 Kim L. Mertens
Kim L. Mertens Andries Kalsbeek
Andries Kalsbeek Maarten R. Soeters2
Maarten R. Soeters2 Hannah M. Eggink
Hannah M. Eggink- 1Master's Program in Biomedical Sciences, University of Amsterdam, Amsterdam, Netherlands
- 2Department of Endocrinology and Metabolism, Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands
- 3Laboratory of Endocrinology, Department Clinical Chemistry, Academic Medical Centre, University of Amsterdam, Amsterdam, Netherlands
- 4Department of Hypothalamic Integration Mechanisms, Netherlands Institute for Neuroscience, Amsterdam, Netherlands
Bile acids are best known as detergents involved in the digestion of lipids. In addition, new data in the last decade have shown that bile acids also function as gut hormones capable of influencing metabolic processes via receptors such as FXR (farnesoid X receptor) and TGR5 (Takeda G protein-coupled receptor 5). These effects of bile acids are not restricted to the gastrointestinal tract, but can affect different tissues throughout the organism. It is still unclear whether these effects also involve signaling of bile acids to the central nervous system (CNS). Bile acid signaling to the CNS encompasses both direct and indirect pathways. Bile acids can act directly in the brain via central FXR and TGR5 signaling. In addition, there are two indirect pathways that involve intermediate agents released upon interaction with bile acids receptors in the gut. Activation of intestinal FXR and TGR5 receptors can result in the release of fibroblast growth factor 19 (FGF19) and glucagon-like peptide 1 (GLP-1), both capable of signaling to the CNS. We conclude that when plasma bile acids levels are high all three pathways may contribute in signal transmission to the CNS. However, under normal physiological circumstances, the indirect pathway involving GLP-1 may evoke the most substantial effect in the brain.
Introduction
Bile acids are synthesized in the liver from cholesterol and released in the intestinal lumen upon food intake. They are predominantly known for their role as nutritional detergents that dissolve lipids and lipid-soluble vitamins. However, a growing body of recent literature describes bile acids as versatile signaling molecules (Houten et al., 2006; de Aguiar Vallim et al., 2013; Kuipers et al., 2014), with a widespread distribution of bile acid receptors throughout the organism. Via these receptors, bile acids are capable of modulating their own synthesis (Chiang, 2009; Lefebvre et al., 2009), lipid, glucose and energy metabolism (Thomas et al., 2008a,b; Lefebvre et al., 2009; Schonewille et al., 2016). In addition, bile acids can signal via intermediate signaling molecules that are released upon activation of bile acid receptors in the intestine. The receptors receptive for these intermediate molecules are also distributed ubiquitously throughout the body.
Bile acids and their associated receptors have been detected in the human and rodent brain (Mano et al., 2004a; Ferdinandusse et al., 2009; Keitel et al., 2010; Huang et al., 2016; McMillin et al., 2016; Zheng et al., 2016), however, it is still not clear whether bile acids are capable of signaling to the central nervous system (CNS) and what this signaling could imply. Two recent reviews discussed the role of bile acids in neurological diseases (Ackerman and Gerhard, 2016; McMillin and DeMorrow, 2016), but did not elaborate on the possible physiological effects of bile acid signaling. Therefore, in this review we discuss the signaling pathways of bile acids implicated in the control of energy metabolism under normal physiological circumstances, involving both direct and indirect pathways to the CNS.
Bile Acid Metabolism and the Enterohepatic Circulation
Bile acid synthesis and enterohepatic cycling have been elaborately reviewed previously (Russell, 2003; Thomas et al., 2008b). In short, bile acids have a cholesterol backbone. Bile acid biosynthesis mainly occurs in hepatocytes (Figure 1), where the classical pathway is initiated by cholesterol 7α-hydroxylase (CYP7A1) which is regulated by the farnesoid X receptor (FXR). The alternative pathway can be initiated by different enzymes that are also expressed outside the liver. De novo synthesized bile acids are called primary bile acids. In humans the primary bile acids are cholic acid (CA) and chenodeoxycholic acid (CDCA); in mice the dominant bile acids are CA and muricholic acid (MCA). Subsequently, these bile acids are conjugated with the amino acids glycine (mainly in humans) or taurine (mainly in mice). Bile acids are transported from the hepatocytes through the bile canaliculi and stored, together with cholesterol and phospholipids, in the gallbladder. Following food intake, the presence of nutrients (especially fats and proteins) in the stomach triggers gallbladder emptying which results in the release of bile acids into the duodenum. When bile acids pass through the intestinal tract, they contribute to the absorption of lipids and fat-soluble vitamins. In the intestine, gut microbiota deconjugate and dehydroxylate the primary bile acids, converting them into secondary bile acids and enhancing the diversity of the bile acid pool (Figure 1). In the jejunum and colon, unconjugated, and uncharged bile acids enter the enterocytes through passive diffusion (Figure 2). In the ileum, active uptake of conjugated bile acids takes place by the apical sodium-dependent bile acid transporter (ASBT). In total about 95% of the bile acids are reabsorbed into intestinal enterocytes. The remaining 5% is excreted via feces, a loss which is compensated for by de novo bile acid synthesis in the liver. Specific transporters in enterocytes make sure that bile acids are redirected to the liver via the portal vein. In the liver, about ~90% of the bile acids are cleared from the hepatic circulation for reuse. Bile acids can be recycled 4–12 times per day between hepatocytes in the liver and enterocytes in the intestine—which is called the enterohepatic circulation (Mok et al., 1977; Figure 2). Only a small portion (< 10%) of the total bile acid pool reaches the systemic circulation. Systemic plasma bile acid concentrations show a postprandial increase, resulting in a daily rhythm associated with food intake that fluctuates between 5 and 15 μM in humans (Angelin and Bjorkhem, 1977; LaRusso et al., 1978; Schalm et al., 1978; Glicksman et al., 2010; Steiner et al., 2011; Sonne et al., 2016). Also in rodents a daily rhythm of plasma bile acid levels has been reported (Ho, 1976a,b; Zhang et al., 2011; Eggink et al., 2017). These feeding-induced changes indicate that circulating bile acids could provide a postprandial signal, transmitting information about the arrival of nutrients and the subsequent availability of energy (Thomas et al., 2008a). In addition, hepatocytes are equipped with a machinery that can actively promote bile acid excretion when hepatic bile acid concentration increase extensively, as accumulating bile acids can be toxic due to their detergent-like function (Zollner et al., 2006). Consequently, many cases of liver failure or liver damage result in an increased efflux of bile acids into the systemic circulation, leading to high levels of plasma bile acids (Neale et al., 1971; Engelking et al., 1980; Benyoub et al., 2011; Tanaka et al., 2012; Quinn et al., 2014; McMillin et al., 2016).
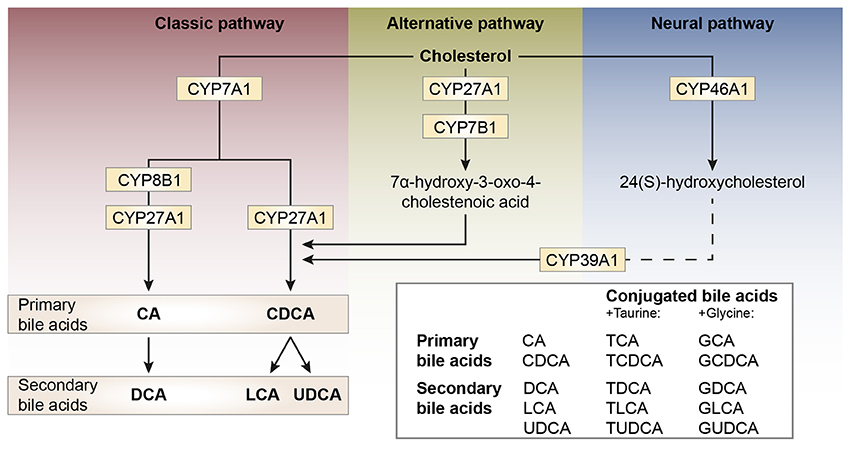
Figure 1. Schematic representation of bile acid synthesis pathways in humans. Bile acid synthesis from cholesterol occurs via different pathways. The classic pathway occurs in the liver and is responsible for the majority of bile acid synthesis. This pathway is initiated by the enzyme cholesterol 7α-hydroxylase (encoded by CYP7A1) and results in the formation of the primary bile acids cholic acid (CA) and chenodeoxycholic acid (CDCA). Key enzymes for the formation of CA or CDCA are sterol 12α-hydroxylase (CYP8B1) and sterol-27 hydroxylase (CYP27A1), respectively. In rodents, the primary bile acids formed are CA and muricholic acid (MCA). The primary bile acids are conjugated to the amino acids glycine (G, mainly in humans) or taurine (T, mainly in rodents) forming conjugated bile acids and bile salts. The formation of secondary bile acids occurs in the intestine under the control of gut flora and when returned to the liver these secondary bile acids can also be conjugated to glycine and taurine. The alternative pathway of bile acid synthesis also occurs in other tissues besides the liver. This pathway is initiated by CYP27A1 and also involves CYP7B1. After several metabolic steps CDCA is formed. The last pathway occurs in the brain and is believed to be important for neuronal cholesterol clearance. Cholesterol is converted to 24(S)-hydroxycholesterol by CYP46A1 and subsequently exits the brain and enters the bloodstream (dotted line). In the liver, bile acid synthesis continues involving CYP39A1 resulting in CDCA after several steps.
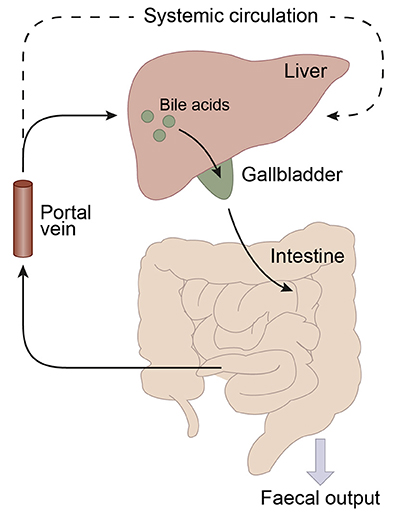
Figure 2. Schematic representation of the enterohepatic circulation of bile acids. Bile acids are synthesized in the liver and stored in the gallbladder. Following food intake, bile acids are released into the duodenum. Traveling down the intestine, the majority of bile acids are taken up by enterocytes. In the jejunum and colon passive diffusion of unconjugated and uncharged bile acids takes place and the ileum is the main site for active uptake of conjugated bile acids by bile salt transporters. About 95% of the bile acids are reabsorbed in the ileum and consequently only a small portion (~5%) of the bile acids is lost through fecal output. The bile acids that are absorbed by the enterocytes are released into the portal vein and redirected to the liver for recycling. Only a small portion escapes the enterohepatic circulation and reaches the systemic circulation. The liver extracts 80–90% of the portal total bile acids.
Bile Acids and the Blood-Brain Barrier
Once in the systemic circulation, bile acids reach the brain via the internal carotid and vertebral arteries that join in an artery ring at the base of the brain—the circle of Willis. From here the arteries arise that ensure blood supply to the brain. In contrast with other capillaries throughout the body, brain capillary endothelial cells are interconnected by tight junctions so substances in the blood need to cross the endothelial cell membranes in order to enter the brain. This blood-brain barrier (BBB) protects the brain from potentially harmful circulating molecules (Bernacki et al., 2008; Abbott et al., 2010).
There are reports that both unconjugated and conjugated bile acids can cross the BBB (Keene et al., 2001; Palmela et al., 2015; McMillin et al., 2016; Figure 3A), however, the involved mechanisms are still uncertain. Unconjugated bile acids might diffuse across the BBB, because CA, CDCA, and deoxycholic acid (DCA) are capable of diffusing across phospholipid bilayers (Kamp and Hamilton, 1993) and their brain levels correlate with their serum levels (Higashi et al., 2017). Indeed, unconjugated ursodeoxycholic acid (UDCA) crossed the BBB in a dose depend manner in orally treated amyotrophic lateral sclerosis patients (Parry et al., 2010). Conjugated bile acids need active transport to cross the BBB due to their larger structure and amphipathic nature (St-Pierre et al., 2001). Indeed, several xenobiotic and bile acid transporters found in the liver, intestine, and kidney are also present at the BBB and choroid plexus providing the machinery for bile acid transport over the BBB. These include members of the solute carrier (SLC) family such as the organic anion transporting polypeptides (OATP) and ASBT, and members of the ATP-binding cassette transporters (ABC) family such as the multidrug resistance protein (MRD) 2 and 4 (Choudhuri et al., 2003; Bernacki et al., 2008; Abbott et al., 2010; Klaassen and Aleksunes, 2010; Ballatori, 2011; Table 1). The main function of these transporters is to protect the brain from potentially toxic molecules by transporting them out of the brain into the bloodstream (Abbott et al., 2010). However, the presence of these transporters on both the basolateral (blood-facing) and apical (brain-facing) side, also facilitates the transport of molecules from the systemic circulation into the CNS (Abe et al., 1998; Klaassen and Aleksunes, 2010). Of interest, an in situ rat brain perfusion with [3H]TCA resulted in no significant uptake of the bile acid in the ipsilateral hemisphere within 2 min, suggesting that the labeled TCA did not cross the BBB (Kitazawa et al., 1998). Direct evidence of in vivo transport of bile acids over the BBB via their transporters is still lacking.
