 Rudimar L. Frozza
Rudimar L. Frozza Mychael V. Lourenco
Mychael V. Lourenco Fernanda G. De Felice
Fernanda G. De Felice- 1Oswaldo Cruz Institute, Fundação Oswaldo Cruz (FIOCRUZ), Rio de Janeiro, Brazil
- 2Institute of Medical Biochemistry Leopoldo de Meis, Rio de Janeiro, Brazil
- 3Institute of Biophysics Carlos Chagas Filho, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 4Department of Biomedical and Molecular Sciences, Centre for Neuroscience Studies, Queen's University, Kingston, ON, Canada
Alzheimer's disease (AD), the most common form of dementia in late life, will become even more prevalent by midcentury, constituting a major global health concern with huge implications for individuals and society. Despite scientific breakthroughs during the past decades that have expanded our knowledge on the cellular and molecular bases of AD, therapies that effectively halt disease progression are still lacking, and focused efforts are needed to address this public health challenge. Because AD is classically recognized as a disease of memory, studies have mainly focused on investigating memory-associated brain defects. However, compelling evidence has indicated that additional brain regions, not classically linked to memory, are also affected in the course of disease. In this review, we outline the current understanding of key pathophysiological mechanisms in AD and their clinical manifestation. We also highlight how considering the complex nature of AD pathogenesis, and exploring repurposed drug approaches can pave the road toward the development of novel therapeutics for AD.
Introduction
Increasing life expectancy has produced a dramatic rise in the number of cases of age-associated diseases, including dementia. Alzheimer's disease (AD) is the most frequent cause of dementia, accounting for 60–80% of all cases (Prince et al., 2016), and epidemiological studies indicate that AD will become even more incident by midcentury, constituting a major personal and societal tragedy. AD is primarily a condition of late life, roughly doubling in prevalence every 5 years after age 65 (Prince et al., 2013), and affects some 47 million people worldwide (Prince et al., 2013). This number is predicted to increase in the next two decades (Prince et al., 2016). The total cost of dementia was estimated around $818 billion in 2010 and has been projected to hit $1 trillion by 2018 worldwide (Prince et al., 2016). This becomes even more dramatic because nearly 60% of people affected by dementia live in low- and middle-income countries.
AD is a complex disorder. While the vast majority of AD cases are sporadic, affecting individuals older than 60 years, genetic mutations cause a rare (<0.5%) familial form of AD, whose symptoms develop earlier, typically between 30 and 50 years of age (Bateman et al., 2010). Further, there is a marked difference in the incidence of AD between women and men. It is estimated that nearly two-thirds of the patients living with AD are women (Alzheimer's Association, 2017), raising the intriguing suggestion that there are biological mechanisms underlying the higher incidence of AD cases in women that still demand to be investigated.
AD is mainly characterized by progressive cognitive impairment. However, as disease progresses, other debilitating non-cognitive symptoms arise, including impaired sleep and appetite, and neuropsychiatric alterations (e.g., depression and apathy) (Ishii and Iadecola, 2015; Lanctôt et al., 2017). In addition, mounting epidemiological studies have supported a link between metabolic disorders and AD (Ott et al., 1996, 1999; Steen et al., 2005; Matsuzaki et al., 2010; Takeda et al., 2010; Crane et al., 2013; De Felice, 2013; De Felice and Lourenco, 2015; Chatterjee et al., 2016). Because AD has been considered a disease of memory, studies on AD pathogenesis have mainly concentrated on how memory and cognitive failure develop, while other symptoms and co-morbidities have remained largely overlooked.
Thus, it is not surprisingly that precise and reliable biomarkers are still lacking for early disease diagnosis. Although conclusive diagnostics has mostly been confirmed through post-mortem examination, it is now widely accepted that pathophysiological changes begin to develop decades prior to initial cognitive symptoms, in a preclinical or presymptomatic stage (Sperling et al., 2011a,b). Further, the addition of novel biomarkers to diagnostic criteria has prompted a shift in how AD is considered as pathological entity, increasing the appreciation that it should not be regarded as having discrete and defined clinical stages, but rather as multifaceted process moving along a continuum (Sperling et al., 2011a; van Maurik et al., 2017; Figure 1). Relatively accurate diagnosis and timely therapies will likely be achieved when neuropsychological, fluid and imaging biomarkers are used in combination (Viola and Klein, 2015; Dubois et al., 2016; Blennow, 2017).
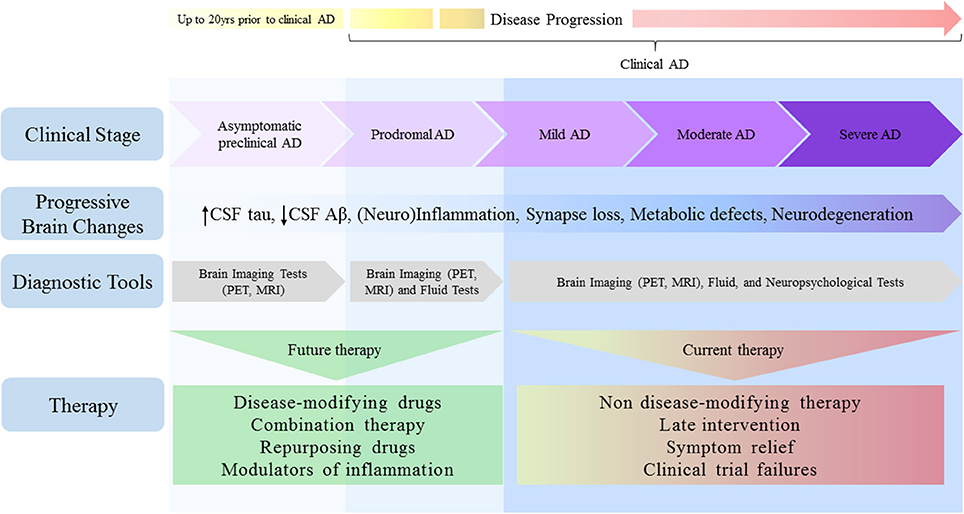
Figure 1. Alzheimer's disease depicted as a continuum: challenges for therapy. Pathophysiological changes in the AD brain begin many years prior to clinical manifestations of disease and move along a continuum, spanning from clinically asymptomatic to severely impaired spectra. Although cognitive symptoms are absent in the preclinical stage, progressive amyloid deposition could drive the patient toward prodromal AD stage, characterized by short-term memory impairment without affecting activity of daily living. As disease progresses, however, many brain areas and their functions become impaired, culminating in severe memory loss and metabolic derangements, both of which affect autonomy. Despite the lack of bona fide biomarkers to date, earlier detection will ensure that treatments reach individuals in a timely manner. Given that therapies initially considered promising have disappointed in clinical trials, current AD research pipeline requires a shift toward the use of disease-modifying approaches, combination and/or repurposing therapies, and the search for agents selectively targeting specific modulators of inflammation.
Although advances in animal and clinical research over the past few decades have improved our knowledge on the pathophysiological course of AD, even drugs with successful preclinical assessment have not been effective in reversing or slowing down AD progression in large clinical trials. These constraints may be due to that clinical trials have predominantly focused on therapies based on anti-amyloid strategies, since the amyloid cascade hypothesis has been placed at the center of therapeutic prospection (Karran et al., 2011; Cummings et al., 2014; Hendrix et al., 2016). Such disappointing outcomes are also suggestive of problems in translating therapies from rodent model species to humans (De Felice and Munoz, 2016). The lack of adequate control for sex differences in animal models adds up to this translational impedance. Therefore, potential therapies that work in a sex of one animal species (usually male rodents) frequently fail to translate to human trials dominated by female participants (often 2:1 female:male in large trials). Furthermore, while neuropathological features of AD are widely recognized, the intricacies of the mechanism involving central and peripheral derangements have not been clearly defined.
Given that AD holds a complex pathology, it has now been believed that more effective treatments could be possible using disease-modifying therapies and drugs targeting multiple molecular pathways (Castellani and Perry, 2012; Cummings et al., 2014; Perry et al., 2014; Stephenson et al., 2014). These should importantly take sex differences into consideration, as recently noticed (Snyder et al., 2016; Zhao et al., 2016). In this review, we discuss recent advances in the AD field, as well as classical and novel mechanisms that might reveal potential new strategies to treat AD.
Molecular Pathogenesis of AD
Tau Phosphorylation, Amyloid Deposition, and Aβ Oligomers
The most distinctive features present in memory-associated brain regions of AD patients are the intracellular neurofibrillary tangles (NFTs) and the extracellular amyloid plaques. The major component of the NFTs is abnormally phosphorylated and aggregated tau protein (Querfurth and LaFerla, 2010; Medeiros et al., 2011; Morris et al., 2011), thereby destabilizing microtubules and compromising axonal transport (Querfurth and LaFerla, 2010; Ittner and Götz, 2011; Medeiros et al., 2011; Morris et al., 2011; Scheltens et al., 2016). It has been recently shown that tangles induce neuronal loss and spatial memory defects (Fu et al., 2017), putatively providing a link between tau pathology and cognitive deficits in early AD. Although pathological alterations of tau were thought to be downstream events of Aβ deposition, it is equally plausible that tau and Aβ act in parallel to enhancing each other's toxic effects and initiate the pathogenic events germane to AD (Small and Duff, 2008; Spires-Jones and Hyman, 2014; Bennett et al., 2017). Fresh evidence has also pointed to soluble, diffusible tau oligomers as important drivers of synaptotoxicity, and possible culprits for the marked progression of tau pathology across the brain (Fá et al., 2016; Carrieri et al., 2017; Piacentini et al., 2017; Puzzo et al., 2017; Reilly et al., 2017).
The amyloid cascade hypothesis suggests that brain accumulation of the amyloid-β peptide (Aβ), produced by sequential cleavage of the amyloid precursor protein (APP) by the β- and γ-secretase enzymes, is a central event in AD (Karran et al., 2011; Selkoe and Hardy, 2016). Soluble Aβ undergoes conformational changes to high β-sheet content, rendering it prone to aggregation into polymeric forms, including soluble oligomers and larger insoluble fibrils. These fibrils ultimately deposit into extracellular amyloid plaques in the AD brains (Stine et al., 2003; Blennow et al., 2006; Finder and Glockshuber, 2007; Lee et al., 2007).
Aβ is physiologically degraded by the peptidases insulin-degrading enzyme, neprilysin, and by endothelin-converting enzyme (Qiu et al., 1998; Iwata et al., 2001; Farris et al., 2003; Leissring et al., 2003). In addition, Aβ can be cleared out by transportation to peripheral circulation across multiple pathways, including the blood-brain barrier, interstitial fluid bulk flow, arachnoid villi, and glymphatic-lymphatic pathways (Tarasoff-Conway et al., 2015). Additionally, Aβ aggregates can be phagocited and degraded by microglia, perivascular macrophages, and astrocytes. Defective clearing systems could thus lead to an imbalance between production and clearance of Aβ in the brain, thereby resulting in subsequent neuronal dysfunction and neurodegeneration (Hardy, 2002).
