 Arnaud Bessac
Arnaud Bessac Patrice D. Cani
Patrice D. Cani Etienne Meunier
Etienne Meunier Gilles Dietrich
Gilles Dietrich Claude Knauf
Claude Knauf
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurosci. , 10 October 2018
Sec. Neuroenergetics and Brain Health
Volume 12 - 2018 | https://doi.org/10.3389/fnins.2018.00725
This article is part of the Research Topic Obesity and Diabetes: Implications for Brain-Immunometabolism View all 16 articles
The gut-brain axis is now considered as a major actor in the control of glycemia. Recent discoveries show that the enteric nervous system (ENS) informs the hypothalamus of the nutritional state in order to control glucose entry in tissues. During type 2 diabetes (T2D), this way of communication is completely disturbed leading to the establishment of hyperglycemia and insulin-resistance. Indeed, the ENS neurons are largely targeted by nutrients (e.g., lipids, peptides) but also by inflammatory factors from different origin (i.e., host cells and gut microbiota). Inflammation, and more particularly in the intestine, contributes to the development of numerous pathologies such as intestinal bowel diseases, Parkinson diseases and T2D. Therefore, targeting the couple ENS/inflammation could represent an attractive therapeutic solution to treat metabolic diseases. In this review, we focus on the role of the crosstalk between intestinal immune cells and ENS neurons in the control of glycemia. In addition, given the growing evidence showing the key role of the gut microbiota in physiology, we will also briefly discuss its potential contribution and role on the immune and neuronal systems.
The population of gut microbiota is estimated to 100 trillion microorganisms. Thus, our body is composed of 50% of bacteria and 50% of human cells (Cani, 2016). Precisely, the human body encloses approximatively 4.1013 bacteria cells with the largest amount inside the large intestine (1011 bacteria cells/g of wet stools) (Sender et al., 2016a). In the last estimation, almost 10 million non-redundant microbial genes have been identified in the human gut (Li et al., 2014). The intestinal tract is subjected to an environmental pressure due to the high microbial load of the luminal content (Sender et al., 2016b). Regarding microbial load, one recent article shows that it is the absolute quantity of microbes and not the proportions of microbes that really matters (Vandeputte et al., 2017). Depending of the mode of analysis of gut microbiota population (classical relative abundance-based profiling vs. quantitative microbiome profiling), the interpretation of data could be completely different, and maybe falsely interpreted. The study of gut microbiota impacts in physiology is also complicated due to the controversial data present in the literature. In fact, results could differ depending of numerous factors that we have to take into account such as for example dietary environments, but also the composition and the activity of the gut microbes (Cani, 2018a). To illustrate this controversy, Prevotella copri and its metabolites could be considered as pro- or anti-diabetic actions depending on the diet or the model used (Cani, 2018a). Therefore, these few examples clearly highlight the fact that we are still at the beginning of the story, and we will need more time to better understand the gut microbiota and its importance in human health.
Nowadays, the impact of gut microbiota in the control of various physiological functions is proposed (Cani, 2018a). Abnormal composition and/or activity of the gut microbiota are associated with the development of numerous pathologies such as cancer, obesity and type 2 diabetes (T2D) (Cani, 2018a; Cani and Jordan, 2018b; Rastelli et al., 2018). Despite the complexity of the crosstalk, a clear link is established between inflammation and modification of the gut microbiota (Stecher, 2015; Cani, 2018a). Here, we will mainly introduce how gut bacteria could modulate the function of intestinal immune cells, and describe the molecular actors involved.
Intestinal bacteria are physically separated from mucosal immune system by a single epithelial cell layer, which the primary function is to absorb nutrients (small intestine) and water (colon). Mucosal immune system prevent microbial invasion and is tightly regulated. One of its major role is to avoid the development of chronic inflammation and the subsequent loss of the intestinal epithelium integrity. Microfold cells or M cells, in the specialized follicle-associated epithelium overlying Peyer Patches (PP), and isolated lymphoid follicles (ILF) are the major cell types that sample bacteria and associated antigens. Processed bacterial-derived antigens are presented locally (i.e., into the PP or ILF) or within the mesenteric lymph nodes that drain dendritic cells to initiate an adaptive immune response (Wells et al., 2017). Both effector- and regulatory-T lymphocytes scattered within the intestinal mucosa are generated in response to commensal bacterial antigens. At steady state, the number and the spectrum of effector-T lymphocytes subsets that are present within the intestinal mucosa are dependent on the host’s microbiota. Any pathogen invasion, disruptions of the mucus barrier or of the intestinal epithelium integrity, and/or failure in the regulatory mechanisms of the immune response may result in mucosal inflammation (Barreau and Hugot, 2014; Al Nabhani et al., 2017). Immune mediators released upon inflammation are largely dependent on the nature of the microbes triggering the immune system (Maloy and Powrie, 2011). Intestinal epithelial cells and resident innate immune cells sense pathogens locally. The interactions between the pathogens and the pattern-recognition receptors (PRRs) expressed both by stromal and immune cells trigger rapid production of immune and microbicide mediators (e.g., cytokines, chemokines, bioactive lipids, and cell-autonomous immune effectors), which restrict pathogen growth. In parallel, dendritic cells will mature upon contact with microbe-associated molecular patterns (MAMPs) or when some factors are released by injured tissues, namely the damage associated molecular patterns (DAMPs) (i.e., a process allowing antigen presentation to T cells) (Maloy and Powrie, 2011; Geginat et al., 2015). Regarding the intrinsic properties of mature dendritic cells and their soluble (e.g., cytokines, chemokines) and cellular (stromal, myeloid and lymphoid cells) immune environment, antigen-primed CD4+ T lymphocytes may acquire different effector functions. Indeed, viral or intracellular bacterial infections drive T lymphocyte commitment toward the Th1 phenotype, a process that relies both on the production of IL-12 and IL-18 by myeloid cells and the subsequent IFNγ released by innate lymphoid cells (ILC)1 (Trinchieri, 2003; Bernink et al., 2013). Th1 CD4+ lymphocytes produce high levels of IFNγ but also TNF-α. The clearance of extracellular bacteria and fungi mainly depends on Th17-polarized lymphocytes that produce IL-17, IL-22, IL-21, TNF-α and GMCSF. The differentiation of naïve CD4+ T lymphocytes into Th17 involves an environment enriched in TGFβ, IL-1β and STAT3-inducing cytokines including IL-6 and IL-23 produced by myeloid cells (e.g., dendritic cells and macrophages). IL-23 plays a key role by reinforcing Th17 functional activity and inducing production of GMCSF and TNF-α. The immune polarization of ILCs from group 2 by IL-25, IL-33 and thymic stromal lymphopoietin is a process specific to helminth parasites. This leads to a potent IL-13 release, crucial immune mediator of the anti-parasitic response (Neill et al., 2010). This frontline type 2 cytokine signaling drives the differentiation of T lymphocytes toward the Th2 phenotype, which is characterized by the synthesis of IL4, IL-5 and IL-13 cytokines, that are key to restrict helminth infections. In addition to cytokines, CD4+ T lymphocytes may also produce neuropeptides including opioids, serotonin and vasoactive intestinal peptide (O’Connell et al., 2006; Boue et al., 2011; Souza-Moreira et al., 2011). Within the site of inflammation, most of these neuropeptides as well as chemokines, proteases and bioactive lipids are also produced by endothelial and epithelial cells, migrant and resident innate immune cells.
