 Changqing Xu
Changqing Xu Sylvia Fitting*
Sylvia Fitting*- Department of Psychology and Neuroscience, University of North Carolina Chapel Hill, Chapel Hill, NC, USA
Due to combined antiretroviral therapy (cART), human immunodeficiency virus type 1 (HIV-1) is considered a chronic disease with high prevalence of mild forms of neurocognitive impairments, also referred to as HIV-associated neurocognitive disorders (HAND). Although opiate drug use can exacerbate HIV-1 Tat-induced neuronal damage, it remains unknown how and to what extent opioids interact with Tat on the GABAergic system. We conducted whole-cell recordings in mouse striatal slices and examined the effects of HIV-1 Tat in the presence and absence of morphine (1 μM) and damgo (1 μM) on GABAergic neurotransmission. Results indicated a decrease in the frequency and amplitude of spontaneous inhibitory postsynaptic currents (sIPSCs) and miniature IPSCs (mIPSCs) by Tat (5–50 nM) in a concentration-dependent manner. The significant Tat-induced decrease in IPSCs was abolished when removing extracellular and/or intracellular calcium. Treatment with morphine or damgo alone significantly decreased the frequency, but not amplitude of IPSCs. Interestingly, morphine but not damgo indicated an additional downregulation of the mean frequency of mIPSCs in combination with Tat. Pretreatment with naloxone (1 μM) and CTAP (1 μM) prevented the Tat-induced decrease in sIPSCs frequency but only naloxone prevented the combined Tat and morphine effect on mIPSCs frequency. Results indicate a Tat- or opioid-induced decrease in GABAergic neurotransmission via μ-opioid receptors with combined Tat and morphine effects involving additional opioid receptor-related mechanisms. Exploring the interactions between Tat and opioids on the GABAergic system may help to guide future research on HAND in the context of opiate drug use.
Introduction
Even though severe cases of human immunodeficiency virus type 1 (HIV-1)-associated dementia have been reduced due to the introduction of combined antiretroviral therapy (cART), 30–50%-infected individuals suffer from HIV-1-associated neurocognitive disorders (HAND) (Bell et al., 1998; Dore et al., 1999; Sacktor et al., 2002; Antinori et al., 2007; Ellis et al., 2007; Heaton et al., 2011; Saylor et al., 2016). HAND is specifically enhanced in the context of drug use, such as opiates, with worsening the consequences of HIV-1 in the central nervous system (CNS) (Bell et al., 1998; Arango et al., 2004; Anthony et al., 2005; Lan et al., 2015). Experimental models of acquired immune deficiency syndrome (AIDS) have further indicated an increase in virus and disease progression by opiates (Pérez-Casanova et al., 2007; Noel et al., 2008; Rivera-Amill et al., 2010; Bokhari et al., 2011; Hu et al., 2012; Masvekar et al., 2015). HIV-1 specifically targets subcortical and cortical areas of the brain, with HIV-1 positive individuals displaying deficits in sensorimotor function, executive function, learning and memory, and attention (Berger and Nath, 1997; Berger and Arendt, 2000; Heaton et al., 2011; Scott et al., 2011). The striatum is one of the regions with the highest viral burden (Kure et al., 1990; Berger and Arendt, 2000; Kumar et al., 2007) and rich in opioid receptor-expressing cells. Animal in vivo and in vitro studies focusing on the striatum have identified exacerbated behavioral and structural changes by opioids in the presence of the HIV-1 transactivator of transcription (Tat) corresponding to a HAND-associated phenotype (Fitting et al., 2010, 2012, 2014).
It has been demonstrated that Tat interacts with different receptors, including chemokine, NMDA, and G protein coupled receptors (Albini et al., 1998; Haughey et al., 2001; Prendergast et al., 2002; Eugenin et al., 2003; Feligioni et al., 2003; Longordo et al., 2006; Shin et al., 2012) leading to deficits in neurotransmission (Sabatier et al., 1991; Behnisch et al., 2004; Brailoiu et al., 2008; Hargus and Thayer, 2013), excitotoxic events (Haughey et al., 1999; Feligioni et al., 2003), synaptodendritic injury (Bertrand et al., 2014), and eventually neuronal death. Although synergism has been demonstrated between opioids and Tat on the glutamatergic system (Gurwell et al., 2001; Gupta et al., 2010; Zou et al., 2011; Fitting et al., 2014; Liu et al., 2016), not much is known about how HIV-1 Tat interacts with opioids on the GABAergic neurotransmitter system and whether the exacerbating effects of opioids in the presence of Tat can also be attributed to its effects on the GABAergic system. It is well known that opioids inhibit GABAergic neurotransmission in the striatum (Johnson and North, 1992; Klitenick et al., 1992; Bergevin et al., 2002) and recently the GABAergic inhibitory system has also been implicated in neuroAIDS, with the GABAergic inhibitory system being downregulated in HIV-1-positive patients with HAND (Gelman et al., 2012; Buzhdygan et al., 2016). In experimental studies it is less clear as Tat has been reported to increase inhibitory synapses and neurotransmission (Brailoiu et al., 2008; Hargus and Thayer, 2013), whereas other studies have reported unchanged or decreased inhibitory synapses or inhibitory release (Musante et al., 2010; Zucchini et al., 2013; Xu et al., 2016).