A growing body of evidence indicates, however, that plaque deposition is not the sole responsible for the impairments observed in AD. On the other hand, the notion that Aβ oligomers (AβOs) are the main toxins responsible for synapse dysfunction and cognitive deficits in AD has attracted considerable attention to improve our understanding of the mechanisms of the disease (Walsh and Selkoe, 2007; Selkoe, 2008; Ferreira and Klein, 2011; Ferreira et al., 2015; Yang et al., 2017). In this context, plaques have been thought to comprise a reservoir from which AβOs diffuse, or may even act sequestering soluble oligomers until they reach a physiological plateau (Selkoe and Hardy, 2016).
A considerable number of studies has reported that AβOs accumulate in the brain and CSF of AD patients (Georganopoulou et al., 2005; Haes et al., 2005; Anker et al., 2009; Xia et al., 2009; Herskovits et al., 2013; Viola et al., 2014; Murakami et al., 2016), and are found in association with synapses in the brains of patients presenting clinical signals of dementia (Koffie et al., 2009; Bjorklund et al., 2012; Perez-Nievas et al., 2013; Bilousova et al., 2016), adding clinical relevance to their role in AD. These studies suggest that synapse-associated AβOs promote detrimental modifications in synapse structure and composition, thereby leading to memory loss. This growing body of evidence props up an early notion that cognitive decline is not only a result of the extracellular accumulation of Aβ and intracellular accumulation of tau but also as a consequence of synapse failure and loss in AD (Terry et al., 1991; Masliah et al., 1992; Selkoe, 2002).
Despite intense research, the exact mechanisms of how AβOs exert their toxicity remains to be fully unveiled. Binding of Aβ aggregates to various receptors may disrupt key neuronal functions. However, the complete identity of receptors to which they bind and the underlying signaling pathways still remain to be fully elucidated (Ferreira et al., 2015).
We now know that AβOs bind to cell surface receptors and trigger multiple aberrant signaling pathways, including calcium signaling (Mattson, 2010; Ferreira et al., 2015), oxidative stress (Smith et al., 1998; Perry et al., 2002; De Felice et al., 2007), derangements in plasticity-related receptors and increased glutamate release from pre-synaptic terminals (Roselli et al., 2005; Shankar et al., 2007; Decker et al., 2010a; Ferreira et al., 2015). In addition, they promote tau hyperphosphorylation (De Felice et al., 2008; Jin et al., 2011), impaired axonal transport (Snyder et al., 2005; Decker et al., 2010b; Miñano-Molina et al., 2011; Bomfim et al., 2012), and drive inhibition of long-term potentiation (LTP) and memory impairment (Rowan et al., 2005; Shankar et al., 2008; Ferreira and Klein, 2011; Ferreira et al., 2015; Yang et al., 2017).
Inflammatory Markers in the Brain
AD pathogenesis appears to include strong interactions with immune mechanisms in the brain. AβOs induce aberrant reactivity of astrocytes and microglia, in the brains of mice and non-human primates (Bomfim et al., 2012; Ledo et al., 2013, 2016; Forny-Germano et al., 2014). Recent studies have further unveiled that disturbances in microglia, as well as interactions with peripheral immune cells, may play key roles in causing synapse loss and neurodegeneration in AD (Browne et al., 2013; Zhang et al., 2013; Baruch et al., 2015, 2016; Guillot-Sestier et al., 2015; Zenaro et al., 2015; Hong et al., 2016a,b). These studies are in line with emerging evidence suggesting that inflammation has a pivotal role in disease pathogenesis, as markers of inflammation, such as TNF-α, IL-1β, IL-6, and other cytokines, have been shown to be increased in the brain, CSF, and plasma of AD patients (Perry et al., 2010; Swardfager et al., 2010; Czirr and Wyss-Coray, 2012; Alcolea et al., 2014; Heneka et al., 2015a; Hong et al., 2016a; Salter and Stevens, 2017).
Increased pro-inflammatory signaling resulting from reactive microglial reduces Aβ clearance, promotes aberrant synaptic pruning (Lee and Landreth, 2010; Mandrekar-Colucci et al., 2012; Heneka et al., 2015a,b; Hong et al., 2016b), prompts Aβ and tau pathologies, and contributes to impaired synapse function (Wang W. Y. et al., 2015). Importantly, TNF-α-dependent mechanisms appear to drive memory defects (Lourenco et al., 2013) and depressive-like behavior in AD mice (Ledo et al., 2016), thereby indicating a causal role of inflammation in deleterious processes linked to AD.
Unfolded Protein Response and Defective Proteostasis
Pro-inflammatory pathways triggered by AβOs, notably via TNF-α, have been reported to induce neuronal stress (Lourenco et al., 2013), likely resulting in defective proteostasis. Furthermore, it has been recently demonstrated that AβOs stimulates eIF2α phosphorylation (Devi and Ohno, 2010, 2013, 2014; Lourenco et al., 2013; Ma et al., 2013; Baleriola et al., 2014). In the brain, eIF2α is a hub that controls protein synthesis-dependent learning and memory and mantain neuronal integrity in health and disease. When phosphorylated, however, eIF2α attenuates the initiation of global protein synthesis (Lourenco et al., 2015).
Aberrant eIF2α phosphorylation and inhibition of protein synthesis have emerged as major molecular pathways driving synapse and memory failure in AD models (Costa-Mattioli et al., 2007; Lourenco et al., 2013, 2015; Ma et al., 2013; Baleriola et al., 2014). In line with this notion, deletion of eIF2α kinases, including PKR, PERK, or GCN2 restores memory and synapse function in mouse models of AD (Lourenco et al., 2013; Ma et al., 2013).
Abnormal accumulation of misfolded proteins in the endoplasmic reticulum triggers the unfolded protein response (UPR), a set of signaling branches aimed at restore cellular homeostasis (Hetz, 2012; Dufey et al., 2014; Hetz and Saxena, 2017). However, when prolonged, UPR signaling might compromise neuronal functions, resulting in neurodegeneration (Lourenco et al., 2015; Freeman and Mallucci, 2016; Hetz and Saxena, 2017). There is now considerable evidence suggesting that AD brain display increased markers of UPR (Hoozemans et al., 2009; Hetz and Saxena, 2017), and that at least the PERK (Ma et al., 2013) and IRE-1a (Lourenco et al., 2013; Duran-Aniotz et al., 2017) branches of UPR are involved in memory defects in AD mice. Further, the chemical chaperone 4-phenylbutyrate alleviates AβO-induced memory defects in mice (Lourenco et al., 2013), thus highlighting the role of UPR in mediating neurotoxicity in AD. The combination of misfolded protein accumulation, activation of brain immune responses and defective proteostasis might thus comprise the very essence of synapse and memory failure in AD.
Novel Pathophysiological Mechanisms in AD
Scientific breakthroughs during the past decades have expanded our knowledge on cellular and molecular aspects of AD. Nevertheless, AD remains largely idiopathic, and therapies that effectively combat disease progression are still lacking. Given that AD largely associates with memory loss, it is not surprising that the vast majority of studies deal with mechanisms implicated in cognitive deterioration. Hence, much less is known about how brain regions that are not directly linked to memory are affected in AD, as well as about mechanisms underlying its major comorbidities.
Numerous studies have investigated how Aβ impacts the hippocampus and the cortex (Ferreira and Klein, 2011; Musiek and Holtzman, 2015), known to be fundamentally involved in acquisition, consolidation, and recollection of new episodic memories. However, early studies indicated that brain regions not necessarily involved in learning and memory might also be affected in AD. It is noteworthy that AD patients exhibit significant non-cognitive deficits (summarized in the Table 1) such as sleep-wake disorders and neuroendocrine alterations attributable to hypothalamic dysfunction (Prinz et al., 1982; White et al., 1996; Csernansky et al., 2006).
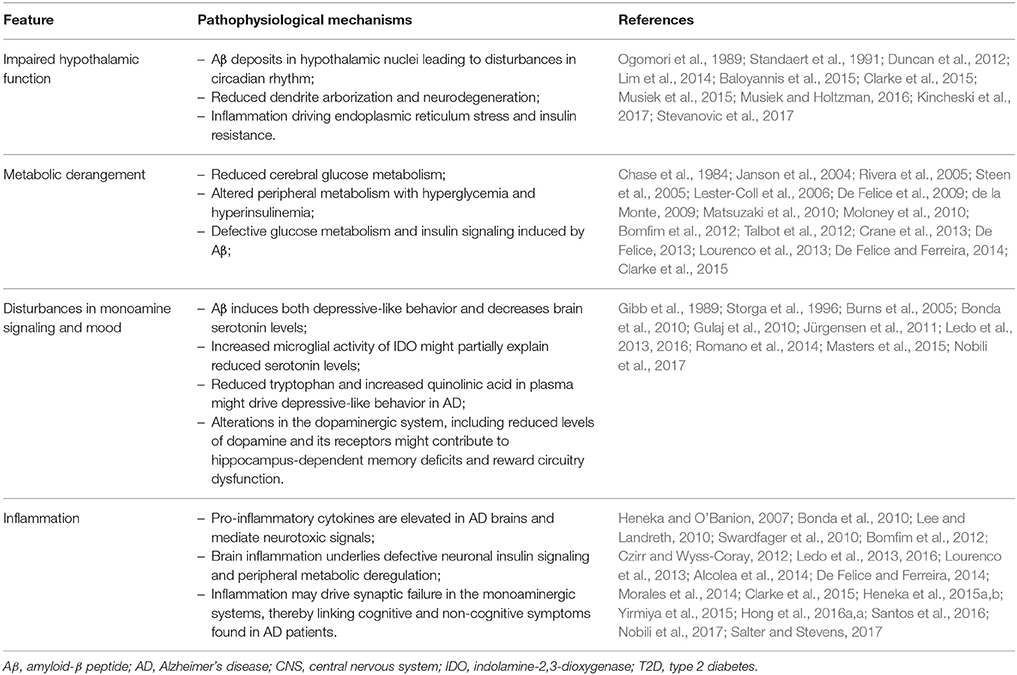
Table 1. Novel pathophysiological mechanisms in AD.