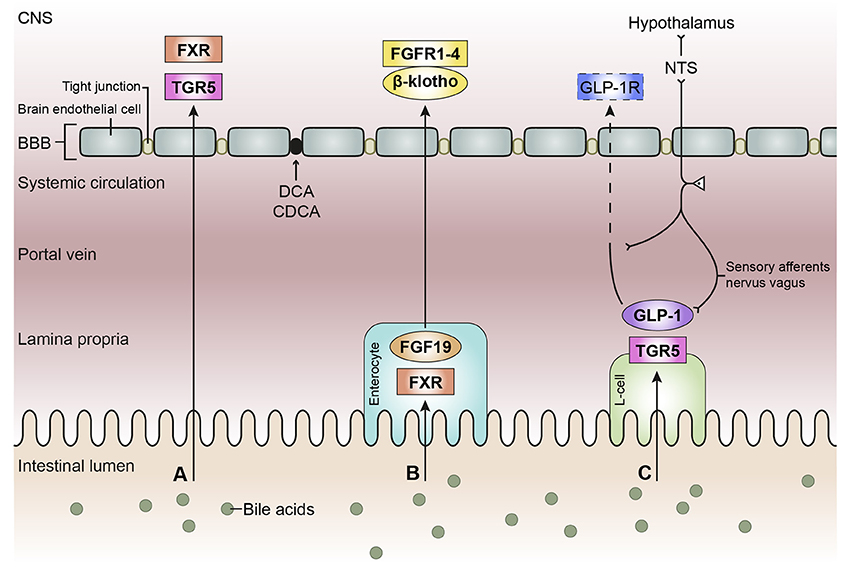
Figure 3. Schematic overview of the bile acid signaling pathways to the central nervous system. Bile acids in the intestinal lumen can signal to the central nervous system (CNS) via different pathways, in this review we focused on the direct pathway (A), the indirect pathway via farnesoid X receptor-fibroblast growth factor 19 (FXR-FGF19) signaling (B), and the indirect pathway via Takeda G protein-coupled receptor-glucagon-like peptide-1 (TGR5-GLP-1) signaling (C). (A) Bile acids in the intestine escape the enterohepatic circulation and reach the systemic circulation. Bile acids need to pass the blood-brain barrier (BBB) in order to interact with receptors in the brain, e.g., FXR and TGR5. Deoxycholic acid (DCA) and chenodeoxy cholic acid (CDCA) have been found to interact with gap junction proteins, resulting in a leaky BBB. (B) Bile acids taken up by enterocytes can activate the nuclear receptor FXR, which results in the release of FGF19. FGF19 is released by the enterocyte and reaches the portal vein, a small portion of FGF19 will not be taken up by the liver and enters the systemic circulation. FGF19 needs to cross the BBB to interact with FGF receptors (1–4) in the brain. The protein β-klotho is necessary for the formation of a stable receptor-complex. (C) in the intestine, a specific group of enteroendocrine cells, L-cells, produces GLP-1 upon the activation of TGR5 which can be triggered by bile acids. GLP-1 is quickly degraded by the enzyme dipeptidyl peptidase-4 (DPP-4, not shown), consequently high concentrations of GLP-1 are only found in the lamina propria of the intestine. A small portion of intact GLP-1 reaches the portal vein and even a smaller portion reaches the systemic circulation. It is questionable whether sufficient intact GLP-1 reaches the brain to interact with GLP-1 receptors, hence the dashed line. GLP-1 receptors are also expressed on afferent terminals of the vagal nerve present in the lamina propria and portal vein. The vagal nerve projects to the nucleus of the solitary tract (NTS) in the brainstem, from where projections are directed toward other brain regions, e.g. the hypothalamus (the vagal-brainstem-hypothalamic pathway).
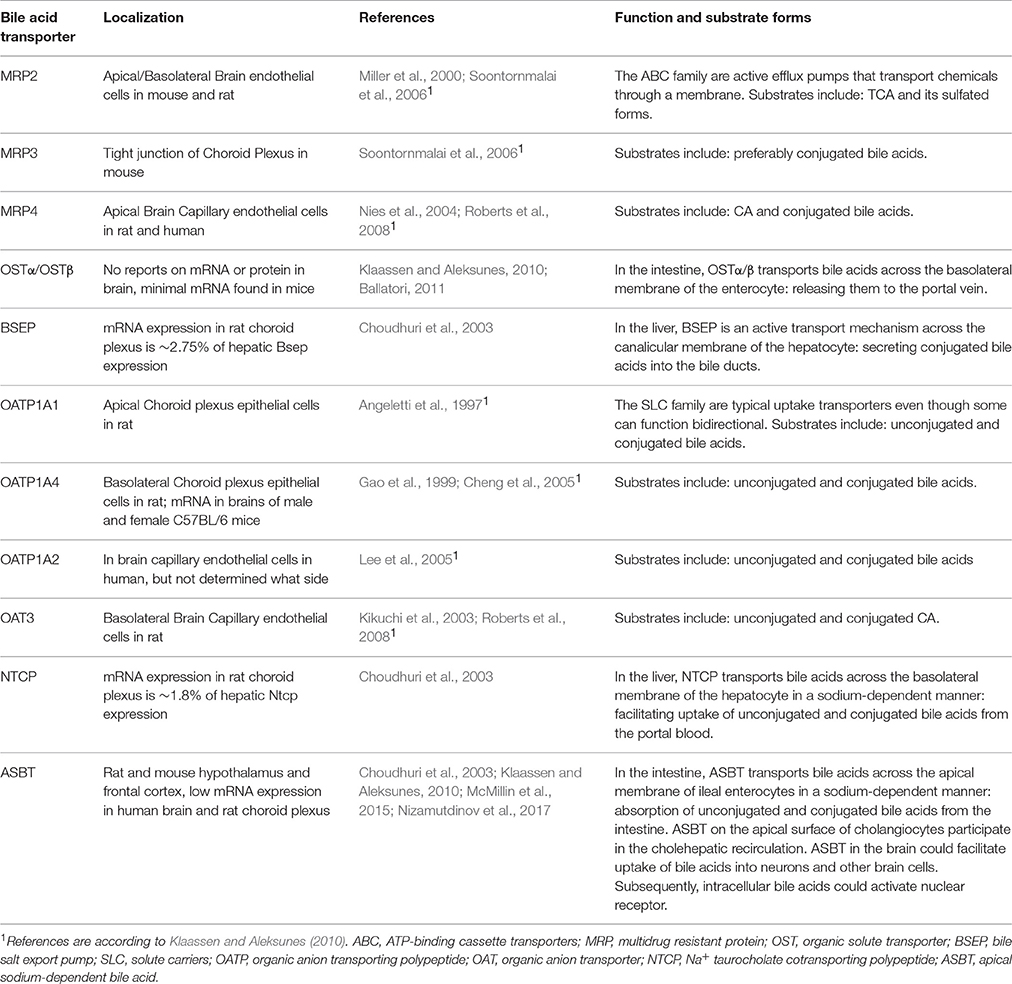
Table 1. Bile acid transporters found in the brain.
Plasma Bile Acid Levels and the Integrity of the Blood-Brain Barrier
During liver failure, plasma bile acid levels can increase dramatically (Benyoub et al., 2011; Tanaka et al., 2012; Quinn et al., 2014; McMillin et al., 2016), sometimes even up to 20-fold in rats (Quinn et al., 2014) and 100-fold in humans (Neale et al., 1971; Engelking et al., 1980). At high concentrations (≥1.5 mM) bile acids are capable of damaging the lipid layers of the BBB (Greenwood et al., 1991), due to their detergent and lytic action on cell membranes (Naqvi et al., 1970; Greenwood et al., 1991). At lower concentrations (0.2–1.5 mM), bile acids may modify the BBB in a more subtle way (Greenwood et al., 1991). The bile acids CDCA and DCA increase phosphorylation of the tight junction protein occludin in a Rac1-dependent mechanism, resulting in the disruption of tight junctions (Quinn et al., 2014) and leading to increased permeability of the BBB (Greenwood et al., 1991; Quinn et al., 2014). Consequently, allowing bile acids and other molecules to diffuse into the brain. UDCA and its glycine-conjugated form glyco-ursodeoxycholic acid (GUDCA) exert a protective effect on brain endothelial cells by reducing apoptosis (Palmela et al., 2015). In addition, a recent study showed that microglial cells express TGR5 and that binding of taurine-conjugated UDCA (TUDCA) to TGR5 has anti-inflammatory effects in a mouse model of acute brain inflammation (Yanguas-Casás et al., 2017). This could explain the neuroprotective effects of TUDCA observed as reduced neuronal apoptosis in several animal models for neurodegenerative diseases, such as Huntington's disease (Keene et al., 2001, 2002), Alzheimer's disease (Sola et al., 2006; Viana et al., 2009), Parkinson's disease (Duan et al., 2002), acute ischemia (Rodrigues et al., 2002), and hemorrhagic stroke (Rodrigues et al., 2003). These findings highlight the physiological differences of bile acid species, where DCA and CDCA interfere and disturb gap junction function in the BBB, but UDCA and its conjugated forms exert a protective effect on brain endothelial cells and neurons. It is still unknown whether these different effects are due to the different affinities of these bile acids for FXR.
Bile Acids in the Central Nervous System
When plasma bile acid concentrations increase during hepatic failure, cerebral bile acid levels also rise excessively in humans and rodents (Bron et al., 1977; Ceryak et al., 1998; Tripodi et al., 2012). Additional reports suggest that these elevated levels of bile acids are derived from the systemic circulation (DeMorrow et al., 2012; Quinn et al., 2014; McMillin et al., 2015, 2016; Palmela et al., 2015). Also in healthy conditions detectable levels of both conjugated and unconjugated bile acids have been reported in the brain, both in rodents and humans (Mano et al., 2004a; Zheng et al., 2016; Higashi et al., 2017; Pan et al., 2017; Table 2). In rats no glycine-conjugated bile acids were detected in the rat brain (Mano et al., 2004a; Higashi et al., 2017). In one study CDCA is the most abundantly present bile acid in the rat brain, making up 92.1% of the total amount of cerebral bile acids and mainly being found in protein-bound form conceivably preventing it from exiting the brain (Mano et al., 2004a). However, a different study found that CA was most abundantly present in the rat brain and did not find the high amounts of protein-bound CDCA (Higashi et al., 2017). The discrepancy in CDCA levels between these studies could not be sufficiently explained. In addition, various bile acid transporters are expressed in the CNS such as ASBT in the hypothalamus and frontal cortex (McMillin et al., 2015; Nizamutdinov et al., 2017; Table 1), providing a mechanism for the neuronal uptake of bile acids.
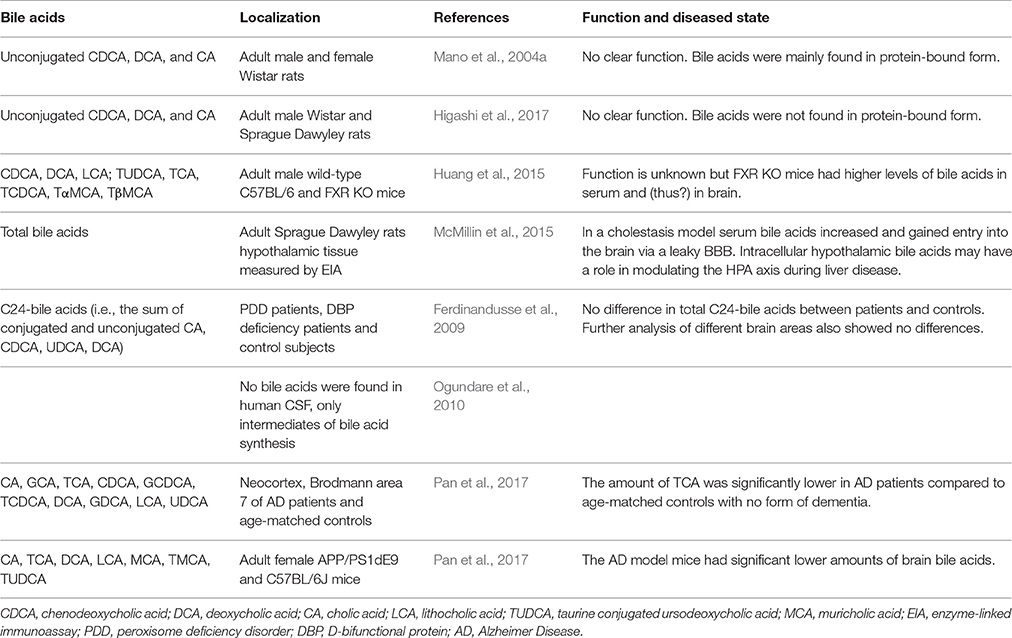
Table 2. Bile acids found in the brain.
The unconjugated bile acids (CA, CDCA, and DCA) seem to be mostly derived from the periphery by passive diffusion as brain levels correlate with serum levels and intraperitoneally injected D4-CA and D4-CDCA are well-detected in the brain (Higashi et al., 2017). There are also indications that at least parts of the biosynthesis pathway for bile acids is present in the brain, because involved enzymes and intermediates have been detected locally (Cali et al., 1991; Björkhem et al., 1998; Lund et al., 1999; Li-Hawkins et al., 2000; Mano et al., 2004a,b; Ogundare et al., 2010). Since only a part of the biosynthesis pathway of bile acids is present in the brain, its main function has been proposed to be cholesterol clearance (McMillin and DeMorrow, 2016).
