 Takafumi Hasegawa
Takafumi Hasegawa Shun Yoshida
Shun Yoshida
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci., 10 January 2018
Sec. Neurodegeneration
Volume 11 - 2017 | https://doi.org/10.3389/fnins.2017.00743
This article is part of the Research TopicBrain Protein Aging and Dementia ControlView all 23 articles
Parkinson's disease (PD) is the second most common devastating neurodegenerative disorder after Alzheimer's disease. The precise molecular and cellular basis underlying PD still remains uncertain; however, accumulating evidence suggests that neuronal cell death is caused by a combination of environmental and genetic factors. Over the previous two decades, more than 20 genes have been identified as the cause of and/or risk for PD. Because sporadic and familial forms of PD have many similarities in clinical and neuropathological features, common molecular pathways, such as aberrant mitochondrial and protein homeostasis, are likely to exist in both conditions. Of the various genes and proteins involved in PD, the versatile DnaJ/Hsp40 co-chaperones have attracted particular attention since several genes encoding this protein family have been successively identified as the cause of the familial forms of PD/Parkinsonism. In this review, we will introduce the current knowledge regarding the integratory and modulatory effect of DnaJ/Hsp40 in various cellular functions and argue how the failure of these proteins may initiate and/or facilitate of the disease.
Newly synthesized proteins are transported to the destination site where they exert distinct functions; however, some proteins fail to properly fold, and misfolded proteins tend to form aggregates that may be potentially harmful to cells. To combat these perpetual threats, cells have evolved dexterous quality control mechanisms that facilitate degradation by the ubiquitin-proteasome system and autophagy-lysosome pathway or refolding of misfolded proteins to normal tertiary structures by molecular chaperones (Oshima et al., 2016; Ciechanover and Kwon, 2017). The most well-known molecular chaperones are heat shock proteins (HSPs), which were named after the phenomenon of puffing in larval salivary gland chromosomes of Drosophila following heat exposure (Pauli et al., 1992). Heat shock and numerous noxious stimuli, including UV, oxidative stress, hypoxia, osmotic stress, and heavy metals, induce the expression of HSPs, which is considered as a fundamental biological defense mechanism (Piano et al., 2004). The family of HSPs are evolutionarily conserved proteins across species and are classified based on their molecular weights; different HSPs have distinct yet partially overlapping functions. For example, Hsp90, which is known to be one of the most abundantly expressed proteins (1–2% of total soluble proteins in an unstressed condition) in the cytosol, forms a complex with its client proteins, such as cell surface receptors, transcription factors, and protein kinases, to modulate their functions (Schopf et al., 2017). Thus, Hsp90 plays pivotal roles in various cellular processes, such as proliferation, differentiation, carcinogenesis, and neurodegeneration. Several Hsp90 inhibitors, including the geldanamycin and its analogs demonstrate anti-tumor effects in various cellular and animal models (Soga et al., 2013). The Hsp70 family is the eukaryotic homolog of the bacterial molecular chaperone DnaK and is widely expressed in various tissues and organs. The members of the Hsp70 family are characterized by their expression levels, activities, and subcellular distribution (Zuiderweg et al., 2017). Under physiological conditions, Hsp70 mainly resides in the cytoplasmic space, whereas in response to stress, it is upregulated and relocates from the cytosol to the nucleus and nucleoli. The heat shock cognate 70 (Hsc70) is, on the other hand, defined by its constitutive expression and cytoplasmic localization. Another Hsp70 member, BiP (also referred to as a 78-kDa of glucose-regulated protein: GRP78), expresses in the endoplasmic reticulum (ER) and acts as a chaperone for secreted or membrane proteins (Dudek et al., 2009). Hsp70 binds and shields the hydrophobic peptides of client proteins in an ATP-dependent manner, thus preventing protein aggregation and proper folding (Zuiderweg et al., 2017). Hsc70 chaperone mediates the lysosomal targeting of substrates with a KFERQ-like sequence (i.e., chaperone-mediated autophagy; Cai et al., 2015). Furthermore, Hsp70 is involved in the intracellular traffic of membranes, including the involvement in regulation of endocytosis and exocytosis mechanisms (Goldfarb et al., 2006).
The complex and multifaceted functions of Hsp70 members are achieved by the assistance of their Hsp40/DnaJ co-chaperones (Qiu et al., 2006; Gorenberg and Chandra, 2017). Hsp40 family proteins were originally identified as mammalian homologs of bacterial DnaJ proteins and are subdivided into three distinct groups, i.e., DnaJA, DnaJB, and DnaJC. Structurally, DnaJ proteins consist of 4 functional domains, namely, an evolutionarily conserved J domain with a stretch of 70 amino acid residues, a glycine/phenylalanine-rich domain (G/F-rich domain), a zinc finger domain that contains CXXCXGXG motifs, and a cysteine-rich region (Qiu et al., 2006). In DnaJA and DnaJB proteins, the J domain is always located at the N-terminus which is followed by the G/F-rich domain. The DnaJC family usually lacks the G/F domain and the cysteine-rich region, and the J domain may be located at any location in the entire sequence. Through the intermediary of the J domain, Hsp40 binds to Hsp70 and promotes its ATPase activity, thus generating the ADP-bound Hsp70, which stably interacts with client proteins (Figure 1; Greene et al., 1998). Both the G/F-rich domain and c-terminal region of Hsp40/DnaJ proteins are likely to be important in their substrate recognition (Perales-Calvo et al., 2010). Although the Hsp40/DnaJ families are ubiquitously expressed, widespread distribution is also present in the central and peripheral nervous systems (Gorenberg and Chandra, 2017). Furthermore, recent genetic and biological studies suggest that Hsp40/DnaJ family genes/proteins directly or cooperatively influence on the initiation of familial Parkinson's disease (PD) and other inherited forms of parkinsonism (Figure 2 and Table 1; Gorenberg and Chandra, 2017; Hasegawa et al., 2017; Puschmann, 2017). It remains obscure how the different mutated genes could result in the progressive loss of striatal dopaminergic innervation as well as the death of nigral neurons. Nevertheless, some relationships, involving the perturbation of cellular systems, now appear to be apparent. The affected systems include synaptic transmission, endosomal trafficking, protein-quality control, and/or mitochondrial systems. In this review, we will summarize the current understandings regarding the functional roles of Hsp40/DnaJ co-chaperones in the familial forms of PD/parkinsonism and discuss how these molecules may influence on the pathological consequence of this disease.