Impaired Hypothalamic Function
Disturbances in hypothalamic nuclei have been reported in patients and animal models of AD (Duncan et al., 2012; Lim et al., 2014; Musiek et al., 2015; Musiek and Holtzman, 2016; Stevanovic et al., 2017). Since the hypothalamus is responsible for controlling circadian rhythm, impairments in its function can at least partially account for sleep disturbances. Nonetheless, although initial results have already shed light on how sleep becomes deregulated in AD (Ju et al., 2014; Musiek and Holtzman, 2016; Kincheski et al., 2017), studies investigating whether hypothalamic defects mediate sleep disturbances in AD are still needed.
Derangements in hypothalamic functions play a central role in peripheral metabolism deregulation and its consequences. For instance, hypothalamic inflammation and impaired proteostasis are critical pathogenic events in the establishment of peripheral insulin resistance in metabolic disorders (Zhang et al., 2008; Milanski et al., 2009; Denis et al., 2010; Arruda et al., 2011; Thaler et al., 2012; Valdearcos et al., 2015). Nonetheless, very few studies so far investigated hypothalamic dysfunction in AD.
Early post-mortem studies identified Aβ deposits in hypothalamic nuclei of AD patients (Ogomori et al., 1989; Standaert et al., 1991), and neurodegeneration with marked retraction of dendrites in early AD (Baloyannis et al., 2015). Further, hypothalamic endoplasmic reticulum stress, inflammation, and insulin resistance were demonstrated in AβO-injected mice and non-human primates (Clarke et al., 2015). Dysfunction triggered by AβOs in the hypothalamus associated with development of persistent peripheral glucose intolerance, which was further demonstrated in several transgenic mouse models of AD (Clarke et al., 2015; Vandal et al., 2015; Stanley et al., 2016), and in human patients (Craft et al., 1992).
Defective Glucose Metabolism and Insulin Signaling
Altered peripheral metabolism with hyperglycemia and hyperinsulinemia, which are cardinal features of type 2 diabetes (T2D), were recently found to positively correlate with development of AD-like brain pathology in humans (Matsuzaki et al., 2010; Crane et al., 2013). Conversely, AD has been associated with increased T2D risk (Janson et al., 2004), suggesting that the connection between AD and T2D may comprise a two-way road. AD progression positively further correlates with reduction of cerebral glucose metabolism in the forebrain, including the posterior parietal lobe and portions of temporal and occipital lobes (Chase et al., 1984).
An important player accounting for impaired glucose metabolism in AD could arise from defects in insulin signaling pathways. AD brains exhibit lower levels of insulin and reduced insulin receptor (IR) expression and sensitivity (Rivera et al., 2005; Steen et al., 2005; Talbot et al., 2012). Further, impairments in insulin signaling downstream machinery have been reported in post-mortem brain tissue and in animal models of AD (Steen et al., 2005; Lester-Coll et al., 2006; de la Monte, 2009; Moloney et al., 2010; Bomfim et al., 2012; Craft, 2012; Talbot et al., 2012; Lourenco et al., 2013; Clarke et al., 2015). Recent studies have shown that AβOs are the toxins linked to impaired hippocampal insulin signaling by promoting internalization and cellular redistribution of insulin receptors, blocking downstream hippocampal insulin signaling (De Felice et al., 2009; Ma et al., 2009; Bomfim et al., 2012). Such body of evidence has established novel molecular parallels between AD and T2D.
The precise molecular mechanisms connecting impaired glucose metabolism and insulin signaling to AD pathogenesis remain to be fully determined. Nonetheless, mounting evidence has pointed to inflammation as a critical player linking AD and metabolic diseases, including T2D (De Felice and Ferreira, 2014; Ferreira et al., 2014; Morales et al., 2014; Heneka et al., 2015b). Overproduction of pro-inflammatory cytokines, notably TNF-α, is a key feature of the pathophysiology of metabolic disorders (Hotamisligil, 2006, 2017). Notably, brain inflammation has recently been proposed to underlie defective neuronal insulin signaling (Bomfim et al., 2012; Lourenco et al., 2013), as well as peripheral metabolic deregulation in AD (Clarke et al., 2015).
Disturbances in Monoamine Signaling and Mood
Mounting evidence supports the notion that microglial activation and brain inflammation could further underlie mood disorders, including depressive behaviors (Yirmiya et al., 2015; Santos et al., 2016). Depression and/or apathy have been reported as frequent comorbidities in AD patients (Lyketsos and Olin, 2002), and have been regarded as risk factors for AD (Green et al., 2003; Ownby et al., 2006; Starkstein and Mizrahi, 2006; Geerlings et al., 2008).
Although clinical and epidemiological studies have revealed a strong connection between AD and depression, the mechanisms connecting these disorders at the molecular and cellular levels have only recently begun to be established. Clues into a mechanistic link between memory and mood disturbances in AD came from recent works showing that AβOs induce both depressive-like behavior and memory deficits in mice and associate with decreased brain serotonin levels (Ledo et al., 2013, 2016) in a similar way to that observed in transgenic mice model of AD (Romano et al., 2014). Reduced serotonin levels may be linked to increased levels and activity of indolamine-2,3-dioxygenase (IDO) follow microglial activation. Interestingly, AD patients were found to have reduced levels of plasma tryptophan and increased quinolinic acid (Gulaj et al., 2010), as well as increased IDO immunoreactivity in microglia (Bonda et al., 2010). Because inflammation plays a significant role in depression, these findings raise the possibility that AβO-induced brain inflammation may constitute a common denominator between cognitive and mood alterations in AD.
Alterations in the dopaminergic system have also been reported in AD patients and experimental models, including reduced levels of dopamine and its receptors (Gibb et al., 1989; Storga et al., 1996; Burns et al., 2005; Jürgensen et al., 2011; Nobili et al., 2017), and are commonly linked to cognitive and non-cognitive symptoms of the disease. It has been recently shown that inflammation and apoptosis take place in the ventral tegmental area, causing selective degeneration of the dopaminergic nuclei before senile plaque deposition, tangles or any sign of neuronal loss in cortical and hippocampal regions in a transgenic mouse model of AD (Nobili et al., 2017).
Given that dopaminergic neurons from ventral tegmental area not only modulate hippocampal synaptic plasticity (Rossato et al., 2009; McNamara et al., 2014; Broussard et al., 2016), but also target the nucleus accumbens and the cerebral cortex (Russo and Nestler, 2013), dopaminergic degeneration in ventral tegmental area might largely contribute to the deficits in hippocampus-dependent memory and reward circuits. These findings may provide an intriguing explanation to recent observations in AD patients indicating that the clinical diagnosis of dementia is associated with early non-cognitive symptoms, such as depression and apathy (Masters et al., 2015). Overall, these recent data suggest that inflammation may drive synaptic failure in the monoaminergic systems, thereby linking the cognitive and non-cognitive symptoms found in AD patients.
Challenges for AD Therapy
Despite intensive investigation of mechanisms of pathogenesis in AD during the past three decades, little has been achieved in terms of effective treatments or approaches to prevent or cure it. Taking into account the dramatic rise in the number of AD cases, huge economic and social hurdle will impact the society if no treatment is developed within the next few years. Additionally, it is noteworthy that advances in therapeutic strategies for AD that lead to even small delays in AD onset or progression would significantly attenuate the global burden of the disease.
Given the conceptual frameshift that occurred in the field in the past few years, AD has not only been viewed with discrete and defined clinical stages, but as a multifaceted process moving along a continuum. Thanks to the evolving biomarker research, it is now recognized that pathophysiological changes begin many years before clinical manifestations of AD. For example, changes in CSF tau levels have been shown to develop ~15 years before the onset of clinical AD, while CSF Aβ42 levels may drop even earlier, up to 20 years before symptom onset (Bateman et al., 2012; Buchhave, 2012; Villemagne et al., 2013; Fagan et al., 2014).
The spectrum of AD spans from clinically asymptomatic to severely impaired (Figure 1). However, these boundaries are challenging, given that separation between healthy aging and preclinical AD is not well-defined in our current understanding. This unmet question will likely be addressed in the future, as early detection biomarkers have become a major research focus.
Sex differences should also be taken into account as a biological variable in AD pathogenesis as women constitute the majority of affected people, accounting for nearly two-thirds of AD patients (Alzheimer's Association, 2017). Reasons for the higher frequency of AD among women could be partly explained by the fact that women live longer. However, late-onset AD risk is greater in women even after controlling for their longer lifespan relative to men (Viña and Lloret, 2010). The biological underpinnings of the increased AD risk in women remain largely unknown.
Nonetheless, it is now accepted that the perimenopause to menopause transition disrupts multiple estrogen-regulated systems, thereby affecting multiple domains of cognitive function (Brinton et al., 2015; Christensen and Pike, 2015). Indeed, recent preclinical studies have implicated that a shift in the bioenergetics system of the brain during menopause onset could serve as an early initiating mechanism for increased AD risk in the female brain (Brinton et al., 2015; Mosconi et al., 2017a,b). These biological variables may lead to increased fatty acid catabolism, Aβ deposition, and impaired synaptic plasticity (Liu et al., 2008; Brinton, 2009; Yao and Brinton, 2012), which could serve as a mechanism that triggers AD (Brinton et al., 2015). As a result, it is conceivable that disappointing outcomes in clinical trials may be partially explained by metabolic differences in women and men. Therefore, recommendations to include both female and male animals in preclinical research should be completely embraced by the research community.
While the amyloid cascade hypothesis has dominated research for the past 20 years, the shift toward disease-modifying drug development in the last decade might be imperative to develop approaches that interrupt the underlying disease processes.
Potential benefits for AD therapy can also emerge from combination pharmacotherapy. This strategy has proven effective for several diseases, including tuberculosis, HIV/AIDS, cardiovascular diseases, and cancer (Perry et al., 2014; Hendrix et al., 2016), and holds potential to enhance the efficacy of drugs that are ineffective on their own, but offer synergistic or additive benefits in combination.
Taking into account the well-known high failure rates in drug development targeting the central nervous system, strategies aimed at repurposing already marketed drugs become an interesting option to speed up drug discovery in AD (Appleby and Cummings, 2013). Given that metabolic derangements seem to play a pivotal role in AD, and that a myriad of drugs for metabolic disease have already been labeled for human use, repurposing such compounds may have the potential to accelerate drug development. That is because preclinical toxicology, human safety, tolerability, and pharmacokinetic assessments could move faster. Impaired brain insulin signaling or brain insulin resistance seems play a central role in the molecular pathogenesis of sporadic AD. Thus, targeting brain insulin signaling through the administration of drugs that have already been previously approved for the treatment of diabetes mellitus, such as insulin and drugs that improve insulin sensitivity, could expedite their development for the treatment of AD (Chen et al., 2016). It worthy to note that anti-diabetic compounds, such as insulin, exenatide, and liraglutide, have already been tested in ongoing clinical trials (clinical trial ID NCT01767909, NCT01255163, and NCT01843075, respectively).