Bile Acid Receptors in the Central Nervous System
The most studied bile acid receptors are FXR (Makishima et al., 1999; Parks et al., 1999; Wang et al., 1999) and the Takeda G protein-coupled receptor 5 (TGR5) (Maruyama et al., 2002; Kawamata et al., 2003). Both receptors are abundantly expressed in the enterohepatic circulation, but also in the brain [FXR: (Huang et al., 2016; McMillin et al., 2016); TGR5: (Maruyama et al., 2002, 2006; Keitel et al., 2010; Yanguas-Casás et al., 2017)]. Other receptors that might bind bile acids and can be found in the CNS are summed in Table 3, their possible functions are reviewed elsewhere (McMillin and DeMorrow, 2016).
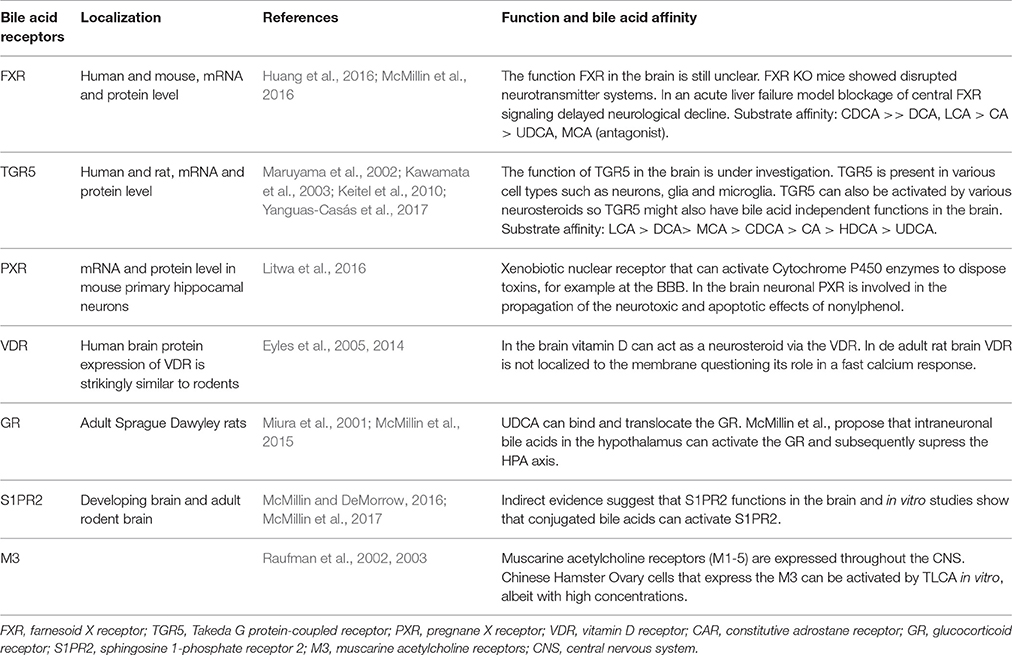
Table 3. Bile acid receptors found in the brain.
Effects of Elevated Plasma Bile Acid Levels on the Central Nervous System
As mentioned above, receptors able to bind bile acids are also expressed in the CNS and thus are capable of mediating the actions of bile acid signaling. Most studies investigated the effects of central bile acid signaling in the pathological state or pharmacologically administered bile acids directly into the brain (reviewed in Ackerman and Gerhard, 2016; McMillin and DeMorrow, 2016). For example a study investigating hepatic encephalopathy induced by acute liver failure in mice found doubled amounts of bile acids in the brain compared to the control situation (McMillin et al., 2016). The elevated plasma and cerebral bile acid levels consequently generate an amplified effect and show what the possible consequences are of pathologic bile acid signaling in the brain. The variety of effects of bile acids in the diseased brain (McMillin and DeMorrow, 2016) illustrates that bile acids cannot be seen as one signal, but different forms have different effects, including their difference in affinity for the receptors. Moreover, they do not reflect on the effects of bile acid signaling to the CNS under normal physiological circumstances caused by the postprandial elevated plasma bile acid levels, of which little is known.
Indirect Bile Acid Signaling to the Central Nervous System via FXR-FGF15/19 Pathway
In addition to the direct signaling pathway described in the previous section, bile acids can also provide a signal to the CNS via the gut-brain axis. After their release into the intestine, bile acids can interact with receptors in the gastrointestinal system and thereby initiate a signal cascade that reaches the CNS. In this section we will discuss the indirect pathway initiated by FXR activation and the release of fibroblast growth factor (FGF) 15/19 (Figure 3B). FXR is primarily activated by CDCA and CA and to a lesser extent by DCA and LCA (Makishima et al., 1999; Parks et al., 1999; Wang et al., 1999). In contrast, UDCA and muricholic acid (MCA) do not seem to activate FXR (Makishima et al., 1999; Parks et al., 1999; Wang et al., 1999) and even seem to antagonize FXR in mice (Sayin et al., 2013; Hu et al., 2014), highlighting an important difference between humans and mice, because MCA is the major bile acid in mice and does not occur in humans (Takahashi et al., 2016). FXR is extensively expressed in hepatocytes and enterocytes. In the enterohepatic circulation FXR functions as a bile acid sensor, providing negative feedback to the bile acid synthesis and transport machinery when bile acid levels rise. For an extensive overview of FXR function in the enterohepatic circulation we recommend (Lefebvre et al., 2009; De Magalhaes Filho et al., 2017).
Intestinal FXR and FGF15/19
In the intestine, activation of FXR can trigger the production of FGF19, a FGF with hormonal characteristics (Holt et al., 2003; Potthoff et al., 2012). The rodent orthologue for human FGF19 is FGF15, which has comparable, but not necessarily identical functions (Inagaki et al., 2005; Jones, 2008). The most abundant expression of FGF15/19 is found in the terminal ileum of the intestine (Holt et al., 2003; Inagaki et al., 2005; Fon Tacer et al., 2010). Bile acids absorbed by enterocytes can activate the nuclear receptor FXR, which leads to the production of FGF15/19 (Kliewer and Mangelsdorf, 2015). The enterocytes release FGF15/19 from their basolateral membrane into the portal vein. Subsequently, FGF15/19 activates the fibroblast growth factor receptor (FGFR) 4 in the liver and this leads to the inhibition of de novo bile acid synthesis by inhibition of Cyp7a1. Liver and intestinal FXR KO models have shown that Cyp7a1 inhibition depends mostly on intestinal FXR activation via FGF15 (Kim et al., 2007). FGF19 mRNA is expressed in the intestine and in hepatocytes in the liver, while in mice FGF15 mRNA is only expressed in the intestine (Song et al., 2009; Fon Tacer et al., 2010). Outside the enterohepatic cycle FGF15/19 can signal in an endocrine manner and is involved in lipid and glucose metabolism (Owen et al., 2015). In both human and mouse FGF15/19 mRNA is widely expressed in the developing brain (Nishimura et al., 1999; Ford-Perriss et al., 2001; Gimeno et al., 2003), but not in the adult brain (Nishimura et al., 1999; Fon Tacer et al., 2010).
Plasma levels of FGF15 in mouse (Katafuchi et al., 2015) and FGF19 in humans (Lundåsen et al., 2006) have been found to follow a daily rhythm. In humans, plasma FGF19 levels respond to food intake and bile acids, showing a postprandial increase following the peak of plasma bile acid levels ~3 h after a meal (Lundåsen et al., 2006; Sonne et al., 2016). In contrast, a different study found that FGF19 levels predominantly respond to carbohydrate intake compared to lipid or protein intake and concluded that this would dissociate the FGF19 response from bile acid signaling (Morton et al., 2013a). Which is an important issue for further research.
FGF15/19 Signaling in the Central Nervous System
FGF19 in the systemic circulation is capable of crossing the BBB and is relatively stable in the brain (Hsuchou et al., 2013). In addition, FGFRs are expressed in the brain (Wanaka et al., 1990; Yazaki et al., 1994; Belluardo et al., 1997; Reuss and von Bohlen und Halbach, 2003), suggesting that FGF19 could signal from the intestine to the CNS. FGF19 binds directly to FGFR4 but a more solid bond is realized when β-Klotho is bound to the FGF19-FGFR4 complex (Xie et al., 1999; Harmer et al., 2004; Wu et al., 2007, 2009). The single-pass transmembrane protein β-Klotho serves as a cofactor for FGF19 activity by physically interacting with FGFRs, increasing the affinity of FGF19 for FGFRs and causing efficient FGF signaling (Kurosu et al., 2007; Ogawa et al., 2007). For successful binding of FGF19 with FGFR1c, 2c, or 3c the presence of β-Klotho is essential (Kurosu et al., 2007; Wu et al., 2009; Yang et al., 2012). Whereas, FGFR1c, 2c, and 3c are highly expressed in the brain, β-Klotho is not and is selectively expressed in particular regions including the suprachiasmatic, arcuate, and paraventricular nucleus of the hypothalamus, the area postrema and solitary nucleus of the dorsal-vagal complex and the nodose ganglia (Bookout et al., 2013; Liang et al., 2014; Owen et al., 2015). These regions also express FGFRs (Fon Tacer et al., 2010; Bookout et al., 2013; Ryan et al., 2013), however, to our knowledge no studies looked at the co-expression of FGFRs and β-Klotho. In the periphery the main target receptor of FGF15/19 is FGFR4. In the brain, the expression of FGFR4 has been detected in the hypothalamus (Ryan et al., 2013) and in cholinergic neurons in the medial habenular nucleus (Itoh et al., 1994; Miyake and Itoh, 1996). Overall the expression of FGFR4 in the CNS is less abundant than FGFR1c-3c (Fon Tacer et al., 2010). Interestingly, intraperitoneal (ip) FGF19 injections in mice resulted in increased FGFR activity in the hypothalamus, more specifically in the arcuate nucleus (ARC) (Marcelin et al., 2014). Staining for pERK1/2 revealed that in the ARC the AGRP/NPY (agouti-related peptide/neuropeptide Y) neurons and not the POMC (pro-opiomelanocortin) neurons were involved in FGF19 signaling. NPY and POMC neurons modulate feeding behavior by stimulating and inhibiting appetite, respectively (van den Heuvel et al., 2011; Gumbs et al., 2016). Intracerebroventricular (icv) administration of FGF19 decreased neural activation in the ARC as measured by c-Fos expression and reduced gene expression of Agrp and Npy (Marcelin et al., 2014), suggesting that central FGF19 signaling inhibits AGRP/NPY neurons in the ARC.
Taken together, FGF15/19 signaling in the CNS can generate a wide spread of effects via the FGFRs that are present in the hypothalamus, medial habenular nucleus and dorsal-vagal complex. Consequently, bile acids in the enterohepatic circulation can extend their signal to these cerebral regions via the FXR-FGF15/19 pathway.
Central FGF15/19 Improves Metabolic Rate and Glucose Metabolism
The effects of central FGF19 are mainly studied in animal models for obesity and diabetics, because overexpression of FGF19 in mice resulted in increased energy expenditure and animals on a high fat diet (HFD) did not become diabetic or obese (Tomlinson et al., 2002). Intravenous (iv) administration of FGF19, also prevented genetic (ob/ob) and diet-induced (HFD) obese mice to develop diabetes by improving glucose metabolism (Fu et al., 2004). This beneficial effect of systemic FGF19 on glucose metabolism was reduced by 50% when an FGFR antagonist was infused in the brain (Morton et al., 2013b). In addition, rats on a HFD showed reduced expression of hypothalamic FGFR1 and 4 compared to chow-fed rats (Ryan et al., 2013). These findings suggest that central FGFR signaling is involved in energy and glucose metabolism.
Icv FGF19 administration increased the metabolic rate in wild type mice (Fu et al., 2004) and in HFD-fed and ob/ob mice reduced weight gain and improved glucose metabolism (Marcelin et al., 2014). A single administration of icv FGF19 had no effect on the energy expenditure, but improved glucose metabolism in ob/ob mice and mice on a HFD (Morton et al., 2013b; Marcelin et al., 2014) as well as in lean and HFD-fed rats (Ryan et al., 2013). Pretreatment with an FGFR inhibitor in the brain blocked the beneficial effects of icv FGF19 on glucose metabolism (Morton et al., 2013b). In a rat model for type 1 diabetes, hyperglycemia could be reversed by icv administration of FGF19 (Perry et al., 2015). An additional observation was that central FGF19 resulted in decreased adrenocorticotropic hormone (ACTH) and corticosterone plasma levels, suggesting the suppression of HPA activity (Perry et al., 2015). The majority of the studies, investigating the effects of central FGF19 on glucose metabolism found no differences in insulin sensitivity or secretion that could explain improved glucose metabolism (Morton et al., 2013b; Ryan et al., 2013; Perry et al., 2015). However, one study did find improved insulin sensitivity in ob/ob and HFD-fed mice treated with icv FGF19 compared to vehicle treated mice (Marcelin et al., 2014). These studies highlight the controversy concerning the involved mechanisms that explain the effects of central FGF19 on glucose metabolism.