How make the link between gut microbiota, immunity and metabolic diseases? The development of metabolic disorders is clearly linked with modification of the gut microbiota population, but there are many other factors in the intestine (e.g., bioactive molecules, metabolites, endocrine cells) that are disturbed in pathological state. The diversity of the gut microbiota may not fully explain the pathologic phenotype. In fact, recent data show the importance of specific alterations that may arise at the level of the crosstalk occurring between gut microbes but also luminal and intestinal factors during metabolic disorders (Aron-Wisnewsky et al., 2018; Cani, 2018). One may also mention the controversy regarding microbial diversity and its potential link with the onset of inflammation. This important topic has been recently reviewed and clearly described the potential interaction between microbes and host cells via immune factors (Cani, 2018a). Modifications of the balance (i.e., gut microbiota and immunity) between healthy and pathological situations participate to the establishment of a type 2 diabetic state. For example in this review (Cani, 2018a), it is discussed that the production of the short-chain fatty acids propionate is altered during diabetes. In physiological condition, propionate can bind to GPR-43 located on intestinal lymphocytes. In pathological situations, the decrease of propionate may lead to the lower abundance of specific T cells (mucosal-associated invariant T cells and Treg) in the lamina propria of the gut. Consequently, the gut barrier function could be altered, and some pathogen-associated molecular patterns (PAMPs) can reach the circulation. This is for instance the case for the levels of lipopolysaccharides (LPS) that are significantly increased in the blood, and trigger systemic low-grade inflammation (Cani et al., 2007; Chu et al., 2018) observed in the whole body and can generate the insulin resistance state observed during diabetes (Cani et al., 2012).
Obesity and associated metabolic disorders such as T2D are closely related to a « low-grade » inflammation state as described above (Wellen and Hotamisligil, 2005). In addition to well-described tissues (liver, adipose tissue,…) (Hotamisligil, 2006), the intestine may presents an alteration of the gut barrier function thereby leading hyper-permeability, which is associated with a dysbiosis and in some cases with a certain degree of inflammation at the level of the gut (for review see Slyepchenko et al., 2016). Indeed, in response to a high-fat diet (HFD) that induces insulin resistance, it has been proposed that the intestinal tract may develop an inflammatory response characterized by an increase in pro-inflammatory cytokines expression such as TNF-α, IL-1β, IL-6 (Ding et al., 2010; Kim et al., 2012). Some studies have also reported an increase in myeloperoxidase activity (de La Serre et al., 2010) and/or a decrease in IL-18 and IL-22 expression (Everard et al., 2014a; Wang et al., 2014), two cytokines involved in the intestinal barrier homeostasis and the defense against pathogens. However, it is important to note that the degree of inflammation observed during obesity and diabetes does not reach the same magnitude as the one observed during intestinal inflammatory bowel diseases or severe infection. Nevertheless, besides HFD, data also support that the genetic deletion of leptin (ob/ob) or leptin receptors (db/db) in mice leads to the development of obesity and T2D with a significant increase in pro-inflammatory cytokines (TNF-α, IL-1β, IFNγ, IL-6) in the portal vein, as well as an altered gut barrier (Brun et al., 2007). In addition to metabolic consequences, deletion of leptin receptors also alters immunological response against Entamoeba histolytica in mice (Guo et al., 2011) as well as in humans (Duggal et al., 2011). The direct link between intestinal inflammation and metabolic disorders is observed in mice knocked-out for TLR5, a component of the innate immune system. TLR5-deficient mice exhibit a diabetic phenotype associated with intestinal inflammation including increase in pro-inflammatory cytokines (IL-1β, TNF-α) and reduction of the length of the colon, one hallmark of intestinal inflammation (Vijay-Kumar et al., 2010; Chassaing et al., 2014).
What are the “local” consequences of this gut inflammation? All mice models above display alterations of the intestinal homeostasis suggesting as close interrelation between “inflammation, microbiota and metabolism” (Geurts et al., 2014). Because of this inflammatory environment, diabetic mice exhibit modifications of intestinal absorptive capacity characterized by an increase of glucose and/or lipids absorption (Ferraris and Vinnakota, 1995; Petit et al., 2007; Mao et al., 2013) that alters the energetic metabolism. Indeed, HFD diabetic mice develop hyperglycemia and hyperinsulinemia (Ding et al., 2010), glucose intolerance and insulin resistance correlated with a decrease in mucus thickness and increased intestinal permeability (Cani et al., 2008; Everard et al., 2013). This is associated with alteration of the gut microbiota composition and activity (Everard et al., 2011, 2014b; Kim et al., 2012; Molinaro et al., 2017) have shown that gut colonization of germ-free mice induces a bi-phasic inflammation that participates to a bi-phasic impairment of glucose metabolism. Interestingly, recent data have also shown in humans a link between gut microbiota and the accumulation of T cells in the gut of obese people following the ingestion of a high-fat diet (Monteiro-Sepulveda et al., 2015). Both the blunted insulin signaling in enterocytes and the altered localization of the glucose transporter GLUT2 in the enterocytes of obese subjects were linked to the nature of cytokines produced by mucosal T cells (Monteiro-Sepulveda et al., 2015). Conversely, in obese and type 2 diabetic patients another study has shown a decreased number of mucosal-associated invariant T cells (MAIT) producing high levels of Th1 and Th17 cytokines (Magalhaes et al., 2015).
