 Curtis Lee Lowery III
Curtis Lee Lowery III Donna Woulfe
Donna Woulfe Fusun Kilic
Fusun Kilic
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 04 March 2019
Sec. Neuropharmacology
Volume 13 - 2019 | https://doi.org/10.3389/fnins.2019.00032
This article is part of the Research Topic Serotonin, Receptors and Transporters: Exploring New and Known Signaling Pathways to Improve the Efficacy of Antidepressant Treatment View all 10 articles
Cigarette smoking is one of the major causes of coronary heart disease with a thirty percent mortality rate in the United States. Cigarette smoking acting on the central nervous system (CNS) to stimulate the sympathetic nervous system (SNS) through, which facilitates the secretion of serotonin (5-HT) and catecholamines to supraphysiological levels in blood. The enhanced levels of 5-HT and catecholamines in smokers’ blood are associated with increases in G protein-coupled receptor signaling and serotonylation of small GTPases, which in turn lead to remodeling of cytoskeletal elements to enhance granule secretion and promote unique expression of sialylated N-glycan structures on smokers’ platelets. These mechanisms enhance aggregation and adhesion of smokers’ platelets relative to those of non-smokers. This review focuses on the known mechanisms by which 5-HT and SERT, in coordinated signaling with catecholamines, impacts cigarette smokers’ platelet biology.
Serotonin (5-HT) is secreted from the enterochromaffin cells of the intestine to the blood and taken into the platelets by a specific transporter, SERT (SLC6A4) via a saturable reuptake mechanism (Rudnick, 1977; Lesch et al., 1993; McNicol and Israels, 1999; McNicol and Israels, 2003). Platelets do not synthesize 5-HT, yet they are the major biological storage pool of circulating 5-HT. Platelet surface located SERT tightly regulates the free 5-HT level in the blood. In platelets, vesicular monoamine transporter (VMAT) on the dense granules takes free 5-HT from the platelet cytoplasm into the dense granule which is sequestered there to millimolar concentrations, while free concentrations in the blood are in the low nanomolar range (McNicol and Israels, 2003; Brenner et al., 2007).
5-HT is a multifunctional signaling molecule, growth factor, and endocrine hormone or paracrine messenger (Maroteaux and Kilic, 2018) while it plays key roles in a variety of psychiatric diseases as a neurotransmitter, it also has extra-cerebral roles as a potent vasoconstrictor as well as a weak agonist of platelet activation, both roles serve to modulate cardiovascular disease (Rapport et al., 1984; Sjoerdsma and Palfreyman, 1990).
In various pathological conditions including hypertension and thrombosis, plasma 5-HT level is elevated (Biondi et al., 1986; Kerr et al., 1999; Vikens et al., 1999; Brenner et al., 2007; Davis et al., 2013; Fraer and Kilic, 2015; Lowery et al., 2017). The cause of 5-HT elevation, its effect on SERT, and the contribution to platelet physiology were studied in preclinical models, rat (Homberg et al., 2006) and mouse (Bengel et al., 1998) with SERT knock-out (KO) gene. These studies demonstrated that platelets were almost completely depleted of 5-HT in SERT-KO rodent models confirming that 5-HT in dense granules are also filled by platelet SERT through an uptake mechanism. Although the source of the initial elevation in plasma 5-HT levels remains controversial it has been proposed that it may originate from an increased rate of 5-HT synthesis or secretion from the enterochromaffin cells of the intestine due to the initial stage of a disease (Gershon and Tack, 2007). Studies with the blood samples collected from hypertensive subjects showed that there is a biphasic relationship with the plasma concentration of 5-HT and the density of SERT on platelet surface (Brenner et al., 2007). The level of SERT proteins on the surface of platelet and the 5-HT uptake rates of platelets initially rise as plasma 5-HT levels are increased, but then fall below normal as the plasma 5-HT level continues to rise (Brenner et al., 2007). These findings indicate a biphasic relationship between plasma 5-HT-level and the platelet SERT density on the plasma membrane, more specifically, down-regulation of platelet SERT in the presence of high level of 5-HT in plasma. We hypothesized that the elevated plasma 5-HT levels downregulate the 5-HT uptake of platelets via decreasing the density of SERT molecules on platelet surface. This hypothesis switches the cellular roles of 5-HT and SERT and proposes that 5-HT controls its own concentration in plasma by modulating the uptake properties of platelet SERT rather than SERT controls the plasma 5-HT levels. Further studies correlated the impact of 5-HT signaling on the membrane trafficking of SERT (Ahmed et al., 2008, 2009; Mercado and Kilic, 2010; Mercado et al., 2011).
In general, translocation of proteins from/to the plasma membrane is mediated by other proteins that facilitate their movement between the surface membrane and intracellular compartments. 5-HT signaling acts on the membrane trafficking of SERT molecules in two independent pathways: (1) on the exocytosis of SERT via acting on small GTPases (Ahmed et al., 2008; Li et al., 2016; Lowery et al., 2017; Mercado and Kilic, 2010; Mercado et al., 2011; Ziu et al., 2012); and (2) on the rate of internalization of SERT via acting on cytoskeletal proteins such as myosin IIa and vimentin (Ozaslan et al., 2003; Ahmed et al., 2009; Mercado and Kilic, 2010). The translocation of SERT from/to platelet plasma membrane is controlled by the 5-HT signaling-dependent pathways.