Thus, the present study investigated the effects of HIV-1 Tat alone and in combination with opioids by focusing on the GABAergic system. Results indicated a Tat and opioid-induced decrease in GABAergic neurotransmission via μ-opioid receptors with the combined effects of Tat and morphine involving more than just μ-opioid receptor-related mechanisms. Exploring the interactions between Tat and opioids on multiple neurotransmitter systems is important in understanding its effects on neuronal function and neuronal network processing.
Materials and Methods
Electrophysiology
Slice Preparation
All animal experiments were performed according to protocols approved by the Animal Care and Use Committee the University of North Carolina at Chapel Hill. Striatal slices were prepared from 14 to 24-day-old male and female C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME). Mice were euthanized using isoflurane, decapitated, and brains were placed into ice-cold sucrose buffer containing (in mM): 254 sucrose, 10 D-glucose, 26 NaHCO3, 2 CaCl2, 2 MgSO4, 3 KCl, and 1.25 NaH2PO4, saturated with 95% O2/5% CO2, at pH 7.4, 300 mOsm. Brains were cut into 300 μM coronal sections through the striatum using a VT 1000S microtome (Leica, Deerfield, IL) and placed into a holding chamber at 33°C for 30 min in a mixture of 50% sucrose saline and 50% artificial cerebrospinal fluid (aCSF) containing (in mM): 128 NaCl, 10 D-glucose, 26 NaHCO3, 2 CaCl2, 2 MgSO4, 3 KCl, and 1.25 NaH2PO4. Slices were then maintained at room temperature in aCSF bubbled continuously with 95% O2/5% CO2.
Recordings and Analyses
Slices were transferred to a recording chamber (Warner Instruments, Hamden, CT) and continuously perfused with aCSF at 2–3 mL/min at ~33°C (Warner SC-20, Hamden, CT). Recordings were made in striatal medium spiny neurons (MSNs) located in the dorsolateral striatum that mainly integrate sensorimotor information (Pennartz et al., 2009) and are specifically affected in HIV-1 positive individuals (Bauer et al., 2005). Striatal MSNs were visualized using an Axio Examiner A1 microscope (Zeiss, Thornwood, NY) equipped with using differential interference contrast (DIC) and Dodt contrast. Patch-clamp recordings in whole-cell mode were obtained using a MultiClamp 700B amplifier (Axon Instruments, Union City, CA) and digitized using Digidata 1550A and pClamp 10.0 software (Molecular Devices, Sunnyvale, CA). Patch pipettes were glass capillaries with a filament (Narishige, Greencale, NY) pulled using a PC-10 puller (Narishige, Greencale, NY, #GD-1.2). The pipette internal solution used for whole-cell patch-clamp experiments consisted of (in mM) unless otherwise stated: 140 KCl, 0.1 CaCl2, 5 EGTA, 10 HEPES, 4 ATP-Mg2+, 0.4 GTP-2Na+, 1 QX314 (Lidocaine N-ethyl bromide), pH 7.2, 290 mOsm. The tip resistance of the patch electrode filled with internal solution was ~5 MΩ. Recordings were conducted immediately following treatment application and recording was conducted over a 5 min time period. Inhibitory postsynaptic currents (IPSCs) were recorded in the presence of the ionotropic glutamate receptor blockers 6,7-dinitroquinoxaline-2,3-dione (DNQX, 20 μM) and 2-amino-5-phosphopentanoate (AP-5; 20 μM), added to the superfusing aCSF. Spontaneous (s) and miniature (m) IPSCs were recorded at a holding potential of −70 mV and collected for 5 min for each treatment. The mIPSC recordings were obtained in the presence of 1 μM tetrodotoxin (TTX). Series resistance was monitored throughout the experiment. If the series resistance was unstable and changed >15% during the experiment, the cell was discarded. Signals were filtered at 2 kHz and digitized at 10 kHz. The Minianalysis software (Version 6.0.8; Synaptosoft, Decatur, GA) was used to perform off-line analysis.