Therefore, the molecular partners linking inflammation, gut microbiota and metabolic disorders could be multiple, but the incretin hormones may represent a real potential of interest. The intestinal tract has also the capacity to produce incretins such as Glucagon-Like Peptide-1 (GLP-1) or Gastric Inhibitory Polypeptide (GIP) in response to nutrients and/or bacterial factors (Cani et al., 2013; Rastelli et al., 2018). HFD and ob/ob mice display an alteration of gut endocrine system including the reduction of GIP- and GLP-1-producing cells and a decrease in proglucagon and GIP expression associated with the alteration of incretins secretion in response to an oral glucose load (Morgan et al., 1988; Anini and Brubaker, 2003; Richards et al., 2016; Shimazu-Kuwahara et al., 2017).
In line with these observations, we have demonstrated that prebiotic treatment in diabetic mice restores the level of GLP-1 to improve glucose metabolism (Cani et al., 2006). Moreover, a chronic exposure to TNF-α alters the release of GLP-1 in HFD mice by acting directly on GLP-1-producing cells (Gagnon et al., 2015). Conversely, a 2 weeks treatment with etanercept, an anti-TNF-α drug, reduces hyperglycemia and hyperinsulinemia of HFD mice. Nowadays, this tripartite concept developed in mice is considered as a potential target to treat metabolic disorders (Geurts et al., 2014).
The gut-brain axis is shown as a major signaling pathway controlling glucose homeostasis, and its disturbance is associated with a T2D phenotype (Abot et al., 2018a,b). In normal conditions, glucose is detected by intestinal glucose sensors (such as SGLT1, GLUT2, TASR1/2) present on intestinal cells (enteroendocrine and brush cells, enteric glial cells and neurons) which inform the whole body from the presence of glucose via the release of various factors (i.e., gut hormones, neurotransmitters, metabolites) (Fournel et al., 2016). Then, intestinal glucose detection generates an afferent nervous message that provokes an increase of hypothalamic nitric oxide (NO) release (Fournel et al., 2017). In turn, this increase of hypothalamic NO release favors the entry of glucose in tissues via an activation of the autonomic nervous system (Fournel et al., 2017). In db/db mice, intestinal inflammation perturbs the detection of glucose by enteric glucose sensors (Duparc et al., 2011). In this case, the intestine sends an aberrant nervous message that fails to increase hypothalamic NO release (Duparc et al., 2011). Accordingly, the insulin resistance state observed during T2D is linked to alteration of gut-brain axis. Recently, we have discovered that the enteric nervous system (ENS) is implicated in the maintenance of glycemia in the whole body via the brain. One of the main functions of ENS is to regulate contractions of intestinal smooth muscle cells. In physiological conditions, the reduction of intestinal contractions provokes a decreased fed hyperglycemia (Fournel et al., 2017; Abot et al., 2018b). The modifications of mechanical contractions are detected by the hypothalamus, which controls glucose utilization (Fournel et al., 2017). During T2D, intestinal inflammation is clearly associated with a dysfunction of ENS neurons (Gonzalez-Correa et al., 2017), which could favor duodenal hyper-contractility (Abot et al., 2018b). Our group has demonstrated that duodenal hyper-contractility participates to the establishment of hyperglycemia of HFD mice by modulating the hypothalamic NO release (Fournel et al., 2017).
Numerous pathologies that link inflammation, gut microbiota and ENS have been described (Greenwood-Van Meerveld et al., 2017). Along those lines, recent data have shown that specific neurodegenerative diseases (i.e., multiple sclerosis, Parkinson disease) were also characterized by gastrointestinal comorbidities. In these patients, the specific alterations in gut motility and other pre-motor symptoms are linked with changes in the gut microbiota composition and activity (Endres and Schafer, 2018). Whether the couple microbiota and metabolites influencing the intestinal neuronal system may explain the onset of the disease remains to be proven, but it is becoming clear that the ENS can be considered as a cellular target to treat inflammatory diseases (T2D, inflammatory bowel diseases and ulcers).
Therefore, targeting the intestinal inflammation and/or the gut microbiota to restore the glucose and mechano-sensors in type 2 diabetic patients presents a real potential of scientific interest, but remains to be determined.
Environmental changes related to the inflammatory processes that include a large spectrum of innate and adaptive immune responses, may affect ENS including myenteric plexus controlling gut motility (Cani et al., 2006).
Intestinal motility is regulated by ENS that includes intrinsic primary afferent neurons (IPAN), interneurons and choline acetyltransferase (ChAT)-producing excitatory and NO synthase (NOS)-producing inhibitory motor neurons (Abot et al., 2018a). In the context of immune-mediated regulation of intestinal motility, muscularis macrophages residing in the myenteric plexus have been largely studied (Mikkelsen, 2010; Muller et al., 2014; De Schepper et al., 2017). However, cytokines, proteases or neuropeptides produced by mucosal immune cells may also modulate the activity of sensory neurons.
IPANs with their axons extending throughout intestinal mucosa and their cell bodies connected with interneurons which synapse with motor neurons may thus constitute central players in the immune-modulation of the intestinal motility. Accordingly, inflammatory cytokines including TNF-α, IL-1β or IL-6 as well as neuromediators released by innate and adaptive immune cells upon intestinal inflammation alter intrinsic and extrinsic enteric sensory neuron activities (Kindt et al., 2010; Basso et al., 2014; Boue et al., 2014; Brierley and Linden, 2014; Buckley et al., 2014; Hughes Moretta et al., 2014; Mawe, 2015). A slower small-bowel transit and reduced smooth muscle contractility have been reported in patients with inflammatory bowel diseases (Rao et al., 1987; Mawe, 2015). In experimental colitis, intestinal motility is either enhanced or reduced depending on the Th1 or Th2 types of inflammation. Th2-driven colitis induces smooth muscle hyper-contractility and augments intestinal transit while Th1-driven colitis is rather associated with hypo-contractility and reduced intestinal transit (Zhao et al., 2003; Shea-Donohue et al., 2012). Moreover, chronic intestinal inflammation often results in increased innervation together with a reduction of the neuronal activation threshold (Villanacci et al., 2008; Brierley and Linden, 2014). The expression of TNF-α receptors (TNFR1, TNFR2) on enteric neurons also suggest that the neuronal activity maybe modulated directly by the inflammatory environment (Chandrasekharan et al., 2013; Gougeon et al., 2013). Actually, TNF-α has the capacity to modulate electrophysiologic properties of neurons via a potentiating effect by increasing the action of cholinergic agonists (carbachol) on gut motility (Rehn et al., 2004). This property of TNF-α could explain the negative impact of cytokines on gut motility. In a molecular point of view, the effect of TNF-α is mediated by an upregulation of the Cyclo-Oxygenase (COX-2), which in turn increases the production of an arachidonic acid derived-molecule, Thromboxane A2 (TXA2) (Rehn et al., 2005). In experimental colitis, Chandrasekharan et al. (2013) have also demonstrated that TNF-α has an effect on the apoptosis rate of enteric neurons. Treatment with TNF inhibitors provokes a significant decrease of inflammatory markers expression such as inducible NOS (iNOS). IL-1β is another cytokine that has its receptors on ENS neurons (Gougeon et al., 2013). Similarly to TNF-α, IL-1β may increase iNOS mRNA expression in myenteric neurons (Valentine et al., 1996). IL-1β could increase excitability of neurons by directly acting through its receptors expressed on cell surface. Mechanisms of IL-1β including depolarization of the membrane potential, decreased membrane conductance, and increased discharge of action potentials could explain the diarrhea phase, which appears in obese/diabetic patient (Acosta and Camilleri, 2014) or during intestinal inflammation (Xia et al., 1999).