Studies in the platelets of mice lacking the gene for tryptophan hydroxylase (TPH1), the rate-limiting enzyme in the synthesis of 5-HT in peripheral cells, demonstrated that intracellular 5-HT acts on the exocytosis of the dense and α-granules during platelet activation (Walther et al., 2003; Ziu et al., 2012, 2014; Lowery et al., 2017). These studies in isolated platelets indicated that 5-HT-stimulation accelerated the exocytosis of granules, which secrete their contents, 5-HT, ADP, and procoagulant molecules, such as fibrinolytic regulators, growth factors, chemokines, immunologic modulators, P-selectin, von Willebrand factor, thrombospondin, fibrinogen, and fibronectin (Shirakawa et al., 2000; Walther et al., 2003; Ziu et al., 2012; Lowery et al., 2017). These findings specifically showed that 5-HT signaling manipulates the exocytosis mechanism of dense and α-granules in platelets. As explained in the following sections, the 5-HT signaling pathway controls the movement of SERT from/to plasma membrane of platelets (Ozaslan et al., 2003; Ahmed et al., 2008, 2009; Mercado and Kilic, 2010; Mercado et al., 2011; Ziu et al., 2012; Li et al., 2016; Lowery et al., 2017). This will elevate plasma 5-HT concentration to supraphysiological levels as seen during smoking cigarette (Lowery et al., 2017). However, smoking also elevates the blood plasma catecholamine levels (Siess et al., 1982; Grassi et al., 1994; Narkiewicz et al., 1998; Parati and Esler, 2012; Lowery et al., 2017). Together with 5-HT circulating catecholamines are associated with the increased risk of arterial thrombosis. Yet, in the absence of cardiovascular disease, elevated blood 5-HT level does not increase blood pressure, suggesting that the elevation in plasma 5-HT level could be a consequence rather than a cause of hypertension (Singh et al., 2013). Thus, the mechanisms by which elevated concentrations of 5-HT may lead to thrombosis in cigarette smokers is an active area of research that may yield improved development and application of anti-thrombotic therapy. In particular, the mechanisms by which 5-HT potentiates platelet aggregation may play a contributing role in the acutely increased risk of arterial thrombosis associated with cigarette smoking. The main goal of this review is to summarize a novel mechanism by which 5-HT and SERT in coordination with catecholamines impact cigarette smokers’ platelet biology.
Cigarette smoking has significant roles in the development of various cardiovascular diseases (CVD) through inhalation exposure of smoked tobacco and the secondary effects of tobacco products on circulating hormone levels. Even passive smoking (second hand smoke), with a smoke exposure about 1/10th that of active smoking, is associated with an approximate 30% increase of coronary artery disease (CAD), compared with an 80% increase in active smokers (Willett et al., 1987; He et al., 1999; Barnoya and Glantz, 2005). Despite increasing social and legal pressure to restrict tobacco use, an estimated 36.5 million adults in the United States still smoke tobacco cigarettes (Crow et al., 2018). Although awareness of these health risks has reduced the prevalence of smoking, a large portion of the population continues to use cigarettes and will suffer adverse cardiovascular events attributable to tobacco use.
While the exact toxic components of cigarette smoke and the mechanisms involved in cigarette-related long term cardiovascular dysfunction have not been fully elucidated, it has been repeatedly demonstrated that the acute effects (minutes to hours) of cigarette smoke are linked to plasma concentrations of nicotine (Powell, 1998; Mendelson et al., 2005; Tweed et al., 2012). Nicotine, a naturally occurring alkaloid found in the tobacco plant, appears to be the primary addictive and bioactive agent in cigarette smoke (Goldberg et al., 1991; Doolittle et al., 1995; Corrigall et al., 2001; Rabinoff et al., 2007). Inhalation results in rapid absorption of nicotine through the lungs into the blood stream. Nicotine crosses the blood-brain barrier and reaches the central nervous system (CNS) within 7 s of inhalation, where it stimulates nicotinic acetylcholine receptors (Doolittle et al., 1995; Mendelson et al., 2005; Sherva et al., 2008). Nicotinic CNS stimulation activates the sympathetic nervous system (SNS) to promote release of many chemical messengers including acetylcholine, adrenocorticotropin hormone (ACTH) norepinephrine (NE), epinephrine (E), arginine vasopressin, 5-HT, and dopamine (DA) (Koob and Le Moal, 2001; Kimes et al., 2003; Sherva et al., 2008; McKee et al., 2011) into blood plasma. Elevations in blood levels of these compounds are associated with the systemic cardiovascular effects of cigarette smoking (Ambrose and Barua, 2004; Mendelson et al., 2005).
Chronic cigarette smoking predisposes the individual to multiple atherosclerotic syndromes as well as peripheral atherosclerosis and aortic aneurysms (Winniford et al., 1987). Long term effects on lungs, blood vessels, and heart are well studied and have repeatedly shown that smoking cessation decreases the risk of all-cause mortality with an exponential decline approaching the risk of non-smokers at 5 years (Rosenberg et al., 1985; Fusegawa et al., 1999) Data also demonstrate an immediate reduction in thrombotic events following smoking cessation indicating important acute effects altering blood and platelet function (FitzGerald et al., 1988; US Department of Health and Human Services, 1990; Samet, 1991).
Thrombosis is the dysregulated formation of a thrombus, comprising the combination of platelet aggregates, and blood clot within blood vessels, such that the flow of blood through the circulatory system is obstructed. Under normal physiological conditions, regulatory mechanisms such as circulating levels of prostacyclin and endothelial expression of ecto-ADPase prevent thrombus formation. Without injury or other insult endothelial cells of intact vessels prevent blood clotting by secreting a heparin-like molecule and thrombomodulin. Additionally, the vascular endothelial cells prevent platelet aggregation and vasospasm by secreting nitric oxide. After vascular damage, thrombus formation is initiated to prevent blood loss after injury to a blood vessel. While acute injury results in hemostasis, which is the localized vascular response to prevent blood loss, thrombosis is a pathological response, typically initiated in the presence of atherosclerotic lesion. While hemostasis and thrombosis share similar initiating mechanisms, thrombosis proceeds to complete vessel occlusion, while physiological hemostasis stems blood loss, but allows maintenance of blood flow.