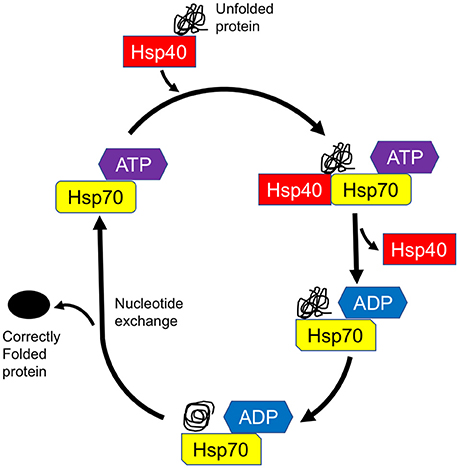
Figure 1. Model of chaperone-assisted protein folding by Hsp70-DnaJ/Hsp40 complex. DnaJ/Hsp40 transiently associates with unfolded protein (substrate) for delivery of the substrate to Hsp70. Through the intermediary of the J domain, Hsp40 binds to Hsp70 and promotes its ATPase activity, thus generating the ADP-bound Hsp70, which stably interacts with client protein. After nucleotide exchange, the substrate is released from Hsp70 and leaves the cycle as a correctly folded protein.
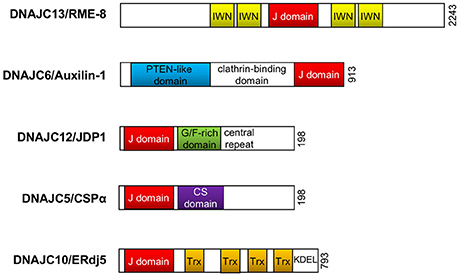
Figure 2. DnaJ/Hsp40 proteins implicated in Parkinson's disease. Schematic illustration of the functional domain organization of DnaJ proteins (DNAJC8/RME-8, DNAJC6/Auxilin-1, DNAJC12/JDP1, DNAJC5/CSPα, and DNAJC10/ERdj5) reviewed in this article. The illustration does not represent the actual molecular size. PTEN, phosphoinositide phosphatase; G/F, glycine/phenylalanine; CS, cysteine-string; Trx, thioredoxin; KDEL, KDEL (Lys-Asp-Glu-Leu) targeting sequence to the ER.
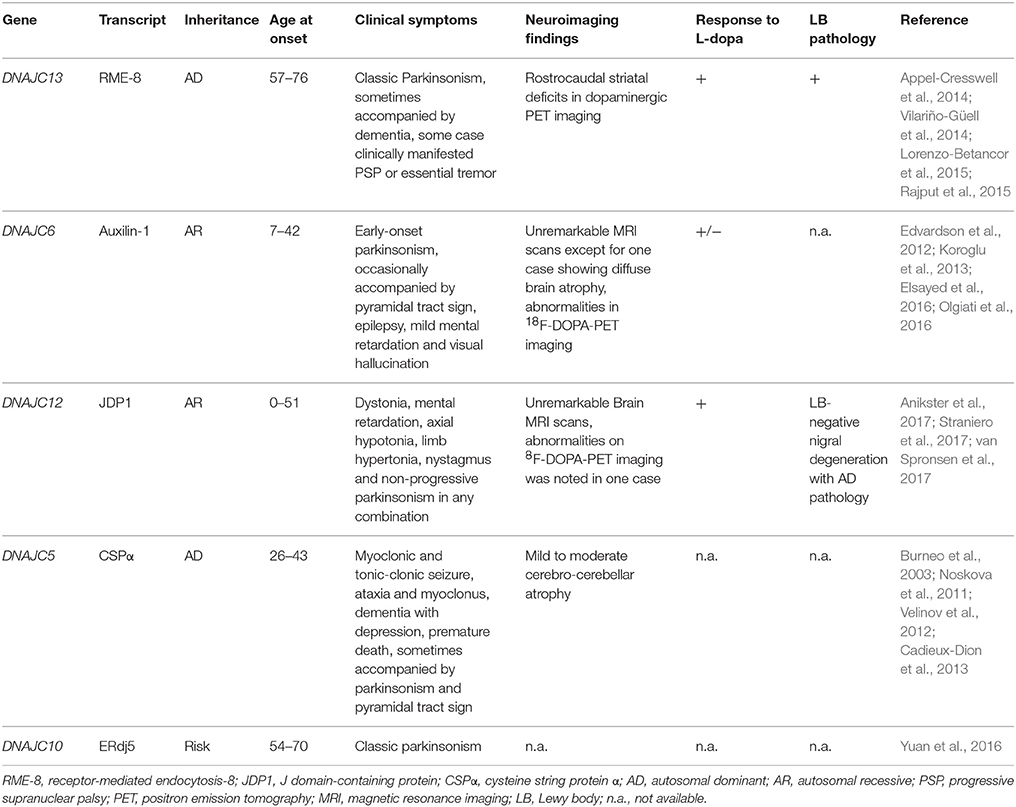
Table 1. Clinicopathological features of DnaJ/Hsp40-linked PD/Parkinsonism.
DNAJC13 is a human homolog of receptor-mediated endocytosis 8 (RME-8), which was originally identified in a genetic screening for mutants defective in the endocytic uptake of yolk protein in nematodes (Zhang et al., 2001). Recently, a p.N855S missense mutation of DNAJC13 was reported in a large Canadian Mennonite PD pedigree with Dutch-German-Russian ancestry (Appel-Cresswell et al., 2014; Vilariño-Güell et al., 2014). The pathogenicity of the N855S mutant is supported by segregation with disease, lack of healthy controls with mutation, and the high level of sequence conservation across species; however, genetic screening in a Caucasian series consisting of 1,938 patients with clinically diagnosed PD and 838 pathologically proven Lewy Body Disease failed to detect any coding variant in exon 24 containing Asn855 in DNAJC13 gene, indicating that mutations in this exon are not a common cause of PD or LBD among Caucasian populations (Lorenzo-Betancor et al., 2015). Subsequent genotyping in a Canadian cohort indicated that missense mutations, such as p.P336A, p.V722L, p.R1266Q, and P.T1895M, are only identified in affected members, and p.E1740Q, and p.L2170W may be associated with the risk variants for PD (Gustavsson et al., 2015). Clinically, DNAJC-linked PD (subsequently designated as PARK21) manifests as slowly progressive, late-onset, dopa-responsive typical parkinsonism with an autosomal dominant inheritance (Vilariño-Güell et al., 2014). Of note, the N855S mutation in DNAJC13 gene has also been observed in patients with essential tremor, a monosymptomatic disorder characterized exclusively by postural- or action-type tremor, suggesting heterogeneity in the clinical manifestation with this mutation (Rajput et al., 2015). Neuropathological examination in three N855S mutation carriers indicated a brainstem or transitional type of Lewy body pathology (Vilariño-Güell et al., 2014). DNAJC13/RME-8 is a large protein with a high molecular mass of 254 kDa; it is widely expressed and is abundant in nervous tissue. Structurally, it consists of four conserved IWN repeats, which are characterized by seven invariant residues, including isoleucine, tryptophan and asparagine, and a J domain in the center region (Fujibayashi et al., 2008). An in vitro pull-down assay demonstrated that the J domain of RME-8 binds to Hsc70 in the presence of ADP (Chang et al., 2004). Cell biological studies have demonstrated that DNAJC13 resides in the endosomal membrane and interacts with a family with sequence similarity 21 (FAM21) tail in the Wiskott–Aldrich syndrome protein and Scar homolog (WASH) complex and sorting nexin 1 (SNX1), thereby controlling the formation of the tubular structure on the endosome surface where retromer-mediated cargo transport occurs (Freeman et al., 2014). From a functional point of view, DNAJC13 plays important roles in the cell surface recycling and lysosomal degradation of transferrin and EGFR, as well as the retrograde transport of cation-independent mannose phosphate receptor (CI-MPR; Girard et al., 2005; Popoff et al., 2009; Shi et al., 2009; Figure 3). The over-expression of p.N855S mutant, but not wild-type, in cultured cells leads to aberrant retention of transferrin in endosomes (Vilariño-Güell et al., 2014), which indicates that a PD-linked DNAJC13 mutation may confer a toxic gain-of-function and hampers endosomal cargo trafficking. Intriguingly, immunofluorescence and co-immunoprecipitation studies demonstrate that vacuolar protein sorting 35 (VPS35), a vital element of retromer that is established as a causal gene for PARK17 PD, is co-localized with DNAJC13 in a primary cortical neuron culture obtained from mice (Vilariño-Güell et al., 2014). Further studies using appropriate animal models are required to better understand the precise molecular mechanisms by which DNAJC13 mutation leads to the dopaminergic degeneration with Lewy body pathology. In an independent study, Deng et al. identified loss-of-function mutations in TMEM230 gene in the same Canadian Mennonite group that Vilariño-Güell and his colleagues previously identified mutations in DNAJC13 (Deng et al., 2016). The TMEM230 transcript localizes to the secretory and recycling vesicle in the neuron and may be involved in synaptic vesicle trafficking and recycling. Another study has shown that TMEM230 is required for Rab8a-mediated transport of secretory vesicles and retromer-mediated cargo trafficking (Kim et al., 2017). Further discussion and reassessment are needed to understand this complex situation.
