 Moataz Abdalkader
Moataz Abdalkader Riikka Lampinen
Riikka Lampinen Katja M. Kanninen
Katja M. Kanninen Tarja M. Malm
Tarja M. Malm Jeffrey R. Liddell
Jeffrey R. Liddell
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurosci., 10 July 2018
Sec. Neurodegeneration
Volume 12 - 2018 | https://doi.org/10.3389/fnins.2018.00466
This article is part of the Research TopicMitochondrial Dysfunction and NeurodegenerationView all 16 articles
Ferroptosis is a newly described form of regulated cell death, distinct from apoptosis, necroptosis and other forms of cell death. Ferroptosis is induced by disruption of glutathione synthesis or inhibition of glutathione peroxidase 4, exacerbated by iron, and prevented by radical scavengers such as ferrostatin-1, liproxstatin-1, and endogenous vitamin E. Ferroptosis terminates with mitochondrial dysfunction and toxic lipid peroxidation. Although conclusive identification of ferroptosis in vivo is challenging, several salient and very well established features of neurodegenerative diseases are consistent with ferroptosis, including lipid peroxidation, mitochondrial disruption and iron dysregulation. Accordingly, interest in the role of ferroptosis in neurodegeneration is escalating and specific evidence is rapidly emerging. One aspect that has thus far received little attention is the antioxidant transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2). This transcription factor regulates hundreds of genes, of which many are either directly or indirectly involved in modulating ferroptosis, including metabolism of glutathione, iron and lipids, and mitochondrial function. This potentially positions Nrf2 as a key deterministic component modulating the onset and outcomes of ferroptotic stress. The minimal direct evidence currently available is consistent with this and indicates that Nrf2 may be critical for protection against ferroptosis. In contrast, abundant evidence demonstrates that enhancing Nrf2 signaling is potently neuroprotective in models of neurodegeneration, although the exact mechanism by which this is achieved is unclear. Further studies are required to determine to extent to which the neuroprotective effects of Nrf2 activation involve the prevention of ferroptosis.
The last few decades have witnessed a surge in the discovery of new forms of regulated cell death that have immense implications for both health and disease (Galluzzi et al., 2018). Ferroptosis is a recently described form of non-apoptotic regulated cell death caused by uncontrolled iron-dependent lipid peroxidation that is distinct in its morphological, biochemical, and genetic profile from other cell death mechanisms (Dixon et al., 2012; Galluzzi et al., 2018). Cells undergoing ferroptosis show none of the classical morphological alterations associated with apoptosis, necroptosis, or autophagy (e.g., cell swelling, nuclear disruption, membrane blebbing, etc.), with the only discernable ultrastructural feature being distinctly altered mitochondrial morphology (Dixon et al., 2012; Stockwell et al., 2017).
A discriminating feature of ferroptosis is the potent capacity of lipid peroxide scavengers (such as ferrostatin-1, liproxstatin-1, and vitamin E) to prevent ferroptotic cell death (Dixon et al., 2012; Friedmann Angeli et al., 2014; Yang et al., 2014). This explicit requirement for lipid peroxidation is a core feature of ferroptosis that distinguishes it from other forms of cell death: ferroptosis inhibitors cannot prevent other forms of cell death (Dixon et al., 2012; Friedmann Angeli et al., 2014; Yang et al., 2014), and conversely classic inhibitors of necrosis, apoptosis, and autophagy do not modulate ferroptosis (Dixon et al., 2012), with the exception of the necroptosis inhibitor, necrostatin-1 (RIPK1 inhibitor), which can inhibit ferroptosis in a necroptosis/RIPK1-independent manner (Friedmann Angeli et al., 2014). The central endogenous suppressor of ferroptosis is the selenoenzyme glutathione peroxidase 4 (Gpx4). Gpx4 detoxifies membrane lipid hydroperoxides, preventing unchecked toxic lipid peroxidation. Gpx4 requires the major cellular antioxidant glutathione as a substrate, and hence ferroptosis is also dependent on glutathione levels (Friedmann Angeli et al., 2014). The precise role of iron in ferroptosis is ironically unclear (see below), however, its involvement is unequivocally indicated by the strong inhibition of cell death associated with iron chelation or limiting iron availability (Figure 1).
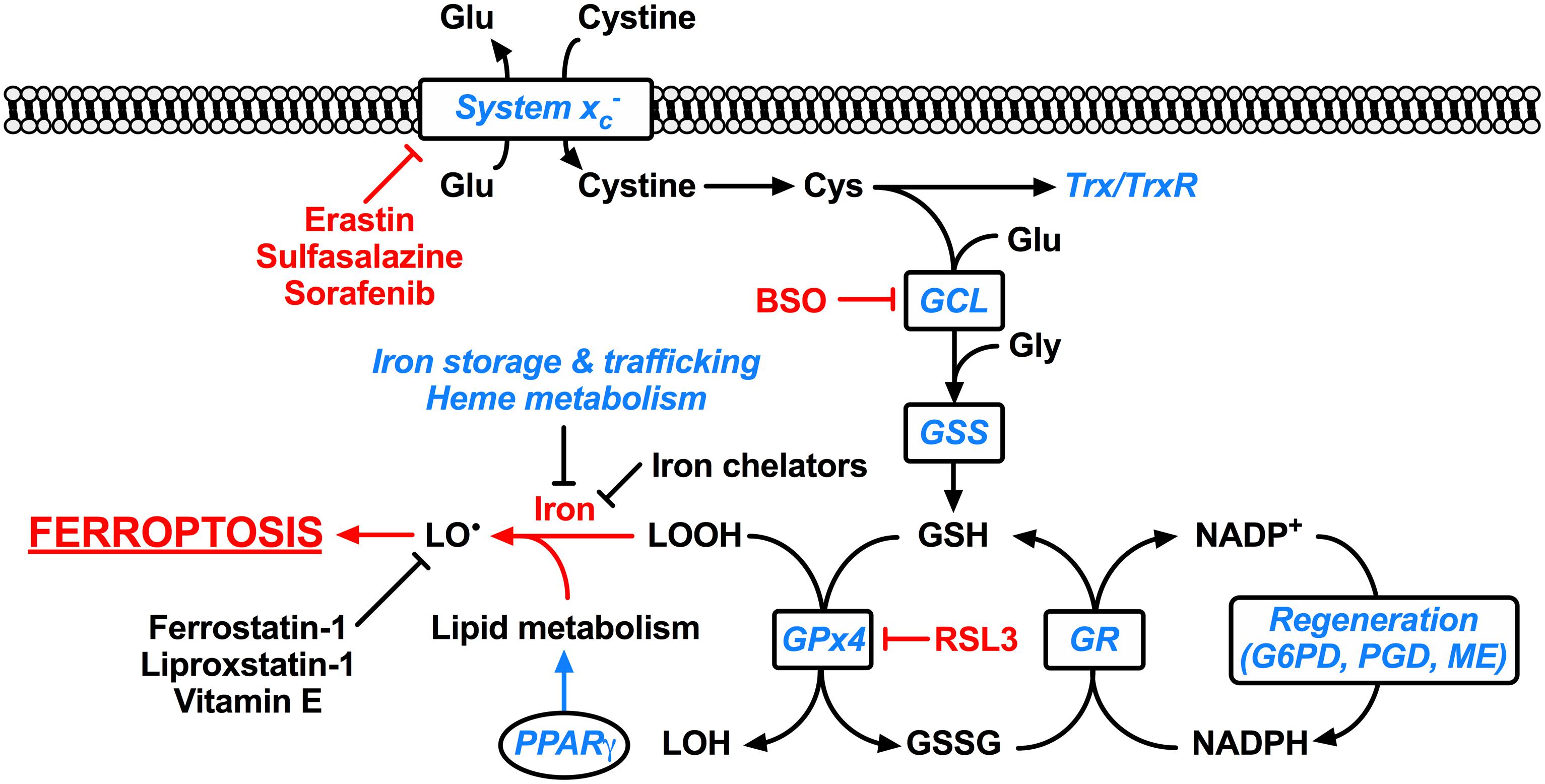