Neuroinflammation, especially at the earliest stages, supports a vicious cycle of microglial activation, release of pro-inflammatory factors, and neuronal damage. Additionally, inflammatory mechanisms, such as those driven by TNF-α, may be orchestrated between the brain and the periphery, providing a likely link between AD and peripheral metabolic deregulation (De Felice and Ferreira, 2014; Ferreira et al., 2014; De Felice and Lourenco, 2015). The important role of neuroinflammation in AD is further supported by findings that gene variants for immune receptors, including TREM2, are associated with altered AD risk (Guerreiro et al., 2013; Heneka et al., 2015a).
A considerable body of evidence supports that inflammation could be a therapeutically relevant target in AD. Nevertheless, trials with anti-inflammatory compounds, such as non-steroidal anti-inflammatory drugs (NSAIDs), peroxisome proliferator-activated receptor-γ (PPAR-γ) activators, minocycline, and TNF-α signaling inhibitors have not yet provided exciting outcomes to date (Calsolaro and Edison, 2016), although lifelong use of NSAIDs has been associated with reduced risk of developing AD (Wang J. et al., 2015).
Additional therapeutic approaches with intravenous immunoglobulins and/or monoclonal antibodies are currently under evaluation, and results have not been conclusive yet. These uncertain results could be, to some extent, due to that anti-inflammatory drugs target generic rather than specific neuroinflammatory components in AD. Thus, specific modulators of inflammation at early disease stages will be essential to understand the potential of targeting inflammation in neurodegeneration.
Concluding Remarks
Although our understanding of AD has considerably increased over recent years, there is a still unmet requirement for effective therapeutics. Properly diagnosing AD is still one of the major hurdles in the field, as reliable biomarkers are lacking. There is fresh and compelling preclinical evidence that brain regions not necessarily involved in learning and memory might also be affected in AD, driving its major comorbidities. As most of therapeutic approaches have had disappointing outcomes so far, it is time to revisit the science underlining our current AD canons, and move toward the search for additional disease mechanisms and keys to treatment. Inflammation plays a critical role in the pathogenesis of AD and seems to drive the metabolic derangements that have been found to positively correlate with disease onset, leading to the emergence of cognitive and non-cognitive symptoms.
A deeper understanding of the complex features underlying major disease symptoms, including behavioral, mood, inflammation, and metabolic disturbances, may contribute to the development of novel and successful therapies. Given the differential prevalence of AD in men and women, sex differences should also be taken into account when studying AD pathophysiology, as they might reveal the need for separate therapeutic approaches. Drugs currently approved for use in AD are not disease-modifying, only confer mild and transient symptomatic management. Intervention at earlier stages using disease-modifying and combination therapy comprised of repourposed drugs and anti-inflammatory agents could pave the road toward successful outcomes in AD therapy.
Author Contributions
RF, ML, and FD: planned, researched and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in the authors' laboratories has been supported by grants from National Institute for Translational Neuroscience (INNT/Brazil), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq/Brazil), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Alzheimer Society Canada and the International Society for Neurochemistry (ISN).
References
Alcolea, D., Carmona-Iragui, M., Suárez-Calvet, M., Sánchez-Saudinós, M. B., Sala, I., Antón-Aguirre, S., et al. (2014). Relationship between β-secretase, inflammation and core cerebrospinal fluid biomarkers for Alzheimer's disease. J. Alzheimer's Dis. 42, 157–167. doi: 10.3233/JAD-140240
Alzheimer's Association (2017). 2017 Alzheimer's disease facts and figures. Alzheimer's Dement. 13, 325–373. doi: 10.1016/j.jalz.2017.02.001
Anker, J. N., Hall, W. P., Lambert, M. P., Velasco, P. T., Mrksich, M., Klein, W. L., et al. (2009). Detection and identification of bioanalytes with high resolution LSPR spectroscopy and MALDI mass spectrometry. J. Phys. Chem. C 113, 5891–5894. doi: 10.1021/jp900266k
Appleby, B. S., and Cummings, J. L. (2013). Discovering new treatments for Alzheimer's disease by repurposing approved medications. Curr. Top. Med. Chem. 13, 2306–2327. doi: 10.2174/15680266113136660162
Arruda, A. P., Milanski, M., Coope, A., Torsoni, A. S., Ropelle, E., Carvalho, D. P., et al. (2011). Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology 152, 1314–1326. doi: 10.1210/en.2010-0659
Baleriola, J., Walker, C. A., Jean, Y. Y., Crary, J. F., Troy, C. M., Nagy, P. L., et al. (2014). Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 158, 1159–1172. doi: 10.1016/j.cell.2014.07.001
Baloyannis, S. J., Mavroudis, I., Mitilineos, D., Baloyannis, I. S., and Costa, V. G. (2015). The hypothalamus in alzheimer's disease. Am. J. Alzheimer's Dis. Other Dementiasr. 30, 478–487. doi: 10.1177/1533317514556876
Baruch, K., Deczkowska, A., Rosenzweig, N., Tsitsou-Kampeli, A., Sharif, A. M., Matcovitch-Natan, O., et al. (2016). PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease. Nat. Med. 22, 135–137. doi: 10.1038/nm.4022
Baruch, K., Rosenzweig, N., Kertser, A., Deczkowska, A., Sharif, A. M., Spinrad, A., et al. (2015). Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer's disease pathology. Nat. Commun. 6, 7967. doi: 10.1038/ncomms8967
Bateman, R. J., Aisen, P. S., De Strooper, B., Fox, N. C., Lemere, C. A., Ringman, J. M., et al. (2010). Autosomal-dominant Alzheimer's disease: a review and proposal for the prevention of Alzheimer's disease. Alzheimers. Res. Ther. 3, 1. doi: 10.1186/alzrt59
Bateman, R. J., Xiong, C., Benzinger, T. L. S., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited alzheimer's disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
Bennett, R. E., DeVos, S. L., Dujardin, S., Corjuc, B., Gor, R., Gonzalez, J., et al. (2017). Enhanced tau aggregation in the presence of amyloid β. Am. J. Pathol. 187, 1601–1612. doi: 10.1016/j.ajpath.2017.03.011
Bilousova, T., Miller, C. A., Poon, W. W., Vinters, H. V., Corrada, M., Kawas, C., et al. (2016). Synaptic Amyloid-β oligomers precede p-Tau and differentiate high pathology control cases. Am. J. Pathol. 186, 185–198. doi: 10.1016/j.ajpath.2015.09.018
Bjorklund, N. L., Reese, L. C., Sadagoparamanujam, V.-M., Ghirardi, V., Woltjer, R. L., and Taglialatela, G. (2012). Absence of amyloid β oligomers at the postsynapse and regulated synaptic Zn2+ in cognitively intact aged individuals with Alzheimer's disease neuropathology. Mol. Neurodegener. 7:23. doi: 10.1186/1750-1326-7-23
Blennow, K. (2017). A review of fluid biomarkers for alzheimer's disease: moving from CSF to blood. Neurol. Ther. 6(Suppl. 1), 15–24. doi: 10.1007/s40120-017-0073-9
Blennow, K., de Leon, M. J., and Zetterberg, H. (2006). Alzheimer's disease. Lancet. 368, 387–403. doi: 10.1016/S0140-6736(06)69113-7
Bomfim, T. R., Forny-Germano, L., Sathler, L. B., Brito-Moreira, J., Houzel, J.-C., Decker, H., et al. (2012). An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer's disease–associated Aβ oligomers. J. Clin. Invest. 122, 1339–1353. doi: 10.1172/JCI57256
Bonda, D., Mailankot, M., Stone, J., Garrett, M. S. M., Castellani, R. J., Siedlak, S. L., et al. (2010). Indoleamine 2,3-dioxygenase and 3-hydroxykynurenine modifications are found in the neuropathology of Alzheimer's disease. Redox Rep. 15, 161–168. doi: 10.1179/174329210X12650506623645
Brinton, R. D. (2009). Estrogen-induced plasticity from cells to circuits: predictions for cognitive function. Trends Pharmacol. Sci. 30, 212–222. doi: 10.1016/j.tips.2008.12.006
Brinton, R. D., Yao, J., Yin, F., Mack, W. J., and Cadenas, E. (2015). Perimenopause as a neurological transition state. Nat. Rev. Endocrinol. 11, 393–405. doi: 10.1038/nrendo.2015.82
Broussard, J. I., Yang, K., Levine, A. T., Tsetsenis, T., Jenson, D., Cao, F., et al. (2016). Dopamine regulates aversive contextual learning and associated in vivo synaptic plasticity in the hippocampus. Cell Rep. 14, 1930–1939. doi: 10.1016/j.celrep.2016.01.070
Browne, T. C., McQuillan, K., McManus, R. M., O'Reilly, J.-A., Mills, K. H. G., and Lynch, M. A. (2013). IFN- production by amyloid -specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of alzheimer's disease. J. Immunol. 190, 2241–2251. doi: 10.4049/jimmunol.1200947
Buchhave, P. (2012). Cerebrospinal fluid levels of β-Amyloid 1-42, but Not of Tau, are fully changed already 5 to 10 years before the onset of alzheimer dementia. Arch. Gen. Psychiatry 69, 98. doi: 10.1001/archgenpsychiatry.2011.155
Burns, J. M., Galvin, J. E., Roe, C. M., Morris, J. C., and McKeel, D. W. (2005). The pathology of the substantia nigra in Alzheimer disease with extrapyramidal signs. Neurology 64, 1397–1403. doi: 10.1212/01.WNL.0000158423.05224.7F
Calsolaro, V., and Edison, P. (2016). Neuroinflammation in Alzheimer's disease: current evidence and future directions. Alzheimer's Dement. 12, 719–732. doi: 10.1016/j.jalz.2016.02.010
Carrieri, C., Cimatti, L., Biagioli, M., Beugnet, A., Zucchelli, S., Fedele, S., et al. (2017). Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Nat. Nanotechnol. 481, 454–457. doi: 10.1126/scitranslmed.aag0481
Castellani, R. J., and Perry, G. (2012). Pathogenesis and disease-modifying therapy in Alzheimer's disease: the flat line of progress. Arch. Med. Res. 43, 694–698. doi: 10.1016/j.arcmed.2012.09.009
Chase, T. N., Foster, N. L., Fedio, P., Brooks, R., Mansi, L., and Di Chiro, G. (1984). Regional cortical dysfunction in Alzheimer's disease as determined by positron emission tomography. Ann. Neurol. 15(Suppl.), S170–S174.