Hemostasis can be conceived as occurring via a three-step process (Levy et al., 2010). The first step is vascular spasm (vasoconstriction), caused by contraction of vascular smooth muscle cells. This local constriction of blood vessels results in decreased blood flow through the area and limits blood loss. At the area of damage, blood is also exposed to collagen in the subendothelial matrix. Collagen promotes adherence and activation of platelets localized at the site of injury. Once activated, platelets secrete dense and alpha granules, releasing platelet-activating factors, such as ADP and P-selectin, and causing the localized production of thromboxane A2 and thrombin on platelet and associated membrane surfaces. These factors activate other nearby platelets causing them to release their contents leading to a cascade effect (O’Connell, 2013). The activated platelets alter their shape through cytoskeletal remodeling, enhancing their adhesion to endothelial and other platelet surfaces and aggregate to form a platelet plug. Cytoskeletal remodeling is achieved predominantly through the modification of small GTP-binding proteins, resulting in downstream shape changes to a more spiny form with projecting filopodia. The increased surface area and alteration of surface proteins causes platelets to adhere to one another and to the exposed collagen in the damaged vessel wall. Signaling interactions then promote the activation of integrin alphaIIb-beta3 and subsequent binding of circulating fibrinogen, allowing formation of a stable platelet plug (Farndale et al., 2004). Coincident with these processes, coagulation is initiated on local membrane surfaces, allowing consequent formation of a clot. There is evidence that both platelet aggregation and clotting are excessively activated after acute exposure to cigarette smoke. Therefore, it is important to elucidate the mechanisms by which cigarette smoking induces platelets to become increasingly activated and increases thrombotic risk.
Cigarette smoking enhances the formation of thrombosis by which studies demonstrate that stopping smoking decreases the risk of the reoccurring of myocardial infarction (US Department of Health and Human Services, 1990). Also, epidemiologic evidence indicates that the acute effects of cigarette smoking produce CNS-mediated activation of the SNS (Siess et al., 1982; Peterson et al., 1983; Porchet et al., 1987; Narkiewicz et al., 1998; Parati and Esler, 2012), which stimulates secretion of pro-thrombotic molecules, such as catecholamines (E, NE, and DA) and 5-HT into the blood at supraphysiological levels (Koob and Le Moal, 2001; Kimes et al., 2003; Sherva et al., 2008; McKee et al., 2011). Recent studies from our laboratories demonstrate that smoking results in several-fold increases in plasma levels of 5-HT and catecholamines (Lowery et al., 2017). In platelets, 5-HT and catecholamine signaling are mediated, respectively, through 5-HT2A, β2-, and α2-adrenergic receptors. Activation of each of these receptors directly act on platelet aggregation (Woulfe, 2005; Millan et al., 2008; Smyth et al., 2009; Dowal and Flaumenhaft, 2010) via stimulating the exocytosis of the granules which are the storages of the several procoagulant molecules as well as 5-HT and catecholamine (McNicol and Israels, 1999, 2003).
Once bound to platelet surfaces, 5-HT activates a 5-HT-specific G protein-coupled receptor, 5-HT2A. Although the Gq-dependent signals leading to platelet activation are well-accepted, less appreciated is the coordinate interaction of these 5-HT and catecholamine-initiated signals with 5-HT uptake into the platelet cytoplasm. In this process, SERT, on the platelet plasma membrane, plays a major role in regulating extracellular vs intracellular 5-HT concentration. When 5-HT is removed from blood plasma into the platelet, it is stored in dense granules through VMAT (Brunk et al., 2006). Upon the saturation of dense granules with 5-HT, transport via VMAT shuts down and 5-HT appears in the cytoplasm in free, unbound form. The elevated plasma 5-HT level also activates platelet surface-localized 5-HT receptors, which initiates signaling through Gq, activates phospholipase C (PLC) results in the hydrolysis of phosphatidylinositol 4,5-biphosphate (PIP2) to inositol-1,4,5-triphosphate (IP3) (Jin and Kunapuli, 1998; McNicol and Israels, 1999; Yang et al., 2002; Quinton et al., 2004; Woulfe, 2005; Millan et al., 2008; Smyth et al., 2009; Dowal and Flaumenhaft, 2010; Bynagari-Settipalli et al., 2012). Formation of IP3 activates the serine/threonine protein kinase C (PKC) family and facilitates the secretion of Ca2+ from intracellular compartments to cytoplasm. Catecholamine (NE/E) activates α2-adrenoceptor which initiates signaling through Gi, inhibits adenylyl cyclase (AC) while activating phosphoinositide 3-kinases (PI3K), leading to the aggregation of platelets in a biphasic manner (Dowal and Flaumenhaft, 2010). Additionally, NE signaling also acts on Src family kinases (SFK) (McNicol and Israels, 2003; Walther et al., 2003; Senis et al., 2014) to allow PLC-dependent hydrolysis of PIP2 to IP3 and the secretion of Ca2+ from the granules (Walther et al., 2003; Ziu et al., 2012). Particularly, related to the role of 5-HT signaling in platelet pathology, elevation of the cytoplasmic free Ca2+ concentration activates transglutaminase (TGase), which transamidates unbound/free cytoplasmic 5-HT to small GTPases; a reaction known as serotonylation (Shirakawa et al., 2000; Walther et al., 2003; Ziu et al., 2012; Senis et al., 2014).
