 Kevin A. Clayton
Kevin A. Clayton Alicia A. Van Enoo
Alicia A. Van Enoo Tsuneya Ikezu
Tsuneya Ikezu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 12 December 2017
Sec. Neurodegeneration
Volume 11 - 2017 | https://doi.org/10.3389/fnins.2017.00680
This article is part of the Research Topic Brain Protein Aging and Dementia Control View all 23 articles
Brain aging is central to late-onset Alzheimer's disease (LOAD), although the mechanisms by which it occurs at protein or cellular levels are not fully understood. Alzheimer's disease is the most common proteopathy and is characterized by two unique pathologies: senile plaques and neurofibrillary tangles, the former accumulating earlier than the latter. Aging alters the proteostasis of amyloid-β peptides and microtubule-associated protein tau, which are regulated in both autonomous and non-autonomous manners. Microglia, the resident phagocytes of the central nervous system, play a major role in the non-autonomous clearance of protein aggregates. Their function is significantly altered by aging and neurodegeneration. This is genetically supported by the association of microglia-specific genes, TREM2 and CD33, and late onset Alzheimer's disease. Here, we propose that the functional characterization of microglia, and their contribution to proteopathy, will lead to a new therapeutic direction in Alzheimer's disease research.
Aging results in a loss of proteostasis that is characteristic of many neurodegenerative disorders. During aging, the mechanisms responsible for protein synthesis, post-translational modifications, and clearance, cumulatively known as “proteostasis,” become dysregulated in the central nervous system (CNS). Impairments in proteostasis result in the accumulation of misfolded proteins as intracellular aggregates, such as neurofibrillary tangles and Lewy bodies, or extracellular plaques, such as senile and prion plaques, which ultimately lead to conditions termed “proteopathies.” Alzheimer's disease (AD) is diagnosed with the development of two unique pathologies, senile plaques and neurofibrillary tangles. A comprehensive understanding of the brain cells responsible for the clearance of protein aggregation due to dysfunction is critical to fully elucidate the etiology of AD. This mechanism is largely understood to be a cell autonomous process, namely executed by neurons. However, non-autonomous processes that may also be involved have recently become the subject of extensive investigation. One representative cell type controlling brain proteostasis is microglia. Interestingly, their functions are largely affected by aging. This is genetically supported by the significant genome-wide association of microglia-specific TREM2 gene with LOAD (Guerreiro et al., 2013; Jonsson et al., 2013). Microglia are the resident phagocytes of the CNS and are implicated in the pathogenesis of many neurocognitive disorders, including neurodevelopmental and neurodegenerative diseases. They are implicated in the phagocytosis and degradation of pathological protein aggregates. Numerous studies have been published recently depicting the changes that cause microglia to become dysfunctional during aging and disease. Once microglia become dysfunctional, they further contribute to CNS destabilization in response to protein aggregates, which ultimately leads to brain degeneration. One prominent aspect of microglial dysfunction is their role in chronic neuroinflammation, a phenomenon in which immune cells recognize and pervade the ailing tissue causing damage through both antigen-specific and non-specific mechanisms. Several groups have recently provided a comprehensive characterization of microglial phenotype in order to elucidate the mechanisms by which microglial dysfunction disrupts the CNS microenvironment (Matcovitch-Natan et al., 2016; Keren-Shaul et al., 2017; Krasemann et al., 2017).
These new concepts considering the causal relationships between microglial dysfunction, neuroinflammation, and protein aging, introduce the question of how novel therapies may halt or reverse the contribution of microglia to the spread of proteopathy and neurodegeneration. Here, we summarize clinical and preclinical studies aimed to prevent protein aggregation or restore homeostatic microglial function. Furthermore, we overview the physiological function of microglia, their changes in response to aging, and their specific neurodegenerative phenotype leading to proteopathy in AD.
Most neurodegenerative disorders are pathologically characterized as proteopathies (Walker and LeVine, 2000). Aging can impact multiple aspects of proteostasis: production, folding, posttranslational modification, and clearance in several pathways, including secretion and autophagosomal, endolysosomal, and proteasomal degradation (Kaushik and Cuervo, 2015), all of which are demonstrated to affect protein aggregation when impaired. Aggregated proteins, such as amyloid-beta peptides (Aβ), are inherently cytotoxic in vitro, causing stress and stimulating synaptic loss, mitochondrial dysfunction, and eventually apoptosis in neurons (Sakono and Zako, 2010). We will mainly describe the metabolism of Aβ and tau in this review.
The production of Aβ from amyloid precursor protein (APP) is the most well-studied component of AD development, especially in early-onset Alzheimer's disease (EOAD) (Rovelet-Lecrux et al., 2006; Sleegers et al., 2006; McNaughton et al., 2012). In addition to the well-known mutations proximal to the α, β, and γ-processing of APP, several mutations in the promoter region of APP have been reported and contribute to enhanced APP gene expression (Athan et al., 2002; Theuns et al., 2006; Hooli et al., 2012; Rodgers et al., 2012). However, the majority of sporadic cases do not show increased expression of APP by aging. Rather, there is a reduction in the amount of Aβ in the cerebrospinal fluid (CSF), suggesting its absorption into amyloid plaques, rather than export to the periphery. Researchers have increasingly argued that sporadic or late-onset AD is more likely caused by a reduction in clearance of Aβ than by its overproduction (Mawuenyega et al., 2010).
There is also a possibility for an age-related shift in APP processing toward the amyloidogenic rather than non-amyloidogenic pathway. This results in the production of pathogenic Aβ, as opposed to the non-pathogenic p3 peptide. One study reported that age did not have an effect on γ-secretase production of Aβ42 (Dewachter et al., 2000). However, the expression of beta-site APP converting enzyme 1 (BACE1), the enzyme designated as β-secretase, is elevated in the AD brain (Li et al., 2004; Zhao et al., 2007). Furthermore, primary cortical neurons were shown to up-regulate BACE1 expression in response to Aβ42 exposure (Mamada et al., 2015). This suggests that Aβ42 production may be self-perpetuating via up-regulation of BACE1 expression in neurons. Given that BACE1 acts on APP primarily in endosomal compartments, age-related increases in early endosome volume could result in increased BACE1 processing of APP (Cataldo et al., 2000). Another possibility is the reduced activity or expression of α-secretase, ADAM10, a component of the non-amyloidogenic pathway. Reduced non-amyloidogenic processing of APP was reported to occur as a result of cellular aging (Kern et al., 2006), although overall activity and expression was increased in cognitively normal subjects (Schuck et al., 2016). This suggests that an age-dependent reduction of ADAM10 function is specific to AD subjects.
Reduced clearance of Aβ has emerged as the central mechanism of amyloid plaque formation in AD. Aβ is thought to be cleared via interstitial fluid (ISF) drainage into the blood vessels, and brought across the blood brain barrier (BBB) into the peripheral bloodstream. There are mutations in APP reported in AD cases that appear to reduce transport of Aβ from the CSF into the blood (Monro et al., 2002). Receptor for advanced glycation end products (RAGE) is proposed to be expressed in the BBB and responsible for shuttling Aβ from the bloodstream into the brain, while LRP1 is responsible for efflux of Aβ42 out of the brain (Deane et al., 2009). However, LRP1 expression in the BBB is reduced in rodents, primates, and humans (Shibata et al., 2000; Deane et al., 2004; Zerbinatti et al., 2004; Donahue et al., 2006). For this reason, RAGE antagonists, which have already been proposed to have effective anti-inflammatory properties, are being further explored for clinical development (see Table 1). In addition to the transport of Aβ out of the ISF to the blood, the CNS is equipped with several other clearance mechanisms, principally mediated via Aβ degrading enzymes. These include enzymes derived from the M13 zinc-binding membrane metalloendopeptidase (such as insulin degrading enzyme), Type II integral membrane bound glycoproteins (NEP-2, ECE-1), membrane-bound zinc metalloproteinase (such as matrix metalloproteinase 2 and 9), thiol-metalloendopeptidase, matrix metalloproteinase, and members of the serine and cysteine protease families (see Miners et al., 2011 for a complete list). One well-investigated protease, neprilysin (NEP), appears to be critical in this pathway, demonstrated by the effectiveness of a combination of NEP inhibitors, over-expression of NEP, and genetic disruption of NEP in increasing proteolytic cleavage and clearance of Aβ (Marr et al., 2003; Dolev and Michaelson, 2004; Nisemblat et al., 2008; Hafez et al., 2011; Takamatsu et al., 2014). Aβ clearance also occurs extracellularly, via microglia-mediated phagocytosis. The effect of aging on this process is further discussed in following sections.
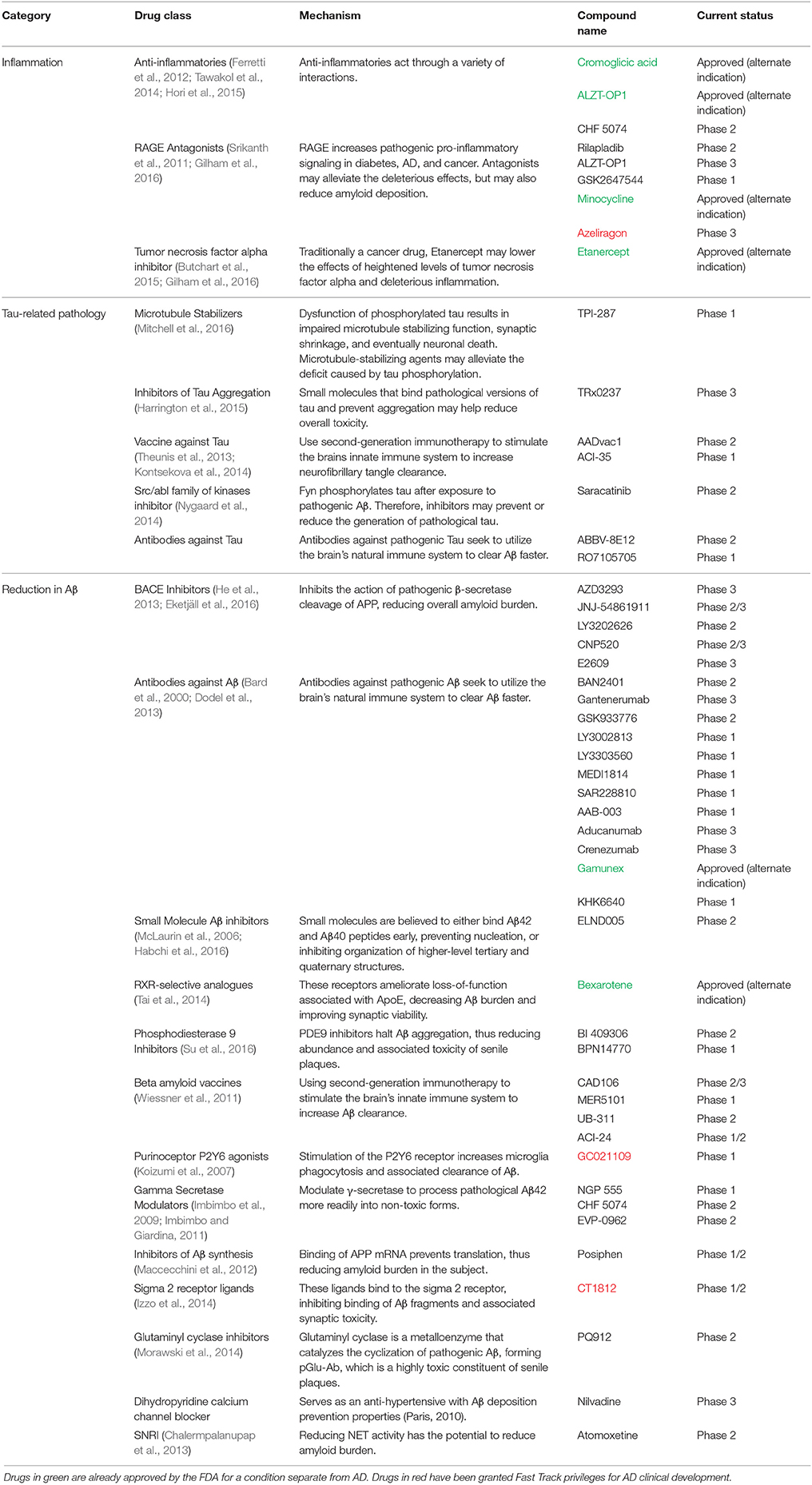
Table 1. A comprehensive list of all of the drugs in clinical development that aim to reduce either Aβ pathology, tau pathology, or inflammation within the past five years categorized by treatment strategy and drug class.
This first reports of cells exhibiting microglia-like phenotypes came from the work of Nissl and Robertson in the late nineteenth century (Gomez-Nicola and Perry, 2015). It wasn't until the late 1930's that these cells were differentiated from other glial cells and received the name of “microglia” by Pio del Rio-Hortega. Hortega used silver staining techniques to describe microglial morphology and introduced the idea of microglia as ramified resting cells (Ginhoux et al., 2013). He pioneered the idea that microglia have the ability to change morphology, migrate, and proliferate in response to their microenvironment and described their basic functional roles as phagocytic cells (Ginhoux et al., 2013). Although Hortega introduced the idea of a mesodermal origin of microglia, a more recent study showed that microglia differentiate from yolk-sac derived myeloid precursor cells (Ginhoux et al., 2010). Utilizing fate mapping technology, Ginhoux et al. have demonstrated that microglia progenitor cells infiltrate into the brain from the yolk sac during early embryonic development and continue to migrate and mature in the early stages of post-natal brain development. Support for the yolk sac hypothesis of microglia origin has prompted scientists to investigate the mechanisms by which microglia maintain homeostatic presence in the CNS throughout the lifetime. Several studies have found that microglia are largely maintained by proliferation, while circulating peripheral monocytes only contribute to the microglia population in disease conditions (Ginhoux et al., 2010; Bruttger et al., 2015). In adulthood, microglia make up approximately 0.5–16.6% of all cells in the brain, depending on brain sub-regional variations (Mittelbronn et al., 2001). They are widely present in the entire CNS, including the brain and spinal cord.
One particularly essential characteristic of microglia is their ability to rapidly change morphology and function in response to changes in their microenvironment (Karperien et al., 2013). Several studies have suggested that microglia morphology falls on a spectrum, ranging from amoeboid to ramified (Stence et al., 2001; Fontainhas et al., 2011). Additionally, newer studies have introduced a third morphological classification; reactive or “alternatively” activated microglia, characterized by thick retracted processes, typically directed toward a lesion or site of protein aggregation (Franco and Fernández-Suárez, 2015). Morphologically, this state falls in between amoeboid and ramified. Ramified microglia, frequently defined as homeostatic or surveying microglia, are characterized by dynamic thin processes extending out from a relatively circular-shaped soma (Kreutzberg, 1996; Fontainhas et al., 2011; Karperien et al., 2013). Surveying microglia are involved in CNS homeostasis by actively making contacts with surrounding synaptic elements. Interestingly, several studies have demonstrated that the most complex microglia appear to be seen in compromised conditions and may be subtly activated, suggesting that ramified microglia may also be slightly reactive (Hinwood et al., 2012; Karperien et al., 2013). Amoeboid microglia display the greatest level of motility, facilitated by a retraction and reduction of processes (Kreutzberg, 1996; Karperien et al., 2013). Amoeboid microglia are most commonly found during the early stages of brain development, before they undergo morphological differentiation to ramified microglia during brain maturation (Harry and Kraft, 2012; Ginhoux et al., 2013). They are also occasionally reported in inflammatory and phagocytic conditions, although their specific function in these states remains unclear (Karperien et al., 2013). Microglia morphology is strongly influenced by neurotransmitter activity. Excitatory neurotransmission significantly increases the ramification of microglia via ATP signaling (Fontainhas et al., 2011). This evidence suggests that microglial processes are highly susceptible to external cues. Although numerous studies have attempted to elucidate the correlation between amoeboid and ramified microglia and their roles in physiological and pathological conditions, the exact functional profiles of different morphological states remain widely debated.
Microglia are known to play an important role in synaptogenesis and synaptic wiring and maintenance, which is crucial for functional brain connectivity (Ginhoux et al., 2010; Paolicelli et al., 2011). Lim et al. have recently shown that microglia-mediated release of IL-10, a pro-inflammatory molecule, led to an increase in dendritic spines (Lim et al., 2013). Concurrently, in a second study, they demonstrated that hippocampal neurons expressed IL-10 receptors during the early stages of brain maturation (Lim et al., 2013). Together, these studies suggest a causal role of microglia in synaptogenesis through microglia-mediated IL-10 signaling. Microglia maintain the ability to modulate synaptic circuits into early adulthood. A study by Parkhurst et al. revealed a microglia-dependent effect on learning-related synaptogenesis (Parkhurst et al., 2013). In addition to their role in synaptic wiring and refinement, microglia are also key modulators of synaptic plasticity. In a study investigating plasticity in the visual system, microglia-mediated synaptic remodeling in layers II/III of the V1 was shown to be activity-dependent (Tremblay et al., 2012). Additional studies have focused on other synaptic functions, such as long term potentiation (LTP), suggesting that microglia actively participate in strengthening neuronal connections through Hebbian plasticity (Penn et al., 1998). Furthermore, several studies have been published highlighting chemokine fractalkine receptor (CX3CR1) signaling and secretion of soluble molecules enhancing NMDA receptor function as key effectors in microglia-mediated modulation of synaptic plasticity (Hayashi et al., 2006; Justin et al., 2011). A study investigating the role of microglia in ocular dominance columns found that microglia modulate experience-driven plasticity in the monocular deprivation model through synaptic pruning (Sipe et al., 2016). Moreover, microglia were shown to have an indirect effect on synaptic strength through upregulation of TNF-α (Lewitus et al., 2016). Together, these findings reinforce the role of microglia in modulating cortical plasticity throughout the lifetime.
Microglia provide crucial supportive functions in the CNS development starting early embryonic stages and persisting into adulthood. In their steady state, microglia play important roles in synaptic maintenance by serving as phagocytic cells, pruning excess or dysfunctional synapses (Tremblay et al., 2010). These processes are controlled by three principal mechanisms: the complement system, chemokine pathway, and activity-dependent signaling.
The complement system, a part of the innate immune response, facilitates phagocytosis in response to antigens. Complement proteins are highly expressed in neurons and glia, but selectively localized to immature synapses (Stevens et al., 2007). Microglia express complement C3 receptor CR3 (aka CD11b, Itgam, and Mac-1), which recognizes activated C3 fragments tagged to excess, immature, or dysfunctional synapses, consequently initiating phagocytosis (Carroll, 2004; Gasque, 2004; Ransohoff and Perry, 2009).
