 E. F. E. Kuiper
E. F. E. Kuiper Eduardo P. de Mattos
Eduardo P. de Mattos Laura B. Jardim2,3,4
Laura B. Jardim2,3,4 Harm H. Kampinga
Harm H. Kampinga Steven Bergink
Steven Bergink- 1Department of Cell Biology, University Medical Center Groningen, University of Groningen, Groningen, Netherlands
- 2Programa de Pós-Graduação em Genética e Biologia Molecular, Department of Genetics, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
- 3Medical Genetics Service, Hospital de Clínicas de Porto Alegre, Porto Alegre, Brazil
- 4Departamento de Medicina Interna, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
Expanded polyglutamine (polyQ) stretches in at least nine unrelated proteins lead to inherited neuronal dysfunction and degeneration. The expansion size in all diseases correlates with age at onset (AO) of disease and with polyQ protein aggregation, indicating that the expanded polyQ stretch is the main driving force for the disease onset. Interestingly, there is marked interpatient variability in expansion thresholds for a given disease. Between different polyQ diseases the repeat length vs. AO also indicates the existence of modulatory effects on aggregation of the upstream and downstream amino acid sequences flanking the Q expansion. This can be either due to intrinsic modulation of aggregation by the flanking regions, or due to differential interaction with other proteins, such as the components of the cellular protein quality control network. Indeed, several lines of evidence suggest that molecular chaperones have impact on the handling of different polyQ proteins. Here, we review factors differentially influencing polyQ aggregation: the Q-stretch itself, modulatory flanking sequences, interaction partners, cleavage of polyQ-containing proteins, and post-translational modifications, with a special focus on the role of molecular chaperones. By discussing typical examples of how these factors influence aggregation, we provide more insight on the variability of AO between different diseases as well as within the same polyQ disorder, on the molecular level.
Introduction
Polyglutaminopathies are a family of diseases characterized by CAG trinucleotide expansions in the coding regions of at least nine unrelated genes, resulting in proteins with an abnormally long polyglutamine (polyQ) stretch, which have a high aggregation propensity. PolyQ aggregates can impede cellular protein homeostasis, loss of which is also observed in many other neurodegenerative diseases (Soto, 2003). These mutant proteins lead to one recessive inherited, X-linked spinal and bulbar muscular atrophy (SBMA), and eight dominantly inherited neuronal dysfunctions, Huntington's disease (HD), dentatorubral pallidoluysian atrophy (DRPLA), and the spinocerebellar ataxias (SCAs) type 1, 2, 3, 6, 7, and 17 (Margolis and Ross, 2001). All known polyglutaminopathies show a strong inverse correlation between expansion size and age at onset (AO) of the disease, with longer repeats significantly correlating with earlier onset of symptoms and higher aggregation proneness of the affected protein, indicating that an expanded polyQ is tightly related to the diseases. There are two main features that are striking in the association between polyQ length and AO. First, there is marked variability between polyQ diseases in expansion thresholds that determines the pathogenicity, indicating that AO has only a partial dependence on the polyQ stretches and their absolute lengths (Figure 1A). Second, there is also CAG-length independent phenotypic variation within a given polyQ disease (Figure 1B). Both these findings imply that factors beyond the polyQ stretch are co-determining disease onset (Ranum et al., 1994; DeStefano et al., 1996; Hayes et al., 2000; Wexler et al., 2004; van de Warrenburg et al., 2005; Kaltenbach et al., 2007; Branco et al., 2008; Lessing and Bonini, 2008; Bettencourt et al., 2011; Tezenas du Montcel et al., 2014; Bečanović et al., 2015). It was hypothesized that the differential effects of distinct polyQ proteins with polyQ tracts of similar lengths could be, at least in part, due to the sequences flanking the polyQ expansion (Nozaki et al., 2001).
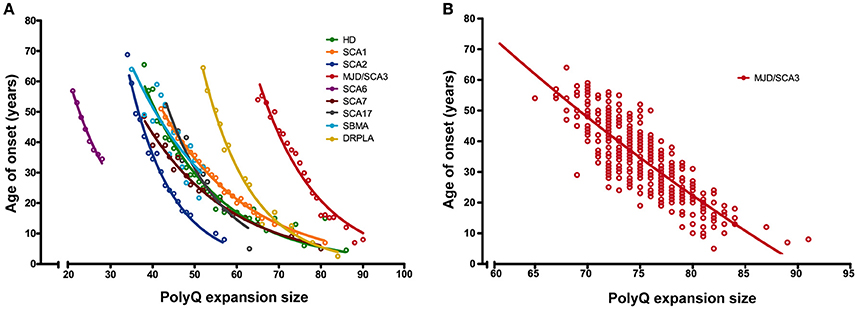
Figure 1. Age of onset of disease inversely correlates with the size of the expanded polyQ tract in all known polyQ diseases. (A) Correlation between age of onset (AO) and CAG expansion size for all nine polyQ diseases identified so far. Circles depict mean AOs for a given expansion size based on multiple reported cohorts of patients. Lines represent the fitted data according to an exponential decay model. (B) Age of onset of disease is not completely determined by the expanded polyQ tract alone. Data on the variability of AO for a particular polyQ expansion size is shown as in (A) and was based on the large cohort of MJD/SCA3 patients reported by Saute and Jardim (2015). Circles represent single patients. Please refer to Supplementary File 1 for a complete list of references of the original cohort descriptions. Note that graph (A,B) are not drawn to the same scale.
Here we discuss that, next to aggregation of the core polyQ stretch, which is common to all polyglutaminopathies (Figure 2A), the context around the cores can modulate aggregation in several ways and may be linked to differential handling of the protein quality control systems, including molecular chaperones, the ubiquitin proteasome system, and autophagy. These degradation processes, and their relationship with the chaperone system, are of importance and greatly influence the aggregation process (Rubinsztein, 2006). Certain chaperones act together with the protein degradation machineries to effectively clear aggregation-prone polypeptides, such as polyQ-containing proteins (Dekker et al., 2015). The molecular details of these downstream events are still unclear and will not be discussed here; instead we will focus on the impact of molecular chaperones on the aggregation process itself. Molecular chaperones are known to influence aggregation of polyQ proteins. This could either be directly by preventing the polyQ stretch from aggregating or via the flanking sequences. For only a few of the molecular chaperones the direct interaction with the polyQ proteins has been shown, although many chaperones are found to co-localize with polyQ inclusions (Cummings et al., 1998; Kazemi-Esfarjani and Benzer, 2000; Schmidt et al., 2002; Helmlinger et al., 2004; Bilen and Bonini, 2007; Hageman et al., 2010; Gao et al., 2011; Kakkar et al., 2014; Matilla-Dueñas et al., 2014; Reis et al., 2016; Zhao et al., 2016). However, co-localization of chaperones does not provide information on their mode of interaction and does not distinguish whether chaperones are truly interacting with the polyQ protein, or whether the presence of chaperones in the aggregates is a mere secondary effect due to a collapse of other cellular components with the inclusions. In this review, we will discuss: first, how polyQ tracts drive aggregation; second, how their flanking sequences could directly affect the aggregation proneness of the polyQ protein; and third, how polyQ proteins can be modified, changed in conformation, or fragmented, inducing aggregation (Figure 2B). We will not focus on the function, or loss of function, of the affected polyQ proteins, since this was so far not shown to be causative for disease, even though the native function of the protein might be important for normal cellular function. Furthermore, we will not go into the discussion on the toxicity of aggregation. For instance, it is still unclear whether the presence of aggregates contributes to SCA2 pathology (Huynh et al., 2000), even though aggregates are found in affected brain areas (Pang et al., 2002; Seidel et al., 2016). Finally, we will highlight the role of chaperones in the aggregation process and include only studies that provide insight in direct interaction of chaperones with the polyQ proteins. Rather than providing a complete overview, molecular mechanisms of typical examples will be discussed, aiming at providing general principles affecting polyQ aggregation on the molecular level that may partially explain the individual differences between patients and steer future studies.