FIGURE 1. Ferroptosis and its molecular regulation by Nrf2. Glutathione peroxidase 4 (Gpx4) utilizes the major cellular antioxidant glutathione (GSH) as a substrate to reduce lipid hydroperoxides (LOOH). Oxidized glutathione (GSSG) generated by Gpx4 is reduced back to glutathione by glutathione reductase (GR) in a reaction requiring NADPH, which can be regenerated by glucose-6-phosphate dehydrogenase (G6PD) and phosphogluconate dehydrogenase (PGD) of the pentose-phosphate pathway, and malic enzyme (ME). The tripeptide glutathione is synthesized by the consecutive action of glutamate-cysteine ligase (GCL) and glutathione synthetase (GSS), where the ligation of cysteine (Cys) and glutamate (Glu) by glutamate-cysteine ligase is the rate-limiting step of glutathione synthesis. The cellular import of cystine by the glutamate-cystine antiporter system xc- constitutes an significant route of cysteine supply for glutathione synthesis. Cysteine is also required for other important cellular antioxidants including thioredoxin (Trx) and thioredoxin reductase (TrxR). Ferroptosis occurs when the Gpx4-catalyzed reduction of lipid hydroperoxides is insufficient to prevent the iron-mediated generation of lipid radicals (LO•). This leads to the propagation of lipid peroxidation and culminates in ferroptosis. Ferroptosis can be experimentally induced by inhibiting Gpx4 via the small molecule inhibitor RSL3, or by limiting glutathione supply to Gpx4. The latter is induced by direct [e.g., buthionine sulfoximine (BSO)] or indirect (e.g., by limiting cysteine availability) inhibition of glutathione synthesis. Cysteine supply is disrupted by inhibitors of system xc- including erastin, sulfasalazine, and sorafenib. Ferroptosis can also be inhibited by iron chelators such as deferoxamine and deferiprone, and lipid radical scavengers such as ferrostatin-1, liproxstatin-1, and vitamin E. Factors transcriptionally regulated by Nrf2 are indicated in blue italics, whereas factors promoting ferroptosis are indicated in red. Clearly Nrf2 signaling is likely to have an integral and pervasive impact on the manifestation of ferroptosis.
Ferroptosis appears to require the presence of specific highly oxidisable phosphatidylethanolamine phospholipids containing the polyunsaturated fatty acids (PUFAs) arachidonic acid and adrenic acid. Acyl-CoA synthetase long-chain family member 4 (ACSL4) is important for the synthesis of phospholipids from these PUFAs, while lysophosphatidylcholine acyltransferase 3 (LPCAT3) is important for their insertion into membrane phospholipids (Kagan et al., 2017). Pharmacological or genetic inhibition of either ACSL4 or LPCAT3 suppresses ferroptosis specifically over other forms of cell death (Dixon et al., 2015; Yuan et al., 2016b; Doll et al., 2017; Kagan et al., 2017). Lipoxygenases can catalyze the formation of lipid hydroperoxides from PUFAs, instigating toxic lipid peroxidation (Yang et al., 2016), although this can occur independently from lipoxygenase activity (Shah et al., 2017).
As mentioned above, the precise role of iron in ferroptosis remains unclear. The ability of the iron chelators including deferoxamine and deferiprone to salvage cells from ferroptotic death in a variety of models underscores the role of iron in triggering ferroptosis (Dixon et al., 2012; Friedmann Angeli et al., 2014; Yang et al., 2014; Do Van et al., 2016). The lipoxygenases involved in generating toxic lipid hydroperoxides require catalytic iron in their active sites (Abeysinghe et al., 1996), hence the protective effect of iron chelation has been proposed to involve inhibition of lipoxygenase activity by removal of the essential catalytic iron from these enzymes. Alternatively, iron has been demonstrated to potentiate ferroptosis in a free radical-mediated process independent from lipoxygenase activity (Shah et al., 2017).
Ferroptosis can be experimentally induced by direct inhibition of Gpx4 via Ras-selective lethal small molecule 3 (RSL3) or by genetic knockdown or deletion of Gpx4 (Friedmann Angeli et al., 2014; Yang et al., 2014). As Gpx4 requires glutathione as a substrate, ferroptosis is also induced by disruption of glutathione supply via inhibition of glutathione synthesis (e.g., buthionine sulfoximine) or inhibiting the supply of cysteine required for glutathione synthesis via inhibition of the cystine-glutamate antiporter system xc- (via erastin, sulfasalazine, or sorafenib) (Dixon et al., 2012; Friedmann Angeli et al., 2014) (Figure 1).
Mitochondria play key roles in regulated cell death (Galluzzi et al., 2018), and ferroptosis is no exception. Cells undergoing ferroptosis exhibit specific mitochondrial morphology. Early studies using the system xc- inhibitor erastin show ferroptosis results in smaller mitochondria with increased mitochondrial membrane density (Yagoda et al., 2007; Dixon et al., 2012). Later studies employing pharmacological or genetic disruption of Gpx4 causes mitochondrial swelling, decreased cristae and outer membrane rupture (Friedmann Angeli et al., 2014; Doll et al., 2017; Maiorino et al., 2017; Ingold et al., 2018). Toxicity induced by inhibition of mitochondrial complex I can be rescued by ferroptosis inhibitors (Basit et al., 2017). Furthermore, lack of ACSL4 prevents the RSL3-mediated rupture of mitochondrial outer membrane (Doll et al., 2017), while knockdown of mitochondrial acyl-CoA synthetase family member 2 (ACSF2; involved in fatty acid metabolism) prevents erastin toxicity, suggesting ACSF2 generates a mitochondrial-specific lipid necessary for ferroptosis (Dixon et al., 2012).
