF is extremely important, as they have the potential to activate different signaling pathways, a behavior completely contingent on the receptors they bind to.
TrkA and p75NTR Receptors and Their Signaling Pathways
Neurotrophins are structurally related and bind similarly to two types of receptors (Figure 2). All neurotrophins bind with the same low affinity to the common neurotrophin receptor, p75NTR, and with high affinity to a family of type I transmembrane receptor tyrosine kinases known as Trks (tropomyosin-related kinases; Barker and Murphy, 1992; Barbacid, 1995). TrkA is the primary receptor for NGF, although there is overlap in binding specificities (Segal, 2003). Trks are generally responsible for signaling survival, differentiation, synapse strengthening, and neurite outgrowth in response to ligand (Kaplan and Miller, 2000; Miller and Kaplan, 2001; Huang and Reichardt, 2003), whereas p75NTR mediates apoptosis as well as survival, synapse weakening, and inhibition/retraction of neurite outgrowth depending on its binding partner (Roux and Barker, 2002; Barker, 2004). The balance of Trks and p75NTR is crucial to the functional outcome of neurotrophin binding; sufficient amounts of activated Trks, for example, can suppress apoptotic pathways activated by p75NTR (Yoon et al., 1998; Segal, 2003), and p75NTR, when complexed with TrkA, increases NGF signaling through TrkA to enhance survival and neurite outgrowth (Epa et al., 2004). In many cell culture systems including rat oligodendrocytes, Schwann cells, embryonic BFCN, and embryonic retinal ganglion cells which express little or no TrkA, p75NTR mediates apoptosis (Roux and Barker, 2002; Volosin et al., 2006). When complexed with sortilin, p75NTR binds proNGF with a higher affinity than NGF, signaling cell death (Nykjaer et al., 2004). p75NTR can also cause growth cone retraction/neurite outgrowth inhibition when complexed with the Nogo-66 receptor (NgR) and its ligand Lingo-1 (Bandtlow and Dechant, 2004; Barker, 2004), Whether p75NTR signals cell death or cell survival depends both on the ligand and on the presence or absence of TrkA and sortilin (Barker, 2004; Ioannou and Fahnestock, 2017). As a result, maintaining the proper balance between TrkA, p75NTR and sortilin is critical for neuronal function and survival.
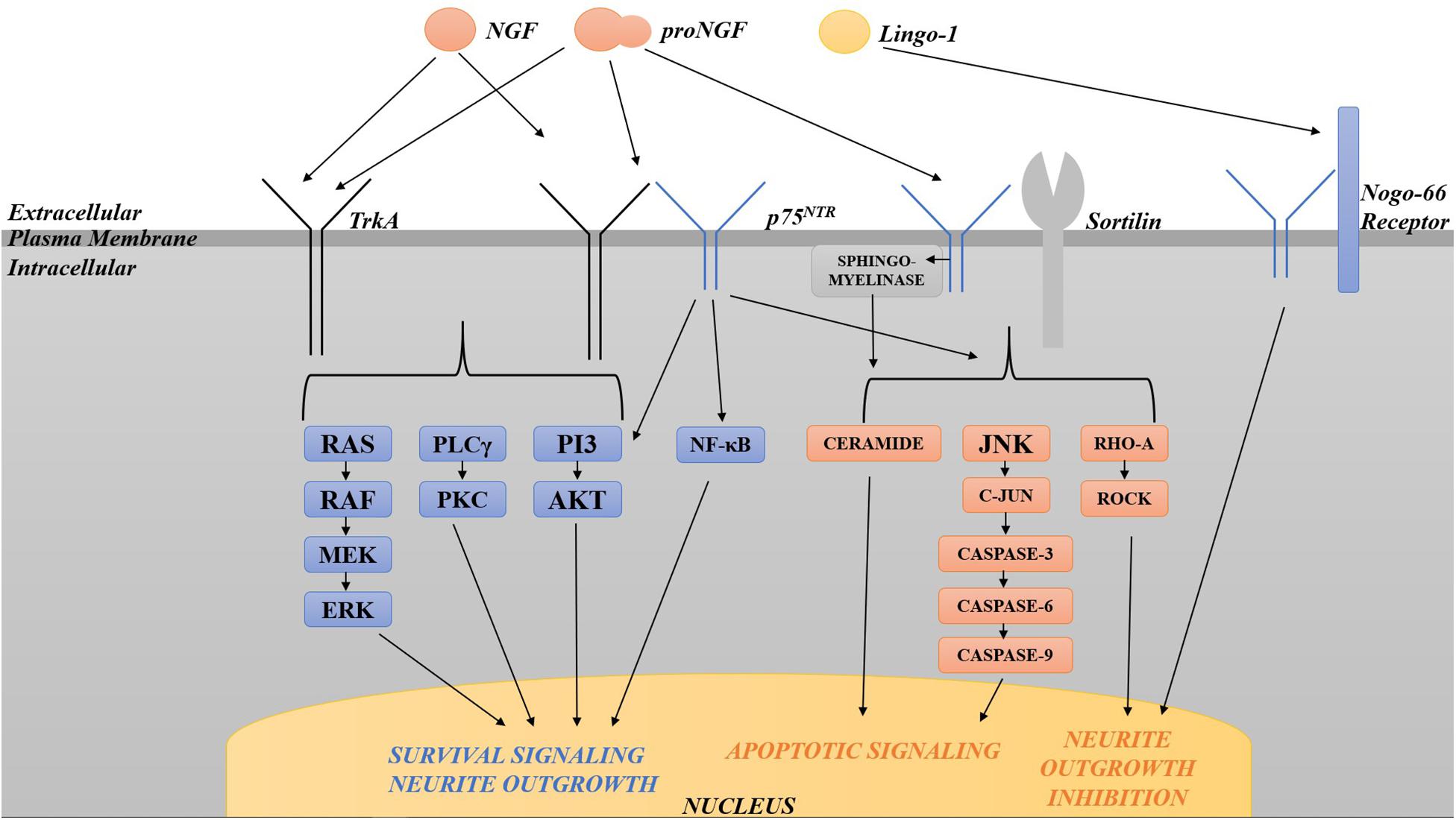
Figure 2. Diagram of the various pathways activated by TrkA and p75NTR. TrkA in isolation activates pro-survival pathways including the Ras-MAP kinase, PLCγ, and PI3 kinase/Akt pathways. p75NTR, when complexed with TrkA, increases pro-survival signaling through TrkA. p75NTR in isolation can also signal survival via the NF-κB and PI3/Akt pathways. p75NTR in isolation activates apoptosis via sphingomyelinase/ceramide, JNK, c-Jun phosphorylation and caspases 3, 6, and 9. When complexed with sortilin, p75NTR binds proNGF with a higher affinity than NGF, signaling cell death through the same pathways. p75NTR can also cause growth cone retraction/neurite outgrowth inhibition when complexed with the Nogo-66 receptor and its ligand Lingo-1. The balance of Trks and p75NTR is crucial to the functional outcome of neurotrophin binding.
Several intracellular signaling pathways are activated by activated Trk receptors and by p75NTR, including the Ras-MAP kinase, PLCγ, and PI3 kinase/Akt pathways for Trks, and the NF-κB and PI3 kinase/Akt, and the sphingomyelinase/ceramide and c-Jun N-terminal kinase (JNK) pathways for p75NTR (Dobrowsky et al., 1994; Kaplan and Miller, 2000; Miller and Kaplan, 2001; Dechant and Barde, 2002; Roux and Barker, 2002; Huang and Reichardt, 2003; Segal, 2003). p75NTR signaling in isolation activates apoptosis via JNK, c-Jun phosphorylation and caspases 3, 6, and 9 (Troy et al., 2002; Bhakar et al., 2003). p75NTR can signal survival through activation of NF-κB (Ladiwala et al., 1998; Foehr et al., 2000; Hughes et al., 2001) or the PI3K/Akt pathway (Roux et al., 2001). Lastly, p75NTR also limits neurite outgrowth via RhoA activation (Roux and Barker, 2002): p75NTR null mice exhibit enhanced sympathetic and sensory sprouting (Kohn et al., 1999; Petrie et al., 2013) and hyper-innervation of the hippocampus by cholinergic neurons (Yeo et al., 1997). These signal transduction pathways downstream of both receptors have been intensely investigated in the case of NGF due to the initial assumption of the biological inactivity of proNGF. Once the biological significance of proNGF was made clear, it was found that many of the same Trk- and p75NTR-induced pathways are activated by proNGF.