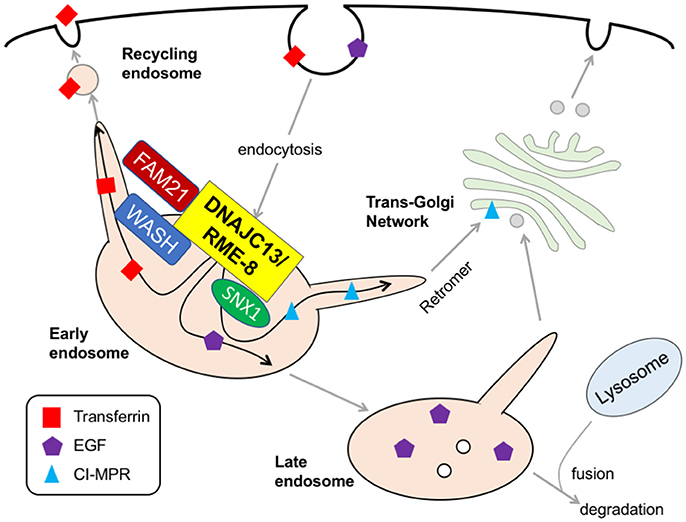
Figure 3. DNAJC13/RME-8. At the surface of early endosome, DNAJC13 interacts with FAM21 tail in the WASH complex and SNX1, thereby controlling the formation of tubular structure on endosome surface where a retromer-mediated cargo transport (e.g., CI-MPR) occurs. DNAJC13 also play important roles in the recycling and the degradation of transferrin and EGFR, respectively. RME-8, receptor-mediated endocytosis-8; FAM21, family with sequence similarity 21; WASH, Wiskott–Aldrich syndrome protein and Scar homolog; SNX1, sorting nexin 1; EGF, epidermal growth factor; CI-MPR, cation-independent mannose 6-phosphate receptor.
In 2012, deleterious (c.801-2 A>G) and truncating (p.Q734X) mutations in the DNAJC6 gene were discovered in two consanguineous families with Palestinian and Turkish origins, respectively (Edvardson et al., 2012; Koroglu et al., 2013). The clinical phenotypes of DNAJC6-linked familial parkinsonism (termed PARK19) are characterized by a juvenile-onset, progressive parkinsonism, which includes bradykinesia, muscle rigidity, resting tremor, hypomimia, and postural instability. In addition to extrapyramidal signs, four members of the Turkish family exhibited hypomentia, pyramidal tract signs and/or seizures (Koroglu et al., 2013). Patients in the Palestinian family had dopa-refractory parkinsonism, whereas affected members in the Turkish family showed a favorable response to levodopa replenishment therapy. Subsequent genetic studies identified 2 different homozygous mutations in the DNAJC6 gene, i.e., a missense mutation (p.R927G) and a putative splice site mutation (c.2223A-T; Olgiati et al., 2016). These families accounted for 2 (2.2%) of 92 probands (mean age at onset: 54.65; range 19–84) with autosomal recessive PD who underwent DNAJC6 genetic screening. These patients showed milder phenotype compared to the patients with truncating mutations probably due to some residual activity of DNAJC6. Another novel non-sense mutation (exon 16, p.Q789*) has also been reported in a consanguineous family with juvenile-onset PD (Elsayed et al., 2016). Of interest, a patient in this family manifested rigid-akinetic form of parkinsonism with visual hallucinations, which are frequently observed non-motor symptom in sporadic PD and dementia with Lewy bodies (Shoji et al., 2014). Because there is variation in the responsiveness of dopamine-replacement therapy, the term “parkinsonism” rather than PD would be suitable in DNAJC6-linked movement disorder. The DNAJC6 encodes the neuron-specific isoform of the co-chaperone auxilin-1, which has a crucial role in the detachment of the clathrin-coat after clathrin-mediated endocytosis (CME, Figure 4). Auxilin-1 is a 100 kDa protein and structurally consists of 3 distinct domains: the N-terminal phosphoinositide phosphatase (PTEN) like domain (residue 40–421), which is required for the recruitment to a clathrin-coated pit, the central clathrin-binding domain, and a C-terminal J domain, which enables its interaction with Hsc70 (Lee et al., 2006). The c.801-2 A>G homozygous splice-site mutation in DNAJC6 yields two misspliced abnormal transcripts that lack either a significant part of the J domain or the PTEN-like domain, which indicates that this pathological mutation results in the defect of functionally active auxilin-1 (Edvardson et al., 2012). The homozygous mutation p.Q734X deletes the 180 amino acid residues of Auxilin-1 at the C-terminus, which are considered important for supporting Hsc70 function in clathrin dissociation from coated vesicles (Koroglu et al., 2013). Moreover, an in silico structural analysis predicted that that the p.R927G missense mutation in the J domain reduces the positive charge on the protein surface, which would be involved in the molecular interaction of auxilin-1 with Hsc70 or other partners (Olgiati et al., 2016). Interestingly, common variants in cyclin-G-associated kinase (GAK), which encodes auxilin-2/DNAJC26, a ubiquitously expressed paralog of auxilin, are also considered a risk factor for idiopathic PD (Pankratz et al., 2009). Auxilin-null mice exhibited a high mortality rate in early life; however, they did not exhibit no obvious neuropathological alterations in the midbrain (Yim et al., 2010). These mice showed higher GAK levels in the brain; however, this compensation appeared to be insufficient for the lack of auxilin in clathrin uncoating since they exhibited an aberrant retention of clathrin-coated vesicles and empty clathrin cages at the synaptic terminals, which may be attributed to the parkinsonian phenotypes. Further investigation using genetically modified animal models is warranted to better understand the role of DNAJC6 in synaptic dopamine function.