Despite these observations, evidence for mitochondrial lipid peroxidation during ferroptosis is mixed. Ferroptosis induced by erastin does not appear to be accompanied by mitochondrial lipid peroxidation in vitro (Dixon et al., 2012; Do Van et al., 2016) or in vivo (Wang et al., 2016). In contrast, disruption of Gpx4 results in mitochondrial lipid peroxidation in vitro and in kidneys (Friedmann Angeli et al., 2014). This discrepancy suggests that the inducing stimuli may be critical for the subcellular localization of lipid peroxidation.
Although mitochondria are clearly impaired in ferroptosis, evidence suggests that they are not driving the cell death process. Cells deficient in mitochondria remain sensitive to ferroptosis (Gaschler et al., 2018). Furthermore, extramitochondrial lipid peroxidation temporally precedes mitochondrial lipid peroxidation, and mitochondrial damage including rupture of the outer mitochondrial membrane is a late event, closely preceding cell lysis (Friedmann Angeli et al., 2014; Jelinek et al., 2018).
Reports on targeting antioxidants to mitochondria are mixed. MitoQ rescues neuronal cells from RSL3 toxicity (Jelinek et al., 2018). However, when compared their non-mitochondrial analogs, mitochondrially targeted radical scavengers are opposingly reported as being less effective (Friedmann Angeli et al., 2014) or more effective (Krainz et al., 2016).
Mitochondrial iron is also implicated in ferroptosis. MitoNEET, also known as CISD1, is an iron-containing outer mitochondrial membrane protein involved in iron export from mitochondria (Mittler et al., 2018). Knockdown of mitoNEET exacerbates erastin toxicity and increases mitochondrial iron content and lipid peroxidation, whereas stabilization of mitoNEET attenuates erastin toxicity and decreases mitochondrial lipid peroxidation (Yuan et al., 2016a). Alternatively, safely sequestering iron within mitochondria via overexpression of mitochondrial ferritin is able to curb erastin-induced cell death, both in vitro and in vivo (Wang et al., 2016).
Explicitly identifying ferroptosis in vivo is hampered by the lack of specific biomarkers. Nevertheless, considerable evidence exists that implicates ferroptosis in neurodegeneration. The association between oxidative stress, lipid peroxidation and neurodegeneration has long been appreciated. Notably, elevated levels of lipid peroxidation are reliably detected in brain tissues and body fluids of Alzheimer’s, Parkinson’s, Huntington’s disease, motor neuron disease and multiple sclerosis patients (Adibhatla and Hatcher, 2010; Shichiri, 2014; Sugiyama and Sun, 2014; Wang et al., 2014; Bradley-Whitman and Lovell, 2015). Iron accumulation is a consistent feature of neurodegeneration (Belaidi and Bush, 2016). The level of iron in brains of individuals with mild cognitive impairment and Alzheimer’s disease correlates with disease progression (Smith et al., 2010; Ayton et al., 2017). Elevated iron is a cardinal feature of Parkinson’s disease substantia nigra (Ayton and Lei, 2014), and increased iron is detected in affected brain regions of patients with motor neuron disease, multiple sclerosis, Huntington’s disease and Friedreich ataxia (Kwan et al., 2012; Li and Reichmann, 2016; Sheykhansari et al., 2018). Reducing brain iron via the chelators deferiprone or deferoxamine is efficacious in clinical trials of Parkinson’s (Devos et al., 2014) and Alzheimer’s patients (Crapper McLachlan et al., 1991), respectively, indicating iron is contributing to the disease process. Further indirect evidence, including diminished glutathione and insufficient Nrf2 signaling (see below), is consistent with the presence of ferroptosis in neurodegeneration (Liddell, 2017; Liddell and White, 2017). Moreover, impaired mitochondrial function is common to many neurodegenerative diseases (Carri et al., 2017; Liddell and White, 2017; Liot et al., 2017; Swerdlow, 2017). Morphologically, mitochondria in brains of mice modeling Huntington’s disease exhibit disrupted cristae (Lee et al., 2011), while those in motor neuron disease human postmortem tissue and model mice feature swollen and vacuolated mitochondria (Jaarsma et al., 2000; Cozzolino and Carri, 2012) reminiscent of the mitochondrial changes evident in ferroptosis.
Since its original characterisation in cancer cells, the concept of ferroptosis has instigated growing efforts to explicitly detect and measure its footprint in neurodegeneration (Guiney et al., 2017; Morris et al., 2018). In this regard, more direct evidence for the role of ferroptosis in neurodegeneration has recently been generated. Neuronal cells are sensitive to erastin and RSL3 toxicity in vitro. This toxicity is associated with mitochondrial impairments, and is rescued by ferroptosis inhibitors or a mitochondrially targeted ROS scavenger (Neitemeier et al., 2017; Jelinek et al., 2018). Genetic models demonstrate the substantial reliance of neurons on Gpx4 to prevent toxic lipid peroxidation. Whereas global deletion of Gpx4 is embryonic lethal (Imai et al., 2003), mice with targeted mutation of Gpx4 selenocysteine to cysteine (sensitive to inactivation), or knockout of Gpx4 specifically in neurons are viable but exhibit selective loss of CA3 hippocampal interneurons, resulting in seizures and early death (Seiler et al., 2008; Ingold et al., 2018). Targeting Gpx4 knockout to photoreceptor neurons results in death of these cells within 21 days of birth (Ueta et al., 2012). Post-development, conditional knockout of Gpx4 in adult mice results in loss of CA1 hippocampal neurons and rapid death, indicating neurons are specifically sensitive to Gpx4 deletion (Yoo et al., 2012). Conditional ablation of Gpx4 targeted to neurons results in dramatic degeneration of motor neurons that rapidly progresses to paralysis and death (Chen et al., 2015). Targeting conditional knockout of Gpx4 to forebrain neurons of adult mice causes cognitive impairments and hippocampal degeneration reminiscent of Alzheimer’s disease (Hambright et al., 2017). These models are all accompanied by lipid peroxidation and mitochondrial impairments, consistent with ferroptosis. Conditional Gpx4 knockout can partially rescued by ferroptosis inhibitors, indicating the involvement of ferroptosis (Chen et al., 2015; Hambright et al., 2017).
Ferroptosis inhibitors have also been investigated in explicit models of neurodegneration. Ferroptosis inhibitors prevent the toxicity of Huntington’s disease-associated mutated Htt in brain slice cultures (Skouta et al., 2014). Ferroptosis inhibitors are also protective in dopaminergic cultured cells, organotypic slice cultures, and in an MPTP mouse model of Parkinson’s disease (Do Van et al., 2016). Furthermore, ferroptosis is implicated in hemorrhagic and ischemic stroke based on the ability of ferroptosis inhibitors to protect against neuronal death in both in vivo and in vitro models (Li et al., 2017; Tuo et al., 2017; Zille et al., 2017). Ischemic stroke is also strongly modulated by brain iron levels (Tuo et al., 2017). Knockout of glutathione synthesis accelerates the disease phenotype in motor neuron disease-model mice, and ultrastructural analysis of spinal cord tissue reveals mitochondrial swelling, rupture and decreased cristae, all consistent with ferroptosis (Vargas et al., 2011).