Chatterjee, S., Peters, S. A. E., Woodward, M., Arango, S. M., Batty, G. D., Beckett, N., et al. (2016). Type 2diabetes as a risk factor for dementia in women compared with men: a pooled analysis of 2.3 million people comprising more than 100,000 cases of dementia. Diabetes Care 39, 300–307. doi: 10.2337/dc15-1588
Chen, Y., Zhang, J., Zhang, B., and Gong, C. X. (2016). Targeting insulin signaling for the treatment of Alzheimer's disease. Curr. Top. Med. Chem. 16, 485–492. doi: 10.2174/1568026615666150813142423
Christensen, A., and Pike, C. J. (2015). Menopause, obesity and inflammation: interactive risk factors for Alzheimer's disease. Front. Aging Neurosci. 7:130. doi: 10.3389/fnagi.2015.00130
Clarke, J. R., Lyra E Silva, N. M., Figueiredo, C. P., Frozza, R. L., Ledo, J. H., Beckman, D., et al. (2015). Alzheimer-associated Aβ oligomers impact the central nervous system to induce peripheral metabolic deregulation. EMBO Mol. Med. 7, 190–210. doi: 10.15252/emmm.201404183
Costa-Mattioli, M., Gobert, D., Stern, E., Gamache, K., Colina, R., Cuello, C., et al. (2007). eIF2α phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell 129, 195–206. doi: 10.1016/j.cell.2007.01.050
Craft, S. (2012). Alzheimer disease: insulin resistance and AD—extending the translational path. Nat. Rev. Neurol. 8, 360–362. doi: 10.1038/nrneurol.2012.112
Craft, S., Zallen, G., and Baker, L. D. (1992). Glucose and memory in mild senile dementia of the alzheimer type. J. Clin. Exp. Neuropsychol. 14, 253–267. doi: 10.1080/01688639208402827
Crane, P. K., Walker, R., Hubbard, R. A., Li, G., Nathan, D. M., Zheng, H., et al. (2013). Glucose levels and risk of dementia. N. Engl. J. Med. 369, 540–548. doi: 10.1056/NEJMoa1215740
Csernansky, J. G., Dong, H., Fagan, A. M., Wang, L., Xiong, C., Holtzman, D. M., et al. (2006). Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am. J. Psychiatry 163, 2164–2169. doi: 10.1176/ajp.2006.163.12.2164
Cummings, J. L., Morstorf, T., and Zhong, K. (2014). Alzheimer's disease drug-development pipeline: few candidates, frequent failures. Alzheimers. Res. Ther. 6, 37. doi: 10.1186/alzrt269
Czirr, E., and Wyss-Coray, T. (2012). The immunology of neurodegeneration. J. Clin. Invest. 122, 1156–1163. doi: 10.1172/JCI58656
Decker, H., Jürgensen, S., Adrover, M. F., Brito-Moreira, J., Bomfim, T. R., Klein, W. L., et al. (2010a). N-Methyl-d-aspartate receptors are required for synaptic targeting of Alzheimer's toxic amyloid-β peptide oligomers. J. Neurochem. 115, 1520–1529. doi: 10.1111/j.1471-4159.2010.07058.x
Decker, H., Lo, K. Y., Unger, S. M., Ferreira, S. T., and Silverman, M. A. (2010b). Amyloid- peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3 in primary cultured hippocampal neurons. J. Neurosci. 30, 9166–9171. doi: 10.1523/JNEUROSCI.1074-10.2010
De Felice, F. G. (2013). Alzheimer' s disease and insulin resistance : translating basic science into clinical applications. J. Clin. Invest. 123, 531–539. doi: 10.1172/JCI64595
De Felice, F. G., and Ferreira, S. T. (2014). Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to alzheimer disease. Diabetes 63, 2262–2272. doi: 10.2337/db13-1954
De Felice, F. G., and Lourenco, M. V. (2015). Brain metabolic stress and neuroinflammation at the basis of cognitive impairment in Alzheimer's disease. Front. Aging Neurosci. 7:94. doi: 10.3389/fnagi.2015.00094
De Felice, F. G., and Munoz, D. P. (2016). Opportunities and challenges in developing relevant animal models for Alzheimer's disease. Ageing Res. Rev. 26, 112–114. doi: 10.1016/j.arr.2016.01.006
De Felice, F. G., Velasco, P. T., Lambert, M. P., Viola, K., Fernandez, S. J., Ferreira, S. T., et al. (2007). Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 282, 11590–11601. doi: 10.1074/jbc.M607483200
De Felice, F. G., Vieira, M. N. N., Bomfim, T. R., Decker, H., Velasco, P. T., Lambert, M. P., et al. (2009). Protection of synapses against Alzheimer's-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. U. S. A. 106, 1971–1976. doi: 10.1073/pnas.0809158106
De Felice, F. G., Wu, D., Lambert, M. P., Fernandez, S. J., Velasco, P. T., Lacor, P. N., et al. (2008). Alzheimer's disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol. Aging 29, 1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029
de la Monte, S. M. (2009). Insulin resistance and Alzheimer's disease. BMB Rep. 42, 475–481. doi: 10.5483/BMBRep.2009.42.8.475
Denis, R. G., Arruda, A. P., Romanatto, T., Milanski, M., Coope, A., Solon, C., et al. (2010). TNF-α transiently induces endoplasmic reticulum stress and an incomplete unfolded protein response in the hypothalamus. Neuroscience 170, 1035–1044. doi: 10.1016/j.neuroscience.2010.08.013
Devi, L., and Ohno, M. (2010). Phospho-eIF2α level is important for determining abilities of BACE1 reduction to rescue cholinergic neurodegeneration and memory defects in 5XFAD mice. PLoS ONE 5:e12974. doi: 10.1371/journal.pone.0012974
Devi, L., and Ohno, M. (2013). Deletion of the eIF2α kinase GCN2 fails to rescue the memory decline associated with alzheimer's disease. PLoS ONE 8:e77335. doi: 10.1371/journal.pone.0077335
Devi, L., and Ohno, M. (2014). PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer's disease. Neurobiol. Aging 35, 2272–2281. doi: 10.1016/j.neurobiolaging.2014.04.031
Dufey, E., Sepúlveda, D., Rojas-Rivera, D., and Hetz, C. (2014). Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 1. An overview. Am. J. Physiol. Physiol. 307, C582–C594. doi: 10.1152/ajpcell.00258.2014
Dubois, B., Hampel, H., Feldman, H. H., Scheltens, P., Aisen, P., Andrieu, S., et al. (2016). Preclinical Alzheimer's disease: definition, natural history, and diagnostic criteria. Alzheimers Dement. 12, 292–323. doi: 10.1016/j.jalz.2016.02.002
Duncan, M. J., Smith, J. T., Franklin, K. M., Beckett, T. L., Murphy, M. P., St. Clair, D. K., et al. (2012). Effects of aging and genotype on circadian rhythms, sleep, and clock gene expression in APPxPS1 knock-in mice, a model for Alzheimer's disease. Exp. Neurol. 236, 249–258. doi: 10.1016/j.expneurol.2012.05.011
Duran-Aniotz, C., Cornejo, V. H., Espinoza, S., Ardiles, Á. O., Medinas, D. B., Salazar, C., et al. (2017). IRE1 signaling exacerbates Alzheimer's disease pathogenesis. Acta Neuropathol. 134, 489–506. doi: 10.1007/s00401-017-1694-x
Fagan, A. M., Xiong, C., Jasielec, M. S., Bateman, R. J., Goate, A. M., Benzinger, T. L. S., et al. (2014). Longitudinal change in CSF biomarkers in autosomal-dominant alzheimer's disease. Sci. Transl. Med. 6:226ra30. doi: 10.1126/scitranslmed.3007901
Fá, M., Puzzo, D., Piacentini, R., Staniszewski, A., Zhang, H., Baltrons, M. A., et al. (2016). Extracellular tau oligomers produce an immediate impairment of LTP and memory. Sci. Rep. 6:19393. doi: 10.1038/srep19393
Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E. A., Frosch, M. P., et al. (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 4162–4167. doi: 10.1073/pnas.0230450100
Ferreira, S. T., Clarke, J. R., Bomfim, T. R., and De Felice, F. G. (2014). Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer's disease. Alzheimer's Dement. 10, S76–S83. doi: 10.1016/j.jalz.2013.12.010
Ferreira, S. T., and Klein, W. L. (2011). The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer's disease. Neurobiol. Learn. Mem. 96, 529–543. doi: 10.1016/j.nlm.2011.08.003
Ferreira, S. T., Lourenco, M. V., Oliveira, M. M., and De Felice, F. G. (2015). Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer's disease. Front. Cell. Neurosci. 9:191. doi: 10.3389/fncel.2015.00191
Finder, V. H., and Glockshuber, R. (2007). Amyloid-beta aggregation. Neurodegener. Dis. 4, 13–27. doi: 10.1159/000100355
Forny-Germano, L., Lyra e Silva, N. M., Batista, A. F., Brito-Moreira, J., Gralle, M., Boehnke, S. E., et al. (2014). Alzheimer's disease-like pathology induced by amyloid- oligomers in nonhuman primates. J. Neurosci. 34, 13629–13643. doi: 10.1523/JNEUROSCI.1353-14.2014
Freeman, O. J., and Mallucci, G. R. (2016). The UPR and synaptic dysfunction in neurodegeneration. Brain Res. 1648, 530–537. doi: 10.1016/j.brainres.2016.03.029
Fu, H., Rodriguez, G. A., Herman, M., Emrani, S., Nahmani, E., Barrett, G., et al. (2017). Tau pathology induces excitatory neuron loss, grid cell dysfunction, and spatial memory deficits reminiscent of early alzheimer's disease. Neuron 93, 533.e5–541.e5. doi: 10.1016/j.neuron.2016.12.023
Geerlings, M. I., Den Heijer, T., Koudstaal, P. J., Hofman, A., and Breteler, M. M. B. (2008). History of depression, depressive symptoms, and medial temporal lobe atrophy and the risk of Alzheimer disease. Neurology 70, 1258–1264. doi: 10.1212/01.wnl.0000308937.30473.d1
Georganopoulou, D. G., Chang, L., Nam, J.-M., Thaxton, C. S., Mufson, E. J., Klein, W. L., et al. (2005). From the cover: nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer's disease. Proc. Natl. Acad. Sci. U. S. A. 102, 2273–2276. doi: 10.1073/pnas.0409336102
Gibb, W. R., Mountjoy, C. Q., Mann, D. M., and Lees, A. J. (1989). The substantia nigra and ventral tegmental area in Alzheimer's disease and Down's syndrome. J. Neurol. Neurosurg. Psychiatry 52, 193–200.