In addition to neurons, the ENS is composed of glial cells that exert a crucial role in intestinal physiology. Suppression of glial cells alters the function of ENS neurons with an increase in excitatory ChAT neurons and a decrease in inhibitory NOS neurons (Aube et al., 2006). Glial cells are exposed to various molecules depending on the environment including immune factors. Proinflammatory cytokines such as IL-1β and/or low IL-10 concentrations have the capacity to block glial cells proliferation (Ruhl et al., 2001a), highlighting links between immune system and enteric nervous system. Moreover, IL-1β has also the capacity to regulate secretory functions of glial cells by increasing IL-6 synthesis and secretion (Ruhl et al., 2001b). In turn, IL-6 may modulate T cell apoptosis or control gut motility to delay gastric emptying (Atreya et al., 2000; Lang Lehrskov et al., 2018).
T2D is associated with modification of the gut physiology including alterations of the function of immune cells and ENS. The crosstalk between the two partners is under the influence of the gut bacteria, which release bioactive factors that could participate as intermediate signaling molecules. Some evidence suggests that the establishment of T2D phenotypes (i.e., hyperglycemia, insulin resistance) may have a potential enteric origin; this is also the case of some neurological disorders (e.g., multiple sclerosis, Parkinson disease, autism). Deciphering the impact of “immune cells-ENS” communication and its impact on “gut-brain” axis could represent a new therapeutic perspective for diabetic patients (Figure 1). Therapeutic strategies have to take into account the importance of intestinal inflammation and its consequence on integrity of ENS. The gut microbiota and its bioactive releasing factors could represent one potential solution.
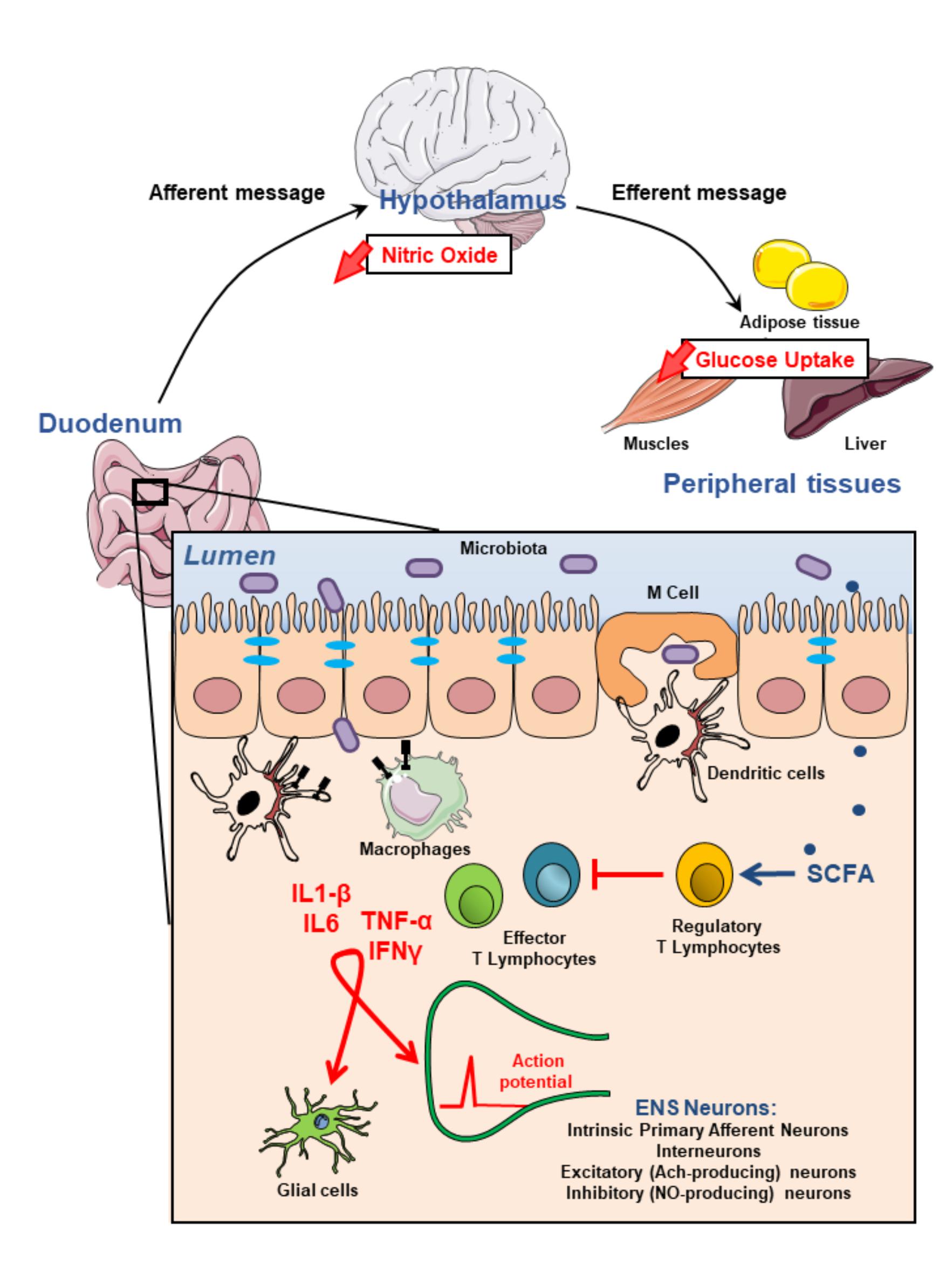
FIGURE 1. Modulation of the enteric nervous system activity by intestinal immune system may impact on glycemia regulation. Most microbiota-derived products activate both epithelial cells and innate immune cells through pathogen recognition receptors such as toll-like receptors. They also initiate, mainly via microbe sampling through M(icrofold) cells, an adaptive immune response. This physiological immune response against intestinal flora generating pro-inflammatory effector T lymhocytes is tighly regulated by regulatory T lymphocytes which induction is associated with the production of short chain fatty acids (SCFAs) by commensal bacteria. Failure of the immune homeostasis may result in the release of higher amounts of proinflammatory cytokines including TNFα, IL1-β, IL6, IFNγ which, in turn, modulate either directly or through glial cells, enteric nervous system (ENS) activity. These alterations of the ENS contribute to impair gut-brain-peripheral axis leading to hyperglycemia and insulin-resistance.