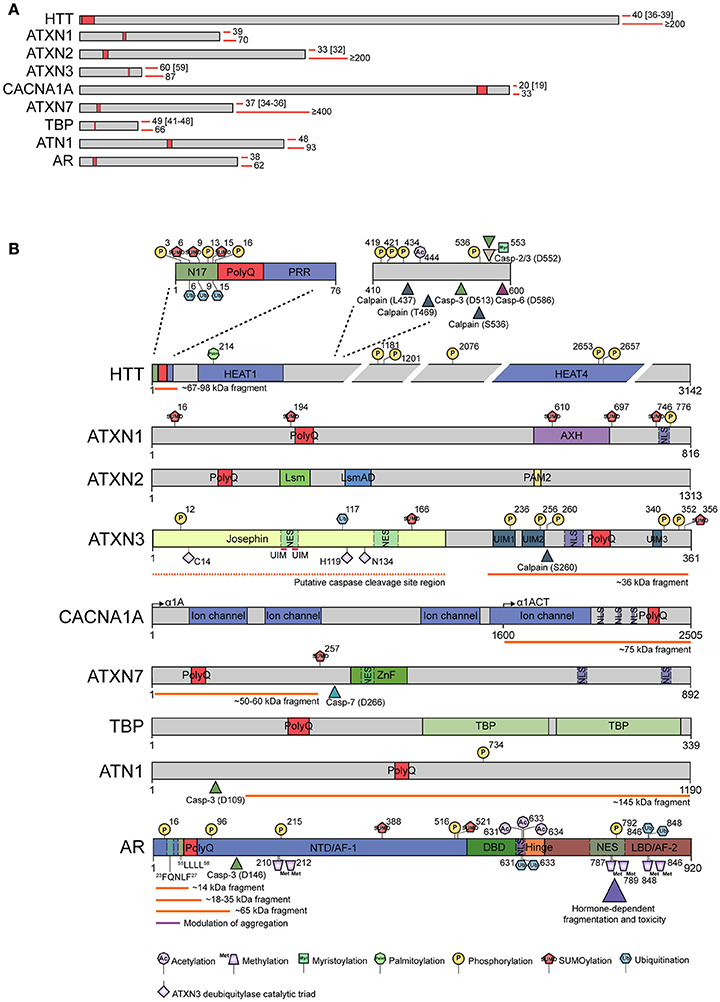
Figure 2. Representation of pathogenic polyQ proteins and known modulating events associated with aggregation. (A) Schematic representation of the nine disease-related polyglutamine proteins drawn to scale. In each case, a polyQ stretch of fixed length is depicted at the approximate position (red boxes). Red bars on the right side of each protein show the smallest and largest number of glutamine repeats identified in patients of each polyQ disease to date. Numbers between brackets represent polyQ expansion sizes that have been reported to behave as incomplete penetrance alleles. (B) Detailed representation of all nine polyQ proteins. Domain organization is indicated. Known post-translational modifications associated with disease, caspase/calpain cleavage sites, and fragments identified are indicated. For ataxin-3, the long isoform with 3 ubiquitin-interacting motifs is shown. Residues C14, H119, and N134 depict the catalytic triad of the deubiquitylase activity of the Josephin domain. The CACNA1A locus encodes two proteins: α1A (full-length α1A) and α1ACT (C-terminal fragment of α1A) using a bicistronic mRNA with a cryptic internal ribosomal entry site. The polyQ is found in both. Many studies report a C-terminal fragment which probably represents α1ACT. For the androgen receptor, the only phosphorylation sites depicted are those with biochemical evidence of modulation of polyQ aggregation, cleavage and/or toxicity. Similarly, amino acid sequences 23FQNLF27 and 55LLLL58 highlight motifs shown to influence polyQ behavior. For simplicity, most huntingtin cleavage products are omitted and only the major N-terminal polyQ containing fragment is indicated. Amino acid numbering is based on Uniprot accession numbers P42858 (HTT), P54253 (ATXN1), Q99700 (ATXN2), P54252 (ATXN3), O00555 (CACNA1A), O15265 (ATXN7), P20226 (TBP), P54259 (ATN1), and P10275 (AR). However, for clarity, some residues are numbered according to their original publication, which might differ from the numbering according to the reference protein sequence (due to the expanding nature of polyQ proteins). AR, androgen receptor; ATN1, atrophin-1; ATXN1, ataxin-1; ATXN2, ataxin-2; ATXN3, ataxin-3; ATXN7, ataxin-7; AXH, ataxin-1/high-mobility group box containing protein-1; CACNA1, α1A subunit of the P/Q-type or CaV2.1 voltage-gated calcium channel; Casp, caspase; DBD, DNA binding domain; HTT, huntingtin; PolyQ, polyglutamine stretch; NTD/AF-1, amino-terminal domain/ activation function-1; LBD/AF-2, ligand-binding domain/ activation function-2; NES, nuclear export signal; NLS, nuclear localization signal; HEAT, huntingtin/elongation factor 3/PR65/A subunit of protein phosphatase 2A/ lipid kinase TOR domain; PRR, proline-rich region; N17, first 17 amino acids of huntingtin; TBP, TATA-binding protein (domain); UIM, ubiquitin-interacting motif; Ub-1/Ub2, ubiquitin-binding sites; Lsm, Like RNA splicing domain Sm and Sm2; LsmAD, Like-Sm-associated domain; PAM2, poly (A)-binding protein interacting motif 2; ZnF, SCA7-like zinc finger domain. For references to specific domains or post-translational modifications, please refer to Supplementary File 1.
Aggregation Properties of the polyQ Stretch
Aggregates formed by polyQ stretches contain identical β-strand-based cores. Already in 1994, Perutz et al. described the ability of elongated polyQ stretches to form β-sheets (Perutz et al., 1994). Like many other amyloidogenic proteins (Sawaya et al., 2007), the polyQ chains can form β-sheets that are connected through interdigitating extended side chains and contain intramolecular β-hairpins (Hoop et al., 2016). Formation of β-hairpins allows for hydrogen bonding between the stacked side chains, providing a strong interaction (Hoop et al., 2016). The β-hairpins play an important role in the aggregation process. Q-stretches with a range up to 25Q are not able to form stable β-hairpins and therefore are not able to induce aggregation, except when mutations known to enhance β-hairpin formation are introduced (Kar et al., 2011, 2013). It is hypothesized that longer polyQ stretches can form more stable intramolecular β-hairpins, providing a critical monomeric nucleus necessary for inducing aggregation (Kar et al., 2011). The high affinity of the β-sheets affects interactions between molecules and might not only do so for the same pathogenic polyQ protein, but also as a secondary effect for other endogenous polyQ containing proteins (Nóbrega et al., 2015). For example, the endogenous, non-expanded TATA-box binding protein (TBP) was found to sequester into aggregates formed by other pathogenic polyQ proteins, such as huntingtin (HTT; Perez et al., 1998; Kim et al., 2002; Matsumoto et al., 2006). Similarly, inclusions containing ataxin-2 (ATXN2), ataxin-3 (ATXN3) and TBP are observed in SCA1, SCA2, SCA3, and DRPLA (Uchihara et al., 2001). Whether these secondary co-aggregating events contribute to disease is currently not clear (Kampinga and Bergink, 2016).
The crucial role for the formation of β-hairpins in the aggregation process is nicely illustrated by findings on missense CAG to CAT mutations. These mutations, coding for histidine, were found in the CAG-repeat in ATXN1, leading to insertion of one or more other amino acids and interrupting the Q-stretch (Sobczak and Krzyzosiak, 2004; Jayaraman et al., 2009; Menon et al., 2013). The AO is in these cases inversely correlated to the longer uninterrupted CAG stretch which, rather than a specific interruption pattern, dictates also the aggregation propensity in vitro (Menon et al., 2013). The structure of the polyQ-stretches is not changed because of the histidine-interruptions but the polyQ aggregation rates are decreased due to the Q-length dependent ability of the protein to form a critical nucleus to initiate aggregation (Jayaraman et al., 2009; Menon et al., 2013).
From all the different intracellular chaperones, so far the only ones described that could act on the β-sheets or β-hairpins formed by the Q-stretch are DNAJB6 and its closest homolog DNAJB8, two members of the DNAJ family of Hsp70 co-chaperones. In a screen for suppressors of aggregation of huntingtin (HTT-119Q) both DNAJB6 and DNAJB8 were superior suppressors of aggregation with a specificity for the polyQ tract, since they were similarly effective in the suppression of aggregation of HTT, ATXN3, the androgen receptor (AR), and polyQ alone (Hageman et al., 2010; Månsson et al., 2013). These DNAJ chaperones have a unique region containing 18 residues of the polar hydroxyl group amino acids serine and threonine, that is exposed on one face of the DNAJB6 monomer where it is predicted to interact with the hydrogen bonds in the polyQ β-hairpins (Månsson et al., 2013; Kakkar et al., 2016).
Aggregation Initiation by Flanking Domains in polyQ-Containing Proteins
A longer Q-stretch not only has a higher aggregation propensity, but also affects the conformation of other parts of the protein. This can cause exposure of other regions in the proteins that have aggregation-prone properties by themselves (Ellisdon et al., 2006; Kelley et al., 2009; Tam et al., 2009). The intrinsic aggregation propensity leads to a two-stage aggregation mechanism (Ellisdon et al., 2006) in which the first aggregation step is actually thought to be a nucleation step of the non-polyQ-containing flanking domains. The formed nucleus can speed up the aggregation of the polyQ-stretch, which is then the second aggregation step. Aggregation of the flanking region and the polyQ stretch may enhance each other in a positive feedback loop accelerating aggregation and AO (Ellisdon et al., 2007; Saunders et al., 2011). The most striking examples of this process are known for HTT and ATXN3.
HTT is a relatively large protein with the polyQ stretch located in the first exon of the protein. The polyQ tract in HTT is flanked by a 17 amino acid long N-terminal (N17) domain and a polyproline domain on its C-terminus (Dehay and Bertolotti, 2006; Rockabrand et al., 2007; Figure 2). The N17 domain is highly soluble by itself and has an intrinsic tendency to collapse into an aggregation-resistant compact coil state (Thakur et al., 2009; Crick et al., 2013). When the Q-stretch is expanded, the N17 domain undergoes a conformational change going into a more α-helical extended state (Tam et al., 2009; Thakur et al., 2009; Sivanandam et al., 2011), exposing a hydrophobic face through which self-association is induced (Kelley et al., 2009; Liebman and Meredith, 2010). Self-association provides an initial nucleus that increases the local concentration of the adjacent polyQ, promoting polyQ aggregation (Kelley et al., 2009; Liebman and Meredith, 2010; Sahoo et al., 2016). Aggregation of HTT can be prevented by modifying the hydrophobic face of the α-helix (Tam et al., 2009), confirming the important role of the N17 domain in initial aggregation. Moreover, synthetic polyQ peptides lacking the N17 domain show much slower aggregation kinetics (Månsson et al., 2013; Monsellier et al., 2015; Sahoo et al., 2016).