Several studies have found that microglia-mediated pruning is dependent on fractalkine signaling, which promotes survival in monocytes (Landsman et al., 2009). CX3CR1 is specific to microglia in the brain and subset of peripheral monocytes, and responds to pruning cues from surrounding neurons. Knock out (KO) of CX3CR1 was associated with a brief reduction in microglia and subsequent deficit in synaptic pruning, which resulted in an excess of excitatory synapses, as well as an increase in spine density and PSD95 expression (Paolicelli et al., 2011). These findings suggest that disruptions in microglia-mediated synaptic pruning are sufficient to induce deficits in brain maturation, resulting in impaired functional connectivity.
Additionally, studies have shown that pruning is activity-dependent and persists into adulthood (Tremblay et al., 2010; Schafer et al., 2012). Recently, one study has found that synaptic pruning is negatively altered in disease conditions and results in microglia-mediated synaptic loss (Hong et al., 2016). This evidence confirms the crucial role microglia play in shaping neuronal circuits throughout the lifetime, in both physiological and pathological conditions.
Microglia are known to phagocytose biological waste and a variety of pathogens, including apoptotic bodies, cellular debris, and exogenous particles, through various well-established phagocytic pathways (Chan et al., 2001; Fu et al., 2014). This phagocytic function is crucial in both health and disease (Aderem and Underhill, 1999). Ravishadran has established a four-step model for the phagocytosis of apoptotic neurons: apoptotic cells first release “find me” signals attracting microglia, followed by the “eat me” process, mediated by specific receptors expressed by targets, the “digest me” phase, resulting in degradation of cellular materials, and finally, the post-phagocytic phase, which involves inflammatory consequences, such as cytokine and chemokine release (Ravichandran, 2010). Additionally, the fifth step, which occurs in proteopathies, has been proposed (Figure 1). Different signaling pathways are implicated in the phagocytosis of various targets (Fu et al., 2014). Extracellular nucleotides, such as ATP and UTP, are the most common “find me” signals released by apoptotic cells. UTP degradation yields UDP, which interacts directly with microglia via P2Y6 receptors (Nimmerjahn and Ravetch, 2006; Koizumi et al., 2007). Fractalkine signaling has also been shown to contribute to microglia-mediated phagocytosis of apoptotic cells (Truman et al., 2008; Noda et al., 2011; Sierra et al., 2013). “Eat me” signals are expressed by targets to initiate recognition by receptors expressed on the microglial cell surface. Toll-like receptors (TLRs) and Fc receptors are commonly implicated in microglia-mediated phagocytosis of α–synuclein (Okun et al., 2010; Hanke and Kielian, 2011; Cao et al., 2012), whereas triggering receptor expressed on myeloid cells 2 (TREM2), is known to control microglia-mediated Aβ compaction, and phagocytosis of apoptotic neurons (Piccio et al., 2007; Takahashi et al., 2007; Yuan et al., 2016). Complement and scavenger receptors are expressed by microglia and astrocytes, specifically in the pathophysiology of AD, multiple sclerosis, and amyotrophic lateral sclerosis (Husemann et al., 2002; Alarcón et al., 2005; Keren-Shaul et al., 2017). Together, these studies indicate that various types of stimulus are able to differentially trigger microglia-mediated phagocytosis via a wide range of signaling pathways. In addition to their role in phagocytosing apoptotic cells, microglia also have an established role in initiating cell death, in a process termed “phagoptosis” (Brown and Neher, 2012). Interestingly, some studies have shown that microglia-mediated “phagoptosis” can have deleterious effects in neurodegenerative diseases by triggering phagocytosis of viable neurons (Kao et al., 2011). These findings suggest that microglia-mediated phagocytosis can have both protective and deleterious effects in disease states (Fu et al., 2014).
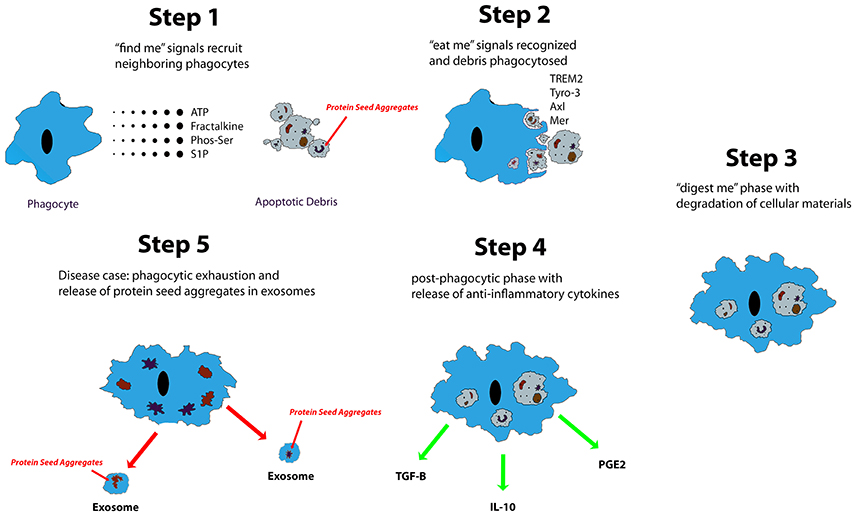
Figure 1. Described are five steps of apoptotic cell clearance via phagocytes. Trace chemicals and molecules associated with cell necrosis and apoptosis stimulate the chemotaxis of phagocytes up their concentration gradient to the source. From there, the phagocyte engulfs the apoptotic debris and begins degradation. Eventually, it will release anti-inflammatory and reparative signals such as TGF-β, IL-10, and PGE2. In neurodegenerative diseases of protein aggregation, protein seed aggregates are packaged into exosomes where they can be shuttled to different regions of the CNS.
In homeostatic physiological conditions, the blood brain barrier (BBB) prevents most peripheral infectious agents, as well as peripheral immune cells, from reaching the CNS. Consequently, the brain depends on its' own immune cells, microglia, to fulfill the roll of peripheral immune cells. As such, microglia have been shown to act as antigen presenting cells (APCs) upon activation in response to immune-related insults (Gottfried-Blackmore et al., 2009). In homeostatic conditions, major histocompatibility complex class II (MHC-II) expression is virtually non-existent in microglial populations (Ford et al., 1995). In inflammatory or neurodegenerative conditions, however, microglial MHC- II expression is quickly upregulated (Perry et al., 1987; Kreutzberg, 1996; Gottfried-Blackmore et al., 2009). One of the largest barriers to comprehending this mechanism comes from the observation that microglia are unable to travel to the lymph nodes, limiting their ability to act as APCs. Several studies have proposed that the CNS fulfills the role of a functional lymphatic system, where antigen-presenting microglia can come into contact with peripheral APCs in the meninges and choroid plexus, effectively playing an active role in T-cell mediated acquired immunity in the CNS (Louveau et al., 2015). However, recently it is speculated that antigens can be delivered to meningeal lymphatic vessels, where meningeal macrophages reside (Raper et al., 2016). There is no evidence of microglial presence in this region, suggesting their limited role for antigen presentation (Mildner et al., 2017).
Resting microglia are known to exhibit dynamic behavior, surveying their environment and making contacts with surrounding cells in order to execute a variety of functions. When activated, microglia become highly motile cells, migrating toward pathological stimuli such as debris and lesions. Chemotaxis from chemo- (chemical) and -taxis (movement), defines the ability of a cell to move in response to chemical triggers. Microglial migration is modulated by microglia-expressed receptors and gradients of different chemoattractant chemokines released by targets in inflammatory conditions (Dijkstra et al., 2004; Wang et al., 2008). The signaling pathways involved in microglia chemotaxis are highly complex. Briefly, studies have highlighted the specific roles of extracellular signal–regulated kinases (ERK1/2), protein kinase A (PKA), phospholipase A2 (PLA2), phosphoinositide 3-kinase (PI3K), and ATP/purinergic receptor signaling pathways in regulating microglial migration (Fan et al., 2017). Monocyte chemoattractant proteins (MCPs) are of particular interest in neuroinflammatory conditions. The expression of CCL2, aka MCP-1, has been associated with microglia activation in the pathogenesis of multiple sclerosis (Simpson et al., 2000) and AD (Conductier et al., 2010). Additionally, neuron-derived Fractalkine (CXCL1) stimulates microglia migration through CX3CR1 receptors (Harrison et al., 1998). Interestingly, CXCL1 is implicated in a variety of neurodegenerative disorders by acting as an anti-inflammatory agent, which may prove to be an interesting therapeutic target (Desforges et al., 2012).
A key function of microglia is their ability to respond rapidly to immune-mediated insults and physical damage in the brain. Microglia modulate the stress response to a variety of pathological triggers in CNS diseases reviewed extensively by Streit and Graeber et al. (Streit et al., 2004; Graeber et al., 2011). It is well-established that physiological responses to infection in the periphery are propagated through microglia in the CNS directly without the help of a cellular messenger (Chen et al., 2012), a response that is largely reduced after microglia depletion (Elmore et al., 2014). The propensity of the brain to propagate an inflammatory response is increased naturally due to aging (Rosczyk et al., 2008). These triggers range from autoimmune damage, such as demyelination from multiple sclerosis (Vowinckel et al., 1997; Ponomarev et al., 2005), CNS infection (Rock et al., 2004), protein aggregation (such as amyloid plaques) (Kamphuis et al., 2012), or cellular debris from neurodegeneration (Fraser et al., 2010), to physical damage from trauma or ischemia. Microglia also contain a number of purinergic receptors that respond to extracellular ATP and ADP, which are signs of possible necrosis and cellular injury (Inoue and Tsuda, 2012). Upon activation, microglial physiological function is altered, characterized by changes in morphology, coupled with upregulation of cell surface receptors and expression of chemokines and cytokines, all dependent on the triggering event (Perry and Teeling, 2013). The have been a subject of intense debate for the past few decades. Studies have demonstrated that microglia activation, independent of other cytotoxic elements, impacts synaptic function (Selkoe, 2002; Di Filippo et al., 2008) and has neurotoxic effects (von Bernhardi et al., 2015b) that correlate with neurodegeneration and decline in cognitive abilities (Cagnin et al., 2001; Kim and de Vellis, 2005). However, it is also widely believed that transient microglia activation is beneficial in neuroinflammatory conditions by promoting neuron survival (Neumann et al., 2006) and repair after brain injury (Kitamura et al., 2004) through anti-inflammatory signaling.
Microglia activation in neuroinflammatory conditions is mediated by a variety of complex signaling pathways, recently reviewed in Kaminska et al. (2016). Pathogen associated molecular patterns (PAMPs) and damage or danger -associated molecular patterns (DAMPs) activate PRRs (pattern recognition receptors) (Janeway, 1992; Kigerl et al., 2014), triggering crucial responses to immune-related insults and physical injury. PAMPs are expressed by microorganisms and play a critical role in innate immunity. DAMPs are produced by damaged cells and trigger a microglial response to brain injury. PAMPs and DAMPs have been reported to mediate microglia activation and immune response via a variety of PRRs, including Toll-like receptors (TLRs) and nucleotide oligomerization domain (NOD)-like receptors (NLRs) (Kigerl et al., 2014). TLRs are commonly activated in CNS injury and promote transcription of pro-inflammatory cytokines via activation of NF-κB and MAP kinase signaling pathways (Akira and Takeda, 2004). Lipopolysaccharide (LPS) is a PAMP of particular relevance to neurodegenerative diseases. It exerts its function through TLR4 and results in the production of several pro-inflammatory cytokines (Lu et al., 2008). TLR4 mutations were shown to reduce microglia activation, while stimulation of TLR4 modulated cytokine expression in AD models (Jin et al., 2008). NLRs trigger the formation of large protein complexes that activate caspace-1, an essential molecule in the production of pro-inflammatory cytokines (Martinon et al., 2002; Kigerl et al., 2014). NLRP3 is expressed on microglia and participates in microglia activation in AD and several prion diseases (Halle et al., 2008; Kigerl et al., 2014). Microglia activation is also mediated by purines, such as ATP and UTP, released by apoptotic cells. These induce chemotaxis and phagocytosis via ubiquitously expressed purinergic receptors on microglial cell surfaces (Koizumi et al., 2013). Additionally, research has shown that in neuroinflammatory conditions, neurotransmitters have the capacity to mediate microglia-neuron interactions (Mead et al., 2012). Microglia express glutamate receptors, which allows them to sense neuron-released extracellular glutamate, a potent neurotoxic factor (Taylor et al., 2005). Interestingly, studies have reported that upon activation, microglia are self-producers of glutamate, which induces excito-neurotoxicity in neurons and contributes to the pathology of several neurodegenerative diseases (Takeuchi et al., 2006). This suggests that microglia are capable of autocrine signaling and supports the idea of a self-renewing neurotoxic cycle in chronic inflammatory conditions. Microglia activation is also initiated by the absence of certain signaling pathways. CD200 is expressed by neurons and interacts via CD200R expressed on microglial cell surfaces. This interaction maintains microglia in a resting, inactive state (Hoek et al., 2000). CD200 expression is downregulated with age and contributes to increased microglia activation and neuroinflammation (Lyons et al., 2007).
Aging is the single most dominant risk factor for all neurodegenerative disorders, resulting in an impairment in protein production, homeostasis, chaperone-mediated folding, trafficking, stability, clearance, and autophagy. It is important to note that the CNS undergoes several changes during aging including, the shrinking of cortical areas (Salat et al., 2004; Raz et al., 2005), restricted neurogenesis (Praag et al., 2005), reduction in synaptic density, reduction in cognitive and psychomotor function in both in humans and in mice (Godbout et al., 2008; Hayashi et al., 2008), and reduction in glucose metabolism in various regions (Salmon et al., 2003; Kalpouzos et al., 2009; Hsieh, 2012). Additionally, aging also typically leads to an overall increase in the level of proinflammatory cytokines, such as IL-1β, IL-6, CD68, CD11b, and Toll-like receptors (TLRs) (Maher et al., 2004; Godbout et al., 2005), and a decrease in anti-inflammatory cytokines, such as IL-10 and IL-4 (Maher et al., 2005; Nolan et al., 2005). Furthermore, overall brain volume normally decreases by approximately 20% by the time a person turns 100 years old. It is also worth noting that the aged brain frequently has impaired vasculature, resulting in reduced oxygen and nutrient delivery to the CNS that may be exacerbated in certain brain regions (Montagne et al., 2015). Moreover, studies have suggested that blood-brain-barrier (BBB) permeability is increased by aging (Blau et al., 2012; Enciu et al., 2013), suggesting a greater susceptibility to external factors.
The natural propensity of the CNS to adopt a more inflammatory microenvironment during aging earned it the nickname “inflamm-aging” (Franceschi et al., 2000; De Martinis et al., 2005). This may be due in part to the increase in reactive oxygen species (ROS) that is evident in the aged brain. Microglia are largely responsible for the production of these species, which include lipid peroxides, superoxide anions, and hydroxyl radicals (Coatrieux et al., 2007). These molecules, in turn, lead to increased oxidative stress and elicit neurotoxic effects. Oxidative stress is known to initiate neuronal cell death in vitro and via a high calorie diet in vivo (Bros et al., 2014; Treviño et al., 2015). Conversely, some anti-inflammatory factors are known to also increase in concentration in the aged CNS. For example, TGF-β1 is a potent anti-inflammatory factor upregulated in the aged brain (Blobe et al., 2000; Tichauer et al., 2014). TGF-β1 has been shown to promote microglial phagocytosis of Aβ (Wyss-Coray et al., 2001). When considering the effect of age on various CNS responses to different stressors, aged mice, in comparison to young mice, appeared to frequently exhibit exaggerated or prolonged release of proinflammatory cytokines, worsened cognitive decline, as well as age-dependent anxiety-like behavior and sociability changes (Shoji et al., 2016).
Microglia undergo changes in their morphology, phagocytic activity, chemotactic activity, surveying activity, and inflammatory responses that could be relevant to their involvement in disease. Research suggests age-related changes prime microglia to polarize and cause damage in response to disease-related insults. When trying to understand the true nature of pathological conditions, it is important to distinguish the changes resulting from the disease itself, vs. normal age-related variations.
Microglia undergo key morphological changes during aging. Microglial surveying processes are reported to be less dynamic, less complex, and to travel more slowly as mice age (Sierra et al., 2007; Damani et al., 2011). This suggests that responses to pathogens, aggregated proteins, or injury will be delayed in aging brains in comparison to younger mouse brains. In the aged brain, microglia appear to have enhanced proliferation in response to injury, shown in a facial nerve axotomy study in rats (Conde and Streit, 2006). Migration velocity of microglia in response to injury also appears to be affected by aging (Damani et al., 2011; Hefendehl et al., 2014). Studies have shown that in aged animals, microglia survey the environment at a lower speed (Hefendehl et al., 2014), possess thinner and fewer distal branches, and contain spheroids within the major processes (Egensperger et al., 1998; Simmons et al., 2007; Streit et al., 2009). It is possible that in these conditions, myelin fragmentation significantly contributes to the formation of these spheroid inclusions in microglia (Safaiyan et al., 2016). Furthermore, aging reduces microglia cell soma volume and results in decreased tissue distribution homogeneity. (Euler and Schuitemaker, 2012; von Bernhardi et al., 2015b). These characteristics are typically referred to as microglial dystrophy, and are considered to be a normal age-dependent phenotypic state. Recently, a new characterization termed “dark microglia” was established (Bisht et al., 2016). These microglia, identified by their extremely electron-dense soma, become more prominent with age and are especially present in disease states. It is suggested that this may represent a senescent state of microglia. Dark microglia are thought to be caused by a build-up of lipofuscin and increased mtDNA mutations (Wong, 2013).
As microglia age, they undergo many changes at the expression level that confer a heightened inflammatory response. For instance, aged microglia express more MHC-II, as well as mouse CD68 (Godbout et al., 2005; Henry et al., 2009). CD200, a membrane glycoprotein expressed on neurons, astrocytes, and oligodendrocytes, acts as a resting or pro-ramification signal for microglia, which express CD200R. Research has shown that CD200 is decreased in the human AD brain (Walker et al., 2009). CX3CL1 is a cytokine present in neurons that appears to have similar functions as CD200 in promoting microglial ramification. CX3CL1 interacts with CX3CR1, which is also widely expressed in microglia (Fuhrmann et al., 2010). Several studies have suggested that Fractalkine signaling appears to be reduced in the aged brain (Lyons et al., 2009; Bachstetter et al., 2011; Vukovic et al., 2012). Smad3, responsible for the canonical signaling pathway for TGFβ and its anti-inflammatory effects, is reduced in the aged brain (von Bernhardi et al., 2015a). Furthermore, IFN-γ, a potent activator of microglia and initiator of pro-inflammatory gene transcription (Rock et al., 2004; Klegeris et al., 2005), is increased in the aged brain. Genes conventionally known to influence microglia maturation were found to be master regulators of age-dependent changes in microglial phenotype (Wehrspaun et al., 2015). In many species, Iba-1 expression is increased in microglia due to age, often accompanied by a less ramified morphology (Streit et al., 2004), suggesting a more proliferative microglial state. It is also important to note age-related changes in astrocytes, which include an increase in glial fibrillary acidic protein (GFAP) expression, indicating a more pro-inflammatory phenotypic state (Godbout et al., 2005).
