 Marco Tulio Núñez
Marco Tulio Núñez Cecilia Hidalgo
Cecilia Hidalgo- 1Iron and Neuroregeneration Laboratory, Department of Biology, Faculty of Sciences, Universidad de Chile, Santiago, Chile
- 2Calcium Signaling Laboratory, Biomedical Research Institute, CEMC, Physiology and Biophysics Program, Institute of Biomedical Sciences, Faculty of Medicine, Universidad de Chile, Santiago, Chile
- 3Department of Neuroscience, Faculty of Medicine, Universidad de Chile, Santiago, Chile
Iron and calcium share the common feature of being essential for normal neuronal function. Iron is required for mitochondrial function, synaptic plasticity, and the development of cognitive functions whereas cellular calcium signals mediate neurotransmitter exocytosis, axonal growth and synaptic plasticity, and control the expression of genes involved in learning and memory processes. Recent studies have revealed that cellular iron stimulates calcium signaling, leading to downstream activation of kinase cascades engaged in synaptic plasticity. The relationship between calcium and iron is Janus-faced, however. While under physiological conditions iron-mediated reactive oxygen species generation boosts normal calcium-dependent signaling pathways, excessive iron levels promote oxidative stress leading to the upsurge of unrestrained calcium signals that damage mitochondrial function, among other downstream targets. Similarly, increases in mitochondrial calcium to non-physiological levels result in mitochondrial dysfunction and a predicted loss of iron homeostasis. Hence, if uncontrolled, the iron/calcium self-feeding cycle becomes deleterious to neuronal function, leading eventually to neuronal death. Here, we review the multiple cell-damaging responses generated by the unregulated iron/calcium self-feeding cycle, such as excitotoxicity, free radical-mediated lipid peroxidation, and the oxidative modification of crucial components of iron and calcium homeostasis/signaling: the iron transporter DMT1, plasma membrane, and intracellular calcium channels and pumps. We discuss also how iron-induced dysregulation of mitochondrial calcium contributes to the generation of neurodegenerative conditions, including Alzheimer’s disease (AD) and Parkinson’s disease (PD).
Introduction
Iron and calcium ions are both essential for maintaining normal brain function. Iron is required for oxidative phosphorylation, Krebs cycle, iron–sulfur cluster and heme synthesis, synaptic plasticity, and the development of cognitive functions (Oexle et al., 1999; Bolton et al., 2000; Hidalgo and Núñez, 2007;Tong and Rouault, 2007; Zorov et al., 2007; Huang et al., 2011; Muñoz et al., 2011; Murray-Kolb, 2013; Mena et al., 2015; Requejo-Aguilar and Bolanos, 2016), while increases in cytoplasmic calcium concentration known as calcium signals mediate the secretion of neurotransmitters, synaptic plasticity, and axonal growth (Berridge, 1998). Moreover, nuclear calcium signals command the expression of neuronal genes involved in learning and memory processes (Bading, 2013).
Recent studies have revealed that cellular iron stimulates neuronal calcium signaling, leading to downstream activation of kinase cascades engaged in synaptic plasticity, a neuronal response associated to memory and learning (Hidalgo and Núñez, 2007; Hidalgo et al., 2007; Muñoz et al., 2011). The relationship between calcium and iron is Janus-faced, however. Dysregulation of iron levels induces calcium dyshomeostasis and abnormal calcium signaling, whereas increased calcium levels enhance redox-active iron levels; neurodegenerative conditions exhibit both types of dysregulation. Thus, current evidence indicates that increased intracellular calcium and aberrant calcium signaling occur in neurodegenerative disorders that include Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) (reviewed in Pchitskaya et al., 2018). Similarly, disruption of neuronal iron homeostasis linked to iron accumulation is a common feature in a plethora of neurodegenerative diseases, which included and other parkinsonisms (Lee et al., 2013), AD (Bulk et al., 2018), Friedrich’s ataxia, pantothenate kinase-associated neurodegeneration (Swaiman, 1991), and other neuronal pathologies that entail brain iron accumulation (Wiethoff and Houlden, 2017).
Here, we will review the evidence that points to a rarely acknowledged relationship between iron homeostasis and calcium homeostasis and signaling, in which dysregulation of one of these processes results in dysregulation of the other, generating a vicious cycle that ends up in neuronal death. The multiple cell-damaging responses generated by the unregulated iron/calcium self-feeding cycle, such as excitotoxicity, free radical-mediated lipid peroxidation, and the oxidative modification of crucial components of iron and calcium homeostasis and signaling, including the iron transporter DMT1, plasma membrane and intracellular calcium channels and pumps will be presented. This review article concludes with a discussion of how dysregulation of calcium homeostasis and signaling contributes to the generation of neurodegenerative diseases with an iron accumulation component, focusing in particular on AD and PD.
Iron Homeostasis in Neuronal Cells
Through the Fenton reaction, redox-active iron is a net producer of the hydroxyl radical, the most reactive radical species in nature (Davies, 2005). In a reductive environment, such as the intracellular milieu, through redox cycling redox-active iron promotes the production of hydroxyl radicals at the expense of O2 and GSH consumption (Núñez et al., 2012). Consequently, under physiological conditions, redox-active iron levels that conform the labile iron pool are very tightly regulated to a cytosolic concentration of 0.5–1.5 μM, which comprises <5% of total intracellular iron (Cabantchik, 2014). In mammals, cellular iron levels are maintained by transcriptional, translational, and vesicular flux mechanisms (Núñez, 2010). The iron regulatory protein (IRPs: IRP1 and IRP2)/iron-responsive element (IRE) is the best-characterized system that regulates at the translation level the expression of iron homeostasis proteins. These proteins include the transferrin receptor 1 (TfR1, involved in iron uptake), the divalent metal transporter 1 (DMT1, involved in iron uptake), ferroportin 1 (FPN1), involved in iron efflux from cells), and ferritin (involved in iron storage) (Wilkinson and Pantopoulos, 2014). The IRP/IRE is a plentiful-oriented system, since it is activated under low iron conditions to ensure an adequate supply of iron for cellular processes. Of note, the IRE–IRP regulatory system is not only regulated by cellular iron status but it is also regulated by reactive oxygen species (ROS), whereby cells elicit a defense mechanism against iron toxicity and iron-catalyzed oxidative stress (Ray et al., 2012).
The iron uptake protein DMT1 (SLC11A2) is a crucial component of cell iron homeostasis. SLC11A2 transcription generates four alternatively spliced mRNAs that differ at their 5′-untranslated region (coding for the DMT1 isoforms 1A and 1B) and at its 3′-untranslated region (coding for isoforms +IRE and –IRE) (Garrick et al., 2006). Thus, the expression of the 1A and 1B isoforms of DMT1 is subjected to differential transcriptional regulation.
The Crucial Relationship Between Iron and the Hypoxia-Inducible Transcription Factor (HIF)
At the systemic level, the hypoxia-inducible transcription factor (HIF) transcription factor family coordinates the cellular response to low oxygen levels by regulating the expression of a large array of target genes during hypoxia, which results in adaptive changes in the hematopoietic, cardiovascular, and respiratory systems (Smith et al., 2008; Mole et al., 2009). The HIF-1 is kept at basal levels by HIF prolyl hydroxylase domain (PHD) enzymes; prolyl-hydroxylation of HIF-1 via PHD signals for its degradation via the ubiquitin-proteasome system (Bruick and McKnight, 2001; Myllyharju, 2013; Yeh et al., 2017). The PHD enzymes are both oxygen- and iron-dependent; thus, hypoxia and iron chelation results in decreased PHD activity and increased HIF-1α activity (Hewitson et al., 2003; Nandal et al., 2011; Flagg et al., 2012).
In recent years, a series of iron chelating agents that exert neuroprotective effects have been developed (Núñez and Chana-Cuevas, 2018). In particular, M30, which is an 8-hydroxyquinoline-based iron chelator developed by the group of Moussa Youdim at Technion-Israel Institute of Technology (Weinreb et al., 2016), stabilizes HIF-1α, most probably by inactivating HIF-1α PHD. In the brain, HIF-1α stabilization by M30 leads to the expression of a broad number of neuroprotective-adaptive mechanisms and pro-survival signaling pathways (Kupershmidt et al., 2011). Real-time RT-PCR revealed that M30 differentially induces the expression of a variety of cellular components, including vascular endothelial growth factor, erythropoietin, enolase-1, TfR1, heme oxygenase-1, inducible nitric oxide synthase (iNOS), glucose transporter 1, brain-derived neurotrophic factor (BDNF), glial cell-derived neurotrophic factor, and the antioxidant enzymes catalase, superoxide dismutase-1, and glutathione peroxidase (Kupershmidt et al., 2009). Further reports have supported the role of iron chelators in inducing neuronal survival pathways (Mechlovich et al., 2014; Guo et al., 2015, 2016; Xiao et al., 2015). It follows that the capacity of iron chelators to induce HIF-1α-mediated neuroprotection adds to the recognized neuroprotective effects of iron chelators, through their ability to prevent hydroxyl radical production via the Fenton reaction. A tempering note comes from the report that treatment of human skin cells with the iron chelator N-(2-hydroxybenzyl)-L-serine (HBSer) does not induce HIF-1α activation, as opposed to desferrioxamine (DFO) and salicylaldehyde isonicotinoyl hydrazone (SIH) used as positive controls (Creighton-Gutteridge and Tyrrell, 2002). The authors conjectured that the lack of HIF-1α activation by HBSer might be related to its lower affinity for iron as compared to DFO and SIH. Of relevance to the theme of this review, however, is the fact that the transcription factor HIF-1α activates the expression of several genes associated with iron homeostasis (Lee and Andersen, 2006), which in non-excitable cells results in an increase in cellular iron content (Qian et al., 2011).
Increased Reactive Oxygen/Nitrogen Species Generation Induces Iron Dyshomeostasis
A significant number of studies have shown that physiological levels of ROS and reactive nitrogen species (RNS) act as signaling molecules in a variety of biological responses (Sen, 2001; Ray et al., 2012; Asiimwe et al., 2016; Lourenco et al., 2017; Moldogazieva et al., 2018; Nemes et al., 2018). The brain is an organ highly susceptible to oxidative stress (Cobley et al., 2018). Hence, neuronal cells have to maintain physiological levels of ROS and RNS to avoid oxidative or nitrosative stress, which arises when excessive ROS/RNS production overcomes the cellular antioxidant systems, which by affecting the redox environment favors excitotoxicity. Henceforth, we will use the broad term oxidative stress to refer to both oxidative and nitrosative stress. Oxidative stress perturbs the function of many cellular processes; it causes glutathione deficiency (Wu et al., 2004) and promotes cell death by inducing mitochondrial depolarization, membrane lipid peroxidation, and DNA damage, among other harmful effects. To counteract the damaging effects of excess ROS/RNS (Duncan and Heales, 2005; Halliwell, 2006; Moncada and Bolaños, 2006), cells make use of enzymatic and non-enzymatic defense mechanisms, some of which engage calcium signals (Gordeeva et al., 2003). Depending on the extent and persistence of the cellular damage, oxidative stress may play a key role as a causative agent of neurodegenerative disease emergence and or progression.
Iron uptake into primary hippocampal neurons stimulates ROS generation (Muñoz et al., 2011), and decreases the redox potential established by the intracellular levels of oxidized and reduced glutathione in neuroblastoma cells (Núñez et al., 2004). The relationship between increased redox-active iron and increased oxidative stress is well accepted (Núñez et al., 2012). Similarly, in cellular models convincing evidence points to a link between increased oxidative tone and iron accumulation. Initial observations indicated that hydrogen peroxide activates IRP1 (Martins et al., 1995; Pantopoulos and Hentze, 1995; Hanson and Leibold, 1999; Caltagirone et al., 2001; Mueller et al., 2001), and that this activation results in increased cellular iron uptake mediated by increased translation of TfR1 and decreased ferritin synthesis (Caltagirone et al., 2001; Sureda et al., 2005). A subsequent study reported that, besides activation of IRP1, exposure of SH-SY5Y cells to H2O2 results in the degradation of the exo-transporter FPN1, which coupled to a decrease in ferritin translation leads to a time-dependent increase in the cellular labile iron pool (Dev et al., 2015), with the ensuing generation of oxidative damage through the production of hydroxyl radicals by the Fenton reaction. In animal models, evidence linking increased ROS as a cause, and iron homeostasis loss as an effect, is still lacking. An early report indicated that vitamin A deficiency results in increased hepcidin mRNA expression and increased iron spleen concentrations, accompanied by increased protein oxidation. These results suggest that vitamin A protects the liver against protein oxidation by maintaining iron homeostasis (Arruda et al., 2009). In a mouse model of experimentally induced hemolysis, excess heme increases intracellular ROS production, which signals for the induction of the unique iron exporter ferroportin, resulting in iron export from macrophages (Jeney et al., 2002; Marro et al., 2010). Of note, decreasing ROS levels by treatment with the antioxidant N-acetylcysteine prevents ferroportin induction and normalizes intracellular iron levels in a mice model of experimentally induced hemolysis (Tangudu et al., 2018). In a separate study using the APPswe/PS1ΔE9 AD mice model, administration for 3 months of antioxidants of plant origin decreased iron levels and malondialdehyde content and increased antioxidant defenses (Zhang et al., 2018). Overall, these results are consistent with the notion that oxidative stress dysregulates iron homeostasis both in cellular and animal models.
Nitric oxide also modifies IRP1 activity; it increases IRP1 binding to IRE and the increase IRE binding activity of IRP1 is coincident with inhibited ferritin synthesis and increased TfR1 mRNA levels (Phillips et al., 1996). Nitric oxide activates IRP1 directly by destabilizing the 4Fe-4S cluster of IRP1 (Drapier et al., 1993; Soum and Drapier, 2003), and may activate IRP1 indirectly by mobilizing intracellular Fe, thus decreasing the intracellular labile iron pool and activating IRP1 (Pantopoulos et al., 1996; Wardrop et al., 2000).
Altogether, these studies point to a robust association between increased ROS, nitric oxide, and increased levels of redox-active iron, which in turn mediate oxidative damage to lipids, protein, and nucleic acids. Yet, a recent study showed that in cultured macrophages, the labile iron pool apparently attenuates peroxynitrite-dependent damage (Damasceno et al., 2018). Whether this protective effect of the labile iron pool is translatable to other cell types and to neurons in particular, remains to be established.
Iron Dyshomeostasis in Neurodegeneration
Increased iron levels and the associated ROS generation are important initiators and mediators of cell death (Dixon and Stockwell, 2014), whereas iron dyshomeostasis has a key role in neurodegeneration. Mutations of genes encoding proteins involved in iron homeostasis are associated with degeneration of central nervous system (CNS) cells (Ponka, 2004; Zecca et al., 2004), while oxidative stress induces cell injury by disrupting cellular iron balance (Carocci et al., 2018; Kerins and Ooi, 2018), thus leading to a vicious circle. Both AD and PD entail increased brain iron content and oxidative stress (Crichton et al., 2011; Ward et al., 2014; Tonnies and Trushina, 2017; Guo et al., 2018), with the implied threat of hydroxyl radical-induced neuronal damage. Of note, binding of Cu or Fe to Aβ peptides generates H2O2 (Huang et al., 1999), which may induce neuronal oxidative stress during AD progression (Cristovao et al., 2016). Moreover, iron overload increases neuronal amyloid-β production and enhances cognitive impairment in a transgenic mice model of AD (Becerril-Ortega et al., 2014) while accumulating evidence suggests that impaired iron homeostasis is an early event in AD progression (Peters et al., 2015).
Parkinson’s disease is a progressive neurodegenerative disease characterized by rigidity, tremor, and slowness of movement as well as by a wide range of debilitating non-motor symptoms. A key feature of PD is the selective loss of the dopaminergic neurons of the substantia nigra; ample evidence shows iron accumulation in these neurons (Jellinger, 1999; Zecca et al., 2004; Berg and Youdim, 2006; Muñoz et al., 2016; An et al., 2018). The observation that pharmacological agents with iron chelation capacity prevent neuronal death in AD or PD experimental models (recently reviewed in Núñez and Chana-Cuevas, 2018) highlights the pivotal role of iron as a mediator of neuronal death in AD and PD.