The exposed hydrophobic face on the N17 domain was identified as an interaction site for several chaperones amongst which the chaperonin TRiC, specifically the subunit CCT1 (Tam et al., 2006). CCT1 can suppress HTT aggregation by binding via its apical substrate-binding domains to the hydrophobic motifs in the N17, preventing the initial step of aggregation (Spiess et al., 2006; Tam et al., 2009; Shahmoradian et al., 2013; Sahl et al., 2015). The constitutively expressed Hsp70 (Hsc70/HSPA8) was found to co-localize, like many other Hsp70s including the prokaryotic DnaK and yeast Ssa1 (Jana et al., 2000; Muchowski et al., 2000; Novoselova et al., 2005; Tam et al., 2006), and interact with the N17 domain of HTT via its client protein binding domain (Monsellier et al., 2015). HSPA8 is not able to delay aggregation of a Q-stretch lacking flanking sequences (Månsson et al., 2013) and acts, similar to CCT1, by disrupting the interaction between N17 domains of HTT, slowing down aggregate formation (Monsellier et al., 2015).
Another example of a polyQ protein that undergoes a similar two-stage aggregation mechanism is ATXN3, causative for SCA3. ATXN3 is involved in proteostasis by editing specific ubiquitin sidechains that are targeting proteins to the proteasome (Kuhlbrodt et al., 2011). ATXN3 has an unstructured C-terminus containing the polyQ expansion and multiple ubiquitin interacting motifs (UIMs), and an N-terminus containing the Josephin domain (JD), which is a structured monomeric domain that folds into a globular conformation (Chow et al., 2004; Masino et al., 2004; Figure 2). The JD is the catalytic domain responsible for the deubiquitinating (DUB) properties of ATXN3 and has a high α-helical content forming a groove with two additional UIMs for recognition of the polyubiquitin chains of different linkages, and positioning them for cleavage (Masino et al., 2004; Nicastro et al., 2009, 2010). Sequence motifs on the helices in the groove are functionally important for binding conjugated ubiquitin but are predicted to be highly amyloidogenic and therefore responsible for the aggregation propensity of the JD itself (Masino et al., 2011; Lupton et al., 2015). Indeed, in vitro the isolated JD shows fibrillogenic behavior even under physiological conditions (Masino et al., 2004, 2011; Ellisdon et al., 2006), but when ubiquitin is added, the aggregation propensity of ATXN3 is lowered (Masino et al., 2011). Expansion of the polyQ stretch influences the conformation of the JD in such a way that the molecular mobility of two α-helices is increased and the amyloidogenic motif gets more exposed (Lupton et al., 2015; Scarff et al., 2015), providing a nucleus through which the first aggregation step of ATXN3 is initiated. This can in turn accelerate aggregation of the polyQ stretch (Gales et al., 2005; Ellisdon et al., 2007). In a dedicated screen, several modifiers of ATXN3 were identified that all fell into the canonical chaperone and ubiquitin pathways (Bilen and Bonini, 2007). Amongst the chaperones was alphaB-crystallin (HSPB5), which was found to interact with the JD in the distorted ubiquitin interacting groove, possibly masking the amyloidogenic motives, and having an effect on the initial nucleation step of ATXN3 (Robertson et al., 2010).
Flanking regions can also suppress aggregation of the polyQ stretch. For example, the proline-rich flanking domain (C38) in HTT has an opposite effect compared to the N17 domain. The C38 is also highly soluble, but actually lowers the rate of aggregation (Bhattacharyya et al., 2006; Dehay and Bertolotti, 2006; Duennwald et al., 2006; Crick et al., 2013). Other polyQ-containing proteins apart from HTT, also have a proline-rich region adjacent to the Q-stretch, like TBP, AR, and ATXN2 (Kim, 2014). It is tempting to speculate that these regions confer an evolutionary benefit and co-evolved with Q stretches to modulate their aggregation.
Binding Partners that Can Influence Aggregation
As we have now seen, the opening up of physiologically needed hydrophobic, aggregation-prone, motifs in non-polyQ-containing parts of the protein, can lead to the unwanted formation of an initial nucleus for aggregation. These motifs are normally buried or in interaction with binding partners (or substrates), like ubiquitin in the case of ATXN3, which prevents exposure of the hydrophobic regions (Masino et al., 2011). Binding partners of polyQ-containing proteins can influence the aggregation to a great extent, also for ataxin-1 (ATXN1). ATXN1 is the protein that underlies SCA1, and has a Q-stretch in the N-terminal part of the protein and an AXH domain in the C-terminus (Figure 2). Just like the JD in ATXN3, the AXH domain in ATXN1 has aggregation-prone properties that are needed for its normal functioning, but therefore can be detrimental in the presence of an expanded polyQ stretch (De Chiara et al., 2013a). The AXH domain is responsible for transcriptional repression, RNA-binding activity, and is necessary for interacting with other proteins, mostly transcriptional regulators. For the domain to be able to bind all its different substrates, it has a remarkable conformational plasticity (Chen et al., 2004; De Chiara et al., 2013b; Deriu et al., 2016). Moreover, the AXH domain is responsible for ATXN1 self-association. Multimerization can bring polyQ stretches together, associated with aggregation and amyloid formation (De Chiara et al., 2005b, 2013a; Lasagna-Reeves et al., 2015). In vivo ATXN1 forms oligomers and interestingly the interaction partner transcriptional repressor Capicua (CIC) is found in these complexes (Lam et al., 2006; Lasagna-Reeves et al., 2015). The interaction of CIC with the AXH domain of ATXN1 stabilizes toxic soluble prefibrillar oligomers of ATXN1. When CIC levels are reduced, ATXN1 forms more fibrillar oligomers that are less toxic (Lasagna-Reeves et al., 2015). Also when the AXH domain is deleted, aggregate formation is reduced (De Chiara et al., 2005a,b). There are chaperones known to prevent ATXN1 aggregation and reduce toxicity, but the exact mechanism of action of the chaperones on ATXN1 is not known (Cummings et al., 1998; Zhai et al., 2008). A possible mechanism of action could be that chaperones bind to the AXH domain of ATXN1 to prevent complex formation or to prevent CIC from binding.
Cleavage/Fragmentation
Fragmented polyQ proteins have been found in patients and proteolytic processing of polyQ proteins into smaller, highly aggregation-prone fragments that are more toxic than the full-length protein has been described for most polyQ diseases, HD (Mangiarini et al., 1996; Martindale et al., 1998), DRPLA (Igarashi et al., 1998; Wellington et al., 1998), SBMA (Butler et al., 1998; Kobayashi et al., 1998; Wellington et al., 1998), and SCAs (Ikeda et al., 1996; Paulson et al., 1997; Zander et al., 2001; Goti et al., 2004; Helmlinger et al., 2004; Kordasiewicz et al., 2006; Matos et al., 2016a; Figure 2B). However, for SCA1, SCA2, and SCA17 the evidence for the presence of fragments is limited (Matos et al., 2016a). Proteases play a key role in the generation of these polyQ fragments, and inhibition of proteases or mutation of their cleavage sites can modulate the disease AO (Ona et al., 1999; Chen et al., 2000; Graham et al., 2006; Aharony et al., 2015). Importantly, expression of these fragments containing the polyQ stretch can already give rise to aggregation and the disease phenotype (Ikeda et al., 1996), although it is still not entirely clear why the polyQ fragments display enhanced toxicity when compared to their respective full-length proteins. Cleavage may lead to changes in aggregation propensity, conformation of the protein, localization, and molecular interactions (Matos et al., 2016a). For SBMA, it has been reported that a conformational change exposing the polyQ tract is already sufficient to drive aggregation (Heine et al., 2015) and cleavage might expose the polyQ stretch in a similar way as such a conformational change does. Protein domains that would otherwise prevent, or enhance, the aggregation may be removed, exposing the Q-stretch itself for aggregation. Finally, recognition sites and binding of molecular chaperones could be changed, exemplifying once more the importance of regions outside the polyQ tract in the modulation of aggregation.