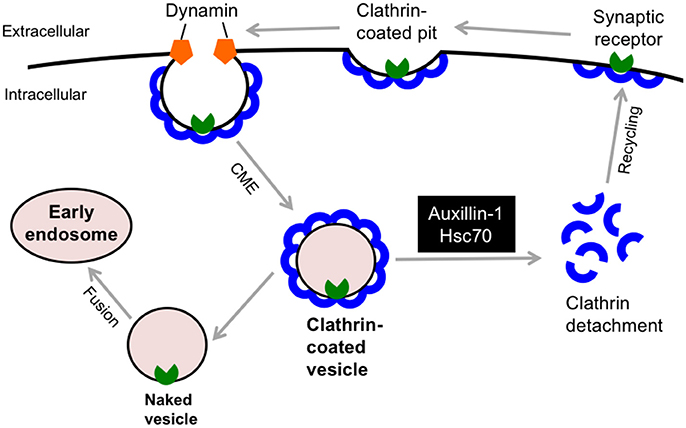
Figure 4. DNAJC6/Auxilin-1 regulates the clathrin-mediated endocytosis. DNAJC6/Auxilin-1 is highly expressed in nerve terminals and plays a crucial role in clathrin uncoating during synaptic receptor endocytosis. CME, clathrin-mediated endocytosis; Hsc70, heat shock cognate 70.
DNAJC12, a member of the Hsp70/Hsc70 co-chaperone proteins, has the J domain and a highly conserved C terminus; however, it lacks other canonical domains present in DnaJ family proteins. This protein is a mammalian ortholog of the J domain-containing protein (JDP) originally identified in Drosophila and is thus termed JDP1 (Hahn et al., 1999). In mammals, two transcript variants of DNAJC12/JDP1 have been identified: the primary isoform is 1.2 kb and encodes 198 amino acids, and the other isoform is 0.7 kb and is composed of 107 amino acids. Northern blot analysis showed that the major transcript of mouse JDP1 is ubiquitously expressed in various organs, including the brain (Hahn et al., 1999). Recently, biallelic mutations [del. 6943 (c.298-968_503-2603del) and p.R72P] of the DNAJC12 gene have been identified as causes for phenylketonuria (PKU), the most frequent inherited metabolic disorder (Anikster et al., 2017). Segregation analysis confirmed autosomal-recessive inheritance in all families. Furthermore, a subsequent study of the genotype-phenotype correlation in DNAJC12 mutations demonstrated that several homozygous null variants (c.187A>T and c.79-2A>G) in DNAJC12 present young-onset, dopa-responsive non-progressive parkinsonism as the cardinal symptom and mild cognitive decline without apparent dystonia (Straniero et al., 2017). The autopsy studies of the proband's brain showed depigmentation in the substantia nigra, and no α-synuclein-positive Lewy pathology was observed. Thus, the PKU with the DNAJC12 mutation should be considered as a differential diagnosis of early-onset parkinsonism, since a strict diet therapy with low phenylalanine may prevent the progression of the disease. Most patients with PKU have mutations in a gene that encodes phenylalanine hydroxylase (PAH), a rate-limiting enzyme of the metabolic pathway that degrades excess phenylalanine to tyrosine (Blau, 2016). Patients with PKU do not show any abnormalities at birth; however, if left untreated, the accumulation of phenylalanine and the insufficient level of tyrosine insidiously lead to psychomotor retardation, microcephaly, epilepsy, generalized dystonia, and hypopigmentation in the skin and hair (Al Hafid and Christodoulou, 2015). A limited number (up to 2%) of PKU cases are caused by a deficiency of tetrahydrobiopterin (BH4), a co-factor of PAH and other members of aromatic amino acid hydroxylases (AAAHs), such as tyrosine hydroxylase (TH) and tryptophan hydroxylase (TPH), which are required for the biosynthesis of dopamine and serotonin, respectively (Blau, 2016). Affinity capture-mass spectrometry (AC-MS) and co-immunoprecipitation demonstrated that DNAJC12 interacts with AAAHs including PAH, TH, and peripheral and neuronal isoforms of TPH (Huttlin et al., 2015; Anikster et al., 2017). A molecular interaction between PAH and JDP, the ortholog of DNAJC12 in fruit fly, was also detected in the two-hybrid-based protein-interaction datasets of Drosophila (the Biological General Repository for Interaction Datasets, BioGRID; Giot et al., 2003). Furthermore, analysis of cerebrospinal fluid from affected individuals showed deficiencies of several catecholamines and their metabolites, including dopamine, serotonin, HVA, and 5-HIAA (van Spronsen et al., 2017). These findings suggest the importance of DNAJC12 for proper function of enzymes involved in catecholamine biosynthesis. The c.158-2A>T splice variant and entire deletion in exon 4 (del. 6943) are considered to be null mutants as the DNAJC12 protein was below the detection level in the fibroblasts obtained from individuals carrying these mutations (Anikster et al., 2017). Moreover, the p.R72P missense variant located in the J domain appears to affect the stability of DNAJC12 as the Arg72 residue is well-conserved across species and is indispensable for maintaining the 3D structure of the J domain through the interactions with the Ser25 residue (Anikster et al., 2017).