In addition, spinal neurons from ALS patients display increased iron and calcium levels (Kasarskis et al., 1995). The success of iron chelation therapy in ALS mouse models sustains the central role of iron in ALS pathogenesis (Jeong et al., 2009; Kupershmidt et al., 2009). Alterations of proteins involved in iron metabolism, inhibition of anterograde axonal transport leading to iron accumulation in ventral motor neurons, and increased mitochondrial iron load in neurons and glia have been proposed as pathogenic mechanisms to explain the abnormal iron accumulation displayed by the neurons and glia of ALS mice (Jeong et al., 2009).
Iron Dyshomeostasis in Inflammation
Inflammation, a condition often found in neurodegenerative environments, is yet another factor that induces iron accumulation (reviewed in Urrutia et al., 2014). Both the p50 and the p65 subunits of the transcription factor NF-κB bind to a NFκB-responsive element present in the 5′ untranslated region of the DMT1-1B promoter, inducing the expression of the 1B isoforms of DMT1 (Paradkar and Roth, 2006a,b). The transcription factor NF-κB is often activated by inflammatory signals (Rothwell and Luheshi, 2000; Hanke and Kielian, 2011; Shih et al., 2015). Accordingly, the possible association between inflammation and iron accumulation has been recently explored (Pelizzoni et al., 2013; Urrutia et al., 2013, 2014; McCarthy et al., 2018). To this purpose, the effects of lipopolysaccharide (LPS) and the pro-inflammatory cytokines TNF-α and IL-6 on mRNA and protein levels of DMT1, FPN1, and hepcidin were investigated in astrocytes, microglia, and neurons isolated from rat brain. A causal association between inflammation and iron accumulation was reported, since in addition to increasing neuronal iron content, treatment with LPS, TNF-α, or IL-6 increases DMT1 expression and protein levels in neurons (Urrutia et al., 2013). Similar results were reported in studies of ventral mesencephalic neurons in primary culture, in which treatment with TNF-α or IL-1β induces an increment in DMT1 and TfR1 protein levels, together with a reduction of FPN1 levels (Wang et al., 2013). Considering that NFκB activation is downstream of the TNF-α, IL-1, and LPS signaling pathways, these results are consistent with a circuit in which inflammatory stimuli induce DMT1 expression and iron accumulation via NFκB activation and hepcidin expression.
An additional link between inflammation and increased oxidative stress resides in the observation that inflammatory stimuli activate the NADPH oxidase (NOX) enzyme, the main regulated source of cellular ROS generation (Green et al., 2001; Urrutia et al., 2014; Sorce et al., 2017). In particular, pro-inflammatory cytokines activate the phagocytic NOX2 enzyme, which is highly expressed in microglia (Sorce and Krause, 2009; Lim et al., 2013; Barrett et al., 2017). In turn, NOX2 activation generates an oxidative extracellular environment that increases ROS levels in neighboring neurons (Schiavone et al., 2009; Gao et al., 2012; Surace and Block, 2012; Zhang et al., 2014).
Of note, NOX activation has also been reported in experimental models of PD and AD. In particular treatment with MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), a prodrug to the Parkinsonian neurotoxin 1-methyl-4-phenylpyridinium (MPP+), results in increased synthesis of the pro-inflammatory cytokine IL-1β, and increased membrane translocation of the cytosolic NOX sub-unit p67phox, which is prevented by minocycline, a tetracycline derivative that exerts multiple anti-inflammatory effects (Wu et al., 2002). In addition, iron supplementation to midbrain neuron-glia cell cultures causes the selective death of dopaminergic neurons. This death is mediated by microglial NOX2 since cells derived from NOX2-/- mice do not present iron-induced neuronal death (Zhang et al., 2014). Similarly, brain tissue from AD patients reveals an elevation in NOX activity coupled to membrane translocation of cytosolic NOX2 subunits and increased NOX1 and NOX3 mRNA transcripts (Shimohama et al., 2000; Ansari and Scheff, 2011). Significantly, the authors found an inverse correlation between postmortem NOX activity and ante-mortem cognitive status (Ansari and Scheff, 2011). Altogether, these results indicate that inflammatory stimuli generate a neuronal phenotype of increased iron influx and increased NOX-produced ROS, with the consequent risk of oxidative damage observed in neurodegenerative diseases.
Role of Iron in Excitotoxicity
Earlier reports indicate that iron plays a role in excitotoxicity, a death process mediated by high levels of intracellular calcium caused by excessive activity of excitatory neurotransmitters. Seminal observations by Cheah and colaborators described a signaling cascade in which stimulation of N-methyl-D-aspartate (NMDA) receptors activates the small GTPase Dexras1 via S-nitrosylation, mediated by activation of neuronal nitric oxide synthase (nNOS). Activated Dexras binds to the peripheral benzodiazepine receptor-associated protein PAP7, which in turn binds to the iron transporter DMT1 (Cheah et al., 2006). Most importantly, treatment with neurotoxic concentrations of NMDA elicits a major increase in iron uptake, resulting in three- to fivefold increase of hydroxyl radical levels and the death of more than 90% of cells, whereas co-treatment with NMDA and a cell-permeant iron chelator largely blocked NMDA-induced cell death. Overall, these results indicate that glutamate-NMDA excitotoxicity elicits DMT1 activation and iron-mediated cell death (Cheah et al., 2006). Although the role of calcium was not explored, the authors hypothesized initial calcium entrance through the NMDA receptor may activate nNOS, thus initiating this neurodegenerative process. As discussed below, crosstalk between NMDA receptor-mediated calcium influx and iron is likely to have a relevant role in neurodegenerative diseases such as AD and PD.
Neuronal Calcium Homeostasis and Signaling
Calcium is a universal second messenger that regulates numerous cellular processes over a wide temporal range (Berridge et al., 2003). In neurons, the free calcium concentration at rest lies in the 70–100 nM range (Clapham, 2007), whereas extracellular calcium concentration is ∼1.5 mM. This large concentration gradient, combined with resting potential values of 70–90 mV (negative inside), generate a significant electrochemical gradient that favors calcium entry. Calcium homeostasis and signaling are vital for cellular function and survival; thus, neuronal cells possess powerful systems to maintain calcium homeostatic and signaling systems (Berridge, 1998; Brini et al., 2014), which regulate the neuronal processes underlying synaptic plasticity and complex brain cognitive functions, such as information processing, learning, and memory. Calcium is stored also in intracellular organelles such as the endoplasmic reticulum (ER), where its free concentration lies in the 0.5 mM range. The main systems in charge of maintaining calcium homeostasis are two plasma membrane calcium (PMCA) transporters, the PMCA pump and the sodium/calcium exchanger (NCX), plus the sarco/ER Ca2+-ATPase (SERCA) pump that maintains the steep concentration gradient between the ER and the cytoplasm (Brini et al., 2014). These transporters utilize different mechanisms and display a range of calcium affinities, transport rates, and capacities to ensure the proper managing of the variety of intracellular increases in free calcium concentration, known as calcium signals, generated by the diverse stimuli neuronal cells receive.
Neuronal activity generates calcium signals that via sequential activation of calcium-dependent signaling cascades and transcription factors promote the expression of genes underlying dendritic development, synaptic plasticity, and neuronal survival (Brini et al., 2014). In response to neuronal stimulation, calcium influx through plasma membrane pathways, including agonist-operated, store-operated, or voltage-gated calcium channels (VGCCs), generates neuronal calcium signals. In addition, the release of calcium from intracellular stores such as the ER also contributes to neuronal calcium signal generation (Brini et al., 2014). All neuronal compartments crucial for neurotransmission have the two types of calcium release channels: the inositol 1,4,5-trisphosphate (IP3) receptor (IP3R) and the ryanodine receptor (RyR) channels (Berridge, 1998). These large channels have complex gating and regulation and mediate a cellular process known as calcium-induced calcium release (CICR), whereby calcium promotes its own release from intracellular calcium stores (Berridge, 1998). This CICR mechanism likely contributes to propagate calcium signals that reach distant targets such as the nucleus (Berridge, 1998), where calcium promotes short or long-lasting changes in neuronal function and structure (Bading, 2013).
ROS Levels Influence Calcium Homeostasis and Signaling
Significant evidence gathered in the last years indicates that increased cellular ROS levels modify the function of key proteins engaged in calcium homeostasis and signaling (Gordeeva et al., 2003; Hidalgo and Donoso, 2008; Gorlach et al., 2015). Redox-dependent regulation of components that maintain neuronal Ca2+ homeostasis may influence the direction and/or the efficiency of Ca2+-signaling pathways. Interactions among ROS and calcium-dependent signaling pathways are bidirectional; while ROS regulate cellular calcium signaling, calcium signals are essential for ROS production because a number of ROS-generating and antioxidant systems of living cells are calcium-dependent (Gordeeva et al., 2003).
Reversible modifications of proteins via ROS-promoted oxidation of their amino acids modify the properties of neuronal proteins engaged in signal transduction, such as protein kinases, protein phosphatases, and transcription factors (Wang et al., 2012). An increase in cellular ROS levels results in the inhibition of NMDA receptor function and in decreased activity of both the PMCA and the SERCA calcium pumps, while it enhances the activity of IP3R and RyR calcium channels (Hidalgo and Donoso, 2008; Joseph et al., 2018). Consequently, ROS generation in neuronal cells increases cellular calcium levels by promoting calcium release mediated by IP3R and RyR channels and by inhibiting both the PMCA and the SERCA pumps: the ROS-mediated increase in calcium levels promotes changes in several calcium-dependent neuronal pathways (Yermolaieva et al., 2000; Riquelme et al., 2011). Depending on the magnitude of the calcium increase, activation of kinases or phosphatases implicated in synaptic plasticity process will occur (reviewed in Hidalgo and Arias-Cavieres, 2016). In addition, activation of the large-conductance Ca2+-activated K+ (BKCa) by oxidation of SH cysteine residues, by decreasing neuronal excitability reduces the vulnerability of hippocampal neurons to hypoxia (Hepp et al., 2005).
Defective Calcium Homeostasis and Signaling in Neurodegeneration
A link between excessive intracellular calcium accumulation and neuronal death is by now firmly established (Demuro et al., 2010; Stefani et al., 2012; Fukui, 2016). Neuronal cells are particularly sensitive to calcium-mediated cytotoxic damage and the failure of neurons to maintain Ca2+ homeostasis is a common feature of aged-linked neurodegenerative pathologies (Berrocal et al., 2015; Brini et al., 2017; Pchitskaya et al., 2018; Strehler and Thayer, 2018). Calcium dyshomeostasis (Pchitskaya et al., 2018) and altered neuronal calcium signaling (Chan et al., 2009; Overk et al., 2015) have been reported in a number of disease conditions, including AD, PD, and ALS (for recent reviews, see Franco-Iborra et al., 2018; Popugaeva et al., 2018; Post et al., 2018; Sirabella et al., 2018). Likewise, defective calcium signaling in glia and autophagy-related pathways has been implicated in neurodegenerative disease (Mustaly-Kalimi et al., 2018). Accruing evidence implicates decreased levels and functional decline of PMCA isoforms in neurodegeneration (Hajieva et al., 2018; Strehler and Thayer, 2018). In addition, in many pathological conditions, disruptions of ER Ca2+ signaling disrupt neuronal function and promote neuronal cell death (Okubo et al., 2018). To date, however, the detailed mechanisms of how neuronal Ca2+ homeostasis and signaling are perturbed in neurodegenerative diseases are not well understood.
The most common neurodegenerative disorder and the leading cause of dementia in the elderly is AD, whereas PD is the most prevalent age-associated movement disorder that entails early and selective degeneration of dopaminergic neurons. The pathological hallmarks of AD are neuronal loss and the presence of amyloid plaques and neurofibrillary tangles in the brain of affected individuals. Yet, before the emergence of plaques and/or neurofibrillary tangles other defects, including Ca2+ and iron dyshomeostasis, mitochondrial dysfunction, increased contacts between the ER and the mitochondria, neuro-inflammation, and alterations in lipid metabolism have been reported (Mattson, 2010; Area-Gomez and Schon, 2017; Lane et al., 2018). How these early defects relate to the emergence and progression of AD remains to be established. Of note, a recent report by the Alzheimer’s Association Calcium Hypothesis Workgroup [AACHW] (2017) analyzed how defective calcium signaling may be an early upstream event in AD progression, and how alterations of the many cellular components that control calcium hemostasis and signaling leads to the decline in the function of more vulnerable neuronal cells. In particular, these more vulnerable neuronal cells present early synaptic dysfunction, upregulation of some calcium signaling related genes and significant decreases in the expression of genes related to synaptic neurotransmission, neurotrophic factor activity, mitochondrial metabolism, and energy production, among others (Alzheimer’s Association Calcium Hypothesis Workgroup [AACHW], 2017).
In PD, dopaminergic neurons of the substantia nigra are especially vulnerable to Ca2+ dyshomeostasis, because their autonomous pace-making activity, through which these neurons generate action potentials in the absence of synaptic input (Guzman et al., 2009). This particular property of dopaminergic neurons imposes a high pressure on adequate handling of activity-generated Ca2+ signals, presumably mediated by the significant calcium influx through plasma membrane L-type calcium channels that markedly surpasses the calcium influx displayed by other neurons (Surmeier et al., 2010).
Dopaminergic neurons are highly vulnerable to oxidants and mitochondrial inhibition compared to other neuronal types (Sherer et al., 2003; Halliwell, 2006; Burbulla et al., 2017). Thus, brain permeant toxins such as rotenone, an inhibitor of NADH dehydrogenase, exclusively affect dopaminergic neurons (Sherer et al., 2003; Cannon et al., 2009). This special vulnerability is likely to produce the mitochondrial Ca2+ imbalance that occurs in PD (Ludtmann and Abramov, 2018). As discussed below, iron-induced ROS increases result in calcium signal generation, which may contribute to the basal calcium burden of dopaminergic neurons.
Several studies have reported defective Ca2+ signaling in other neurodegenerative conditions such as ALS (Siklos et al., 1998; Grosskreutz et al., 2010; Jaiswal, 2013; Muhling et al., 2014) and multiple sclerosis (MS; Gonsette, 2008). The death of motor neuron is a characteristic feature of ALS. Excitotoxicity, which involves glutamate and calcium overload, or cell death caused by high levels of intracellular calcium caused by excessive activity of excitatory neurotransmitters, may contribute to ALS pathology. Calcium dysregulation is also present in MS, where the associated disability results from neuronal and axonal loss initiated by microglia activation and mediated by oxidative stress, excitotoxicity, calcium dysregulation, and mitochondrial dysfunction, leading to proteolytic enzyme production and apoptosis (Gonsette, 2008).
Additionally, calcium dysregulation has been associated with neuroinflammation, a CNS response that comprises biochemical and cellular responses to injury, infection, or neurodegenerative diseases (DiSabato et al., 2016). Neuroinflammation markers increase in some neurodegenerative conditions; microglia, the innate CNS immune cells, play key roles as mediators of these neuroinflammatory responses. Of note, Ca2+ and neuroinflammatory signaling mechanisms exhibit extensive crosstalk and bidirectional interactions. As an example, chronic brain inflammation leads to a feedback reduction in NMDA receptors caused by excess synaptic glutamate activity during microglial activation (Rosi et al., 2004).
Despite the abundant literature that points to defective calcium homeostasis and signaling in neurodegeneration, scarce information is available in the literature regarding the protective effects of calcium modulators in preventing these defects. Yet, all recent reports on this subject highlight the fact that this is an urgent task. The non-competitive NMDA receptor antagonist Memantine, which reduces excitoxicity by inhibiting neuronal calcium entry (Bezprozvanny and Mattson, 2008), has been the most common drug for treating moderate to severe AD; nevertheless, Memantine provides only limited benefits for AD patients. A recent report showed that immunotherapy with a murine analog of the anti-Aβ antibody Aducanumab restores calcium homeostasis in a transgenic AD mice model (Kastanenka et al., 2016), whereas a recent review addresses the evidence linking neurodegeneration to RyR dysfunction and describes novel therapeutic approaches to target abnormal RyR function (Kushnir et al., 2018). There are no reports, however, on the efficacy of these strategies for treating AD patients.