For ATXN3, a cleavage product containing the C-terminal fragment from amino acid 221 with the 71Q expansion was found in mice showing the disease phenotype, but rarely in mice not showing the phenotype (Goti et al., 2004). This polypeptide was also found in SCA3 patients (Goti et al., 2004) indicating that fragmentation of the polyQ protein ATXN3 has a strong correlation with disease. Interestingly, while full-length ATXN3 with an expanded polyQ was mostly non-aggregating, co-expression with truncated ATXN3 makes the full-length protein co-localize with the truncated version in perinuclear aggregates (Paulson et al., 1997). More putative cleavage sites in ATXN3 were identified (Haacke et al., 2006; Colomer Gould et al., 2007) and it was shown that caspases are not the sole contributors to the fragmentation of ATXN3, but also the activity of calpains, such as calpain-2, is involved (Simões et al., 2012; Hübener et al., 2013). ATXN3 cleavage and translocation to the nucleus, and thus also aggregation, can be prevented by inhibiting calpains through overexpression of calpastatin in mice (Simões et al., 2012). Conversely, knocking down calpastatin worsened aggregation (Hübener et al., 2013). These data clearly show that under non-stressed conditions in vivo, fragmentation is both required and sufficient for aggregation of polyQ containing ATXN3. Similar data has been found for HTT. In almost all studies on HD, a fragment containing the first exon of HTT with the polyQ stretch is being used, since this fragment already gives rise to the HD phenotype. Toxic N-terminal fragments are found to be generated through cleavage by caspases, both in animal models and in patients (Wellington et al., 2002; Sawa et al., 2005; Graham et al., 2006; Maglione et al., 2006). Like in SCA3, fragmentation of HTT is crucial for disease progression, since the HD disease phenotype can be rescued by either mutating the cleavage site of caspase-6 in exon 13 (Graham et al., 2006), genetically ablating caspase-6 (Wong et al., 2015), or pharmacologically inhibiting caspases 1, 3, or 6 (Ona et al., 1999; Chen et al., 2000; Aharony et al., 2015). We have already discussed the ability of certain chaperones to bind to the N17 domain, which is present in the cleaved fragments.
Post-Translational Modifications
Post translational modifications (PTMs) like phosphorylation, ubiquitination, and SUMOylation, can affect the aggregation propensity of many polyQ proteins (Humbert et al., 2001; Steffan et al., 2004; Luo et al., 2005; Warby et al., 2005; Menon et al., 2012; Matos et al., 2016b; Figure 2). The transient nature of the PTMs usually indicates differential regulation of proteins and they can provide an interesting extra layer of modulation, possibly influencing all of the above-mentioned features of polyQ aggregation. PTMs can create alternative binding surfaces, affecting the affinity to binding partners like proteases and chaperones, and can lead to conformational changes to expose the Q-stretch. Therefore, either increased or decreased PTMs are associated with aggregation.
For most of the polyQ proteins there are several residues known to be modified (see Figure 2B for PTMs that impact aggregation). For ATXN3 six phosphorylation sites have been described, in the catalytic JD and in the UIMs (Fei et al., 2007; Mueller et al., 2009; Matos et al., 2016b; Figure 2). Phosphorylation of serine (S)340 and S352 in the third UIM did not change aggregation propensity, but shifted the localization of the aggregates from the cytoplasm to the nucleus (Mueller et al., 2009). Phosphorylation of S256 in the second UIM was shown to inhibit the formation of large insoluble polyQ complexes (Fei et al., 2007), and phosphorylation of S12 in the JD also reduces aggregation (Matos et al., 2016b). The protective effect of constitutive phosphorylation of S12 might be dependent on its close proximity to the catalytic sites in the JD, causing hindrance of the intramolecular aggregation. Phosphorylation of HTT on S421 (Humbert et al., 2001) and S434 (Luo et al., 2005), leads to a decrease in polyQ aggregation due to a reduction in caspase-mediated cleavage thus preventing the formation of fragments (Luo et al., 2005; Warby et al., 2009). For ATXN1, S776 is the most studied phosphorylation site since it leads to reduced aggregate formation (Emamian et al., 2003; Orr, 2012). Another interesting PTM on ATXN1 is ubiquitination of K589 in the AXH domain. Mutating this residue leads to reduced degradation and, hence, more aggregation of ATXN1 (Kang et al., 2015), suggesting that PTMs may also affect the degradation of polyQ proteins resulting in a higher concentration of proteins at risk for aggregation.
Chaperone-dependent degradation of still soluble polyQ proteins could therefore be another important aspect in ameliorating disease. Interestingly, the co-chaperone CHIP (C-terminus of Hsp70-interacting protein), an E3 ligase that can interact with and modulate Hsp70 activity (Ballinger et al., 1999; Scheufler et al., 2000), has been implicated as a modulator in many polyQ diseases (Jana et al., 2005; Choi et al., 2007; Gao et al., 2011). CHIP interacts with ATXN1 via the phosphorylated S776 and the phospho-dead S776A mutation reduced this interaction. The CHIP-ATXN1 interaction is likely mediated via Hsp70, since the tetratricopeptide repeat (TPR) domain of CHIP, with which it interacts with Hsp70, is needed for the interaction and for promotion of ATXN1 degradation (Choi et al., 2007). A similar model of CHIP and Hsp70 interaction with HTT and ATXN3 was proposed, although no single modified residue was identified as a recognition site (Jana et al., 2005).
Members of DNAJ family of Hsp70 co-chaperones were also shown to play a role in the PTM dependent degradation of polyQ proteins, like in ATXN3 (Gao et al., 2011). DNAJB1 was identified to suppress aggregate formation of ATXN3 (Chai et al., 1999), but aggregation of the S256A mutant of ATXN3 could not be prevented by DNAJB1 (Fei et al., 2007), it is still unclear whether DNAJB1 has preferential affinity for phosphorylated ATXN3. Interestingly, Hsp70 can prevent S256A aggregation (Fei et al., 2007). Next to DNAJB1, DNAJB2 was found to suppress polyQ protein aggregation via two UIMs that were shown to be crucial for its interaction with K63-linked ubiquitination of HTT (Labbadia et al., 2012). Intriguingly, all the PTMs on HTT are less present in polyQ-expanded HTT, especially in the regions in the brain that are mostly affected, abolishing the possible protective effect of the modifications (Luo et al., 2005; Warby et al., 2005; Aiken et al., 2009). Currently it is unclear whether the drop in modification is causal or a consequence of aggregation.
Perspectives
The expanded polyQ stretches in the different disease-associated proteins are the determining factor of disease onset and progression in all of the polyglutaminopathies. Above a certain threshold, Q-stretches are prone to aggregate. However, more often than not, the Q-stretch and its aggregation propensities are modulated by secondary events that we categorized here; flanking regions, which have modulating capacity due to intrinsic stability issues, binding of partners (including chaperones), modification by PTMs, and cleavage of the Q-stretch. The examples of molecular interactions described, clearly indicate that polyQ protein aggregation is a multifactorial and likely multistep process that not always has to go through the same sequence of events toward aggregate formation. For example, the intrinsic fibrillogenic behavior of the JD and cleavage of ATXN3 (leading to a fragment not containing the JD) can both trigger aggregation independently. It could very well be that initial aggregation can be triggered via different mechanisms leading to secondary events that stimulate aggregation further. Thus, in vivo aggregation of the JD might stimulate ATXN3 cleavage and, vice versa, cleavage might destabilize the JD domain resulting in a fast forward feedback loop of aggregation. Modulating events, together with the unique expression pattern and level of each polyQ protein, could explain the variation in AO between the nine diseases.
Moreover, the modulating events acting on the flanking regions might also explain the variation of AO among patients with a similar Q length within a given polyQ disease. By combining information on Q length (CAG repeat), expression levels of the chaperone DNAJB6, which modulates Q aggregation directly, and the expression levels of chaperones that act on the disease-specific flanking regions, with the PTM and fragmentation status, perhaps a better predication of AO could be made. A strategy targeting chaperones acting on the Q-stretch with those acting on the flanking regions might provide a synergistic approach for delaying AO, benefiting individuals diagnosed with an expanded polyQ tract. There is little information on the factors influencing progression of disease after onset and it would also be of interest to know whether progression of disease is influenced by the same factors that modulate aggregation propensity. If so, these could be used as a therapeutic modality as well.