Treatments
HIV-1 Tat1−86 (5–50 nM, rtat HIV-1 IIIB, ImmunoDX, Woburn, MA), morphine sulfate (1 μM, NIDA Drug Supply System, Baltimore, MD) damgo (1 μM, Tocris, Ellisville, MO), naloxone (1 μM, Sigma-Aldrich, St. Louis, MO), and CTAP (D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2; 1 μM, Tocris, Ellisville, MO) were dissolved in distilled water and administered by bath application. For the control we used bath application of distilled water by itself. As an additional control for Tat, we used heat-inactivated Tat1−86. Tat1−86 (50 nM) was heat-inactivated by incubation at 85°C for 30 min. Even though the concentration of Tat in the cerebral spinal fluid (CSF) has been reported at 1.14 nM (16 ng/mL) (Westendorp et al., 1995), the Tat concentration utilized in this study is generally accepted (Mayne et al., 2000; Haughey et al., 2001; Prendergast et al., 2002; Speth et al., 2002; Singh et al., 2004; Wallace et al., 2006; Brailoiu et al., 2008; Fitting et al., 2014). Opioid concentrations were based on previous studies (Johnson and North, 1992; Bergevin et al., 2002; McQuiston, 2007). DNQX (6,7-dinitroquinoxaline-2,3-dione, 20 μM), AP-5 (DL-2-amino-5-phosphonovaleric acid, 20 μM), and TTX (tetrodotoxin, 1 μM) were purchased from Tocris (Ellisville, MO). To manipulate levels we used three different experimental conditions: (1) aCSF without calcium, (2) aCSF with cadmium chloride (CdCl2, 200 μM, Sigma-Aldrich, St. Louis, MO), which blocks high and low threshold voltage-dependent calcium channels, and (3) endoplasmic reticulum calcium pump inhibitor thapsigargin (1 μM, Sigma-Aldrich, St. Louis, MO) that depletes the intracellular calcium stores.
Statistics
All numerical values are expressed as mean ± standard error of the mean (SEM). Data were analyzed using paired Student's t-tests. An alpha level of p < 0.05 was considered significant.
Results
Tat Concentration Dependent Effects on IPSCs in Striatal Slices
To explore the effects of Tat on GABergic neurotransmission patch-clamp recordings were performed on striatal MSNs (Figure 1). sIPSCs and mIPSCs were confirmed by the application of GABAA receptor antagonist bicuculline (data not shown). The representative traces of sIPSCs before and after Tat application (5–50 nM) are shown in Figure 1A. As shown in Figure 1B, the mean (±SEM) frequency of sIPSCs (Hz) were as follows - for control: 1.34 ± 0.39, for Tat (5 nM): 1.03 ± 0.28, for Tat (10 nM): 0.91 ± 0.22, for Tat (50 nM): 0.81 ± 0.18; n = 13. A significant Tat effect was noted on sIPSCs, with all Tat concentrations demonstrating decreased mean frequency of sIPSCs compared to control [control vs. Tat (5 nM): t(12) = 2.6, p = 0.023, control vs. Tat (10 nM): t(12) = 2.5, p = 0.027, control vs. Tat (50 nM): t(12) = 2.6, p = 0.023] and Tat (10 nM) significantly differing from Tat [50 nM; t(12) = 2.3 p = 0.039]. As depicted in Figure 1C, the mean (±SEM) amplitude of sIPSCs (pA) were as follows - for control: 37.40 ± 4.09, for Tat (5 nM): 41.30 ± 3.94, for Tat (10 nM): 43.82 ± 4.00, for Tat (50 nM): 45.98 ± 3.74; n = 13. In contrast to mean sIPSC frequency, the mean amplitude of sIPSCs indicated only a significant increase by Tat (50 nM) compared to control [t(12) = 2.7, p = 0.018] and compared to Tat (5 nM) [t(12) = 2.5, p = 0.027]. To assess mIPSCs, TTX was added to the bath to eliminate large-amplitude, action potential-dependent IPSCs. As shown in Figure 1D, the mean (±SEM) frequency of mIPSCs (Hz) were as follows - for control: 1.10 ± 0.36, for Tat (5 nM): 1.06 ± 0.35, for Tat (10 nM): 0.92 ± 0.32, for Tat (50 nM): 0.79 ± 0.30; n = 15. A concentration dependent decrease in the frequency of mIPSCs by Tat was noted in the presence of TTX, with all conditions indicating significance from each other [control vs. Tat (10 nM): t(14) = 2.7, p = 0.018, control vs. Tat (50 nM): t(14) = 2.7, p = 0.016, Tat (5 nM) vs. Tat (10 nM): t(14) = 2.3, p = 0.040, Tat (5 nM) vs. Tat (50 nM): t(14) = 2.4, p = 0.031, Tat (10 nM) vs. Tat (50 nM): t(14) = 2.3, p = 0.039], except for Tat (5 nM) compared to control. As depicted in Figure 1E, the mean (±SEM) amplitude of mIPSCs (pA) were as follows - for control: 34.18 ± 2.46, for Tat (5 nM): 32.14 ± 2.68, for Tat (10 nM): 30.72 ± 3.37, for Tat (50 nM): 29.23 ± 3.52; n = 15. The mean amplitude of mIPSCs indicated a significant decrease by Tat (50 nM) compared to control [t(14) = 2.2, p = 0.049]. Heat-inactivated Tat (50 nM) revealed no significant effects on sIPSCs (n = 9 neurons, Figure 1F) and mIPSCs (n = 10 neurons, Figure 1G). Data were normalized to 100% for control and indicated following percent values for heat-inactivated Tat (50 nM): sIPSC frequency: 84.61 ± 5.97, sIPSC amplitude: 93.71 ± 3.73, mIPSC frequency: 96.54 ± 5.17, mIPSC amplitude 92.22 ± 4.04. Overall, Tat produced a significant decrease in action potential-dependent IPSCs (sIPSCs) and action potential-independent IPSCs (mIPSCs), with altering GABAergic neurotransmission presynaptically and partially postsynaptically, depending on Tat concentration. In the subsequent experiments Tat (10 nM) was used over Tat (50 nM) as 10 nM is closer to mimicking the physiological condition compared to 50 nM of Tat.