The HIF-1–Calcium Relationship
In SH-SY5Y neuroblastoma cells, chelation of calcium by 1,2-bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid (BAPTA) induces HIF-1α protein accumulation and nuclear localization, whereas the cytosolic calcium elevation produced by inhibition of the SERCA pump attenuates these effects (Berchner-Pfannschmidt et al., 2004). Further experiments indicated that calcium chelation attenuates the interaction of HIF-1α with the von-Hippel-Lindau protein, thus decreasing the proteasomal degradation of HIF-1α (Berchner-Pfannschmidt et al., 2004). Control experiments indicated that BAPTA did not complex intracellular iron ions, thus the above effects were caused by calcium chelation and not by stimulation of iron-activated PHD enzymes (Berchner-Pfannschmidt et al., 2004). Subsequent studies showed that intermittent hypoxia causes HIF-1α accumulation through a mechanism that involves increased cell calcium and increased generation of ROS by NOX, which induces a complex signaling pathway that results in increased HIF-1α synthesis and decreased hydroxylase-dependent HIF-1α degradation (Yuan et al., 2008).
Although brain levels of HIF-1α are considered neuroprotective (Kietzmann et al., 2001; Siddiq et al., 2005; Guo et al., 2016; Lee et al., 2017; Merelli et al., 2018), under certain conditions, HIF-1α acts as a pro-apoptotic factor (Piret et al., 2002; Zhang et al., 2007; Merelli et al., 2018). For example, the anesthetic isoflurane induces apoptotic neurodegeneration in a process mediated by HIF-1α, since knockdown of HIF-1α expression attenuates isoflurane-induced neurotoxicity (Jiang et al., 2012). In addition, isoflurane induces a significant elevation of cytoplasmic calcium levels in cultured neurons. Based on previous observations, showing that calcium mediates HIF-1α expression (Yuan et al., 2005; Riganti et al., 2009), the authors hypothesized that the high calcium levels induced by isofluorane promote HIF-1α-mediated apoptotic neurodegeneration. Studies discerning the calcium concentrations and other cell physiological condition, which elicit each of the above responses, should shed light on these apparent contradictory responses.
Overall, the emerging picture from the above results is consistent with the idea that low calcium concentrations stabilize HIF-1α protein levels by decreasing its proteasomal degradation. The effect of calcium concentrations above homeostatic levels is not clear and may depend on cell type: high calcium levels may induce a protective response consisting in increased HIF-1α synthesis coupled to decreased degradation or may mediate HIF-1α degradation.
Iron Modifies Neuronal Calcium Homeostasis and Calcium Signaling Pathways
Iron overload increases intracellular calcium levels, which in turn activate calcineurin (Lee et al., 2016), a calcium-dependent phosphatase which has a key role in synaptic plasticity and memory processes (Baumgartel and Mansuy, 2012). Increases in cytoplasmic calcium levels via the NMDA receptor stimulate the neuronal NOS enzyme (Ghosh and Salerno, 2003) and generate superoxide anion in hippocampal neurons (Brennan et al., 2009), while in cortical neurons, this calcium increase induces a signaling cascade that enhances superoxide generation through NOX activation (Girouard et al., 2009).
Both synaptic plasticity and memory processes require activity-dependent hippocampal ROS generation (Bindokas et al., 1996; Kahlert et al., 2005; Kishida and Klann, 2007). In primary hippocampal neurons, the ROS increase induced by iron promotes calcium release from the ER mediated by redox-sensitive RyR channels; the resulting increase in intracellular calcium levels link NMDA receptor stimulation to ERK1/2 activation and nuclear translocation (Muñoz, 2012). Since several reports indicate that CREB-dependent transcription of synaptic plasticity related genes entails ERK1/2-mediated long-term CREB phosphorylation (Wu et al., 2001; Lynch, 2004; Thomas and Huganir, 2004). Accordingly, these findings indicate that iron-induced, RyR-mediated, calcium release contributes to this activity-dependent neuronal response.
In summary, the current evidence suggests that crosstalk between ROS and calcium plays an essential role in many pathophysiological conditions including neurodegenerative diseases such as PD and AD. Here we complement this idea by adding the component of iron overload, and the ensuing oxidative stress, as a significant factor in promoting calcium dysregulation in neurodegeneration.
The Synergism Between Iron and Calcium in Lipid Peroxidation
A link between iron-mediated lipid peroxidation and calcium dyshomeostasis was reported more than 30 years ago. Peroxidation of rat brain synaptosomes by Fe2+ and H2O2 generates rapid and large uptake of Ca2+ by isolated synaptosomes; Fe2+ also enhances Ca2+ uptake by spinal cord neurons in culture, an effect that is coupled to lipid peroxidation and the release of arachidonic acid from cells, in a process prevented by the iron chelator DFO (Braughler et al., 1985). The authors concluded that Fe2+ and Ca2+ act synergistically to produce lipid peroxidation and damage to neuronal membranes (Braughler et al., 1985). A further report proposed that this is not a direct effect of calcium but the result of independently initiated processes of lipid peroxidation and Ca2+ translocation, which interact subsequently in a synergistic manner (Bors et al., 1990).
The relationship between iron-induced lipid peroxidation and dysregulation of Ca2+-ATPase is long-dated (Pelizzoni et al., 2008; Mata and Sepulveda, 2010; Zaidi, 2010). Plasma membrane lipid peroxidation initiated by the Fenton reaction results in increased calcium content, derived from the inhibition of the plasma membrane and ER Ca2+-ATPases (Pereira et al., 1996). This inhibitory effect is shared by other radical species, in particular by peroxynitrite (Zaidi and Michaelis, 1999). Oxidants also inhibit SERCA activity while promoting calcium release from the ER (Gordeeva et al., 2003; Hidalgo and Donoso, 2008). Arguably, the observed upsurge in intracellular Ca+2 induced by iron-mediated lipid peroxidation could be compounded by an oxidant-induced loss of PMCA and SERCA activity, which combined with ROS-mediated activation of IP3R/RyR channels would result in further and deleterious increases in the intracellular Ca2+ concentration.
Iron, Calcium, and HIF-1 Activity
Considering the overlapping effects of iron and calcium on HIF-1 activity detailed above, a Fe/Ca bipartisan regulation of HIF-1 can be hypothesized (Figure 1). In neurodegenerative diseases with an iron accumulation component, decreased HIF-1 activity is predicted by a mechanism that involves the maintenance of maximal HIF-1 hydrolase activities and the increase of intracellular calcium mediated by dysregulation of redox-sensitive calcium homeostatic and signaling pathways. In neurons, decreased HIF-1 activity results in decreased neuroprotective, neuroregenerative, and antioxidant responses associated to HIF-1 activity (see above).
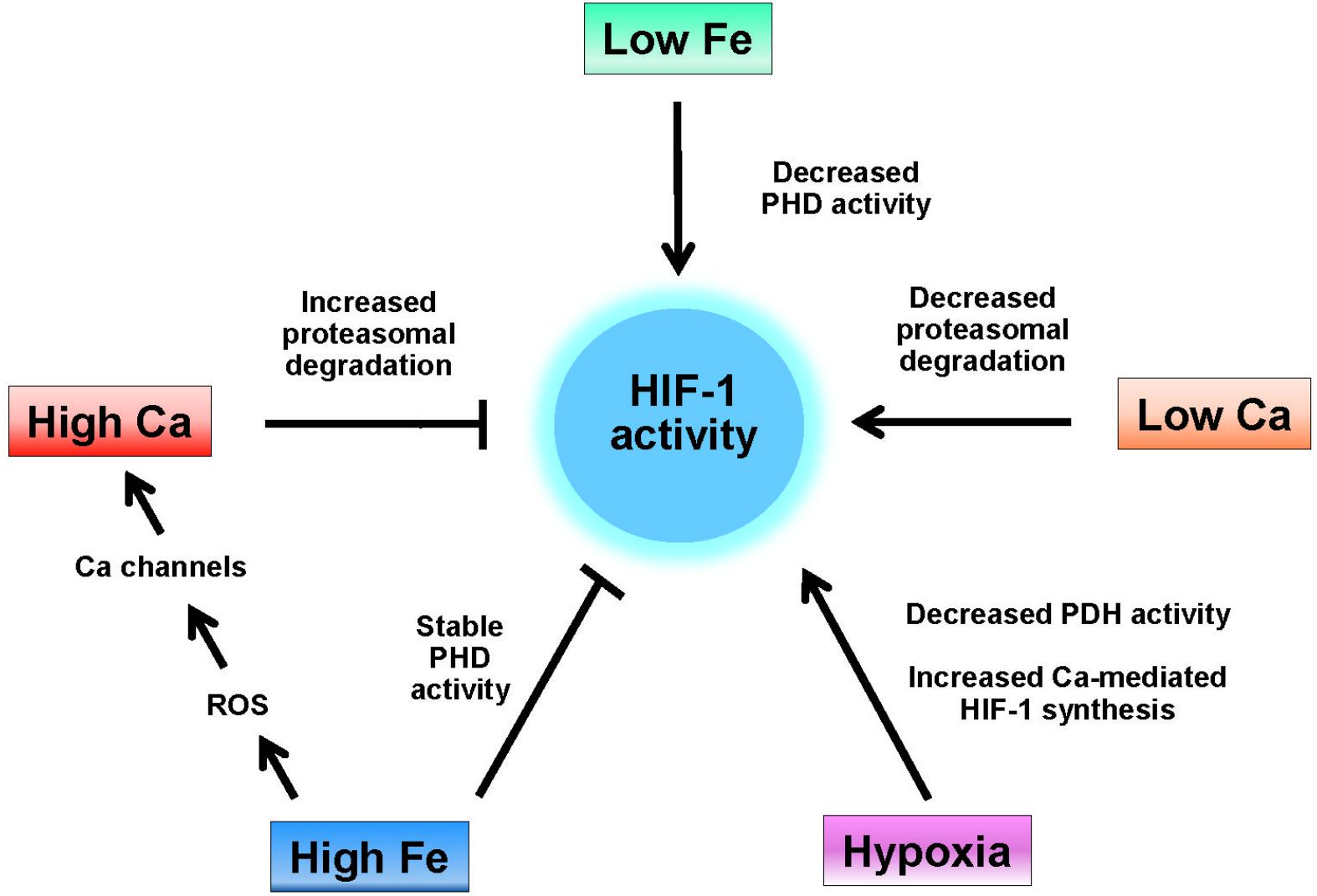
Figure 1. Regulation of HIF-1 activity by iron and calcium. HIF-1 activity is induced by hypoxia, which decreases the activity of HIF-1 hydrolases (PHD), by low iron (e.g., iron chelation), which also decreases the activity of HIF-1 hydrolases, and by low intracellular calcium, which decreases the interaction of HIF-1α with the von-Hippel-Lindau protein (see text). High intracellular calcium levels increase the rate of HIF-1 proteasomal degradation while high levels of iron both maintain the activity of HIF-1 hydrolases and, through enhanced ROS production increase the cellular levels of calcium by activation of RyR, IP3R, and L-type calcium channels.
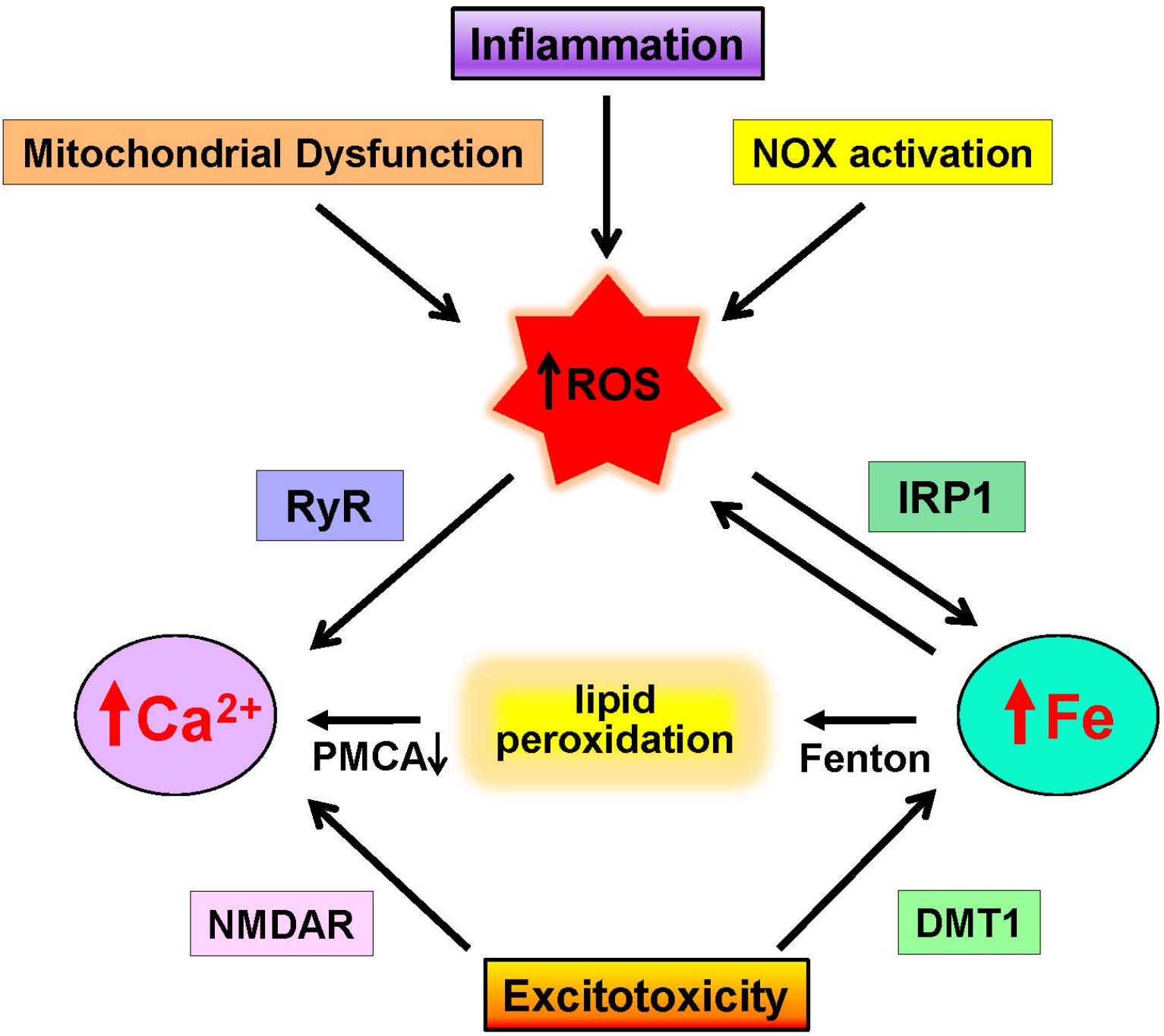
Figure 2. Neuronal pathways that increase calcium and iron levels. Increases in ROS due to mitochondrial dysfunction, inflammation, and NOX activation promote excessive ROS generation that promotes calcium release via RyR channels and increases redox-active iron by IRP1 activation. Through the Fenton reaction, iron induces lipid peroxidation that inhibits the PMCA calcium pump. In addition, excitotoxic conditions over-activate NMDA receptors (NMDAR), which results in increased cytoplasmic calcium levels and increases iron levels via DMT1 activation.
Calcium-Induced Iron Dyshomeostasis: Possible Role in Neurodegeneration
Excessive intracellular calcium levels, such as those associated with neurodegenerative conditions, are likely to produce high H2O2 levels via NOX2 and to enhance mitochondrial calcium uptake and ROS production. These two ROS sources, NOX2, and mitochondria should affect jointly iron homeostasis by increasing the cellular labile iron pool, as occurs in SH-SY5Y cells exposed to H2O2 (Dev et al., 2015). These findings raise the possibility that conditions that increase postsynaptic calcium levels following NMDA receptor activation, which promote H2O2 generation in neocortical neurons via a cascade engaging the NOS and NOX2 enzymes (Girouard et al., 2009), may lead to an upsurge of the labile iron pool.
The role of voltage-gated Ca2+ channels as mediators of iron transport into neuronal cells is well established (Gaasch et al., 2007; Lockman et al., 2012). Another link between iron and calcium during neurodegeneration was provided by the observation that the hippocampal and nigral neuron loss induced by intra-cerebroventricular FeCl3 injection in rats is largely prevented by pre-treatment with nicardipine, a blocker of L-type VGCCs (Bostanci and Bagirici, 2013). The authors concluded that these channels mediate the neurotoxic effects of extracellular iron by inducing either calcium and/or iron uptake. The question remains as to whether the protective effect of nicardipine is due to decreased iron influx, decreased calcium influx, or to a calcium-mediated process leading to neurodegeneration downstream of iron-induced oxidative stress. Other studies have also provided evidence of iron influx through NMDA receptors or voltage-operated calcium channels in primary hippocampal neurons (Pelizzoni et al., 2011) and the dopaminergic MES23.5 cell line (Wang et al., 2017).