Ferroptosis also appears to mechanistically overlap with other cell death modalities in neurodegeneration. Consistent with a distinct form of cell death, it was initially reported that ferroptosis induced by erastin does not involve mitochondrial genes implicated in other cell death pathways, nor the release of cytochrome c (Dixon et al., 2012). However, recent studies show that ferroptosis induces the pro-apoptotic translocation of BH3 interacting-domain death agonist (BID) to mitochondria in neuronal cells (Neitemeier et al., 2017; Jelinek et al., 2018). Oxytosis is described as glutamate-induced inhibition of cystine uptake via system xc- leading to glutathione depletion and subsequent lipoxygenase-dependent toxic lipid peroxidation (Tan et al., 2001). This is clearly very similar to ferroptosis and has led to the recent proposal that perhaps ferroptosis and oxytosis are in fact the same pathway (Lewerenz et al., 2018). Accordingly, oxytosis in neurons causes mitochondrial morphological changes similar to ferroptosis (Lee et al., 2011), and the toxicity of glutamate mirrors that of erastin or RSL3 in neuronal cells, and are all amenable to prevention by ferroptosis inhibitors (Liu et al., 2015; Neitemeier et al., 2017; Jelinek et al., 2018) or necrostatin-1 (Xu et al., 2007). Furthermore, oxytosis/ferroptosis in neurons involves the translocation of apoptosis inducing factor (AIF) from mitochondria to nucleus, prevention of which alleviates cell death (Xu et al., 2010; Neitemeier et al., 2017; Jelinek et al., 2018). To further complicate the mechanisms of cell death, mitochondrial AIF release is the penultimate stage of parthanatos, a poly ADP-ribose polymerase 1 (PARP-1)-mediated form of cell death (Fricker et al., 2018; Galluzzi et al., 2018). Parthanatos can be induced by oxidative stress, and is also implicated in neurodegeneration (Fricker et al., 2018; Galluzzi et al., 2018). Hence both initiating events and late stages are shared by ferroptosis/oxytosis and parthanatos.
Together these studies show that many features facilitating ferroptosis are present in neurodegeneration, and mounting evidence indicates that targeting ferroptosis with specific inhibitors is a valid therapeutic strategy. However, overlapping mechanisms highlight the need for more comprehensive delineation of the cellular mechanisms of cell death, and the potential for new or repurposed treatments. An alternate approach to attenuate ferroptosis is to augment the endogenous anti-ferroptotic mechanisms within cells. To this end, the role of the transcription factor Nrf2 will now be discussed.
In terms of endogenous cellular mechanisms preventing ferroptosis, the antioxidant transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) is exquisitely positioned to modulate the onset and outcomes of ferroptosis. Nrf2 is responsible for regulating hundreds of antioxidant genes (Gao et al., 2014). Under normal conditions, Nrf2 resides in the cytosol bound to its negative regulator Kelch-like ECH-associated protein 1 (Keap1). Keap1 constitutively targets Nrf2 for ubiquitination and proteasomal degradation, thereby maintaining Nrf2 signaling capacity at a low level. However, states of increased oxidative stress facilitate the dissociation of Nrf2 from Keap1, and promote the nuclear translocation of Nrf2. In the nucleus, Nrf2 interacts with antioxidant response elements (AREs) in the promoter region of target genes resulting in their transcriptional activation (Yamamoto et al., 2018).
Importantly in the context of ferroptosis, almost all genes thus far implicated in ferroptosis are transcriptionally regulated by Nrf2 (Table 1). These include genes for glutathione regulation (synthesis, cysteine supply via system xc-, glutathione reductase, glutathione peroxidase 4), NADPH regeneration which is critical for Gpx4 activity (glucose 6-phosphate dehydrogenase, phosphogluconate dehydrogenase, malic enzyme), and iron regulation (including iron export and storage, heme synthesis and catabolism) (Sasaki et al., 2002; Lee et al., 2003; Wu et al., 2011; Kerins and Ooi, 2017). In addition, Nrf2 is involved in the regulation of lipids via the ligand-mediated transcription factor peroxisome proliferator-activated receptor gamma (PPARγ). Nrf2 and PPARγ are reciprocally regulated, with activation of either upregulating the other (Cai et al., 2017; Lee, 2017). PPARγ is a major regulator of lipid metabolism (Cai et al., 2017), and can be activated by oxidized lipids relevant to the initiation of ferroptosis (Itoh et al., 2008). Hence Nrf2 indirectly modulates the lipids whose abundance contributes to the sensitivity to ferroptosis (Doll et al., 2017) (Figure 1).

TABLE 1. Ferroptosis-related genes that are transcriptionally regulated by Nrf2.
Nrf2 also plays important roles in modulating mitochondrial function. Nrf2 can be physically bound to mitochondria and thus monitor and respond to changes in mitochondrial function (Lo and Hannink, 2008). Nrf2 also regulates mitochondrial dynamics, including biogenesis (Piantadosi et al., 2008; Merry and Ristow, 2016) via interaction with peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) (Dinkova-Kostova and Abramov, 2015; Navarro et al., 2017), and mitophagy via a P62-dependent, PINK1/Parkin-independent mechanism (East et al., 2014). Mitochondria in Nrf2 knockout mice have impaired function, whereas activation of Nrf2 enhances mitochondrial function and resistance to stressors (Greco and Fiskum, 2010; Neymotin et al., 2011; Holmstrom et al., 2016).
Therefore, by virtue of its direct and indirect regulation of many genes central to ferroptosis and its regulation of mitochondrial function, Nrf2 is positioned to be a key player in ferroptosis. Despite this, the role of Nrf2 in ferroptosis has received very little attention thus far, limited mainly to in vitro studies using cancer cells. As expected, Nrf2 activation confers resistance to ferroptosis in cancer cells (Sun et al., 2016b; Chen et al., 2017a,b; Roh et al., 2017). Genetic modulation of Nrf2 expression, including knockdown and overexpression, can fine-tune the sensitivity of glioma cells to the ferroptosis inducers erastin and RSL3 (Fan et al., 2017). This may contribute to the cancer-promoting and chemoresistance effects of elevated Nrf2 present in many cancers (Fan et al., 2017; Taguchi and Yamamoto, 2017).
Nrf2 activation is sensitive to shifts in cellular redox status, hence it follows that the lipid peroxidation accompanying ferroptosis should activate Nrf2. Indeed, the ferroptosis inducers erastin and sorafenib are sufficient to activate Nrf2 in hepatocellular carcinoma cells (Houessinon et al., 2016; Sun et al., 2016b). Furthermore, the protective action of the mitochondrially targeted antioxidant, MitoQ, involves Nrf2 activation (Hu et al., 2018).