Researchers have sought to further elucidate age-associated changes by investigating the number and dynamics of existing microglia in the CNS. There does not appear to be a change in the overall number of microglia as the brain ages. However, during aging, as microglia generally become dysfunctional, they remain in the brain for longer periods of time (Mosher and Wyss-Coray, 2014). A recent study suggested that some microglia can live to be as much as 40 years old, with an average lifespan of 4.2 years and typical yearly turnover rate of 28% (Réu et al., 2017). Furthermore, the effect of peripheral monocyte infiltration on microglia phenotype must be taken into account, given that the cytokine-release profile can differ between the two (Ritzel et al., 2015). A previous study in rats has shown an age-associated increase in blood-derived monocytes, identified as CD11b+ CD45high cells (Blau et al., 2012).
Research investigating the effect of long-term intraperitoneal LPS injection has suggested that some of these changes may be more prominent in certain brain regions (Hart et al., 2012). Indeed, recent studies have revealed that the effect of aging on the transcriptome of microglia is highly dependent upon location within the CNS (Grabert et al., 2016). Given that neurodegenerative diseases frequently follow a region-specific onset, it is important to consider the idea that region-specific microglia priming could contribute to this phenomenon. The fact that the cerebellum is significantly less susceptible to amyloid deposition (Johnson-Wood et al., 1997), while microglia appear to exhibit a hyper immune-alert phenotype (Grabert et al., 2016) during aging in this region, supports this notion.
During aging, microglia generally seem to exhibit an enhanced response to both CNS and peripheral insults. In vivo, aged mice appear to undergo an exaggerated inflammatory response to peripheral LPS injection, defined as an increased release of pro-inflammatory cytokine IL-1β (Godbout et al., 2005). Although immunoreactivity of microglia seems to be increased, age also appears to promote a senescent phenotype in microglia that reduces their functional capabilities, which appears to be accentuated in vitro. One study pointed out the propensity of isolated microglia to be more ramified, and exhibit a reduction in chemotaxis, phagocytosis, autophagic capacity, and overall reactivity (Caldeira et al., 2014) after aging in vitro. These findings were recently reproduced in the context of amyloid pathology, where microglia exhibited reduced phagocytic capabilities after 2 weeks in culture (Caldeira et al., 2017). Primary microglia isolated from 15 month old C57BL/6 were shown to have increased secretion of IL-6 in response to LPS, as well as a reduced ability to phagocytose Aβ oligomers, when compared to younger mice (Njie et al., 2012). A decrease in the ability to migrate was also noted, along with a more senescent phenotype, and less complicated inflammatory response. Furthermore, primary microglia isolated from aged mice exhibit increased release of pro-inflammatory TNF-α, IL-1β, IL-6, and IL-10 in response to challenge with LPS (Sierra et al., 2007). Aged microglia also tend to secrete more ROS, while those from young animals predominantly secrete NO (Tichauer et al., 2014). It can be difficult to distinguish whether these established changes in microglia in vitro are due to age or due to the effects of changing from the in vivo to in vitro environment. Nonetheless, these findings present strong evidence that aged microglia are more readily primed for activation, may be easily triggered by pathological elements in neurodegenerative diseases.
Microglia activation in vitro is often classified into two categories, M1 pro-inflammatory classical activation and M2 anti-inflammatory alternative activation (Colton, 2009). This bipolar model has evolved and is now understood to represent a spectrum, where activation status can fall anywhere between M1 and M2 (Mantovani et al., 2005; Martinez and Gordon, 2014). Pro-inflammatory molecules, such as IFN-γ, TNFα, and LPS, induce the M1 classical activation of microglia (Delgado and Ganea, 2003; Martinez and Gordon, 2014). Microglia in the pro-inflammatory state secrete a variety of inflammatory cytokines, including TNF, IL-6, IL-12, IL-23, IL-1β, as well as other cytotoxic molecules, such as ROS and NO, all of which promote neurotoxicity and reinforce the inflammatory response (Delgado and Ganea, 2003; Cherry et al., 2014). Oxidative stress is implicated in nearly all neurodegenerative disorders (Gandhi and Abramov, 2012). Studies have suggested that the accumulation of reactive oxidative species results in neuronal damage and triggers apoptosis (Gilgun-Sherki et al., 2001). LPS has been show to mediate activation-induced production and secretion of ROS and reactive nitrogen species (RNS) in microglia (Dimayuga et al., 2007). Using a co-culture system of microglia and fetal neuronal cells, Chao and collaborators revealed that both LPS and IFN-γ stimulation resulted in the production of NO, which induced neurotoxicity in neuronal cells. The production of ROS and NOS was later shown to be dependent on nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity in microglia (Qin et al., 2004; Block and Hong, 2007). In vivo, mice deficient of NADPH oxidase were demonstrated to experience reduced nigrostriatal degeneration in response to systemic LPS injection (Qin et al., 2013), suggesting that microglia are capable of being modulated to prevent harmful activation.
The M1 pro-inflammatory profile of microglia is counter to the M2, anti-inflammatory activation state. Stein and collaborators first reported the ability of microglia to adopt anti-inflammatory properties upon stimulation by IL-4 (Stein et al., 1992). Microglia activation by IL-4 has been shown to upregulate IGF-1 production, leading to neuroprotective and regenerative effects (Butovsky et al., 2006). Additionally, other Th2-associated cytokines, such as, IL-10 and TGFα, as well as glucocorticoids, have been reported to promote M2 activation in microglia (Goerdt et al., 1999). M2 microglia have further been subdivided into three functional subclasses: M2a, M2b, and M2c (Mantovani et al., 2004; Chhor et al., 2013). M2a is induced in microglia via IL-13 and IL-4 signaling and is primarily responsible for Arg-1 production, a molecule known to participate in collagen formation facilitating tissue repair (Chhor et al., 2013). Arg1 was also recently shown to increase uptake of Aβ (Cherry et al., 2015). Microglia M2b phenotype is triggered by TLR agonists. Interestingly, the M2b subtype is capable of producing both pro- and anti-inflammatory cytokines (Bell-Temin et al., 2015). The M2c subtype, induced by IL-10, TGFα, and glucocorticoids, have two key functions following brain injury; termination of the pro-inflammatory immune response (Bell-Temin et al., 2015) and repair and regeneration after brain injury (Mantovani et al., 2004). A more recent study validated microglia's regenerative role in disease states by showing that TGFα derived from M2 microglia encourages proliferation and maturation of neural precursor cells in tissue damaged from ischemic stroke (Choi et al., 2017).
The M1/M2 classification scheme contributes to a basic understanding of well-defined microglia-mediated immunological responses in vitro. Several experts, however, have questioned its comprehensiveness in describing in vivo processes and its validity in disease states (Butovsky et al., 2014; Martinez and Gordon, 2014; Ransohoff, 2016). Microglia are thought to be particularly hard to research, given that their gene expression profiles can change fairly dramatically when taken from the CNS and placed into the in vitro environment (Gosselin et al., 2017). Particularly, there appears to be an increase in the expression of genes associated with inflammation and stress. SORL1, the receptor for APOE protein whose deficiency has been noted in AD patients (Scherzer et al., 2004), appears to be under-expressed in the in vitro environment (Gosselin et al., 2017). Isolation of primary microglia is a tenuous and complicated process. Immunohistochemistry in microglia is also noted amongst researchers to be challenging, due to issues with granular staining and autofluorescence (Koellhoffer et al., 2017). Microglia are identified by a variety of markers to distinguish them from other glial cells and neurons. Unfortunately, they share a number of these markers with peripheral macrophages, making them hard to distinguish. Recent research suggests a third, new classification of microglia, may better reflect their in vivo phenotypes, specifically in disease states.
Beyond normal aging, microglia in neurodegenerative conditions experience a specific change in phenotypic state, which researchers have struggled to characterize. Given that there are clear distinctions between microglia that promote neurogenesis and reverse atrophy, and those that release ROS and pro-inflammatory cytokines, understanding the phenotypic state responsible for mediating neuroinflammatory damage is of paramount importance. Potential therapeutic interventions should target the specific deleterious activities of harmful microglia, while leaving beneficial neuroprotective mechanisms unhindered.
Microglia activation is a necessary and beneficial function in response to acute neuro-inflammatory events and aids in sustaining brain homeostasis. Chronic activation, however, can occur from excessive neuronal or immune-related damage in various CNS diseases (Polazzi and Monti, 2010). This can lead to the sustained release of pro-inflammatory molecules and harmful production of ROS which results in detrimental effects. Moderate increases in these cytokines are generally considered a normal part of aging. However, large increases, as observed in AD, lead to excessive neurotoxicity (Giunta et al., 2008; Glass et al., 2010). In turn, increased neurotoxicity triggers additional microglial activation, initiating a harmful loop of inflammation and neuronal damage termed “reactive microgliosis” (Streit et al., 1999). Microglia have been shown to be activated in nearly all neurological disorders (Neumann et al., 2009). Signs of microglia activation have been reported in autoimmune diseases, such as multiple sclerosis (Goldmann and Prinz, 2013; Luo et al., 2017), prion diseases, such as Creutzfeldt-Jakob Disease (CJD) (Aguzzi and Zhu, 2017), neurodegenerative diseases, such as Parkinson's Disease (PD) and AD, as well as traumatic brain injury (TBI) and ischemia (Jassam et al., 2017; Liu et al., 2017). Their implication in the pathophysiology of such a wide variety of neurological disorders has made them an interesting target for potential therapeutic approaches.
With regards to AD, some observations can be confusing. As mentioned in the previous section, aged microglia frequently become “dystrophic” and are highly immunoreactive. However, studies have reported reduced phagocytosis of Aβ in older AD mice (Floden and Combs, 2011), which may be caused by decreased expression of CD36, an Aβ interacting protein. In AD patients, as well as mouse models of AD, Smad3 signaling appears to be reduced (Tesseur et al., 2006; Ueberham et al., 2006), likely resulting in the pathological activation of microglia. Overall microglia number in both Alzheimer's patients and AD mouse models is increased and correlates with disease severity (Olmos-Alonso et al., 2016). This suggests that disease pathology promotes microglia proliferation. Microglia behavior in AD may also depend heavily upon the stage of the disease. One study reported that Aβ fibrils enhance microglia phagocytosis, while Aβ oligomers attenuate phagocytosis (Pan et al., 2011). This finding suggests that microglia presence is more significant after considerable protein deposition.
A recent study expanded current microglia classification beyond the typical M1/M2 scheme. Researchers in this study named this third category of microglia, “disease-associated” microglia (MGnD), using a gene expression profile revealed through K-means clustering. These microglia revealed a reduction in the expression of 68 homeostatic microglial genes and upregulation of 28 inflammatory molecules (Krasemann et al., 2017). A large portion of these responses were eliminated due to microglia-specific KO of APOE, suggesting that APOE potently induces phenotypic changes in disease-associated microglia and is up-regulated in the presence of plaques (Krasemann et al., 2017). MGnD microglia also exhibited a significant increase in miR-155 expression. MiR-155, a significantly up-regulated microRNA in microglia after challenge with an insult, is largely responsible for the release of pro-inflammatory cytokines IL-6, IL-1β, NOS2, and TNFα (Woodbury et al., 2015). Beyond microglia interaction, APOE is important for maintaining hippocampal neurogenesis and suppressing astrogenesis in mice, both of which are reduced via APOE4 mutation (Li et al., 2009). TREM2 KO was shown to have very similar effects on the gene expression profile as the APOE KO, suggesting these two molecules work concurrently to determine microglia phenotype. Another large study utilizing single-cell RNA-seq to identify unique microglia subpopulations present in 5XFAD mice found gene expression profiles associated with increase in APOE, TREM2, and Cst7 expression, amongst others, in disease-associated microglia, referred to as “DAM.” These same microglia exhibited decreased expression of homeostatic genes P2RY12 and CX3CR1 (Keren-Shaul et al., 2017). Lpl was selected as a consistent marker for the disease associated microglia subtype, and found to be present on phagocytic plaque-associated microglia positive for Thioflavin-S. The expression of genes known to influence microglia maturation and ramification such as RUNX1, SALL 1, TAL1, and IRF8 is also affected by AD pathology (Olmos-Alonso et al., 2016). It is important to recognize the differences between MGnD and classically activated M1 or M2 microglia (Figure 2). MGnD are a result of chronic exposure to disease pathology and can be distinguished from M1 microglia by the presence APOE, TREM2, and M2-associated anti-inflammatory markers such as arginase 1 (Arg1) and chitinase-3-like protein (Ym1), as well as the absence of homeostatic transcription factor Egr1 (Krasemann et al., 2017). In contrast, M1 microglia activated through LPS down-regulate TREM2 expression (Kleinberger et al., 2017; Zhong et al., 2017b). Furthermore, plaque-associated microglia exhibit a hyperactive immune response to LPS injection in comparison to non-plaque-associated microglia (Yin et al., 2017), suggesting their contribution to neuroinflammation in disease states is more detrimental.
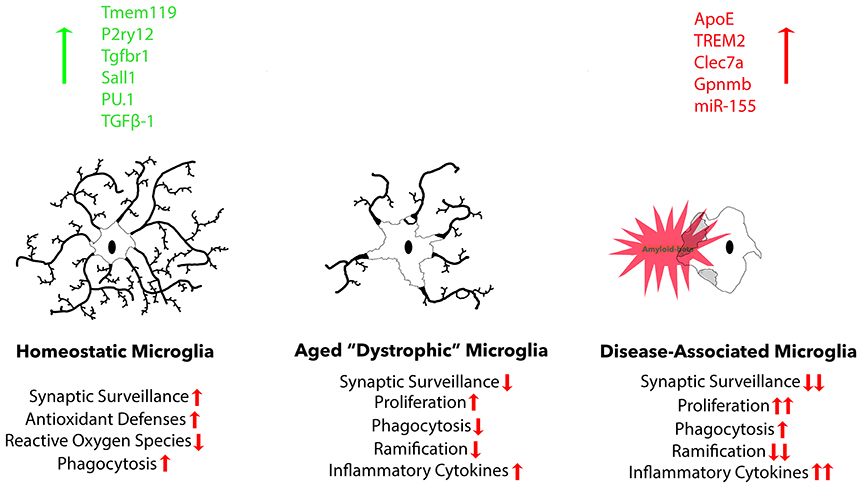
Figure 2. Three common microglia phenotypes are described. Homeostatic microglia are found in the adult brain under non-infectious, non-diseased, and non-aged conditions, exhibiting robust expression of homeostatic microglial markers: Tmem119, P2ry12, Tgfbr1, and transcription factor Sall1. During normal aging, homeostatic markers gradually decline, resulting in reduced functional aspects, including proliferation, phagocytosis, ramification, and cytokine secretion. Finally, there is a distinct microglia phenotype that is associated with neurodegeneration that possesses a more exacerbated dystrophic phenotype, but is specifically associated with plaques and dystrophic neurites that cause neurodegeneration.
The MGnD and DAM phenotypes are largely dependent on TREM2 expression, which is up-regulated in the microglia of diseased brains and MGnD microglia specifically (Ofengeim et al., 2017; Yin et al., 2017). This evidence suggests that the APOE-TREM2 signaling cascade is responsible for the changes in gene expression profile which induce the MGnD phenotype in microglia (Keren-Shaul et al., 2017; Krasemann et al., 2017; Yin et al., 2017). Two papers recently reported conflicting results of TREM2 KO in different mouse models of tauopathy. In hTau mice, prior to the time of expected significant neuronal loss, TREM2 KO was reported to increase the deposition of hyperphosphorylated tau and promote a less homeostatic microglial phenotype (Bemiller et al., 2017). However, in a study investigating PS19 mice with TREM2 KO, researchers found a reduction in brain atrophy in the absence of any changes in tauopathy or a reduction of MGnD-associated markers APOE and Cst7 (Krasemann et al., 2017; Leyns et al., 2017). Collectively, these recent studies support the idea that the APOE-TREM2 signaling pathway shifts microglia toward an MGnD phenotype, which actively contributes to the tauopathy-induced reduction of neuropil space in the entorhinal cortex of PS19 mice. During aging in the CNS, microglia become more dystrophic, suffer a reduction in functional characteristics, and begin to exhibit a gene expression signature similar to that of MGnD microglia. Although age is not required for the generation of the MGnD phenotype, aged microglia are primed to make this transition. Further research is necessary to generate a more concrete and time-dependent understanding of the APOE-TREM2 signaling complex as it relates to the MGnD phenotype.
Several techniques exist allowing researchers to design pharmacological agents aimed at reducing the buildup of pathological Aβ. One validated method is to focus on developing inhibitors of BACE1, which is the membrane protease responsible for the beta-site cleavage of APP (Eketjäll et al., 2016). Several of these compounds have been in clinical trials in recent years, such as the Merck EPOCH trial in patients with mild to moderate AD. This trial was speculated to have failed because the drug was administered too late to substantially address the pathology. Another obstacle in clinical trials is heterogeneous patient enrollment, which introduces the possibility of including patients that do not have AD, but suffer from dementia due to a different disease. Instead of inhibiting Aβ production, some compounds are being designed to address the prion-like properties of amyloid aggregation itself. This concept uses a small molecule, which binds to nascent amyloid fibrils or aggregates, preventing nucleation or further accumulation (McLaurin et al., 2006; Habchi et al., 2016). Additionally, there are also antibodies designed to bind pathological Aβ protofibrils, a passive immunization technique in which the antigen, Aβ, is bound, leading to complement activation and phagocytosis by neighboring phagocytes (Bard et al., 2000). However, considerable evidence has suggested this pursuit may be clinically ineffective—decreasing amyloid burden in patients with an already significant degree of cognitive deficits, and hence neurodegeneration, does not appear to dampen the rate of decline (Holmes et al., 2008). This was most recently found during Phase 3 of a clinical trial for Solanezumab (Doody et al., 2014). Aducanumab, a fully human IgG isolated from cognitively normal donors that binds a conformational epitope of Aβ, is currently in Phase 3 trials (Sevigny et al., 2016). Neurofibrillary tangles (NFTs), the second pathological hallmark of AD, are another promising target for pharmacological intervention. Several classes of pharmacologic agents may be effective in preventing both the aggregation and spread of NFTs in AD. Small molecule drugs, that work by binding to tau to inhibit its aggregation, are currently being developed (Harrington et al., 2015). Additionally, several tau vaccines are also currently in development, with one entering Phase II of clinical trial (Theunis et al., 2013; Kontsekova et al., 2014).