Green, R. C., Cupples, L. A., Kurz, A., Auerbach, S., Go, R., Sadovnick, D., et al. (2003). Depression as a risk factor for Alzheimer disease: the MIRAGE Study. Arch. Neurol. 60, 753–759. doi: 10.1001/archneur.60.5.753
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer's Disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Guillot-Sestier, M. V., Doty, K. R., Gate, D., Rodriguez, J., Leung, B. P., Rezai-Zadeh, K., et al. (2015). Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron. 85, 534–548. doi: 10.1016/j.neuron.2014.12.068
Gulaj, E., Pawlak, K., Bien, B., and Pawlak, D. (2010). Kynurenine and its metabolites in Alzheimer's disease patients. Adv. Med. Sci. 55, 204–211. doi: 10.2478/v10039-010-0023-6
Haes, A. J., Chang, L., Klein, W. L., and Van Duyne, R. P. (2005). Detection of a biomarker for Alzheimer's disease from synthetic and clinical samples using a nanoscale optical biosensor. J. Am. Chem. Soc. 127, 2264–2271. doi: 10.1021/ja044087q
Hardy, J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hendrix, J. A., Bateman, R. J., Brashear, H. R., Duggan, C., Carrillo, M. C., Bain, L. J., et al. (2016). Challenges, solutions, and recommendations for Alzheimer's disease combination therapy. Alzheimer's Dement. 12, 623–630. doi: 10.1016/j.jalz.2016.02.007
Heneka, M. T., Carson, M. J., Khoury, J., El, Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015a). Neuroinflammation in Alzheimer's disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Heneka, M. T., and O'Banion, M. K. (2007). Inflammatory processes in Alzheimer's disease. J. Neuroimmunol. 184, 69–91. doi: 10.1016/j.jneuroim.2006.11.017
Heneka, M. T., Golenbock, D. T., and Latz, E. (2015b). Innate immunity in Alzheimer's disease. Nat. Immunol. 16, 229–236. doi: 10.1038/ni.3102
Herskovits, A. Z., Locascio, J. J., Peskind, E. R., Li, G., and Hyman, B. T. (2013). A luminex assay detects amyloid β oligomers in alzheimer's disease cerebrospinal fluid. PLoS ONE 8:e67898. doi: 10.1371/journal.pone.0067898
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
Hetz, C., and Saxena, S. (2017). ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 13, 477–491. doi: 10.1038/nrneurol.2017.99
Hoozemans, J. J. M., van Haastert, E. S., Nijholt, D. A. T., Rozemuller, A. J. M., Eikelenboom, P., and Scheper, W. (2009). The unfolded protein response is activated in pretangle neurons in Alzheimer's disease hippocampus. Am. J. Pathol. 174, 1241–1251. doi: 10.2353/ajpath.2009.080814
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016a). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hong, S., Dissing-Olesen, L., and Stevens, B. (2016b). New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 36, 128–134. doi: 10.1016/j.conb.2015.12.004
Hotamisligil, G. S. (2006). Inflammation and metabolic disorders. Nature 444, 860–867. doi: 10.1038/nature05485
Hotamisligil, G. S. (2017). Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185. doi: 10.1038/nature21363
Ishii, M., and Iadecola, C. (2015). Metabolic and non-cognitive manifestations of Alzheimers disease: the hypothalamus as both culprit and target of pathology. Cell Metab. 22, 761–776. doi: 10.1016/j.cmet.2015.08.016
Ittner, L. M., and Götz, J. (2011). Amyloid-β and tau — a toxic pas de deux in Alzheimer's disease. Nat. Rev. Neurosci. 12, 65–72. doi: 10.1038/nrn2967
Iwata, N., Tsubuki, S., Takaki, Y., Shirotani, K., Lu, B., Gerard, N. P., et al. (2001). Metabolic regulation of brain Abeta by neprilysin. Science 292, 1550–1552. doi: 10.1126/science.1059946
Janson, J., Laedtke, T., Parisi, J. E., O'Brien, P., Petersen, R. C., and Butler, P. C. (2004). Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 53, 474–481. doi: 10.2337/diabetes.53.2.474
Jin, M., Shepardson, N., Yang, T., Chen, G., Walsh, D., and Selkoe, D. J. (2011). Soluble amyloid -protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 5819–5824. doi: 10.1073/pnas.1017033108
Jürgensen, S., Antonio, L. L., Mussi, G. E. A., Brito-Moreira, J., Bomfim, T. R., De Felice, F. G., et al. (2011). Activation of D1/D5 dopamine receptors protects neurons from synapse dysfunction induced by amyloid-β oligomers. J. Biol. Chem. 286, 3270–3276. doi: 10.1074/jbc.M110.177790
Ju, Y.-E. S., Lucey, B. P., and Holtzman, D. M. (2014). Sleep and Alzheimer disease pathology–a bidirectional relationship. Nat. Rev. Neurol. 10, 115–119. doi: 10.1038/nrneurol.2013.269
Karran, E., Mercken, M., Strooper, B., and De (2011). The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 10, 698–712. doi: 10.1038/nrd3505
Kincheski, G. C., Valentim, I. S., Clarke, J. R., Cozachenco, D., Castelo-Branco, M. T. L., Ramos-Lobo, A. M., et al. (2017). Chronic sleep restriction promotes brain inflammation and synapse loss, and potentiates memory impairment induced by amyloid-β oligomers in mice. Brain Behav. Immun. 64, 140–151. doi: 10.1016/j.bbi.2017.04.007
Koffie, R. M., Meyer-Luehmann, M., Hashimoto, T., Adams, K. W., Mielke, M. L., Garcia-Alloza, M., et al. (2009). Oligomeric amyloid associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. U.S.A. 106, 4012–4017. doi: 10.1073/pnas.0811698106
Lanctôt, K. L., Amatniek, J., Ancoli-Israel, S., Arnold, S. E., Ballard, C., Cohen-Mansfield, J., et al. (2017). Neuropsychiatric signs and symptoms of Alzheimer's disease: new treatment paradigms. Alzheimer's Dement. Transl. Res. Clin. Interv. 3, 440–449. doi: 10.1016/j.trci.2017.07.001
Ledo, J. H., Azevedo, E. P., Beckman, D., Ribeiro, F. C., Santos, L. E., Razolli, D. S., et al. (2016). Cross talk between brain innate immunity and serotonin signaling underlies depressive-like behavior induced by Alzheimer's amyloid- oligomers in mice. J. Neurosci. 36, 12106–12116. doi: 10.1523/JNEUROSCI.1269-16.2016
Ledo, J. H., Azevedo, E. P., Clarke, J. R., Ribeiro, F. C., Figueiredo, C. P., Foguel, D., et al. (2013). Amyloid-β oligomers link depressive-like behavior and cognitive deficits in mice. Mol. Psychiatry 18, 1053–1054. doi: 10.1038/mp.2012.168
Lee, C. Y. D., and Landreth, G. E. (2010). The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 117, 949–960. doi: 10.1007/s00702-010-0433-4
Lee, S., Fernandez, E. J., and Good, T. A. (2007). Role of aggregation conditions in structure, stability, and toxicity of intermediates in the Abeta fibril formation pathway. Protein Sci. 16, 723–732. doi: 10.1110/ps.062514807
Leissring, M. A., Farris, W., Chang, A. Y., Walsh, D. M., Wu, X., Sun, X., et al. (2003). Enhanced proteolysis of β-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 40, 1087–1093. doi: 10.1016/S0896-6273(03)00787-6
Lester-Coll, N., Rivera, E. J., Soscia, S. J., Doiron, K., Wands, J. R., and de la Monte, S. M. (2006). Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer's disease. J. Alzheimers. Dis. 9, 13–33. doi: 10.3233/JAD-2006-9102
Lim, A. S. P., Ellison, B. A., Wang, J. L., Yu, L., Schneider, J. A., Buchman, A. S., et al. (2014). Sleep is related to neuron numbers in the ventrolateral preoptic/intermediate nucleus in older adults with and without Alzheimer's disease. Brain 137, 2847–2861. doi: 10.1093/brain/awu222
Liu, F., Day, M., Muñiz, L. C., Bitran, D., Arias, R., Revilla-Sanchez, R., et al. (2008). Activation of estrogen receptor-β regulates hippocampal synaptic plasticity and improves memory. Nat. Neurosci. 11, 334–343. doi: 10.1038/nn2057
Lourenco, M. V., Clarke, J. R., Frozza, R. L., Bomfim, T. R., Forny-Germano, L., Batista, A. F., et al. (2013). TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer's β-amyloid oligomers in mice and monkeys. Cell Metab. 18, 831–843. doi: 10.1016/j.cmet.2013.11.002
Lourenco, M. V., Ferreira, S. T., and De Felice, F. G. (2015). Neuronal stress signaling and eIF2α phosphorylation as molecular links between Alzheimer's disease and diabetes. Prog. Neurobiol. 129, 37–57. doi: 10.1016/j.pneurobio.2015.03.003
Lyketsos, C. G., and Olin, J. (2002). Depression in Alzheimer's disease: overview and treatment. Biol. Psychiatry 52, 243–252. doi: 10.1016/S0006-3223(02)01348-3
Ma, Q.-L., Yang, F., Rosario, E. R., Ubeda, O. J., Beech, W., Gant, D. J., et al. (2009). Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-jun n-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J. Neurosci. 9, 9078–9089. doi: 10.1523/JNEUROSCI.1071-09.2009
Ma, T., Trinh, M. A., Wexler, A. J., Bourbon, C., Gatti, E., Pierre, P., et al. (2013). Suppression of eIF2α kinases alleviates Alzheimer's disease–related plasticity and memory deficits. Nat. Neurosci. 16, 1299–1305. doi: 10.1038/nn.3486
Mandrekar-Colucci, S., Karlo, J. C., and Landreth, G. E. (2012). Mechanisms underlying the rapid peroxisome proliferator-activated receptor- -mediated amyloid clearance and reversal of cognitive deficits in a murine model of alzheimer's disease. J. Neurosci. 32, 10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012
Masliah, E., Ellisman, M., Carragher, B., Mallory, M., Young, S., Hansen, L., et al. (1992). Three-dimensional analysis of the relationship between synaptic pathology and neuropil threads in Alzheimer disease. J. Neuropathol. Exp. Neurol. 51, 404–414. doi: 10.1097/00005072-199207000-00003
Masters, M. C., Morris, J. C., and Roe, C. M. (2015). “Noncognitive” symptoms of early Alzheimer disease : a longitudinal analysis. Neurology 84, 617–622. doi: 10.1212/WNL.0000000000001238
Matsuzaki, T., Sasaki, K., Tanizaki, Y., Hata, J., Fujimi, K., Matsui, Y., et al. (2010). Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology 75, 764–770. doi: 10.1212/WNL.0b013e3181eee25f
Mattson, M. P. (2010). ER calcium and Alzheimer's disease: in a state of flux. Sci. Signal. 3:pe10. doi: 10.1126/scisignal.3114pe10
McNamara, C. G., Tejero-Cantero, Á., Trouche, S., Campo-Urriza, N., and Dupret, D. (2014). Dopaminergic neurons promote hippocampal reactivation and spatial memory persistence. Nat. Neurosci. 17, 1658–1660. doi: 10.1038/nn.3843
Medeiros, R., Baglietto-Vargas, D., and Laferla, F. M. (2011). The role of Tau in Alzheimer's disease and related disorders. CNS Neurosci. Ther. 17, 514–524. doi: 10.1111/j.1755-5949.2010.00177.x