Biological Activity of ProNGF
We and others have shown that both native and recombinant proNGF, with or without amino acid substitutions or tags, has neurite outgrowth and neuronal survival activity (Saboori and Young, 1986; Chen et al., 1997; Rattenholl et al., 2001; Fahnestock et al., 2004b; Clewes et al., 2008; Masoudi et al., 2009; Ioannou and Fahnestock, 2017). Many investigators (Lee et al., 2001; Fahnestock et al., 2004b) use mutated proNGFs that are resistant to processing into mature NGF to rule out any contribution of mature NGF. This cleavage-resistant proNGF binds to and activates TrkA and its downstream signal transducers, Erk1/2 (Fahnestock et al., 2004b; Masoudi et al., 2009) and Akt (Ioannou and Fahnestock, 2017), but with less affinity than mature NGF. Although proNGF has a lower affinity for TrkA than does NGF (Clewes et al., 2008), proNGF produces the same maximal activity as NGF (Fahnestock et al., 2004b); the low affinity is outweighed by the relative abundance of proNGF in the adult brain. In addition, endogenously expressed proNGF is similarly neurotrophic in vivo: transgenic mice over-expressing wild-type proNGF exhibit robust sympathetic and sensory axonal sprouting into brain parenchyma with no cell loss in peripheral ganglia (Buttigieg et al., 2007).
In contrast to these data, Lee et al. (2001) reported that proNGF exhibits apoptotic activity. Neither the many structural differences between the different cleavage-resistant proNGFs nor different expression systems or purification methods accounted for their differences in biological activity (Masoudi et al., 2009). However, cell culture conditions, which influence receptor complement, explain why some labs report apoptotic activity of proNGF whereas others report neurotrophic activity. ProNGF exhibits neurite outgrowth and survival activity on newborn rodent superior cervical ganglion neurons, which maintain relatively high TrkA levels compared to little or no p75NTR (Fahnestock et al., 2004b; Masoudi et al., 2009). ProNGF is also neurotrophic for rat pheochromocytoma (PC12) cells differentiated (primed) by growth in NGF, which express high levels of TrkA vs. p75NTR, whereas unprimed PC12 cells (grown in serum), which express high levels of p75NTR compared to TrkA, respond to proNGF with increased apoptosis (Fahnestock et al., 2004b; Masoudi et al., 2009; Ioannou and Fahnestock, 2017). Importantly, this contrasts with NGF, which is neurotrophic under both growth conditions (Ioannou and Fahnestock, 2017). Supporting the importance of relative TrkA vs. p75NTR levels, apoptotic activity of proNGF in vivo has been shown only in situations where TrkA is missing or down-regulated and p75NTR is up-regulated (Beattie et al., 2002; Harrington et al., 2004; Volosin et al., 2006; Provenzano et al., 2008). Furthermore, knocking down TrkA in PC12 cells changes proNGF’s neurotrophic activity to apoptotic activity (Ioannou and Fahnestock, 2017). Lastly, proNGF is apoptotic for TrkA-deficient PC12nnr5 cells but is neurotrophic for TrkA-over-expressing PC12nnr5-B5 cells (Ioannou and Fahnestock, 2017).
It was reported that proNGF requires intracellular proteolysis to activate TrkA (Boutilier et al., 2008), a finding that contrasts with the fact that little to no mature NGF is present in both human and rodent brains. However, the MAPK and Akt assays that were carried out to establish this occurred for 1 h, a longer time period than usual for these assays and sufficient to allow proNGF endocytosis and cleavage. In contrast, we demonstrated that cleavage-resistant proNGF robustly activates MAPK after 5 min. Furthermore, proNGF activates TrkA even when incubated for 1 h in the presence of a furin inhibitor which allows uptake but not cleavage (Masoudi et al., 2009). These results demonstrated that cleavage to mature NGF is not required for proNGF signaling and implicate proNGF as the form of NGF that normally signals and is endocytosed. Thus, proNGF binds to TrkA and is internalized without cleavage, and endocytosed proNGF activates MAPK. Furthermore, this internalized proNGF associates with signaling endosomes in PC12 cells (Di Matteo et al., 2017) and is retrogradely transported in dorsal root ganglion neurons (De Nadai et al., 2016).
In sum, proNGF can signal without cleavage, and the balance between TrkA and p75NTR determines whether proNGF is neurotrophic or apoptotic. This is of critical importance because TrkA is lost in AD (Mufson et al., 1996; Counts et al., 2004), as discussed below.
Retrograde Signaling and Transport
Axonal transport defects are common characteristics of many neurodegenerative diseases, and mutations in components of the axonal transport machinery have demonstrated that impaired axonal transport can cause neurodegeneration (Perlson et al., 2010). Lack of retrograde neurotrophic support has been postulated to cause neurodegeneration and death of BFCNs in AD (Appel, 1981). BFCN cell bodies may receive a neurotrophic signal from their distal axon terminals in the cortex and hippocampus by binding of ligand to receptors at axon terminals to produce local signals within axon terminals and retrograde signals to the cell body (Zhang et al., 2000; MacInnis and Campenot, 2002; MacInnis et al., 2003), internalization of the receptor-ligand complex and retrograde transport to cell bodies via signaling endosomes containing activated TrkA complexed with ligand (Grimes et al., 1996; Watson et al., 1999; Delcroix et al., 2003; Ye et al., 2003), or both (Campenot and MacInnis, 2003; Ascano et al., 2012). Both mechanisms of neurotrophic signaling may be compromised in AD.
Reduced proNGF signaling and transport may be due to disrupted transport machinery or to reduced TrkA, or both. In animal models, unilateral fimbrial transection, which severs cholinergic projections from the medial septum to the hippocampus, and colchicine, which disrupts axonal transport, increase hippocampal NGF-immunoreactivity but not NGF mRNA (Korsching et al., 1986; Ginn and Peterson, 1992), consistent with interrupted retrograde transport and with our findings in human brain. Reduced retrograde transport of NGF has been demonstrated in animal models of aging and disease (Cooper et al., 1994; Salehi et al., 2006), and abnormal tau phosphorylation and aggregation were shown to inhibit both anterograde and retrograde transport in the squid giant axon (Tiernan et al., 2016).
TrkA mRNA is lost in the basal forebrain of persons with AD (Mufson et al., 1996, 1997; Counts et al., 2004; Ginsberg et al., 2006). This decreases the amount of TrkA protein destined for anterograde transport to BFCN distal axon terminals (Scott-Solomon and Kuruvilla, 2018). TrkA protein levels are reduced in the cortex (BFCN terminal axons) of AD patients (Mufson et al., 1997; Counts et al., 2004; Ginsberg et al., 2006), while many studies, but not all, report no change in p75NTR levels (Counts et al., 2004; Mufson et al., 2005, 2007; Ginsberg et al., 2006). Internalization and retrograde transport of mature NGF requires TrkA (Eveleth and Bradshaw, 1992; Loeb and Greene, 1993), but whether this is true of proNGF is unknown. p75NTR is not required for NGF transport (Curtis et al., 1995) but may be capable of retrograde transport of NGF in the absence of TrkA (von Bartheld et al., 1996). Interestingly, the PI3K pathway may be required for TrkA-mediated retrograde transport, since inhibition of TrkA-mediated PI3K activity blocks retrograde transport of NGF in sympathetic neurons (Bartlett et al., 1998; Reynolds et al., 1998).
Recently, TrkA has been shown to bind APP, the precursor to amyloid-β, resulting in its non-pathological processing (Matrone et al., 2008a,b; Calissano et al., 2010). Thus, loss of TrkA may be an early event in AD underlying toxic amyloid buildup, consistent with the idea of basal forebrain degeneration being a possible initiating occurrence in AD. In addition, both aggregated Aβ and tau impede the bidirectional transport of TrkA in hippocampal neurons (Vossel et al., 2010). Tau reduction prevents Aβ-induced impairment of transport of TrkA but not of BDNF (Ramser et al., 2013). Thus, both Aβ and tau may regulate neurotrophin and neurotrophin receptor axonal transport.