Pharmacological modulation targeting the intracellular trafficking of APP via intervention on the enzymes involved in its cleavage may also be successful in regulating Aβ accumulation. APP is not exclusively found on the cell surface; it is most frequently localized to the trans-Golgi network (TGN) (Caporaso et al., 1994; Hartmann et al., 1997; Xu et al., 1997). After shuttling to the plasma membrane, APP is reinternalized into endosomes and, eventually, lysosomes (Haass et al., 1993a,b; Koo and Squazzo, 1994), where Aβ production primarily occurs (Huse et al., 2000, 2002). Therefore, enhanced sequestering of APP to the plasma membrane and out of the TGN is a potential therapeutic venue for AD. Currently, there are no drugs in development targeting this mechanism. TPI-287, a microtubule-stabilizing agent, is a unique drug, which influences APP intracellular trafficking, and may also facilitate kinesin-mediated axonal transport of APP (Zempel and Mandelkow, 2014).
Given the overwhelming evidence supporting the role of microglia in the pathogenesis of neurodegenerative functions, it is of interest to discuss potential therapeutic interventions targeting the neurotoxic effects of microglia.
One strategy is to pharmacologically enhance microglial clearance of protein aggregates. Sargramostim is a synthetic form of the hematopoietic growth factor granulocyte-macrophage colony-stimulating factor (GM-CSF), which is FDA approved to increase white blood cell count after chemotherapy (Markovic et al., 2008). Evidence suggests that GM-CSF stimulates phagocytosis of Aβ via bone-marrow derived macrophages and microglia (Mitrasinovic et al., 2001), although GM-CSF may also potentially exacerbate their inflammatory response. Sargramostim was entered into two Phase 2 clinical trials to assess treatment and safety in patients with mild cognitive impairment or AD. One of these trials has since been withdrawn. Valproic acid, an anticonvulsant medication which exerts its function via inhibition of voltage-gated sodium channels and increases levels of gamma-aminobutyric acid (Löscher, 2002), can enhance microglial phagocytosis of Aβ, although it has not yet been tested in clinical trials. Additionally, vasoactive intestinal peptide, a multifunctional neuropeptide, which can enhance microglial phagocytosis, while suppressing the production of TNF-α and ROS (Song et al., 2012), could also be effective for restoring homeostatic microglial functions.
Gotz et al. previously demonstrates the acceleration of NFT formation by Aβ injection into the mouse brain (Götz et al., 2001). This finding was validated in a double transgenic mouse model. The model was created by crossing P301L tau mice with APP mice expressing the familial Swedish mutations of APP (K670N and M671L), resulting in a marked acceleration in tau pathology development (Lewis et al., 2001). The same result was reported in a different double transgenic model, created by crossed PS19 mice with APPV717F mice (Hurtado et al., 2010). The potential contribution of microglia to accelerate tau pathology in the presence of Aβ accumulation has yet to be tested. The spread of pathogenic tau protein may be facilitated by phagocytic microglia, which are activated by Aβ accumulation in the brain.
Immune cells, including microglia, are highly efficient in secreting extracellular vesicles, such as exosomes and ectosomes (Robbins and Morelli, 2014; Greening et al., 2015). Research has shown that microglia promote the spread of pathological protein aggregates through exosomes (Sarko and McKinney, 2017; Soria et al., 2017). Exosomes are extracellular vesicles that are between 30 and 150 nm in size (Zomer et al., 2010; Vlassov et al., 2012; Thompson et al., 2016) and possess a myriad of roles in the CNS and periphery (Budnik et al., 2016). They are frequently excreted by antigen-presenting cells, such as microglia (Nair-Gupta et al., 2014), but are also secreted by all other cells of the CNS (Frühbeis et al., 2012). In AD, microglia have been shown to congregate around senile plaques in order to phagocytose them, consequently secreting Aβ oligomer-containing exosomes (Rajendran et al., 2006). Exosomes isolated from AD brains also contain hyper-phosphorylated tau oligomers (Saman et al., 2012), which are interestingly found in the CSF of AD patients (Saman et al., 2012; Fiandaca et al., 2015). In ALS, exosomal TDP-43 is increased, suggesting that pathological protein-containing exosomes play a role in several neurodegenerative disorders (Iguchi et al., 2016). Exosomal spread is the proposed mechanism through which microglia enhance tau propagation in mouse models of AD (Asai et al., 2015). In this study, depletion of microglia was demonstrated to reduce the spread of AAV-induced tau pathology from the medial entorhinal cortex to the dentate gyrus.
Inhibiting microglia-mediated exosome excretion may show promising results in halting disease progression in neurodegenerative disorders. Pharmacological inhibition, via GW4869, of neutral sphingomyelinase 2 (nSMase2), which synthesizes ceramide from sphingomyelin and is critical for exosome synthesis, successfully halted the packaging of human tau into exosomes in microglia (Asai et al., 2015). In addition to preventing exosome-mediated tau propagation, GW4869 also attenuates the release of pro-inflammatory cytokines in macrophages in response to LPS, while not appearing to have potent cytotoxic affects (Essandoh et al., 2015). Increased exosome secretion was demonstrated to enhance spread of prion protein in cell culture (Guo et al., 2016), which was prevented via treatment with GW4869. Evidence for the role of exosomes in prion disease, including ALS, have been presented (Vella et al., 2007; Guo et al., 2016; Iguchi et al., 2016). However, administration of GW4869 in a mouse model of ALS expressing human mutant TDP-43A315T (autosomal mutation of ALS), appeared to worsen disease phenotypes, increase levels of insoluble TDP-43, and cause cytoplasmic accumulation of TDP-43 in neurons (Iguchi et al., 2016). GW4869 was also reported to disrupt synaptic connections (Tabatadze et al., 2010), suggesting potential CNS damage with this compound. The compound impaired spatial memory, as assessed via the Morris Water maze, but not spatial recognition memory, tested in the Y maze. Chronic exposure of GW4869 was also reported to have no effect on novel object recognition (Iguchi et al., 2016), suggesting inconclusive results. An alternative strategy is necessary for regulating exosomes synthesis and secretion.
Preventing the pathogenic change in microglial phenotype and infiltration of peripheral monocytes in neurodegenerative conditions is another promising strategy. Long-term use of non-steroidal anti-inflammatory drugs (NSAIDs) was classically recognized to reduce the risk of developing AD and PD by two meta-analysis studies (Chen et al., 2005; Vlad et al., 2008). However, this was not fully reproduced in clinical trials (Vlad et al., 2008; Imbimbo et al., 2010; Breitner et al., 2011).
Cumulatively, these studies have led scientists to question whether microglia are insufficient for clearing proteinopathies, or if they engage in a mechanism that progresses the pathology. This question is complex and must incorporate the findings of cutting-edge studies, which have used the depletion of microglia via CSF1R inhibitors (PLX3397 and PLX5622 among others), to assess the role of microglia in proteinopathies and disease pathogenesis. These compounds are able to deplete virtually all microglia in the CNS after as little as 1 week of oral administration without significant side effects (Elmore et al., 2014; Dagher et al., 2015; Olmos-Alonso et al., 2016). No abnormal behaviors were observed in wild type mice, even after two months of treatment with PLX3397, as evaluated by contextual fear conditioning, elevated plus maze, open field, and Barnes maze (Elmore et al., 2014). Microglia depletion appeared to block disease-mediated increases in exploratory activity of mice, suggesting this may have a beneficial effect on cognition without significantly affecting amyloid burden (Grathwohl et al., 2009; Dagher et al., 2015; Olmos-Alonso et al., 2016; Spangenberg et al., 2016). CSF1R inhibitors are currently on clinical trials for oncology and joint neoplasm indications, but not for AD or other neurodegenerative disorders. No apparent side effects were detected in treated groups. Cognitive evaluations of patients in these trials will provide us with better information on the effects of microglia have on mental capacity.
Interestingly, replacement of microglia was tested as a means of protecting against excitotoxic injury with success (Vinet et al., 2012). In this study, microglia-deprived hippocampal slice cultures where infiltrated by exogenous microglia. Given that ROS production in microglia is deleterious, one therapeutic strategy may be to inhibit NOX (Altenhöfer et al., 2015). NADPH oxidase is largely responsible for ROS production in microglia in response to LPS (Qin et al., 2004). Furthermore, NOX activity was found to correlate with cognitive impairment in AD, while KO of NOX was associated with a reduction in cognitive impairment (Bordt and Polster, 2014). Studies have reported that treatment with the NOX inhibitor apocymin leads to a reduction in amyloid burden, reduced overall microglia number, and reduced cerebral amyloid angiopathy in an AD mouse model (Han et al., 2015). In addition, delivery of adeno-associated virus (AAV) containing CD200, a glycoprotein involved in maintaining a quiescent state in microglia, into the hippocampus of 6-month old Tg2576 APP mice resulted in increased neurogenesis and reduced amyloidosis (Varnum et al., 2015). Microglia were also treated with CD200 in vitro, which enhanced their phagocytosis and promoted an M2-like phenotype. These studies support the notion that aged microglia are largely ineffective in the brain and are incapable of significantly reducing amyloid burden. Furthermore, their inflammatory presence serves only to exacerbate cytotoxicity of neurons, leading to increased neuronal loss and cognitive impairment. More thorough assessment of these findings may be necessary, but ample evidence exists to suggest that microglia presence in the CNS is detrimental in the context of neurodegenerative disease.
This notion should be considered when interpreting AD research assessing microglial dysfunction, such as studies examining the effect of TREM2 disruption in animal models of amyloidosis. Results of these studies were controversial, with some stipulating that TREM2 in microglia increase phagocytosis and clearance of Aβ (Wang et al., 2015; Xiang et al., 2016; Yuan et al., 2016), while others have suggested that disruption of TREM2 reduces amyloid plaque load (Ulrich et al., 2014; Jay et al., 2015; Krasemann et al., 2017). These differences could be caused by the different functional role of TREM2 at the various stages of Aβ accumulation in the brain. TREM2 is up-regulated in the brains of AD patients and several mutations have been identified risk factors for late-onset AD (Frischmeyer-Guerrerio et al., 2013). TREM2 expression is increased in plaque-associated microglia (Guerreiro et al., 2013) and in particular, in the processes of microglia which are in direct contact with plaques (Yuan et al., 2016). TREM2 disruption has also been shown to reduce the overall amount of plaque-associated microglia (Krasemann et al., 2017). These were unexpected results since phagocytosis of not only Aβ, but a number of other molecules recognized by phagocytes, is dependent on TREM2 (Kleinberger et al., 2014; Xiang et al., 2016; Yuan et al., 2016; Varnum et al., 2017). However, it is important to note that complete depletion of microglia has repeatedly failed to influence overall amyloid deposition (Grathwohl et al., 2009; Dagher et al., 2015; Olmos-Alonso et al., 2016). If TREM2 KO confers a dysfunctional microglia phenotype, which results in increased amyloid burden, then this effect is likely to be caused by the functional activity of microglia (Krasemann et al., 2017). One possibility is the anti-inflammatory effect of TREM2 signaling. Expression of pro-inflammatory cytokines IL-1β, IL-6, and TNF-α in response to challenge with Aβ42 was shown to be drastically increased due to knockdown with DAP12/TYROBP and synergized with TREM2 KO (Zhong et al., 2017a). This also supports the notion that Aβ42 is a ligand for TREM2, although one study reported that the TREM2-TYROBP association is not affected by Aβ presence in cell culture. However, it has also been shown that TREM2 is down-regulated in primary microglia cultures in response to Aβ42 (Zheng et al., 2016), which was prevented with treatment with a JNK inhibitor (Zhong et al., 2017a). JNK inhibitors are proposed as a possible therapeutic target for AD (Yarza et al., 2015) and have entered clinical trials for a number of indications, but have no approvals to date. Nonetheless, research has repeatedly suggested that TREM2 is upregulated in disease-associated microglia (Frank et al., 2008; Frischmeyer-Guerrerio et al., 2013; Krasemann et al., 2017). These findings support the notion that dysfunctional microglia in disease contribute to a cytotoxic environment through the release of pro-inflammatory cytokines and ROS. The connection between APOE, TREM2, and microglia will continue to be investigated.
The contribution of circulating peripheral monocytes to the microglia population in the CNS has been thoroughly investigated and shown to function through parabiotic mechanisms. In these experiments, the blood streams of two mice are connected, where one mouse contains a reporter in circulating monocytes that allows them to be distinguished from those of the partner. One study found low levels of monocyte infiltration after full body irradiation in mice (Hess et al., 2004), while another study attempted the same experiment and did not find evidence of any CNS penetration (Ajami et al., 2007). Two mouse models of microgliosis, facial nerve axotomy and mSOD, did not provide any evidence of CNS penetration either. Reports were also released with evidence suggesting that bone-marrow transplanted myeloid cells can penetrate the CNS mainly when the BBB is compromised (Eglitis and Mezey, 1997; Brazelton et al., 2000; Ajami et al., 2007; Mildner et al., 2009). To test the effect of peripheral lymphocytes on amyloid clearance in brain, parabiosis experiments have been conducted in which the blood supply of young WT mice was connected to that of older APP mice. These experiments did not result in a reduction in amyloid burden, suggesting that immune infiltration of circulating monocytes is not a significant factor (Middeldorp et al., 2016). If circulating monocytes are unable to penetrate the BBB without significant damage to the BBB, this is unlikely to be a factor in neurodegenerative diseases, but could be applicable to other neurologic disorders with BBB impairment, such as traumatic brain injury or stroke.
Microglia are an important aspect of CNS homeostasis, damage repair, proteinostasis, and proteopathy in aging. The aged brain shows changes in microglial phenotype, which are associated with changes in protein clearance, misfolding, aggregation, and spread. Proteopathy and neuronal cell loss in neurodegenerative conditions also contribute to a shift in microglial phenotype from homeostatic to pathological, which permanently leads to harmful inflammatory responses and further promotes cortical degeneration. This mechanism provides us with novel therapeutic targets for AD and related proteopathies.
KC and AV contributed equally to the manuscript. TI wrote and edited the manuscript. All authors listed have approved this work for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work is in funded in part by National Institute of Health grants 5T32GM008541 (KC), 1RF1AG05419 and 1R01AG054672 (TI), Alzheimer's Association DVT-14-320835 (TI), BrightFocus Foundation A2016551S (TI), and Massachusetts Neuroscience Consortium (TI).
Aderem, A., and Underhill, D. M. (1999). Mechanisms of Phagocytosis in Macrophages. Annu. Rev. Immunol. 17, 593–623. doi: 10.1146/annurev.immunol.17.1.593
Aguzzi, A., and Zhu, C. (2017). Microglia in prion diseases. J. Clin. Invest. 127, 3230–3239. doi: 10.1172/JCI90605
Ajami, B., Bennett, J. L., Krieger, C., Tetzlaff, W., and Rossi, F. M. V. (2007). Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 10, 1538–1543. doi: 10.1038/nn2014
Akira, S., and Takeda, K. (2004). Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511. doi: 10.1038/nri1391
Alarcón, R., Fuenzalida, C., Santibá-ez, M., and von Bernhardi, R. (2005). Expression of scavenger receptors in glial cells: comparing the adhesion of astrocytes and microglia from neonatal rats to surface-bound β-amyloid. J. Biol. Chem. 280, 30406–30415. doi: 10.1074/jbc.M414686200
Altenhöfer, S., Radermacher, K. A., Kleikers, P. W. M., Wingler, K., and Schmidt, H. H. (2015). Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid. Redox Signal. 23, 406–427. doi: 10.1089/ars.2013.5814
Asai, H., Ikezu, S., Tsunoda, S., Medalla, M., Luebke, J., Haydar, T., et al. (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593. doi: 10.1038/nn.4132
Athan, E. S., Lee, J. H., Arriaga, A., Mayeux, R. P., and Tycko, B. (2002). Polymorphisms in the promoter of the human app gene: functional evaluation and allele frequencies in Alzheimer disease. Arch. Neurol. 59, 1793–1799. doi: 10.1001/archneur.59.11.1793
Bachstetter, A. D., Morganti, J. M., Jernberg, J., Schlunk, A., Mitchell, S. H., Brewster, K. W., et al. (2011). Fractalkine and CX(3)CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol. Aging 32, 2030–2044. doi: 10.1016/j.neurobiolaging.2009.11.022
Bard, F., Cannon, C., Barbour, R., Burke, R.-L., Games, D., Grajeda, H., et al. (2000). Peripherally administered antibodies against amyloid [beta]-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 6, 916–919. doi: 10.1038/78682
Bell-Temin, H., Culver-Cochran, A. E., Chaput, D., Carlson, C. M., Kuehl, M., Burkhardt, B. R., et al. (2015). Novel molecular insights into classical and alternative activation states of microglia as revealed by stable isotope labeling by Amino Acids in Cell Culture (SILAC)-based Proteomics. Mol. Cell. Proteom. 14, 3173–3184. doi: 10.1074/mcp.M115.053926
Bemiller, S. M., McCray, T. J., Allan, K., Formica, S. V., Xu, G., Wilson, G., et al. (2017). TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegener. 12:74. doi: 10.1186/s13024-017-0216-6
Bisht, K., Sharma, K. P., Lecours, C., Gabriela Sánchez, M., El Hajj, H., Milior, G., et al. (2016). Dark microglia: a new phenotype predominantly associated with pathological states. Glia 64, 826–839. doi: 10.1002/glia.22966
Blau, C. W., Cowley, T. R., O'Sullivan, J., Grehan, B., Browne, T. C., Kelly, L., et al. (2012). The age-related deficit in LTP is associated with changes in perfusion and blood-brain barrier permeability. Neurobiol. Aging 33, 1005.e23–1005.e35. doi: 10.1016/j.neurobiolaging.2011.09.035
Blobe, G. C., Schiemann, W. P., and Lodish, H. F. (2000). Role of Transforming Growth Factor β in Human Disease. N. Eng. J. Med. 342, 1350–1358. doi: 10.1056/NEJM200005043421807
Block, M. L., and Hong, J. S. (2007). Chronic microglial activation and progressive dopaminergic neurotoxicity. Biochem. Soc. Trans. 35:1127. doi: 10.1042/BST0351127
Bordt, E. A., and Polster, B. M. (2014). NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: a bipartisan affair? Free Radic. Biol. Med. 76, 34–46. doi: 10.1016/j.freeradbiomed.2014.07.033
Brazelton, T. R., Rossi, F. M. V., Keshet, G. I., and Blau, H. M. (2000). From marrow to brain: expression of neuronal phenotypes in adult mice. Science 290:1775. doi: 10.1126/science.290.5497.1775
Breitner, J. C., Baker, L. D., Montine, T. J., Meinert, C. L., Lyketsos, C. G., Ashe, K. H., et al. (2011). Extended results of the Alzheimer disease anti-inflammatory prevention trial (ADAPT). Alzheim. Dement. 7, 402–411. doi: 10.1016/j.jalz.2010.12.014
Bros, H., Millward, J. M., Paul, F., Niesner, R., and Infante-Duarte, C. (2014). Oxidative damage to mitochondria at the nodes of Ranvier precedes axon degeneration in ex vivo transected axons. Exp. Neurol. 261, 127–135. doi: 10.1016/j.expneurol.2014.06.018
Brown, G. C., and Neher, J. J. (2012). Eaten alive! Cell death by primary phagocytosis: 'phagoptosis'. Trends Biochem. Sci. 37, 325–332. doi: 10.1016/j.tibs.2012.05.002
Bruttger, J., Karram, K., Wörtge, S., Regen, T., Marini, F., Hoppmann, N., et al. (2015). Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity 43, 92–106. doi: 10.1016/j.immuni.2015.06.012
Budnik, V., Ruiz-Canada, C., and Wendler, F. (2016). Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 17, 160–172. doi: 10.1038/nrn.2015.29
Butchart, J., Brook, L., Hopkins, V., Teeling, J., Püntener, U., Culliford, D., et al. (2015). Etanercept in Alzheimer disease: a randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 84, 2161–2168. doi: 10.1212/WNL.0000000000001617
Butovsky, O., Jedrychowski, M. P., Moore, C. S., Cialic, R., Lanser, A. J., Gabriely, G., et al. (2014). Identification of a unique TGF-β dependent molecular and functional signature in Microglia. Nat. Neurosci. 17, 131–143. doi: 10.1038/nn0914-1286d
Butovsky, O., Ziv, Y., Schwartz, A., Landa, G., Talpalar, A. E., Pluchino, S., et al. (2006). Microglia activated by IL-4 or IFN-γ differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol. Cell. Neurosci. 31, 149–160. doi: 10.1016/j.mcn.2005.10.006
Cagnin, A., Brooks, D. J., Kennedy, A. M., Gunn, R. N., Myers, R., Turkheimer, F. E., et al. (2001). In-vivo measurement of activated microglia in dementia. Lancet 358, 461–467. doi: 10.1016/S0140-6736(01)05625-2
Caldeira, C., Cunha, C., Vaz, A. R., Falcão, A. S., Barateiro, A., Seixas, E., et al. (2017). Key aging-associated alterations in primary Microglia response to beta-amyloid stimulation. Front. Aging Neurosci. 9:277. doi: 10.3389/fnagi.2017.00277
Caldeira, C., Oliveira, A. F., Cunha, C., Vaz, A. R., Falcão, A. S., Fernandes, A., et al. (2014). Microglia change from a reactive to an age-like phenotype with the time in culture. Front. Cell. Neurosci. 8:152. doi: 10.3389/fncel.2014.00152
Cao, T., Thomas, T. C., Ziebell, J. M., Pauly, J. R., and Lifshitz, J. (2012). Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience 225, 65–75. doi: 10.1016/j.neuroscience.2012.08.058
Caporaso, G. L., Takei, K., Gandy, S. E., Matteoli, M., Mundigl, O., Greengard, P., et al. (1994). Morphologic and biochemical analysis of the intracellular trafficking of the Alzheimer beta/A4 amyloid precursor protein. J. Neurosci. 14, 3122.