Author Contributions
EK and ED compiled all the data and contributed equally to this work. EK, ED, LJ, HK, and SB gave intellectual feedback and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
EK was awarded a Topmaster fellowship from the Groningen University Institute for Drug Exploration (GUIDE). ED was awarded a Science without Borders fellowship from the Brazilian Ministry of Education. LJ and ED are supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (National Counsel of Technological and Scientific Development, CNPq). SB has received a grant from the Nederlandse Organisatie voor Wetenschappelijk Onderzoek Aard-en Levenswetenschappen.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnins.2017.00145/full#supplementary-material
References
Aharony, I., Ehrnhoefer, D. E., Shruster, A., Qiu, X., Franciosi, S., Hayden, M. R., et al. (2015). A Huntingtin-based peptide inhibitor of caspase-6 provides protection from mutant Huntingtin-induced motor and behavioral deficits. Hum. Mol. Genet. 24, 2604–2614. doi: 10.1093/hmg/ddv023
Aiken, C. T., Steffan, J. S., Guerrero, C. M., Khashwji, H., Lukacsovich, T., Simmons, D., et al. (2009). Phosphorylation of threonine 3: implications for huntingtin aggregation and neurotoxicity. J. Biol. Chem. 284, 29427–29436. doi: 10.1074/jbc.M109.013193
Ballinger, C. A., Connell, P., Wu, Y., Hu, Z., Thompson, L. J., Yin, L. Y., et al. (1999). Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 19, 4535–4545. doi: 10.1128/MCB.19.6.4535
Bečanović, K., Nørremølle, A., Neal, S. J., Kay, C., Collins, J. A., Arenillas, D., et al. (2015). A SNP in the HTT promoter alters NF-κB binding and is a bidirectional genetic modifier of Huntington disease. Nat. Neurosci. 18, 807–816. doi: 10.1038/nn.4014
Bettencourt, C., Raposo, M., Kazachkova, N., Cymbron, T., Santos, C., Kay, T., et al. (2011). The APOE ε2 allele increases the risk of earlier age at onset in Machado-Joseph disease. Arch. Neurol. 68, 1580–1583. doi: 10.1001/archneurol.2011.636
Bhattacharyya, A., Thakur, A. K., Chellgren, V. M., Thiagarajan, G., Williams, A. D., Chellgren, B. W., et al. (2006). Oligoproline effects on polyglutamine conformation and aggregation. J. Mol. Biol. 355, 524–535. doi: 10.1016/j.jmb.2005.10.053
Bilen, J., and Bonini, N. M. (2007). Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet. 3:e177. doi: 10.1371/journal.pgen.0030177
Branco, J., Al-Ramahi, I., Ukani, L., Pérez, A. M., Fernandez-Funez, P., Rincón-Limas, D., et al. (2008). Comparative analysis of genetic modifiers in Drosophila points to common and distinct mechanisms of pathogenesis among polyglutamine diseases. Hum. Mol. Genet. 17, 376–390. doi: 10.1093/hmg/ddm315
Butler, R., Leigh, P. N., McPhaul, M. J., and Gallo, J. M. (1998). Truncated forms of the androgen receptor are associated with polyglutamine expansion in X-linked spinal and bulbar muscular atrophy. Hum. Mol. Genet. 7, 121–127. doi: 10.1093/hmg/7.1.121
Chai, Y., Koppenhafer, S. L., Bonini, N. M., and Paulson, H. L. (1999). Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease. J. Neurosci. 19, 10338–10347.
Chen, M., Ona, V. O., Li, M., Ferrante, R. J., Fink, K. B., Zhu, S., et al. (2000). Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat. Med. 6, 797–801. doi: 10.1038/80538
Chen, Y. W., Allen, M. D., Veprintsev, D. B., Löwe, J., and Bycroft, M. (2004). The structure of the AXH domain of spinocerebellar ataxin-1. J. Biol. Chem. 279, 3758–3765. doi: 10.1074/jbc.M309817200
Choi, J. Y., Ryu, J. H., Kim, H. S., Park, S. G., Bae, K. H., Kang, S., et al. (2007). Co-chaperone CHIP promotes aggregation of ataxin-1. Mol. Cell. Neurosci. 34, 69–79. doi: 10.1016/j.mcn.2006.10.002
Chow, M. K. M., MacKay, J. P., Whisstock, J. C., Scanlon, M. J., and Bottomley, S. P. (2004). Structural and functional analysis of the Josephin domain of the polyglutamine protein ataxin-3. Biochem. Biophys. Res. Commun. 322, 387–394. doi: 10.1016/j.bbrc.2004.07.131
Colomer Gould, V. F., Goti, D., Pearce, D., Gonzalez, G. A., Gao, H., Bermudez de Leon, M., et al. (2007). A mutant ataxin-3 fragment results from processing at a site N-terminal to amino acid 190 in brain of Machado-Joseph disease-like transgenic mice. Neurobiol. Dis. 27, 362–369. doi: 10.1016/j.nbd.2007.06.005
Crick, S. L., Ruff, K. M., Garai, K., Frieden, C., and Pappu, R., V. (2013). Unmasking the roles of N- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. U.S.A. 110, 20075–20080. doi: 10.1073/pnas.1320626110
Cummings, C. J., Mancini, M. A., Antalffy, B., DeFranco, D. B., Orr, H. T., and Zoghbi, H. Y. (1998). Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 19, 148–154. doi: 10.1038/502
De Chiara, C., Menon, R. P., Adinolfi, S., De Boer, J., Ktistaki, E., Kelly, G., et al. (2005a). The AXH domain adopts alternative folds: the solution structure of HBP1 AXH. Structure 13, 743–753. doi: 10.1016/j.str.2005.02.016
De Chiara, C., Menon, R. P., Dal Piaz, F., Calder, L., and Pastore, A. (2005b). Polyglutamine is not all: the functional role of the AXH domain in the ataxin-1 protein. J. Mol. Biol. 354, 883–893. doi: 10.1016/j.jmb.2005.09.083
De Chiara, C., Menon, R. P., Kelly, G., and Pastore, A. (2013a). Protein-protein interactions as a strategy towards protein-specific drug design: the example of ataxin-1. PLoS ONE 8:e76456. doi: 10.1371/journal.pone.0076456
De Chiara, C., Rees, M., Menon, R. P., Pauwels, K., Lawrence, C., Konarev, P. V., et al. (2013b). Self-assembly and conformational heterogeneity of the AXH domain of ataxin-1: an unusual example of a chameleon fold. Biophys. J. 104, 1304–1313. doi: 10.1016/j.bpj.2013.01.048
Dehay, B., and Bertolotti, A. (2006). Critical role of the proline-rich region in Huntingtin for aggregation and cytotoxicity in yeast. J. Biol. Chem. 281, 35608–35615. doi: 10.1074/jbc.M605558200
Dekker, S. L., Kampinga, H. H., and Bergink, S. (2015). DNAJs: more than substrate delivery to HSPA. Front. Mol. Biosci. 2:35. doi: 10.3389/fmolb.2015.00035
Deriu, M. A., Grasso, G., Tuszynski, J. A., Massai, D., Gallo, D., Morbiducci, U., et al. (2016). Characterization of the AXH domain of Ataxin-1 using enhanced sampling and functional mode analysis. Proteins 84, 666–673. doi: 10.1002/prot.25017
DeStefano, A. L., Cupples, L. A., Maciel, P., Gaspar, C., Radvany, J., Dawson, D. M., et al. (1996). A familial factor independent of CAG repeat length influences age at onset of Machado-Joseph disease. Am. J. Hum. Genet. 59, 119–127.
Duennwald, M. L., Jagadish, S., Muchowski, P. J., and Lindquist, S. (2006). Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc. Natl. Acad. Sci. U.S.A. 103, 11045–11050. doi: 10.1073/pnas.0604547103
Ellisdon, A. M., Pearce, M. C., and Bottomley, S. P. (2007). Mechanisms of ataxin-3 misfolding and fibril formation: kinetic analysis of a disease-associated polyglutamine protein. J. Mol. Biol. 368, 595–605. doi: 10.1016/j.jmb.2007.02.058
Ellisdon, A. M., Thomas, B., and Bottomley, S. P. (2006). The two-stage pathway of ataxin-3 fibrillogenesis involves a polyglutamine-independent step. J. Biol. Chem. 281, 16888–16896. doi: 10.1074/jbc.M601470200
Emamian, E. S., Kaytor, M. D., Duvick, L. A., Zu, T., Tousey, S. K., Zoghbi, H. Y., et al. (2003). Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 38, 375–387. doi: 10.1016/S0896-6273(03)00258-7
Fei, E., Jia, N., Zhang, T., Ma, X., Wang, H., Liu, C., et al. (2007). Phosphorylation of ataxin-3 by glycogen synthase kinase 3β at serine 256 regulates the aggregation of ataxin-3. Biochem. Biophys. Res. Commun. 357, 487–492. doi: 10.1016/j.bbrc.2007.03.160
Gales, L., Cortes, L., Almeida, C., Melo, C. V., Costa, M. D. C., Maciel, P., et al. (2005). Towards a structural understanding of the fibrillization pathway in Machado-Joseph's disease: trapping early oligomers of non-expanded ataxin-3. J. Mol. Biol. 353, 642–654. doi: 10.1016/j.jmb.2005.08.061
Gao, X. C., Zhou, C. J., Zhou, Z. R., Zhang, Y. H., Zheng, X. M., Song, A. X., et al. (2011). Co-chaperone HSJ1a dually regulates the proteasomal degradation of ataxin-3. PLoS ONE 6:e19763. doi: 10.1371/journal.pone.0019763
Goti, D., Katzen, S. M., Mez, J., Kurtis, N., Kiluk, J., Ben-Haïem, L., et al. (2004). A mutant ataxin-3 putative-cleavage fragment in brains of Machado-Joseph disease patients and transgenic mice is cytotoxic above a critical concentration. J. Neurosci. 24, 10266–10279. doi: 10.1523/JNEUROSCI.2734-04.2004
Graham, R. K., Deng, Y., Slow, E. J., Haigh, B., Bissada, N., Lu, G., et al. (2006). Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 125, 1179–1191. doi: 10.1016/j.cell.2006.04.026
Haacke, A., Broadley, S. A., Boteva, R., Tzvetkov, N., Hartl, F. U., and Breuer, P. (2006). Proteolytic cleavage of polyglutamine-expanded ataxin-3 is critical for aggregation and sequestration of non-expanded ataxin-3. Hum. Mol. Genet. 15, 555–568. doi: 10.1093/hmg/ddi472
Hageman, J., Rujano, M. A., van Waarde, M. A. W. H., Kakkar, V., Dirks, R. P., Govorukhina, N., et al. (2010). A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell. 37, 355–369. doi: 10.1016/j.molcel.2010.01.001
Hayes, S., Turecki, G., Brisebois, K., Lopes-Cendes, I., Gaspar, C., Riess, O., et al. (2000). CAG repeat length in RAI1 is associated with age at onset variability in spinocerebellar ataxia type 2 (SCA2). Hum. Mol. Genet. 9, 1753–1758. doi: 10.1093/hmg/9.12.1753