AB, PDC, EM, GD, and CK have wrote the manuscript.
PDC is a senior research associate at FRS-FNRS (Fonds de la Recherche Scientifique). PDC is a recipient of grants from FRS-FNRS such as FRFS-WELBIO (WELBIO-CR-2017C-02) and The Excellence Of Science (EOS 30770923). This work is supported in part by the Funds Baillet Latour (Grant for Medical Research 2015) and ERC Starting Grant 2013 (Starting grant 336452-ENIGMO).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abot, A., Cani, P. D., and Knauf, C. (2018a). Impact of intestinal peptides on the enteric nervous system: novel approaches to control glucose metabolism and food intake. Front. Endocrinol. 9:328. doi: 10.3389/fendo.2018.00328
Abot, A., Lucas, A., Bautzova, T., Bessac, A., Fournel, A., Le-Gonidec, S., et al. (2018b). Galanin enhances systemic glucose metabolism through enteric nitric oxide synthase-expressed neurons. Mol. Metab. 10, 100–108. doi: 10.1016/j.molmet.2018.01.020
Acosta, A., and Camilleri, M. (2014). Gastrointestinal morbidity in obesity. Ann. N. Y. Acad. Sci. 1311, 42–56. doi: 10.1111/nyas.12385
Al Nabhani, Z., Dietrich, G., Hugot, J. P., and Barreau, F. (2017). Nod2: the intestinal gate keeper. PLoS Pathog. 13:e1006177. doi: 10.1371/journal.ppat.1006177
Anini, Y., and Brubaker, P. L. (2003). Role of leptin in the regulation of glucagon-like peptide-1 secretion. Diabetes Metab. Res. Rev. 52, 252–259.
Aron-Wisnewsky, J., Prifti, E., Belda, E., Ichou, F., Kayser, B. D., Dao, M. C., et al. (2018). Major microbiota dysbiosis in severe obesity: fate after bariatric surgery. Gut doi: 10.1136/gutjnl-2018-316103 [Epub ahead of print].
Atreya, R., Mudter, J., Finotto, S., Mullberg, J., Jostock, T., Wirtz, S., et al. (2000). Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat. Med. 6, 583–588. doi: 10.1038/75068
Aube, A. C., Cabarrocas, J., Bauer, J., Philippe, D., Aubert, P., Doulay, F., et al. (2006). Changes in enteric neurone phenotype and intestinal functions in a transgenic mouse model of enteric glia disruption. Gut 55, 630–637. doi: 10.1136/gut.2005.067595
Barreau, F., and Hugot, J. P. (2014). Intestinal barrier dysfunction triggered by invasive bacteria. Curr. Opin. Microbiol. 17, 91–98. doi: 10.1016/j.mib.2013.12.003
Basso, L., Boue, J., Bourreille, A., and Dietrich, G. (2014). Endogenous regulation of inflammatory pain by T-cell-derived opioids: when friend turns to foe. Inflamm. Bowel Dis. 20, 1870–1877. doi: 10.1097/MIB.0000000000000073
Bernink, J. H., Peters, C. P., Munneke, M., te Velde, A. A., Meijer, S. L., Weijer, K., et al. (2013). Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 14, 221–229. doi: 10.1038/ni.2534
Boue, J., Basso, L., Cenac, N., Blanpied, C., Rolli-Derkinderen, M., Neunlist, M., et al. (2014). Endogenous regulation of visceral pain via production of opioids by colitogenic CD4( + ) T cells in mice. Gastroenterology 146, 166–175. doi: 10.1053/j.gastro.2013.09.020
Boue, J., Blanpied, C., Brousset, P., Vergnolle, N., and Dietrich, G. (2011). Endogenous opioid-mediated analgesia is dependent on adaptive T cell response in mice. J. Immunol. 186, 5078–5084. doi: 10.4049/jimmunol.1003335
Brierley, S. M., and Linden, D. R. (2014). Neuroplasticity and dysfunction after gastrointestinal inflammation. Nat. Rev. Gastroenterol. Hepatol. 11, 611–627. doi: 10.1038/nrgastro.2014.103
Brun, P., Castagliuolo, I., Di Leo, V., Buda, A., Pinzani, M., Palu, G., et al. (2007). Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G518–G525. doi: 10.1152/ajpgi.00024.2006
Buckley, M. M., O’Halloran, K. D., Rae, M. G., Dinan, T. G., and O’Malley, D. (2014). Modulation of enteric neurons by interleukin-6 and corticotropin-releasing factor contributes to visceral hypersensitivity and altered colonic motility in a rat model of irritable bowel syndrome. J. Physiol. 592, 5235–5250. doi: 10.1113/jphysiol.2014.279968
Cani, P. D. (2016). Interactions between gut microbes and host cells control gut barrier and metabolism. Int. J. Obes. Suppl. 6(Suppl. 1), S28–S31. doi: 10.1038/ijosup.2016.6
Cani, P. D. (2018). Severe obesity and gut microbiota: does bariatric surgery really reset the system? Gut doi: 10.1136/gutjnl-2018-316815 [Epub ahead of print].
Cani, P. D. (2018a). Human gut microbiome: hopes, threats and promises. Gut 67, 1716–1725. doi: 10.1136/gutjnl-2018-316723
Cani, P. D., Amar, J., Iglesias, M. A., Poggi, M., Knauf, C., Bastelica, D., et al. (2007). Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes Metab. Res. Rev. 56, 1761–1772.