Consequently, these studies provide different explanations concerning the mechanisms that drive the beneficial effect of central FGF19 action on glucose and energy metabolism. This highlights that further research is necessary to reveal the underlying mechanisms that mediate central FGF19 action. Altogether, little is known about the neurocircuitry involved in FGF19-FGFR signaling that could be instigated by bile acid binding to FXR in the intestine. In addition, it should be studied to which extend the different FGFs contribute to the FGFRs signaling in the CNS, because FGF21 binds to the same FGFR and β-Klotho complexes as FGF19 and generates similar effects when administered in the brain (Owen et al., 2015; Degirolamo et al., 2016). However, above all the question remains whether the postprandial increase in plasma FGF19 is sufficient to elicit a substantial effect in the CNS.
Indirect Bile Acid Signaling to Central Nervous System via TGR5-GLP-1 Pathway
In this section we will discuss the indirect pathway involving signaling via intestinal TGR5 (Figure 3C), which is the other well-studied bile acid receptor that is expressed abundantly in the enterohepatic circulation (Thomas et al., 2008a). The TGR5 receptor can be activated by both conjugated and unconjugated bile acids, with litocholic acid (LCA) and taurolitocholic acid (TLCA) being the most potent bile acids (Kawamata et al., 2003). In the brain, TGR5 can also be activated by other endogenous ligands, such as neurosteroids (Keitel et al., 2010). In the intestine, stimulation of TGR5 by bile acids can also result in the release of the gut hormone GLP-1, which is capable of extending the bile acid signal from the intestine to other parts of the body, including the CNS (Figure 3C; Katsuma et al., 2005; Thomas et al., 2009; Ullmer et al., 2013).
Intestinal TGR5 and GLP-1
GLP-1 is an incretin that influences energy homeostasis by reducing appetite and food intake and inhibiting gastric emptying (Drucker and Nauck, 2006). In the gut a particular group of entero-endocrine cells, L-cells, are responsible for the production of GLP-1 and are predominantly found in the terminal ileum and colon (Drucker and Nauck, 2006; Lim and Brubaker, 2006). The action of bile acids on GLP-1 release is predominantly regulated via TGR5 receptors located at the basolateral membrane of L-cells, thus not facing the lumen of the intestine (Brighton et al., 2015). This means that bile acids first need to cross both the apical and the basolateral membrane of intestinal cells in order to activate TGR5 and provoke a GLP-1 response. L-cell GLP-1 release follows a circadian rhythm suggesting it is also under control of the molecular clock system (Gil-Lozano et al., 2014). In addition, GLP-1 release by L-cells can also be triggered via different routes not involving bile acid-induced TGR-5 activation (Lim and Brubaker, 2006). These include indirect routes via endocrine and neural signals induced by the presence of food in the stomach and upper intestine (Lim and Brubaker, 2006; Holst, 2007), stimuli thought to be responsible for the rapid postprandial release of GLP-1 (Holst, 2007). When food reaches the ileum, the GLP-1 producing L cells are directly stimulated by glucose, fat (Lim and Brubaker, 2006; Ezcurra et al., 2013), and bile acids (Katsuma et al., 2005; Thomas et al., 2009). The amplitude of the evoked GLP-1 response depends on meal size (Vilsboll et al., 2003). It is difficult to differentiate between the effects induced by GLP-1 in general and the effects that are particularly induced by GLP-1 as a consequence of TGR5 activation by bile acids. Research using TGR5 knockout (TGR5−/−) mice showed that these mice still produce GLP-1 and seemed not different from wild type mice (Thomas et al., 2009). However, TGR5−/− mice fed a HFD displayed impaired glucose tolerance compared to wild types (Thomas et al., 2009). This might indicate that under normal circumstances sufficient GLP-1 is released via signaling routes not involving TGR5-activation. However, this TGR5-independent GLP-1 signal might not be proficient under more extreme circumstances e.g., when high amounts of fat are digested.
Intestinal GLP-1 Signaling to the Central Nervous System via Systemic Circulation
Intestinal GLP-1 can reach the brain via two major routes, one being via the systemic circulation and interacting with GLP-1 receptors in the brain (Orskov et al., 1996; Yamamoto et al., 2003) and the other route being through signaling via the vagus nerve (Abbott et al., 2005; Rüttimann et al., 2009). When GLP-1 is released from the basolateral membrane of the L-cells, GLP-1 is taken up by capillaries and transported to the portal vein and subsequently the liver (Holst, 2007). Nonetheless, only a fraction of intestinal GLP-1 reaches the liver in its active form, because the endothelial membranes of the capillaries express the enzyme dipeptidyl peptidase-4 (DPP-4), which degrades GLP-1 rapidly (Holst and Deacon, 2005). Due to the rapid decay only 25% of the intestinal GLP-1 reaches the hepatic portal vein (Holst, 2007). Of this portion only half reaches the systemic circulation via the liver. DDP-4 is also present in plasma, therefore the small amounts of GLP-1 reaching the systemic circulation have a half-life of only 1–2 min (Holst, 2007). In rats, a regular chow meal led to a transient increase in GLP-1 levels in the hepatic portal vein but not in the vena cava, showing that the postprandial GLP-1 increase is not substantial in the systemic circulation (Punjabi et al., 2014). Contrasting, in humans a postprandial increase in plasma GLP-1 levels was evident, lasting several minutes (Vilsbøll et al., 2001; Calanna et al., 2013; Sonne et al., 2014). The human subjects used for plasma GLP-1 measurements (Sonne et al., 2014) also showed a postprandial increase in plasma bile acid levels (Sonne et al., 2016).
The GLP-1 receptor is expressed in various tissues including the CNS (Richards et al., 2014; Cork et al., 2015). GLP-1 is capable of crossing the BBB (Kastin et al., 2002), but it is questionable whether sufficient intact GLP-1 reaches the BBB and other distal tissues to elicit a substantial effect. Therefore, only the area postrema and subfornical area—circumventricular organs—may be plausible brain regions that could gate peripheral GLP-1 signaling via its GLP-1 receptors (Göke et al., 1995; Orskov et al., 1996; Yamamoto et al., 2003). This pathway was established by iv administration of GLP-1 in rats (Orskov et al., 1996; Yamamoto et al., 2003; Punjabi et al., 2014), however, this pathway might not be substantial under normal physiological circumstances when GLP-1 release is triggered by food intake and bile acids.
Intestinal GLP-1 Signaling to the Central Nervous System via Vagal Nerve Afferents
The other pathway through which intestinal GLP-1 could signal to the CNS is via activation of vagal afferent fibers. These sensory fibers originate in the nodose ganglion and provide terminals into peripheral tissues, including liver tissue (Dardevet et al., 2004, 2005), hepatic portal vein (Balkan and Li, 2000; Vahl et al., 2007), and lamina propria of the intestine (Berthoud et al., 2004; Nakagawa et al., 2004). These terminals express GLP-1 receptors and are therefore responsive to local GLP-1 levels (Holst, 2007). The nodose ganglion projects to the nucleus of the solitary tract (NTS) in the hindbrain (Nakagawa et al., 2004; Holst, 2007). NTS neurons are bidirectional connected with other brain regions, including the hypothalamus (Ricardo and Koh, 1978; van der Kooy et al., 1984). In animal models the signal transmission after ip GLP-1 administration was abolished following subdiaphragmatic vagal deafferentation or after transection of the brainstem-hypothalamic pathway (Abbott et al., 2005; Rüttimann et al., 2009). This established the importance of the vagal-brainstem-hypothalamic pathway for the signal transmission of GLP-1 from the gastrointestinal tract to the CNS. Subsequently, the brainstem and hypothalamus are connected with brain regions involved in autonomic function, metabolic processing, and cognitive and emotional functioning (Rogers et al., 2016). These findings raise the question whether bile acids themselves could directly interact with the vagal nerve and relay a signal to the CNS. However, we have not found reports that show the expression of bile acid receptors on the vagal nerve.
GLP-1 Signaling via the Vagal Nerve Afferents Is Involved in Glucose Metabolism and Energy Homeostasis
Via the vagal-brainstem-hypothalamic pathway peripheral GLP-1 can affect many brain regions and consequently many processes. However, the most studied effects of peripheral GLP-1 are its inhibitory effect on food intake and increased perception of satiety (Tang-Christensen et al., 1996; Turton et al., 1996; Abbott et al., 2005; Talsania et al., 2005; Williams et al., 2006, 2009; Scott and Moran, 2007; Hayes et al., 2008; Rüttimann et al., 2009; Punjabi et al., 2014), which are both believed to be mediated predominantly by the CNS (Turton et al., 1996). These data suggest that vagal nerve terminals in the lamina propria of the intestine are involved in regulating appetite.
Postprandial, the highest GLP-1 concentrations are found in the lamina propria of the intestine and second in the hepatic portal vein (Holst and Deacon, 2005; Holst, 2007). GLP-1 signaling via vagal afferents in the hepatic portal vein does not modulate food intake (Rüttimann et al., 2009), but is involved in modulating glucose metabolism by interacting with hepatoportal glucose sensors (Balkan and Li, 2000; Burcelin et al., 2001; Vahl et al., 2007). This reveals a pathway through which bile acids may be capable to modulate glucose metabolism: TGR5-mediated GLP-1 secretion acting upon hepatoportal glucose sensors. Indeed, TGR5−/− mice on a HFD have impaired glucose tolerance and TGR5 over expression in transgenic mice improved glucose tolerance in combination with increased GLP-1 and insulin secretion (Thomas et al., 2009). The above results indicate a differentiation in GLP-1 pathways: glucose homeostasis is mediated via the vagal afferents in the hepatic portal vein and energy homeostasis is mediated via the vagal afferents in the lamina propria (Rüttimann et al., 2009).
Taken together, bile acids in the intestine can signal to the brain by using GLP-1 as an intermediate molecule to activate vagal nerve afferents in the lamia propria and hepatic portal vein that project to the NTS in the hindbrain and subsequently to the hypothalamus. What the exact contribution of bile acids is in the overall GLP-1 signal is difficult to determine, because other nutrient and indirect signals could trigger GLP-1 release.
GLP-1 Released from Neurons in the Hindbrain
An important consideration is that in addition to GLP-1 release from the intestine, GLP-1 is also produced in the brain. GLP-1 is secreted from a population of preproglucagon (PPG) cells in the NTS and in the intermediate reticular nucleus within the hindbrain (Han et al., 1986; Drucker, 1990; Larsen et al., 1997; Trapp and Cork, 2015). PPG neurons project to a variety of brain regions involved in energy homeostasis and autonomic control including the hypothalamus, thalamus, and amygdala (Merchenthaler et al., 1999; Llewellyn-Smith et al., 2011; Trapp and Cork, 2015). Central GLP-1 signaling is generally linked to energy homeostasis (Cabou and Burcelin, 2011) and glucose metabolism (Sandoval, 2008; Sandoval et al., 2008). The presence of GLP-1-releasing neurons in the brain adds an extra difficulty to elucidating the effects of peripheral GLP-1 in the brain. Electrophysiological findings indicate that PPG cells receive monosynaptic input from vagal afferent fibers (Hisadome et al., 2010). This could mean that peripheral GLP-1 from the intestine could modulate the activity of PPG cells via vagal nerve afferents and consequently stimulate GLP-1 release in the CNS (Hisadome et al., 2010). However, direct evidence is lacking.
Glucagon-Like Peptide-2
Together with GLP-1 also glucagon-like peptide-2 (GLP-2) is released from intestinal L-cells in response to nutrients and bile acids. In addition, also in the brain GLP-2 is released from the preproglucagonergic neurons in the brainstem together with GLP-1. GLP-2 acts via its own G protein-coupled receptor, GLP-2R which is mainly expressed in the gastro-intestinal tract and CNS. In the gut, GLP-2 functions in intestinal mucosal health and stimulates nutrient absorption, and in this way influences energy homeostasis (Baldassano et al., 2016). Recently, it has been shown that GLP-2 also stimulates gall bladder filling via GLP-2R and in a TGR5 independent manner in mice (Yusta et al., 2017). In contrast to GLP-1 and the GLP-1R, the functions of GLP-2 and GLP-2R in the brain have not been studied much, but it is thought that GLP-2 has anorexic effects and reduces appetite by activating the GLP-2R in the ARC of the hypothalamus (reviewed in Guan, 2014; Baldassano et al., 2016). However, in humans, peripheral GLP-2 administration had no effect on satiety or food intake (Schmidt et al., 2003; Sørensen et al., 2003). GLP-2 is not an incretin and does not receive as much attention as GLP-1 with respect to research on glucose metabolism and diabetes (Janssen et al., 2013). We are not aware of any studies that investigated the effects of bile acids or postprandial intestinal GLP-2 release on central GLP-2 functioning. The blood half-life of GLP-2 is a few minutes longer than that of GLP-1, but both are efficiently cleared by the kidneys.
Concluding Remarks
In this review we discussed three different pathways via which bile acids could signal to the CNS. In the direct pathway (Figure 3A), bile acids reach the brain via the systemic circulation. In the brain, the machinery for bile acid signaling is present, i.e., receptors able to bind bile acids and transporters to transport bile acids into neurons (Tables 1–3). However, it remains uncertain whether this pathway is substantial under normal physiological circumstances. More research is required to determine whether the postprandial increase in plasma bile acids is also translated into increased bile acid levels in the brain and whether these amounts are sufficient to activate bile acid receptors expressed in the brain. Considering the current information, we believe that this pathway does not exert a prominent route for bile acid signaling to the CNS.