Heine, E. M., Berger, T. R., Pluciennik, A., Orr, C. R., Zboray, L., and Merry, D. E. (2015). Proteasome-mediated proteolysis of the polyglutamine-expanded androgen receptor is a late event in spinal and bulbar muscular atrophy (SBMA) pathogenesis. J. Biol. Chem. 290, 12572–12584. doi: 10.1074/jbc.M114.617894
Helmlinger, D., Bonnet, J., Mandel, J. L., Trottier, Y., and Devys, D. (2004). Hsp70 and Hsp40 chaperones do not modulate retinal phenotype in SCA7 mice. J. Biol. Chem. 279, 55969–55977. doi: 10.1074/jbc.M409062200
Hoop, C. L., Lin, H.-K., Kar, K., Magyarfalvi, G., Lamley, J. M., Boatz, J. C., et al. (2016). Huntingtin exon 1 fibrils feature an interdigitated β-hairpin–based polyglutamine core. Proc. Natl. Acad. Sci. U.S.A. 113, 1546–1551. doi: 10.1073/pnas.1521933113
Hübener, J., Weber, J. J., Richter, C., Honold, L., Weiss, A., Murad, F., et al. (2013). Calpain-mediated ataxin-3 cleavage in the molecular pathogenesis of spinocerebellar ataxia type 3 (SCA3). Hum. Mol. Genet. 22, 508–518. doi: 10.1093/hmg/dds449
Humbert, S., Bryson, E. A., Cordelie, F. P., Connors, N. C., Datta, S. R., Finkbeiner, S., et al. (2001). The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves huntingtin phosphorylation by Akt. Dev. Cell. 2, 831–837. doi: 10.1016/S1534-5807(02)00188-0
Huynh, D. P., Figueroa, K., Hoang, N., and Pulst, S. M. (2000). Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat. Genet. 26, 44–50. doi: 10.1038/79162
Igarashi, S., Koide, R., Shimohata, T., Yamada, M., Hayashi, Y., Takano, H., et al. (1998). Suppression of aggregate formation and apoptosis by transglutaminase inhibitors in cells expressing truncated DRPLA protein with an expanded polyglutamine stretch. Nat. Genet. 18, 111–117. doi: 10.1038/ng0298-111
Ikeda, H., Yamaguchi, M., Sugai, S., Aze, Y., Narumiya, S., and Kakizuka, A. (1996). Expanded polyglutamine in the Machado-Joseph disease protein induces cell death in vitro and in vivo. Nat. Genet. 13, 196–202. doi: 10.1038/ng0696-196
Jana, N. R., Dikshit, P., Goswami, A., Kotliarova, S., Murata, S., Tanaka, K., et al. (2005). Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 280, 11635–11640. doi: 10.1074/jbc.M412042200
Jana, N. R., Tanaka, M., Wang, G. H., and Nukina, N. (2000). Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum. Mol. Genet. 9, 2009–2018. doi: 10.1093/hmg/9.13.2009
Jayaraman, M., Kodali, R., and Wetzel, R. (2009). The impact of ataxin-1-like histidine insertions on polyglutamine aggregation. Protein Eng. Des. Sel. 22, 469–478. doi: 10.1093/protein/gzp023
Kakkar, V., Månsson, C., de Mattos, E. P., Bergink, S., van der Zwaag, M., van Waarde, M. A. W. H., et al. (2016). The S/T-rich motif in the DNAJB6 chaperone delays polyglutamine aggregation and the onset of disease in a mouse model. Mol. Cell. 62, 272–283. doi: 10.1016/j.molcel.2016.03.017
Kakkar, V., Meister-Broekema, M., Minoia, M., Carra, S., and Kampinga, H. H. (2014). Barcoding heat shock proteins to human diseases: looking beyond the heat shock response. Dis. Model. Mech. 7, 421–434. doi: 10.1242/dmm.014563
Kaltenbach, L. S., Romero, E., Becklin, R. R., Chettier, R., Bell, R., Phansalkar, A., et al. (2007). Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 3:e82. doi: 10.1371/journal.pgen.0030082
Kampinga, H. H., and Bergink, S. (2016). Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 15, 748–759. doi: 10.1016/S1474-4422(16)00099-5
Kang, A. R., Park, S. H., Lee, S., Choi, D. Y., Kim, K. P., Song, H. K., et al. (2015). A key lysine residue in the AXH domain of ataxin-1 is essential for its ubiquitylation. Biochim. Biophys. Acta 1854, 356–364. doi: 10.1016/j.bbapap.2015.01.012
Kar, K., Hoop, C. L., Drombosky, K. W., Baker, M. A., Kodali, R., Arduini, I., et al. (2013). β-Hairpin-mediated nucleation of polyglutamine amyloid formation. J. Mol. Biol. 425, 1183–1197. doi: 10.1016/j.jmb.2013.01.016
Kar, K., Jayaraman, M., Sahoo, B., Kodali, R., and Wetzel, R. (2011). Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat. Struct. Mol. Biol. 18, 328–336. doi: 10.1038/nsmb.1992
Kazemi-Esfarjani, P., and Benzer, S. (2000). Genetic suppression of polyglutamine toxicity in Drosophila. Science 287, 1837–1840. doi: 10.1126/science.287.5459.1837
Kelley, N. W., Huang, X., Tam, S., Spiess, C., Frydman, J., and Pande, V. S. (2009). The predicted structure of the headpiece of the huntingtin protein and its implications on huntingtin aggregation. J. Mol. Biol. 388, 919–927. doi: 10.1016/j.jmb.2009.01.032
Kim, M. (2014). Pathogenic polyglutamine expansion length correlates with polarity of the flanking sequences. Mol. Neurodegener. 9:45. doi: 10.1186/1750-1326-9-45
Kim, S., Nollen, E. A. A., Kitagawa, K., Bindokas, V. P., and Morimoto, R. I. (2002). Polyglutamine protein aggregates are dynamic. Nat. Cell Biol. 4, 826–831. doi: 10.1038/ncb863
Kobayashi, Y., Miwa, S., Merry, D. E., Kume, A., Mei, L., Doyu, M., et al. (1998). Caspase-3 cleaves the expanded androgen receptor protein of spinal and bulbar muscular atrophy in a polyglutamine repeat length-dependent manner. Biochem. Biophys. Res. Commun. 252, 145–150. doi: 10.1006/bbrc.1998.9624
Kordasiewicz, H. B., Thompson, R. M., Clark, H. B., and Gomez, C. M. (2006). C-termini of P/Q-type Ca2+ channel α1A subunits translocate to nuclei and promote polyglutamine-mediated toxicity. Hum. Mol. Genet. 15, 1587–1599. doi: 10.1093/hmg/ddl080
Kuhlbrodt, K., Janiesch, P. C., Kevei, É., Segref, A., Barikbin, R., and Hoppe, T. (2011). The Machado-Joseph disease deubiquitylase ATX-3 couples longevity and proteostasis. Nat. Cell Biol. 13, 273–281. doi: 10.1038/ncb2200
Labbadia, J., Novoselov, S. S., Bett, J. S., Weiss, A., Paganetti, P., Bates, G. P., et al. (2012). Suppression of protein aggregation by chaperone modification of high molecular weight complexes. Brain 135, 1180–1196. doi: 10.1093/brain/aws022
Lam, Y. C., Bowman, A. B., Jafar-Nejad, P., Lim, J., Richman, R., Fryer, J. D., et al. (2006). ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell 127, 1335–1347. doi: 10.1016/j.cell.2006.11.038
Lasagna-Reeves, C. A., Rousseaux, M. W. C., Guerrero-Munoz, M. J., Park, J., Jafar-Nejad, P., Richman, R., et al. (2015). A native interactor scaffolds and stabilizes toxic Ataxin-1 oligomers in SCA1. Elife 4:e07558. doi: 10.7554/eLife.07558
Lessing, D., and Bonini, N. M. (2008). Polyglutamine genes interact to modulate the severity and progression of neurodegeneration in Drosophila. PLoS Biol. 6:e29. doi: 10.1371/journal.pbio.0060029
Liebman, S. W., and Meredith, S. C. (2010). Protein folding: sticky N17 speeds huntingtin pile-up. Nat. Chem. Biol. 6, 7–8. doi: 10.1038/nchembio.279
Luo, S., Vacher, C., Davies, J. E., and Rubinsztein, D. C. (2005). Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implications for mutant huntingtin toxicity. J. Cell Biol. 169, 647–656. doi: 10.1083/jcb.200412071
Lupton, C. J., Steer, D. L., Bottomley, S. P., Hughes, V. A., Wintrode, P. L., and Ellisdon, A. M. (2015). Enhanced molecular mobility of ordinarily structured regions drives polyglutamine disease. J. Biol. Chem. 290, 24190–24200. doi: 10.1074/jbc.M115.659532
Maglione, V., Cannella, M., Gradini, R., Cislaghi, G., and Squitieri, F. (2006). Huntingtin fragmentation and increased caspase 3, 8 and 9 activities in lymphoblasts with heterozygous and homozygous Huntington's disease mutation. Mech. Ageing Dev. 127, 213–216. doi: 10.1016/j.mad.2005.09.011
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., et al. (1996). Exon I of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506. doi: 10.1016/S0092-8674(00)81369-0
Månsson, C., Kakkar, V., Monsellier, E., Sourigues, Y., Härmark, J., Kampinga, H. H., et al. (2013). DNAJB6 is a peptide-binding chaperone which can suppress amyloid fibrillation of polyglutamine peptides at substoichiometric molar ratios. Cell Stress Chaperones 19, 227–239. doi: 10.1007/s12192-013-0448-5
Margolis, R. L., and Ross, C. A. (2001). Expansion explosion: new clues to the pathogenesis of repeat expansion neurodegenerative diseases. Trends Mol. Med. 7, 479–482. doi: 10.1016/S1471-4914(01)02179-7
Martindale, D., Hackam, A., Wieczorek, A., Ellerby, L., Wellington, C., McCutcheon, K., et al. (1998). Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat. Genet. 18, 150–154. doi: 10.1038/ng0298-150
Masino, L., Nicastro, G., Calder, L., Vendruscolo, M., and Pastore, A. (2011). Functional interactions as a survival strategy against abnormal aggregation. FASEB J. 25, 45–54. doi: 10.1096/fj.10-161208
Masino, L., Nicastro, G., Menon, R. P., Dal Piaz, F., Calder, L., and Pastore, A. (2004). Characterization of the structure and the amyloidogenic properties of the Josephin domain of the polyglutamine-containing protein ataxin-3. J. Mol. Biol. 344, 1021–1035. doi: 10.1016/j.jmb.2004.09.065
Matilla-Dueñas, A., Ashizawa, T., Brice, A., Magri, S., McFarland, K. N., Pandolfo, M., et al. (2014). Consensus paper: pathological mechanisms underlying neurodegeneration in spinocerebellar ataxias. Cerebellum 13, 269–302. doi: 10.1007/s12311-013-0539-y
Matos, C. A., de Almeida, L. P., and Nóbrega, C. (2016a). Proteolytic cleavage of polyglutamine disease-causing proteins: revisiting the toxic fragment hypothesis. Curr. Pharm. Des. 22. doi: 10.2174/1381612822666161227121912. [Epub ahead of print].