Milanski, M., Degasperi, G., Coope, A., Morari, J., Denis, R., Cintra, D. E., et al. (2009). Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J. Neurosci. 29, 359–370. doi: 10.1523/JNEUROSCI.2760-08.2009
Miñano-Molina, A. J., España, J., Martín, E., Barneda-Zahonero, B., Fadó, R., Solé, M., et al. (2011). Soluble oligomers of amyloid-β peptide disrupt membrane trafficking of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor contributing to early synapse dysfunction. J. Biol. Chem. 286, 27311–27321. doi: 10.1074/jbc.M111.227504
Moloney, A. M., Griffin, R. J., Timmons, S., O'Connor, R., Ravid, R., and O'Neill, C. (2010). Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer's disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 31, 224–243. doi: 10.1016/j.neurobiolaging.2008.04.002
Morales, I., Guzmán-MartÃ-nez, L., Cerda-Troncoso, C., FarÃ-as, G. A., and Maccioni, R. B. (2014). Neuroinflammation in the pathogenesis of Alzheimer's disease. A rational framework for the search of novel therapeutic approaches. Front. Cell. Neurosci. 8:112. doi: 10.3389/fncel.2014.00112
Morris, M., Maeda, S., Vossel, K., and Mucke, L. (2011). The many faces of Tau. Neuron 70, 410–426. doi: 10.1016/j.neuron.2011.04.009
Mosconi, L., Berti, V., Guyara-Quinn, C., McHugh, P., Petrongolo, G., Osorio, R. S., et al. (2017a). Perimenopause and emergence of an Alzheimer's bioenergetic phenotype in brain and periphery. PLoS ONE 12:e0185926. doi: 10.1371/journal.pone.0185926
Mosconi, L., Berti, V., Quinn, C., McHugh, P., Petrongolo, G., Varsavsky, I., et al. (2017b). Sex differences in Alzheimer risk. Neurology 89, 1382–1390. doi: 10.1212/WNL.0000000000004425
Murakami, K., Tokuda, M., Suzuki, T., Irie, Y., Hanaki, M., Izuo, N., et al. (2016). Monoclonal antibody with conformational specificity for a toxic conformer of amyloid β42 and its application toward the Alzheimer's disease diagnosis. Sci. Rep. 6:29038. doi: 10.1038/srep29038
Musiek, E. S., and Holtzman, D. M. (2015). Three dimensions of the amyloid hypothesis: time, space and “wingmen.” Nat. Neurosci. 18, 800–806. doi: 10.1038/nn.4018
Musiek, E. S., and Holtzman, D. M. (2016). Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 354, 1004–1008. doi: 10.1126/science.aah4968
Musiek, E. S., Xiong, D. D., and Holtzman, D. M. (2015). Sleep, circadian rhythms, and the pathogenesis of Alzheimer Disease. Exp. Mol. Med. 47, e148. doi: 10.1038/emm.2014.121
Nobili, A., Latagliata, E. C., Viscomi, M. T., Cavallucci, V., Cutuli, D., Giacovazzo, G., et al. (2017). Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer's disease. Nat. Commun. 8:14727. doi: 10.1038/ncomms14727
Ogomori, K., Kitamoto, T., Tateishi, J., Sato, Y., Suetsugu, M., and Abe, M. (1989). Beta-protein amyloid is widely distributed in the central nervous system of patients with Alzheimer's disease. Am. J. Pathol. 134, 243–251.
Ott, A., Stolk, R. P., Hofman, A., Van Harskamp, F., Grobbee, D. E., and Breteler, M. M. B. (1996). Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia 39, 1392–1397. doi: 10.1007/s001250050588
Ott, A., Stolk, R. P., van Harskamp, F., Pols, H. A. P., Hofman, A., and Breteler, M. M. B. (1999). Diabetes mellitus and the risk of dementia: the Rotterdam Study. Neurology 53, 1937–1937. doi: 10.1212/WNL.53.9.1937
Ownby, R. L., Crocco, E., Acevedo, A., John, V., and Loewenstein, D. (2006). Depression and risk for Alzheimer disease. Arch. Gen. Psychiatry 63, 530. doi: 10.1001/archpsyc.63.5.530
Perez-Nievas, B. G., Stein, T. D., Tai, H. C., Dols-Icardo, O., Scotton, T. C., Barroeta-Espar, I., et al. (2013). Dissecting phenotypic traits linked to human resilience to Alzheimer's pathology. Brain 136, 2510–2526. doi: 10.1093/brain/awt171
Perry, D., Sperling, R., Katz, R., Berry, D., Dilts, D., Hanna, D., et al. (2014). Building a roadmap for developing combination therapies for Alzheimer's disease. Expert Rev. Neurother. 15, 327–333. doi: 10.1586/14737175.2015.996551
Perry, G., Cash, A. D., and Smith, M. A. (2002). Alzheimer disease and oxidative stress. J. Biomed. Biotechnol. 23, 120–123. doi: 10.1155/S1110724302203010
Perry, V. H., Nicoll, J. A. R., and Holmes, C. (2010). Microglia in neurodegenerative disease. Nat. Rev. Neurol. 6, 193–201. doi: 10.1038/nrneurol.2010.17
Piacentini, R., Li Puma, D. D., Mainardi, M., Lazzarino, G., Tavazzi, B., Arancio, O., et al. (2017). Reduced gliotransmitter release from astrocytes mediates tau-induced synaptic dysfunction in cultured hippocampal neurons. Glia 65, 1302–1316. doi: 10.1002/glia.23163
Prince, M., Bryce, R., Albanese, E., Wimo, A., Ribeiro, W., and Ferri, C. P. (2013). The global prevalence of dementia: a systematic review and metaanalysis. Alzheimer's Dement. 9, 63–75. doi: 10.1016/j.jalz.2012.11.007
Prince, M., Comas-Herrera, A., Knapp, M., Guerchet, M., and Karagiannidou, M. (2016). World Alzheimer Report 2016 Improving Healthcare for People Living with Dementia. Coverage, Quality and Costs Now and in the Future. Available online at: https://www.alz.co.uk/research/world-report-2016
Prinz, P., Vitaliano, P. P., Vitiello, M. V., Bokan, J., Raskind, M., Peskind, E., et al. (1982). Sleep, EEG and mental function changes in senile dementia of the Alzheimer's type. Neurobiol. Aging 3, 361–370.
Puzzo, D., Piacentini, R., Fá, M., Gulisano, W., Li Puma, D. D., Staniszewski, A., et al. (2017). LTP and memory impairment caused by extracellular Aβ and tau oligomers is APP- dependent. Elife 6:e26991. doi: 10.7554/eLife.26991
Qiu, W. Q., Walsh, D. M., Ye, Z., Vekrellis, K., Zhang, J., Podlisny, M. B., et al. (1998). Insulin-degrading enzyme regulates extracellular levels of amyloid β- protein by degradation. J. Biol. Chem. 273, 32730–32738. doi: 10.1074/jbc.273.49.32730
Querfurth, H. W., and LaFerla, F. M. (2010). Alzheimer's disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Reilly, P., Winston, C. N., Baron, K. R., Trejo, M., Rockenstein, E. M., Akers, J. C., et al. (2017). Novel human neuronal tau model exhibiting neurofibrillary tangles and transcellular propagation. Neurobiol. Dis. 106, 222–234. doi: 10.1016/j.nbd.2017.06.005
Rivera, E. J., Goldin, A., Fulmer, N., Tavares, R., Wands, J. R., and de la Monte, S. M. (2005). Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer's disease: link to brain reductions in acetylcholine. J. Alzheimers Dis. 8, 247–268. doi: 10.3233/JAD-2005-8304
Romano, A., Pace, L., Tempesta, B., Lavecchia, A. M., Macheda, T., Bedse, G., et al. (2014). Depressive-like behavior is paired to monoaminergic alteration in a murine model of Alzheimer's disease. Int. J. Neuropsychopharmacol. 18, 1–12. doi: 10.1093/ijnp/pyu020
Roselli, F., Tirard, M., Lu, J., Hutzler, P., Lamberti, P., Livrea, P., et al. (2005). Soluble β-amyloid 1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J. Neurosci. 25, 11061–11070. doi: 10.1523/JNEUROSCI.3034-05.2005
Rossato, J. I., Bevilaqua, L. R. M., Izquierdo, I., Medina, J. H., and Cammarota, M. (2009). Dopamine controls persistence of long-term memory storage. Science 325, 1017–1020. doi: 10.1126/science.1172545
Rowan, M. J., Klyubin, I., Wang, Q., and Anwyl, R. (2005). Synaptic plasticity disruption by amyloid beta protein: modulation by potential Alzheimer's disease modifying therapies. Biochem. Soc. Trans. 33, 563–567. doi: 10.1042/BST0330563
Russo, S. J., and Nestler, E. J. (2013). The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 14, 609–625. doi: 10.1038/nrn3381
Salter, M. W., and Stevens, B. (2017). Microglia emerge as central players in brain disease. Nat. Med. 23, 1018–1027. doi: 10.1038/nm.4397
Santos, L. E., Beckman, D., and Ferreira, S. T. (2016). Microglial dysfunction connects depression and Alzheimer's disease. Brain. Behav. Immun. 55, 151–165. doi: 10.1016/j.bbi.2015.11.011
Scheltens, P., Blennow, K., Breteler, M. M. B., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer's disease. Lancet 388, 505–517. doi: 10.1016/S0140-6736(15)01124-1
Selkoe, D. J. (2002). Alzheimer's disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Selkoe, D. J. (2008). Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res. 192, 106–113. doi: 10.1007/978-3-540-76330-7_8
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 8, 1–14. doi: 10.15252/emmm.201606210
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., and Sabatini, B. L. (2007). Natural oligomers of the alzheimer amyloid- protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866-75. doi: 10.1523/JNEUROSCI.4970-06.2007
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Small, S. A., and Duff, K. (2008). Linking Aβ and Tau in late-onset Alzheimer's disease: a dual pathway hypothesis. Neuron 60, 534–542. doi: 10.1016/j.neuron.2008.11.007
Smith, M. A., Hirai, K., Hsiao, K., Pappolla, M. A., Harris, P. L., Siedlak, S. L., et al. (1998). Amyloid-beta deposition in Alzheimer transgenic mice is associated with oxidative stress. J. Neurochem. 70, 2212–2215. doi: 10.1046/j.1471-4159.1998.70052212.x
Snyder, E. M., Nong, Y., Almeida, C. G., Paul, S., Moran, T., Choi, E. Y., et al. (2005). Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 8, 1051–1058. doi: 10.1038/nn1503
Snyder, H. M., Asthana, S., Bain, L., Brinton, R., Craft, S., Dubal, D. B., et al. (2016). Sex biology contributions to vulnerability to Alzheimer's disease: a think tank convened by the Women's Alzheimer's Research Initiative. Alzheimer's Dement. 12, 1186–1196. doi: 10.1016/j.jalz.2016.08.004
Sperling, R. A., Aisen, P. S., Beckett, L. A., Bennett, D. A., Craft, S., Fagan, A. M., et al. (2011a). Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's Dement. 7, 280–292. doi: 10.1016/j.jalz.2011.03.003
Sperling, R. A., Jack, C. R., and Aisen, P. S. (2011b). Testing the right target and right drug at the right stage. Sci. Transl. Med. 3:111cm33. doi: 10.1126/scitranslmed.3002609
Spires-Jones, T. L., and Hyman, B. T. (2014). The intersection of amyloid beta and tau at synapses in Alzheimer's disease. Neuron 82, 756–771. doi: 10.1016/j.neuron.2014.05.004
Standaert, D. G., Lee, V. M., Greenberg, B. D., Lowery, D. E., and Trojanowski, J. Q. (1991). Molecular features of hypothalamic plaques in Alzheimer's disease. Am. J. Pathol. 139, 681–691.