Reduced retrograde transport of NGF has been demonstrated in animal models of aging and Down’s syndrome (Cooper et al., 1994; Salehi et al., 2006). Animal models of AD exhibit reduced TrkA as well as defective NGF retrograde transport (Cooper et al., 2001; Salehi et al., 2006). Reduced TrkA and/or retrograde transport may lead to neurodegeneration and cell death in the presence of increased proNGF (Masoudi et al., 2009; Ioannou and Fahnestock, 2017; Allard et al., 2018). Thus, multiple mechanisms may be at work to reduce the trophic support of BFCNs in AD.
Summary
ProNGF is abundant in the CNS and can have both neurotrophic and apoptotic activities, depending on the receptor complement. ProNGF binds strongly to p75NTR, but it also binds to TrkA and elicits survival signaling through the MAPK and Akt pathways. ProNGF controls survival and death of NGF-dependent neurons in the basal forebrain, depending on the balance of TrkA and p75NTR. This is particularly important because TrkA receptors are lost in AD.
Axonal transport defects are common characteristics of many neurodegenerative diseases, and mutations in components of the axonal transport machinery have demonstrated that impaired axonal transport can cause neurodegeneration. BFCNs are uniquely dependent upon a constant supply of target-derived proNGF for their survival and function, and therefore reduced retrograde axonal transport of proNGF may cause degeneration and death of BFCNs. These neurons are crucial for learning, memory and attention, they are among the most vulnerable neurons in aging and AD, and they depend upon target-derived proNGF for their survival and function.
Multiple mechanisms may limit survival of BFCNs in AD. ProNGF accumulates in cortex and hippocampus in AD but is reduced in basal forebrain, suggesting a retrograde transport defect. The transport deficit may be the result of AD pathology or loss of TrkA, or both. The accumulation of proNGF, along with loss of TrkA, reduces retrograde survival signals and switches proNGF signaling to p75NTR apoptotic pathways, thus initiating a neurodegenerative cascade. Rescue of axonal transport of signaling endosomes carrying cargoes such as BDNF, which supports motor neurons, ameliorates disease progression in a mouse model of ALS (Kieran et al., 2005). Thus, restoring retrograde transport of proNGF, for example by increasing TrkA levels, may inhibit disease progression in AD. Understanding proNGF biological activity and metabolism will aid in designing treatments to prevent degeneration of BFCNs.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
Funding from the Canadian Institutes of Health Research, grant numbers MOP-102723 and PJT-159493 to MF.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Allard, S., Jacobs, M. L., Do Carmo, S., and Cuello, A. C. (2018). Compromise of cortical proNGF maturation causes selective retrograde atrophy in cholinergic nucleus basalis neurons. Neurobiol. Aging 67, 10–20. doi: 10.1016/j.neurobiolaging.2018.03.002
Apfel, S. C. (2002). Nerve growth factor for the treatment of diabetic neuropathy: what went wrong, what went right, and what does the future hold? Int. Rev. Neurobiol. 50, 393–413. doi: 10.1016/S0074-7742(02)50083-0
Appel, S. H. (1981). A unifying hypothesis for the cause of amyotrophic lateral sclerosis, Parkinsonism, and Alzheimer disease. Ann. Neurol. 10, 499–505. doi: 10.1002/ana.410100602
Ascano, M., Bodmer, D., and Kuruvilla, R. (2012). Endocytic trafficking of neurotrophins in neural development. Trends Cell Biol. 22, 266–273. doi: 10.1016/j.tcb.2012.02.005
Auld, D. S., Mennicken, F., Day, J. C., and Quirion, R. (2001a). Neurotrophins differentially enhance acetylcholine release, acetylcholine content and choline acetyltransferase activity in basal forebrain neurons. J. Neurochem. 77, 253–262.
Auld, D. S., Mennicken, F., and Quirion, R. (2001b). Nerve growth factor rapidly induces prolonged acetylcholine release from cultured basal forebrain neurons: differentiation between neuromodulatory and neurotrophic influences. J. Neurosci. 21, 3375–3382.
Bandtlow, C., and Dechant, G. (2004). From cell death to neuronal regeneration, effects of the p75 neurotrophin receptor depend on interactions with partner subunits. Sci. Stke 235:e24.
Barbacid, M. (1995). Structural and functional properties of the TRK family of neurotrophin receptors. Ann. N. Y. Acad. Sci. 766, 442–458. doi: 10.1111/j.1749-6632.1995.tb26693.x
Barker, P. A. (2004). p75NTR is positively promiscuous: novel partners and new insights. Neuron 42, 529–533. doi: 10.1016/j.neuron.2004.04.001
Barker, P. A., and Murphy, R. A. (1992). The nerve growth factor receptor: a multicomponent system that mediates the actions of the neurotrophin family of proteins. Mol. Cell. Biochem. 110, 1–15. doi: 10.1007/BF02385000
Bartlett, S. E., Reynolds, A. J., and Hendry, I. A. (1998). Retrograde axonal transport of neurotrophins: differences between neuronal populations and implications for motor neuron disease. Immunol. Cell Biol. 76, 419–423. doi: 10.1046/j.1440-1711.1998.00767.x
Bartus, R. T., Dean, R. L. III, Beer, B., and Lippa, A. S. (1982). The cholinergic hypothesis of geriatric memory dysfunction. Science 217, 408–414. doi: 10.1126/science.7046051
Baxter, M. G., and Chiba, A. A. (1999). Cognitive functions of the basal forebrain. Curr. Opin. Neurobiol. 9, 178–183. doi: 10.1016/S0959-4388(99)80024-5
Beattie, M. S., Harrington, A. W., Lee, R., Kim, J. Y., Boyce, S. L., Longo, F. M., et al. (2002). ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron 36, 375–386. doi: 10.1016/S0896-6273(02)01005-X
Belrose, J. C., Masoudi, R., Michalski, B., and Fahnestock, M. (2014). Increased pro-nerve growth factor and decreased brain-derived neurotrophic factor in non-Alzheimer’s disease tauopathies. Neurobiol. Aging 35, 926–933. doi: 10.1016/j.neurobiolaging.2013.08.029
Berger, E. A., and Shooter, E. M. (1977). Evidence for pro-beta-nerve growth factor, a biosynthetic precursor to beta-nerve growth factor. Proc. Natl. Acad. Sci. U.S.A. 74, 3647-3651. doi: 10.1073/pnas.74.9.3647
Bhakar, A. L., Howell, J. L., Paul, C. E., Salehi, A. H., Becker, E. B., Said, F., et al. (2003). Apoptosis induced by p75NTR overexpression requires Jun kinase-dependent phosphorylation of Bad. J. Neurosci. 23, 11373–11381. doi: 10.1523/JNEUROSCI.23-36-11373.2003
Bierer, L. M., Haroutunian, V., Gabriel, S., Knott, P. J., Carlin, L. S., Purohit, D. P., et al. (1995). Neurochemical correlates of dementia severity in Alzheimer’s disease: relative importance of the cholinergic deficits. J. Neurochem. 64, 749–760. doi: 10.1046/j.1471-4159.1995.64020749.x