Given the pervasive lipid peroxidation evident in neurodegeneration, it would be expected that Nrf2 would be elevated in neurodegenerative diseases. While there is some evidence for Nrf2 activation in neurodegeneration, it appears to be relatively mild and clearly insufficient to prevent neuronal dysfunction (Liddell, 2017). In some cases, particularly for motor neuron disease, Nrf2 signaling appears to be impaired (Moujalled et al., 2017). This is in contrast to the strong activation of Nrf2 evident when genetic or pharmacological Nrf2 inducers are applied to models of neurodegeneration (Liddell, 2017). These treatments are robustly neuroprotective in animal models of disease. While several drugs targeting Nrf2 are currently under clinical investigation, to date, dimethyl fumarate (Tecfidera) remains the only clinically approved drug for the treatment of a neurodegenerative disease (relapsing-remitting multiple sclerosis) in which Nrf2 activation clearly contributes to its mechanism of action (Havrdova et al., 2017).
The above mentioned studies in cancer cells provide empirical in vitro support for the anti-ferroptotic action of Nrf2. However, direct in vivo evidence for an anti-ferroptotic effect of Nrf2 induction does not yet exist, and it is currently unknown whether and to what extent the demonstrated neuroprotective efficacy of Nrf2 activation in models of neurodegeneration involve attenuation of ferroptosis. Evaluation of ferroptosis in vivo is currently hindered by the lack of specific markers of ferroptosis. Studies examining the effect of Nrf2 activation in explicit in vivo models of ferroptosis (e.g., Gpx4 knockout) would provide some insight.
Ferroptosis is an iron-dependent form of regulated cell death instigated by impaired glutathione metabolism, culminating in mitochondrial failure and toxic lipid peroxidation. Emerging evidence implicates ferroptosis in neurodegeneration, both in the molecular and biochemical signatures of neurodegeneration, and in terms of functional abrogation of neuron death via specific ferroptosis inhibitors. Elucidating how ferroptosis provokes neurodegeneration will expose new therapeutic opportunities to treat these diseases. To this end, targeting the antioxidant transcription factor Nrf2 is an attractive option. Nrf2 signaling is involved in regulating mitochondrial function and impacts almost all identified molecular aspects of ferroptosis. Treatments targeting Nrf2 have been demonstrated to exert anti-ferroptotic effects in the context of cancer cells, and are beneficial in many models of neurodegeneration. The protective mechanism of Nrf2 activation in these models may involve attenuating ferroptosis via upregulation of the endogenous anti-ferroptotic machinery, however, direct evidence for this is currently lacking. The myriad of Nrf2 actions described here suggest that targeting Nrf2 is an exciting therapeutic option to attenuate ferroptosis.
Several questions remain unanswered. The discovery of bonafide biomarkers of ferroptosis will be invaluable to unequivocally probe its involvement in neurodegeneration and facilitate the development of therapeutic treatments targeting ferroptosis. The continued evaluation of ferroptosis inhibitors and comparison to inhibitors of other cell death modalities in further models of neurodegeneration will help elucidate the key pathways involved. Finally, whether Nrf2 activation is directly alleviating ferroptotic stress.
Taken together, the arguments presented in this review elucidate a coherent network that links Nrf2 signaling to mitochondrial function and ferroptotic cell death, and proposes the targeting of Nrf2 as a rational line of therapy for ferroptotic neurodegeneration.
JL conceived the review. MA prepared the first draft. RL, KK, and TM reviewed literature and contributed to writing. JL prepared the final manuscript.
JL was supported by an Australian National Health and Medical Research Council Peter Doherty Fellowship. We gratefully acknowledge funding from the Academy of Finland and The Sigrid Juselius Foundation, Finland.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abeysinghe, R. D., Roberts, P. J., Cooper, C. E., Maclean, K. H., Hider, R. C., and Porter, J. B. (1996). The environment of the lipoxygenase iron binding site explored with novel hydroxypyridinone iron chelators. J. Biol. Chem. 271, 7965–7972. doi: 10.1074/jbc.271.14.7965
Adibhatla, R. M., and Hatcher, J. F. (2010). Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 12, 125–169. doi: 10.1089/ars.2009.2668
Agyeman, A. S., Chaerkady, R., Shaw, P. G., Davidson, N. E., Visvanathan, K., Pandey, A., et al. (2012). Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res. Treat. 132, 175–187. doi: 10.1007/s10549-011-1536-9
Alam, J., Stewart, D., Touchard, C., Boinapally, S., Choi, A. M., and Cook, J. L. (1999). Nrf2, a Cap‘n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 274, 26071–26078. doi: 10.1074/jbc.274.37.26071
Ayton, S., Fazlollahi, A., Bourgeat, P., Raniga, P., Ng, A., Lim, Y. Y., et al. (2017). Cerebral quantitative susceptibility mapping predicts amyloid-beta-related cognitive decline. Brain 140, 2112–2119. doi: 10.1093/brain/awx137
Ayton, S., and Lei, P. (2014). Nigral iron elevation is an invariable feature of Parkinson’s disease and is a sufficient cause of neurodegeneration. BioMed Res. Int. 2014:581256. doi: 10.1155/2014/581256
Basit, F., Van Oppen, L. M., Schockel, L., Bossenbroek, H. M., Van Emst-De Vries, S. E., Hermeling, J. C., et al. (2017). Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 8:e2716. doi: 10.1038/cddis.2017.133
Belaidi, A. A., and Bush, A. I. (2016). Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J. Neurochem. 139(Suppl. 1), 179–197. doi: 10.1111/jnc.13425
Bradley-Whitman, M. A., and Lovell, M. A. (2015). Biomarkers of lipid peroxidation in Alzheimer disease (AD): an update. Arch. Toxicol. 89, 1035–1044. doi: 10.1007/s00204-015-1517-6
Cai, W., Yang, T., Liu, H., Han, L., Zhang, K., Hu, X., et al. (2017). Peroxisome proliferator-activated receptor gamma (PPARgamma): a master gatekeeper in CNS injury and repair. Prog. Neurobiol. 163–164, 27–58. doi: 10.1016/j.pneurobio.2017.10.002
Campbell, M. R., Karaca, M., Adamski, K. N., Chorley, B. N., Wang, X., and Bell, D. A. (2013). Novel hematopoietic target genes in the NRF2-mediated transcriptional pathway. Oxid. Med. Cell. Longev. 2013:120305. doi: 10.1155/2013/120305