DNAJC5, also referred to as cysteine string protein α (CSPα), is a 34 kDa protein and synaptic co-chaperone of the DnaJ/Hsp40 family (Burgoyne and Morgan, 2015). The name originates from the existence of a cysteine string domain that consists of 13–15 heavily palmitoylated cysteine residues within 25 amino acids. Structurally, DNAJC5/CSPα is divided into the following domains: a phosphorylation site for protein kinase A at an N-terminus, a conserved J domain, an adjacent linker region, the cysteine string domain, and a less conserved C-terminal domain (Burgoyne and Morgan, 2015). Mutations (p.L116del and p.L115R) in the cysteine string domain of DNAJC5 have previously been identified as causes for an autosomal-dominant, adult-onset neuronal ceroid lipofuscinosis (ANCL, also referred to as Kufs disease), a neurodegenerative disorder characterized by abnormal accumulation of fluorogenic lipids, granular substances not only in the neuronal cells in the brain but also in some other tissues (Noskova et al., 2011; Velinov et al., 2012; Cadieux-Dion et al., 2013). Sanger sequencing revealed that p.L116del mutation was the only indel completely co-segregating with the phenotype in the pedigree. The clinical picture of individuals who carry pathogenic DNAJC5 mutations consists of tonic-clonic and myoclonic epileptic seizure, ataxia, and dementia with premature death. In some cases, patients subsequently began to manifest parkinsonism (Burneo et al., 2003; Cadieux-Dion et al., 2013). The over-expression of EGFP-tagged DNAJC5 in the subclone of murine Cath.a differentiated neuronal cell line (CAD5) demonstrated cell surface localization of wild-type DNAJC5, whereas the p.L116del and p.L115R mutant proteins showed diffuse cytoplasmic localization in addition to aberrant accumulation in the ER and Golgi apparatus (Noskova et al., 2011). As expected, these mutants were less palmitoylated than wild-type DNAJC5, which may lead to impaired membrane tethering of DNAJC5/CSPα and a reduced protein level of DNAJC5/CSPα in the brains of affected individuals. Furthermore, p.L116del and p.L115R mutations in the cysteine-string domain cause DNAJC5/CSPα to form high molecular weight SDS-resistant aggregates, which are also present in post-mortem brain tissue from patients, suggesting a cluster of palmitoylated cysteines are essential for aggregation of CSPα (Diez-Ardanuy et al., 2017). In agreement with this, transgenic zebrafish model expressing the human mutant DNAJC5 gene under the control of a zebrafish neuron-specific promoter demonstrated mutant DNAJC5 protein aggregates in the affected neurons (Yao et al., 2017). However, the precise molecular mechanisms by which the mutant DNAJC5 leads to neurodegeneration accompanied by abnormal accumulation of autofluorescence materials in neuronal tissues still remain obscure. Accumulating evidence suggests that the DNAJC5/CSPα behaves as a critical regulator of synaptic proteostasis. Together with a small glutamine-rich tetratricopeptide repeat domain protein (SGT), DNAJC5/CSPα forms an enzymatically active chaperone complex and binds misfolded client proteins on synaptic vesicles, thereby preventing the buildup of misfolded proteins in the nerve terminal (Donnelier and Braun, 2014). For example, the DNAJC5-Hsc70-SGT chaperone complex stabilizes synaptosomal-associated protein 25 (SNAP-25) and facilitates its ability to assemble into the soluble NSF attachment protein receptor (SNARE) complex (Sharma et al., 2011). Intriguingly, the over-expression of α-synuclein, a culprit protein in PD, protects DNAJC5/CSPα-deficient mice from neurodegeneration (Chandra et al., 2005), which suggests that α-synuclein may compensate for the loss of DNAJC5/CSPα in the nervous system. This finding has been corroborated by a recent study showing that DNAJC5/CSPα removes neurodegeneration-related toxic proteins (e.g., abnormally expanded huntingtin and mutant superoxide dismutase 1) from neurons via extracellular vesicles (Deng et al., 2017).
A recent comprehensive analysis of genetic variants in a well-characterized Han-Chinese cohort with sporadic PD demonstrated that the DNAJC10 gene variant rs13414223 decreased the risk of PD (Yuan et al., 2016). The statistical differences in genotypic and allelic frequencies between the PD and the controls were p = 0.004 and 0.002 (odds ratio = 0.652), respectively. The gene of DNAJC10 encodes the 90 kDa ER-resident co-chaperone ERdj5, which is a component of ER-associated degradation (ERAD), a quality-control machinery by which unfolded/misfolded proteins are degraded in eukaryotic cells (Ushioda et al., 2008). The ERdj family comprises five members, and each member contains a conserved N-terminal J domain; ERdj5 is the only member that has thioredoxin (Trx)-like domains with catalytic active CXXC motifs (Cunnea et al., 2003). A KDEL tetrapeptide sequence is present at the C terminus of DNAJC10/ERdj5, possibly mediating the targeting to the ER (Stornaiuolo et al., 2003). Via its reductase activity, DNAJC10/ERdj5 cleaves the disulfide bonds of misfolded proteins and thereby accelerates ERAD through the associations with EDEM (ER degradation-enhancing alpha-mannosidase-like protein) and the ER chaperone BiP/GRP78 (Ushioda et al., 2008). DNAJC10/ERdj5 is ubiquitously expressed across brain areas, including the cerebral cortex, striatum, hippocampus, hypothalamus, cerebellar cortex, and brainstem (Cunnea et al., 2003), which are the anatomical regions of neuronal cell loss and Lewy body formation in PD (Braak and Braak, 2000). Intriguingly, C. elegans DJ-27, an ortholog of mammalian ERdj5, showed a protective effect against aggregate formation, behavior abnormalities, and mitochondrial fragmentation in worm models of human Alzheimer, Parkinson, and Huntington diseases (Munoz-Lobato et al., 2014); these findings suggest that DNAJC5 may counteract protein misfolding/aggregation toxicity in neuronal cells. Studies in higher model organisms, such as vertebrates, would be useful and advantageous to figure out how the DNAJC10/ERdj5 mutation can be involved in the pathogenesis of PD.
Although more than 90% of PD cases occur sporadically, both familial and sporadic PD share common pathological features, such as cytoplasmic inclusions and dopaminergic cell loss (Hasegawa et al., 2004; Takeda et al., 2006). Evidence from recent genetic studies in rare familial forms of PD indicates that DnaJ/Hsp40 molecular chaperones are profoundly involved in the pathogenesis of PD. The diverse functions of the DnaJ/Hsp40 family in protein folding/unfolding, membrane trafficking, synaptic modulation and mitochondrial function are considered to not only affect the dopaminergic neurotransmission but also concomitantly influence on the PD-related neuropathological changes, such as nigral cell loss and Lewy body formation. HSPs are known to be localized with noxious proteinaceous aggregates in various neurodegenerative diseases including PD, Alzheimer's disease and prion disease (Matsuzaki et al., 2004; San Gil et al., 2017). Interestingly, several studies have shown that selective upregulation of HSPs such as DnaJ/Hsp40 and Hsp70 prevents clinicopathological progression in a variety of cellular and animal models (Popiel et al., 2012; Gao et al., 2015; Takeuchi et al., 2015). Thus, genetic as well as pharmacological manipulation of specific DnaJ/Hsp40 function may provide beneficial effects on protein and cellular homeostasis in neurodegenerative conditions. Deciphering the precise modes of DnaJ/Hsp40 functions in the pathological cascades in PD will shed light on the pathogenic mechanisms involved and may provide important clues regarding the disease-modifying strategies for this devastating neurodegenerative disease.
All authors (TH, SY, NS, JK, and MA) participated in the discussion of the paper. TH and SY mainly wrote the manuscript.