The indirect pathway mediated by FXR-FGF15/19 (Figure 3B) could exert an effect via the CNS through the presence of FGFRs in the brain. FGF15/19 signaling in the brain is associated with energy and glucose homeostasis. However, it is questionable whether the postprandial increase of plasma FGF15/19 is sufficient for substantial signaling in the CNS. We believe that under normal physiological circumstances the peripheral mediated consequences of FGF15/19 signaling exceed the effects that are possibly mediated via the CNS.
The indirect pathway mediated by TGR5-GLP-1 (Figure 3C) can signal to the CNS via two routes, through the systemic circulation and via the vagal nerve. The latter route is the most significant signaling route, because postprandial GLP-1 levels are high in the lamina propria of the intestine and hepatic portal vein, where vagal nerve terminals are present. The vagal nerve signals to the brainstem and subsequently to other brain regions. Via this pathway bile acids could influence glucose and energy homeostasis, among other things. Currently this seems to be the only noteworthy signaling route to the CNS initiated by bile acids under normal physiological circumstances. However, the exact implications of bile acids for this signaling route and their contribution to the whole-body postprandial response remains an interesting subject for future research.
Author Contributions
All authors contributed to the design and concept of the review. KM drafted the manuscript and provided the figures. AK, MS, and HE critically reviewed the manuscript and attributed with important intellectual content. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work.
Funding
MS is funded by the Dutch Diabetes Research Foundation (grant 2011.80.1423).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbott, C. R., Monteiro, M., Small, C. J., Sajedi, A., Smith, K. L., Parkinson, J. R., et al. (2005). The inhibitory effects of peripheral administration of peptide YY 3–36 and glucagon-like peptide-1 on food intake are attenuated by ablation of the vagal–brainstem–hypothalamic pathway. Brain Res. 1044, 127–131. doi: 10.1016/j.brainres.2005.03.011
Abbott, N. J., Patabendige, A. A., Dolman, D. E., Yusof, S. R., and Begley, D. J. (2010). Structure and function of the blood–brain barrier. Neurobiol. Dis. 37, 13–25. doi: 10.1016/j.nbd.2009.07.030
Abe, T., Kakyo, M., Sakagami, H., Tokui, T., Nishio, T., Tanemoto, M., et al. (1998). Molecular characterization and tissue distribution of a new organic anion transporter subtype (oatp3) that transports thyroid hormones and taurocholate and comparison with oatp2. J. Biol. Chem. 273, 22395–22401. doi: 10.1074/jbc.273.35.22395
Ackerman, H. D., and Gerhard, G. S. (2016). Bile acids in neurodegenerative disorders. Front. Aging Neurosci. 8:263. doi: 10.3389/fnagi.2016.00263
Angeletti, R. H., Novikoff, P. M., Juvvadi, S. R., Fritschy, J. M., Meier, P. J., and Wolkoff, A. W. (1997). The choroid plexus epithelium is the site of the organic anion transport protein in the brain. Proc. Natl. Acad. Sci. U.S.A. 94, 283–286. doi: 10.1073/pnas.94.1.283
Angelin, B., and Bjorkhem, I. (1977). Postprandial serum bile acids in healthy man. Evidence for differences in absorptive pattern between individual bile acids. Gut 18, 606–609. doi: 10.1136/gut.18.8.606
Baldassano, S., Amato, A., and Mule, F. (2016). Influence of glucagon-like peptide 2 on energy homeostasis. Peptides 86, 1–5. doi: 10.1016/j.peptides.2016.09.010
Balkan, B., and Li, X. (2000). Portal GLP-1 administration in rats augments the insulin response to glucose via neuronal mechanisms. Am. J. Physiol. Regul. Integrat. Comp. Physiol. 279, r1449–r1454.
Ballatori, N. (2011). Pleiotropic functions of the organic solute transporter Ostalpha-Ostbeta. Dig. Dis. 29, 13–17. doi: 10.1159/000324123
Belluardo, N., Wu, G., Mudo, G., Hansson, A. C., Pettersson, R., and Fuxe, K. (1997). Comparative localization of fibroblast growth factor receptor-1,-2, and-3 mRNAs in the rat brain: in situ hybridization analysis. J. Comp. Neurol. 379, 226–246. doi: 10.1002/(SICI)1096-9861(19970310)379:2<226::AID-CNE5>3.0.CO;2-5
Benyoub, K., Muller, M., Bonnet, A., Simon, R., Gazon, M., Duperret, S., et al. (2011). Amounts of bile acids and bilirubin removed during single-pass albumin dialysis in patients with liver failure. Ther. Apher. Dial. 15, 504–506. doi: 10.1111/j.1744-9987.2011.00980.x
Bernacki, J., Dobrowolska, A., Nierwinska, K., and Malecki, A. (2008). Physiology and pharmacological role of the blood-brain barrier. Pharmacol. Rep. 60, 600–622.
Berthoud, H. R., Blackshaw, L. A., Brookes, S. J., and Grundy, D. (2004). Neuroanatomy of extrinsic afferents supplying the gastrointestinal tract. Neurogastroenterol. Motil. 16, 28–33. doi: 10.1111/j.1743-3150.2004.00471.x
Björkhem, I., Lutjohann, D., Diczfalusy, U., Stahle, L., Ahlborg, G., and Wahren, J. (1998). Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 39, 1594–1600.
Bookout, A. L., de Groot, M. H., Owen, B. M., Lee, S., Gautron, L., Lawrence, H. L., et al. (2013). FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat. Med. 19, 1147–1152. doi: 10.1038/nm.3249
Brighton, C. A., Rievaj, J., Kuhre, R. E., Glass, L. L., Schoonjans, K., Holst, J. J., et al. (2015). Bile acids trigger GLP-1 release predominantly by accessing basolaterally located G protein–coupled bile acid receptors. Endocrinology 156, 3961–3970. doi: 10.1210/en.2015-1321
Bron, B., Waldram, R., Silk, D. B., and Williams, R. (1977). Serum, cerebrospinal fluid, and brain levels of bile acids in patients with fulminant hepatic failure. Gut 18, 692–696. doi: 10.1136/gut.18.9.692
Burcelin, R., Da Costa, A., Drucker, D., and Thorens, B. (2001). Glucose competence of the hepatoportal vein sensor requires the presence of an activated glucagon-like peptide-1 receptor. Diabetes 50, 1720–1728. doi: 10.2337/diabetes.50.8.1720
Cabou, C., and Burcelin, R. (2011). GLP-1, the gut-brain, and brain-periphery axes. Rev. Diabet. Stud. 8, 418–431. doi: 10.1900/RDS.2011.8.418
Calanna, S., Christensen, M., Holst, J. J., Laferrere, B., Gluud, L. L., Vilsboll, T., et al. (2013). Secretion of glucagon-like peptide-1 in patients with type 2 diabetes mellitus: systematic review and meta-analyses of clinical studies. Diabetologia 56, 965–972. doi: 10.1007/s00125-013-2841-0
Cali, J. J., Hsieh, C. L., Francke, U., and Russell, D. W. (1991). Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J. Biol. Chem. 266, 7779–7783.
Ceryak, S., Bouscarel, B., Malavolti, M., and Fromm, H. (1998). Extrahepatic deposition and cytotoxicity of lithocholic acid: studies in two hamster models of hepatic failure and in cultured human fibroblasts. Hepatology 27, 546–556. doi: 10.1002/hep.510270232
Cheng, X., Maher, J., Chen, C., and Klaassen, C. D. (2005). Tissue distribution and ontogeny of mouse organic anion transporting polypeptides (Oatps). Drug Metab. Dispos. 33, 1062–1073. doi: 10.1124/dmd.105.003640
Chiang, J. Y. (2009). Bile acids: regulation of synthesis. J. Lipid Res. 50, 1955–1966. doi: 10.1194/jlr.R900010-JLR200
Choudhuri, S., Cherrington, N. J., Li, N., and Klaassen, C. D. (2003). Constitutive expression of various xenobiotic and endobiotic transporter mRNAs in the choroid plexus of rats. Drug Metab. Dispos. 31, 1337–1345. doi: 10.1124/dmd.31.11.1337
Cork, S. C., Richards, J. E., Holt, M. K., Gribble, F. M., Reimann, F., and Trapp, S. (2015). Distribution and characterisation of glucagon-like peptide-1 receptor expressing cells in the mouse brain. Mol. Metab. 4, 718–731. doi: 10.1016/j.molmet.2015.07.008
Dardevet, D., Moore, M. C., DiCostanzo, C. A., Farmer, B., Neal, D. W., Snead, W., et al. (2005). Insulin secretion-independent effects of GLP-1 on canine liver glucose metabolism do not involve portal vein GLP-1 receptors. Am. J. Physiol. Gastrointest. Liver Physiol. 289, g806–g814. doi: 10.1152/ajpgi.00121.2005
Dardevet, D., Moore, M. C., Neal, D., DiCostanzo, C. A., Snead, W., and Cherrington, A. D. (2004). Insulin-independent effects of GLP-1 on canine liver glucose metabolism: duration of infusion and involvement of hepatoportal region. Am. J. Physiol. Endocrinol. Metabol. 287, e75–e81. doi: 10.1152/ajpendo.00035.2004
de Aguiar Vallim, T. Q., Tarling, E. J., and Edwards, P. A. (2013). Pleiotropic roles of bile acids in metabolism. Cell Metab. 17, 657–669. doi: 10.1016/j.cmet.2013.03.013
De Magalhaes Filho, C. D., Downes, M., and Evans, R. M. (2017). Farnesoid X receptor an emerging target to combat obesity. Dig. Dis. 35, 185–190. doi: 10.1159/000450909
Degirolamo, C., Sabba, C., and Moschetta, A. (2016). Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat. Rev. Drug Discov. 15, 51–69. doi: 10.1038/nrd.2015.9
DeMorrow, S., Frampton, G., Galindo, C., Pae, H. Y., and Quinn, M. (2012). Increased serum bile acids after extrahepatic biliary obstruction causes leakiness to the blood brain barrier via the disruption of tight junctions. FASEB J. 26, 1117. doi: 10.1096/fj.1530-6860
Drucker, D. J. (1990). Glucagon and the glucagon-like peptides. Pancreas 5, 484–488. doi: 10.1097/00006676-199007000-00018
Drucker, D. J., and Nauck, M. A. (2006). The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 368, 1696–1705. doi: 10.1016/S0140-6736(06)69705-5
Duan, W.-M., Rodrigures, C. M. P., Zhao, L.-R., Steer, C. J., and Low, W. C. (2002). Tauroursodeoxycholic acid improves the survival and function of nigral transplants in a rat model of Parkinson's disease. Cell Transplant. 11, 195–205. doi: 10.0000/096020198389960
Eggink, H. M., Oosterman, J. E., de Goede, P., de Vries, E. M., Foppen, E., Koehorst, M., et al. (2017). Complex interaction between circadian rhythm and diet on bile acid homeostasis in male rats. Chronobiol. Int. doi: 10.1080/07420528.2017.1363226. [Epub ahead of print].