Carri, M. T., D’ambrosi, N., and Cozzolino, M. (2017). Pathways to mitochondrial dysfunction in ALS pathogenesis. Biochem. Biophys. Res. Commun. 483, 1187–1193. doi: 10.1016/j.bbrc.2016.07.055
Chen, D., Rauh, M., Buchfelder, M., Eyupoglu, I. Y., and Savaskan, N. (2017a). The oxido-metabolic driver ATF4 enhances temozolamide chemo-resistance in human gliomas. Oncotarget 8, 51164–51176. doi: 10.18632/oncotarget.17737
Chen, D., Tavana, O., Chu, B., Erber, L., Chen, Y., Baer, R., et al. (2017b). NRF2 is a major target of ARF in p53-independent tumor suppression. Mol. Cell 68, 224–232. doi: 10.1016/j.molcel.2017.09.009
Chen, L., Hambright, W. S., Na, R., and Ran, Q. (2015). Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J. Biol. Chem. 290, 28097–28106. doi: 10.1074/jbc.M115.680090
Cho, H. Y., Gladwell, W., Wang, X., Chorley, B., Bell, D., Reddy, S. P., et al. (2010). Nrf2-regulated PPAR{gamma} expression is critical to protection against acute lung injury in mice. Am. J. Res. Crit. Care Med. 182, 170–182. doi: 10.1164/rccm.200907-1047OC
Cozzolino, M., and Carri, M. T. (2012). Mitochondrial dysfunction in ALS. Prog. Neurobiol. 97, 54–66. doi: 10.1016/j.pneurobio.2011.06.003
Crapper McLachlan, D. R., Dalton, A. J., Kruck, T. P., Bell, M. Y., Smith, W. L., Kalow, W., et al. (1991). Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337, 1304–1308. doi: 10.1016/0140-6736(91)92978-B
Devos, D., Moreau, C., Devedjian, J. C., Kluza, J., Petrault, M., Laloux, C., et al. (2014). Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 21, 195–210. doi: 10.1089/ars.2013.5593
Dinkova-Kostova, A. T., and Abramov, A. Y. (2015). The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 88, 179–188. doi: 10.1016/j.freeradbiomed.2015.04.036
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Dixon, S. J., Winter, G. E., Musavi, L. S., Lee, E. D., Snijder, B., Rebsamen, M., et al. (2015). Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609. doi: 10.1021/acschembio.5b00245
Do Van, B., Gouel, F., Jonneaux, A., Timmerman, K., Gele, P., Petrault, M., et al. (2016). Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 94, 169–178. doi: 10.1016/j.nbd.2016.05.011
Doll, S., Proneth, B., Tyurina, Y. Y., Panzilius, E., Kobayashi, S., Ingold, I., et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98. doi: 10.1038/nchembio.2239
East, D. A., Fagiani, F., Crosby, J., Georgakopoulos, N. D., Bertrand, H., Schaap, M., et al. (2014). PMI: a DeltaPsim independent pharmacological regulator of mitophagy. Chem. Biol. 21, 1585–1596. doi: 10.1016/j.chembiol.2014.09.019
Fan, Z., Wirth, A. K., Chen, D., Wruck, C. J., Rauh, M., Buchfelder, M., et al. (2017). Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 6:e371. doi: 10.1038/oncsis.2017.65
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., and Brown, G. C. (2018). Neuronal cell death. Physiol. Rev. 98, 813–880. doi: 10.1152/physrev.00011.2017
Friedmann Angeli, J. P., Schneider, M., Proneth, B., Tyurina, Y. Y., Tyurin, V. A., Hammond, V. J., et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191. doi: 10.1038/ncb3064
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25, 486–541. doi: 10.1038/s41418-017-0012-4
Gao, B., Doan, A., and Hybertson, B. M. (2014). The clinical potential of influencing Nrf2 signaling in degenerative and immunological disorders. Clin. Pharmacol. 6, 19–34.
Gaschler, M. M., Hu, F., Feng, H., Linkermann, A., Min, W., and Stockwell, B. R. (2018). Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem. Biol. 13, 1013–1020. doi: 10.1021/acschembio.8b00199
Greco, T., and Fiskum, G. (2010). Brain mitochondria from rats treated with sulforaphane are resistant to redox-regulated permeability transition. J. Bioenerg. Biomembr. 42, 491–497. doi: 10.1007/s10863-010-9312-9
Guiney, S. J., Adlard, P. A., Bush, A. I., Finkelstein, D. I., and Ayton, S. (2017). Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 104, 34–48. doi: 10.1016/j.neuint.2017.01.004
Hambright, W. S., Fonseca, R. S., Chen, L., Na, R., and Ran, Q. (2017). Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 12, 8–17. doi: 10.1016/j.redox.2017.01.021
Havrdova, E., Giovannoni, G., Gold, R., Fox, R. J., Kappos, L., Phillips, J. T., et al. (2017). Effect of delayed-release dimethyl fumarate on no evidence of disease activity in relapsing-remitting multiple sclerosis: integrated analysis of the phase III DEFINE and CONFIRM studies. Eur. J. Neurol. 24, 726–733. doi: 10.1111/ene.13272
Holmstrom, K. M., Kostov, R. V., and Dinkova-Kostova, A. T. (2016). The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 1, 80–91. doi: 10.1016/j.cotox.2016.10.002
Houessinon, A., Francois, C., Sauzay, C., Louandre, C., Mongelard, G., Godin, C., et al. (2016). Metallothionein-1 as a biomarker of altered redox metabolism in hepatocellular carcinoma cells exposed to sorafenib. Mol. Cancer 15:38. doi: 10.1186/s12943-016-0526-2
Hu, Q., Ren, J., Li, G., Wu, J., Wu, X., Wang, G., et al. (2018). The mitochondrially targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis. 9:403. doi: 10.1038/s41419-018-0436-x
Imai, H., Hirao, F., Sakamoto, T., Sekine, K., Mizukura, Y., Saito, M., et al. (2003). Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem. Biophys. Res. Commun. 305, 278–286. doi: 10.1016/S0006-291X(03)00734-4
Ingold, I., Berndt, C., Schmitt, S., Doll, S., Poschmann, G., Buday, K., et al. (2018). Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409.e421–422.e421. doi: 10.1016/j.cell.2017.11.048
Itoh, T., Fairall, L., Amin, K., Inaba, Y., Szanto, A., Balint, B. L., et al. (2008). Structural basis for the activation of PPARgamma by oxidized fatty acids. Nat. Struct. Mol. Biol. 15, 924–931. doi: 10.1038/nsmb.1474
Jaarsma, D., Haasdijk, E. D., Grashorn, J. A., Hawkins, R., Van Duijn, W., Verspaget, H. W., et al. (2000). Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol. Dis. 7, 623–643. doi: 10.1006/nbdi.2000.0299
Jelinek, A., Heyder, L., Daude, M., Plessner, M., Krippner, S., Grosse, R., et al. (2018). Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 117, 45–57. doi: 10.1016/j.freeradbiomed.2018.01.019
Kagan, V. E., Mao, G., Qu, F., Angeli, J. P., Doll, S., Croix, C. S., et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90. doi: 10.1038/nchembio.2238
Kerins, M. J., and Ooi, A. (2017). The roles of NRF2 in modulating cellular iron homeostasis. Antioxid. Redox Signal. doi: 10.1089/ars.2017.7176 [Epub ahead of print].