This work was supported in part by a Grant-in-Aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control) [grant number 17H05683] and a Grant-in-Aid for Scientific Research (C) [grant number 17K09744] from the Ministry of Education, Culture, Sports, Science and Technology, MEXT; the Strategic Research Program for Brain Sciences from the Japan Agency for Medical Research and Development, AMED; Novartis Pharma Research Grants.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Al Hafid, N., and Christodoulou, J. (2015). Phenylketonuria: a review of current and future treatments. Transl. Pediatr. 4, 304–317. doi: 10.3978/j.issn.2224-4336.2015.10.07
Anikster, Y., Haack, T. B., Vilboux, T., Pode-Shakked, B., Thony, B., Shen, N., et al. (2017). Biallelic mutations in DNAJC12 cause hyperphenylalaninemia, dystonia, and intellectual disability. Am. J. Hum. Genet. 100, 257–266. doi: 10.1016/j.ajhg.2017.01.002
Appel-Cresswell, S., Rajput, A. H., Sossi, V., Thompson, C., Silva, V., McKenzie, J., et al. (2014). Clinical, positron emission tomography, and pathological studies of DNAJC13 p.N855S Parkinsonism. Mov. Disord. 29, 1684–1687. doi: 10.1002/mds.26019
Blau, N. (2016). Genetics of phenylketonuria: then and now. Hum. Mutat. 37, 508–515. doi: 10.1002/humu.22980
Braak, H., and Braak, E. (2000). Pathoanatomy of Parkinson's disease. J. Neurol. 247(Suppl. 2), II3–II10. doi: 10.1007/PL00007758
Burgoyne, R. D., and Morgan, A. (2015). Cysteine string protein (CSP) and its role in preventing neurodegeneration. Semin. Cell Dev. Biol. 40, 153–159. doi: 10.1016/j.semcdb.2015.03.008
Burneo, J. G., Arnold, T., Palmer, C. A., Kuzniecky, R. I., Oh, S. J., and Faught, E. (2003). Adult-onset neuronal ceroid lipofuscinosis (Kufs disease) with autosomal dominant inheritance in Alabama. Epilepsia 44, 841–846. doi: 10.1046/j.1528-1157.2003.39802.x
Cadieux-Dion, M., Andermann, E., Lachance-Touchette, P., Ansorge, O., Meloche, C., Barnabe, A., et al. (2013). Recurrent mutations in DNAJC5 cause autosomal dominant Kufs disease. Clin. Genet. 83, 571–575. doi: 10.1111/cge.12020
Cai, Z., Zeng, W., Tao, K., E, Z., Wang, B., and Yang, Q. (2015). Chaperone-mediated autophagy: roles in neuroprotection. Neurosci. Bull. 31, 452–458. doi: 10.1007/s12264-015-1540-x
Chandra, S., Gallardo, G., Fernandez-Chacon, R., Schluter, O. M., and Sudhof, T. C. (2005). Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396. doi: 10.1016/j.cell.2005.09.028
Chang, H. C., Hull, M., and Mellman, I. (2004). The J-domain protein Rme-8 interacts with Hsc70 to control clathrin-dependent endocytosis in Drosophila. J. Cell Biol. 164, 1055–1064. doi: 10.1083/jcb.200311084
Ciechanover, A., and Kwon, Y. T. (2017). Protein quality control by molecular chaperones in neurodegeneration. Front. Neurosci. 11:185. doi: 10.3389/fnins.2017.00185
Cunnea, P. M., Miranda-Vizuete, A., Bertoli, G., Simmen, T., Damdimopoulos, A. E., Hermann, S., et al. (2003). ERdj5, an endoplasmic reticulum (ER)-resident protein containing DnaJ and thioredoxin domains, is expressed in secretory cells or following ER stress. J. Biol. Chem. 278, 1059–1066. doi: 10.1074/jbc.M206995200
Deng, H. X., Shi, Y., Yang, Y., Ahmeti, K. B., Miller, N., Huang, C., et al. (2016). Identification of TMEM230 mutations in familial Parkinson's disease. Nat. Genet. 48, 733–739. doi: 10.1038/ng.3589
Deng, J., Koutras, C., Donnelier, J., Alshehri, M., Fotouhi, M., Girard, M., et al. (2017). Neurons export extracellular vesicles enriched in cysteine string protein and misfolded protein cargo. Sci. Rep. 7:956. doi: 10.1038/s41598-017-01115-6
Diez-Ardanuy, C., Greaves, J., Munro, K. R., Tomkinson, N. C., and Chamberlain, L. H. (2017). A cluster of palmitoylated cysteines are essential for aggregation of cysteine-string protein mutants that cause neuronal ceroid lipofuscinosis. Sci. Rep. 7:10. doi: 10.1038/s41598-017-00036-8
Donnelier, J., and Braun, J. E. (2014). CSPα-chaperoning presynaptic proteins. Front. Cell Neurosci. 8:116. doi: 10.3389/fncel.2014.00116
Dudek, J., Benedix, J., Cappel, S., Greiner, M., Jalal, C., Muller, L., et al. (2009). Functions and pathologies of BiP and its interaction partners. Cell Mol. Life Sci. 66, 1556–1569. doi: 10.1007/s00018-009-8745-y
Edvardson, S., Cinnamon, Y., Ta-Shma, A., Shaag, A., Yim, Y. I., Zenvirt, S., et al. (2012). A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE 7:e36458. doi: 10.1371/journal.pone.0036458
Elsayed, L. E., Drouet, V., Usenko, T., Mohammed, I. N., Hamed, A. A., Elseed, M. A., et al. (2016). A novel nonsense mutation in DNAJC6 expands the phenotype of autosomal-recessive juvenile-onset Parkinson's disease. Ann. Neurol. 79, 335–337. doi: 10.1002/ana.24591
Freeman, C. L., Hesketh, G., and Seaman, M. N. (2014). RME-8 coordinates the activity of the WASH complex with the function of the retromer SNX dimer to control endosomal tubulation. J. Cell Sci. 127, 2053–2070. doi: 10.1242/jcs.144659
Fujibayashi, A., Taguchi, T., Misaki, R., Ohtani, M., Dohmae, N., Takio, K., et al. (2008). Human RME-8 is involved in membrane trafficking through early endosomes. Cell Struct. Funct. 33, 35–50. doi: 10.1247/csf.07045
Gao, X., Carroni, M., Nussbaum-Krammer, C., Mogk, A., Nillegoda, N. B., Szlachcic, A., et al. (2015). Human Hsp70 disaggregase reverses Parkinson's-linked alpha-synuclein amyloid fibrils. Mol. Cell 59, 781–793. doi: 10.1016/j.molcel.2015.07.012
Giot, L., Bader, J. S., Brouwer, C., Chaudhuri, A., Kuang, B., Li, Y., et al. (2003). A protein interaction map of Drosophila melanogaster. Science 302, 1727–1736. doi: 10.1126/science.1090289