Carroll, M. C. (2004). The complement system in B cell regulation. Mol. Immunol. 41, 141–146. doi: 10.1016/j.molimm.2004.03.017
Cataldo, A. M., Peterhoff, C. M., Troncoso, J. C., Gomez-Isla, T., Hyman, B. T., and Nixon, R. A. (2000). Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer's disease and down syndrome : differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 157, 277–286. doi: 10.1016/S0002-9440(10)64538-5
Chalermpalanupap, T., Kinkead, B., Hu, W. T., Kummer, M. P., Hammerschmidt, T., Heneka, M. T., et al. (2013). Targeting norepinephrine in mild cognitive impairment and Alzheimer's disease. Alzheim. Res. Ther. 5:21. doi: 10.1186/alzrt175
Chan, A., Magnus, T., and Gold, R. (2001). Phagocytosis of apoptotic inflammatory cells by microglia and modulation by different cytokines: mechanism for removal of apoptotic cells in the inflamed nervous system. Glia 33, 87–95. doi: 10.1002/1098-1136(20010101)33:1<87::AID-GLIA1008>3.0.CO;2-S
Chen, H., Cúrdia, V., and Ferrero, A. (2012). The macroeconomic effects of large-scale asset purchase programmes*. Econ. J. 122, F289–F315. doi: 10.1111/j.1468-0297.2012.02549.x
Chen, H., Jacobs, E., Schwarzschild, M. A., McCullough, M. L., Calle, E. E., Thun, M. J., et al. (2005). Nonsteroidal antiinflammatory drug use and the risk for Parkinson's disease. Ann. Neurol. 58, 963–967. doi: 10.1002/ana.20682
Cherry, J. D., Olschowka, J. A., and O'Banion, M. K. (2014). Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J. Neuroinflam. 11:98. doi: 10.1186/1742-2094-11-98
Cherry, J. D., Olschowka, J. A., and O'Banion, M. K. (2015). Arginase 1+ microglia reduce Aβ plaque deposition during IL-1β-dependent neuroinflammation. J. Neuroinflam. 12:203. doi: 10.1186/s12974-015-0411-8
Chhor, V., Le Charpentier, T., Lebon, S., Oré, M.-V., Celador, I. L., Josserand, J., et al. (2013). Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 32, 70–85. doi: 10.1016/j.bbi.2013.02.005
Choi, J. Y., Kim, J. Y., Kim, J. Y., Park, J., Lee, W. T., and Lee, J. E. (2017). M2 phenotype microglia-derived cytokine stimulates proliferation and neuronal differentiation of endogenous stem cells in ischemic brain. Exp. Neurobiol. 26, 33–41. doi: 10.5607/en.2017.26.1.33
Coatrieux, C., Sanson, M., Negre-Salvayre, A., Parini, A., Hannun, Y., Itohara, S., et al. (2007). MAO-A-induced mitogenic signaling is mediated by reactive oxygen species, MMP-2, and the sphingolipid pathway. Free Rad. Biol. Med. 43, 80–89. doi: 10.1016/j.freeradbiomed.2007.03.036
Colton, C. A. (2009). Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 4, 399–418. doi: 10.1007/s11481-009-9164-4
Conde, J. R., and Streit, W. J. (2006). Effect of aging on the microglial response to peripheral nerve injury. Neurobiol. Aging. 27, 1451–1461. doi: 10.1016/j.neurobiolaging.2005.07.012
Conductier, G., Blondeau, N., Guyon, A., Nahon, J.-L., and Rovère, C. (2010). The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J. Neuroimmunol. 224, 93–100. doi: 10.1016/j.jneuroim.2010.05.010
Dagher, N. N., Najafi, A. R., Kayala, K. M. N., Elmore, M. R. P., White, T. E., Medeiros, R., et al. (2015). Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J. Neuroinflam. 12:139. doi: 10.1186/s12974-015-0366-9
Damani, M. R., Zhao, L., Fontainhas, A. M., Amaral, J., Fariss, R. N., and Wong, W. T. (2011). Age-related alterations in the dynamic behavior of Microglia. Aging Cell 10, 263–276. doi: 10.1111/j.1474-9726.2010.00660.x
De Martinis, M., Franceschi, C., Monti, D., and Ginaldi, L. (2005). Inflamm-ageing and lifelong antigenic load as major determinants of ageing rate and longevity. FEBS Lett. 579, 2035–2039. doi: 10.1016/j.febslet.2005.02.055
Deane, R., Bell, R. D., Sagare, A., and Zlokovic, B. V. (2009). Clearance of amyloid-β peptide across the blood-brain barrier: implication for therapies in Alzheimer's disease. CNS Neurol. Disord. Drug Targets 8, 16–30. doi: 10.2174/187152709787601867
Deane, R., Wu, Z., Sagare, A., Davis, J., Du Yan, S., Hamm, K., et al. (2004). LRP/Amyloid β-Peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron 43, 333–344. doi: 10.1016/j.neuron.2004.07.017
Delgado, M., and Ganea, D. (2003). Vasoactive intestinal peptide prevents activated microglia-induced neurodegeneration under inflammatory conditions: potential therapeutic role in brain trauma. FASEB J. 17, 1922–1924. doi: 10.1096/fj.02-1029fje
Desforges, N. M., Hebron, M. L., Algarzae, N. K., Lonskaya, I., and Moussa, C. E. H. (2012). Fractalkine mediates communication between pathogenic proteins and microglia: implications of anti-inflammatory treatments in different stages of neurodegenerative diseases. Int. J. Alzheim. Dis. 2012:345472. doi: 10.1155/2012/345472
Dewachter, I., Van Dorpe, J., Smeijers, L., Gilis, M., Kuipéri, C., Laenen, I., et al. (2000). Aging increased amyloid peptide and caused amyloid plaques in brain of old APP/V717I transgenic mice by a different mechanism than mutant Presenilin1. J. Neurosci. 20, 6452–6458.
Di Filippo, M., Tozzi, A., Costa, C., Belcastro, V., Tantucci, M., Picconi, B., et al. (2008). Plasticity and repair in the post-ischemic brain. Neuropharmacology 55, 353–362. doi: 10.1016/j.neuropharm.2008.01.012
Dijkstra, F. A., Hobbie, S. E., Knops, J. M. H., and Reich, P. B. (2004). Nitrogen deposition and plant species interact to influence soil carbon stabilization. Ecol. Lett. 7, 1192–1198. doi: 10.1111/j.1461-0248.2004.00679.x
Dimayuga, F. O., Wang, C., Clark, J. M., Dimayuga, E. R., Dimayuga, V. M., and Bruce-Keller, A. J. (2007). SOD1 overexpression alters ROS production and reduces neurotoxic inflammatory signaling in microglial cells. J. Neuroimmunol. 182, 89–99. doi: 10.1016/j.jneuroim.2006.10.003
Dodel, R., Rominger, A., Bartenstein, P., Barkhof, F., Blennow, K., Förster, S., et al. (2013). Intravenous immunoglobulin for treatment of mild-to-moderate Alzheimer's disease: a phase 2, randomised, double-blind, placebo-controlled, dose-finding trial. Lancet Neurol. 12, 233–243. doi: 10.1016/S1474-4422(13)70014-0
Dolev, I., and Michaelson, D. M. (2004). A nontransgenic mouse model shows inducible amyloid-β (Aβ) peptide deposition and elucidates the role of apolipoprotein E in the amyloid cascade. Proc. Natl. Acad. Sci. U.S.A. 101, 13909–13914. doi: 10.1073/pnas.0404458101
Donahue, J. E., Flaherty, S. L., Johanson, C. E., Duncan, J. A., Silverberg, G. D., Miller, M. C., et al. (2006). RAGE, LRP-1, and amyloid-beta protein in Alzheimer's disease. Acta Neuropathol. 112, 405–415. doi: 10.1007/s00401-006-0115-3
Doody, R. S., Thomas, R. G., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., et al. (2014). Phase 3 Trials of Solanezumab for Mild-to-Moderate Alzheimer's Disease. N. Engl. J. Med. 370, 311–321. doi: 10.1056/NEJMoa1312889
Egensperger, R., Kösel, S., von Eitzen, U., and Graeber, M. B. (1998). Microglial activation in Alzheimer disease: association with APOE genotype. Brain Pathol. 8, 439–447. doi: 10.1111/j.1750-3639.1998.tb00166.x
Eglitis, M. A., and Mezey, É. (1997). Hematopoietic cells differentiate into both microglia and macroglia in the brains of adult mice. Proc. Natl. Acad. Sci. U.S.A. 94, 4080–4085. doi: 10.1073/pnas.94.8.4080
Eketjäll, S., Janson, J., Kaspersson, K., Bogstedt, A., Jeppsson, F., Fälting, J., et al. (2016). AZD3293: a novel, orally active BACE1 inhibitor with high potency and permeability and markedly slow off-rate kinetics. J. Alzheim. Dis. 50, 1109–1123. doi: 10.3233/JAD-150834
Elmore, M. R. P., Najafi, A. R., Koike, M. A., Dagher, N. N., Spangenberg, E. E., Rice, R. A., et al. (2014). CSF1 receptor signaling is necessary for microglia viability, which unmasks a cell that rapidly repopulates the microglia-depleted adult brain. Neuron 82, 380–397. doi: 10.1016/j.neuron.2014.02.040
Enciu, A.-M., Gherghiceanu, M., and Popescu, B. O. (2013). Triggers and effectors of oxidative stress at blood-brain barrier level: relevance for brain ageing and neurodegeneration. Oxid. Med. Cell. Longev. 2013:297512. doi: 10.1155/2013/297512
Essandoh, K., Yang, L., Wang, X., Huang, W., Qin, D., Hao, J., et al. (2015). Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim. Biophys. Acta 1852, 2362–2371. doi: 10.1016/j.bbadis.2015.08.010
Euler, Z., and Schuitemaker, H. (2012). Cross-reactive broadly neutralizing antibodies: timing is everything. Front. Immunol. 3:215. doi: 10.3389/fimmu.2012.00215
Fan, Y., Xie, L., and Chung, C. Y. (2017). Signaling pathways controlling Microglia chemotaxis. Mol. Cells 40, 163–168. doi: 10.14348/molcells.2017.0011
Ferretti, M. T., Allard, S., Partridge, V., Ducatenzeiler, A., and Cuello, A. C. (2012). Minocycline corrects early, pre-plaque neuroinflammation and inhibits BACE-1 in a transgenic model of Alzheimer's disease-like amyloid pathology. J. Neuroinflammation 9, 1–16. doi: 10.1186/1742-2094-9-62
Fiandaca, M. S., Kapogiannis, D., Mapstone, M., Boxer, A., Eitan, E., Schwartz, J. B., et al. (2015). Identification of pre-clinical Alzheimer's disease by a profile of pathogenic proteins in neurally-derived blood exosomes: a case-control study. Alzheim. Dement. 11, 600–607.e1. doi: 10.1016/j.jalz.2014.06.008
Floden, A. M., and Combs, C. K. (2011). Microglia demonstrate age-dependent interaction with Beta-amyloid fibrils. J. Alzheim. Dis. 25, 279–293. doi: 10.3233/JAD-2011-101014
Fontainhas, A. M., Wang, M., Liang, K. J., Chen, S., Mettu, P., Damani, M., et al. (2011). Microglial morphology and dynamic behavior is regulated by ionotropic glutamatergic and GABAergic neurotransmission. PLoS ONE 6:e15973. doi: 10.1371/journal.pone.0015973
Ford, A. L., Goodsall, A. L., Hickey, W. F., and Sedgwick, J. D. (1995). Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J. Immunol. 154:4309.
Franceschi, C., BonafÈ, M., Valensin, S., Olivieri, F., De Luca, M., Ottaviani, E., et al. (2000). Inflamm-aging: an evolutionary perspective on immunosenescence. Ann. N.Y. Acad. Sci. 908, 244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x
Franco, R., and Fernández-Suárez, D. (2015). Alternatively activated microglia and macrophages in the central nervous system. Progr. Neurobiol. 131(Suppl. C), 65–86. doi: 10.1016/j.pneurobio.2015.05.003
Frank, S., Burbach, G. J., Bonin, M., Walter, M., Streit, W., Bechmann, I., et al. (2008). TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 56, 1438–1447. doi: 10.1002/glia.20710
Fraser, H. B., Moses, A. M., and Schadt, E. E. (2010). Evidence for widespread adaptive evolution of gene expression in budding yeast. Proc. Natl. Acad. Sci. U.S.A. 107, 2977–2982. doi: 10.1073/pnas.0912245107
Frischmeyer-Guerrerio, P. A., Guerrerio, A. L., Oswald, G., Chichester, K., Myers, L., Halushka, M. K., et al. (2013). TGFβ receptor mutations impose a strong predisposition for Human Allergic Disease. Sci. Transl. Med. 5:195ra94. doi: 10.1126/scitranslmed.3006448
Frühbeis, C., Fröhlich, D., and Krämer-Albers, E.-M. (2012). Emerging roles of exosomes in Neuron–Glia communication. Front. Physiol. 3:119. doi: 10.3389/fphys.2012.00119
Fu, R., Shen, Q., Xu, P., Luo, J. J., and Tang, Y. (2014). Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 49, 1422–1434. doi: 10.1007/s12035-013-8620-6
Fuhrmann, M., Bittner, T., Jung, C. K. E., Burgold, S., Page, R. M., Mitteregger, G., et al. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat. Neurosci. 13, 411–413. doi: 10.1038/nn.2511
Gandhi, S., and Abramov, A. Y. (2012). Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012:428010. doi: 10.1155/2012/428010
Gasque, P. (2004). Complement: a unique innate immune sensor for danger signals. Mol. Immunol. 41, 1089–1098. doi: 10.1016/j.molimm.2004.06.011
Gilgun-Sherki, Y., Melamed, E., and Offen, D. (2001). Oxidative stress induced-neurodegenerative diseases: the need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 40, 959–975. doi: 10.1016/S0028-3908(01)00019-3
Gilham, D., Wasiak, S., Tsujikawa, L. M., Halliday, C., Norek, K., Patel, R. G., et al. (2016). RVX-208, a BET-inhibitor for treating atherosclerotic cardiovascular disease, raises ApoA-I/HDL and represses pathways that contribute to cardiovascular disease. Atherosclerosis 247, 48–57. doi: 10.1016/j.atherosclerosis.2016.01.036
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Ginhoux, F., Lim, S., Hoeffel, G., Low, D., and Huber, T. (2013). Origin and differentiation of microglia. Front. Cell. Neurosci. 7:45. doi: 10.3389/fncel.2013.00045
Giunta, B., Fernandez, F., Nikolic, W. V., Obregon, D., Rrapo, E., Town, T., et al. (2008). Inflammaging as a prodrome to Alzheimer's disease. J. Neuroinflam. 5:51. doi: 10.1186/1742-2094-5-51
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms Underlying Inflammation in Neurodegeneration. Cell 140, 918–934. doi: 10.1016/j.cell.2010.02.016
Godbout, J. P., Chen, J., Abraham, J., Richwine, A. F., Berg, B. M., Kelley, K. W., et al. (2005). Exaggerated neuroinflammation and sickness behavior in aged mice after activation of the peripheral innate immune system. FASEB J. 19, 1329–1331. doi: 10.1096/fj.05-3776fje
Godbout, J. P., Moreau, M., Lestage, J., Chen, J., Sparkman, N. L., O'Connor, J., et al. (2008). Aging exacerbates depressive-like behavior in mice in response to activation of the peripheral innate immune system. Neuropsychopharmacology 33, 2341–2351. doi: 10.1038/sj.npp.1301649
Goerdt, S., Politz, O., Schledzewski, K., Birk, R., Gratchev, A., Guillot, P., et al. (1999). Alternative versus classical activation of macrophages. Pathobiology 67, 222–226. doi: 10.1159/000028096
Goldmann, T., and Prinz, M. (2013). Role of Microglia in CNS Autoimmunity. Clin. Develop. Immunol. 2013:208093. doi: 10.1155/2013/208093
Gomez-Nicola, D., and Perry, V. H. (2015). Microglial dynamics and role in the healthy and diseased brain: a paradigm of functional plasticity. Neuroscientist 21, 169–184. doi: 10.1177/1073858414530512
Gosselin, D., Skola, D., Coufal, N. G., Holtman, I. R., Schlachetzki, J. C. M., Sajti, E., et al. (2017). An environment-dependent transcriptional network specifies human microglia identity. Science 356:eaal3222. doi: 10.1126/science.aal3222
Gottfried-Blackmore, A., Kaunzner, U. W., Idoyaga, J., Felger, J. C., McEwen, B. S., and Bulloch, K. (2009). Acute in vivo exposure to interferon-γ enables resident brain dendritic cells to become effective antigen presenting cells. Proc. Natl. Acad. Sci. U.S.A. 106, 20918–20923. doi: 10.1073/pnas.0911509106
Götz, J., Chen, F., van Dorpe, J., and Nitsch, R. M. (2001). Formation of neurofibrillary tangles in P301L Tau Transgenic mice induced by Aβ42 fibrils. Science 293:1491. doi: 10.1126/science.1062097
Grabert, K., Michoel, T., Karavolos, M. H., Clohisey, S., Baillie, J. K., Stevens, M. P., et al. (2016). Microglial brain region-dependent diversity and selective regional sensitivities to ageing. Nat. Neurosci. 19, 504–516. doi: 10.1038/nn.4222
Graeber, M. B., Li, W., and Rodriguez, M. L. (2011). Role of microglia in CNS inflammation. FEBS Lett. 585, 3798–3805. doi: 10.1016/j.febslet.2011.08.033
Grathwohl, S. A., Kälin, R. E., Bolmont, T., Prokop, S., Winkelmann, G., Kaeser, S. A., et al. (2009). Formation and maintenance of Alzheimer's disease β-amyloid plaques in the absence of microglia. Nat. Neurosci. 12, 1361–1363. doi: 10.1038/nn.2432
Greening, D. W., Gopal, S. K., Xu, R., Simpson, R. J., and Chen, W. (2015). Exosomes and their roles in immune regulation and cancer. Sem. Cell Develop. Biol. 40(Suppl. C), 72–81. doi: 10.1016/j.semcdb.2015.02.009
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 Variants in Alzheimer's Disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Guo, B. B., Bellingham, S. A., and Hill, A. F. (2016). Stimulating the release of exosomes increases the intercellular transfer of prions. J. Biol. Chem. 291, 5128–5137. doi: 10.1074/jbc.M115.684258
Haass, C., Hung, A. Y., Schlossmacher, M. G., Oltersdorf, T., Teplow, D. B., and Selkoe, D. J. (1993a). Normal cellular processing of the β-Amyloid precursor protein results in the secretion of the Amyloid β Peptide and related moleculesa. Ann. N.Y. Acad. Sci. 695, 109–116. doi: 10.1111/j.1749-6632.1993.tb23037.x
Haass, C., Hung, A. Y., Schlossmacher, M. G., Teplow, D. B., and Selkoe, D. J. (1993b). beta-Amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J. Biol. Chem. 268, 3021–3024.