Bierl, M. A., Jones, E. E., Crutcher, K. A., and Isaacson, L. G. (2005). ’Mature’ nerve growth factor is a minor species in most peripheral tissues. Neurosci. Lett. 380, 133–137. doi: 10.1016/j.neulet.2005.01.029
Boutilier, J., Ceni, C., Pagdala, P. C., Forgie, A., Neet, K. E., and Barker, P. A. (2008). Proneurotrophins require endocytosis and intracellular proteolysis to induce TrkA activation. J. Biol. Chem. 283, 12709–12716. doi: 10.1074/jbc.M710018200
Bresnahan, P. A., Leduc, R., Thomas, L., Thorner, J., Gibson, H. L., Brake, A. J., et al. (1990). Human fur gene encodes a yeast KEX2-like endoprotease that cleaves pro-ß-NGF in vivo. J. Cell Biol. 111, 2851–2859. doi: 10.1083/jcb.111.6.2851
Bruno, M. A., and Cuello, A. C. (2006). Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc. Natl. Acad. Sci. U.S.A. 103, 6735–6740. doi: 10.1073/pnas.0510645103
Bruno, M. A., Leon, W. C., Fragoso, G., Mushynski, W. E., Almazan, G., and Cuello, A. C. (2009a). Amyloid beta-induced nerve growth factor dysmetabolism in Alzheimer disease. J. Neuropathol. Exp. Neurol. 68, 857–869. doi: 10.1097/NEN.0b013e3181aed9e6
Bruno, M. A., Mufson, E. J., Wuu, J., and Cuello, A. C. (2009b). Increased matrix metalloproteinase 9 activity in mild cognitive impairment. J. Neuropathol. Exp. Neurol. 68, 1309–1318. doi: 10.1097/NEN.0b013e3181c22569
Buttigieg, H., Kawaja, M. J., and Fahnestock, M. (2007). Neurotrophic activity of proNGF in vivo. Exp. Neurol. 204, 832–835. doi: 10.1016/j.expneurol.2006.12.011
Calissano, P., Amadoro, G., Matrone, C., Ciafrè, S., Marolda, R., Corsetti, V., et al. (2010). Does the term “trophic” actually mean antiamyloidogenic? The case of NGF. Cell Death Differ. 17, 1126–1133. doi: 10.1038/cdd.2010.38
Campenot, R. B., and MacInnis, B. L. (2003). Retrograde transport of neurotrophins: fact and function. J. Neurobiol. 58, 217–229. doi: 10.1002/neu.10322
Chen, Y., Dicou, E., and Djakiew, D. (1997). Characterization of nerve growth factor precursor expression in rat spermatids and the trophic effects of nerve growth factor in the maintenance of Sertoli cell viability. Mol. Cell. Endocrinol. 127, 129–136. doi: 10.1016/S0303-7207(96)04001-4
Clewes, O., Fahey, M. S., Tyler, S. J., Watson, J. J., Seok, H., Catania, C., et al. (2008). Human ProNGF: biological effects and binding profiles at TrkA, P75NTR and sortilin. J. Neurochem. 107, 1124–1135. doi: 10.1111/j.1471-4159.2008.05698.x
Coleman, P. D., and Flood, D. G. (1987). Neuron numbers and dendritic extent in normal aging and Alzheimer’s disease. Neurobiol. Aging 8, 521–545. doi: 10.1016/0197-4580(87)90127-8
Cooper, J. D., Lindholm, D., and Sofroniew, M. V. (1994). Reduced transport of [125I]nerve growth factor by cholinergic neurons and down-regulated TrkA expression in the medial septum of aged rats. Neuroscience 62, 625–629. doi: 10.1016/0306-4522(94)90462-6
Cooper, J. D., Salehi, A., Delcroix, J. D., Howe, C. L., Belichenko, P. V., Chua-Couzens, J., et al. (2001). Failed retrograde transport of NGF in a mouse model of Down’s syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proc. Natl. Acad. Sci. U.S.A. 98, 10439–10444. doi: 10.1073/pnas.181219298
Counts, S. E., Nadeem, M., Wuu, J., Ginsberg, S. D., Saragovi, H. U., and Mufson, E. J. (2004). Reduction of cortical TrkA but not p75(NTR) protein in early-stage Alzheimer’s disease. Ann. Neurol. 56, 520–531. doi: 10.1002/ana.20233
Coyle, J. T., Price, D. L., and DeLong, M. R. (1983). Alzheimer’s disease: a disorder of cortical cholinergic innervations. Science 219, 1184–1190. doi: 10.1126/science.6338589
Crutcher, K. A., Scott, S. A., Liang, S., Everson, W. V., and Weingartner, J. (1993). Detection of NGF-like activity in human brain tissue: increased levels in Alzheimer’s disease. J. Neurosci. 13, 2540–2550. doi: 10.1523/JNEUROSCI.13-06-02540.1993
Cuello, A. C., and Bruno, M. A. (2007). The failure in NGF maturation, and its increased degradation as the probable cause for the vulnerability of cholinergic neurons in Alzheimer’s disease. Neurochem. Res. 32, 1041–1045. doi: 10.1007/s11064-006-9270-0
Curtis, R., Adryan, K. M., Stark, J. L., Park, J., Compton, D. L., Weskamp, G., et al. (1995). Differential role of the low affinity neurotrophin receptor (p75) in retrograde transport of the neurotrophins. Neuron 14, 1201–1211. doi: 10.1016/0896-6273(95)90267-8
De Nadai, T., Marchetti, L., Di Rienzo, C., Calvello, M., Signore, G., Di Matteo, P., et al. (2016). Precursor and mature NGF live tracking: one versus many at a time in the axons. Sci. Rep. 6:20272. doi: 10.1038/srep20272
Dechant, G., and Barde, Y.-A. (2002). The neurotrophin receptor p75(NTR): novel functions and implications for diseases of the nervous system. Nat. Neurosci. 5, 1131–1136. doi: 10.1038/nn1102-1131
Delcroix, J. D., Valletta, J. S., Wu, C., Hunt, S. J., Kowal, A. S., and Mobley, W. C. (2003). NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron 39, 69–84. doi: 10.1016/S0896-6273(03)00397-0
Delsite, R., and Djakiew, D. (1999). Characterization of nerve growth factor precursor protein expression by human prostate stromal cells: a role in selective neurotrophin stimulation of prostate epithelial cell growth. Prostate 41, 39-48. doi: 10.1002/(SICI)1097-0045(19990915)41:1<39::AID-PROS6>3.0.CO;2-E
Di Matteo, P., Calvello, M., Luin, S., Marchetti, L., and Cattaneo, A. (2017). An optimized procedure for the site-directed labeling of NGF and proNGF for imaging purposes. Front. Mol. Biosci. 4:4. doi: 10.3389/fmolb.2017.00004
Dicou, E. (1992). Nerve growth factor precursors in the rat thyroid and hippocampus. Mol. Brain Res. 14, 136-138. doi: 10.1016/0169-328X(92)90021-3
DiStefano, P. S., Friedman, B., Radziejewski, C., Alexander, C., Boland, P., Schick, C. M., et al. (1992). The neurotrophins BDNF, NT-3, and NGF display distinct patterns of retrograde axonal transport in peripheral and central neurons. Neuron 8, 983–993. doi: 10.1016/0896-6273(92)90213-W
Dobrowsky, R. T., Werner, M. H., Castellino, A. M., Chao, M. V., and Hannun, Y. A. (1994). Activation of the sphingomyelin cycle through the low-affinity neurotrophin receptor. Science 265, 1596–1599. doi: 10.1126/science.8079174
Duman, R. S., and Monteggia, L. M. (2006). A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 59, 1116–1127. doi: 10.1016/j.biopsych.2006.02.013
Edwards, R. H., Selby, M. J., Garcia, P. D., and Rutter, W. J. (1988a). Processing of the native nerve growth factor precursor to form biologically active nerve growth factor. J. Biol. Chem. 263, 6810-6815.