The Mitochondrion Melting POT: Calcium, Iron, and Apoptotic Cell Death
Mitochondria, by taking up and releasing calcium, act as important regulators of cellular calcium levels. The large mitochondrial membrane potential difference (negative inside) provides the driving force for calcium entry through the mitochondrial calcium uniporter (MCU), a component of the inner mitochondrial membrane; in physiological conditions, calcium antiporters rapidly extrude calcium from the mitochondria and restore the basal calcium levels (Giorgi et al., 2018). Mitochondrial calcium concentration modulates ATP production and mitochondrial metabolism, whereas mitochondrial calcium overload induced by deregulated intracellular calcium levels promotes apoptotic cell death, necrosis, and autophagy (Giorgi et al., 2012; Rimessi et al., 2013; Marchi et al., 2018). In pathological conditions, the MCU has been implicated in excitotoxicity, iron overload, inflammation, and oxidative stress-induced mitochondrial dysfunction and cell death (Liao et al., 2017). The ROS generated by mitochondrial enzymes or the electron transport chain presumably regulate diverse cellular physiological pathways, whereas dysregulated ROS generation may contribute to the development of human diseases (Angelova and Abramov, 2017).
Neurodegenerative disorders such as AD, PD, and ALS display dysregulation of calcium homeostasis and mitochondrial dysfunction; in addition, excitotoxicity leads to mitochondrial calcium overload and promotes necrotic or apoptotic-like excitotoxic cell death (Hansson Petersen et al., 2008; Rosales-Corral et al., 2012; Bose and Beal, 2016; Gibson and Thakkar, 2017; Giorgi et al., 2018). An increase in mitochondrial calcium is of particular importance in dopaminergic neurons, which are constantly exposed to calcium influx (Surmeier et al., 2011). The interaction between calcium overload and excessive ROS production (Chinopoulos and Adam-Vizi, 2006) causes further damage to the electron transport chain and promotes oxidative stress, which induces the oxidation of mitochondrial lipids, proteins, and DNA leading to cytotoxicity (Muravchick and Levy, 2006). The vicious cycle between calcium overload and oxidative stress favors the sustained opening of the mitochondrial permeability transition pore (mPTP), leading to the collapse of the mitochondrial membrane potential and mitochondrial swelling, which in turn promote the apoptotic response (Di Lisa and Bernardi, 2009). Opening of the mPTP has a central role in the pathogenesis of neurodegenerative disorders (Vila and Przedborski, 2003; Perier et al., 2005).
Excessive iron levels promote oxidative stress leading to the upsurge of unrestrained calcium signals that damage mitochondrial function. Iron accumulation in the cytoplasm induces excessive iron influx into mitochondria through mitoferrin1/2 (Paradkar et al., 2009), which causes mitochondrial dysfunction by increased ROS production leading to oxidative damage. Hence, if uncontrolled, this calcium/iron self-feeding cycle becomes deleterious to mitochondrial and neuronal function, leading eventually to neuronal death. In this regard, it is of note that, by preserving mitochondrial calcium homeostasis, the iron-binding protein lactoferrin protects vulnerable dopamine neurons from degeneration (Rousseau et al., 2013).
In primary hippocampal neurons, iron-induced stimulation of redox-sensitive RyR-mediated calcium release causes significant mitochondrial fragmentation (Sanmartin et al., 2014). Iron overload induces oxidative stress in the ER earlier than in the mitochondria, thereby increasing ER stress and calcium levels, and consequently causing mitochondrial fragmentation and neuronal cell death (Lee et al., 2016). Mitochondrial fragmentation presumably contributes to the impairment of neuronal function produced by iron overload, since inhibition of mitochondrial division protects neuronal cells from excitotoxicity (Ruiz et al., 2018). Moreover, iron overload of HT-22 hippocampal neuron cells increases intracellular calcium levels, causes mitochondrial fragmentation via dephosphorylation of Drp1, and increases apoptotic neuronal death; of note, calcium chelation and inhibition of calcineurin prevented mitochondrial fragmentation and cell death (Lee et al., 2016). Based on these findings, the authors suggested that calcium-mediated calcineurin signaling, by regulating mitochondrial dynamics, has a key role in iron-induced neurotoxicity.
In summary, mitochondrial calcium overload combined with excessive iron-mediated ROS generation and mPTP opening compose a cell death-inducing sequence implicated in neuronal dysfunction and neurodegeneration.
Oxytosis/Ferroptosis a Possible Meeting Point for Calcium and Iron-Induced Cell Death
Oxytosis was first described as a glutamate-induced, calcium-dependent form of cell death (Murphy et al., 1988). Initial studies on the molecular mechanisms of oxytosis pointed to a decrease in intracellular glutathione levels, enhanced mitochondrial ROS production, lipid peroxidation, and massive calcium influx as causes of death (Miyamoto et al., 1989). The mechanistic link between glutamate exposure and glutathione depletion was the inhibition of the cysteine/glutamate antiporter xc- by glutamate, which deprived cells of cystine, a precursor in glutamate synthesis (Murphy et al., 1989). As glutathione levels decrease, ROS levels increase exponentially (Tan et al., 1998), which gives rise to the activation of signaling pathways that culminate in large influx of calcium and cell death (Maher et al., 2018). Thus, the increased calcium influx is an essential mediator of the cell death program, as cells do not die in calcium-free medium.
Ferroptosis is a form of regulated cell death that occurs because of lethal lipid peroxidation. Ferroptotic cell death is blocked by iron chelators, lipophilic antioxidants, and by agents that block propagation of lipid peroxidation (Stockwell et al., 2017; Fricker et al., 2018). Ferroptosis is induced by inhibition of the cystine-glutamate antiporter xc- or by inhibition of glutathione peroxidase 4 (Gpx4). Induction of ferroptosis by erastin (an inhibitor of the antiporter xc-) results in cell death, which is morphologically characterized by mitochondrial shrinkage, with no overt signs of apoptosis or necrosis (Dixon et al., 2012). Ferroptotic death derives from depletion of cell glutathione, increased oxidative stress, and iron-mediated lipid peroxidation to lethal levels (Stockwell et al., 2017). Inhibition of Gpx4 also causes ferroptosis (Yang et al., 2014). Gpx4 is a lipid peroxidase that directly reduces phospholipid hydroperoxides at the expense of reduced glutathione. In mice, conditional deletion in forebrain neurons of Gpx4 resulted in hippocampal neurodegeneration and behavior dysfunction, associated with markers of ferroptosis such as elevated lipid peroxidation, ERK1/2 activation, and neuroinflammation (Hambright et al., 2017). Given the large similarity in their cell death mechanisms, the current view is that oxytosis and ferroptosis are similar forms of cell death (Fricker et al., 2018; Lewerenz et al., 2018; Maher et al., 2018).
Although a lethal influx of calcium is an intrinsic feature of oxytosis, the participation of calcium in ferroptosis is still poorly defined. Initial work by Dixon et al. (2012) revealed that calcium chelators do not inhibit erastin-induced HT-1080 cell death. Nevertheless, as mentioned above, an increase in cellular ROS levels enhances the activity of IP3R and RyR calcium release channels and decreases the activity of both the PMCA and the SERCA calcium pumps. These effects increase cellular calcium levels by promoting calcium release from the ER while inhibiting its removal by the calcium pumps.
Lipid peroxidation-derived modification of some calcium channels could also be intermediates in ferroptotic death (Maher et al., 2018). In particular, 4-hydroxynonenal, a product of hydroxyl radical-mediated lipid peroxidation, induces the opening of NMDA receptor and VGCCs (Lu et al., 2002). Indeed, the store-operated calcium entry (SOCE) system, which activates calcium influx channels in the plasma membrane upon the release of intracellular calcium from the ER participates in the neuronal death induced by MPP+, a Parkinsonian toxin that inhibits mitochondria electron transport chain complex I. Inhibition of the SOCE response decreased apoptotic cell death, reduced ROS production, and decreased lipid peroxidation induced by MPP+ (Li et al., 2013). In the same vein, a recent report shows that the pharmacological or transcriptional inhibition of SOCE proteins protects against oxidative glutamate toxicity and against MPP+ injury (Maher et al., 2018).
Overall, these results point to a possible participation of calcium in ferroptosis, a participation driven by raises in cytoplasmic calcium because of oxidative- and lipid peroxidation-derived modifications of the calcium homeostasis machinery (Figure 3). This participation may be specific to cells with weak antioxidant defenses, which could allow for the oxidative activation of calcium channels.
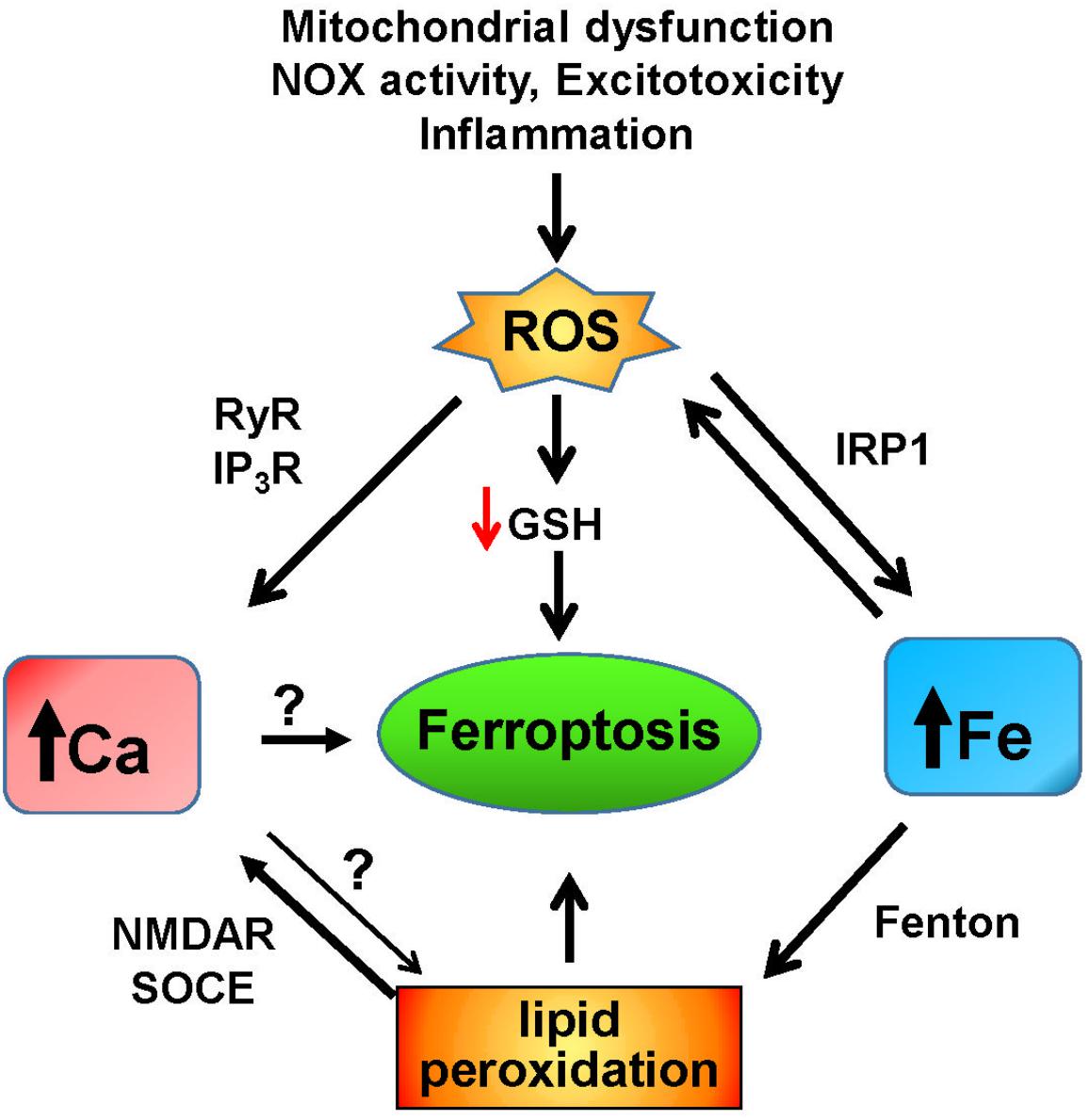
Figure 3. Iron and calcium-mediated cell death by ferroptosis. Increases in ROS due to mitochondrial dysfunction, non-physiological activation of NOX, excitotoxicity, inflammation, or other factors result in the increase of redox-active iron mediated by activation of IRP1. Through the Fenton reaction, iron induces lipid peroxidation that activates NMDAR and the SOCE system, which results in increased cytoplasmic calcium. Increased iron levels and lipid peroxidation mediate ferroptotic cell death, which may also be induced by unregulated calcium levels. Increased ROS levels decrease the intracellular GSH content, leading to ferroptosis, and activate the intracellular calcium channels RyR and IP3R, which adds to the increase in cytoplasmic calcium caused by iron-induced lipid peroxidation.
Protection of Iron Overload by Nrf2; the Calcium Connection
Under oxidative stress conditions, the transcription factor known as nuclear factor (erythroid-derived 2)-like 2 (Nrf2) directs the expression of a large array of cytoprotective genes (Tonelli et al., 2018). In particular, Nrf2 protects cells from the injurious effects of iron overload by regulating the expression of genes involved in iron storage and iron export (Chorley et al., 2012; Kerins and Ooi, 2018). These findings raise the possibility that Nrf2 may counteract the oxidative stress induced in neuronal cells by iron overload. Thus, Nrf2 may restore the intracellular labile iron pool by inducing the expression of the iron exporter FPN1 and ferritin, which oxidizes Fe2+ to Fe3+ and stores it within its structure making it unavailable for the Fenton reaction (Orino et al., 2001). In addition, intracellular iron levels act as a link between HIF-1α and Nrf2: Nrf2-induced ferritin upregulation activates HIF-1α by making iron unavailable to HIF prolyl-hydroxylase (Siegert et al., 2015), whereas Nrf2 inhibition results in decreased activation of HIF-1α by hypoxia (Kim et al., 2011; Ji et al., 2014).
Recent studies point to a connection between ROS, cellular calcium signals, and Nrf2. The neurotrophin BDNF, besides inducing complex neuronal signaling cascades underlying synaptic plasticity (Bolton et al., 2000), also induces neuronal antioxidant responses through BDNF-induced Nrf2 nuclear translocation (Bouvier et al., 2017; Bruna et al., 2018). In this regard, a recent study showed that BDNF-mediated activation of Nrf2 in astrocytes, which is under circadian control, protects dopaminergic neurons from ferroptosis (Ishii et al., 2018). Of note, in primary hippocampal neurons, ROS and RyR-mediated calcium signals are required for BDNF-induced nuclear translocation of Nrf2 (Bruna et al., 2018). It remains to be established if iron overload in hippocampal neurons engages BDNF and RyR-mediated calcium signals to promote Nrf2-induced expression of protective genes.
Conclusion
This article reviews the little acknowledged relationship between iron and calcium in neurodegeneration. The main premise of this article is that increased ROS production generated by iron accumulation profoundly influences calcium homeostasis and signaling (Figure 4).