Habchi, J., Arosio, P., Perni, M., Costa, A. R., Yagi-Utsumi, M., Joshi, P., et al. (2016). An anticancer drug suppresses the primary nucleation reaction that initiates the production of the toxic Aβ42 aggregates linked with Alzheimer's disease. Sci. Adv. 2:e1501244. doi: 10.1126/sciadv.1501244
Hafez, D., Huang, J. Y., Huynh, A. M., Valtierra, S., Rockenstein, E., Bruno, A. M., et al. (2011). Neprilysin-2 Is an important β-Amyloid degrading enzyme. Am. J. Pathol. 178, 306–312. doi: 10.1016/j.ajpath.2010.11.012
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., Reinheckel, T., et al. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 9, 857–865. doi: 10.1038/ni.1636
Han, B. H., Zhou, M.-L. Johnson, A. W., Singh, I., Liao, F., Vellimana, A. K., et al. (2015). Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc. Natl. Acad. Sci. U.S.A. 112, E881–E890. doi: 10.1073/pnas.1414930112
Hanke, M. L., and Kielian, T. (2011). Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin. Sci. 121, 367–387. doi: 10.1042/CS20110164
Harrington, C. R., Storey, J. M. D., Clunas, S., Harrington, K. A., Horsley, D., Ishaq, A., et al. (2015). Cellular models of aggregation-dependent template-directed proteolysis to characterize tau aggregation inhibitors for treatment of Alzheimer disease. J. Biol. Chem. 290, 10862–10875. doi: 10.1074/jbc.M114.616029
Harrison, J. K., Jiang, Y., Chen, S., Xia, Y., Maciejewski, D., McNamara, R. K., et al. (1998). Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. U.S.A. 95, 10896–10901. doi: 10.1073/pnas.95.18.10896
Harry, G. J., and Kraft, A. D. (2012). Microglia in the developing brain: a potential target with lifetime effects. Neurotoxicology 33, 191–206. doi: 10.1016/j.neuro.2012.01.012
Hart, A. D., Wyttenbach, A., Hugh Perry, V., and Teeling, J. L. (2012). Age related changes in microglial phenotype vary between CNS regions: grey versus white matter differences. Brain Behav. Immun. 26, 754–765. doi: 10.1016/j.bbi.2011.11.006
Hartmann, T., Bieger, S. C., Bruhl, B., Tienari, P. J., Ida, N., Allsop, D., et al. (1997). Distinct sites of intracellular production for Alzheimer's disease A[beta]40/42 amyloid peptides. Nat. Med. 3, 1016–1020. doi: 10.1038/nm0997-1016
Hayashi, Y., Ishibashi, H., Hashimoto, K., and Nakanishi, H. (2006). Potentiation of the NMDA receptor-mediated responses through the activation of the glycine site by microglia secreting soluble factors. Glia 53, 660–668. doi: 10.1002/glia.20322
Hayashi, Y., Yoshida, M., Yamato, M., Ide, T., Wu, Z., Ochi-Shindou, M., et al. (2008). Reverse of age-dependent memory impairment and mitochondrial DNA damage in microglia by an overexpression of human mitochondrial transcription factor A in mice. J. Neurosci. 28:8624. doi: 10.1523/JNEUROSCI.1957-08.2008
He, P., Cheng, X., Staufenbiel, M., Li, R., and Shen, Y. (2013). Long-term treatment of thalidomide ameliorates amyloid-like pathology through inhibition of β-secretase in a mouse model of Alzheimer's Disease. PLoS ONE 8:e55091. doi: 10.1371/journal.pone.0055091
Hefendehl, J. K., Neher, J. J., Sühs, R. B., Kohsaka, S., Skodras, A., and Jucker, M. (2014). Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 13, 60–69. doi: 10.1111/acel.12149
Henry, C. J., Huang, Y., Wynne, A. M., and Godbout, J. P. (2009). Peripheral Lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain Behav. Immun. 23, 309–317. doi: 10.1016/j.bbi.2008.09.002
Hess, D. C., Abe, T., Hill, W. D., Studdard, A. M., Carothers, J., Masuya, M., et al. (2004). Hematopoietic origin of microglial and perivascular cells in brain. Exp. Neurol. 186, 134–144. doi: 10.1016/j.expneurol.2003.11.005
Hinwood, M., Morandini, J., Day, T. A., and Walker, F. R. (2012). Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb. Cortex 22, 1442–1454. doi: 10.1093/cercor/bhr229
Hoek, R. M., Ruuls, S. R., Murphy, C. A., Wright, G. J., Goddard, R., Zurawski, S. M., et al. (2000). Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 290, 1768–1771. doi: 10.1126/science.290.5497.1768
Holmes, C., Boche, D., Wilkinson, D., Yadegarfar, G., Hopkins, V., Bayer, A., et al. (2008). Long-term effects of Aβ42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 372, 216–223. doi: 10.1016/S0140-6736(08)61075-2
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hooli, B. V., Mohapatra, G., Mattheisen, M., Parrado, A. R., Roehr, J. T., Shen, Y., et al. (2012). Role of common and rare APP DNA sequence variants in Alzheimer disease. Neurology 78, 1250–1257. doi: 10.1212/WNL.0b013e3182515972
Hori, Y., Takeda, S., Cho, H., Wegmann, S., Shoup, T. M., Takahashi, K., et al. (2015). A Food and Drug Administration-approved Asthma Therapeutic Agent Impacts Amyloid β in the Brain in a Transgenic Model of Alzheimer Disease. J. Biol. Chem. 290, 1966–1978. doi: 10.1074/jbc.M114.586602
Hsieh, J. (2012). Orchestrating transcriptional control of adult neurogenesis. Genes Dev. 26, 1010–1021. doi: 10.1101/gad.187336.112
Hurtado, D. E., Molina-Porcel, L., Iba, M., Aboagye, A. K., Paul, S. M., Trojanowski, J. Q., et al. (2010). Aβ accelerates the spatiotemporal progression of tau pathology and Augments Tau Amyloidosis in an Alzheimer Mouse Model. Am. J. Pathol. 177, 1977–1988. doi: 10.2353/ajpath.2010.100346
Huse, J. T., Liu, K., Pijak, D. S., Carlin, D., Lee, V. M. Y., and Doms, R. W. (2002). β-Secretase processing in the trans-golgi network preferentially generates truncated amyloid species that accumulate in Alzheimer's disease brain. J. Biol. Chem. 277, 16278–16284. doi: 10.1074/jbc.M111141200
Huse, J. T., Pijak, D. S., Leslie, G. J., Lee, V. M. Y., and Doms, R. W. (2000). Maturation and endosomal targeting of β-Site amyloid precursor protein-cleaving enzyme: the Alzheimer's disease β-secretase. J. Biol. Chem. 275, 33729–33737. doi: 10.1074/jbc.M004175200
Husemann, J., Loike, J. D., Anankov, R., Febbraio, M., and Silverstein, S. C. (2002). Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia 40, 195–205. doi: 10.1002/glia.10148
Iguchi, Y., Eid, L., Parent, M., Soucy, G., Bareil, C., Riku, Y., et al. (2016). Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 139, 3187–3201. doi: 10.1093/brain/aww237
Imbimbo, B. P., and Giardina, G. A. (2011). γ-Secretase Inhibitors and Modulators for the Treatment of Alzheimer's Disease: disappointments and hopes. Curr. Top. Med. Chem. 11, 1555–1570. doi: 10.2174/156802611795860942
Imbimbo, B. P., Hutter-Paier, B., Villetti, G., Facchinetti, F., Cenacchi, V., Volta, R., et al. (2009). CHF5074, a novel γ-secretase modulator, attenuates brain β-amyloid pathology and learning deficit in a mouse model of Alzheimer's disease. Br. J. Pharmacol. 156, 982–993. doi: 10.1111/j.1476-5381.2008.00097.x
Imbimbo, B. P., Solfrizzi, V., and Panza, F. (2010). Are NSAIDs useful to treat Alzheimer's disease or mild cognitive impairment? Front. Aging Neurosci. 2:19. doi: 10.3389/fnagi.2010.00019
Inoue, K., and Tsuda, M. (2012). Purinergic systems, neuropathic pain and the role of microglia. Exp. Neurol. 234, 293–301. doi: 10.1016/j.expneurol.2011.09.016
Izzo, N. J., Xu, J., Zeng, C., Kirk, M. J., Mozzoni, K., Silky, C., et al. (2014). Alzheimer's Therapeutics Targeting Amyloid Beta 1–42 Oligomers II: sigma-2/PGRMC1 Receptors Mediate Abeta 42 Oligomer Binding and Synaptotoxicity. PLoS ONE 9:e111899. doi: 10.1371/journal.pone.0111899
Janeway, C. A. (1992). The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol. Tod. 13, 11–16. doi: 10.1016/0167-5699(92)90198-G
Jassam, Y. N., Izzy, S., Whalen, M., McGavern, D. B., and El Khoury, J. (2017). Neuroimmunology of traumatic brain injury: time for a paradigm shift. Neuron 95, 1246–1265. doi: 10.1016/j.neuron.2017.07.010
Jay, T. R., Miller, C. M., Cheng, P. J., Graham, L. C., Bemiller, S., and Broihier, M. L. (2015). TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J. Exp. Med. 212. doi: 10.1084/jem.20142322
Jin, J.-J., Kim, H.-D., Maxwell, J. A., Li, L., and Fukuchi, K.-I. (2008). Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer's disease. J. Neuroinflam. 5, 23–23. doi: 10.1186/1742-2094-5-23
Johnson-Wood, K., Lee, M., Motter, R., Hu, K., Gordon, G., Barbour, R., et al. (1997). Amyloid precursor protein processing and Aβ(42) deposition in a transgenic mouse model of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 94, 1550–1555. doi: 10.1073/pnas.94.4.1550
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Justin, T. R., Josh, M. M., Adam, D. B., Charles, E. H., Melinda, M. P., Bethany, A. G., et al. (2011). CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci. 31, 16241–16250. doi: 10.1523/JNEUROSCI.3667-11.2011
Kalpouzos, G., Chételat, G., Baron, J.-C., Landeau, B., Mevel, K., Godeau, C., et al. (2009). Voxel-based mapping of brain gray matter volume and glucose metabolism profiles in normal aging. Neurobiol. Aging 30, 112–124. doi: 10.1016/j.neurobiolaging.2007.05.019
Kaminska, B., Mota, M., and Pizzi, M. (2016). Signal transduction and epigenetic mechanisms in the control of microglia activation during neuroinflammation. Biochim. Biophys. Acta 1862, 339–351. doi: 10.1016/j.bbadis.2015.10.026
Kamphuis, W., Mamber, C., Moeton, M., Kooijman, L., Sluijs, J. A., Jansen, A. H. P., et al. (2012). GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS ONE 7:e42823. doi: 10.1371/journal.pone.0042823
Kao, A. W., Eisenhut, R. J., Martens, L. H., Nakamura, A., Huang, A., Bagley, J. A., et al. (2011). A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc. Natl. Acad. Sci. U.S.A. 108, 4441–4446. doi: 10.1073/pnas.1100650108
Karperien, A., Ahammer, H., and Jelinek, H. (2013). Quantitating the subtleties of microglial morphology with fractal analysis. Front. Cell. Neurosci. 7:3. doi: 10.3389/fncel.2013.00003
Kaushik, S., and Cuervo, A. M. (2015). Proteostasis and aging. Nat. Med. 21, 1406–1415. doi: 10.1038/nm.4001
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer's disease. Cell 169, 1276–1290.e17. doi: 10.1016/j.cell.2017.05.018
Kern, A., Roempp, B., Prager, K., Walter, J., and Behl, C. (2006). Down-regulation of endogenous amyloid precursor protein processing due to cellular aging. J. Biol. Chem. 281, 2405–2413. doi: 10.1074/jbc.M505625200
Kigerl, K. A., de Rivero Vaccari, J. P., Dietrich, W. D., Popovich, P. G., and Keane, R. W. (2014). Pattern recognition receptors and central nervous system repair. Exp. Neurol. 258, 5–16. doi: 10.1016/j.expneurol.2014.01.001
Kim, S. U., and de Vellis, J. (2005). Microglia in health and disease. J. Neurosci. Res. 81, 302–313. doi: 10.1002/jnr.20562
Kitamura, R., Tsukamoto, K., Harada, K., Shimizu, A., Shimada, S., Kobayashi, T., et al. (2004). Mechanisms underlying the dysfunction of melanocytes in vitiligo epidermis: role of SCF/KIT protein interactions and the downstream effector, MITF-M. J. Pathol. 202, 463–475. doi: 10.1002/path.1538
Klegeris, A., Bissonnette, C. J., and McGeer, P. L. (2005). Modulation of human microglia and THP-1 cell toxicity by cytokines endogenous to the nervous system. Neurobiol. Aging 26, 673–682. doi: 10.1016/j.neurobiolaging.2004.06.012
Kleinberger, G., Brendel, M., Mracsko, E., Wefers, B., Groeneweg, L., Xiang, X., et al. (2017). The FTD-like syndrome causing TREM2 T66M mutation impairs microglia function, brain perfusion, and glucose metabolism. EMBO J. 36, 1837–1853. doi: 10.15252/embj.201796516
Kleinberger, G., Yamanishi, Y., Suárez-Calvet, M., Czirr, E., Lohmann, E., Cuyvers, E., et al. (2014). TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 6:243ra86. doi: 10.1126/scitranslmed.3009093
Koellhoffer, E. C., McCullough, L. D., and Ritzel, R. M. (2017). Old maids: aging and its impact on Microglia function. Int. J. Mol. Sci. 18:769. doi: 10.3390/ijms18040769
Koizumi, S., Ohsawa, K., Inoue, K., and Kohsaka, S. (2013). Purinergic receptors in microglia: functional modal shifts of microglia mediated by P2 and P1 receptors. Glia 61, 47–54. doi: 10.1002/glia.22358
Koizumi, S., Shigemoto-Mogami, Y., Nasu-Tada, K., Shinozaki, Y., Ohsawa, K., Tsuda, M., et al. (2007). UDP acting at P2Y(6) receptors is a mediator of microglial phagocytosis. Nature 446, 1091–1095. doi: 10.1038/nature05704
Kontsekova, E., Zilka, N., Kovacech, B., Novak, P., and Novak, M. (2014). First-in-man tau vaccine targeting structural determinants essential for pathological tau–tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer's disease model. Alzheim. Res. Ther. 6, 44–44. doi: 10.1186/alzrt278
Koo, E. H., and Squazzo, S. L. (1994). Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 269, 17386–17389.
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e9. doi: 10.1016/j.immuni.2017.08.008.
Kreutzberg, G. W. (1996). Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 19, 312–318. doi: 10.1016/0166-2236(96)10049-7
Landsman, L., Bar-On, L., Zernecke, A., Kim, K.-W., Krauthgamer, R., Shagdarsuren, E., et al. (2009). CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood 113:963. doi: 10.1182/blood-2008-07-170787
Lewis, J., Dickson, D. W., Lin, W.-L., Chisholm, L., Corral, A., Jones, G., et al. (2001). Enhanced Neurofibrillary Degeneration in Transgenic Mice Expressing Mutant Tau and APP. Science 293:1487. doi: 10.1126/science.1058189
Lewitus, G. M., Konefal, S. C., Greenhalgh, A. D., Pribiag, H., Augereau, K., and Stellwagen, D. (2016). Microglial TNFα suppresses cocaine-induced plasticity and behavioral sensitization. Neuron 90, 483–491. doi: 10.1016/j.neuron.2016.03.030
Leyns, C. E. G., Ulrich, J. D., Finn, M. B., Stewart, F. R., Koscal, L. J., Remolina Serrano, J., et al. (2017). TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. U.S.A. 114, 11524–11529. doi: 10.1073/pnas.1710311114
Li, G., Bien-Ly, N., Andrews-Zwilling, Y., Xu, Q., Bernardo, A., Ring, K., et al. (2009). GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knock-in mice. Cell Stem Cell 5, 634–645. doi: 10.1016/j.stem.2009.10.015
Li, R., Lindholm, K., Yang, L. B., Yue, X., Citron, M., Yan, R., et al. (2004). Amyloid β peptide load is correlated with increased β-secretase activity in sporadic Alzheimer's disease patients. Proc. Natl. Acad. Sci. U.S.A. 101, 3632–3637. doi: 10.1073/pnas.0205689101
Lim, S.-H., Park, E., You, B., Jung, Y., Park, A. R., Park, S. G., et al. (2013). Neuronal Synapse Formation Induced by Microglia and Interleukin 10. PLoS ONE 8:e81218. doi: 10.1371/journal.pone.0081218
Liu, Z., Ran, Y., Huang, S., Wen, S., Zhang, W., Liu, X., et al. (2017). Curcumin protects against ischemic stroke by Titrating microglia/macrophage polarization. Front. Aging Neurosci. 9:233. doi: 10.3389/fnagi.2017.00233
Löscher, W. (2002). Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epil. Res. 50, 105–123.