The action of E/NE signaling on platelet via β2-receptor is a controversial topic. Yet, E/NE activates AC through the Gs, upregulates the intracellular concentration of the second messenger cAMP, a powerful inhibitor of platelet aggregation. The cellular act of cAMP reorganizes the actin/myosin cytoskeletal network in a Rho-GTP dependent manner via stimulating Protein kinase A (PKA) (Woulfe, 2005; Millan et al., 2008; Smyth et al., 2009). Thus, transamidation of Rho- and Rab-GTPases with 5-HT are important for platelet cytoskeletal reorganization and secretory behavior, respectively. We hypothesize that following cigarette smoking, IP3 is activated by 5-HT2A and α2-receptor signals to alter the cytoskeletal network in platelets and counteract the tonic inhibitory effect of β2-receptor signaling elevated cAMP level in platelets (Figure 1).
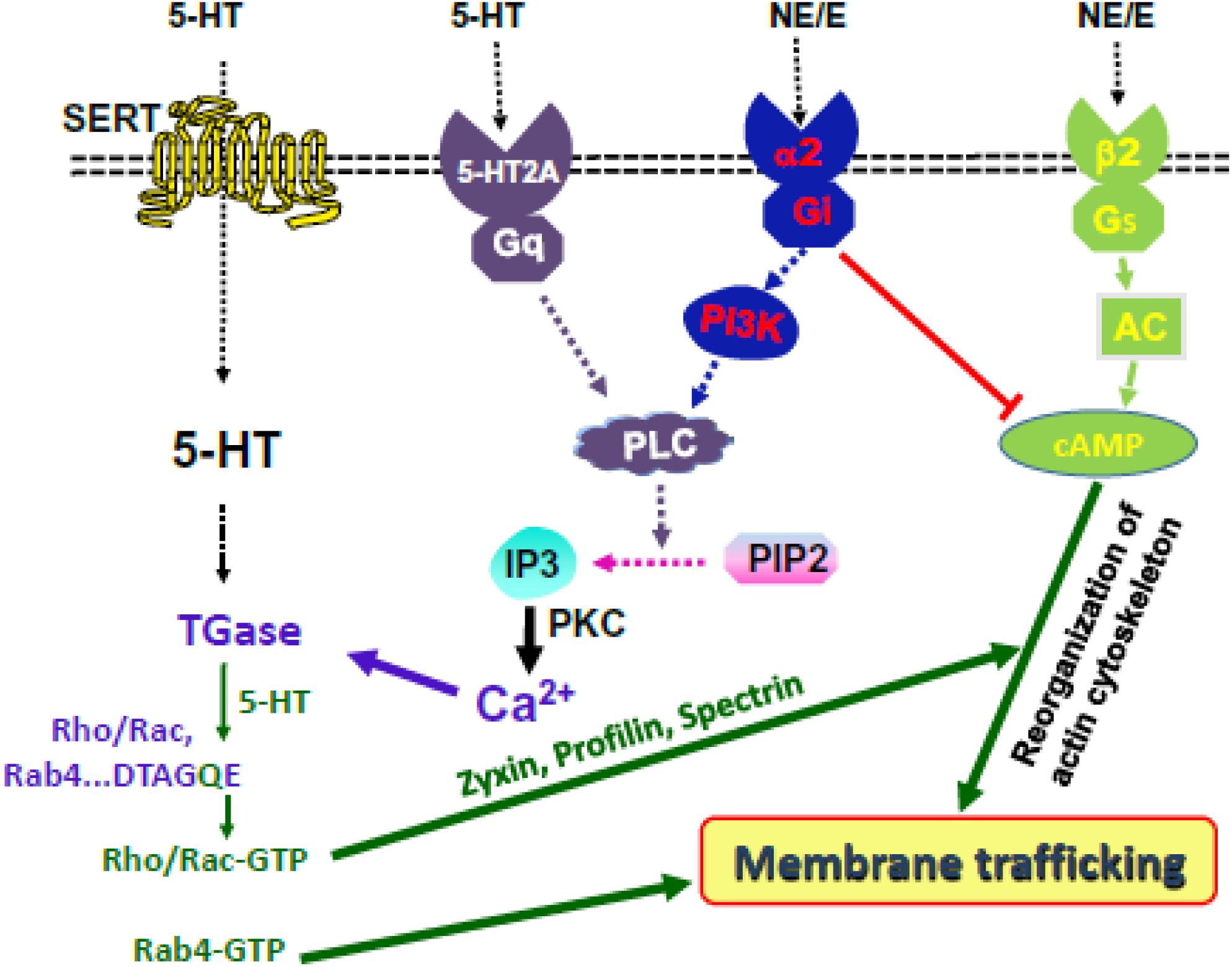
Figure 1. Hypothesis at a glance. 5-HT and NE signaling activate IP3 and elevate the level of cytoplasmic Ca2+. TGase serotonylates small GTPases. 5-HT and NE control platelet membrane-trafficking dynamics by enabling Rab4-GTP formation and Rho/Rac-GTP-mediated cytoskeletal rearrangement, downstream of cAMP/PKA.
In the last decade, we reported several studies related with the impact of SNS-activated secretion of 5-HT into the blood (Brenner et al., 2007; Mercado and Kilic, 2010; Fraer and Kilic, 2015; Lowery et al., 2017), on platelet aggregation via activation of the 5-HT receptor (5-HT2A) (Ziu et al., 2014), on the secretion of granules as a result of serotonylation of small GTPases (Ahmed et al., 2008; Mercado et al., 2011; Ziu et al., 2012; Li et al., 2016), alteration of the cytoskeletal network (Ahmed et al., 2009; Li et al., 2016), and alteration of the structure and composition of glycans on the surface of mouse platelets (Mercado et al., 2013). Based on these studies, we proposed that 5-HT-mediated signaling and serotonylation of small GTPases accelerates the secretion rates of granule components such as ADP and P-selectin, remodeling of cytoskeletal network also permits altered localization of enzymes regulating glycosylation. The structures of glycans, specifically the terminal positions, act as ligands to the receptors. The best examples are P- and E-selectins which regulate various cell–cell adhesion in occurrence of vascular pathophysiology (Kelm and Schauer, 1997). Together, these studies show that the platelet surface glycans’ structures and distributions were altered by 5-HT treatment.