Engelking, L. R., Dasher, C. A., and Hirschowitz, B. I. (1980). Within-day fluctuations in serum bile-acid concentrations among normal control subjects and patients with hepatic disease. Am. J. Clin. Pathol. 73, 196–201. doi: 10.1093/ajcp/73.2.196
Eyles, D. W., Liu, P. Y., Josh, P., and Cui, X. (2014). Intracellular distribution of the vitamin D receptor in the brain: comparison with classic target tissues and redistribution with development. Neuroscience 268, 1–9. doi: 10.1016/j.neuroscience.2014.02.042
Eyles, D. W., Smith, S., Kinobe, R., Hewison, M., and McGrath, J. J. (2005). Distribution of the vitamin D receptor and 1α-hydroxylase in human brain. J. Chem. Neuroanat. 29, 21–30. doi: 10.1016/j.jchemneu.2004.08.006
Ezcurra, M., Reimann, F., Gribble, F. M., and Emery, E. (2013). Molecular mechanisms of incretin hormone secretion. Curr. Opin. Pharmacol. 13, 922–927. doi: 10.1016/j.coph.2013.08.013
Ferdinandusse, S., Denis, S., Faust, P. L., and Wanders, R. J. (2009). Bile acids: the role of peroxisomes. J. Lipid Res. 50, 2139–2147. doi: 10.1194/jlr.R900009-JLR200
Fon Tacer, K., Bookout, A. L., Ding, X., Kurosu, H., John, G. B., Wang, L., et al. (2010). Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol. Ndocrinol. 24, 2050–2064. doi: 10.1210/me.2010-0142
Ford-Perriss, M., Abud, H., and Murphy, M. (2001). Fibroblast growth factors in the developing central nervous system. Clin. Exp. Pharmacol. Physiol. 28, 493–503. doi: 10.1046/j.1440-1681.2001.03477.x
Fu, L., John, L. M., Adams, S. H., Yu, X. X., Tomlinson, E., Renz, M., et al. (2004). Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 145, 2594–2603. doi: 10.1210/en.2003-1671
Gao, B., Stieger, B., Noe, B., Fritschy, J. M., and Meier, P. J. (1999). Localization of the organic anion transporting polypeptide 2 (Oatp2) in capillary endothelium and choroid plexus epithelium of rat brain. J. Histochem. Cytochem. 47, 1255–1264. doi: 10.1177/002215549904701005
Gil-Lozano, M., Mingomataj, E. L., Wu, W. K., Ridout, S. A., and Brubaker, P. L. (2014). Circadian secretion of the intestinal hormone GLP-1 by the Rodent L Cell. Diabetes 63, 3674–3685. doi: 10.2337/db13-1501
Gimeno, L., Brulet, P., and Marti, S. (2003). Study of Fgf15 gene expression in developing mouse brain. Gene Exp. Patterns 3, 473–481. doi: 10.1016/S1567-133X(03)00059-0
Glicksman, C., Pournaras, D. J., Wright, M., Roberts, R., Mahon, D., Welbourn, R., et al. (2010). Postprandial plasma bile acid responses in normal weight and obese subjects. Ann. Clin. Biochem. 47(Pt. 5), 482–484. doi: 10.1258/acb.2010.010040
Göke, R., Larsen, P. J., Mikkelsen, J. D., and Sheikh, S. P. (1995). Distribution of GLP-1 binding sites in the rat brain: evidence that exendin-4 is a ligand of brain GLP-1 binding sites. Eur. J. Neurosci. 7, 2294–2300. doi: 10.1111/j.1460-9568.1995.tb00650.x
Greenwood, J., Adu, J., Davey, A. J., Abbott, N. J., and Bradbury, M. W. (1991). The effect of bile salts on the permeability and ultrastructure of the perfused, energy-depleted, rat blood-brain barrier. J. Cereb. Blood Flow Metab. 11, 644–654. doi: 10.1038/jcbfm.1991.116
Guan, X. (2014). The CNS glucagon-like peptide-2 receptor in the control of energy balance and glucose homeostasis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 307, r585–r596. doi: 10.1152/ajpregu.00096.2014
Gumbs, M. C., van den Heuvel, J. K., and la Fleur, S. E. (2016). The effect of obesogenic diets on brain Neuropeptide, Y. Physiol. Behav. 162, 161–173. doi: 10.1016/j.physbeh.2016.04.049
Han, V. K., Hynes, M. A., Jin, C., Towle, A. C., Lauder, J. M., and Lund, P. K. (1986). Cellular localization of proglucagon/glucagon-like peptide I messenger RNAs in rat brain. J. Neurosci. Res. 16, 97–107. doi: 10.1002/jnr.490160110
Harmer, N. J., Pellegrini, L., Chirgadze, D., Fernandez-Recio, J., and Blundell, T. L. (2004). The crystal structure of fibroblast growth factor (FGF) 19 reveals novel features of the FGF family and offers a structural basis for its unusual receptor affinity. Biochemistry 43, 629–640. doi: 10.1021/bi035320k
Hayes, M. R., Skibicka, K. P., and Grill, H. J. (2008). Caudal brainstem processing is sufficient for behavioral, sympathetic, and parasympathetic responses driven by peripheral and hindbrain glucagon-like-peptide-1 receptor stimulation. Endocrinology 149, 4059–4068. doi: 10.1210/en.2007-1743
Higashi, T., Watanabe, S., Tomaru, K., Yamazaki, W., Yoshizawa, K., Ogawa, S., et al. (2017). Unconjugated bile acids in rat brain: analytical method based on LC/ESI-MS/MS with chemical derivatization and estimation of their origin by comparison to serum levels. Steroids 125, 107–113. doi: 10.1016/j.steroids.2017.07.001
Hisadome, K., Reimann, F., Gribble, F. M., and Trapp, S. (2010). Leptin directly depolarizes preproglucagon neurons in the nucleus tractus solitarius: electrical properties of glucagon-like Peptide 1 neurons. Diabetes 59, 1890–1898. doi: 10.2337/db10-0128
Ho, K. J. (1976a). Circadian distribution of bile acid in the enterohepatic circulatory system in hamsters. J. Lipid Res. 17, 600–604.
Ho, K. J. (1976b). Circadian distribution of bile acids in the enterohepatic circulatory system in rats. Am. J. Physiol. 230, 1331–1335.
Holst, J. J. (2007). The physiology of glucagon-like peptide 1. Physiol. Rev. 87, 1409–1439. doi: 10.1152/physrev.00034.2006
Holst, J. J., and Deacon, C. F. (2005). Glucagon-like peptide-1 mediates the therapeutic actions of DPP-IV inhibitors. Diabetologia 48, 612–615. doi: 10.1007/s00125-005-1705-7
Holt, J. A., Luo, G., Billin, A. N., Bisi, J., McNeill, Y. Y., Kozarsky, K. F., et al. (2003). Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 17, 1581–1591. doi: 10.1101/gad.1083503
Houten, S. M., Watanabe, M., and Auwerx, J. (2006). Endocrine functions of bile acids. EMBO J. 25, 1419–1425. doi: 10.1038/sj.emboj.7601049
Hsuchou, H., Pan, W., and Kastin, A. J. (2013). Fibroblast growth factor 19 entry into brain. Fluids Barriers CNS 10:1. doi: 10.1186/2045-8118-10-32
Hu, X., Bonde, Y., Eggertsen, G., and Rudling, M. (2014). Muricholic bile acids are potent regulators of bile acid synthesis via a positive feedback mechanism. J. Intern. Med. 275, 27–38. doi: 10.1111/joim.12140
Huang, C., Wang, J., Hu, W., Wang, C., Lu, X., Tong, L., et al. (2016). Identification of functional farnesoid X receptors in brain neurons. FEBS Lett. 18, 3233–3242. doi: 10.1002/1873-3468.12373
Huang, F., Wang, T., Lan, Y., Yang, L., Pan, W., Zhu, Y., et al. (2015). Deletion of mouse FXR gene disturbs multiple neurotransmitter systems and alters neurobehavior. Front. Behav. Neurosci. 9:70. doi: 10.3389/fnbeh.2015.00070
Inagaki, T., Choi, M., Moschetta, A., Peng, L., Cummins, C. L., McDonald, J. G., et al. (2005). Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2, 217–225. doi: 10.1016/j.cmet.2005.09.001
Itoh, N., Yazaki, N., Tagashira, S., Miyake, A., Ozaki, K., Minami, M., et al. (1994). Rat FGF receptor-4 mRNA in the brain is expressed preferentially in the medial habenular nucleus. Mol. Brain Res. 21, 344–348. doi: 10.1016/0169-328X(94)90265-8
Janssen, P., Rotondo, A., Mule, F., and Tack, J. (2013). Review article: a comparison of glucagon-like peptides 1 and 2. Aliment. Pharmacol. Ther. 37, 18–36. doi: 10.1111/apt.12092
Jones, S. (2008). Mini-review: endocrine actions of fibroblast growth factor 19. Mol. Pharm. 5, 42–48. doi: 10.1021/mp700105z
Kamp, F., and Hamilton, J. A. (1993). Movement of fatty acids, fatty acid analogs, and bile acids across phospholipid bilayers. Biochemistry 32, 11074–11085. doi: 10.1021/bi00092a017
Kastin, A. J., Akerstrom, V., and Pan, W. (2002). Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J. Mol. Neurosc. 18, 7–14. doi: 10.1385/JMN:18:1-2:07
Katafuchi, T., Esterházy, D., Lemoff, A., Ding, X., Sondhi, V., Kliewer, S. A., et al. (2015). Detection of FGF15 in plasma by stable isotope standards and capture by anti-peptide antibodies and targeted mass spectrometry. Cell Metab. 21, 898–904. doi: 10.1016/j.cmet.2015.05.004
Katsuma, S., Hirasawa, A., and Tsujimoto, G. (2005). Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 329, 386–390. doi: 10.1016/j.bbrc.2005.01.139
Kawamata, Y., Fujii, R., Hosoya, M., Harada, M., Yoshida, H., Miwa, M., et al. (2003). A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 278, 9435–9440. doi: 10.1074/jbc.M209706200
Keene, C. D., Rodrigues, C. M., Eich, T., Linehan-Stieers, C., Abt, A., Kren, B. T., et al. (2001). A bile acid protects against motor and cognitive deficits and reduces striatal degeneration in the 3-nitropropionic acid model of Huntington's disease. Exp. Neurol. 171, 351–360. doi: 10.1006/exnr.2001.7755
Keene, C. D., Rodrigues, C. M., Eich, T., Chhabra, M. S., Steer, C. J., and Low, W. C. (2002). Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington's disease. Proc. Natl. Acad. Sci. U.S.A. 99, 10671–10676. doi: 10.1073/pnas.162362299
Keitel, V., Görg, B., Bidmon, H. J., Zemtsova, I., Spomer, L., Zilles, K., et al. (2010). The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain. Glia 58, 1794–1805. doi: 10.1002/glia.21049
Kikuchi, R., Kusuhara, H., Sugiyama, D., and Sugiyama, Y. (2003). Contribution of organic anion transporter 3 (Slc22a8) to the elimination of p-aminohippuric acid and benzylpenicillin across the blood-brain barrier. J. Pharmacol. Exp. Ther. 306, 51–58. doi: 10.1124/jpet.103.049197
Kim, I., Ahn, S. H., Inagaki, T., Choi, M., Ito, S., Guo, G. L., et al. (2007). Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 48, 2664–2672. doi: 10.1194/jlr.M700330-JLR200
Kitazawa, T., Terasaki, T., Suzuki, H., Kakee, A., and Sugiyama, Y. (1998). Efflux of taurocholic acid across the blood-brain barrier: interaction with cyclic peptides. J. Pharmacol. Exp. Ther. 286, 890–895.
Klaassen, C. D., and Aleksunes, L. M. (2010). Xenobiotic, bile acid, and cholesterol transporters: function and regulation. Pharmacol. Rev. 62, 1–96. doi: 10.1124/pr.109.002014
Kliewer, S. A., and Mangelsdorf, D. J. (2015). Bile acids as hormones: the FXR-FGF15/19 Pathway. Dig. Dis. 33, 327–331. doi: 10.1159/000371670
Kuipers, F., Bloks, V. W., and Groen, A. K. (2014). Beyond intestinal soap—bile acids in metabolic control. Nat. Rev. Endocrinol. 10, 488–498. doi: 10.1038/nrendo.2014.60
Kurosu, H., Choi, M., Ogawa, Y., Dickson, A. S., Goetz, R., Eliseenkova, A. V., et al. (2007). Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 282, 26687–26695. doi: 10.1074/jbc.M704165200
Larsen, P. J., Tang-Christensen, M., Holst, J. J., and Ørskov, C. (1997). Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in the rat hypothalamus and brainstem. Neuroscience 77, 257–270. doi: 10.1016/S0306-4522(96)00434-4
LaRusso, N. F., Hoffman, N. E., Korman, M. G., Hofmann, A. F., and Cowen, A. E. (1978). Determinants of fasting and postprandial serum bile acid levels in healthy man. Am. J. Dig. Dis. 23, 385–391. doi: 10.1007/BF01072919
Lee, W., Glaeser, H., Smith, L. H., Roberts, R. L., Moeckel, G. W., Gervasini, G., et al. (2005). Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): implications for altered drug disposition and central nervous system drug entry. J. Biol. Chem. 280, 9610–9617. doi: 10.1074/jbc.M411092200
Lefebvre, P., Cariou, B., Lien, F., Kuipers, F., and Staels, B. (2009). Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 89, 147–191. doi: 10.1152/physrev.00010.2008
Liang, Q., Zhong, L., Zhang, J., Wang, Y., Bornstein, S. R., Triggle, C. R., et al. (2014). FGF21 maintains glucose homeostasis by mediating the cross talk between liver and brain during prolonged fasting. Diabetes 63, 4064–4075. doi: 10.2337/db14-0541
Li-Hawkins, J., Lund, E. G., Bronson, A. D., and Russell, D. W. (2000). Expression cloning of an oxysterol 7alpha-hydroxylase selective for 24-hydroxycholesterol. J. Biol. Chem. 275, 16543–16549. doi: 10.1074/jbc.M001810200
Lim, G. E., and Brubaker, P. L. (2006). Glucagon-like peptide 1 secretion by the L-cell the view from within. Diabetes 55(Suppl. 2), S70–S77. doi: 10.2337/db06-S020
Litwa, E., Rzemieniec, J., Wnuk, A., Lason, W., Krzeptowski, W., and Kajta, M. (2016). RXRα, PXR and CAR xenobiotic receptors mediate the apoptotic and neurotoxic actions of nonylphenol in mouse hippocampal cells. J. Steroid Biochem. Mol. Biol. 156, 43–52. doi: 10.1016/j.jsbmb.2015.11.018
Llewellyn-Smith, I. J., Reimann, F., Gribble, F. M., and Trapp, S. (2011). Preproglucagon neurons project widely to autonomic control areas in the mouse brain. Neuroscience 180, 111–121. doi: 10.1016/j.neuroscience.2011.02.023
Lund, E. G., Guileyardo, J. M., and Russell, D. W. (1999). cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc. Natl. Acad. Sci. U.S.A. 96, 7238–7243. doi: 10.1073/pnas.96.13.7238
Lundåsen, T., Gälman, C., Angelin, B., and Rudling, M. (2006). Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J. Intern. Med. 260, 530–536. doi: 10.1111/j.1365-2796.2006.01731.x
Makishima, M., Okamoto, A. Y., Repa, J. J., Tu, H., Learned, R. M., Luk, A., et al. (1999). Identification of a nuclear receptor for bile acids. Science 284, 1362–1365.