Edwards, R. H., Selby, M. J., Mobley, W. C., Weinrich, S. L., Hruby, D. E., and Rutter, W. J. (1988b). Processing and secretion of nerve growth factor: expression in mammalian cells with a Vaccinia virus vector. Mol. Cell. Biol. 8, 2456–2464. doi: 10.1128/MCB.8.6.2456
Edwards, R. H., Selby, M. J., and Rutter, W. J. (1986). Differential RNA splicing predicts two distinct nerve growth factor precursors. Nature 319, 784–787. doi: 10.1038/319784a0
Epa, W. R., Markovska, K., and Barrett, G. L. (2004). The p75 neurotrophin receptor enhances TrkA signalling by binding to Shc and augmenting its phosphorylation. J. Neurochem. 89, 344–353. doi: 10.1111/j.1471-4159.2004.02344.x
Eveleth, D. D., and Bradshaw, R. A. (1992). Nerve growth factor non-responsive pheochromocytoma cells: altered internalization results in signaling dysfunction. J. Cell Biol. 117, 291–299. doi: 10.1083/jcb.117.2.291
Fahnestock, M. (1991). Structure and biosynthesis of nerve growth factor. Curr. Top. Microbiol. Immunol. 165, 1–26. doi: 10.1007/978-3-642-75747-1_1
Fahnestock, M. (2011). BDNF: the link between beta-amyloid and memory loss. Future Neurol. 6, 627–639. doi: 10.2217/fnl.11.44
Fahnestock, M., Garzon, D., Holsinger, R. M., and Michalski, B. (2002). Neurotrophic factors and Alzheimer’s disease: are we focusing on the wrong molecule? J. Neural Transm. Suppl. 62, 241–252. doi: 10.1007/978-3-7091-6139-5_22
Fahnestock, M., Michalski, B., Xu, B., and Coughlin, M. D. (2001). The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Mol. Cell. Neurosci. 18, 210–220. doi: 10.1006/mcne.2001.1016
Fahnestock, M., Scott, S. A., Jetté, N., Weingartner, J. A., and Crutcher, K. A. (1996). Nerve growth factor mRNA and protein levels measured in the same tissue from normal and Alzheimer’s disease parietal cortex. Brain Res. Mol. Brain Res. 42, 175–178. doi: 10.1016/S0169-328X(96)00193-3
Fahnestock, M., Yu, G., and Coughlin, M. D. (2004a). ProNGF: a neurotrophic or an apoptotic molecule? Prog. Brain Res. 146, 101–110.
Fahnestock, M., Yu, G., Michalski, B., Mathew, S., Colquhoun, A., Ross, G. M., et al. (2004b). The nerve growth factor precursor proNGF exhibits neurotrophic activity but is less active than mature nerve growth factor. J. Neurochem. 89, 581–592.
Foehr, E. D., Lin, X., O’Mahony, A., Geleziunas, R., Bradshaw, R. A., and Greene, W. C. (2000). NF-kappa B signaling promotes both cell survival and neurite process formation in nerve growth factor-stimulated PC12 cells. J. Neurosci. 20, 7556–7563. doi: 10.1523/JNEUROSCI.20-20-07556.2000
Friedman, W. J., Ibáñez, C. F., Hallböök, F., Persson, H., Cain, L. D., Dreyfus, C. F., et al. (1993). Differential actions of neurotrophins in the locus coeruleus and basal forebrain. Exp. Neurol. 119, 72–78. doi: 10.1006/exnr.1993.1007
Ginn, S. R., and Peterson, G. M. (1992). Studies related to the use of colchicine as a neurotoxin in the septohippocampal cholinergic system. Brain Res. 590, 144–152. doi: 10.1016/0006-8993(92)91090-2
Ginsberg, S. D., Che, S., Wuu, J., Counts, S. E., and Mufson, E. J. (2006). Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J. Neurochem. 97, 475–487. doi: 10.1111/j.1471-4159.2006.03764.x
Greene, L. A., Shooter, E. M., and Varon, S. (1968). Enzymatic activities of mouse nerve growth factor and its subunits. Proc. Natl. Acad. Sci. U.S.A. 60, 1383-1388. doi: 10.1073/pnas.60.4.1383
Grimes, M. L., Zhou, J., Beattie, E. C., Yuen, E. C., Hall, D. E., Valletta, J. S., et al. (1996). Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signaling endosomes. J. Neurosci. 16, 7950–7964. doi: 10.1523/JNEUROSCI.16-24-07950.1996
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Harrington, A. W., Leiner, B., Blechschmitt, C., Arevalo, J. C., Lee, R., Morl, K., et al. (2004). Secreted proNGF is a pathophysiological death-inducing ligand after adult CNS injury. Proc. Natl. Acad. Sci. U.S.A. 101, 6226–6230. doi: 10.1073/pnas.0305755101
Hartikka, J., and Hefti, F. (1988). Development of septal cholinergic neurons in culture: plating density and glial cells modulate effect of NGF on survival, fiber growth, and expression of transmitter-specific enzymes. J. Neurosci. 8, 2967–2985. doi: 10.1523/JNEUROSCI.08-08-02967.1988
Hatanaka, H., Tsukui, H., and Nihonmatsu, I. (1988). Developmental change in the nerve growth factor action from induction of choline acetyltransferase to promotion of cell survival in cultured basal forebrain cholinergic neurons from postnatal rats. Dev. Brain Res. 39, 85–95. doi: 10.1016/0165-3806(88)90069-7
Hefti, F. (1986). Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J. Neurosci. 6, 2155–2162. doi: 10.1523/JNEUROSCI.06-08-02155.1986
Hefti, F. (1994). Neurotrophic factor therapy for nervous system degenerative diseases. J. Neurobiol. 25, 1418-1435.
Hefti, F., Hartikka, J., and Knusel, B. (1989). Function of neurotrophic factors in the adult and aging brain and their possible use in the treatment of neurodegenerative diseases. Neurobiol. Aging 10, 515–533. doi: 10.1016/0197-4580(89)90118-8
Hefti, F., and Weiner, W. J. (1986). Nerve growth factor and Alzheimer’s disease. Ann. Neurol. 20, 275–281. doi: 10.1002/ana.410200302
Holtzman, D. M., Li, Y., Parada, L. F., Kinsman, S., Chen, C. K., Valletta, J. S., et al. (1992). p140trk mRNA marks NGF-responsive forebrain neurons: evidence that trk gene expression is induced by NGF. Neuron 9, 465–478. doi: 10.1016/0896-6273(92)90184-F
Huang, E. J., and Reichardt, L. F. (2003). Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642. doi: 10.1146/annurev.biochem.72.121801.161629
Hughes, A. L., Messineo-Jones, D., Lad, S. P., and Neet, K. E. (2001). Distinction between differentiation, cell cycle, and apoptosis signals in PC12 cells by the nerve growth factor mutant delta9/13, which is selective for the p75 neurotrophin receptor. J. Neurosci. Res. 63, 10–19. doi: 10.1002/1097-4547(20010101)63:1<10::AID-JNR2>3.0.CO;2-R
Hyman, B. T., Van Hoesen, G. W., Damasio, A. R., and Barnes, C. L. (1984). Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science 225, 1168–1170. doi: 10.1126/science.6474172
Ioannou, M. S., and Fahnestock, M. (2017). ProNGF, but not NGF, switches from neurotrophic to apoptotic activity in response to reductions in TrkA receptor levels. Int. J. Mol. Sci. 18:E599. doi: 10.3390/ijms18030599
Ip, N. Y., Ibanez, C. F., Nye, S. H., McClain, J., Jones, P. F., Gies, D. R., et al. (1992). Mammalian neurotrophin-4: structure, chromosomal localization, tissue distribution, and receptor specificity. Proc. Natl. Acad. Sci. U.S.A. 89, 3060-3064. doi: 10.1073/pnas.89.7.3060
Iqbal, K., and Grundke-Iqbal, I. (2008). Alzheimer neurofibrillary degeneration: significance, etiopathogenesis, therapeutics and prevention. J. Cell. Mol. Med. 12, 38–55. doi: 10.1111/j.1582-4934.2008.00225.x
Ittner, L. M., and Götz, J. (2011). Amyloid-β, and tau–a toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 12, 65–72. doi: 10.1038/nrn2967
Iulita, M. F., Do Carmo, S., Ower, A. K., Fortress, A. M., Flores Aguilar, L., Hanna, M., et al. (2014). Nerve growth factor metabolic dysfunction in Down’s syndrome brains. Brain 137(Pt 3), 860–872. doi: 10.1093/brain/awt372
Jetté, N., Cole, M. S., and Fahnestock, M. (1994). NGF mRNA is not decreased in frontal cortex from Alzheimer’s disease patients. Brain Res. Mol. Brain Res. 25, 242–250. doi: 10.1016/0169-328X(94)90159-7
Jongstra-Bilen, J., Coblentz, L., and Shooter, E. M. (1989). The in vitro processing of the NGF precursors by the gamma-subunit of the 7S NGF complex. Mol. Brain Res. 5, 159-169.