Matos, C. A., Nóbrega, C., Louros, S. R., Almeida, B., Ferreiro, E., Valero, J., et al. (2016b). Ataxin-3 phosphorylation decreases neuronal defects in spinocerebellar ataxia type 3 models. J. Cell Biol. 212, 465–480. doi: 10.1083/jcb.201506025
Matsumoto, G., Kim, S., and Morimoto, R. I. (2006). Huntingtin and mutant SOD1 form aggregate structures with distinct molecular properties in human cells. J. Biol. Chem. 281, 4477–4485. doi: 10.1074/jbc.M509201200
Menon, R. P., Nethisinghe, S., Faggiano, S., Vannocci, T., Rezaei, H., Pemble, S., et al. (2013). The role of interruptions in polyQ in the pathology of SCA1. PLoS Genet. 9:e1003648. doi: 10.1371/journal.pgen.1003648
Menon, R. P., Soong, D., de Chiara, C., Holt, M. R., Anilkumar, N., and Pastore, A. (2012). The importance of serine 776 in Ataxin-1 partner selection: a FRET analysis. Sci. Rep. 2:919. doi: 10.1038/srep00919
Monsellier, E., Redeker, V., Ruiz-Arlandis, G., Bousset, L., and Melki, R. (2015). Molecular interaction between the chaperone Hsc70 and the N-terminal flank of huntingtin exon 1 modulates aggregation. J. Biol. Chem. 290, 2560–2576. doi: 10.1074/jbc.M114.603332
Muchowski, P. J., Schaffar, G., Sittler, A., Wanker, E. E., Hayer-Hartl, M. K., and Hartl, F. U. (2000). Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. U.S.A. 97, 7841–7846. doi: 10.1073/pnas.140202897
Mueller, T., Breuer, P., Schmitt, I., Walter, J., Evert, B. O., and Wüllner, U. (2009). CK2-dependent phosphorylation determines cellular localization and stability of ataxin-3. Hum. Mol. Genet. 18, 3334–3343. doi: 10.1093/hmg/ddp274
Nicastro, G., Masino, L., Esposito, V., Menon, R. P., De Simone, A., Fraternali, F., et al. (2009). Josephin domain of ataxin-3 contains two distinct ubiquitin-binding sites. Biopolymers 91, 1203–1214. doi: 10.1002/bip.21210
Nicastro, G., Todi, S. V., Karaca, E., Bonvin, A. M. J. J., Paulson, H. L., and Pastore, A. (2010). Understanding the role of the josephin domain in the polyub binding and cleavage properties of ataxin-3. PLoS ONE 5:e12430. doi: 10.1371/journal.pone.0012430
Nóbrega, C., Carmo-Silva, S., Albuquerque, D., Vasconcelos-Ferreira, A., Vijayakumar, U. G., Mendonça, L., et al. (2015). Re-establishing ataxin-2 downregulates translation of mutant ataxin-3 and alleviates Machado-Joseph disease. Brain 138, 3537–3554. doi: 10.1093/brain/awv298
Novoselova, T. V., Margulis, B. A., Novoselov, S. S., Sapozhnikov, A. M., Van Der Spuy, J., Cheetham, M. E., et al. (2005). Treatment with extracellular HSP70/HSC70 protein can reduce polyglutamine toxicity and aggregation. J. Neurochem. 94, 597–606. doi: 10.1111/j.1471-4159.2005.03119.x
Nozaki, K., Onodera, O., Takano, H., and Tsuji, S. (2001). Amino acid sequences flanking polyglutamine stretches influence their potential for aggregate formation. Neuroreport 12, 3357–3364. doi: 10.1097/00001756-200110290-00042
Ona, V. O., Li, M., Vonsattel, J. P., Andrews, L. J., Khan, S. Q., Chung, W. M., et al. (1999). Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature 399, 263–267. doi: 10.1038/20446
Orr, H. T. (2012). SCA1-phosphorylation, a regulator of ataxin-1 function and pathogenesis. Prog. Neurobiol. 99, 179–185. doi: 10.1016/j.pneurobio.2012.04.003
Pang, J. T., Giunti, P., Chamberlain, S., An, S. F., Vitaliani, R., Scaravilli, T., et al. (2002). Neuronal intranuclear inclusions in SCA2: a genetic, morphological and immunohistochemical study of two cases. Brain 125, 656–663. doi: 10.1093/brain/awf060
Paulson, H. L., Perez, M. K., Trottier, Y., Trojanowski, J. Q., Subramony, S. H., Das, S. S., et al. (1997). Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 19, 333–344. doi: 10.1016/S0896-6273(00)80943-5
Perez, M. K., Paulson, H. L., Pendse, S. J., Saionz, S. J., Bonini, N. M., and Pittman, R. N. (1998). Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J. Cell Biol. 143, 1457–1470. doi: 10.1083/jcb.143.6.1457
Perutz, M., Johnson, T., and Suzuki, M. (1994). Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc. Natl. Acad. Sci. U.S.A. 91, 5355–5358. doi: 10.1073/pnas.91.12.5355
Ranum, L. P., Chung, M. Y., Banfi, S., Bryer, A., Schut, L. J., Ramesar, R., et al. (1994). Molecular and clinical correlations in spinocerebellar ataxia type I: evidence for familial effects on the age at onset. Am. J. Hum. Genet. 55, 244–252.
Reis, S. D., Pinho, B. R., and Oliveira, J. M. A. (2016). Modulation of molecular chaperones in Huntington's disease and other polyglutamine disorders. Mol. Neurobiol. doi: 10.1007/s12035-016-0120-z. [Epub ahead of print].