Girard, M., Poupon, V., Blondeau, F., and McPherson, P. S. (2005). The DnaJ-domain protein RME-8 functions in endosomal trafficking. J. Biol. Chem. 280, 40135–40143. doi: 10.1074/jbc.M505036200
Goldfarb, S. B., Kashlan, O. B., Watkins, J. N., Suaud, L., Yan, W., Kleyman, T. R., et al. (2006). Differential effects of Hsc70 and Hsp70 on the intracellular trafficking and functional expression of epithelial sodium channels. Proc. Natl. Acad. Sci. U.S.A. 103, 5817–5822. doi: 10.1073/pnas.0507903103
Gorenberg, E. L., and Chandra, S. S. (2017). The role of co-chaperones in synaptic proteostasis and neurodegenerative disease. Front. Neurosci. 11:248. doi: 10.3389/fnins.2017.00248
Greene, M. K., Maskos, K., and Landry, S. J. (1998). Role of the J-domain in the cooperation of Hsp40 with Hsp70. Proc. Natl. Acad. Sci. U.S.A. 95, 6108–6113. doi: 10.1073/pnas.95.11.6108
Gustavsson, E. K., Trinh, J., Guella, I., Vilarino-Guell, C., Appel-Cresswell, S., Stoessl, A. J., et al. (2015). DNAJC13 genetic variants in parkinsonism. Mov. Disord. 30, 273–278. doi: 10.1002/mds.26064
Hahn, Y., Lee, J., Seong, C., Yoon, J., and Chung, J. H. (1999). Structural analysis of phylogenetically conserved J domain protein gene. Biochim. Biophys. Acta 1447, 325–333. doi: 10.1016/S0167-4781(99)00162-1
Hasegawa, T., Matsuzaki, M., Takeda, A., Kikuchi, A., Akita, H., Perry, G., et al. (2004). Accelerated alpha-synuclein aggregation after differentiation of SH-SY5Y neuroblastoma cells. Brain Res. 1013, 51–59. doi: 10.1016/j.brainres.2004.04.018
Hasegawa, T., Sugeno, N., Kikuchi, A., Baba, T., and Aoki, M. (2017). Membrane trafficking illuminates a path to Parkinson's disease. Tohoku J. Exp. Med. 242, 63–76. doi: 10.1620/tjem.242.63
Huttlin, E. L., Ting, L., Bruckner, R. J., Gebreab, F., Gygi, M. P., Szpyt, J., et al. (2015). The BioPlex network: a systematic exploration of the human interactome. Cell 162, 425–440. doi: 10.1016/j.cell.2015.06.043
Kim, M. J., Deng, H. X., Wong, Y. C., Siddique, T., and Krainc, D. (2017). The Parkinson's disease-linked protein TMEM230 is required for Rab8a-mediated secretory vesicle trafficking and retromer trafficking. Hum. Mol. Genet. 26, 729–741. doi: 10.1093/hmg/ddw413
Koroglu, C., Baysal, L., Cetinkaya, M., Karasoy, H., and Tolun, A. (2013). DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinson. Relat. Disord. 19, 320–324. doi: 10.1016/j.parkreldis.2012.11.006
Lee, D. W., Wu, X., Eisenberg, E., and Greene, L. E. (2006). Recruitment dynamics of GAK and auxilin to clathrin-coated pits during endocytosis. J. Cell Sci. 119, 3502–3512. doi: 10.1242/jcs.03092
Lorenzo-Betancor, O., Ogaki, K., Soto-Ortolaza, A. I., Labbe, C., Walton, R. L., Strongosky, A. J., et al. (2015). DNAJC13 p.Asn855Ser mutation screening in Parkinson's disease and pathologically confirmed Lewy body disease patients. Eur. J. Neurol. 22, 1323–1325. doi: 10.1111/ene.12770
Matsuzaki, M., Hasegawa, T., Takeda, A., Kikuchi, A., Furukawa, K., Kato, Y., et al. (2004). Histochemical features of stress-induced aggregates in alpha-synuclein overexpressing cells. Brain Res. 1004, 83–90. doi: 10.1016/j.brainres.2004.01.017
Munoz-Lobato, F., Rodriguez-Palero, M. J., Naranjo-Galindo, F. J., Shephard, F., Gaffney, C. J., Szewczyk, N. J., et al. (2014). Protective role of DNJ-27/ERdj5 in Caenorhabditis elegans models of human neurodegenerative diseases. Antioxid. Redox Signal. 20, 217–235. doi: 10.1089/ars.2012.5051
Noskova, L., Stranecky, V., Hartmannova, H., Pristoupilova, A., Baresova, V., Ivanek, R., et al. (2011). Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am. J. Hum. Genet. 89, 241–252. doi: 10.1016/j.ajhg.2011.07.003
Olgiati, S., Quadri, M., Fang, M., Rood, J. P., Saute, J. A., Chien, H. F., et al. (2016). DNAJC6 mutations associated with early-onset Parkinson's disease. Ann. Neurol. 79, 244–256. doi: 10.1002/ana.24553
Oshima, R., Hasegawa, T., Tamai, K., Sugeno, N., Yoshida, S., Kobayashi, J., et al. (2016). ESCRT-0 dysfunction compromises autophagic degradation of protein aggregates and facilitates ER stress-mediated neurodegeneration via apoptotic and necroptotic pathways. Sci. Rep. 6:24997. doi: 10.1038/srep24997
Pankratz, N., Wilk, J. B., Latourelle, J. C., Destefano, A. L., Halter, C., Pugh, E. W., et al. (2009). Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum. Genet. 124, 593–605. doi: 10.1007/s00439-008-0582-9
Pauli, D., Arrigo, A. P., and Tissieres, A. (1992). Heat shock response in Drosophila. Experientia 48, 623–629. doi: 10.1007/BF02118306
Perales-Calvo, J., Muga, A., and Moro, F. (2010). Role of DnaJ G/F-rich domain in conformational recognition and binding of protein substrates. J. Biol. Chem. 285, 34231–34239. doi: 10.1074/jbc.M110.144642
Piano, A., Valbonesi, P., and Fabbri, E. (2004). Expression of cytoprotective proteins, heat shock protein 70 and metallothioneins, in tissues of Ostrea edulis exposed to heat and heavy metals. Cell Stress Chaperones 9, 134–142. doi: 10.1379/483.1
Popiel, H. A., Takeuchi, T., Fujita, H., Yamamoto, K., Ito, C., Yamane, H., et al. (2012). Hsp40 gene therapy exerts therapeutic effects on polyglutamine disease mice via a non-cell autonomous mechanism. PLoS ONE 7:e51069. doi: 10.1371/journal.pone.0051069
Popoff, V., Mardones, G. A., Bai, S. K., Chambon, V., Tenza, D., Burgos, P. V., et al. (2009). Analysis of articulation between clathrin and retromer in retrograde sorting on early endosomes. Traffic 10, 1868–1880. doi: 10.1111/j.1600-0854.2009.00993.x