Our recently reported studies demonstrate that cigarette smoking induces specific changes to surface glycans and that these changes are associated with a potentiating effect on platelet aggregation (Lowery et al., 2017). Proteomic and glycomic analyses by Dr. Richard Jones at MS Bioworks and Dr. Parastoo Azadi at CCRC identified differences in both the number and specification of core proteins and glycans eluted from platelet plasma membranes isolated from nonsmokers versus smokers’ blood. Cigarette smoking acutely changes the glycan structure from high mannose to sialylated N-glycan structures expressed on platelet surfaces. Of particular functional relevance, removing the N-glycans from the surfaces of smokers’ platelets counteracted the smoking-mediated enhancements in platelet aggregation (Lowery et al., 2017), indicating that remodeling the platelet surface with N-glycan may establish a more adhesive environment and provide a mechanism by which smoking contributes to platelet activation and thrombosis. However, it remains to be investigated whether the high mannose structures present exclusively on resting (nonsmokers’) platelets protect against platelet aggregation. Specifically, proteins involved in the GTPase-activating, and associated with the actin cytoskeletal network were differentially and significantly altered on the surface of the smokers’ platelet compared to those of non-smokers.
These findings suggest that plasma 5-HT and catecholamine levels influence the alteration of glycans on platelet surfaces; however, the mechanisms by which 5-HT/catecholamine specifically induce surface glycan alteration is still not known. The abundance of oligomannose glycans on platelets suggests an attenuated N-glycan maturation pathway at the megakaryocyte level; however, the alteration in glycans on smokers’ platelet surfaces occurs within 15 min. This glycan structural alteration is too rapid to be explained by traditional glycan modification in Golgi apparatus after protein synthesis and subsequent completion of the membrane trafficking process (Lowery et al., 2017). Therefore, we propose that the involvement of 5-HT/catecholamine signaling in alteration of surface glycans could be through the membrane trafficking of several existing glycoproteins to the plasma membrane. In support of this proposal, in vitro studies of 5-HT/catecholamine-exposed nonsmokers’ platelets show an elevation in plasma glycan level and percent platelet aggregation rates to the levels found in smokers’ plasma, indicating a rapid additive effect of 5-HT and adrenergic receptors’ signaling. Furthermore, pharmacologic blockade of 5-HT and adrenergic receptors reduces both plasma glycan and platelet aggregation levels to that of non-smokers’ plasma (Lowery et al., 2017).
We propose that following cigarette smoking-activated 5-HT as well as α2-receptors downstream elements, such as IP3 hydrolysis-associated cytoplasmic level of free Ca and the cytoskeletal network in a cooperative manner to counteract the inhibitory effect of cAMP in platelet activation. Therefore, we propose that smoking-associated high levels of 5-HT and catecholamine in blood plasma make platelets prone to aggregation, in part, by changing the cytoskeletal network to accelerate the movement of N-glycan to the platelet surface.
This review article is a short summary of the studies performed by CL in FK’s laboratories during his Ph.D. program. DW has involved in the project through her expertise in G-protein coupled receptors on platelet.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ahmed, B., Bukhari, I. A., Jeffus, B. C., Harney, J. T., Thyparambil, S., Ziu, E., et al. (2009). The cellular distribution of serotonin transporter is impeded on serotonin-altered vimentin network. PLoS One 4:e4730. doi: 10.1371/journal.pone.0004730
Ahmed, B. A., Jeffus, B., Harney, J. T., Bukhari, I. S., Unal, R., Lupashin, V. V., et al. (2008). Serotonin transamidates Rab4 and facilitates binding to the C-terminus of hSERT. J. Biol. Chem. 283, 9388–9398. doi: 10.1074/jbc.M706367200
Ambrose, J., and Barua, R. (2004). The pathophysiology of cigarette smoking and cardiovascular disease. J. Am. Coll. Cardiol. 43, 1731–1737. doi: 10.1016/j.jacc.2003.12.047
Barnoya, J., and Glantz, S. A. (2005). Cardiovascular effects of secondhand smoke: nearly as large as smoking. Circulation 111, 2684–2698. doi: 10.1161/CIRCULATIONAHA.104.492215
Bengel, D., Murphy, D. L., Andrews, A. M., Wichems, C. H., Feltner, D., Heils, A., et al. (1998). Altered brain serotonin homeostasis and locomotor insensitivity to 3,4-methylenedioxymethamphetamine (‘ecstasy’) in serotonin transporter deficient mice. Mol. Pharmacol. 53, 649–655. doi: 10.1124/mol.53.4.649
Biondi, M. L., Agostoni, A., and Marasini, B. (1986). Serotonin levels in hypertension. J. Hypertens. 4, S39–S41.