Kaplan, D. R., and Miller, F. D. (2000). Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 10, 381–391. doi: 10.1016/S0959-4388(00)00092-1
Kieran, D., Hafezparast, M., Bohnert, S., Dick, J. R., Martin, J., Schiavo, G., et al. (2005). A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J. Cell Biol. 169, 561–567. doi: 10.1083/jcb.200501085
Klein, R. L., Hirko, A. C., Meyers, C. A., Grimes, J. R., Muzyczka, N., and Meyer, E. M. (2000). NGF gene transfer to intrinsic basal forebrain neurons increases cholinergic cell size and protects from age-related, spatial memory deficits in middle-aged rats. Brain Res. 875, 144–151. doi: 10.1016/S0006-8993(00)02634-2
Kohn, J., Aloyz, R. S., Toma, J. G., Haak-Frendscho, M., and Miller, F. D. (1999). Functionally antagonistic interactions between the TrkA and p75 neurotrophin receptors regulate sympathetic neuron growth and target innervation. J. Neurosci. 19, 5393–5408. doi: 10.1523/JNEUROSCI.19-13-05393.1999
Kojima, M., Ikeuchi, T., and Hatanaka, H. (1994). Nerve growth factor induces trkA mRNA expression in cultured basal forebrain cholinergic neurons from 17-day fetal rats. Neurosci. Lett. 169, 47–50. doi: 10.1016/0304-3940(94)90353-0
Kojima, M., Ikeuchi, T., and Hatanaka, H. (1995). Role of nerve growth factor in the expression of trkA mRNA in cultured embryonic rat basal forebrain cholinergic neurons. J. Neurosci. Res. 42, 775–783. doi: 10.1002/jnr.490420606
Koliatsos, V. E., Applegate, M. D., Knusel, B., Junard, E. O., Burton, L. E., Mobley, W. C., et al. (1991). Recombinant human nerve growth factor prevents retrograde degeneration of axotomized basal forebrain cholinergic neurons in the rat. Exp. Neurol. 112, 161-173. doi: 10.1016/0014-4886(91)90066-L
Koliatsos, V. E., Price, D. L., Gouras, G. K., Cayouette, M. H., Burton, L. E., and Winslow, J. W. (1994). Highly selective effects of nerve growth factor, brain-derived neurotrophic factor, and neurotrophin-3 on intact and injured basal forebrain magnocellular neurons. J. Comp. Neurol. 343, 247–262. doi: 10.1002/cne.903430206
Korsching, S., Heumann, R., Thoenen, H., and Hefti, E. (1986). Cholinergic denervation of the rat hippocampus by fimbrial transection leads to a transient accumulation of nerve growth factor (NGF) without change in mRNA content. Neurosci. Lett. 66, 175–180. doi: 10.1016/0304-3940(86)90186-2
Ladiwala, U., Lachance, C., Simoneau, S., Bhakar, A., Barker, P. A., and Antel, J. P. (1998). p75 neurotrophin receptor expression on adult human oligodendrocytes: signaling without cell death in response to NGF. J. Neurosci. 18, 1297–1304. doi: 10.1523/JNEUROSCI.18-04-01297.1998
Lambiase, A., Manni, L., Rama, P., and Bonini, S. (2003). Clinical application of nerve growth factor on human corneal ulcer. Arch. Ital. Biol. 141, 141–148.
Lapchak, P. A., Araujo, D. M., Carswell, S., and Hefti, F. (1993). Distribution of [125I]nerve growth factor in the rat brain following a single intraventricular injection: correlation with the topographical distribution of trkA messenger RNA-expressing cells. Neuroscience 54, 445–460. doi: 10.1016/0306-4522(93)90265-H
Lapchak, P. A., and Hefti, F. (1991). Effect of recombinant human nerve growth factor on presynaptic cholinergic function in rat hippocampal slices following partial septohippocampal lesions: measures of [3H]acetylcholine synthesis, [3H]acetylcholine release and choline acetyltransferase activity. Neuroscience 42, 639–649. doi: 10.1016/0306-4522(91)90033-K
Lee, R., Kermani, P., Teng, K. K., and Hempstead, B. L. (2001). Regulation of cell survival by secreted proneurotrophins. Science 294, 1945-1948. doi: 10.1126/science.1065057
Levi-Montalcini, R. (1987). The nerve growth factor 35 years later. Science 4, 1154-1162. doi: 10.1126/science.3306916
Levi-Montalcini, R., and Angeletti, P. U. (1968). Nerve growth factor. Physiol. Rev. 48, 534-569. doi: 10.1152/physrev.1968.48.3.534
Li, Y., Holtzman, D. M., Kromer, L. F., Kaplan, D. R., Chua-Couzens, J., Clary, D. O., et al. (1995). Regulation of TrkA and ChAT expression in developing rat basal forebrain: evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. J. Neurosci. 15, 2888–2905. doi: 10.1523/JNEUROSCI.15-04-02888.1995
Lindsay, R. M., Wiegand, S., Altar, C. A., and DiStefano, P. (1994). Neurotrophic factors: from molecule to man. Trends Neurol. Sci. 17, 182–190. doi: 10.1016/0166-2236(94)90099-X
Loeb, D. M., and Greene, L. A. (1993). Transfection with trk restores “slow” NGF binding, efficient NGF uptake, and multiple NGF responses to NGF-non-responsive PC12 cell mutants. J. Neurosci. 13, 2919–2929. doi: 10.1523/JNEUROSCI.13-07-02919.1993
Lorenzi, M. V., Knusel, B., Hefti, F., and Strauss, W. L. (1992). Nerve growth factor regulation of choline acetyltransferase gene expression in rat embryo basal forebrain cultures. Neurosci. Lett. 140, 185–188. doi: 10.1016/0304-3940(92)90098-R
MacInnis, B. L., and Campenot, R. B. (2002). Retrograde support of neuronal survival without retrograde transport of nerve growth factor. Science 295, 1536–1539. doi: 10.1126/science.1064913
MacInnis, B. L., Senger, D. L., and Campenot, R. B. (2003). Spatial requirements for TrkA kinase activity in the support of neuronal survival and axon growth in rat sympathetic neurons. Neuropharmacology 45, 995–1010. doi: 10.1016/S0028-3908(03)00273-9
Maisonpierre, P. C., Belluscio, L., Squinto, S., Ip, N. Y., Furth, M. E., Lindsay, R. M., et al. (1990). Neurotrophin-3: a neurotrophic factor related to NGF and BDNF. Science 247, 1446-1451. doi: 10.1126/science.2321006
Masoudi, R., Ioannou, M., Coughlin, M. D., Pagadala, P., Neet, K. E., Clewes, O., et al. (2009). Biological activity of nerve growth factor precursor is dependent upon relative levels of its receptors. J. Biol. Chem. 284, 18424–18433. doi: 10.1074/jbc.M109.007104
Matrone, C., Ciotti, M. T., Mercanti, D., Marolda, R., and Calissano, P. (2008a). NGF and BDNF signaling control amyloidogenic route and a production in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 13139–13144. doi: 10.1073/pnas.0806133105
Matrone, C., Di Luzio, A., Meli, G., D’Aguanno, S., Severini, C., Ciotti, M. T., et al. (2008b). Activation of the amyloidogenic route by NGF deprivation induces apoptotic death in PC12 cells. J. Alzheimers Dis. 13, 81–96.
Miller, F. D., and Kaplan, D. R. (2001). Neurotrophin signalling pathways regulating neuronal apoptosis. Cell. Mol. Life Sci. 58, 1045–1053. doi: 10.1007/PL00000919
Mount, H. T., Dreyfus, C. F., and Black, I. B. (1994). Neurotrophin-3 selectively increases cultured Purkinje cell survival. Neuroreport 5, 2497-2500. doi: 10.1097/00001756-199412000-00023
Mowla, S. J., Pareek, S., Farhadi, H. F., Petrecca, K., Fawcett, J. P., Seidah, N. G., et al. (1999). Differential sorting of nerve growth factor and brain-derived neurotrophic factor in hippocampal neurons. J. Neurosci. 19, 2069-2080.