Cani, P. D., Bibiloni, R., Knauf, C., Waget, A., Neyrinck, A. M., Delzenne, N. M., et al. (2008). Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes Metab. Res. Rev. 57, 1470–1481. doi: 10.2337/db07-1403
Cani, P. D., Everard, A., and Duparc, T. (2013). Gut microbiota, enteroendocrine functions and metabolism. Curr. Opin. Pharmacol. 13, 935–940. doi: 10.1016/j.coph.2013.09.008
Cani, P. D., and Jordan, B. F. (2018b). Gut microbiota-mediated inflammation in obesity: a link with gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. doi: 10.1038/s41575-018-0025-6 [Epub ahead of print].
Cani, P. D., Knauf, C., Iglesias, M., Drucker, D., Delzenne, N., and Burcelin, R. (2006). Improvement of glucose tolerance and hepatic insulin sensitivity by oligofructose requires a functional GLP-1 receptor. Diabetes Metab. Res. Rev. 55, 1484–1490.
Cani, P. D., Osto, M., Geurts, L., and Everard, A. (2012). Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes 3, 279–288. doi: 10.4161/gmic.19625
Chandrasekharan, B., Jeppsson, S., Pienkowski, S., Belsham, D. D., Sitaraman, S. V., Merlin, D., et al. (2013). Tumor necrosis factor-neuropeptide Y cross talk regulates inflammation, epithelial barrier functions, and colonic motility. Inflamm. Bowel Dis. 19, 2535–2546. doi: 10.1097/01.MIB.0000437042.59208.9f
Chassaing, B., Ley, R. E., and Gewirtz, A. T. (2014). Intestinal epithelial cell toll-like receptor 5 regulates the intestinal microbiota to prevent low-grade inflammation and metabolic syndrome in mice. Gastroenterology 147, 1363.e17–1377.e17. doi: 10.1053/j.gastro.2014.08.033
Chu, H., Duan, Y., Yang, L., and Schnabl, B. (2018). Small metabolites, possible big changes: a microbiota-centered view of non-alcoholic fatty liver disease. Gut doi: 10.1136/gutjnl-2018-316307 [Epub ahead of print].
de La Serre, C. B., Ellis, C. L., Lee, J., Hartman, A. L., Rutledge, J. C., and Raybould, H. E. (2010). Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G440–G448. doi: 10.1152/ajpgi.00098.2010
De Schepper, S., Stakenborg, N., Matteoli, G., Verheijden, S., and Boeckxstaens, G. E. (2017). Muscularis macrophages: key players in intestinal homeostasis and disease. Cell. Immunol. 330, 142–150. doi: 10.1016/j.cellimm.2017.12.009
Ding, S., Chi, M. M., Scull, B. P., Rigby, R., Schwerbrock, N. M., Magness, S., et al. (2010). High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One 5:e12191. doi: 10.1371/journal.pone.0012191
Duggal, P., Guo, X., Haque, R., Peterson, K. M., Ricklefs, S., Mondal, D., et al. (2011). A mutation in the leptin receptor is associated with Entamoeba histolytica infection in children. J. Clin. Invest. 121, 1191–1198. doi: 10.1172/JCI45294
Duparc, T., Naslain, D., Colom, A., Muccioli, G. G., Massaly, N., Delzenne, N. M., et al. (2011). Jejunum inflammation in obese and diabetic mice impairs enteric glucose detection and modifies nitric oxide release in the hypothalamus. Antioxid. Redox. Signal. 14, 415–423. doi: 10.1089/ars.2010.3330
Endres, K., and Schafer, K. H. (2018). Influence of commensal microbiota on the enteric nervous system and its role in neurodegenerative diseases. J. Innate. Immun. 10, 172–180. doi: 10.1159/000488629
Everard, A., Belzer, C., Geurts, L., Ouwerkerk, J. P., Druart, C., Bindels, L. B., et al. (2013). Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. U.S.A. 110, 9066–9071. doi: 10.1073/pnas.1219451110
Everard, A., Geurts, L., Caesar, R., Van Hul, M., Matamoros, S., Duparc, T., et al. (2014a). Intestinal epithelial MyD88 is a sensor switching host metabolism towards obesity according to nutritional status. Nat. Commun. 5:5648. doi: 10.1038/ncomms6648
Everard, A., Lazarevic, V., Gaia, N., Johansson, M., Stahlman, M., Backhed, F., et al. (2014b). Microbiome of prebiotic-treated mice reveals novel targets involved in host response during obesity. ISME J. 8, 2116–2130. doi: 10.1038/ismej.2014.45
Everard, A., Lazarevic, V., Derrien, M., Girard, M., Muccioli, G. G., Neyrinck, A. M., et al. (2011). Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes Metab. Res. Rev. 60, 2775–2786. doi: 10.2337/db11-0227
Ferraris, R. P., and Vinnakota, R. R. (1995). Intestinal nutrient transport in genetically obese mice. Am. J. Clin. Nutr. 62, 540–546. doi: 10.1093/ajcn/62.3.540
Fournel, A., Drougard, A., Duparc, T., Marlin, A., Brierley, S. M., Castro, J., et al. (2017). Apelin targets gut contraction to control glucose metabolism via the brain. Gut 66, 258–269. doi: 10.1136/gutjnl-2015-310230
Fournel, A., Marlin, A., Abot, A., Pasquio, C., Cirillo, C., Cani, P. D., et al. (2016). Glucosensing in the gastrointestinal tract: impact on glucose metabolism. Am. J. Physiol. Gastrointest. Liver Physiol. 310, G645–G658. doi: 10.1152/ajpgi.00015.2016
Gagnon, J., Sauve, M., Zhao, W., Stacey, H. M., Wiber, S. C., Bolz, S. S., et al. (2015). Chronic exposure to TNFalpha impairs secretion of glucagon-like peptide-1. Endocrinology 156, 3950–3960. doi: 10.1210/en.2015-1361
Geginat, J., Nizzoli, G., Paroni, M., Maglie, S., Larghi, P., Pascolo, S., et al. (2015). Immunity to pathogens taught by specialized human dendritic cell subsets. Front. Immunol. 6:527. doi: 10.3389/fimmu.2015.00527
Geurts, L., Neyrinck, A. M., Delzenne, N. M., Knauf, C., and Cani, P. D. (2014). Gut microbiota controls adipose tissue expansion, gut barrier and glucose metabolism: novel insights into molecular targets and interventions using prebiotics. Benef. Microbes 5, 3–17. doi: 10.3920/BM2012.0065
Gonzalez-Correa, C. A., Mulett-Vasquez, E., Miranda, D. A., Gonzalez-Correa, C. H., and Gomez-Buitrago, P. A. (2017). The colon revisited or the key to wellness, health and disease. Med. Hypotheses 108, 133–143. doi: 10.1016/j.mehy.2017.07.032
Gougeon, P. Y., Lourenssen, S., Han, T. Y., Nair, D. G., Ropeleski, M. J., and Blennerhassett, M. G. (2013). The pro-inflammatory cytokines IL-1beta and TNFalpha are neurotrophic for enteric neurons. J. Neurosci. 33, 3339–3351. doi: 10.1523/JNEUROSCI.3564-12.2013