Brenner, B., Harney, J. T., Ahmed, B. A., Jeffus, B. C., Unal, R., Mehta, J. L., et al. (2007). Plasma serotonin level and the platelet serotonin transporter. J. Neurochem. 102, 206–216. doi: 10.1111/j.1471-4159.2007.04542.x
Brunk, I., Blex, C., Rachakonda, S., Höltje, M., Winter, S., Pahner, I., et al. (2006). The first luminal domain of vesicular monoamine transporters mediates G-protein-dependent regulation of transmitter uptake. J. Biol. Chem. 281, 33373–33385. doi: 10.1074/jbc.M603204200
Bynagari-Settipalli, Y. S., Lakhani, P., Jin, J., Bhavaraju, K., Rico, M. C., Kim, S., et al. (2012). Protein Kinase C isoform epsilon negatively regulates ADP-induced calcium mobilization and thromboxane generation in platelets. Arterioscler. Thromb. Vasc. Biol. 32, 1211–1219. doi: 10.1161/ATVBAHA.111.242388
Corrigall, W. A., Zack, M., Eissenberg, T., Belsito, L., and Scher, R. (2001). Acute subjective and physiological responses to smoking in adolescents. Addiction 96, 1409–1417. doi: 10.1046/j.1360-0443.2001.961014095.x
Crow, R. S., Lohman, M. C., Titus, A. J., Bruce, M. L., Mackenzie, T. A., Bartels, S. J., et al. (2018). Mortality risk along the frailty spectrum: data from the National Health and Nutrition Examination Survey 1999 to 2004. J. Am. Geriatr. Soc. 66, 496–502. doi: 10.1111/jgs.15220
Davis, R. P., Szasz, T., Garver, H., Burnett, R., Tykocki, N. R., and Watts, S. W. (2013). One-month serotonin infusion results in a prolonged fall in blood pressure in the deoxycorticosterone acetate (DOCA) salt hypertensive rat. ACS Chem. Neurosci. 4, 141–148. doi: 10.1021/cn300114a
Doolittle, D. J., Winegar, R., Lee, C. K., Caldwell, W. S., Hayes, A. W., and deBethizy, J. D. (1995). The genotoxic potential of nicotine and its major metabolites. Mutat. Res. 344, 95–102. doi: 10.1016/0165-1218(95)00037-2
Dowal, L., and Flaumenhaft, R. (2010). Targeting platelet G-protein coupled receptors (GPCRs): looking beyond conventional GPCR antagonism. Curr. Vasc. Pharmacol. 8, 140–154. doi: 10.2174/157016110790886938
Farndale, R. W., Sixma, J. J., Barnes, M. J., and De Groot, P. G. (2004). The role of collagen in thrombosis and hemostasis. J. Thromb. Haemost. 2, 561–573. doi: 10.1111/j.1538-7836.2004.00665.x
FitzGerald, G. A., Oates, J. A., and Nowak, J. (1988). Cigarette smoking and hemostatic function. Am. Heart J. 115(1 Pt 2), 267–271. doi: 10.1016/0002-8703(88)90648-5
Fraer, M., and Kilic, F. (2015). Serotonin: a different player in hypertension-induced thrombosis. Hypertension 65, 942–948. doi: 10.1161/HYPERTENSIONAHA.114.05061
Fusegawa, Y., Goto, S., Handa, S., Kawada, T., and Ando, Y. (1999). Platelet spontaneous aggregation in platelet-rich plasma is increased in habitual smokers. Thromb. Res. 93, 271–278. doi: 10.1016/S0049-3848(98)00184-4
Gershon, M. D., and Tack, J. (2007). The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology 132, 397–414. doi: 10.1053/j.gastro.2006.11.002
Goldberg, R. J., Bengtson, J., Chen, Z. Y., Anderson, K. M., Locati, E., and Levy, D. (1991). Duration of the QT interval and total and cardiovascular mortality in healthy persons (The Framingham Heart Study experience). Am. J. Cardiol. 67, 55–58. doi: 10.1016/0002-9149(91)90099-7
Grassi, G., Seravalle, G., Calhoun, D. A., Bolla, G. B., Giannattasio, C., Marabini, M., et al. (1994). Mechanisms responsible for sympathetic activation by cigarette smoking in humans. Circulation 90, 248–253. doi: 10.1161/01.CIR.90.1.248
He, J., Vupputuri, S., Allen, K., Prerost, M. R., Hughes, J., and Whelton, P. K. (1999). Passive smoking and the risk of coronary heart disease–a meta-analysis of epidemiologic studies. N. Engl. J. Med. 340, 920–926. doi: 10.1056/NEJM199903253401204
Homberg, J. H., Mudde, J. M., Braam, B., Ellenbroek, B., and Cuppen, E. (2006). Blood pressure in mutant rats lacking the 5-hydroxytryptamine transporter. Hypertension 48, e115–e116. doi: 10.1161/01.HYP.0000246306.61289.d8
Jin, J., and Kunapuli, S. P. (1998). Coactivation of two different G protein-coupled receptors is essential for ADP-induced plateletaggregation. Proc. Natl. Acad. Sci. U.S.A. 95, 8070–8074. doi: 10.1073/pnas.95.14.8070
Kelm, S., and Schauer, R. (1997). Sialic acids in molecular and cellular interactions. Int. Rev. Cytol. 175, 137–240. doi: 10.1016/S0074-7696(08)62127-0
Kerr, S., Brosnan, M. J., McIntyre, M., Reid, J. L., Dominiczak, A. F., and Hamilton, C. A. (1999). Superoxide anion production is increased in a model of genetic hypertension: role of the endothelium. Hypertension 33, 1353–1358. doi: 10.1161/01.HYP.33.6.1353
Kimes, A. S., Horti, A. G., London, E. D., Chefer, I., Contoreggi, C., Ernst, M., et al. (2003). 2-[18F]F-A-85380: PET imaging of brain nicotinic acetylcholine receptors and whole body distribution in humans. FASEB J. 10, 1331–1333. doi: 10.1096/fj.02-0492fje
Koob, G. F., and Le Moal, M. (2001). Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 24, 97–129. doi: 10.1016/S0893-133X(00)00195-0
Lesch, K. P., Wolozin, B. L., Murphy, D. L., and Riderer, P. (1993). Primary structure of the human platelet 5-HT uptake site: identity with the brain 5-HT transporter. J. Neurochem. 60, 2319–2322. doi: 10.1111/j.1471-4159.1993.tb03522.x
Levy, J. H., Dutton, R. P., Hemphill, J. C., Shander, A., Cooper, D., Paidas, M. J., et al. (2010). Multidisciplinary approach to the challenge of hemostasis. Anesth. Analg. 110, 354–364. doi: 10.1213/ANE.0b013e3181c84ba5
Li, Y., Hadden, C., Cooper, A., Ahmed, A., Wu, H., Lupashin, V. V., et al. (2016). Sepsis-induced elevation in plasma serotonin facilitates endothelial hyperpermeability. Sci. Rep. 9:22747. doi: 10.1038/srep22747
Lowery, C. L. I. I. I., Elliott, C., Cooper, A., Hadden, C., Sonon, R. N., Azadi, P., et al. (2017). Cigarette smoking-associated alterations in serotonin/adrenalin signaling pathways of platelets. J. Am. Heart Assoc. 6:e005465. doi: 10.1161/JAHA.116.005465
Maroteaux, L., and Kilic, F. (2018). Frontiers of serotonin beyond the brain. Pharmacol. Res. doi: 10.1016/j.phrs.2018.10.022 [Epub ahead of print].