Louveau, A., Smirnov, I., Keyes, T. J., Eccles, J. D., Rouhani, S. J., Peske, J. D., et al. (2015). Structural and functional features of central nervous system lymphatics. Nature 523, 337–341. doi: 10.1038/nature14432
Lu, Y.-C., Yeh, W.-C., and Ohashi, P. S. (2008). LPS/TLR4 signal transduction pathway. Cytokine 42, 145–151. doi: 10.1016/j.cyto.2008.01.006
Luo, C., Jian, C., Liao, Y., Huang, Q., Wu, Y., Liu, X., et al. (2017). The role of microglia in multiple sclerosis. Neuropsychiatr. Dis. Treat. 13, 1661–1667. doi: 10.2147/NDT.S140634
Lyons, A., Downer, E. J., Crotty, S., Nolan, Y. M., Mills, K. H. G., and Lynch, M. A. (2007). CD200 ligand–receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J. Neurosci. 27:8309. doi: 10.1523/JNEUROSCI.1781-07.2007
Lyons, A., Lynch, A. M., Downer, E. J., Hanley, R., O'Sullivan, J. B., Smith, A., et al. (2009). Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J. Neurochem. 110, 1547–1556. doi: 10.1111/j.1471-4159.2009.06253.x
Maccecchini, M. L., Chang, M. Y., Pan, C., John, V., Zetterberg, H., and Greig, N. H. (2012). Posiphen as a candidate drug to lower CSF amyloid precursor protein, amyloid-β peptide and τ levels: target engagement, tolerability and pharmacokinetics in humans. J. Neurol. Neurosurg. Psychiatr. 83, 894–902. doi: 10.1136/jnnp-2012-302589
Maher, F. O., Martin, D. S. D., and Lynch, M. A. (2004). Increased IL-1beta in cortex of aged rats is accompanied by downregulation of ERK and PI-3 kinase. Neurobiol. Aging 25, 795–806. doi: 10.1016/j.neurobiolaging.2003.08.007
Maher, F. O., Nolan, Y., and Lynch, M. A. (2005). Downregulation of IL-4-induced signalling in hippocampus contributes to deficits in LTP in the aged rat. Neurobiol. Aging 26, 717–728. doi: 10.1016/j.neurobiolaging.2004.07.002
Mamada, N., Tanokashira, D., Hosaka, A., Kametani, F., Tamaoka, A., and Araki, W. (2015). Amyloid β-protein oligomers upregulate the β-secretase, BACE1, through a post-translational mechanism involving its altered subcellular distribution in neurons. Mol. Brain 8:73. doi: 10.1186/s13041-015-0163-5
Mantovani, A., Sica, A., and Locati, M. (2005). Macrophage polarization comes of age. Immunity 23, 344–346. doi: 10.1016/j.immuni.2005.10.001
Mantovani, A., Sica, A., Sozzani, S., Allavena, P., Vecchi, A., and Locati, M. (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677–686. doi: 10.1016/j.it.2004.09.015
Markovic, S. N., Suman, V. J., Nevala, W. K., Geeraerts, L., Creagan, E. T., Erickson, L. A., et al. (2008). A dose-escalation study of aerosolized sargramostim in the treatment of metastatic melanoma: an NCCTG study. Am. J. Clin. Oncol. 31, 573–579. doi: 10.1097/COC.0b013e318173a536
Marr, R. A., Rockenstein, E., Mukherjee, A., Kindy, M. S., Hersh, L. B., Gage, F. H., et al. (2003). Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J. Neurosci. 23, 1992–1996.
Martinez, F. O., and Gordon, S. (2014). The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6:13. doi: 10.12703/P6-13
Martinon, F., Burns, K., and Tschopp, J. (2002). The Inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426.
Matcovitch-Natan, O., Winter, D. R., Giladi, A., Vargas Aguilar, S., Spinrad, A., Sarrazin, S., et al. (2016). Microglia development follows a stepwise program to regulate brain homeostasis. Science 353:aad8670. doi: 10.1126/science.aad8670
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., et al. (2010). Decreased clearance of CNS amyloid-β in Alzheimer's disease. Science 330, 1774–1774. doi: 10.1126/science.1197623
McLaurin, J., Kierstead, M. E., Brown, M. E., Hawkes, C. A., Lambermon, M. H. L., Phinney, A. L., et al. (2006). Cyclohexanehexol inhibitors of A[beta] aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nat. Med. 12, 801–808. doi: 10.1038/nm1423
McNaughton, D., Knight, W., Guerreiro, R., Ryan, N., Lowe, J., Poulter, M., et al. (2012). Duplication of amyloid precursor protein (APP), but not prion protein (PRNP) gene is a significant cause of early onset dementia in a large UK series. Neurobiol. Aging 33:426.e13-426.e21. doi: 10.1016/j.neurobiolaging.2010.10.010
Mead, E. L., Mosley, A., Eaton, S., Dobson, L., Heales, S. J., and Pocock, J. M. (2012). Microglial neurotransmitter receptors trigger superoxide production in microglia; consequences for microglial–neuronal interactions. J. Neurochem. 121, 287–301. doi: 10.1111/j.1471-4159.2012.07659.x
Middeldorp, J., Lehallier, B., Villeda, S. A., Miedema, S. S., Evans, E., Czirr, E., et al. (2016). Preclinical assessment of young blood plasma for alzheimer disease. JAMA Neurol. 73, 1325–1333. doi: 10.1001/jamaneurol.2016.3185
Mildner, A., Huang, H., Radke, J., Stenzel, W., and Priller, J. (2017). P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia 65, 375–387. doi: 10.1002/glia.23097
Mildner, A., Mack, M., Schmidt, H., Brück, W., Djukic, M., Zabel, M. D., et al. (2009). CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain 132, 2487–2500. doi: 10.1093/brain/awp144
Miners, J. S., Barua, N., Kehoe, P. G., Gill, S., and Love, S. (2011). Aβ-degrading enzymes: potential for treatment of Alzheimer disease. J. Neuropathol.Exp. Neurol. 70, 944–959. doi: 10.1097/NEN.0b013e3182345e46
Mitchell, D., Bergendahl, G., Ferguson, W., Roberts, W., Higgins, T., Ashikaga, T., et al. (2016). A phase 1 trial of TPI 287 as a single agent and in combination with temozolomide in patients with refractory or recurrent neuroblastoma or medulloblastoma. Pediatr. Blood Cancer 63, 39–46. doi: 10.1002/pbc.25687
Mitrasinovic, O. M., Perez, G. V., Zhao, F., Lee, Y. L., Poon, C., and Murphy, G. M. (2001). Overexpression of Macrophage Colony-stimulating Factor Receptor on Microglial Cells Induces an Inflammatory Response. J. Biol. Chem. 276, 30142–30149. doi: 10.1074/jbc.M104265200
Mittelbronn, M., Dietz, K. J., Schluesener, H., and Meyermann, R. (2001). Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 101, 249–255.
Monro, O. R., Mackic, J. B., Yamada, S., Segal, M. B., Ghiso, J., Maurer, C., et al. (2002). Substitution at codon 22 reduces clearance of Alzheimer's amyloid-β peptide from the cerebrospinal fluid and prevents its transport from the central nervous system into blood. Neurobiol. Aging 23, 405–412. doi: 10.1016/S0197-4580(01)00317-7
Montagne, A., Barnes, S. R., Sweeney, M. D., Halliday, M. R., Sagare, A. P., Zhao, Z., et al. (2015). Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302. doi: 10.1016/j.neuron.2014.12.032
Morawski, M., Schilling, S., Kreuzberger, M., Waniek, A., Jäger, C., Koch, B., et al. (2014). Glutaminyl cyclase in human cortex: correlation with (pGlu)-Amyloid-β Load and cognitive decline in Alzheimer's disease. J. Alzheim. Dis. 39, 385–400. doi: 10.3233/JAD-131535
Mosher, K. I., and Wyss-Coray, T. (2014). Microglial dysfunction in brain aging and Alzheimer's disease. Biochem. Pharmacol. 88, 594–604. doi: 10.1016/j.bcp.2014.01.008
Nair-Gupta, P., Baccarini, A., Tung, N., Seyffer, F., Florey, O., Huang, Y., et al. (2014). TLR signals induce phagosomal MHC-I delivery from the endosomal recycling compartment to allow cross-presentation. Cell 158, 506–521. doi: 10.1016/j.cell.2014.04.054
Neumann, G., Noda, T., and Kawaoka, Y. (2009). Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459, 931–939. doi: 10.1038/nature08157
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130. doi: 10.1126/science.1134108
Nimmerjahn, F., and Ravetch, J. V. (2006). Fcγ receptors: old friends and new family members. Immunity 24, 19–28. doi: 10.1016/j.immuni.2005.11.010
Nisemblat, Y., Belinson, H., Dolev, I., and Michaelson, D. M. (2008). Activation of the Amyloid cascade by intracerebroventricular injection of the protease inhibitor phosphoramidon. Neurodegener. Dis. Manag. 5, 166–169. doi: 10.1159/000113692
Njie, e. G., Boelen, E., Stassen, F. R., Steinbusch, H. W. M., Borchelt, D. R., and Streit, W. J. (2012). Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol. Aging 33, 195.e1–195.12. doi: 10.1016/j.neurobiolaging.2010.05.008
Noda, M., Doi, Y., Liang, J., Kawanokuchi, J., Sonobe, Y., Takeuchi, H., et al. (2011). Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. J. Biol. Chem. 286, 2308–2319. doi: 10.1074/jbc.M110.169839
Nolan, Y., Maher, F. O., Martin, D. S., Clarke, R. M., Brady, M. T., Bolton, A. E., et al. (2005). Role of Interleukin-4 in regulation of age-related inflammatory changes in the Hippocampus. J. Biol. Chem. 280, 9354–9362. doi: 10.1074/jbc.M412170200
Nygaard, H. B., van Dyck, C. H., and Strittmatter, S. M. (2014). Fyn kinase inhibition as a novel therapy for Alzheimer's disease. Alzheim. Res. Ther. 6:8. doi: 10.1186/alzrt238
Ofengeim, D., Mazzitelli, S., Ito, Y., DeWitt, J. P., Mifflin, L., Zou, C., et al. (2017). RIPK1 mediates a disease-associated microglial response in Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 114, E8788–E8797. doi: 10.1073/pnas.1714175114
Okun, E., Griffioen, K. J., Gen Son, T., Lee, J.-H., Roberts, N. J., Mughal, M. R., et al. (2010). TLR2 activation inhibits embryonic neural progenitor cell proliferation. J. Neurochem. 114, 462–474. doi: 10.1111/j.1471-4159.2010.06778.x
Olmos-Alonso, A., Schetters, S. T. T., Sri, S., Askew, K., Mancuso, R., Vargas-Caballero, M., et al. (2016). Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer's-like pathology. Brain 139, 891–907. doi: 10.1093/brain/awv379
Pan, X.-D., Zhu, Y.-G., Lin, N., Zhang, J., Ye, Q.-Y., Huang, H.-P., et al. (2011). Microglial phagocytosis induced by fibrillar β-amyloid is attenuated by oligomeric β-amyloid: implications for Alzheimer's disease. Mol. Neurodegener. 6:45. doi: 10.1186/1750-1326-6-45
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333:1456. doi: 10.1126/science.1202529
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R., Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through BDNF. Cell 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Penn, A. A., Riquelme, P. A., Feller, M. B., and Shatz, C. J. (1998). Competition in retinogeniculate patterning driven by spontaneous activity. Science 279:2108. doi: 10.1126/science.279.5359.2108
Perry, V. H., and Teeling, J. (2013). Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 35, 601–612. doi: 10.1007/s00281-013-0382-8
Perry, V. H., Brown, M. C., and Gordon, S. (1987). The macrophage response to central and peripheral nerve injury. A possible role for macrophages in regeneration. J. Exp. Med. 165, 1218–1223.
Piccio, L., Buonsanti, C., Mariani, M., Cella, M., Gilfillan, S., Cross, A. H., et al. (2007). Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur. J. Immunol. 37, 1290–1301. doi: 10.1002/eji.200636837
Polazzi, E., and Monti, B. (2010). Microglia and neuroprotection: from in vitro studies to therapeutic applications. Progr. Neurobiol. 92, 293–315. doi: 10.1016/j.pneurobio.2010.06.009
Ponomarev, E. D., Shriver, L. P., Maresz, K., and Dittel, B. N. (2005). Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J. Neurosci. Res. 81, 374–389. doi: 10.1002/jnr.20488
Praag, H. V., Shubert, T., Zhao, C., and Gage, F. H. (2005). Exercise enhances learning and hippocampal neurogenesis in aged mice. J. Neurosci. 25, 8680–8685. doi: 10.1523/JNEUROSCI.1731-05.2005
Qin, L., Liu, Y., Hong, J.-S., and Crews, F. T. (2013). NADPH oxidase and aging drive microglial activation, oxidative stress and dopaminergic neurodegeneration following systemic LPS administration. Glia 61, 855–868. doi: 10.1002/glia.22479
Qin, L., Liu, Y., Wang, T., Wei, S.-J., Block, M. L., Wilson, B., et al. (2004). NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated Microglia. J. Biol. Chem. 279, 1415–1421. doi: 10.1074/jbc.M307657200
Rajendran, L., Honsho, M., Zahn, T. R., Keller, P., Geiger, K. D., Verkade, P., et al. (2006). Alzheimer's disease β-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 103, 11172–11177. doi: 10.1073/pnas.0603838103
Ransohoff, R. M. (2016). A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991. doi: 10.1038/nn.4338
Ransohoff, R. M., and Perry, V. H. (2009). Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 27, 119–145. doi: 10.1146/annurev.immunol.021908.132528
Raper, D., Louveau, A., and Kipnis, J. (2016). How do meningeal lymphatic vessels drain the CNS? Trends Neurosci. 39, 581–586. doi: 10.1016/j.tins.2016.07.001
Ravichandran, K. S. (2010). Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J. Exp. Med. 207, 1807–1817. doi: 10.1084/jem.20101157
Raz, N., Lindenberger, U., Rodrigue, K. M., Kennedy, K. M., Head, D., Williamson, A., et al. (2005). Regional brain changes in aging healthy adults: general trends, individual differences and modifiers. Cereb. Cortex 15, 1676–1689. doi: 10.1093/cercor/bhi044
Réu, P., Khosravi, A., Bernard, S., Mold, J. E., Salehpour, M., Alkass, K., et al. (2017). The lifespan and turnover of microglia in the human brain. Cell Rep. 20, 779–784. doi: 10.1016/j.celrep.2017.07.004
Ritzel, R. M., Patel, A. R., Pan, S., Crapser, J., Hammond, M., Jellison, E., et al. (2015). Age- and location-related changes in microglial function. Neurobiol. Aging 36, 2153–2163. doi: 10.1016/j.neurobiolaging.2015.02.016
Robbins, P. D., and Morelli, A. E. (2014). Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 14, 195–208. doi: 10.1038/nri3622
Rock, R. B., Gekker, G., Hu, S., Sheng, W. S., Cheeran, M., Lokensgard, J. R., et al. (2004). Role of Microglia in central nervous system infections. Clin. Microbiol. Rev. 17, 942–964. doi: 10.1128/CMR.17.4.942-964.2004
Rodgers, S. P., Born, H. A., Das, P., and Jankowsky, J. L. (2012). Transgenic APP expression during postnatal development causes persistent locomotor hyperactivity in the adult. Mol. Neurodegener. 7:28. doi: 10.1186/1750-1326-7-28
Rosczyk, H. A., Sparkman, N. L., and Johnson, R. W. (2008). Neuroinflammation and cognitive function in aged mice following minor surgery. Exp. Gerontol. 43, 840–846. doi: 10.1016/j.exger.2008.06.004
Rovelet-Lecrux, A., Hannequin, D., Raux, G., Meur, N. L., Laquerriere, A., Vital, A., et al. (2006). APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 38, 24–26. doi: 10.1038/ng1718
Safaiyan, S., Kannaiyan, N., Snaidero, N., Brioschi, S., Biber, K., Yona, S., et al. (2016). Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 19, 995–998. doi: 10.1038/nn.4325
Sakono, M., and Zako, T. (2010). Amyloid oligomers: formation and toxicity of Aβ oligomers. FEBS J. 277, 1348–1358. doi: 10.1111/j.1742-4658.2010.07568.x
Salat, D. H., Buckner, R. L., Snyder, A. Z., Greve, D. N., Desikan, R. S. R., Busa, E., et al. (2004). Thinning of the cerebral cortex in aging. Cereb. Cortex 14, 721–730. doi: 10.1093/cercor/bhh032
Salmon, E., Garraux, G., Delbeuck, X., Collette, F., Kalbe, E., Zuendorf, G., et al. (2003). Predominant ventromedial frontopolar metabolic impairment in frontotemporal dementia. Neuroimage 20, 435–440. doi: 10.1016/S1053-8119(03)00346-X
Saman, S., Kim, W., Raya, M., Visnick, Y., Miro, S., Saman, S., et al. (2012). Exosome-associated Tau Is Secreted in Tauopathy Models and Is Selectively Phosphorylated in Cerebrospinal Fluid in Early Alzheimer Disease. J. Biol. Chem. 287, 3842–3849. doi: 10.1074/jbc.M111.277061
Sarko, D. K., and McKinney, C. E. (2017). Exosomes: origins and therapeutic potential for neurodegenerative disease. Front. Neurosci. 11:82. doi: 10.3389/fnins.2017.00082
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Scherzer, C. R., Offe, K., Gearing, M., Rees, H. D., Fang, G., Heilman, C. J., et al. (2004). Loss of apolipoprotein e receptor lr11 in alzheimer disease. Arch. Neurol. 61, 1200–1205. doi: 10.1001/archneur.61.8.1200
Schuck, F., Wolf, D., Fellgiebel, A., and Endres, K. (2016). Increase of α-secretase ADAM10 in platelets along cognitively healthy aging. J. Alzheimers Dis. 50, 817–826. doi: 10.3233/JAD-150737
Selkoe, D. J. (2002). Alzheimer's disease is a synaptic failure. Science, 298:789. doi: 10.1126/science.1074069