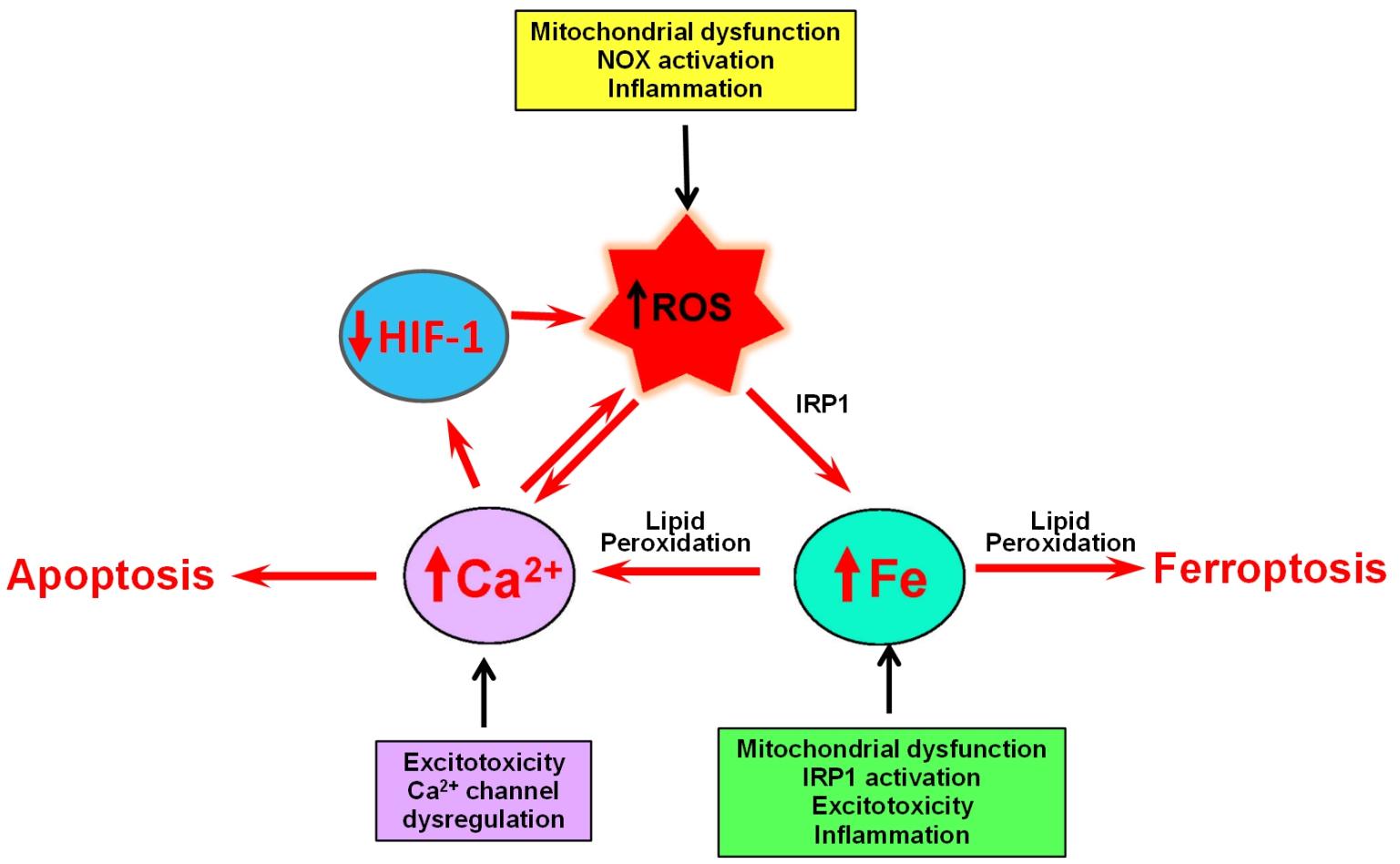
Figure 4. The self-feeding cycle ROS–Fe–Ca. Mitochondrial dysfunction promotes in increased ROS and activation IRP1, which results in increased iron accumulation. Excess iron promotes lipid peroxidation by hydroxyl radicals derived from the Fenton reaction. Lipid peroxidation modifies the activity of a variety of proteins involved in calcium homeostasis that results in massive calcium influx. Increased cytoplasmic calcium levels result in increased mitochondrial calcium and accentuated mitochondrial dysfunction, oxidative stress, and damage. If uncontrolled, this ROS–iron–calcium self-feeding cycle becomes deleterious to mitochondrial and neuronal function, leading eventually to apoptotic or ferroptotic cell death. In yet another aspect of this cycle, high intracellular calcium levels increase the rate of HIF-1 proteasome degradation, which results in increased ROS levels, product of decreased expression of antioxidant proteins.
The influence of increased ROS is clearly shown in the process of oxytosis, in which lipid peroxidation initiated by Fenton-derived hydroxyl radicals results in the modification of a variety of proteins involved in calcium homeostasis that brings to calcium upsurges and cell death. Increased cytoplasmic calcium levels caused by iron dysregulation result in increased mitochondrial calcium and accentuated oxidative stress and damage. If uncontrolled, this calcium/iron self-feeding cycle becomes deleterious to mitochondrial and neuronal function, leading eventually to neuronal death. In yet another aspect of neurodegeneration, high intracellular calcium levels increase the rate of HIF-1 proteasomal degradation while high levels of iron both maintain the activity of HIF-1 hydrolases and, through oxidative stress increase the cellular levels of calcium by activation of ER and PMCA channels. The main conclusion we draw from this review is that dysregulated iron or calcium levels promote deleterious crosstalk between iron and calcium that can result in neuronal dysfunction and death. To our knowledge, this is the first attempt in literature to systemize this important relationship.
Author Contributions
MN and CH wrote this article.
Funding
This work was supported by FONDEF project 17I10095 from the National Commission of Science and Technology (CONICYT) of Chile; Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT; Grants Nos. 1170053 and 11140580), and by Biomedical Neuroscience Institute (Grant No. BNI P09-015-F).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alzheimer’s Association Calcium Hypothesis Workgroup [AACHW] (2017). Calcium hypothesis of Alzheimer’s disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 13, 178.e17–182.e17. doi: 10.1016/j.jalz.2016.12.006
An, H., Zeng, X., Niu, T., Li, G., Yang, J., Zheng, L., et al. (2018). Quantifying iron deposition within the substantia nigra of Parkinson’s disease by quantitative susceptibility mapping. J. Neurol. Sci. 386, 46–52. doi: 10.1016/j.jns.2018.01.008
Angelova, P. R., and Abramov, A. Y. (2017). Alpha-synuclein and beta-amyloid - different targets, same players: calcium, free radicals and mitochondria in the mechanism of neurodegeneration. Biochem. Biophys. Res. Commun. 483, 1110–1115. doi: 10.1016/j.bbrc.2016.07.103
Ansari, M. A., and Scheff, S. W. (2011). NADPH-oxidase activation and cognition in Alzheimer disease progression. Free Radic. Biol. Med. 51, 171–178. doi: 10.1016/j.freeradbiomed.2011.03.025
Area-Gomez, E., and Schon, E. A. (2017). On the pathogenesis of Alzheimer’s Disease: the MAM hypothesis. FASEB J. 31, 864–867. doi: 10.1096/fj.201601309
Arruda, S. F., Siqueira, E. M., and De Valencia, F. F. (2009). Vitamin A deficiency increases hepcidin expression and oxidative stress in rat. Nutrition 25, 472–478. doi: 10.1016/j.nut.2008.11.030
Asiimwe, N., Yeo, S. G., Kim, M. S., Jung, J., and Jeong, N. Y. (2016). Nitric oxide: exploring the contextual link with alzheimer’s disease. Oxid. Med. Cell. Longev. 2016:7205747. doi: 10.1155/2016/7205747
Bading, H. (2013). Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci. 14, 593–608. doi: 10.1038/nrn3531
Barrett, J. P., Henry, R. J., Villapol, S., Stoica, B. A., Kumar, A., Burns, M. P., et al. (2017). NOX2 deficiency alters macrophage phenotype through an IL-10/STAT3 dependent mechanism: implications for traumatic brain injury. J. Neuroinflammation 14:65. doi: 10.1186/s12974-017-0843-4
Baumgartel, K., and Mansuy, I. M. (2012). Neural functions of calcineurin in synaptic plasticity and memory. Learn. Mem. 19, 375–384. doi: 10.1101/lm.027201.112
Becerril-Ortega, J., Bordji, K., Freret, T., Rush, T., and Buisson, A. (2014). Iron overload accelerates neuronal amyloid-beta production and cognitive impairment in transgenic mice model of Alzheimer’s disease. Neurobiol. Aging 35, 2288–2301. doi: 10.1016/j.neurobiolaging.2014.04.019
Berchner-Pfannschmidt, U., Petrat, F., Doege, K., Trinidad, B., Freitag, P., Metzen, E., et al. (2004). Chelation of cellular calcium modulates hypoxia-inducible gene expression through activation of hypoxia-inducible factor-1alpha. J. Biol. Chem. 279, 44976–44986. doi: 10.1074/jbc.M313995200
Berg, D., and Youdim, M. B. (2006). Role of iron in neurodegenerative disorders. Top. Magn. Reson. Imaging 17, 5–17. doi: 10.1097/01.rmr.0000245461.90406.ad
Berridge, M. J. (1998). Neuronal calcium signaling. Neuron 21, 13–26. doi: 10.1016/S0896-6273(00)80510-3
Berridge, M. J., Bootman, M. D. and Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529. doi: 10.1038/nrm1155
Berrocal, M., Corbacho, I., Vazquez-Hernandez, M., Avila, J., Sepulveda, M. R., and Mata, A. M. (2015). Inhibition of PMCA activity by tau as a function of aging and Alzheimer’s neuropathology. Biochim. Biophys. Acta 1852, 1465–1476. doi: 10.1016/j.bbadis.2015.04.007
Bezprozvanny, I., and Mattson, M. P. (2008). Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 31, 454–463. doi: 10.1016/j.tins.2008.06.005
Bindokas, V. P., Jordan, J., Lee, C. C., and Miller, R. J. (1996). Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J. Neurosci. 16, 1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996
Bolton, M. M., Pittman, A. J., and Lo, D. C. (2000). Brain-derived neurotrophic factor differentially regulates excitatory and inhibitory synaptic transmission in hippocampal cultures. J. Neurosci. 20, 3221–3232. doi: 10.1523/JNEUROSCI.20-09-03221.2000
Bors, W., Buettner, G. R., Michel, C., and Saran, M. (1990). Calcium in lipid peroxidation: does calcium interact with superoxide? Arch. Biochem. Biophys. 278, 269–272. doi: 10.1016/0003-9861(90)90258-Z
Bose, A., and Beal, M. F. (2016). Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 139(Suppl. 1), 216–231. doi: 10.1111/jnc.13731
Bostanci, M. O., and Bagirici, F. (2013). Blocking of L-type calcium channels protects hippocampal and nigral neurons against iron neurotoxicity. The role of L-type calcium channels in iron-induced neurotoxicity. Int. J. Neurosci. 123, 876–882. doi: 10.3109/00207454.2013.813510
Bouvier, E., Brouillard, F., Molet, J., Claverie, D., Cabungcal, J. H., Cresto, N., et al. (2017). Nrf2-dependent persistent oxidative stress results in stress-induced vulnerability to depression. Mol. Psychiatry 22, 1701–1713. doi: 10.1038/mp.2016.211
Braughler, J. M., Duncan, L. A., and Chase, R. L. (1985). Interaction of lipid peroxidation and calcium in the pathogenesis of neuronal injury. Cent. Nerv. Syst. Trauma 2, 269–283. doi: 10.1089/cns.1985.2.269
Brennan, A. M., Suh, S. W., Won, S. J., Narasimhan, P., Kauppinen, T. M., Lee, H., et al. (2009). NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 12, 857–863. doi: 10.1038/nn.2334
Brini, M., Cali, T., Ottolini, D., and Carafoli, E. (2014). Neuronal calcium signaling: function and dysfunction. Cell. Mol. Life Sci. 71, 2787–2814. doi: 10.1007/s00018-013-1550-7
Brini, M., Leanza, L., and Szabo, I. (2017). Lipid-mediated modulation of intracellular ion channels and redox state: physiopathological implications. Antioxid. Redox Signal. doi: 10.1089/ars.2017.7215 [Epub ahead of print].
Bruick, R. K., and McKnight, S. L. (2001). A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294, 1337–1340. doi: 10.1126/science.1066373
Bruna, B., Lobos, P., Herrera-Molina, R., Hidalgo, C., Paula-Lima, A., and Adasme, T. (2018). The signaling pathways underlying BDNF-induced Nrf2 hippocampal nuclear translocation involve ROS, RyR-Mediated Ca(2+) signals, ERK and PI3K. Biochem. Biophys. Res. Commun. 505, 201–207. doi: 10.1016/j.bbrc.2018.09.080
Bulk, M., Van Der Weerd, L., Breimer, W., Lebedev, N., Webb, A., Goeman, J. J., et al. (2018). Quantitative comparison of different iron forms in the temporal cortex of Alzheimer patients and control subjects. Sci. Rep. 8:6898. doi: 10.1038/s41598-018-25021-7
Burbulla, L. F., Song, P., Mazzulli, J. R., Zampese, E., Wong, Y. C., Jeon, S., et al. (2017). Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 357, 1255–1261. doi: 10.1126/science.aam9080
Cabantchik, Z. I. (2014). Labile iron in cells and body fluids: physiology, pathology, and pharmacology. Front. Pharmacol. 5:45. doi: 10.3389/fphar.2014.00045
Caltagirone, A., Weiss, G., and Pantopoulos, K. (2001). Modulation of cellular iron metabolism by hydrogen peroxide. Effects of H2O2 on the expression and function of iron-responsive element-containing mRNAs in B6 fibroblasts. J. Biol. Chem. 276, 19738–19745. doi: 10.1074/jbc.M100245200
Cannon, J. R., Tapias, V., Na, H. M., Honick, A. S., Drolet, R. E., and Greenamyre, J. T. (2009). A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 34, 279–290. doi: 10.1016/j.nbd.2009.01.016
Carocci, A., Catalano, A., Sinicropi, M. S., and Genchi, G. (2018). Oxidative stress and neurodegeneration: the involvement of iron. Biometals 31, 715–735. doi: 10.1007/s10534-018-0126-2
Chan, C. S., Gertler, T. S., and Surmeier, D. J. (2009). Calcium homeostasis, selective vulnerability and Parkinson’s disease. Trends Neurosci. 32, 249–256. doi: 10.1016/j.tins.2009.01.006
Cheah, J. H., Kim, S. F., Hester, L. D., Clancy, K. W., Patterson, S. E. III, Papadopoulos, V., et al. (2006). NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron 51, 431–440. doi: 10.1016/j.neuron.2006.07.011
Chinopoulos, C., and Adam-Vizi, V. (2006). Calcium, mitochondria and oxidative stress in neuronal pathology. Novel aspects of an enduring theme. FEBS J. 273, 433–450. doi: 10.1111/j.1742-4658.2005.05103.x
Chorley, B. N., Campbell, M. R., Wang, X., Karaca, M., Sambandan, D., Bangura, F., et al. (2012). Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 40, 7416–7429. doi: 10.1093/nar/gks409
Cobley, J. N., Fiorello, M. L., and Bailey, D. M. (2018). 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 15, 490–503. doi: 10.1016/j.redox.2018.01.008
Creighton-Gutteridge, M., and Tyrrell, R. M. (2002). A novel iron chelator that does not induce HIF-1 activity. Free Radic. Biol. Med. 33, 356–363. doi: 10.1016/S0891-5849(02)00884-5
Crichton, R. R., Dexter, D. T., and Ward, R. J. (2011). Brain iron metabolism and its perturbation in neurological diseases. J. Neural Transm. 118, 301–314. doi: 10.1007/s00702-010-0470-z
Cristovao, J. S., Santos, R., and Gomes, C. M. (2016). Metals and neuronal metal binding proteins implicated in Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2016:9812178. doi: 10.1155/2016/9812178
Damasceno, F. C., Condeles, A. L., Lopes, A. K. B., Facci, R. R., Linares, E., Truzzi, D. R., et al. (2018). The labile iron pool attenuates peroxynitrite-dependent damage and can no longer be considered solely a pro-oxidative cellular iron source. J. Biol. Chem. 293, 8530–8542. doi: 10.1074/jbc.RA117.000883
Davies, M. J. (2005). The oxidative environment and protein damage. Biochim. Biophys. Acta 1703, 93–109. doi: 10.1016/j.bbapap.2004.08.007
Demuro, A., Parker, I., and Stutzmann, G. E. (2010). Calcium signaling and amyloid toxicity in Alzheimer disease. J. Biol. Chem. 285, 12463–12468. doi: 10.1074/jbc.R109.080895
Dev, S., Kumari, S., Singh, N., Kumar Bal, S., Seth, P., and Mukhopadhyay, C. K. (2015). Role of extracellular hydrogen peroxide in regulation of iron homeostasis genes in neuronal cells: implication in iron accumulation. Free Radic. Biol. Med. 86, 78–89. doi: 10.1016/j.freeradbiomed.2015.05.025
Di Lisa, F., and Bernardi, P. (2009). A CaPful of mechanisms regulating the mitochondrial permeability transition. J. Mol. Cell Cardiol. 46, 775–780. doi: 10.1016/j.yjmcc.2009.03.006
DiSabato, D. J., Quan, N., and Godbout, J. P. (2016). Neuroinflammation: the devil is in the details. J. Neurochem. 139(Suppl. 2), 136–153. doi: 10.1111/jnc.13607
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Dixon, S. J., and Stockwell, B. R. (2014). The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10, 9–17. doi: 10.1038/nchembio.1416
Drapier, J. C., Hirling, H., Wietzerbin, J., Kaldy, P., and Kuhn, L. C. (1993). Biosynthesis of nitric oxide activates iron regulatory factor in macrophages. EMBO J. 12, 3643–3649. doi: 10.1002/j.1460-2075.1993.tb06038.x
Duncan, A. J., and Heales, S. J. (2005). Nitric oxide and neurological disorders. Mol. Aspects Med. 26, 67–96. doi: 10.1016/j.mam.2004.09.004
Flagg, S. C., Martin, C. B., Taabazuing, C. Y., Holmes, B. E., and Knapp, M. J. (2012). Screening chelating inhibitors of HIF-prolyl hydroxylase domain 2 (PHD2) and factor inhibiting HIF (FIH). J. Inorg. Biochem. 113, 25–30. doi: 10.1016/j.jinorgbio.2012.03.002
Franco-Iborra, S., Vila, M., and Perier, C. (2018). Mitochondrial quality control in neurodegenerative diseases: focus on parkinson’s disease and huntington’s disease. Front. Neurosci. 12:342. doi: 10.3389/fnins.2018.00342
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., and Brown, G. C. (2018). Neuronal cell death. Physiol. Rev. 98, 813–880. doi: 10.1152/physrev.00011.2017
Fukui, K. (2016). Reactive oxygen species induce neurite degeneration before induction of cell death. J. Clin. Biochem. Nutr. 59, 155–159. doi: 10.3164/jcbn.16-34
Gaasch, J. A., Geldenhuys, W. J., Lockman, P. R., Allen, D. D., and Van Der Schyf, C. J. (2007). Voltage-gated calcium channels provide an alternate route for iron uptake in neuronal cell cultures. Neurochem. Res. 32, 1686–1693. doi: 10.1007/s11064-007-9313-1
Gao, H. M., Zhou, H., and Hong, J. S. (2012). NADPH oxidases: novel therapeutic targets for neurodegenerative diseases. Trends Pharmacol. Sci. 33, 295–303. doi: 10.1016/j.tips.2012.03.008
Garrick, M. D., Singleton, S. T., Vargas, F., Kuo, H. C., Zhao, L., Knopfel, M., et al. (2006). DMT1: which metals does it transport? Biol. Res. 39, 79–85. doi: 10.4067/S0716-97602006000100009
Ghosh, D. K., and Salerno, J. C. (2003). Nitric oxide synthases: domain structure and alignment in enzyme function and control. Front. Biosci. 8, D193–D209. doi: 10.2741/959
Gibson, G. E., and Thakkar, A. (2017). Interactions of mitochondria/metabolism and calcium regulation in alzheimer’s disease: a calcinist point of view. Neurochem. Res. 42, 1636–1648. doi: 10.1007/s11064-017-2182-3
Giorgi, C., Baldassari, F., Bononi, A., Bonora, M., De Marchi, E., Marchi, S., et al. (2012). Mitochondrial Ca(2+) and apoptosis. Cell Calcium 52, 36–43. doi: 10.1016/j.ceca.2012.02.008
Giorgi, C., Marchi, S., and Pinton, P. (2018). The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 19, 713–730. doi: 10.1038/s41580-018-0052-8
Girouard, H., Wang, G., Gallo, E. F., Anrather, J., Zhou, P., Pickel, V. M., et al. (2009). NMDA receptor activation increases free radical production through nitric oxide and NOX2. J. Neurosci. 29, 2545–2552. doi: 10.1523/JNEUROSCI.0133-09.2009
Gonsette, R. E. (2008). Neurodegeneration in multiple sclerosis: the role of oxidative stress and excitotoxicity. J. Neurol. Sci. 274, 48–53. doi: 10.1016/j.jns.2008.06.029
Gordeeva, A. V., Zvyagilskaya, R. A., and Labas, Y. A. (2003). Cross-talk between reactive oxygen species and calcium in living cells. Biochemistry 68, 1077–1080.