McKee, S. A., Sinha, R., Weinberger, A. H., Sofuoglu, M., Harrison, E. L., Lavery, M., et al. (2011). Stress decreases the ability to resist smoking and potentiates smoking intensity and reward. J. Psychopharmacol. 25, 490–502. doi: 10.1177/0269881110376694
McNicol, A., and Israels, S. J. (1999). Platelet dense granules: structure, function and implications for haemostasis. Thromb. Res. 95, 1–18. doi: 10.1016/S0049-3848(99)00015-8
McNicol, A., and Israels, S. J. (2003). Platelets and anti-platelet therapy. J. Pharmacol. Sci. 93, 381–396. doi: 10.1254/jphs.93.381
Mendelson, J. H., Sholar, M. B., Goletiani, N., Siegel, A. J., and Mello, N. K. (2005). Effects of low- and high-nicotine cigarette smoking on mood states and the HPA axis in men. Neuropsychopharmacology 30, 1751–1763. doi: 10.1038/sj.npp.1300753
Mercado, C., and Kilic, F. (2010). Molecular mechanisms of SERT in platelets: regulation of plasma serotonin levels. Mol. Interv. 10, 231–241. doi: 10.1124/mi.10.4.6
Mercado, C., Ziu, E., and Kilic, F. (2011). Communication between 5-HT and small GTPases. Curr. Opin. Pharmacol. 11, 23–28. doi: 10.1016/j.coph.2011.01.006
Mercado, C. P., Quintero, M. V., Li, Y., Singh, P., Byrd, A. K., Talabnin, K., et al. (2013). A serotonin-induced N-glycan switch regulates platelet aggregation. Sci. Rep. 30:2795. doi: 10.1038/srep02795
Millan, M. J., Marin, P., Bockaert, J., and Mannoury la Cour, C. (2008). Signaling at G-protein-coupled serotonin receptors: recent advances and future research directions. Trends Pharmacol. Sci. 29, 454–464. doi: 10.1016/j.tips.2008.06.007
Narkiewicz, K., van de Borne, P. J., Hausberg, M., Cooley, R. L., Winniford, M. D., Davison, D. E., et al. (1998). Cigarette smoking increases sympathetic outflow in humans. Circulation 98, 528–534. doi: 10.1161/01.CIR.98.6.528
Ozaslan, D., Wang, S., Ahmed, B., Bene, A., Kocabas, A. M., and Kilic, F. (2003). Glycosyl modification facilitates homo- and hetero-oligomerization of serotonin transporter. A specific role for the sialic acid residues. J. Biol. Chem. 278, 43991–44000. doi: 10.1074/jbc.M306360200
Parati, G., and Esler, M. (2012). The human sympathetic nervous system: its relevance in hypertension and heart failure. Eur. Heart J. 33, 1058–1066. doi: 10.1093/eurheartj/ehs041
Peterson, D. F., Coote, J. H., Gilbey, M. P., and Futuro-Neto, H. A. (1983). Differential pattern of sympathetic outflow during upper airway stimulation with smoke. Am. J. Physiol. 245, 433–437. doi: 10.1152/ajpregu.1983.245.3.R433
Porchet, H. C., Benowitz, N. L., Sheiner, L. B., and Copeland, J. R. (1987). Apparent tolerance to the acute effect of nicotine results in part from distribution kinetics. J. Clin. Invest. 80, 1466–1471. doi: 10.1172/JCI113227
Powell, J. T. (1998). Vascular damage from smoking: disease mechanisms at the arterial wall. Vasc. Med. 3, 21–28. doi: 10.1177/1358836X9800300105
Quinton, T. M., Murugappan, S., Kim, S., Jin, J., and Kunapuli, S. P. (2004). Different G protein-coupled signaling pathways are involved in alpha granule release from human platelets. J. Thromb. Haemost. 2, 978–984. doi: 10.1111/j.1538-7836.2004.00741.x
Rabinoff, M., Caskey, N., Rissling, A., and Park, C. (2007). Pharmacological and chemical effects of cigarette additives. Am. J. Public Health 97, 1981–1991. doi: 10.2105/AJPH.2005.078014
Rapport, M. M., Green, A. A., and Page, I. H. (1984). Serum vasoconstrictor, serotonin; isolation and characterization. J. Biol. Chem. 176, 1243–1251.