Stanley, M., Macauley, S. L., Caesar, E. E., Koscal, L. J., Moritz, W., Robinson, G. O., et al. (2016). The effects of peripheral and central high insulin on brain insulin signaling and amyloid-β in young and old APP/PS1 mice. J. Neurosci. 36, 11704–11715. doi: 10.1523/JNEUROSCI.2119-16.2016
Starkstein, S. E., and Mizrahi, R. (2006). Depression in Alzheimer's disease. Exp. Rev. Neurother. 6, 887–895. doi: 10.1586/14737175.6.6.887
Steen, E., Terry, B. M., Rivera, E. J., Cannon, J. L., Neely, T. R., Tavares, R., et al. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease–is this type 3 diabetes? J. Alzheimers. Dis. 7, 63–80. doi: 10.3233/JAD-2005-7107
Stephenson, D., Perry, D., Bens, C., Bain, L. J., Berry, D., Krams, M., et al. (2014). Charting a path toward combination therapy for Alzheimer's disease. Exp. Rev. Neurother. 15, 1–7. doi: 10.1586/14737175.2015.1002173
Stevanovic, K., Yunus, A., Joly-Amado, A., Gordon, M., Morgan, D., Gulick, D., et al. (2017). Disruption of normal circadian clock function in a mouse model of tauopathy. Exp. Neurol. 294, 58–67. doi: 10.1016/j.expneurol.2017.04.015
Stine, W. B., Dahlgren, K. N., Krafft, G. A., and LaDu, M. J. (2003). In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J. Biol. Chem. 278, 11612–11622. doi: 10.1074/jbc.M210207200
Storga, D., Vrecko, K., Birkmayer, J. G. D., and Reibnegger, G. (1996). Monoaminergic neurotransmitters, their precursors and metabolites in brains of Alzheimer patients. Neurosci. Lett. 203, 29–32. doi: 10.1016/0304-3940(95)12256-7
Swardfager, W., Lanctt, K., Rothenburg, L., Wong, A., Cappell, J., and Herrmann, N. (2010). A meta-analysis of cytokines in Alzheimer's disease. Biol. Psychiatry 68, 930–941. doi: 10.1016/j.biopsych.2010.06.012
Takeda, S., Sato, N., Uchio-Yamada, K., Sawada, K., Kunieda, T., Takeuchi, D., et al. (2010). Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and A deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. U.S.A. 107, 7036–7041. doi: 10.1073/pnas.1000645107
Talbot, K., Wang, H. Y., Kazi, H., Han, L. Y., Bakshi, K. P., Stucky, A., et al. (2012). Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Invest. 122, 1316–1338. doi: 10.1172/JCI59903
Tarasoff-Conway, J. M., Carare, R. O., Osorio, R. S., Glodzik, L., Butler, T., Fieremans, E., et al. (2015). Clearance systems in the brain—implications for Alzheimer disease. Nat. Rev. Neurol. 11, 457–470. doi: 10.1038/nrneurol.2015.119
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., et al. (1991). Physical basis of cognitive alterations in alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580.
Thaler, J. P., Yi, C. X., Schur, E. A., Guyenet, S. J., Hwang, B. H., Dietrich, M. O., et al. (2012). Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Invest. 122, 153–162. doi: 10.1172/JCI59660
Valdearcos, M., Xu, A. W., and Koliwad, S. K. (2015). Hypothalamic inflammation in the control of metabolic function. Annu. Rev. Physiol. 77, 131–160. doi: 10.1146/annurev-physiol-021014-071656
Vandal, M., White, P., Chevrier, G., Tremblay, C., St-Amour, I., Planel, E., et al. (2015). Age-dependent impairment of glucose tolerance in the 3xTg-AD mouse model of Alzheimer's disease. FASEB J. 29, 4273–4284. doi: 10.1096/fj.14-268482
van Maurik, I. S., Zwan, M. D., Tijms, B. M., Bouwman, F. H., Teunissen, C. E., Scheltens, P., et al. (2017). Interpreting biomarker results in individual patients with mild cognitive impairment in the Alzheimer's biomarkers in daily Practice (ABIDE) Project. JAMA Neurol. 74, 1481–1491. doi: 10.1001/jamaneurol.2017.2712
Villemagne, V. L., Burnham, S., Bourgeat, P., Brown, B., Ellis, K. A., Salvado, O., et al. (2013). Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 12, 357–367. doi: 10.1016/S1474-4422(13)70044-9
Viña, J., and Lloret, A. (2010). Why women have more Alzheimer's disease than men: gender and mitochondrial toxicity of amyloid-beta peptide. J. Alzheimers. Dis. 20(Suppl. 2), S527–S533. doi: 10.3233/JAD-2010-100501
Viola, K. L., and Klein, W. L. (2015). Amyloid β oligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 129, 183–206. doi: 10.1007/s00401-015-1386-3
Viola, K. L., Sbarboro, J., Sureka, R., De, M., Bicca, M. A., Wang, J., et al. (2014). Towards non-invasive diagnostic imaging of early-stage Alzheimer's disease. Nat. Nanotechnol. 10, 91–98. doi: 10.1038/nnano.2014.254
Walsh, D. M., and Selkoe, D. J. (2007). Aβ oligomers - A decade of discovery. J. Neurochem. 101, 1172–1184 doi: 10.1111/j.1471-4159.2006.04426.x
Wang, J., Tan, L., Wang, H.-F., Tan, C.-C., Meng, X.-F., Wang, C., et al. (2015). Anti-inflammatory drugs and risk of Alzheimer's disease: an updated systematic review and meta-analysis. J. Alzheimers Dis. 44, 385–396. doi: 10.3233/JAD-141506
Wang, W.-Y., Tan, M.-S., Yu, J.-T., and Tan, L. (2015). Role of pro-inflammatory cytokines released from microglia in Alzheimer's disease. Ann. Transl. Med. 3, 136. doi: 10.3978/j.issn.2305-5839.2015.03.49
White, H., Pieper, C., Schmader, K., and Fillenbaum, G. (1996). Weight change in Alzheimer's disease. J. Am. Geriatr. Soc. 44, 265–272. doi: 10.1111/j.1532-5415.1996.tb00912.x
Xia, W., Yang, T., Shankar, G., Smith, I. M., Shen, Y., Walsh, D. M., et al. (2009). A specific enzyme-linked immunosorbent assay for measuring β-amyloid protein oligomers in human plasma and brain tissue of patients with alzheimer disease. Arch. Neurol. 66, 190–199. doi: 10.1001/archneurol.2008.565
Yang, T., Li, S., Xu, H., Walsh, D. M., and Selkoe, D. J. (2017). Large soluble oligomers of amyloid β-protein from alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J. Neurosci. 37, 152–163. doi: 10.1523/JNEUROSCI.1698-16.2016
Yao, J., and Brinton, R. D. (2012). Estrogen regulation of mitochondrial bioenergetics. Adv. Pharmacol. 64, 327–371. doi: 10.1016/B978-0-12-394816-8.00010-6
Yirmiya, R., Rimmerman, N., and Reshef, R. (2015). Depression as a microglial disease. Trends Neurosci. 38, 637–658. doi: 10.1016/j.tins.2015.08.001
Zenaro, E., Pietronigro, E., Della Bianca, V., Piacentino, G., Marongiu, L., Budui, S., et al. (2015). Neutrophils promote Alzheimer's disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 21, 880–886. doi: 10.1038/nm.3913
Zhang, J., Ke, K. F., Liu, Z., Qiu, Y. H., and Peng, Y. P. (2013). Th17 Cell-mediated neuroinflammation is involved in neurodegeneration of Aβ1-42-Induced Alzheimer's disease model rats. PLoS ONE 8:e75786. doi: 10.1371/journal.pone.0075786
Zhang, X., Zhang, G., Zhang, H., Karin, M., Bai, H., and Cai, D. (2008). Hypothalamic β/NF-κB and ER stress link overnutrition to energy imbalance and obesity. Cell 135, 61–73. doi: 10.1016/j.cell.2008.07.043
Keywords: Alzheimer's disease, inflammation, metabolic derangements, memory defects, preclinical, therapy
Citation: Frozza RL, Lourenco MV and De Felice FG (2018) Challenges for Alzheimer's Disease Therapy: Insights from Novel Mechanisms Beyond Memory Defects. Front. Neurosci. 12:37. doi: 10.3389/fnins.2018.00037
Received: 27 November 2017; Accepted: 16 January 2018;
Published: 06 February 2018.
Edited by:
Athanasios Alexiou, Novel Global Community Educational Foundation (NGCEF), Hebersham, AustraliaReviewed by:
Marzia Perluigi, Sapienza Università di Roma, ItalyCheng-Xin Gong, Institute for Basic Research in Developmental Disabilities (IBR), United States
Copyright © 2018 Frozza, Lourenco and De Felice. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rudimar L. Frozza, rudimar.frozza@ioc.fiocruz.br
Fernanda G. De Felice, felice@bioqmed.ufrj.br