Gorlach, A., Bertram, K., Hudecova, S., and Krizanova, O. (2015). Calcium and ROS: a mutual interplay. Redox Biol. 6, 260–271. doi: 10.1016/j.redox.2015.08.010
Green, S. P., Cairns, B., Rae, J., Errett-Baroncini, C., Hongo, J. A., Erickson, R. W., et al. (2001). Induction of gp91-phox, a component of the phagocyte NADPH oxidase, in microglial cells during central nervous system inflammation. J. Cereb. Blood Flow Metab. 21, 374–384. doi: 10.1097/00004647-200104000-00006
Grosskreutz, J., Van Den Bosch, L., and Keller, B. U. (2010). Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 47, 165–174. doi: 10.1016/j.ceca.2009.12.002
Guo, C., Hao, L. J., Yang, Z. H., Chai, R., Zhang, S., Gu, Y., et al. (2016). Deferoxamine-mediated up-regulation of HIF-1alpha prevents dopaminergic neuronal death via the activation of MAPK family proteins in MPTP-treated mice. Exp. Neurol. 280, 13–23. doi: 10.1016/j.expneurol.2016.03.016
Guo, C., Zhang, Y. X., Wang, T., Zhong, M. L., Yang, Z. H., Hao, L. J., et al. (2015). Intranasal deferoxamine attenuates synapse loss via up-regulating the P38/HIF-1alpha pathway on the brain of APP/PS1 transgenic mice. Front. Aging Neurosci. 7:104. doi: 10.3389/fnagi.2015.00104
Guo, J. D., Zhao, X., Li, Y., Li, G. R., and Liu, X. L. (2018). Damage to dopaminergic neurons by oxidative stress in Parkinson’s disease (Review). Int. J. Mol. Med. 41, 1817–1825. doi: 10.3892/ijmm.2018.3406
Guzman, J. N., Sanchez-Padilla, J., Chan, C. S., and Surmeier, D. J. (2009). Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci. 29, 11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009
Hajieva, P., Baeken, M. W., and Moosmann, B. (2018). The role of plasma membrane calcium atpases (PMCAS) in neurodegenerative disorders. Neurosci. Lett. 663, 29–38. doi: 10.1016/j.neulet.2017.09.033
Halliwell, B. (2006). Oxidative stress and neurodegeneration: where are we now? J. Neurochem. 97, 1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x
Hambright, W. S., Fonseca, R. S., Chen, L., Na, R., and Ran, Q. (2017). Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 12, 8–17. doi: 10.1016/j.redox.2017.01.021
Hanke, M. L., and Kielian, T. (2011). Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin. Sci. 121, 367–387. doi: 10.1042/CS20110164
Hanson, E. S., and Leibold, E. A. (1999). Regulation of the iron regulatory proteins by reactive nitrogen and oxygen species. Gene Expr. 7, 367–376.
Hansson Petersen, C. A., Alikhani, N., Behbahani, H., Wiehager, B., Pavlov, P. F., Alafuzoff, I., et al. (2008). The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. U.S.A. 105, 13145–13150. doi: 10.1073/pnas.0806192105
Hepp, S., Gerich, F. J., and Muller, M. (2005). Sulfhydryl oxidation reduces hippocampal susceptibility to hypoxia-induced spreading depression by activating BK channels. J. Neurophysiol. 94, 1091–1103. doi: 10.1152/jn.00291.2005
Hewitson, K. S., Mcneill, L. A., Elkins, J. M., and Schofield, C. J. (2003). The role of iron and 2-oxoglutarate oxygenases in signalling. Biochem. Soc. Trans. 31, 510–515. doi: 10.1042/bst0310510
Hidalgo, C., and Arias-Cavieres, A. (2016). Calcium, reactive oxygen species, and synaptic plasticity. Physiology 31, 201–215. doi: 10.1152/physiol.00038.2015
Hidalgo, C., Carrasco, M. A., Muñoz, P., and Núñez, M. T. (2007). A role for reactive oxygen/nitrogen species and iron on neuronal synaptic plasticity. Antioxid. Redox. Signal. 9, 245–255. doi: 10.1089/ars.2007.9.245
Hidalgo, C., and Donoso, P. (2008). Crosstalk between calcium and redox signaling: from molecular mechanisms to health implications. Antioxid. Redox Signal. 10, 1275–1312. doi: 10.1089/ars.2007.1886
Hidalgo, C., and Núñez, M. T. (2007). Calcium, iron and neuronal function. IUBMB Life 59, 280–285. doi: 10.1080/15216540701222906
Huang, M. L., Lane, D. J., and Richardson, D. R. (2011). Mitochondrial mayhem: the mitochondrion as a modulator of iron metabolism and its role in disease. Antioxid. Redox Signal. 15, 3003–3019. doi: 10.1089/ars.2011.3921
Huang, X., Atwood, C. S., Hartshorn, M. A., Multhaup, G., Goldstein, L. E., Scarpa, R. C., et al. (1999). The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 38, 7609–7616. doi: 10.1021/bi990438f
Ishii, T., Warabi, E., and Mann, G. E. (2018). Circadian control of BDNF-mediated Nrf2 activation in astrocytes protects dopaminergic neurons from ferroptosis. Free Radic. Biol. Med. doi: 10.1016/j.freeradbiomed.2018.09.002 [Epub ahead of print].
Jaiswal, M. K. (2013). Calcium, mitochondria, and the pathogenesis of ALS: the good, the bad, and the ugly. Front. Cell. Neurosci. 7:199. doi: 10.3389/fncel.2013.00199
Jellinger, K. A. (1999). The role of iron in neurodegeneration: prospects for pharmacotherapy of Parkinson’s disease. Drugs Aging 14, 115–140. doi: 10.2165/00002512-199914020-00004
Jeney, V., Balla, J., Yachie, A., Varga, Z., Vercellotti, G. M., Eaton, J. W., et al. (2002). Pro-oxidant and cytotoxic effects of circulating heme. Blood 100, 879–887. doi: 10.1182/blood.V100.3.879
Jeong, S. Y., Rathore, K. I., Schulz, K., Ponka, P., Arosio, P., and David, S. (2009). Dysregulation of iron homeostasis in the CNS contributes to disease progression in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 29, 610–619. doi: 10.1523/JNEUROSCI.5443-08.2009
Ji, X., Wang, H., Zhu, J., Zhu, L., Pan, H., Li, W., et al. (2014). Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Int. J. Cancer 135, 574–584. doi: 10.1002/ijc.28699
Jiang, L., Bechtel, M. D., Galeva, N. A., Williams, T. D., Michaelis, E. K., and Michaelis, M. L. (2012). Decreases in plasma membrane Ca(2)(+)-ATPase in brain synaptic membrane rafts from aged rats. J. Neurochem. 123, 689–699. doi: 10.1111/j.1471-4159.2012.07918.x
Joseph, S. K., Young, M. P., Alzayady, K., Yule, D. I., Ali, M., Booth, D. M., et al. (2018). Redox regulation of type-I inositol trisphosphate receptors in intact mammalian cells. J. Biol. Chem. 293, 17464–17476. doi: 10.1074/jbc.RA118.005624
Kahlert, S., Zundorf, G., and Reiser, G. (2005). Glutamate-mediated influx of extracellular Ca2+ is coupled with reactive oxygen species generation in cultured hippocampal neurons but not in astrocytes. J. Neurosci. Res. 79, 262–271. doi: 10.1002/jnr.20322
Kasarskis, E. J., Tandon, L., Lovell, M. A., and Ehmann, W. D. (1995). Aluminum, calcium, and iron in the spinal cord of patients with sporadic amyotrophic lateral sclerosis using laser microprobe mass spectroscopy: a preliminary study. J. Neurol. Sci. 130, 203–208. doi: 10.1016/0022-510X(95)00037-3
Kastanenka, K. V., Bussiere, T., Shakerdge, N., Qian, F., Weinreb, P. H., Rhodes, K., et al. (2016). Immunotherapy with aducanumab restores calcium homeostasis in Tg2576 Mice. J. Neurosci. 36, 12549–12558. doi: 10.1523/JNEUROSCI.2080-16.2016
Kerins, M. J., and Ooi, A. (2018). The roles of NRF2 in modulating cellular iron homeostasis. Antioxid. Redox Signal. 29, 1756–1773. doi: 10.1089/ars.2017.7176
Kietzmann, T., Knabe, W., and Schmidt-Kastner, R. (2001). Hypoxia and hypoxia-inducible factor modulated gene expression in brain: involvement in neuroprotection and cell death. Eur. Arch. Psychiatry Clin. Neurosci. 251, 170–178. doi: 10.1007/s004060170037
Kim, T. H., Hur, E. G., Kang, S. J., Kim, J. A., Thapa, D., Lee, Y. M., et al. (2011). NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Cancer Res. 71, 2260–2275. doi: 10.1158/0008-5472.CAN-10-3007
Kishida, K. T., and Klann, E. (2007). Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid. Redox Signal. 9, 233–244. doi: 10.1089/ars.2007.9.233
Kupershmidt, L., Weinreb, O., Amit, T., Mandel, S., Bar-Am, O., and Youdim, M. B. (2011). Novel molecular targets of the neuroprotective/neurorescue multimodal iron chelating drug M30 in the mouse brain. Neuroscience 189, 345–358. doi: 10.1016/j.neuroscience.2011.03.040
Kupershmidt, L., Weinreb, O., Amit, T., Mandel, S., Carri, M. T., and Youdim, M. B. (2009). Neuroprotective and neuritogenic activities of novel multimodal iron-chelating drugs in motor-neuron-like NSC-34 cells and transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. 23, 3766–3779. doi: 10.1096/fj.09-130047
Kushnir, A., Wajsberg, B., and Marks, A. R. (2018). Ryanodine receptor dysfunction in human disorders. Biochim. Biophys. Acta Mol. Cell. Res. doi: 10.1016/j.bbamcr.2018.07.011 [Epub ahead of print].