Mufson, E. J., Counts, S. E., Fahnestock, M., and Ginsberg, S. D. (2007). Cholinotrophic molecular substrates of mild cognitive impairment in the elderly. Curr. Alzheimer Res. 4, 340–350. doi: 10.2174/156720507781788855
Mufson, E. J., Counts, S. E., Peng, S., and Fahnestock, M. (2005). “Shift in the balance of TrkA and pro-NGF in prodromal Alzheimer’s disease,” in Alzheimer’s and Parkinson’s Diseases: Insights, Progress and Perspectives, eds I. Hanin, M. Memo, F. Stocchi, and A. Fischer (Bologna: Medimond), 203–209.
Mufson, E. J., Lavine, N., Jaffar, S., Kordower, J. H., Quirion, R., and Saragovi, H. U. (1997). Reduction in p140-TrkA receptor protein within the nucleus basalis and cortex in Alzheimer’s disease. Exp. Neurol. 146, 91–103. doi: 10.1006/exnr.1997.6504
Mufson, E. J., Li, J., Sobreviela, T., and Kordower, J. H. (1996). Decreased trkA gene expression within basal forebrain neurons in Alzheimer’s disease. Neuroreport 8, 25–29. doi: 10.1097/00001756-199612200-00006
Narisawa-Saito, M., Wakabayashi, K., Tsuji, S., Takahashi, H., and Nawa, H. (1996). Regional specificity of alterations in NGF, BDNF and NT-3 levels in Alzheimer’s disease. Neuroreport 7, 2925–2928. doi: 10.1097/00001756-199611250-00024
Nykjaer, A., Lee, R., Teng, K. K., Jansen, P., Madsen, P., Nielsen, M. S., et al. (2004). Sortilin is essential for proNGF-induced neuronal cell death. Nature 427, 843–848. doi: 10.1038/nature02319
Oosawa, H., Fujii, T., and Kawashima, K. (1999). Nerve growth factor increases the synthesis and release of acetylcholine and the expression of vesicular acetylcholine transporter in primary cultured rat embryonic septal cells. J. Neurosci. Res. 57, 381–387. doi: 10.1002/(SICI)1097-4547(19990801)57:3<381::AID-JNR10>3.0.CO;2-C
Parikh, V., Howe, W. M., Welchko, R. M., Naughton, S. X., D’Amore, D. E., Han, D. H., et al. (2013). Diminished trkA receptor signaling reveals cholinergic-attentional vulnerability of aging. Eur. J. Neurosci. 37, 278–293. doi: 10.1111/ejn.12090
Peng, S., Wuu, J., Mufson, E. J., and Fahnestock, M. (2004). Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J. Neuropathol. Exp. Neurol. 63, 641–649. doi: 10.1093/jnen/63.6.641
Peng, S., Wuu, J., Mufson, E. J., and Fahnestock, M. (2005). Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 93, 1412–1421. doi: 10.1111/j.1471-4159.2005.03135.x
Perez, S. E., He, B., Muhammad, N., Oh, K. J., Fahnestock, M., Ikonomovic, M. D., et al. (2011). Cholinotrophic basal forebrain system alterations in 3xTg-AD transgenic mice. Neurobiol. Dis. 41, 338–352. doi: 10.1016/j.nbd.2010.10.002
Perlson, E., Maday, S., Fu, M. M., Moughamian, A. J., and Holzbaur, E. L. F. (2010). Retrograde axonal transport: pathways to cell death? Trends Neurosci. 33, 335–344. doi: 10.1016/j.tins.2010.03.006
Petrie, C. N., Smithson, L. J., Crotty, A. M., Michalski, B., Fahnestock, M., and Kawaja, M. D. (2013). Overexpression of nerve growth factor by murine smooth muscle cells: role of the p75 neurotrophin receptor on sympathetic and sensory sprouting. J. Comp. Neurol. 521, 2621–2643. doi: 10.1002/cne.23302
Pongrac, J. L., and Rylett, R. J. (1996). Differential effects of nerve growth factor on expression of choline acetyltransferase and sodium-coupled choline transport in basal forebrain cholinergic neurons in culture. J. Neurochem. 66, 804–810. doi: 10.1046/j.1471-4159.1996.66020804.x
Price, D. L. (1986). New perspectives on Alzheimer’s disease. Annu. Rev. Neurosci. 9, 489–512. doi: 10.1146/annurev.ne.09.030186.002421
Provenzano, M. J., Xu, N., Ver Meer, M. R., Clark, J. J., and Hansen, M. R. (2008). p75NTR and sortilin increase after facial nerve injury. Laryngoscope 118, 87–93. doi: 10.1097/MLG.0b013e31814b8d9f
Ramser, E. M., Gan, K. J., Decker, H., Fan, E. Y., Suzuki, M. M., Ferreira, S. T., et al. (2013). Amyloid-β oligomers induce tau-independent disruption of BDNF axonal transport via calcineurin activation in cultured hippocampal neurons. Mol. Biol. Cell 24, 2494–2505. doi: 10.1091/mbc.E12-12-0858
Randolph, C. L., Bierl, M. A., and Isaacson, L. G. (2007). Regulation of NGF, and NT-3 protein expression in peripheral targets by sympathetic input. Brain Res. 1144, 59–69. doi: 10.1016/j.brainres.2007.01.099
Rattenholl, A., Lilie, H., Grossmann, A., Stern, A., Schwarz, E., and Rudolph, R. (2001). The pro-sequence facilitates folding of human nerve growth factor from Escherichia coli inclusion bodies. Eur. J. Biochem. 268, 3296-3303.
Reinshagen, M., Geerling, I., Eysselein, V. E., Adler, G., Huff, K. R., Moore, G. P., et al. (2000). Commercial recombinant human beta-nerve growth factor and adult rat dorsal root ganglia contain an identical molecular species of nerve growth factor prohormone. J. Neurochem. 74, 2127-2133.
Reynolds, A. J., Bartlett, S. E., and Hendry, I. A. (1998). Signalling events regulating the retrograde axonal transport of 125I-beta nerve growth factor in vivo. Brain Res. 798, 67–74. doi: 10.1016/S0006-8993(98)00396-5
Rice, F. L., Albers, K. M., Davis, B. M., Silos-Santiago, I., Wilkinson, G. A., LeMaster, A. M., et al. (1998). Differential dependency of unmyelinated and A delta epidermal and upper dermal innervation on neurotrophins, trk receptors, and p75LNGFR. Dev. Biol. 198, 57-81.
Roux, P. P., and Barker, P. A. (2002). Neurotrophin signaling through the p75 neurotrophin receptor. Prog. Neurobiol. 67, 203–233. doi: 10.1016/S0301-0082(02)00016-3
Roux, P. P., Bhakar, A. L., Kennedy, T. E., and Barker, P. A. (2001). The p75 neurotrophin receptor activates Akt (protein kinase B) through a phosphatidylinositol 3-kinase-dependent pathway. J. Biol. Chem. 276, 23097–23104. doi: 10.1074/jbc.M011520200
Ruit, K. G., Elliott, J. L., Osborne, P. A., Yan, Q., and Snider, W. D. (1992). Selective dependence of mammalian dorsal root ganglion neurons on nerve growth factor during embryonic development. Neuron 8, 573-587. doi: 10.1016/0896-6273(92)90284-K
Rylett, R. J., Goddard, S., Schmidt, B. M., and Williams, L. R. (1993). Acetylcholine synthesis and release following continuous intracerebral administration of NGF in adult and aged Fischer-344 rats. J. Neurosci. 13, 3956–3963. doi: 10.1523/JNEUROSCI.13-09-03956.1993
Saboori, A. M., and Young, M. (1986). Nerve growth factor: biosynthetic products of the mouse salivary glands. Characterization of stable high molecular weight and 32,000-dalton nerve growth factors. Biochemistry 25, 5565-5571.
Salehi, A., Delcroix, J. D., Belichenko, P. V., Zhan, K., Wu, C., Valletta, J. S., et al. (2006). Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 51, 29–42. doi: 10.1016/j.neuron.2006.05.022
Scheff, S. W., DeKosky, S. T., and Price, D. A. (1990). Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol. Aging 11, 29–37. doi: 10.1016/0197-4580(90)90059-9
Schmitz, T. W., and Nathan Spreng, R. (2016). Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat. Commun. 7:13249. doi: 10.1038/ncomms13249
Schober, A., Wolf, N., Huber, K., Hertel, R., Krieglstein, K., Minichiello, L., et al. (1998). TrkB and neurotrophin-4 are important for development and maintenance of sympathetic preganglionic neurons innervating the adrenal medulla. J. Neurosci. 18, 7272-7284.