Mano, N., Goto, T., Uchida, M., Nishimura, K., Ando, M., Kobayashi, N., et al. (2004a). Presence of protein-bound unconjugated bile acids in the cytoplasmic fraction of rat brain. J. Lipid Res. 45, 295–300. doi: 10.1194/jlr.M300369-JLR200
Mano, N., Sato, Y., Nagata, M., Goto, T., and Goto, J. (2004b). Bioconversion of 3beta-hydroxy-5-cholenoic acid into chenodeoxycholic acid by rat brain enzyme systems. J. Lipid Res. 45, 1741–1748. doi: 10.1194/jlr.M400157-JLR200
Marcelin, G., Jo, Y.-H., Li, X., Schwartz, G. J., Zhang, Y., Dun, N. J., et al. (2014). Central action of FGF19 reduces hypothalamic AGRP/NPY neuron activity and improves glucose metabolism. Mol. Metabol. 3, 19–28. doi: 10.1016/j.molmet.2013.10.002
Maruyama, T., Miyamoto, Y., Nakamura, T., Tamai, Y., Okada, H., Sugiyama, E., et al. (2002). Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 298, 714–719. doi: 10.1016/S0006-291X(02)02550-0
Maruyama, T., Tanaka, K., Suzuki, J., Miyoshi, H., Harada, N., Nakamura, T., et al. (2006). Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J. Endocrinol. 191, 197–205. doi: 10.1677/joe.1.06546
McMillin, M., and DeMorrow, S. (2016). Effects of bile acids on neurological function and disease. FASEB J. 30, 3658–3668. doi: 10.1096/fj.201600275R
McMillin, M., Frampton, G., Grant, S., Khan, S., Diocares, J., Petrescu, A., et al. (2017). Bile acid-mediated Sphingosine-1-Phosphate Receptor 2 signaling promotes neuroinflammation during hepatic encephalopathy in mice. Front. Cell. Neurosci. 11:191. doi: 10.3389/fncel.2017.00191
McMillin, M., Frampton, G., Quinn, M., Ashfaq, S., de los Santos, M. III., Grant, S., et al. (2016). Bile acid signaling is involved in the neurological decline in a murine model of acute liver failure. Am. J. Pathol. 186, 312–323. doi: 10.1016/j.ajpath.2015.10.005
McMillin, M., Frampton, G., Quinn, M., Divan, A., Grant, S., Patel, N., et al. (2015). Suppression of the HPA axis during cholestasis can be attributed to hypothalamic bile acid signaling. Mol. Endocrinol. 29, 1720–1730. doi: 10.1210/me.2015-1087
Merchenthaler, I., Lane, M., and Shughrue, P. (1999). Distribution of pre-pro-glucagon and glucagon-like peptide-1 receptor messenger RNAs in the rat central nervous system. J. Comp. Neurol. 403, 261–280. doi: 10.1002/(SICI)1096-9861(19990111)403:2<261::AID-CNE8>3.0.CO;2-5
Miller, D. S., Nobmann, S. N., Gutmann, H., Toeroek, M., Drewe, J., and Fricker, G. (2000). Xenobiotic transport across isolated brain microvessels studied by confocal microscopy. Mol. Pharmacol. 58, 1357–1367. doi: 10.1124/mol.58.6.1357
Miura, T., Ouchida, R., Yoshikawa, N., Okamoto, K., Makino, Y., Nakamura, T., et al. (2001). Functional modulation of the glucocorticoid receptor and suppression of NF-kappaB-dependent transcription by ursodeoxycholic acid. J. Biol. Chem. 276, 47371–47378. doi: 10.1074/jbc.M107098200
Miyake, A., and Itoh, N. (1996). Rat fibroblast growth factor receptor-4 mRNA in the brain is preferentially expressed in cholinergic neurons in the medial habenular nucleus. Neurosci. Lett. 203, 101–104. doi: 10.1016/0304-3940(95)12272-9
Mok, H. Y., Von Bergmann, K., and Grundy, S. M. (1977). Regulation of pool size of bile acids in man. Gastroenterology 73(4 Pt. 1), 684–690.
Morton, G. J., Kaiyala, K. J., Foster-Schubert, K. E., Cummings, D. E., and Schwartz, M. W. (2013a). Carbohydrate feeding dissociates the postprandial FGF19 response from circulating bile acid levels in humans. J. Clin. Endocrinol. Metab. 99, e241–e245. doi: 10.1210/jc.2013-3129
Morton, G. J., Matsen, M. E., Bracy, D. P., Meek, T. H., Nguyen, H. T., Stefanovski, D., et al. (2013b). FGF19 action in the brain induces insulin-independent glucose lowering. J. Clin. Invest. 123, 4799–4808. doi: 10.1172/JCI70710
Nakagawa, A., Satake, H., Nakabayashi, H., Nishizawa, M., Furuya, K., Nakano, S., et al. (2004). Receptor gene expression of glucagon-like peptide-1, but not glucose-dependent insulinotropic polypeptide, in rat nodose ganglion cells. Autonom. Neurosci. 110, 36–43. doi: 10.1016/j.autneu.2003.11.001
Naqvi, S. H., Herndon, B. L., Del Rosario, L., and Nicholas, H. J. (1970). Intracerebrally injected monohydroxy and other C24 steroid acids as demyelinating agents in the guinea pig. Lipids 5, 964–969. doi: 10.1007/BF02533198
Neale, G., Lewis, B., Weaver, V., and Panveliwalla, D. (1971). Serum bile acids in liver disease. Gut 12, 145–152. doi: 10.1136/gut.12.2.145
Nies, A. T., Jedlitschky, G., Konig, J., Herold-Mende, C., Steiner, H. H., Schmitt, H. P., et al. (2004). Expression and immunolocalization of the multidrug resistance proteins, MRP1-MRP6 (ABCC1-ABCC6), in human brain. Neuroscience 129, 349–360. doi: 10.1016/j.neuroscience.2004.07.051
Nishimura, T., Utsunomiya, Y., Hoshikawa, M., Ohuchi, H., and Itoh, N. (1999). Structure and expression of a novel human FGF, FGF-19, expressed in the fetal brain. Biochim. Biophys. Acta 1444, 148–151. doi: 10.1016/S0167-4781(98)00255-3
Nizamutdinov, D., DeMorrow, S., McMillin, M., Kain, J., Mukherjee, S., Zeitouni, S., et al. (2017). Hepatic alterations are accompanied by changes to bile acid transporter-expressing neurons in the hypothalamus after traumatic brain injury. Sci. Rep. 7:40112. doi: 10.1038/srep40112
Ogawa, Y., Kurosu, H., Yamamoto, M., Nandi, A., Rosenblatt, K. P., Goetz, R., et al. (2007). BetaKlotho is required for metabolic activity of fibroblast growth factor 21. Proc. Natl. Acad. Sci. U.S.A. 104, 7432–7437. doi: 10.1073/pnas.0701600104
Ogundare, M., Theofilopoulos, S., Lockhart, A., Hall, L. J., Arenas, E., Sjovall, J., et al. (2010). Cerebrospinal fluid steroidomics: are bioactive bile acids present in brain? J. Biol. Chem. 285, 4666–4679. doi: 10.1074/jbc.M109.086678
Orskov, C., Poulsen, S. S., Moller, M., and Holst, J. J. (1996). Glucagon-like peptide I receptors in the subfornical organ and the area postrema are accessible to circulating glucagon-like peptide I. Diabetes 45, 832–835. doi: 10.2337/diab.45.6.832
Owen, B. M., Mangelsdorf, D. J., and Kliewer, S. A. (2015). Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol. Metab. 26, 22–29. doi: 10.1016/j.tem.2014.10.002
Palmela, I., Correia, L., Silva, R. F., Sasaki, H., Kim, K. S., Brites, D., et al. (2015). Hydrophilic bile acids protect human blood-brain barrier endothelial cells from disruption by unconjugated bilirubin: an in vitro study. Front. Neurosci. 9:80. doi: 10.3389/fnins.2015.00080
Pan, X., Elliott, C. T., McGuinness, B., Passmore, P., Kehoe, P. G., Holscher, C., et al. (2017). Metabolomic profiling of bile acids in clinical and experimental samples of Alzheimer's disease. Metabolites 17:7. doi: 10.3390/metabo7020028
Parks, D. J., Blanchard, S. G., Bledsoe, R. K., Chandra, G., Consler, T. G., Kliewer, S. A., et al. (1999). Bile acids: natural ligands for an orphan nuclear receptor. Science 284, 1365–1368.
Parry, G. J., Rodrigues, C. M., Aranha, M. M., Hilbert, S. J., Davey, C., Kelkar, P., et al. (2010). Safety, tolerability, and cerebrospinal fluid penetration of ursodeoxycholic acid in patients with amyotrophic lateral sclerosis. Clin. Neuropharmacol. 33, 17–21. doi: 10.1097/WNF.0b013e3181c47569
Perry, R. J., Lee, S., Ma, L., Zhang, D., Schlessinger, J., and Shulman, G. I. (2015). FGF1 and FGF19 reverse diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nat. Commun. 6:7980. doi: 10.1038/ncomms7980
Potthoff, M. J., Kliewer, S. A., and Mangelsdorf, D. J. (2012). Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev. 26, 312–324. doi: 10.1101/gad.184788.111
Punjabi, M., Arnold, M., Rüttimann, E., Graber, M., Geary, N., Pacheco-López, G., et al. (2014). Circulating glucagon-like peptide-1 (GLP-1) inhibits eating in male rats by acting in the hindbrain and without inducing avoidance. Endocrinology 155, 1690–1699. doi: 10.1210/en.2013-1447
Quinn, M., McMillin, M., Galindo, C., Frampton, G., Pae, H. Y., and DeMorrow, S. (2014). Bile acids permeabilize the blood brain barrier after bile duct ligation in rats via Rac1-dependent mechanisms. Dig. Liver Dis. 46, 527–534. doi: 10.1016/j.dld.2014.01.159
Raufman, J. P., Chen, Y., Cheng, K., Compadre, C., Compadre, L., and Zimniak, P. (2002). Selective interaction of bile acids with muscarinic receptors: a case of molecular mimicry. Eur. J. Pharmacol. 457, 77–84. doi: 10.1016/S0014-2999(02)02690-0
Raufman, J.-P., Cheng, K., and Zimniak, P. (2003). Review: activation of muscarinic receptor signaling by bile acids: physiological and medical implications. Dig. Dis. Sci. 48, 1431–1444. doi: 10.1023/A:1024733500950
Reuss, B., and von Bohlen und Halbach, O. (2003). Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 313, 139–157. doi: 10.1007/s00441-003-0756-7
Ricardo, J. A., and Koh, E. T. (1978). Anatomical evidence of direct projections from the nucleus of the solitary tract to the hypothalamus, amygdala, and other forebrain structures in the rat. Brain Res. 153, 1–26. doi: 10.1016/0006-8993(78)91125-3
Richards, P., Parker, H. E., Adriaenssens, A. E., Hodgson, J. M., Cork, S. C., Trapp, S., et al. (2014). Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes 63, 1224–1233. doi: 10.2337/db13-1440
Roberts, L. M., Black, D. S., Raman, C., Woodford, K., Zhou, M., Haggerty, J. E., et al. (2008). Subcellular localization of transporters along the rat blood-brain barrier and blood-cerebral-spinal fluid barrier by in vivo biotinylation. Neuroscience 155, 423–438. doi: 10.1016/j.neuroscience.2008.06.015
Rodrigues, C. M., Spellman, S. R., Solá, S., Grande, A. W., Linehan-Stieers, C., Low, W. C., et al. (2002). Neuroprotection by a bile acid in an acute stroke model in the rat. J. Cereb. Blood Flow Metab. 22, 463–471. doi: 10.1097/00004647-200204000-00010
Rodrigues, C. M., Sola, S., Nan, Z., Castro, R. E., Ribeiro, P. S., Low, W. C., et al. (2003). Tauroursodeoxycholic acid reduces apoptosis and protects against neurological injury after acute hemorrhagic stroke in rats. Proc. Natl. Acad. Sci. U.S.A. 100, 6087–6092. doi: 10.1073/pnas.1031632100
Rogers, G. B., Keating, D. J., Young, R. L., Wong, M. L., Licinio, J., and Wesselingh, S. (2016). From gut dysbiosis to altered brain function and mental illness: mechanisms and pathways. Mol. Psychiatry 21, 738–748. doi: 10.1038/mp.2016.50
Russell, D. W. (2003). The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 72, 137–174. doi: 10.1146/annurev.biochem.72.121801.161712
Rüttimann, E. B., Arnold, M., Hillebrand, J. J., Geary, N., and Langhans, W. (2009). Intrameal hepatic portal and intraperitoneal infusions of glucagon-like peptide-1 reduce spontaneous meal size in the rat via different mechanisms. Endocrinology 150, 1174–1181. doi: 10.1210/en.2008-1221
Ryan, K. K., Kohli, R., Gutierrez-Aguilar, R., Gaitonde, S. G., Woods, S. C., and Seeley, R. J. (2013). Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats. Endocrinology 154, 9–15. doi: 10.1210/en.2012-1891
Sandoval, D. (2008). CNS GLP-1 regulation of peripheral glucose homeostasis. Physiol. Behav. 94, 670–674. doi: 10.1016/j.physbeh.2008.04.018
Sandoval, D. A., Bagnol, D., Woods, S. C., D'Alessio, D. A., and Seeley, R. J. (2008). Arcuate glucagon-like peptide 1 receptors regulate glucose homeostasis but not food intake. Diabetes 57, 2046–2054. doi: 10.2337/db07-1824
Sayin, S. I., Wahlstrom, A., Felin, J., Jantti, S., Marschall, H. U., Bamberg, K., et al. (2013). Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 17, 225–235. doi: 10.1016/j.cmet.2013.01.003
Schalm, S. W., LaRusso, N. F., Hofmann, A. F., Hoffman, N. E., van Berge-Henegouwen, G. P., and Korman, M. G. (1978). Diurnal serum levels of primary conjugated bile acids. Assessment by specific radioimmunoassays for conjugates of cholic and chenodeoxycholic acid. Gut 19, 1006–1014. doi: 10.1136/gut.19.11.1006
Schmidt, P. T., Naslund, E., Gryback, P., Jacobsson, H., Hartmann, B., Holst, J. J., et al. (2003). Peripheral administration of GLP-2 to humans has no effect on gastric emptying or satiety. Regul. Pept. 116, 21–25. doi: 10.1016/S0167-0115(03)00175-7
Schonewille, M., de Boer, J. F., and Groen, A. K. (2016). Bile salts in control of lipid metabolism. Curr. Opin. Lipidol. 27, 295–301. doi: 10.1097/MOL.0000000000000303
Scott, K. A., and Moran, T. H. (2007). The GLP-1 agonist exendin-4 reduces food intake in nonhuman primates through changes in meal size. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, r983–r987. doi: 10.1152/ajpregu.00323.2007
Sola, S., Amaral, J. D., Borralho, P. M., Ramalho, R. M., Castro, R. E., Aranha, M. M., et al. (2006). Functional modulation of nuclear steroid receptors by tauroursodeoxycholic acid reduces amyloid β-peptide-induced apoptosis. Mol. Endocrinol. 20, 2292–2303. doi: 10.1210/me.2006-0063
Song, K. H., Li, T., Owsley, E., Strom, S., and Chiang, J. Y. (2009). Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology 49, 297–305. doi: 10.1002/hep.22627
Sonne, D. P., Rehfeld, J. F., Holst, J. J., Vilsboll, T., and Knop, F. K. (2014). Postprandial gallbladder emptying in patients with type 2 diabetes: potential implications for bile-induced secretion of glucagon-like peptide 1. Eur. J. Endocrinol. 171, 407–419. doi: 10.1530/EJE-14-0309
Sonne, D. P., van Nierop, F. S., Kulik, W., Soeters, M. R., Vilsbøll, T., and Knop, F. K. (2016). Postprandial plasma concentrations of individual bile acids and FGF-19 in patients with Type 2 diabetes. J. Clin. Endocrinol. Metab. 101, 3002–3009. doi: 10.1210/jc.2016-1607
Soontornmalai, A., Vlaming, M. L., and Fritschy, J. M. (2006). Differential, strain-specific cellular and subcellular distribution of multidrug transporters in murine choroid plexus and blood-brain barrier. Neuroscience 138, 159–169. doi: 10.1016/j.neuroscience.2005.11.011
Sørensen, L. B., Flint, A., Raben, A., Hartmann, B., Holst, J. J., and Astrup, A. (2003). No effect of physiological concentrations of glucagon-like peptide-2 on appetite and energy intake in normal weight subjects. Int. J. Obes. Relat. Metab. Disord. 27, 450–456. doi: 10.1038/sj.ijo.0802247
Steiner, C., Othman, A., Saely, C. H., Rein, P., Drexel, H., von Eckardstein, A., et al. (2011). Bile acid metabolites in serum: intraindividual variation and associations with coronary heart disease, metabolic syndrome and diabetes mellitus. PLoS ONE 6:e25006. doi: 10.1371/journal.pone.0025006
St-Pierre, M. V., Kullak-Ublick, G. A., Hagenbuch, B., and Meier, P. J. (2001). Transport of bile acids in hepatic and non-hepatic tissues. J. Exp. Biol. 204(Pt. 10), 1673–1686.