Lane, D. J. R., Ayton, S., and Bush, A. I. (2018). Iron and Alzheimer’s disease: an update on emerging mechanisms. J. Alzheimers Dis. 64, S379–S395. doi: 10.3233/JAD-179944
Lee, D. G., Park, J., Lee, H. S., Lee, S. R., and Lee, D. S. (2016). Iron overload-induced calcium signals modulate mitochondrial fragmentation in HT-22 hippocampal neuron cells. Toxicology 365, 17–24. doi: 10.1016/j.tox.2016.07.022
Lee, D. W., and Andersen, J. K. (2006). Role of HIF-1 in iron regulation: potential therapeutic strategy for neurodegenerative disorders. Curr. Mol. Med. 6, 883–893. doi: 10.2174/156652406779010849
Lee, J. C., Tae, H. J., Kim, I. H., Cho, J. H., Lee, T. K., Park, J. H., et al. (2017). Roles of HIF-1alpha, VEGF, and NF-kappaB in ischemic preconditioning-mediated neuroprotection of hippocampal CA1 pyramidal neurons against a subsequent transient cerebral ischemia. Mol. Neurobiol. 54, 6984–6998. doi: 10.1007/s12035-016-0219-2
Lee, J. H., Han, Y. H., Kang, B. M., Mun, C. W., Lee, S. J., and Baik, S. K. (2013). Quantitative assessment of subcortical atrophy and iron content in progressive supranuclear palsy and parkinsonian variant of multiple system atrophy. J. Neurol. 260, 2094–2101. doi: 10.1007/s00415-013-6951-x
Lewerenz, J., Ates, G., Methner, A., Conrad, M., and Maher, P. (2018). Oxytosis/ferroptosis-(Re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front. Neurosci. 12:214. doi: 10.3389/fnins.2018.00214
Li, X., Chen, W., Zhang, L., Liu, W. B., and Fei, Z. (2013). Inhibition of store-operated calcium entry attenuates MPP(+)-induced oxidative stress via preservation of mitochondrial function in PC12 cells: involvement of Homer1a. PLoS One 8:e83638. doi: 10.1371/journal.pone.0083638
Liao, Y., Dong, Y., and Cheng, J. (2017). The function of the mitochondrial calcium uniporter in neurodegenerative disorders. Int. J. Mol. Sci. 18:E248. doi: 10.3390/ijms18020248
Lim, H., Kim, D., and Lee, S. J. (2013). Toll-like receptor 2 mediates peripheral nerve injury-induced NADPH oxidase 2 expression in spinal cord microglia. J. Biol. Chem. 288, 7572–7579. doi: 10.1074/jbc.M112.414904
Lockman, J. A., Geldenhuys, W. J., Bohn, K. A., Desilva, S. F., Allen, D. D., and Van Der Schyf, C. J. (2012). Differential effect of nimodipine in attenuating iron-induced toxicity in brain- and blood-brain barrier-associated cell types. Neurochem. Res. 37, 134–142. doi: 10.1007/s11064-011-0591-2
Lourenco, C. F., Ledo, A., Barbosa, R. M., and Laranjinha, J. (2017). Neurovascular-neuroenergetic coupling axis in the brain: master regulation by nitric oxide and consequences in aging and neurodegeneration. Free Radic. Biol. Med. 108, 668–682. doi: 10.1016/j.freeradbiomed.2017.04.026
Lu, C., Chan, S. L., Fu, W., and Mattson, M. P. (2002). The lipid peroxidation product 4-hydroxynonenal facilitates opening of voltage-dependent Ca2+ channels in neurons by increasing protein tyrosine phosphorylation. J. Biol. Chem. 277, 24368–24375. doi: 10.1074/jbc.M201924200
Ludtmann, M. H. R., and Abramov, A. Y. (2018). Mitochondrial calcium imbalance in Parkinson’s disease. Neurosci. Lett. 663, 86–90. doi: 10.1016/j.neulet.2017.08.044
Lynch, M. A. (2004). Long-term potentiation and memory. Physiol. Rev. 84, 87–136. doi: 10.1152/physrev.00014.2003
Maher, P., Van Leyen, K., Dey, P. N., Honrath, B., Dolga, A., and Methner, A. (2018). The role of Ca(2+) in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell Calcium 70, 47–55. doi: 10.1016/j.ceca.2017.05.007
Marchi, S., Patergnani, S., Missiroli, S., Morciano, G., Rimessi, A., Wieckowski, M. R., et al. (2018). Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 69, 62–72. doi: 10.1016/j.ceca.2017.05.003
Marro, S., Chiabrando, D., Messana, E., Stolte, J., Turco, E., Tolosano, E., et al. (2010). Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position -7007 of the FPN1 promoter. Haematologica 95, 1261–1268. doi: 10.3324/haematol.2009.020123
Martins, E. A., Robalinho, R. L., and Meneghini, R. (1995). Oxidative stress induces activation of a cytosolic protein responsible for control of iron uptake. Arch. Biochem. Biophys. 316, 128–134. doi: 10.1006/abbi.1995.1019
Mata, A. M., and Sepulveda, M. R. (2010). Plasma membrane Ca-ATPases in the nervous system during development and ageing. World J. Biol. Chem. 1, 229–234. doi: 10.4331/wjbc.v1.i7.229
Mattson, M. P. (2010). ER calcium and Alzheimer’s disease: in a state of flux. Sci. Signal. 3:e10. doi: 10.1126/scisignal.3114pe10
McCarthy, R. C., Sosa, J. C., Gardeck, A. M., Baez, A. S., Lee, C. H., and Wessling-Resnick, M. (2018). Inflammation-induced iron transport and metabolism by brain microglia. J. Biol. Chem. 293, 7853–7863. doi: 10.1074/jbc.RA118.001949
Mechlovich, D., Amit, T., Bar-Am, O., Mandel, S., Youdim, M. B., and Weinreb, O. (2014). The novel multi-target iron chelator, M30 modulates HIF-1alpha-related glycolytic genes and insulin signaling pathway in the frontal cortex of APP/PS1 Alzheimer’s disease mice. Curr. Alzheimer Res. 11, 119–127. doi: 10.2174/1567205010666131212112529
Mena, N. P., Urrutia, P. J., Lourido, F., Carrasco, C. M., and Núñez, M. T. (2015). Mitochondrial iron homeostasis and its dysfunctions in neurodegenerative disorders. Mitochondrion 21, 92–105. doi: 10.1016/j.mito.2015.02.001
Merelli, A., Rodriguez, J. C. G., Folch, J., Regueiro, M. R., Camins, A., and Lazarowski, A. (2018). Understanding the role of hypoxia inducible factor during neurodegeneration for new therapeutics opportunities. Curr. Neuropharmacol. 16, 1484–1498. doi: 10.2174/1570159X16666180110130253
Miyamoto, M., Murphy, T. H., Schnaar, R. L., and Coyle, J. T. (1989). Antioxidants protect against glutamate-induced cytotoxicity in a neuronal cell line. J. Pharmacol. Exp. Ther. 250, 1132–1140.
Moldogazieva, N. T., Lutsenko, S. V., and Terentiev, A. A. (2018). Reactive oxygen and nitrogen species-induced protein modifications: implication in carcinogenesis and anticancer therapy. Cancer Res. 78, 6040–6047. doi: 10.1158/0008-5472.CAN-18-0980
Mole, D. R., Blancher, C., Copley, R. R., Pollard, P. J., Gleadle, J. M., Ragoussis, J., et al. (2009). Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 284, 16767–16775. doi: 10.1074/jbc.M901790200
Moncada, S., and Bolaños, J. P. (2006). Nitric oxide, cell bioenergetics and neurodegeneration. J. Neurochem. 97, 1676–1689. doi: 10.1111/j.1471-4159.2006.03988.x
Mueller, S., Pantopoulos, K., Hubner, C. A., Stremmel, W., and Hentze, M. W. (2001). IRP1 activation by extracellular oxidative stress in the perfused rat liver. J. Biol. Chem. 276, 23192–23196. doi: 10.1074/jbc.M100654200
Muhling, T., Duda, J., Weishaupt, J. H., Ludolph, A. C., and Liss, B. (2014). Elevated mRNA-levels of distinct mitochondrial and plasma membrane Ca(2+) transporters in individual hypoglossal motor neurons of endstage SOD1 transgenic mice. Front. Cell. Neurosci. 8:353. doi: 10.3389/fncel.2014.00353
Muñoz, P. (2012). Iron-mediated redox modulation in neural plasticity. Commun. Integr. Biol. 5, 166–168. doi: 10.4161/cib.18710
Muñoz, P., Humeres, A., Elgueta, C., Kirkwood, A., Hidalgo, C., and Núñez, M. T. (2011). Iron mediates N-methyl-D-aspartate receptor-dependent stimulation of calcium-induced pathways and hippocampal synaptic plasticity. J. Biol. Chem. 286, 13382–13392. doi: 10.1074/jbc.M110.213785
Muñoz, Y., Carrasco, C. M., Campos, J. D., Aguirre, P., and Núñez, M. T. (2016). Parkinson’s Disease: the mitochondria-iron link. Parkinsons Dis. 2016:7049108. doi: 10.1155/2016/7049108
Muravchick, S., and Levy, R. J. (2006). Clinical implications of mitochondrial dysfunction. Anesthesiology 105, 819–837. doi: 10.1097/00000542-200610000-00029
Murphy, T. H., Malouf, A. T., Sastre, A., Schnaar, R. L., and Coyle, J. T. (1988). Calcium-dependent glutamate cytotoxicity in a neuronal cell line. Brain Res. 444, 325–332. doi: 10.1016/0006-8993(88)90941-9
Murphy, T. H., Miyamoto, M., Sastre, A., Schnaar, R. L., and Coyle, J. T. (1989). Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2, 1547–1558. doi: 10.1016/0896-6273(89)90043-3
Murray-Kolb, L. E. (2013). Iron and brain functions. Curr. Opin. Clin. Nutr. Metab. Care 16, 703–707. doi: 10.1097/MCO.0b013e3283653ef8
Mustaly-Kalimi, S., Littlefield, A. M., and Stutzmann, G. E. (2018). Calcium signaling deficits in glia and autophagic pathways contributing to neurodegenerative disease. Antioxid. Redox Signal. 29, 1158–1175. doi: 10.1089/ars.2017.7266
Myllyharju, J. (2013). Prolyl 4-hydroxylases, master regulators of the hypoxia response. Acta Physiol. 208, 148–165. doi: 10.1111/apha.12096
Nandal, A., Ruiz, J. C., Subramanian, P., Ghimire-Rijal, S., Sinnamon, R. A., Stemmler, T. L., et al. (2011). Activation of the HIF prolyl hydroxylase by the iron chaperones PCBP1 and PCBP2. Cell Metab. 14, 647–657. doi: 10.1016/j.cmet.2011.08.015
Nemes, R., Koltai, E., Taylor, A. W., Suzuki, K., Gyori, F., and Radak, Z. (2018). Reactive oxygen and nitrogen species regulate key metabolic, anabolic, and catabolic pathways in skeletal muscle. Antioxidants 7:E85. doi: 10.3390/antiox7070085
Núñez, M. T. (2010). Regulatory mechanisms of intestinal iron absorption-uncovering of a fast-response mechanism based on DMT1 and ferroportin endocytosis. Biofactors 36, 88–97. doi: 10.1002/biof.84
Núñez, M. T., and Chana-Cuevas, P. (2018). New perspectives in iron chelation therapy for the treatment of neurodegenerative diseases. Pharmaceuticals 11:E109. doi: 10.3390/ph11040109
Núñez, M. T., Gallardo, V., Muñoz, P., Tapia, V., Esparza, A., Salazar, J., et al. (2004). Progressive iron accumulation induces a biphasic change in the glutathione content of neuroblastoma cells. Free Radic. Biol. Med. 37, 953–960. doi: 10.1016/j.freeradbiomed.2004.06.005
Núñez, M. T., Urrutia, P., Mena, N., Aguirre, P., Tapia, V., and Salazar, J. (2012). Iron toxicity in neurodegeneration. Biometals 25, 761–776. doi: 10.1007/s10534-012-9523-0
Oexle, H., Gnaiger, E., and Weiss, G. (1999). Iron-dependent changes in cellular energy metabolism: influence on citric acid cycle and oxidative phosphorylation. Biochim. Biophys. Acta 1413, 99–107. doi: 10.1016/S0005-2728(99)00088-2
Okubo, Y., Mikami, Y., Kanemaru, K., and Iino, M. (2018). Role of endoplasmic reticulum-mediated Ca(2+) signaling in neuronal cell death. Antioxid. Redox Signal. 29, 1147–1157. doi: 10.1089/ars.2018.7498
Orino, K., Lehman, L., Tsuji, Y., Ayaki, H., Torti, S. V., and Torti, F. M. (2001). Ferritin and the response to oxidative stress. Biochem. J. 357, 241–247. doi: 10.1042/bj3570241
Overk, C. R., Rockenstein, E., Florio, J., Cheng, Q., and Masliah, E. (2015). Differential calcium alterations in animal models of neurodegenerative disease: reversal by FK506. Neuroscience 310, 549–560. doi: 10.1016/j.neuroscience.2015.08.068
Pantopoulos, K., and Hentze, M. W. (1995). Rapid responses to oxidative stress mediated by iron regulatory protein. EMBO J. 14, 2917–2924. doi: 10.1002/j.1460-2075.1995.tb07291.x
Pantopoulos, K., Weiss, G., and Hentze, M. W. (1996). Nitric oxide and oxidative stress (H2O2) control mammalian iron metabolism by different pathways. Mol. Cell. Biol. 16, 3781–3788. doi: 10.1128/MCB.16.7.3781
Paradkar, P. N., and Roth, J. A. (2006a). Nitric oxide transcriptionally down-regulates specific isoforms of divalent metal transporter (DMT1) via NF-kappaB. J. Neurochem. 96, 1768–1777.
Paradkar, P. N., and Roth, J. A. (2006b). Post-translational and transcriptional regulation of DMT1 during P19 embryonic carcinoma cell differentiation by retinoic acid. Biochem. J. 394, 173–183.
Paradkar, P. N., Zumbrennen, K. B., Paw, B. H., Ward, D. M., and Kaplan, J. (2009). Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell. Biol. 29, 1007–1016. doi: 10.1128/MCB.01685-08
Pchitskaya, E., Popugaeva, E., and Bezprozvanny, I. (2018). Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 70, 87–94. doi: 10.1016/j.ceca.2017.06.008
Pelizzoni, I., Macco, R., Morini, M. F., Zacchetti, D., Grohovaz, F., and Codazzi, F. (2011). Iron handling in hippocampal neurons: activity-dependent iron entry and mitochondria-mediated neurotoxicity. Aging Cell 10, 172–183. doi: 10.1111/j.1474-9726.2010.00652.x
Pelizzoni, I., Macco, R., Zacchetti, D., Grohovaz, F., and Codazzi, F. (2008). Iron and calcium in the central nervous system: a close relationship in health and sickness. Biochem. Soc. Trans. 36, 1309–1312. doi: 10.1042/BST0361309
Pelizzoni, I., Zacchetti, D., Campanella, A., Grohovaz, F., and Codazzi, F. (2013). Iron uptake in quiescent and inflammation-activated astrocytes: a potentially neuroprotective control of iron burden. Biochim. Biophys. Acta 1832, 1326–1333. doi: 10.1016/j.bbadis.2013.04.007
Pereira, C., Ferreira, C., Carvalho, C., and Oliveira, C. (1996). Contribution of plasma membrane and endoplasmic reticulum Ca(2+)-ATPases to the synaptosomal [Ca2+]i increase during oxidative stress. Brain Res. 713, 269–277. doi: 10.1016/0006-8993(95)01554-X
Perier, C., Tieu, K., Guegan, C., Caspersen, C., Jackson-Lewis, V., Carelli, V., et al. (2005). Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proc. Natl. Acad. Sci. U.S.A. 102, 19126–19131. doi: 10.1073/pnas.0508215102
Peters, D. G., Connor, J. R., and Meadowcroft, M. D. (2015). The relationship between iron dyshomeostasis and amyloidogenesis in Alzheimer’s disease: two sides of the same coin. Neurobiol. Dis. 81, 49–65. doi: 10.1016/j.nbd.2015.08.007
Phillips, J. D., Kinikini, D. V., Yu, Y., Guo, B., and Leibold, E. A. (1996). Differential regulation of IRP1 and IRP2 by nitric oxide in rat hepatoma cells. Blood 87, 2983–2992.
Piret, J. P., Mottet, D., Raes, M., and Michiels, C. (2002). Is HIF-1alpha a pro- or an anti-apoptotic protein? Biochem. Pharmacol. 64, 889–892.
Ponka, P. (2004). Hereditary causes of disturbed iron homeostasis in the central nervous system. Ann. N. Y. Acad. Sci. 1012, 267–281. doi: 10.1196/annals.1306.022
Popugaeva, E., Pchitskaya, E., and Bezprozvanny, I. (2018). Dysregulation of intracellular calcium signaling in alzheimer’s disease. Antioxid. Redox Signal. 29, 1176–1188. doi: 10.1089/ars.2018.7506
Post, M. R., Lieberman, O. J., and Mosharov, E. V. (2018). Can interactions between alpha-synuclein, dopamine and calcium explain selective neurodegeneration in Parkinson’s disease? Front. Neurosci. 12:161. doi: 10.3389/fnins.2018.00161
Qian, Z. M., Wu, X. M., Fan, M., Yang, L., Du, F., Yung, W. H., et al. (2011). Divalent metal transporter 1 is a hypoxia-inducible gene. J. Cell Physiol. 226, 1596–1603. doi: 10.1002/jcp.22485.