Robertson, A. L., Headey, S. J., Saunders, H. M., Ecroyd, H., Scanlon, M. J., Carver, J. A., et al. (2010). Small heat-shock proteins interact with a flanking domain to suppress polyglutamine aggregation. Proc. Natl. Acad. Sci. U.S.A. 107, 10424–10429. doi: 10.1073/pnas.0914773107
Rockabrand, E., Slepko, N., Pantalone, A., Nukala, V. N., Kazantsev, A., Marsh, J. L., et al. (2007). The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum. Mol. Genet. 16, 61–77. doi: 10.1093/hmg/ddl440
Rubinsztein, D. C. (2006). The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786. doi: 10.1038/nature05291
Sahl, S. J., Lau, L., Vonk, W. I. M., Weiss, L. E., Frydman, J., and Moerner, W. E. (2015). Delayed emergence of subdiffraction-sized mutant huntingtin fibrils following inclusion body formation. Q. Rev. Biophys. 49, e2. doi: 10.1017/s0033583515000219
Sahoo, B., Arduini, I., Drombosky, K. W., Kodali, R., Sanders, L. H., Greenamyre, J. T., et al. (2016). Folding landscape of mutant huntingtin Exon1: diffusible multimers, oligomers and fibrils, and no detectable monomer. PLoS ONE 11:e0155747. doi: 10.1371/journal.pone.0155747
Saunders, H. M., Gilis, D., Rooman, M., Dehouck, Y., Robertson, A. L., and Bottomley, S. P. (2011). Flanking domain stability modulates the aggregation kinetics of a polyglutamine disease protein. Protein Sci. 20, 1675–1681. doi: 10.1002/pro.698
Saute, J. A. M., and Jardim, L. B. (2015). Machado Joseph disease: clinical and genetic aspects, and current treatment. Expert Opin. Orphan Drugs 3, 517–535. doi: 10.1517/21678707.2015.1025747
Sawa, A., Nagata, E., Sutcliffe, S., Dulloor, P., Cascio, M. B., Ozeki, Y., et al. (2005). Huntingtin is cleaved by caspases in the cytoplasm and translocated to the nucleus via perinuclear sites in Huntington's disease patient lymphoblasts. Neurobiol. Dis. 20, 267–274. doi: 10.1016/j.nbd.2005.02.013
Sawaya, M. R., Sambashivan, S., Nelson, R., Ivanova, M. I., Sievers, S. A., Apostol, M. I., et al. (2007). Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 447, 453–457. doi: 10.1038/nature05695
Scarff, C. A., Almeida, B., Fraga, J., Macedo-Ribeiro, S., Radford, S. E., and Ashcroft, A. E. (2015). Examination of ataxin-3 aggregation by structural mass spectrometry techniques: a rationale for expedited aggregation upon polyglutamine expansion. Mol. Cell. Proteomics 14, 1241–1253. doi: 10.1074/mcp.M114.044610
Scheufler, C., Brinker, A., Bourenkov, G., Pegoraro, S., Moroder, L., Bartunik, H., et al. (2000). Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 101, 199–210. doi: 10.1016/S0092-8674(00)80830-2
Schmidt, T., Lindenberg, K. S., Krebs, A., Schöls, L., Laccone, F., Herms, J., et al. (2002). Protein surveillance machinery in brains with spinocerebellar ataxia type 3: redistribution and differential recruitment of 26S proteasome subunits and chaperones to neuronal intranuclear inclusions. Ann. Neurol. 51, 302–310. doi: 10.1002/ana.10101
Seidel, K., Siswanto, S., Fredrich, M., Bouzrou, M., den Dunnen, W. F. A., Özerden, I., et al. (2016). On the distribution of intranuclear and cytoplasmic aggregates in the brainstem of patients with Spinocerebellar Ataxia Type 2 and 3. Brain Pathol. doi: 10.1111/bpa.12412. [Epub ahead of print].
Shahmoradian, S. H., Galaz-Montoya, J. G., Schmid, M. F., Cong, Y., Ma, B., Spiess, C., et al. (2013). TRiC's tricks inhibit huntingtin aggregation. Elife 2:e00710. doi: 10.7554/eLife.00710
Simões, A. T., Gonçalves, N., Koeppen, A., Déglon, N., Kügler, S., Duarte, C. B., et al. (2012). Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado-Joseph disease. Brain 135, 2428–2439. doi: 10.1093/brain/aws177
Sivanandam, V. N., Jayaraman, M., Hoop, C. L., Kodali, R., Wetzel, R., and Van Der Wel, P. C. A. (2011). The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J. Am. Chem. Soc. 133, 4558–4566. doi: 10.1021/ja110715f
Sobczak, K., and Krzyzosiak, W. J. (2004). Patterns of CAG repeat interruptions in SCA1 and SCA2 genes in relation to repeat instability. Hum. Mutat. 24, 236–247. doi: 10.1002/humu.20075
Soto, C. (2003). Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 49–60. doi: 10.1038/nrn1007
Spiess, C., Miller, E. J., McClellan, A. J., and Frydman, J. (2006). Identification of the TRiC/CCT substrate binding sites uncovers the function of subunit diversity in eukaryotic chaperonins. Mol. Cell. 24, 25–37. doi: 10.1016/j.molcel.2006.09.003
Steffan, J. S., Agrawal, N., Pallos, J., Rockabrand, E., Trotman, L. C., Slepko, N., et al. (2004). SUMO modification of Huntingtin and Huntington's disease pathology. Science 304, 100–104. doi: 10.1126/science.1092194
Tam, S., Geller, R., Spiess, C., and Frydman, J. (2006). The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol. 8, 1155–1162. doi: 10.1038/ncb1477
Tam, S., Spiess, C., Auyeung, W., Joachimiak, L., Chen, B., Poirier, M. A., et al. (2009). The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 16, 1279–1285. doi: 10.1038/nsmb.1700
Tezenas du Montcel, S., Durr, A., Bauer, P., Figueroa, K. P., Ichikawa, Y., Brussino, A., et al. (2014). Modulation of the age at onset in spinocerebellar ataxia by CAG tracts in various genes. Brain 137, 2444–2455. doi: 10.1093/brain/awu174
Thakur, A. K., Jayaraman, M., Mishra, R., Thakur, M., Chellgren, V. M., Byeon, I.-J. L., et al. (2009). Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 16, 380–389. doi: 10.1038/nsmb.1570
Uchihara, T., Fujigasaki, H., Koyano, S., Nakamura, A., Yagishita, S., and Iwabuchi, K. (2001). Non-expanded polyglutamine proteins in intranuclear inclusions of hereditary ataxias - triple-labeling immunofluorescence study. Acta Neuropathol. 102, 149–152. doi: 10.1007/s004010100364
van de Warrenburg, B. P. C., Hendriks, H., Dürr, A., van Zuijlen, M. C. A., Stevanin, G., Camuzat, A., et al. (2005). Age at onset variance analysis in spinocerebellar ataxias: a study in a Dutch-French cohort. Ann. Neurol. 57, 505–512. doi: 10.1002/ana.20424
Warby, S. C., Chan, E. Y., Metzler, M., Gan, L., Singaraja, R. R., Crocker, S. F., et al. (2005). Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum. Mol. Genet. 14, 1569–1577. doi: 10.1093/hmg/ddi165
Warby, S. C., Doty, C. N., Graham, R. K., Shively, J., Singaraja, R. R., and Hayden, M. R. (2009). Phosphorylation of huntingtin reduces the accumulation of its nuclear fragments. Mol. Cell. Neurosci. 40, 121–127. doi: 10.1016/j.mcn.2008.09.007
Wellington, C. L., Ellerby, L. M., Gutekunst, C.-A., Rogers, D., Warby, S., Graham, R. K., et al. (2002). Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington's disease. J. Cell Biol. 22, 749–759. doi: 10.1038/35096019
Wellington, C. L., Ellerby, L. M., Hackam, A. S., Margolis, R. L., Trifiro, M. A., Singaraja, R., et al. (1998). Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J. Biol. Chem. 273, 9158–9167. doi: 10.1074/jbc.273.15.9158
Wexler, N. S., Lorimer, J., Porter, J., Gomez, F., Moskowitz, C., Shackell, E., et al. (2004). Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc. Natl. Acad. Sci. U.S.A. 101, 3498–3503. doi: 10.1073/pnas.0308679101
Wong, B. K. Y., Ehrnhoefer, D. E., Graham, R. K., Martin, D. D. O., Ladha, S., Uribe, V., et al. (2015). Partial rescue of some features of Huntington Disease in the genetic absence of caspase-6 in YAC128 mice. Neurobiol. Dis. 76, 24–36. doi: 10.1016/j.nbd.2014.12.030
Zander, C., Takahashi, J., El Hachimi, K. H., Fujigasaki, H., Albanese, V., Lebre, A. S., et al. (2001). Similarities between spinocerebellar ataxia type 7 (SCA7) cell models and human brain: proteins recruited in inclusions and activation of caspase-3. Hum. Mol. Genet. 10, 2569–2579. doi: 10.1093/hmg/10.22.2569
Zhai, R. G., Zhang, F., Hiesinger, P. R., Cao, Y., Haueter, C. M., and Bellen, H. J. (2008). NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature 452, 887–891. doi: 10.1038/nature06721
Keywords: aggregation, Huntington's disease, Machado-Joseph disease, molecular chaperones, polyglutamine disease
Citation: Kuiper EFE, de Mattos EP, Jardim LB, Kampinga HH and Bergink S (2017) Chaperones in Polyglutamine Aggregation: Beyond the Q-Stretch. Front. Neurosci. 11:145. doi: 10.3389/fnins.2017.00145
Received: 20 January 2017; Accepted: 08 March 2017;
Published: 23 March 2017.
Edited by:
Tiago Fleming Outeiro, University Medical Center Goettingen, GermanyReviewed by:
Clevio Nobrega, University of the Algarve, PortugalPedro Domingos, Instituto de Tecnologia Quimica e Biológica—Universidade Nova de Lisboa, Portugal
Tatiana Rosado Rosenstock, Santa Casa de São Paulo School of Medical Sciences, Brazil
Copyright © 2017 Kuiper, de Mattos, Jardim, Kampinga and Bergink. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven Bergink, cy5iZXJnaW5rQHVtY2cubmw=
†These authors have contributed equally to this work.