Greenwood-Van Meerveld, B., Johnson, A. C., and Grundy, D. (2017). Gastrointestinal physiology and function. Handb. Exp. Pharmacol. 239, 1–16. doi: 10.1007/164_2016_118
Guo, X., Roberts, M. R., Becker, S. M., Podd, B., Zhang, Y., Chua, S. C. Jr., et al. (2011). Leptin signaling in intestinal epithelium mediates resistance to enteric infection by Entamoeba histolytica. Mucosal Immunol. 4, 294–303. doi: 10.1038/mi.2010.76
Hotamisligil, G. S. (2006). Inflammation and metabolic disorders. Nature 444, 860–867. doi: 10.1038/nature05485
Hughes, P. A., Moretta, M., Lim, A., Grasby, D. J., Bird, D., Brierley, S. M., et al. (2014). Immune derived opioidergic inhibition of viscerosensory afferents is decreased in irritable bowel syndrome patients. Brain Behav. Immun. 42, 191–203. doi: 10.1016/j.bbi.2014.07.001
Kim, K. A., Gu, W., Lee, I. A., Joh, E. H., and Kim, D. H. (2012). High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS One 7:e47713. doi: 10.1371/journal.pone.0047713
Kindt, S., Vanden Berghe, P., Boesmans, W., Roosen, L., and Tack, J. (2010). Prolonged IL-1beta exposure alters neurotransmitter and electrically induced Ca(2 + ) responses in the myenteric plexus. Neurogastroenterol. Motil. 22:321-e85. doi: 10.1111/j.1365-2982.2009.01414.x
Lang Lehrskov, L., Lyngbaek, M. P., Soederlund, L., Legaard, J., Ehses, A., Heywood, S. E., et al. (2018). Interleukin-6 delays gastric emptying in humans with direct effects on glycemic control. Cell Metab. 27, 1201.e3–1211.e3. doi: 10.1016/j.cmet.2018.04.008
Li, J., Jia, H., Cai, X., Zhong, H., Feng, Q., Sunagawa, S., et al. (2014). An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 32, 834–841. doi: 10.1038/nbt.2942
Magalhaes, I., Pingris, K., Poitou, C., Bessoles, S., Venteclef, N., Kiaf, B., et al. (2015). Mucosal-associated invariant T cell alterations in obese and type 2 diabetic patients. J. Clin. Invest. 125, 1752–1762. doi: 10.1172/JCI78941
Maloy, K. J., and Powrie, F. (2011). Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474, 298–306. doi: 10.1038/nature10208
Mao, J., Hu, X., Xiao, Y., Yang, C., Ding, Y., Hou, N., et al. (2013). Overnutrition stimulates intestinal epithelium proliferation through beta-catenin signaling in obese mice. Diabetes Metab. Res. Rev. 62, 3736–3746. doi: 10.2337/db13-0035
Mawe, G. M. (2015). Colitis-induced neuroplasticity disrupts motility in the inflamed and post-inflamed colon. J. Clin. Invest. 125, 949–955. doi: 10.1172/JCI76306
Mikkelsen, H. B. (2010). Interstitial cells of Cajal, macrophages and mast cells in the gut musculature: morphology, distribution, spatial and possible functional interactions. J. Cell Mol. Med. 14, 818–832. doi: 10.1111/j.1582-4934.2010.01025.x
Molinaro, A., Caesar, R., Holm, L. M., Tremaroli, V., Cani, P. D., and Backhed, F. (2017). Host-microbiota interaction induces bi-phasic inflammation and glucose intolerance in mice. Mol. Metab. 6, 1371–1380. doi: 10.1016/j.molmet.2017.08.016
Monteiro-Sepulveda, M., Touch, S., Mendes-Sa, C., Andre, S., Poitou, C., Allatif, O., et al. (2015). Jejunal T Cell inflammation in human obesity correlates with decreased enterocyte insulin signaling. Cell Metab. 22, 113–124. doi: 10.1016/j.cmet.2015.05.020
Morgan, L. M., Hampton, S. M., Tredger, J. A., Cramb, R., and Marks, V. (1988). Modifications of gastric inhibitory polypeptide (GIP) secretion in man by a high-fat diet. Br. J. Nutr. 59, 373–380. doi: 10.1079/BJN19880046
Muller, P. A., Koscso, B., Rajani, G. M., Stevanovic, K., Berres, M. L., Hashimoto, D., et al. (2014). Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell 158:1210. doi: 10.1016/j.cell.2014.08.002
Neill, D. R., Wong, S. H., Bellosi, A., Flynn, R. J., Daly, M., Langford, T. K., et al. (2010). Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370. doi: 10.1038/nature08900
O’Connell, P. J., Wang, X., Leon-Ponte, M., Griffiths, C., Pingle, S. C., and Ahern, G. P. (2006). A novel form of immune signaling revealed by transmission of the inflammatory mediator serotonin between dendritic cells and T cells. Blood 107, 1010–1017. doi: 10.1182/blood-2005-07-2903
Petit, V., Arnould, L., Martin, P., Monnot, M. C., Pineau, T., Besnard, P., et al. (2007). Chronic high-fat diet affects intestinal fat absorption and postprandial triglyceride levels in the mouse. J. Lipid Res. 48, 278–287. doi: 10.1194/jlr.M600283-JLR200
Rao, S. S., Read, N. W., Davison, P. A., Bannister, J. J., and Holdsworth, C. D. (1987). Anorectal sensitivity and responses to rectal distention in patients with ulcerative colitis. Gastroenterology 93, 1270–1275. doi: 10.1016/0016-5085(87)90255-1
Rastelli, M., Knauf, C., and Cani, P. D. (2018). Gut microbes and health: a focus on the mechanisms linking microbes, obesity, and related disorders. Obesity 26, 792–800. doi: 10.1002/oby.22175
Rehn, M., Hild, D., and Diener, M. (2005). Upregulation of cyclooxygenase-2 and thromboxane A2 production mediate the action of tumor necrosis factor-alpha in isolated rat myenteric ganglia. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G586–G591. doi: 10.1152/ajpgi.00020.2005
Rehn, M., Hubschle, T., and Diener, M. (2004). TNF-alpha hyperpolarizes membrane potential and potentiates the response to nicotinic receptor stimulation in cultured rat myenteric neurones. Acta Physiol. Scand. 181, 13–22. doi: 10.1111/j.1365-201X.2004.01269.x
Richards, P., Pais, R., Habib, A. M., Brighton, C. A., Yeo, G. S., Reimann, F., et al. (2016). High fat diet impairs the function of glucagon-like peptide-1 producing L-cells. Peptides 77, 21–27. doi: 10.1016/j.peptides.2015.06.006
Ruhl, A., Franzke, S., and Stremmel, W. (2001a). IL-1beta and IL-10 have dual effects on enteric glial cell proliferation. Neurogastroenterol. Motil. 13, 89–94.