Ray, P. D., Huang, B. W., and Tsuji, Y. (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 24, 981–990. doi: 10.1016/j.cellsig.2012.01.008
Requejo-Aguilar, R., and Bolanos, J. P. (2016). Mitochondrial control of cell bioenergetics in Parkinson’s disease. Free Radic. Biol. Med. 100, 123–137. doi: 10.1016/j.freeradbiomed.2016.04.012
Riganti, C., Doublier, S., Viarisio, D., Miraglia, E., Pescarmona, G., Ghigo, D., et al. (2009). Artemisinin induces doxorubicin resistance in human colon cancer cells via calcium-dependent activation of HIF-1alpha and P-glycoprotein overexpression. Br. J. Pharmacol. 156, 1054–1066. doi: 10.1111/j.1476-5381.2009.00117.x
Rimessi, A., Bonora, M., Marchi, S., Patergnani, S., Marobbio, C. M., Lasorsa, F. M., et al. (2013). Perturbed mitochondrial Ca2+ signals as causes or consequences of mitophagy induction. Autophagy 9, 1677–1686. doi: 10.4161/auto.24795
Riquelme, D., Alvarez, A., Leal, N., Adasme, T., Espinoza, I., Valdes, J. A., et al. (2011). High-frequency field stimulation of primary neurons enhances ryanodine receptor-mediated Ca2+ release and generates hydrogen peroxide, which jointly stimulate NF-kappaB activity. Antioxid. Redox Signal. 14, 1245–1259. doi: 10.1089/ars.2010.3238
Rosales-Corral, S., Acuna-Castroviejo, D., Tan, D. X., Lopez-Armas, G., Cruz-Ramos, J., Muñoz, R., et al. (2012). Accumulation of exogenous amyloid-beta peptide in hippocampal mitochondria causes their dysfunction: a protective role for melatonin. Oxid. Med. Cell. Longev. 2012:843649. doi: 10.1155/2012/843649
Rosi, S., Ramirez-Amaya, V., Hauss-Wegrzyniak, B., and Wenk, G. L. (2004). Chronic brain inflammation leads to a decline in hippocampal NMDA-R1 receptors. J Neuroinflammation 1:12. doi: 10.1186/1742-2094-1-12
Rothwell, N. J., and Luheshi, G. N. (2000). Interleukin 1 in the brain: biology, pathology and therapeutic target. Trends Neurosci. 23, 618–625. doi: 10.1016/S0166-2236(00)01661-1
Rousseau, E., Michel, P. P., and Hirsch, E. C. (2013). The iron-binding protein lactoferrin protects vulnerable dopamine neurons from degeneration by preserving mitochondrial calcium homeostasis. Mol. Pharmacol. 84, 888–898. doi: 10.1124/mol.113.087965
Ruiz, A., Alberdi, E., and Matute, C. (2018). Mitochondrial division inhibitor 1 (mdivi-1) protects neurons against excitotoxicity through the modulation of mitochondrial function and intracellular Ca(2+) signaling. Front. Mol. Neurosci. 11:3. doi: 10.3389/fnmol.2018.00003
Sanmartin, C. D., Paula-Lima, A. C., Garcia, A., Barattini, P., Hartel, S., Núñez, M. T., et al. (2014). Ryanodine receptor-mediated Ca(2+) release underlies iron-induced mitochondrial fission and stimulates mitochondrial Ca(2+) uptake in primary hippocampal neurons. Front. Mol. Neurosci. 7:13. doi: 10.3389/fnmol.2014.00013
Schiavone, S., Sorce, S., Dubois-Dauphin, M., Jaquet, V., Colaianna, M., Zotti, M., et al. (2009). Involvement of NOX2 in the development of behavioral and pathologic alterations in isolated rats. Biol. Psychiatry 66, 384–392. doi: 10.1016/j.biopsych.2009.04.033
Sen, C. K. (2001). Antioxidants in exercise nutrition. Sports Med. 31, 891–908. doi: 10.2165/00007256-200131130-00001
Sherer, T. B., Betarbet, R., Testa, C. M., Seo, B. B., Richardson, J. R., Kim, J. H., et al. (2003). Mechanism of toxicity in rotenone models of Parkinson’s disease. J. Neurosci. 23, 10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003
Shih, R. H., Wang, C. Y., and Yang, C. M. (2015). NF-kappaB signaling pathways in neurological inflammation: a mini review. Front. Mol. Neurosci. 8:77. doi: 10.3389/fnmol.2015.00077
Shimohama, S., Tanino, H., Kawakami, N., Okamura, N., Kodama, H., Yamaguchi, T., et al. (2000). Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem. Biophys. Res. Commun. 273, 5–9. doi: 10.1006/bbrc.2000.2897
Siddiq, A., Ayoub, I. A., Chavez, J. C., Aminova, L., Shah, S., Lamanna, J. C., et al. (2005). Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J. Biol. Chem. 280, 41732–41743. doi: 10.1074/jbc.M504963200
Siegert, I., Schodel, J., Nairz, M., Schatz, V., Dettmer, K., Dick, C., et al. (2015). Ferritin-Mediated iron sequestration stabilizes hypoxia-inducible factor-1alpha upon LPS activation in the presence of ample oxygen. Cell Rep. 13, 2048–2055. doi: 10.1016/j.celrep.2015.11.005
Siklos, L., Engelhardt, J. I., Alexianu, M. E., Gurney, M. E., Siddique, T., and Appel, S. H. (1998). Intracellular calcium parallels motoneuron degeneration in SOD-1 mutant mice. J. Neuropathol. Exp. Neurol. 57, 571–587. doi: 10.1097/00005072-199806000-00005
Sirabella, R., Valsecchi, V., Anzilotti, S., Cuomo, O., Vinciguerra, A., Cepparulo, P., et al. (2018). Ionic homeostasis maintenance in ALS: focus on new therapeutic targets. Front. Neurosci. 12:510. doi: 10.3389/fnins.2018.00510
Smith, T. G., Robbins, P. A., and Ratcliffe, P. J. (2008). The human side of hypoxia-inducible factor. Br. J. Haematol. 141, 325–334. doi: 10.1111/j.1365-2141.2008.07029.x
Sorce, S., and Krause, K. H. (2009). NOX enzymes in the central nervous system: from signaling to disease. Antioxid. Redox Signal. 11, 2481–2504. doi: 10.1089/ARS.2009.2578
Sorce, S., Stocker, R., Seredenina, T., Holmdahl, R., Aguzzi, A., Chio, A., et al. (2017). NADPH oxidases as drug targets and biomarkers in neurodegenerative diseases: what is the evidence? Free Radic. Biol. Med. 112, 387–396. doi: 10.1016/j.freeradbiomed.2017.08.006
Soum, E., and Drapier, J. C. (2003). Nitric oxide and peroxynitrite promote complete disruption of the [4Fe-4S] cluster of recombinant human iron regulatory protein 1. J. Biol. Inorg. Chem. 8, 226–232. doi: 10.1007/s00775-002-0412-9
Stefani, I. C., Wright, D., Polizzi, K. M., and Kontoravdi, C. (2012). The role of ER stress-induced apoptosis in neurodegeneration. Curr. Alzheimers Res. 9, 373–387. doi: 10.2174/156720512800107618
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Strehler, E. E., and Thayer, S. A. (2018). Evidence for a role of plasma membrane calcium pumps in neurodegenerative disease: recent developments. Neurosci. Lett. 663, 39–47. doi: 10.1016/j.neulet.2017.08.035
Surace, M. J., and Block, M. L. (2012). Targeting microglia-mediated neurotoxicity: the potential of NOX2 inhibitors. Cell. Mol. Life Sci. 69, 2409–2427. doi: 10.1007/s00018-012-1015-4
Sureda, A., Hebling, U., Pons, A., and Mueller, S. (2005). Extracellular H2O2 and not superoxide determines the compartment-specific activation of transferrin receptor by iron regulatory protein 1. Free Radic. Res. 39, 817–824. doi: 10.1080/10715760500164045
Surmeier, D. J., Guzman, J. N., and Sanchez-Padilla, J. (2010). Calcium, cellular aging, and selective neuronal vulnerability in Parkinson’s disease. Cell. Calcium 47, 175–182. doi: 10.1016/j.ceca.2009.12.003
Surmeier, D. J., Guzman, J. N., Sanchez-Padilla, J., and Goldberg, J. A. (2011). The origins of oxidant stress in Parkinson’s disease and therapeutic strategies. Antioxid. Redox Signal. 14, 1289–1301. doi: 10.1089/ars.2010.3521
Swaiman, K. F. (1991). Hallervorden-Spatz syndrome and brain iron metabolism. Arch. Neurol. 48, 1285–1293. doi: 10.1001/archneur.1991.00530240091029
Tan, S., Sagara, Y., Liu, Y., Maher, P., and Schubert, D. (1998). The regulation of reactive oxygen species production during programmed cell death. J. Cell. Biol. 141, 1423–1432. doi: 10.1083/jcb.141.6.1423
Tangudu, N. K., Alan, B., Vinchi, F., Worle, K., Lai, D., Vettorazzi, S., et al. (2018). Scavenging reactive oxygen species production normalizes ferroportin expression and ameliorates cellular and systemic iron disbalances in hemolytic mouse model. Antioxid. Redox Signal. 29, 484–499. doi: 10.1089/ars.2017.7089
Thomas, G. M., and Huganir, R. L. (2004). MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183. doi: 10.1038/nrn1346
Tonelli, C., Chio, I. I. C., and Tuveson, D. A. (2018). Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 29, 1727–1745. doi: 10.1089/ars.2017.7342
Tong, W. H., and Rouault, T. A. (2007). Metabolic regulation of citrate and iron by aconitases: role of iron-sulfur cluster biogenesis. Biometals 20, 549–564. doi: 10.1007/s10534-006-9047-6
Tonnies, E., and Trushina, E. (2017). Oxidative stress, synaptic dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 57, 1105–1121. doi: 10.3233/JAD-161088
Urrutia, P., Aguirre, P., Esparza, A., Tapia, V., Mena, N. P., Arredondo, M., et al. (2013). Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 126, 541–549. doi: 10.1111/jnc.12244
Urrutia, P. J., Mena, N. P., and Núñez, M. T. (2014). The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front. Pharmacol. 5:38. doi: 10.3389/fphar.2014.00038
Vila, M., and Przedborski, S. (2003). Targeting programmed cell death in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 365–375. doi: 10.1038/nrn1100
Wang, J., Song, N., Jiang, H., Wang, J., and Xie, J. (2013). Pro-inflammatory cytokines modulate iron regulatory protein 1 expression and iron transportation through reactive oxygen/nitrogen species production in ventral mesencephalic neurons. Biochim. Biophys. Acta 1832, 618–625. doi: 10.1016/j.bbadis.2013.01.021
Wang, Q. M., Xu, Y. Y., Liu, S., and Ma, Z. G. (2017). Isradipine attenuates MPTP-induced dopamine neuron degeneration by inhibiting up-regulation of L-type calcium channels and iron accumulation in the substantia nigra of mice. Oncotarget 8, 47284–47295. doi: 10.18632/oncotarget.17618
Wang, Y., Yang, J., and Yi, J. (2012). Redox sensing by proteins: oxidative modifications on cysteines and the consequent events. Antioxid. Redox Signal. 16, 649–657. doi: 10.1089/ars.2011.4313
Ward, R. J., Zucca, F. A., Duyn, J. H., Crichton, R. R., and Zecca, L. (2014). The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 13, 1045–1060. doi: 10.1016/S1474-4422(14)70117-6
Wardrop, S. L., Watts, R. N., and Richardson, D. R. (2000). Nitrogen monoxide activates iron regulatory protein 1 RNA-binding activity by two possible mechanisms: effect on the [4Fe-4S] cluster and iron mobilization from cells. Biochemistry 39, 2748–2758. doi: 10.1021/bi991099t
Weinreb, O., Amit, T., Bar-Am, O., and Youdim, M. B. (2016). Neuroprotective effects of multifaceted hybrid agents targeting MAO, cholinesterase, iron and beta-amyloid in ageing and Alzheimer’s disease. Br. J. Pharmacol. 173, 2080–2094. doi: 10.1111/bph.13318
Wiethoff, S., and Houlden, H. (2017). Neurodegeneration with brain iron accumulation. Handb. Clin. Neurol. 145, 157–166. doi: 10.1016/B978-0-12-802395-2.00011-0
Wilkinson, N., and Pantopoulos, K. (2014). The IRP/IRE system in vivo: insights from mouse models. Front. Pharmacol. 5:176. doi: 10.3389/fphar.2014.00176
Wu, D. C., Jackson-Lewis, V., Vila, M., Tieu, K., Teismann, P., Vadseth, C., et al. (2002). Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. 22, 1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002
Wu, G., Fang, Y. Z., Yang, S., Lupton, J. R., and Turner, N. D. (2004). Glutathione metabolism and its implications for health. J. Nutr. 134, 489–492. doi: 10.1093/jn/134.3.489
Wu, G. Y., Deisseroth, K., and Tsien, R. W. (2001). Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. U.S.A. 98, 2808–2813. doi: 10.1073/pnas.051634198
Xiao, J., Lv, Y., Lin, B., Tipoe, G. L., Youdim, M. B., Xing, F., et al. (2015). A novel antioxidant multitarget iron chelator M30 protects hepatocytes against ethanol-induced injury. Oxid. Med. Cell. Longev. 2015:607271. doi: 10.1155/2015/607271
Yang, W. S., Sriramaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi: 10.1016/j.cell.2013.12.010
Yeh, T. L., Leissing, T. M., Abboud, M. I., Thinnes, C. C., Atasoylu, O., Holt-Martyn, J. P., et al. (2017). Molecular and cellular mechanisms of HIF prolyl hydroxylase inhibitors in clinical trials. Chem. Sci. 8, 7651–7668. doi: 10.1039/c7sc02103h
Yermolaieva, O., Brot, N., Weissbach, H., Heinemann, S. H., and Hoshi, T. (2000). Reactive oxygen species and nitric oxide mediate plasticity of neuronal calcium signaling. Proc. Natl. Acad. Sci. U.S.A. 97, 448–453. doi: 10.1073/pnas.97.1.448
Yuan, G., Nanduri, J., Bhasker, C. R., Semenza, G. L., and Prabhakar, N. R. (2005). Ca2+/calmodulin kinase-dependent activation of hypoxia inducible factor 1 transcriptional activity in cells subjected to intermittent hypoxia. J. Biol. Chem. 280, 4321–4328. doi: 10.1074/jbc.M407706200
Yuan, G., Nanduri, J., Khan, S., Semenza, G. L., and Prabhakar, N. R. (2008). Induction of HIF-1alpha expression by intermittent hypoxia: involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J. Cell. Physiol. 217, 674–685. doi: 10.1002/jcp.21537
Zaidi, A. (2010). Plasma membrane Ca-ATPases: targets of oxidative stress in brain aging and neurodegeneration. World J. Biol. Chem. 1, 271–280. doi: 10.4331/wjbc.v1.i9.271
Zaidi, A., and Michaelis, M. L. (1999). Effects of reactive oxygen species on brain synaptic plasma membrane Ca(2+)-ATPase. Free Radic. Biol. Med. 27, 810–821. doi: 10.1016/S0891-5849(99)00128-8
Zecca, L., Youdim, M. B., Riederer, P., Connor, J. R., and Crichton, R. R. (2004). Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 5, 863–873. doi: 10.1038/nrn1537
Zhang, W., Yan, Z. F., Gao, J. H., Sun, L., Huang, X. Y., Liu, Z., et al. (2014). Role and mechanism of microglial activation in iron-induced selective and progressive dopaminergic neurodegeneration. Mol. Neurobiol. 49, 1153–1165. doi: 10.1007/s12035-013-8586-4
Zhang, X., Zhou, K., Wang, R., Cui, J., Lipton, S. A., Liao, F. F., et al. (2007). Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J. Biol. Chem. 282, 10873–10880. doi: 10.1074/jbc.M608856200
Zhang, Y., Kong, W. N., and Chai, X. Q. (2018). Compound of icariin, astragalus, and puerarin mitigates iron overload in the cerebral cortex of Alzheimer’s disease mice. Neural Regen. Res. 13, 731–736. doi: 10.4103/1673-5374.230302
Keywords: neurodegenerative diseases, reactive oxygen species, mitochondria, HIF-1, Nrf-2, inflammation, ferroptosis
Citation: Núñez MT and Hidalgo C (2019) Noxious Iron–Calcium Connections in Neurodegeneration. Front. Neurosci. 13:48. doi: 10.3389/fnins.2019.00048
Received: 16 November 2018; Accepted: 18 January 2019;
Published: 12 February 2019.
Edited by:
Giorgio Biasiotto, Università degli Studi di Brescia, ItalyReviewed by:
Veronica Perez de la Cruz, Instituto Nacional de Neurología y Neurocirugía (INNN), MexicoNicola B. Mercuri, University of Rome Tor Vergata, Italy
Copyright © 2019 Núñez and Hidalgo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Tulio Núñez, bW51bmV6QHVjaGlsZS5jbA==