Rosenberg, L., Kaufman, D. W., Helmrich, S. P., Miller, D. R., Stolley, P. D., and Shapiro, S. (1985). Myocardial infarction and cigarette smoking in women younger than 50 years of age. JAMA 253, 2965–2969. doi: 10.1001/jama.1985.03350440043028
Rudnick, G. (1977). Active transport of 5-hydroxytryptamine by plasma membrane vesicles isolated from human blood platelets. J. Biol. Chem. 252, 2170–2174.
Senis, Y. A., Mazharian, A., and Mori, J. (2014). Src family kinases: at the forefront of platelet activation. Blood 124, 2013–2024. doi: 10.1182/blood-2014-01-453134
Sherva, R., Wilhelmsen, K., Pomerleau, C. S., Chasse, S. A., Rice, J. P., Snedecor, S. M., et al. (2008). Association of a single nucleotide polymorphism in neuronal acetylcholine receptor subunit alpha 5 (CHRNA5) with smoking status and with ‘pleasurable buzz’ during early experimentation with smoking. Addiction 103, 1544–1552. doi: 10.1111/j.1360-0443.2008.02279.x
Shirakawa, R., Yoshioka, A., Horiuchi, H., Nishioka, H., Tabuchi, A., and Kita, T. (2000). Small GTPase Rab4 regulates Ca2+-induced α-granule secretion in platelets. J. Biol. Chem. 275, 33844–33849. doi: 10.1074/jbc.M002834200
Siess, W., Lorenz, R., Roth, P., and Weber, P. C. (1982). Plasma catecholamines, platelet aggregation and associated thromboxane formation after physical exercise, smoking or norepinephrine infusion. Circulation 66, 44–48. doi: 10.1161/01.CIR.66.1.44
Singh, P., Fletcher, T. W., Li, Y., Rusch, N. J., and Kilic, F. (2013). Serotonin uptake rates in platelets from angiotension II-induced hypertensive mice. Health 5, 31–39. doi: 10.4236/health.2013.54A005
Sjoerdsma, A., and Palfreyman, M. G. (1990). History of serotonin and serotonin disorders. Ann. N. Y. Acad. Sci. 600, 1–7. doi: 10.1111/j.1749-6632.1990.tb16869.x
Smyth, S. S., Smyth, S. S., Woulfe, D. S., Weitz, J. I., Gachet, C., Conley, P. B., et al. (2009). G-protein-coupled receptors as signaling targets for antiplatelet therapy. Arterioscler. Thromb. Vasc. Biol. 29, 449–457. doi: 10.1161/ATVBAHA.108.176388
Tweed, J. O., Hsia, S. H., Lutfy, K., and Friedman, T. C. (2012). The endocrine effects of nicotine and cigarette smoke. Trends Endocrinol. Metab. 7, 334–342. doi: 10.1016/j.tem.2012.03.006
US Department of Health and Human Services (1990). The Health Benefits of Smoking Cessation A Report of the Surgeon General. Atlanta: US Department of Health and Human Services.
Vikens, K., Farstad, M., and Nordrehaug, J. E. (1999). Serotonin is associated with coronary artery disease and cardiac events. Circulation 100, 483–489. doi: 10.1161/01.CIR.100.5.483
Walther, D. J., Peter, J. U., Winter, S., Holtje, M., Paulmann, N., Grohmann, M., et al. (2003). Serotonylation of small GTPases is a signal transduction pathway that triggers platel etalpha-granule release. Cell 26, 851–862. doi: 10.1016/S0092-8674(03)01014-6
Willett, W. C., Green, A., Stampfer, M. J., Speizer, F. E., Colditz, G. A., Rosner, B., et al. (1987). Relative and absolute excess risks of coronary heart disease among women who smoke cigarettes. New Engl. J. Med. 317, 1303–1309. doi: 10.1056/NEJM198711193172102
Winniford, M. D., Jansen, D. E., Reynolds, G. A., Apprill, P., Black, W. H., and Hillis, L. D. (1987). Cigarette smoking-induced coronary vasoconstriction in atherosclerotic coronary artery disease and prevention by calcium antagonists and nitroglycerin. Am. J. Cardiol. 59, 203–207. doi: 10.1016/0002-9149(87)90785-5
Woulfe, D. S. (2005). Platelet G protein-coupled receptors in hemostasis and thrombosis. J. Thromb. Haemost. 310, 2193–2200. doi: 10.1111/j.1538-7836.2005.01338.x
Yang, J., Wu, J., Jiang, H., Mortensen, R., Austin, S., Manning, D. R., et al. (2002). Signaling through Gi family members in platelets. Redundancy and specific regulation of adenylyl cyclase and other effectors. J. Biol. Chem. 277, 46035–46042. doi: 10.1074/jbc.M208519200
Ziu, E., Freyaldenhoven, S., Mercado, C. P., Ahmed, B. A., Preeti, P., Lensing, S., et al. (2012). Down-regulation of the serotonin transporter in hyperreactive platelets counteracts the pro-thrombotic effect of serotonin. J. Mol. Cell. Cardiol. 52, 1112–1121. doi: 10.1016/j.yjmcc.2012.02.004
Keywords: cigarette smoking, serotonin, catecholamine, platelet aggregation, serotonin transporter
Citation: Lowery CL III, Woulfe D and Kilic F (2019) Responses of Plasma Catecholamine, Serotonin, and the Platelet Serotonin Transporter to Cigarette Smoking. Front. Neurosci. 13:32. doi: 10.3389/fnins.2019.00032
Received: 30 October 2018; Accepted: 14 January 2019;
Published: 04 March 2019.
Edited by:
Nasser Haddjeri, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Clinton Canal, Mercer University, United StatesCopyright © 2019 Lowery, Woulfe and Kilic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fusun Kilic, ZnVzdW5raWxpY0B5YWhvby5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.