Puschmann, A. (2017). New genes causing hereditary Parkinson's disease or Parkinsonism. Curr. Neurol. Neurosci. Rep. 17:66. doi: 10.1007/s11910-017-0780-8
Qiu, X. B., Shao, Y. M., Miao, S., and Wang, L. (2006). The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol. Life Sci. 63, 2560–2570. doi: 10.1007/s00018-006-6192-6
Rajput, A., Ross, J. P., Bernales, C. Q., Rayaprolu, S., Soto-Ortolaza, A. I., Ross, O. A., et al. (2015). VPS35 and DNAJC13 disease-causing variants in essential tremor. Eur. J. Hum. Genet. 23, 887–888. doi: 10.1038/ejhg.2014.164
San Gil, R., Ooi, L., Yerbury, J. J., and Ecroyd, H. (2017). The heat shock response in neurons and astroglia and its role in neurodegenerative diseases. Mol. Neurodegener. 12:65. doi: 10.1186/s13024-017-0208-6
Schopf, F. H., Biebl, M. M., and Buchner, J. (2017). The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 18, 345–360. doi: 10.1038/nrm.2017.20
Sharma, M., Burre, J., and Sudhof, T. C. (2011). CSPα promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol. 13, 30–39. doi: 10.1038/ncb2131
Shi, A., Sun, L., Banerjee, R., Tobin, M., Zhang, Y., and Grant, B. D. (2009). Regulation of endosomal clathrin and retromer-mediated endosome to Golgi retrograde transport by the J-domain protein RME-8. EMBO J. 28, 3290–3302. doi: 10.1038/emboj.2009.272
Shoji, Y., Nishio, Y., Baba, T., Uchiyama, M., Yokoi, K., Ishioka, T., et al. (2014). Neural substrates of cognitive subtypes in Parkinson's disease: a 3-year longitudinal study. PLoS ONE 9:e110547. doi: 10.1371/journal.pone.0110547
Soga, S., Akinaga, S., and Shiotsu, Y. (2013). Hsp90 inhibitors as anti-cancer agents, from basic discoveries to clinical development. Curr. Pharm. Des. 19, 366–376. doi: 10.2174/138161213804143617
Stornaiuolo, M., Lotti, L. V., Borgese, N., Torrisi, M. R., Mottola, G., Martire, G., et al. (2003). KDEL and KKXX retrieval signals appended to the same reporter protein determine different trafficking between endoplasmic reticulum, intermediate compartment, and Golgi complex. Mol. Biol. Cell 14, 889–902. doi: 10.1091/mbc.E02-08-0468
Straniero, L., Guella, I., Cilia, R., Parkkinen, L., Rimoldi, V., Young, A., et al. (2017). DNAJC12 and dopa-responsive nonprogressive parkinsonism. Ann. Neurol. 82, 640–646. doi: 10.1002/ana.25048
Takeda, A., Hasegawa, T., Matsuzaki-Kobayashi, M., Sugeno, N., Kikuchi, A., Itoyama, Y., et al. (2006). Mechanisms of neuronal death in synucleinopathy. J. Biomed. Biotechnol. 2006:19365. doi: 10.1155/JBB/2006/19365
Takeuchi, T., Suzuki, M., Fujikake, N., Popiel, H. A., Kikuchi, H., Futaki, S., et al. (2015). Intercellular chaperone transmission via exosomes contributes to maintenance of protein homeostasis at the organismal level. Proc. Natl. Acad. Sci. U.S.A. 112, E2497–E2506. doi: 10.1073/pnas.1412651112
Ushioda, R., Hoseki, J., Araki, K., Jansen, G., Thomas, D. Y., and Nagata, K. (2008). ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 321, 569–572. doi: 10.1126/science.1159293
van Spronsen, F. J., Himmelreich, N., Rüfenacht, V., Shen, N., Vliet, D. V., Al-Owain, M., et al. (2017). Heterogeneous clinical spectrum of DNAJC12-deficient hyperphenylalaninemia: from attention deficit to severe dystonia and intellectual disability. J. Med. Genet. doi: 10.1136/jmedgenet-2017-104875. [Epub ahead of print].
Velinov, M., Dolzhanskaya, N., Gonzalez, M., Powell, E., Konidari, I., Hulme, W., et al. (2012). Mutations in the gene DNAJC5 cause autosomal dominant Kufs disease in a proportion of cases: study of the Parry family and 8 other families. PLoS ONE 7:e29729. doi: 10.1371/annotation/26d7eb64-ccd2-41db-b1aa-7cdc8c1eff95
Vilariño-Güell, C., Rajput, A., Milnerwood, A. J., Shah, B., Szu-Tu, C., Trinh, J., et al. (2014). DNAJC13 mutations in Parkinson disease. Hum. Mol. Genet. 23, 1794–1801. doi: 10.1093/hmg/ddt570
Yao, X., Liu, X., Zhang, Y., Li, Y., Zhao, C., Yao, S., et al. (2017). Gene therapy of adult neuronal ceroid lipofuscinoses with CRISPR/Cas9 in Zebrafish. Hum. Gene Ther. 28, 588–597. doi: 10.1089/hum.2016.190
Yim, Y. I., Sun, T., Wu, L. G., Raimondi, A., De Camilli, P., Eisenberg, E., et al. (2010). Endocytosis and clathrin-uncoating defects at synapses of auxilin knockout mice. Proc. Natl. Acad. Sci. U.S.A. 107, 4412–4417. doi: 10.1073/pnas.1000738107
Yuan, L., Song, Z., Deng, X., Zheng, W., Guo, Y., Yang, Z., et al. (2016). Systematic analysis of genetic variants in Han Chinese patients with sporadic Parkinson's disease. Sci. Rep. 6:33850. doi: 10.1038/srep33850
Zhang, Y., Grant, B., and Hirsh, D. (2001). RME-8, a conserved J-domain protein, is required for endocytosis in Caenorhabditis elegans. Mol. Biol. Cell 12, 2011–2021. doi: 10.1091/mbc.12.7.2011
Keywords: DnaJ protein, Hsp40, co-chaperones, Parkinson's disease, neurodegeneration
Citation: Hasegawa T, Yoshida S, Sugeno N, Kobayashi J and Aoki M (2018) DnaJ/Hsp40 Family and Parkinson's Disease. Front. Neurosci. 11:743. doi: 10.3389/fnins.2017.00743
Received: 21 October 2017; Accepted: 20 December 2017;
Published: 10 January 2018.
Edited by:
Naruhiko Sahara, National Institute of Radiological Sciences (NIRS), JapanReviewed by:
Takashi Nonaka, Tokyo Metropolitan Institute of Medical Science, JapanCopyright © 2018 Hasegawa, Yoshida, Sugeno, Kobayashi and Aoki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takafumi Hasegawa, dGhhc2VnYXdhQG1lZC50b2hva3UuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.