Krainz, T., Gaschler, M. M., Lim, C., Sacher, J. R., Stockwell, B. R., and Wipf, P. (2016). A mitochondrial-targeted nitroxide is a potent inhibitor of ferroptosis. ACS Cent. Sci. 2, 653–659. doi: 10.1021/acscentsci.6b00199
Kwak, M. K., Wakabayashi, N., Itoh, K., Motohashi, H., Yamamoto, M., and Kensler, T. W. (2003). Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 278, 8135–8145. doi: 10.1074/jbc.M211898200
Kwan, J. Y., Jeong, S. Y., Van Gelderen, P., Deng, H. X., Quezado, M. M., Danielian, L. E., et al. (2012). Iron accumulation in deep cortical layers accounts for MRI signal abnormalities in ALS: correlating 7 tesla MRI and pathology. PLoS One 7:e35241. doi: 10.1371/journal.pone.0035241
Lee, C. (2017). Collaborative power of Nrf2 and PPARgamma activators against metabolic and drug-induced oxidative injury. Oxid. Med. Cell. Longev. 2017:1378175. doi: 10.1155/2017/1378175
Lee, J., Kosaras, B., Del Signore, S. J., Cormier, K., Mckee, A., Ratan, R. R., et al. (2011). Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol. 121, 487–498. doi: 10.1007/s00401-010-0788-5
Lee, J. M., Calkins, M. J., Chan, K., Kan, Y. W., and Johnson, J. A. (2003). Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 278, 12029–12038. doi: 10.1074/jbc.M211558200
Lewerenz, J., Ates, G., Methner, A., Conrad, M., and Maher, P. (2018). Oxytosis/ferroptosis-(Re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front. Neurosci. 12:214. doi: 10.3389/fnins.2018.00214
Li, K., and Reichmann, H. (2016). Role of iron in neurodegenerative diseases. J. Neural Transm. 123, 389–399. doi: 10.1007/s00702-016-1508-7
Li, Q., Han, X., Lan, X., Gao, Y., Wan, J., Durham, F., et al. (2017). Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2:e90777. doi: 10.1172/jci.insight.90777
Liddell, J. R. (2017). Are astrocytes the predominant cell type for activation of Nrf2 in aging and neurodegeneration? Antioxidants 6:E65. doi: 10.3390/antiox6030065
Liddell, J. R., and White, A. R. (2017). Nexus between mitochondrial function, iron, copper and glutathione in Parkinson’s disease. Neurochem. Int. 117, 126–138. doi: 10.1016/j.neuint.2017.05.016
Liot, G., Valette, J., Pepin, J., Flament, J., and Brouillet, E. (2017). Energy defects in Huntington’s disease: why “in vivo” evidence matters. Biochem. Biophys. Res. Commun. 483, 1084–1095. doi: 10.1016/j.bbrc.2016.09.065
Liu, Y., Wang, W., Li, Y., Xiao, Y., Cheng, J., and Jia, J. (2015). The 5-lipoxygenase inhibitor zileuton confers neuroprotection against glutamate oxidative damage by inhibiting ferroptosis. Biol. Pharm. Bull. 38, 1234–1239. doi: 10.1248/bpb.b15-00048
Lo, S. C., and Hannink, M. (2008). PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp. Cell Res. 314, 1789–1803. doi: 10.1016/j.yexcr.2008.02.014
Maiorino, M., Conrad, M., and Ursini, F. (2017). GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid. Redox Signal. 29, 61–74. doi: 10.1089/ars.2017.7115
Marro, S., Chiabrando, D., Messana, E., Stolte, J., Turco, E., Tolosano, E., et al. (2010). Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position -7007 of the FPN1 promoter. Haematologica 95, 1261–1268. doi: 10.3324/haematol.2009.020123
Merry, T. L., and Ristow, M. (2016). Nuclear factor erythroid-derived 2-like 2 (NFE2L2, Nrf2) mediates exercise-induced mitochondrial biogenesis and the anti-oxidant response in mice. J. Physiol. 594, 5195–5207. doi: 10.1113/JP271957
Mittler, R., Darash-Yahana, M., Sohn, Y. S., Bai, F., Song, L., Cabantchik, I. Z., et al. (2018). NEET proteins: a new link between iron metabolism, ROS and cancer. Antioxid. Redox Signal. doi: 10.1089/ars.2018.7502 [Epub ahead of print].
Morris, G., Berk, M., Carvalho, A. F., Maes, M., Walker, A. J., and Puri, B. K. (2018). Why should neuroscientists worry about iron? The emerging role of ferroptosis in the pathophysiology of neuroprogressive diseases. Behav. Brain Res. 341, 154–175. doi: 10.1016/j.bbr.2017.12.036
Moujalled, D., Grubman, A., Acevedo, K., Yang, S., Ke, Y. D., Moujalled, D. M., et al. (2017). TDP-43 mutations causing amyotrophic lateral sclerosis are associated with altered expression of RNA-binding protein hnRNP K and affect the Nrf2 antioxidant pathway. Hum. Mol. Genet. 26, 1732–1746. doi: 10.1093/hmg/ddx093
Navarro, E., Gonzalez-Lafuente, L., Perez-Liebana, I., Buendia, I., Lopez-Bernardo, E., Sanchez-Ramos, C., et al. (2017). Heme-Oxygenase I and PCG-1alpha regulate mitochondrial biogenesis via microglial activation of alpha7 nicotinic acetylcholine receptors using PNU282987. Antioxid. Redox Signal. 27, 93–105. doi: 10.1089/ars.2016.6698
Neitemeier, S., Jelinek, A., Laino, V., Hoffmann, L., Eisenbach, I., Eying, R., et al. (2017). BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 12, 558–570. doi: 10.1016/j.redox.2017.03.007
Neymotin, A., Calingasan, N. Y., Wille, E., Naseri, N., Petri, S., Damiano, M., et al. (2011). Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 51, 88–96. doi: 10.1016/j.freeradbiomed.2011.03.027
Piantadosi, C. A., Carraway, M. S., Babiker, A., and Suliman, H. B. (2008). Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 103, 1232–1240. doi: 10.1161/01.RES.0000338597.71702.ad
Pietsch, E. C., Chan, J. Y., Torti, F. M., and Torti, S. V. (2003). Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J. Biol. Chem. 278, 2361–2369. doi: 10.1074/jbc.M210664200
Roh, J. L., Kim, E. H., Jang, H., and Shin, D. (2017). Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 11, 254–262. doi: 10.1016/j.redox.2016.12.010
Salazar, M., Rojo, A. I., Velasco, D., De Sagarra, R. M., and Cuadrado, A. (2006). Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 281, 14841–14851. doi: 10.1074/jbc.M513737200
Sasaki, H., Sato, H., Kuriyama-Matsumura, K., Sato, K., Maebara, K., Wang, H., et al. (2002). Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 277, 44765–44771. doi: 10.1074/jbc.M208704200