Ruhl, A., Franzke, S., Collins, S. M., and Stremmel, W. (2001b). Interleukin-6 expression and regulation in rat enteric glial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 280, G1163–G1171.
Sender, R., Fuchs, S., and Milo, R. (2016a). Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell 164, 337–340. doi: 10.1016/j.cell.2016.01.013
Sender, R., Fuchs, S., and Milo, R. (2016b). Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 14:e1002533. doi: 10.1371/journal.pbio.1002533
Shea-Donohue, T., Notari, L., Sun, R., and Zhao, A. (2012). Mechanisms of smooth muscle responses to inflammation. Neurogastroenterol. Motil. 24, 802–811. doi: 10.1111/j.1365-2982.2012.01986.x
Shimazu-Kuwahara, S., Harada, N., Yamane, S., Joo, E., Sankoda, A., Kieffer, T. J., et al. (2017). Attenuated secretion of glucose-dependent insulinotropic polypeptide (GIP) does not alleviate hyperphagic obesity and insulin resistance in ob/ob mice. Mol. Metab. 6, 288–294. doi: 10.1016/j.molmet.2017.01.006
Slyepchenko, A., Maes, M., Machado-Vieira, R., Anderson, G., Solmi, M., Sanz, Y., et al. (2016). Intestinal dysbiosis, gut hyperpermeability and bacterial translocation: missing links between depression, obesity and type 2 diabetes. Curr. Pharm. Des. 22, 6087–6106. doi: 10.2174/1381612822666160922165706
Souza-Moreira, L., Campos-Salinas, J., Caro, M., and Gonzalez-Rey, E. (2011). Neuropeptides as pleiotropic modulators of the immune response. Neuroendocrinology 94, 89–100. doi: 10.1159/000328636
Stecher, B. (2015). The roles of inflammation, nutrient availability and the commensal microbiota in enteric pathogen infection. Microbiol. Spectr. 3, 1–17. doi: 10.1128/microbiolspec.MBP-0008-2014
Trinchieri, G. (2003). Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 3, 133–146. doi: 10.1038/nri1001
Valentine, J. F., Tannahill, C. L., Stevenot, S. A., Sallustio, J. E., Nick, H. S., and Eaker, E. Y. (1996). Colitis and interleukin 1beta up-regulate inducible nitric oxide synthase and superoxide dismutase in rat myenteric neurons. Gastroenterology 111, 56–64. doi: 10.1053/gast.1996.v111.pm8698225
Vandeputte, D., Kathagen, G., D’Hoe, K., Vieira-Silva, S., Valles-Colomer, M., Sabino, J., et al. (2017). Quantitative microbiome profiling links gut community variation to microbial load. Nature 551, 507–511. doi: 10.1038/nature24460
Vijay-Kumar, M., Aitken, J. D., Carvalho, F. A., Cullender, T. C., Mwangi, S., Srinivasan, S., et al. (2010). Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328, 228–231. doi: 10.1126/science.1179721
Villanacci, V., Bassotti, G., Nascimbeni, R., Antonelli, E., Cadei, M., Fisogni, S., et al. (2008). Enteric nervous system abnormalities in inflammatory bowel diseases. Neurogastroenterol. Motil. 20, 1009–1016. doi: 10.1111/j.1365-2982.2008.01146.x
Wang, X., Ota, N., Manzanillo, P., Kates, L., Zavala-Solorio, J., Eidenschenk, C., et al. (2014). Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature 514, 237–241. doi: 10.1038/nature13564
Wellen, K. E., and Hotamisligil, G. S. (2005). Inflammation, stress, and diabetes. J. Clin. Invest. 115, 1111–1119. doi: 10.1172/JCI25102
Wells, J. M., Brummer, R. J., Derrien, M., MacDonald, T. T., Troost, F., Cani, P. D., et al. (2017). Homeostasis of the gut barrier and potential biomarkers. Am. J. Physiol. Gastrointest. Liver Physiol. 312, G171–G193. doi: 10.1152/ajpgi.00048.2015
Xia, Y., Hu, H. Z., Liu, S., Ren, J., Zafirov, D. H., and Wood, J. D. (1999). IL-1beta and IL-6 excite neurons and suppress nicotinic and noradrenergic neurotransmission in guinea pig enteric nervous system. J. Clin. Invest. 103, 1309–1316. doi: 10.1172/JCI5823
Keywords: enteric nervous system, immune cells, microbiota, gut-brain axis, diabetes
Citation: Bessac A, Cani PD, Meunier E, Dietrich G and Knauf C (2018) Inflammation and Gut-Brain Axis During Type 2 Diabetes: Focus on the Crosstalk Between Intestinal Immune Cells and Enteric Nervous System. Front. Neurosci. 12:725. doi: 10.3389/fnins.2018.00725
Received: 27 June 2018; Accepted: 21 September 2018;
Published: 10 October 2018.
Edited by:
Joana M. Gaspar, Universidade Federal de Santa Catarina, BrazilReviewed by:
Fátima Oliveira Martins, Centro de Estudos de Doenças Crónicas (CEDOC), PortugalCopyright © 2018 Bessac, Cani, Meunier, Dietrich and Knauf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claude Knauf, Y2xhdWRlLmtuYXVmQGluc2VybS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.