Sevigny, J., Chiao, P., Bussière, T., Weinreb, P. H., Williams, L., Maier, M., et al. (2016). The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature 537, 50–56. doi: 10.1038/nature19323
Shibata, M., Yamada, S., Kumar, S. R., Calero, M., Bading, J., Frangione, B., et al. (2000). Clearance of Alzheimer's amyloid-β(1-40) peptide from brain by LDL receptor–related protein-1 at the blood-brain barrier. J. Clin. Invest. 106, 1489–1499. doi: 10.1172/JCI10498
Shoji, H., Takao, K., Hattori, S., and Miyakawa, T. (2016). Age-related changes in behavior in C57BL/6J mice from young adulthood to middle age. Mol. Brain 9:11. doi: 10.1186/s13041-016-0191-9
Sierra, A., Abiega, O., Shahraz, A., and Neumann, H. (2013). Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front. Cell. Neurosci. 7:6. doi: 10.3389/fncel.2013.00006
Sierra, A., Gottfried-Blackmore, A. C., McEwen, B. S., and Bulloch, K. (2007). Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 55, 412–424. doi: 10.1002/glia.20468
Simmons, D. A., Casale, M., Alcon, B., Pham, N., Narayan, N., and Lynch, G. (2007). Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington's disease. Glia 55, 1074–1084. doi: 10.1002/glia.20526
Simpson, J., Rezaie, P., Newcombe, J., Cuzner, M. L., Male, D., and Woodroofe, M. N. (2000). Expression of the beta-chemokine receptors CCR2, CCR3 and CCR5 in multiple sclerosis central nervous system tissue. J. Neuroimmunol. 108, 192–200. doi: 10.1016/S0165-5728(00)00274-5
Sipe, G. O., Lowery, R. L., Tremblay, M. È., Kelly, E. A., Lamantia, C. E., and Majewska, A. K. (2016). Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 7:10905. doi: 10.1038/ncomms10905
Sleegers, K., Brouwers, N., Gijselinck, I., Theuns, J., Goossens, D., Wauters, J., et al. (2006). APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain 129, 2977–2983. doi: 10.1093/brain/awl203
Song, J., Shu, L., Zhang, Z., Tan, X., Sun, E., Jin, X., et al. (2012). Reactive oxygen species-mediated mitochondrial pathway is involved in Baohuoside I-induced apoptosis in human non-small cell lung cancer. Chem. Biol. Interact. 199, 9–17. doi: 10.1016/j.cbi.2012.05.005
Soria, F. N., Pampliega, O., Bourdenx, M., Meissner, W. G., Bezard, E., and Dehay, B. (2017). Exosomes, an unmasked culprit in neurodegenerative diseases. Front. Neurosci. 11:26. doi: 10.3389/fnins.2017.00026
Spangenberg, E. E., Lee, R. J., Najafi, A. R., Rice, R. A., Elmore, M. R. P., Blurton-Jones, M., et al. (2016). Eliminating microglia in Alzheimer's mice prevents neuronal loss without modulating amyloid-β pathology. Brain 139, 1265–1281. doi: 10.1093/brain/aww016
Srikanth, V., Maczurek, A., Phan, T., Steele, M., Westcott, B., Juskiw, D., et al. (2011). Advanced glycation endproducts and their receptor RAGE in Alzheimer's disease. Neurobiol. Aging 32, 763–777. doi: 10.1016/j.neurobiolaging.2009.04.016
Stein, M., Keshav, S., Harris, N., and Gordon, S. (1992). Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J. Exp. Med. 176, 287–292. doi: 10.1084/jem.176.1.287
Stence, N., Waite, M., and Dailey, M. E. (2001). Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia 33, 256–266. doi: 10.1002/1098-1136(200103)33:3<256::AID-GLIA1024>3.0.CO;2-J
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi: 10.1016/j.cell.2007.10.036
Streit, W. J., Braak, H., Xue, Q.-S., and Bechmann, I. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 118, 475–485. doi: 10.1007/s00401-009-0556-6
Streit, W. J., Mrak, R. E., and Griffin, W. S. T. (2004). Microglia and neuroinflammation: a pathological perspective. J. Neuroinflammation 1:14. doi: 10.1186/1742-2094-1-14
Streit, W. J., Walter, S. A., and Pennell, N. A. (1999). Reactive microgliosis. Progr. Neurobiol. 57, 563–581. doi: 10.1016/S0301-0082(98)00069-0
Su, T., Zhang, T., Xie, S., Yan, J., Wu, Y., Li, X., et al. (2016). Discovery of novel PDE9 inhibitors capable of inhibiting Aβ aggregation as potential candidates for the treatment of Alzheimer's disease. Sci. Rep. 6:21826. doi: 10.1038/srep21826
Tabatadze, N., Savonenko, A., Song, H., Bandaru, V. V. R., Chu, M., and Haughey, N. J. (2010). Inhibition of neutral sphingomyelinase-2 perturbs brain sphingolipid balance and spatial memory in mice. J. Neurosci. Res. 88, 2940–2951. doi: 10.1002/jnr.22438
Tai, L. M., Koster, K. P., Luo, J., Lee, S. H., Wang, Y.-T., Collins, N. C., et al. (2014). Amyloid-β pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J. Biol. Chem. 289, 30538–30555. doi: 10.1074/jbc.M114.600833
Takahashi, K., Prinz, M., Stagi, M., Chechneva, O., and Neumann, H. (2007). TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med. 4:e124. doi: 10.1371/journal.pmed.0040124
Takamatsu, K., Ikeda, T., Haruta, M., Matsumura, K., Ogi, Y., Nakagata, N., et al. (2014). Degradation of amyloid beta by human induced pluripotent stem cell-derived macrophages expressing Neprilysin-2. Stem Cell Res. 13, 442–453. doi: 10.1016/j.scr.2014.10.001
Takeuchi, H., Jin, S., Wang, J., Zhang, G., Kawanokuchi, J., Kuno, R., et al. (2006). Tumor necrosis factor-α induces neurotoxicity via glutamate release from hemichannels of activated Microglia in an autocrine manner. J. Biol. Chem. 281, 21362–21368. doi: 10.1074/jbc.M600504200
Tawakol, A., Singh, P., Rudd, J. H. F., Soffer, J., Cai, G., Vucic, E., et al. (2014). Effect of treatment for 12 weeks with Rilapladib, a lipoprotein-associated phospholipase A2 inhibitor, on arterial inflammation as assessed with 18F-fluorodeoxyglucose-positron emission tomography imaging. J. Am. Coll. Cardiol. 63, 86–88. doi: 10.1016/j.jacc.2013.07.050
Taylor, D. L., Jones, F., Kubota, E. S. F. C. S., and Pocock, J. M. (2005). Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor α-induced neurotoxicity in concert with microglial-derived fas ligand. J. Neurosci. 25:2952. doi: 10.1523/JNEUROSCI.4456-04.2005
Tesseur, I., Zou, K., Esposito, L., Bard, F., Berber, E., Can, J. V., et al. (2006). Deficiency in neuronal TGF-β signaling promotes neurodegeneration and Alzheimer's pathology. J. Clin. Invest. 116, 3060–3069. doi: 10.1172/JCI27341
Theunis, C., Crespo-Biel, N., Gafner, V., Pihlgren, M., López-Deber, M. P., Reis, P., et al. (2013). Efficacy and Safety of A Liposome-Based Vaccine against Protein Tau, Assessed in Tau.P301L Mice That Model Tauopathy. PLoS ONE 8:e72301. doi: 10.1371/journal.pone.0072301
Theuns, J., Marjaux, E., Vandenbulcke, M., Van Laere, K., Kumar-Singh, S., Bormans, G., et al. (2006). Alzheimer dementia caused by a novel mutation located in the APP C-terminal intracytosolic fragment. Hum. Mutat. 27, 888–896. doi: 10.1002/humu.20402
Thompson, A. G., Gray, E., Heman-Ackah, S. M., Mager, I., Talbot, K., Andaloussi, S. E., et al. (2016). Extracellular vesicles in neurodegenerative disease [mdash] pathogenesis to biomarkers. Nat. Rev. Neurol. 12, 346–357. doi: 10.1038/nrneurol.2016.68
Tichauer, J. E., Flores, B., Soler, B., Bernhardi, L. E.-V., Ramírez, G., and von Bernhardi, R. (2014). Age-dependent changes on TGFβ1 Smad3 pathway modify the pattern of microglial cell activation. Brain Behav. Immun. 37, 187–196. doi: 10.1016/j.bbi.2013.12.018
Tremblay, M. S., Colley, R. C., Saunders, T. J., Healy, G. N., and Owen, N. (2010). Physiological and health implications of a sedentary lifestyle. Appl. Physiol. Nutr. Metab. 35, 725–740. doi: 10.1139/H10-079
Tremblay, M.-È., Zettel, M. L., Ison, J. R., Allen, P. D., and Majewska, A. K. (2012). Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia 60, 541–558. doi: 10.1002/glia.22287
Treviño, S., Aguilar-Alonso, P., Flores Hernandez, J. A., Brambila, E., Guevara, J., Flores, G., et al. (2015). A high calorie diet causes memory loss, metabolic syndrome and oxidative stress into hippocampus and temporal cortex of rats. Synapse 69, 421–433. doi: 10.1002/syn.21832
Truman, L. A., Ford, C. A., Pasikowska, M., Pound, J. D., Wilkinson, S. J., Dumitriu, I. E., et al. (2008). CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 112:5026. doi: 10.1182/blood-2008-06-162404
Ueberham, U., Ueberham, E., Gruschka, H., and Arendt, T. (2006). Altered subcellular location of phosphorylated Smads in Alzheimer's disease. Euro. J. Neurosci. 24, 2327–2334. doi: 10.1111/j.1460-9568.2006.05109.x
Ulrich, J. D., Finn, M. B., Wang, Y., Shen, A., Mahan, T. E., Jiang, H., et al. (2014). Altered microglial response to Aβ plaques in APPPS1-21 mice heterozygous for TREM2. Mol. Neurodegener. 9, 20–20. doi: 10.1186/1750-1326-9-20
Varnum, M. M., Clayton, K. A., Yoshii-Kitahara, A., Yonemoto, G., Koro, L., Ikezu, S., et al. (2017). A split-luciferase complementation, real-time reporting assay enables monitoring of the disease-associated transmembrane protein TREM2 in live cells. J. Biol. Chem. 292, 10651–10663. doi: 10.1074/jbc.M116.759159
Varnum, M. M., Kiyota, T., Ingraham, K. L., Ikezu, S., and Ikezu, T. (2015). The Anti-inflammatory glycoprotein, cd200, restores neurogenesis and enhances amyloid phagocytosis in a mouse model of Alzheimer's disease. Neurobiol. Aging 36, 2995–3007. doi: 10.1016/j.neurobiolaging.2015.07.027
Vella, L. J., Sharples, R. A., Lawson, V. A., Masters, C. L., Cappai, R., and Hill, A. F. (2007). Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J. Pathol. 211, 582–590. doi: 10.1002/path.2145
Vinet, J., van Weering, H. R. J., Heinrich, A., Kälin, R. E., Wegner, A., Brouwer, N., et al. (2012). Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J. Neuroinflam. 9, 27–27. doi: 10.1186/1742-2094-9-27
Vlad, S. C., Miller, D. R., Kowall, N. W., and Felson, D. T. (2008). Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 70, 1672–1677. doi: 10.1212/01.wnl.0000311269.57716.63
Vlassov, A. V., Magdaleno, S., Setterquist, R., and Conrad, R. (2012). Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 1820, 940–948. doi: 10.1016/j.bbagen.2012.03.017
von Bernhardi, R., Cornejo, F., Parada, G. E., and Eugenín, J. (2015a). Role of TGFβ signaling in the pathogenesis of Alzheimer's disease. Front. Cell. Neurosci. 9:426. doi: 10.3389/fncel.2015.00426
von Bernhardi, R., Eugenín-von Bernhardi, L., and Eugenín, J. (2015b). Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 7:124. doi: 10.3389/fnagi.2015.00124
Vowinckel, E., Reutens, D., Becher, B., Verge, G., Evans, A., Owens, T., et al. (1997). PK11195 binding to the peripheral benzodiazepine receptor as a marker of microglia activation in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neurosci. Res. 50, 345–353. doi: 10.1002/(SICI)1097-4547(19971015)50:2<345::AID-JNR22>3.0.CO;2-5
Vukovic, J., Colditz, M. J., Blackmore, D. G., Ruitenberg, M. J., and Bartlett, P. F. (2012). Microglia modulate hippocampal neural precursor activity in response to exercise and aging. J. Neurosci. 32:6435. doi: 10.1523/JNEUROSCI.5925-11.2012
Walker, D. G., Dalsing-Hernandez, J. E., Campbell, N. A., and Lue, L.-F. (2009). Decreased expression of CD200 and CD200 receptor in Alzheimer's disease: a potential mechanism leading to chronic inflammation. Exp. Neurol. 215, 5–19. doi: 10.1016/j.expneurol.2008.09.003
Walker, L. C., and LeVine, H. (2000). The cerebral proteopathies. Mol. Neurobiol. 21, 83–95. doi: 10.1385/MN:21:1-2:083
Wang, J., Shiozawa, Y., Wang, J., Wang, Y., Jung, Y., Pienta, K. J., et al. (2008). The role of CXCR7/RDC1 as a chemokine receptor for CXCL12/SDF-1 in prostate cancer. J. Biol. Chem. 283, 4283–4294. doi: 10.1074/jbc.M707465200
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., and Robinette, M. L. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Wehrspaun, C. C., Haerty, W., and Ponting, C. P. (2015). Microglia recapitulate a hematopoietic master regulator network in the aging human frontal cortex. Neurobiol. Aging 36, 2443.e9–2443.e20. doi: 10.1016/j.neurobiolaging.2015.04.008
Wiessner, C., Wiederhold, K. H., Tissot, A. C., Frey, P., Danner, S., Jacobson, L. H., et al. (2011). The second-generation active Aβ immunotherapy CAD106 reduces amyloid accumulation in APP transgenic mice while minimizing potential side effects. J. Neurosci. 31, 9323–9331. doi: 10.1523/JNEUROSCI.0293-11.2011
Wong, W. T. (2013). Microglial aging in the healthy CNS: phenotypes, drivers, and rejuvenation. Front. Cell. Neurosci. 7:22. doi: 10.3389/fncel.2013.00022
Woodbury, M. E., Freilich, R. W., Cheng, C. J., Asai, H., Ikezu, S., Boucher, J. D., et al. (2015). miR-155 is essential for inflammation-induced hippocampal neurogenic dysfunction. J. Neurosci. 35, 9764–9781. doi: 10.1523/JNEUROSCI.4790-14.2015
Wyss-Coray, T., Lin, C., Yan, F., Yu, G. Q., Rohde, M., McConlogue, L., et al. (2001). TGF-[beta]1 promotes microglial amyloid-[beta] clearance and reduces plaque burden in transgenic mice. Nat. Med. 7, 612–618. doi: 10.1038/87945
Xiang, X., Werner, G., Bohrmann, B., Liesz, A., Mazaheri, F., Capell, A., et al. (2016). TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol. Med. 8, 992–1004. doi: 10.15252/emmm.201606370
Xu, H., Sweeney, D., Wang, R., Thinakaran, G., Lo, A. C. Y., Sisodia, S. S., et al. (1997). Generation of Alzheimer β-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formation. Proc. Natl. Acad. Sci. U.S.A. 94, 3748–3752. doi: 10.1073/pnas.94.8.3748
Yarza, R., Vela, S., Solas, M., and Ramirez, M. J. (2015). c-Jun N-terminal Kinase (JNK) signaling as a therapeutic target for Alzheimer's disease. Front. Pharmacol. 6:321. doi: 10.3389/fphar.2015.00321
Yin, Z., Raj, D., Saiepour, N., Van Dam, D., Brouwer, N., Holtman, I. R., et al. (2017). Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer's disease. Neurobiol. Aging 55, 115–122. doi: 10.1016/j.neurobiolaging.2017.03.021
Yuan, P., Condello, C., Keene, C. D., Wang, Y., Bird, T. D., Paul, S. M., et al. (2016). TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90, 724–739. doi: 10.1016/j.neuron.2016.05.003
Zempel, H., and Mandelkow, E. (2014). Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 37, 721–732. doi: 10.1016/j.tins.2014.08.004
Zerbinatti, C. V., Wozniak, D. F., Cirrito, J., Cam, J. A., Osaka, H., Bales, K. R., et al. (2004). Increased soluble amyloid-β peptide and memory deficits in amyloid model mice overexpressing the low-density lipoprotein receptor-related protein. Proc. Natl. Acad. Sci. U.S.A. 101, 1075–1080. doi: 10.1073/pnas.0305803101
Zhao, J., Fu, Y., Yasvoina, M., Shao, P., Hitt, B., and O'Connor, T. (2007). Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J. Neurosci. 27, 3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007
Zheng, H., Liu, C.-C., Atagi, Y., Chen, X.-F., Jia, L., Yang, L., et al. (2016). Opposing roles of the triggering receptor expressed on Myeloid Cells 2 (TREM2) and TREM-like Transcript 2 (TREML2) in Microglia Activation. Neurobiol. Aging 42, 132–141. doi: 10.1016/j.neurobiolaging.2016.03.004
Zhong, L., Chen, X.-F., Wang, T., Wang, Z., Liao, C., Wang, Z., et al. (2017a). Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 214, 597–607. doi: 10.1084/jem.20160844
Zhong, L., Zhang, Z.-L., Li, X., Liao, C., Mou, P., Wang, T., et al. (2017b). TREM2/DAP12 complex regulates inflammatory responses in Microglia via the JNK signaling pathway. Front. Aging Neurosci. 9:204. doi: 10.3389/fnagi.2017.00204
Keywords: Alzheimer's disease, microglia, neurodegeneration, amyloid-beta peptide, tau protein, neuroinflammation, proteopathy
Citation: Clayton KA, Van Enoo AA and Ikezu T (2017) Alzheimer's Disease: The Role of Microglia in Brain Homeostasis and Proteopathy. Front. Neurosci. 11:680. doi: 10.3389/fnins.2017.00680
Received: 01 October 2017; Accepted: 21 November 2017;
Published: 12 December 2017.
Edited by:
Naruhiko Sahara, National Institute of Radiological Sciences (NIRS), JapanReviewed by:
Takahisa Kanekiyo, Mayo Clinic Florida, United StatesCopyright © 2017 Clayton, Van Enoo and Ikezu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tsuneya Ikezu, dGlrZXp1QGJ1LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.