Seiler, A., Schneider, M., Forster, H., Roth, S., Wirth, E. K., Culmsee, C., et al. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 8, 237–248. doi: 10.1016/j.cmet.2008.07.005
Shah, R., Shchepinov, M. S., and Pratt, D.A. (2017). Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent. Sci. 4, 387–396. doi: 10.1021/acscentsci.7b00589
Sheykhansari, S., Kozielski, K., Bill, J., Sitti, M., Gemmati, D., Zamboni, P., et al. (2018). Redox metals homeostasis in multiple sclerosis and amyotrophic lateral sclerosis: a review. Cell Death Dis. 9:348. doi: 10.1038/s41419-018-0379-2
Shichiri, M. (2014). The role of lipid peroxidation in neurological disorders. J. Clin. Biochem. Nutr. 54, 151–160. doi: 10.3164/jcbn.14-10
Skouta, R., Dixon, S. J., Wang, J., Dunn, D. E., Orman, M., Shimada, K., et al. (2014). Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 136, 4551–4556. doi: 10.1021/ja411006a
Smith, M. A., Zhu, X., Tabaton, M., Liu, G., Mckeel, D. W. Jr., et al. (2010). Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J. Alzheimers Dis. 19, 363–372. doi: 10.3233/JAD-2010-1239
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Sugiyama, A., and Sun, J. (2014). Immunochemical detection of lipid hydroperoxide- and aldehyde-modified proteins in diseases. Subcell. Biochem. 77, 115–125. doi: 10.1007/978-94-007-7920-4_10
Sun, X., Niu, X., Chen, R., He, W., Chen, D., Kang, R., et al. (2016a). Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 64, 488–500. doi: 10.1002/hep.28574
Sun, X., Ou, Z., Chen, R., Niu, X., Chen, D., Kang, R., et al. (2016b). Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184. doi: 10.1002/hep.28251
Swerdlow, R. H. (2017). Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimers Dis. 62, 1403–1416. doi: 10.3233/JAD-170585
Taguchi, K., and Yamamoto, M. (2017). The KEAP1-NRF2 system in cancer. Front. Oncol. 7:85. doi: 10.3389/fonc.2017.00085
Tan, S., Schubert, D., and Maher, P. (2001). Oxytosis: a novel form of programmed cell death. Curr. Top. Med. Chem. 1, 497–506. doi: 10.2174/1568026013394741
Thimmulappa, R. K., Mai, K. H., Srisuma, S., Kensler, T. W., Yamamoto, M., and Biswal, S. (2002). Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 62, 5196–5203.
Tuo, Q. Z., Lei, P., Jackman, K. A., Li, X. L., Xiong, H., Li, X. L., et al. (2017). Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 22, 1520–1530. doi: 10.1038/mp.2017.171
Ueta, T., Inoue, T., Furukawa, T., Tamaki, Y., Nakagawa, Y., Imai, H., et al. (2012). Glutathione peroxidase 4 is required for maturation of photoreceptor cells. J. Biol. Chem. 287, 7675–7682. doi: 10.1074/jbc.M111.335174
Vargas, M. R., Johnson, D. A., and Johnson, J. A. (2011). Decreased glutathione accelerates neurological deficit and mitochondrial pathology in familial ALS-linked hSOD1(G93A) mice model. Neurobiol. Dis. 43, 543–551. doi: 10.1016/j.nbd.2011.04.025
Wang, P., Xie, K., Wang, C., and Bi, J. (2014). Oxidative stress induced by lipid peroxidation is related with inflammation of demyelination and neurodegeneration in multiple sclerosis. Eur. Neurol. 72, 249–254. doi: 10.1159/000363515
Wang, Y. Q., Chang, S. Y., Wu, Q., Gou, Y. J., Jia, L., Cui, Y. M., et al. (2016). The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front. Aging Neurosci. 8:308. doi: 10.3389/fnagi.2016.00308
Wu, K. C., Cui, J. Y., and Klaassen, C. D. (2011). Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 123, 590–600. doi: 10.1093/toxsci/kfr183
Xu, X., Chua, C. C., Kong, J., Kostrzewa, R. M., Kumaraguru, U., Hamdy, R. C., et al. (2007). Necrostatin-1 protects against glutamate-induced glutathione depletion and caspase-independent cell death in HT-22 cells. J. Neurochem. 103, 2004–2014. doi: 10.1111/j.1471-4159.2007.04884.x
Xu, X., Chua, C. C., Zhang, M., Geng, D., Liu, C. F., Hamdy, R. C., et al. (2010). The role of PARP activation in glutamate-induced necroptosis in HT-22 cells. Brain Res. 1343, 206–212. doi: 10.1016/j.brainres.2010.04.080
Yagoda, N., Von Rechenberg, M., Zaganjor, E., Bauer, A. J., Yang, W. S., Fridman, D. J., et al. (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868. doi: 10.1038/nature05859
Yamamoto, M., Kensler, T. W., and Motohashi, H. (2018). The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 98, 1169–1203. doi: 10.1152/physrev.00023.2017
Yang, W. S., Kim, K. J., Gaschler, M. M., Patel, M., Shchepinov, M. S., and Stockwell, B. R. (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. U.S.A. 113, E4966–E4975. doi: 10.1073/pnas.1603244113
Yang, W. S., Sriramaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi: 10.1016/j.cell.2013.12.010
Yoo, S. E., Chen, L., Na, R., Liu, Y., Rios, C., Van Remmen, H., et al. (2012). Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic. Biol. Med. 52, 1820–1827. doi: 10.1016/j.freeradbiomed.2012.02.043
Yuan, H., Li, X., Zhang, X., Kang, R., and Tang, D. (2016a). CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 478, 838–844. doi: 10.1016/j.bbrc.2016.08.034
Yuan, H., Li, X., Zhang, X., Kang, R., and Tang, D. (2016b). Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 478, 1338–1343. doi: 10.1016/j.bbrc.2016.08.124
Keywords: Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, motor neuron disease, RSL3, erastin, Keap1, system xc-
Citation: Abdalkader M, Lampinen R, Kanninen KM, Malm TM and Liddell JR (2018) Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front. Neurosci. 12:466. doi: 10.3389/fnins.2018.00466
Received: 21 March 2018; Accepted: 19 June 2018;
Published: 10 July 2018.
Edited by:
Victor Tapias, Cornell University, United StatesReviewed by:
Zezong Gu, University of Missouri, United StatesCopyright © 2018 Abdalkader, Lampinen, Kanninen, Malm and Liddell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeffrey R. Liddell, amxpZGRlbGxAdW5pbWVsYi5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.