Takahashi, S., Fukami, T., Masuo, Y., Brocker, C. N., Xie, C., Krausz, K. W., et al. (2016). Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J. Lipid Res. 57, 2130–2137. doi: 10.1194/jlr.M071183
Talsania, T., Anini, Y., Siu, S., Drucker, D. J., and Brubaker, P. L. (2005). Peripheral exendin-4 and peptide YY3–36 synergistically reduce food intake through different mechanisms in mice. Endocrinology 146, 3748–3756. doi: 10.1210/en.2005-0473
Tanaka, N., Matsubara, T., Krausz, K. W., Patterson, A. D., and Gonzalez, F. J. (2012). Disruption of phospholipid and bile acid homeostasis in mice with nonalcoholic steatohepatitis. Hepatology 56, 118–129. doi: 10.1002/hep.25630
Tang-Christensen, M., Larsen, P. J., Goke, R., Fink-Jensen, A., Jessop, D. S., Moller, M., et al. (1996). Central administration of GLP-1-(7-36) amide inhibits food and water intake in rats. Am. J. Physiol. 271(4 Pt. 2), r848–r856.
Thomas, C., Auwerx, J., and Schoonjans, K. (2008a). Bile acids and the membrane bile acid receptor TGR5–connecting nutrition and metabolism. Thyroid 18, 167–174. doi: 10.1089/thy.2007.0255
Thomas, C., Gioiello, A., Noriega, L., Strehle, A., Oury, J., Rizzo, G., et al. (2009). TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 10, 167–177. doi: 10.1016/j.cmet.2009.08.001
Thomas, C., Pellicciari, R., Pruzanski, M., Auwerx, J., and Schoonjans, K. (2008b). Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Disc. 7, 678–693. doi: 10.1038/nrd2619.
Tomlinson, E., Fu, L., John, L., Hultgren, B., Huang, X., Renz, M., et al. (2002). Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 143, 1741–1747. doi: 10.1210/endo.143.5.8850
Trapp, S., and Cork, S. C. (2015). PPG neurons of the lower brain stem and their role in brain GLP-1 receptor activation. Am. J. Physiol. Regul. 309, R795–804. doi: 10.1152/ajpregu.00333.2015
Tripodi, V., Contin, M., Fernández, M. A., and Lemberg, A. (2012). Bile acids content in brain of common duct ligated rats. Ann. Hepatol. 11, 930–934.
Turton, M. D., O'Shea, D., Gunn, I., Beak, S. A., Edwards, C. M. B., Meeran, K., et al. (1996). A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 379, 69–72.
Ullmer, C., Alvarez Sanchez, R., Sprecher, U., Raab, S., Mattei, P., Dehmlow, H., et al. (2013). Systemic bile acid sensing by G protein-coupled bile acid receptor 1 (GPBAR1) promotes PYY and GLP-1 release. Br. J. Pharmacol. 169, 671–684. doi: 10.1111/bph.12158
Vahl, T. P., Tauchi, M., Durler, T. S., Elfers, E. E., Fernandes, T. M., Bitner, R. D., et al. (2007). Glucagon-like peptide-1 (GLP-1) receptors expressed on nerve terminals in the portal vein mediate the effects of endogenous GLP-1 on glucose tolerance in rats. Endocrinology 148, 4965–4973. doi: 10.1210/en.2006-0153
van den Heuvel, J. K., van Rozen, A. J., Adan, R. A., and la Fleur, S. E. (2011). An overview on how components of the melanocortin system respond to different high energy diets. Eur. J. Pharmacol. 660, 207–212. doi: 10.1016/j.ejphar.2010.12.019
van der Kooy, D., Koda, L. Y., McGinty, J. F., Gerfen, C. R., and Bloom, F. E. (1984). The organization of projections from the cortes, amygdala, and hypothalamus to the nucleus of the solitary tract in rat. J. Comp. Neurol. 224, 1–24. doi: 10.1002/cne.902240102
Viana, R. J., Nunes, A. F., Castro, R. E., Ramalho, R. M., Meyerson, J., Fossati, S., et al. (2009). Tauroursodeoxycholic acid prevents E22Q Alzheimer's Abeta toxicity in human cerebral endothelial cells. Cell. Mol. Life Sci. 66, 1094–1104. doi: 10.1007/s00018-009-8746-x
Vilsbøll, T., Krarup, T., Deacon, C. F., Madsbad, S., and Holst, J. J. (2001). Reduced postprandial concentrations of intact biologically active gluccagon-like peptide 1 in type 2 diabetic patients. Diabetes 50, 609–613. doi: 10.2337/diabetes.50.3.609
Vilsboll, T., Krarup, T., Sonne, J., Madsbad, S., Volund, A., Juul, A. G., et al. (2003). Incretin secretion in relation to meal size and body weight in healthy subjects and people with type 1 and type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 88, 2706–2713. doi: 10.1210/jc.2002-021873
Wanaka, A., Johnson, E. M., and Milbrand, J. (1990). Localization of FGF receptor mRNA in the adult rat central nervous system by in situ hybridization. Neuron 5, 267–281. doi: 10.1016/0896-6273(90)90164-B
Wang, H., Chen, J., Hollister, K., Sowers, L. C., and Forman, B. M. (1999). Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 3, 543–553. doi: 10.1016/S1097-2765(00)80348-2
Williams, D. L., Baskin, D. G., and Schwartz, M. W. (2006). Leptin regulation of the anorexic response to glucagon-like peptide-1 receptor stimulation. Diabetes 55, 3387–3393. doi: 10.2337/db06-0558
Williams, D. L., Baskin, D. G., and Schwartz, M. W. (2009). Evidence that intestinal glucagon-like peptide-1 plays a physiological role in satiety. Endocrinology 150, 1680–1687. doi: 10.1210/en.2008-1045
Wu, X., Ge, H., Gupte, J., Weiszmann, J., Shimamoto, G., Stevens, J., et al. (2007). Co-receptor requirements for fibroblast growth factor-19 signaling. J. Biol. Chem. 282, 29069–29072. doi: 10.1074/jbc.C700130200
Wu, X., Ge, H., Lemon, B., Weiszmann, J., Gupte, J., Hawkins, N., et al. (2009). Selective activation of FGFR4 by an FGF19 variant does not improve glucose metabolism in ob/ob mice. Proc. Natl. Acad. Sci. U.S.A. 106, 14379–14384. doi: 10.1073/pnas.0907812106
Xie, M.-H., Holcomb, I., Deuel, B., Dowd, P., Huang, A., Vagts, A., et al. (1999). FGF-19, a novel fibroblast growth factor with unique specificity for FGFR4. Cytokine 11, 729–735. doi: 10.1006/cyto.1999.0485
Yamamoto, H., Kishi, T., Lee, C. E., Choi, B. J., Fang, H., Hollenberg, A. N., et al. (2003). Glucagon-like peptide-1-responsive catecholamine neurons in the area postrema link peripheral glucagon-like peptide-1 with central autonomic control sites. J. Neurosci. 23, 2939–2946.
Yang, C., Jin, C., Li, X., Wang, F., McKeehan, W. L., and Luo, Y. (2012). Differential specificity of endocrine FGF19 and FGF21 to FGFR1 and FGFR4 in complex with KLB. PLoS ONE 7:e33870. doi: 10.1371/journal.pone.0033870
Yanguas-Casás, N., Barreda-Manso, M. A., Nieto-Sampedro, M., and Romero-Ramirez, L. (2017). TUDCA: an agonist of the bile acid receptor GPBAR1/TGR5 with anti-inflammatory effects in microglial cells. J. Cell. Physiol. 232, 2231–2245. doi: 10.1002/jcp.25742
Yazaki, N., Hosoi, Y., Kawabata, K., Miyake, A., Minami, M., Satoh, M., et al. (1994). Differential expression patterns of mRNAs for members of the fibroblast growth factor receptor family, FGFR-1–FGFR-4, in rat brain. J. Neurosci. Res. 37, 445–452. doi: 10.1002/jnr.490370403
Yusta, B., Matthews, D., Flock, G. B., Ussher, J. R., Lavoie, B., Mawe, G. M., et al. (2017). Glucagon-like peptide-2 promotes gallbladder refilling via a TGR5-independent, GLP-2R-dependent pathway. Mol. Metab. 6, 503–511. doi: 10.1016/j.molmet.2017.03.006
Zhang, Y. K., Guo, G. L., and Klaassen, C. D. (2011). Diurnal variations of mouse plasma and hepatic bile acid concentrations as well as expression of biosynthetic enzymes and transporters. PLoS ONE 6:e16683. doi: 10.1371/journal.pone.0016683
Zheng, X., Chen, T., Zhao, A., Wang, X., Xie, G., Huang, F., et al. (2016). The brain metabolome of male rats across the lifespan. Sci. Rep. 6:24125. doi: 10.1038/srep24125
Keywords: bile acids, CNS, brain, FXR, FGF19, TGR5, GLP-1
Citation: Mertens KL, Kalsbeek A, Soeters MR and Eggink HM (2017) Bile Acid Signaling Pathways from the Enterohepatic Circulation to the Central Nervous System. Front. Neurosci. 11:617. doi: 10.3389/fnins.2017.00617
Received: 07 July 2017; Accepted: 23 October 2017;
Published: 07 November 2017.
Edited by:
Miguel López, Universidade de Santiago de Compostela, SpainReviewed by:
Angel Nadal, Universidad Miguel Hernández de Elche, SpainJoseph Pisegna, University of California, Los Angeles, United States
Copyright © 2017 Mertens, Kalsbeek, Soeters and Eggink. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hannah M. Eggink, aC5tLmVnZ2lua0BhbWMudXZhLm5s