Scott, S. A., Mufson, E. J., Weingartner, J. A., Skau, K. A., and Crutcher, K. A. (1995). Nerve growth factor in Alzheimer’s disease: increased levels throughout the brain coupled with declines in nucleus basalis. J. Neurosci. 15, 6213–6221. doi: 10.1523/JNEUROSCI.15-09-06213.1995
Scott-Solomon, E., and Kuruvilla, R. (2018). Mechanisms of neurotrophin trafficking via Trk receptors. Mol. Cell. Neurosci. 91, 25–33. doi: 10.1016/j.mcn.2018.03.013
Segal, R. A. (2003). Selectivity in neurotrophin signaling: theme and variations. Annu. Rev. Neurosci. 26, 299–330. doi: 10.1146/annurev.neuro.26.041002.131421
Seidah, N. G., Benjannet, S., Pareek, S., Chrétien, M., and Murphy, R. A. (1996a). Cellular processing of the neurotrophin precursors of NT3 and BDNF by the mammalian proprotein convertases. FEBS Lett. 379, 247–250.
Seidah, N. G., Benjannet, S., Pareek, S., Savaria, D., Hamelin, J., Goulet, B., et al. (1996b). Cellular processing of the nerve growth factor precursor by the mammalian pro-protein convertases. Biochem. J. 314, 951–960. doi: 10.1042/bj3140951
Seiler, M., and Schwab, M. E. (1984). Specific retrograde transport of nerve growth factor (NGF) from neocortex to nucleus basalis in the rat. Brain Res. 300, 33-39. doi: 10.1016/0006-8993(84)91338-6
Selby, M. J., Edwards, R. H., Sharp, F., and Rutter, W. J. (1987). Mouse nerve growth factor gene: structure and expression. Mol. Cell Biol. 7, 3057–3064. doi: 10.1128/MCB.7.9.3057
Suter, U., Heymach, J. V. Jr., and Shooter, E. M. (1991). Two conserved domains in the NGF propeptide are necessary and sufficient for the biosynthesis of correctly processed and biologically active NGF. EMBO J. 10, 2395-2400. doi: 10.1002/j.1460-2075.1991.tb07778.x
Takei, N., Kuramoto, H., Endo, Y., and Hatanaka, H. (1997). NGF and BDNF increase the immunoreactivity of vesicular acetylcholine transporter in cultured neurons from the embryonic rat septum. Neurosci. Lett. 226, 207–209. doi: 10.1016/S0304-3940(97)00284-X
Takei, N., Tsukui, H., and Hatanaka, H. (1989). Intracellular storage and evoked release of acetylcholine from postnatal rat basal forebrain cholinergic neurons in culture with nerve growth factor. J. Neurochem. 53, 1405–1410. doi: 10.1111/j.1471-4159.1989.tb08531.x
Thoenen, H., and Barde, Y.-A. (1980). Physiology of nerve growth factor. Physiol. Rev. 60, 1284-1335. doi: 10.1152/physrev.1980.60.4.1284
Tiernan, C. T., Combs, B., Cox, K., Morfini, G., Brady, S. T., Counts, S. E., et al. (2016). Pseudophosphorylation of tau at S422 enhances SDS-stable dimer formation and impairs both anterograde and retrograde fast axonal transport. Exp. Neurol. 283(Pt A), 318–329. doi: 10.1016/j.expneurol.2016.06.030
Troy, C. M., Friedman, J. E., and Friedman, W. J. (2002). Mechanisms of p75-mediated death of hippocampal neurons. Role of caspases. J. Biol. Chem. 277, 34295–34302. doi: 10.1074/jbc.M205167200
Volosin, M., Song, W., Almeida, R. D., Kaplan, D. R., Hempstead, B. L., and Friedman, W. J. (2006). Interaction of survival and death signaling in basal forebrain neurons: roles of neurotrophins and proneurotrophins. J. Neurosci. 26, 7756–7766. doi: 10.1523/JNEUROSCI.1560-06.2006
von Bartheld, C. S., Williams, R., Lefcort, F., Clary, D. O., Reichardt, L. F., and Bothwell, M. (1996). Retrograde transport of neurotrophins fm the eye to the brain in chick embryos—roles of the p75NTR and TrkB receptors. J. Neurosci. 16, 2995–3008. doi: 10.1523/JNEUROSCI.16-09-02995.1996
Vossel, K. A., Zhang, K., Brodbeck, J., Daub, A. C., Sharma, P., Finkbeiner, S., et al. (2010). Tau reduction prevents Abeta-induced defects in axonal transport. Science 330:198. doi: 10.1126/science.1194653
Watson, F. L., Heerssen, H. M., Moheban, D. B., Lin, M. Z., Sauvageot, C. M., Bhattacharyya, A., et al. (1999). Rapid nuclear responses to target-derived neurotrophins require retrograde transport of ligand-receptor complex. J. Neurosci. 19, 7889–7900. doi: 10.1523/JNEUROSCI.19-18-07889.1999
Whitehouse, P. J. (1998). The cholinergic deficit in Alzheimer’s disease. J. Clin. Psychiatry 59(Suppl. 13), 19-22.
Wilcock, G. K., Esiri, M. M., Bowen, D. M., and Smith, C. C. (1982). Alzheimer’s disease. Correlation of cortical choline acetyltransferase activity with the severity of dementia, and histological abnormalities. J. Neurol. Sci. 57, 407–417. doi: 10.1016/0022-510X(82)90045-4
Williams, L. R., and Rylett, R. J. (1990). Exogenous nerve growth factor increases the activity of high-affinity choline uptake and choline acetyltransferase in brain of Fisher 344 male rats. J. Neurochem. 55, 1042–1049. doi: 10.1111/j.1471-4159.1990.tb04594.x
Williams, L. R., Varon, S., Peterson, G. M., Wictorin, K., Fischer, W., Bjorklund, A., et al. (1986). Continuous infusion of nerve growth factor prevents basal forebrain neuronal death after fimbria fornix transection. Proc. Natl. Acad. Sci. U.S.A. 83, 9231–9235. doi: 10.1073/pnas.83.23.9231
Yardley, G., Relf, B., Lakshmanan, J., Reinshagen, M., and Moore, G. P. (2000). Expression of nerve growth factor mRNA and its translation products in the anagen hair follicle. Exp. Dermatol. 9, 283-289. doi: 10.1034/j.1600-0625.2000.009004283.x
Ye, H., Kuruvilla, R., Zweifel, L. S., and Ginty, D. D. (2003). Evidence in support of signaling endosome-based retrograde survival of sympathetic neurons. Neuron 39, 57–68. doi: 10.1016/S0896-6273(03)00266-6
Yeo, T. T., Chuacouzens, J., Butcher, L. L., Bredesen, D. E., Cooper, J. D., Valletta, J. S., et al. (1997). Absence of p75NTR causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. J. Neurosci. 17, 7594–7605. doi: 10.1523/JNEUROSCI.17-20-07594.1997
Yoon, S. O., Casaccia-Bonnefil, P., Carter, B., and Chao, M. V. (1998). Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J. Neurosci. 18, 3273–3281. doi: 10.1523/JNEUROSCI.18-09-03273.1998
Keywords: nerve growth factor, TrkA, p75NTR, neurotrophins, basal forebrain cholinergic neurons, survival, apoptosis, retrograde transport
Citation: Fahnestock M and Shekari A (2019) ProNGF and Neurodegeneration in Alzheimer’s Disease. Front. Neurosci. 13:129. doi: 10.3389/fnins.2019.00129
Received: 04 December 2018; Accepted: 05 February 2019;
Published: 22 February 2019.
Edited by:
Natalia N. Nalivaeva, University of Leeds, United KingdomReviewed by:
Elizabeth J. Coulson, The University of Queensland, AustraliaChristine Gall, University of California, Irvine, United States
Copyright © 2019 Fahnestock and Shekari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margaret Fahnestock, ZmFobmVzdEBtY21hc3Rlci5jYQ==