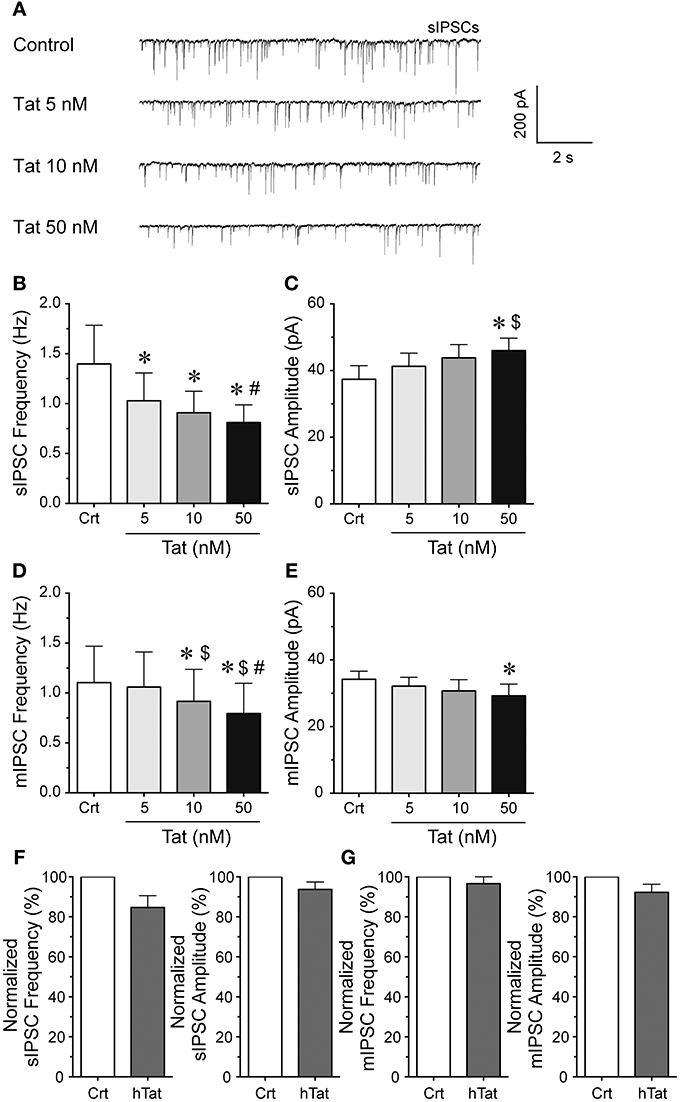
Figure 1. Tat concentration-dependently decreased the frequency of sIPSCs and mIPSC in striatal MSNs. (A) Representative traces show sIPSCs before and after application of Tat concentrations (5–50 nM). (B) Tat (5–50 nM) concentration-dependently decreased the mean frequency of sIPSCs (n = 13 neurons). (C) For the mean amplitude of sIPSCs only the highest Tat concentration (50 nM) increased the mean amplitude of sIPSCs (n = 13 neurons). (D) For mIPSCs, the mean frequency was concentration-dependently decreased by Tat (5–50 nM) (n = 15 neurons), whereas (E) the mean amplitude of mIPSCs showed no Tat effects except for a decrease by the highest Tat (50 nM) concentration (n = 15 neurons). (F) No effects for heat-inactivated Tat (50 nM) were noted on the normalized data of the mean frequency and mean amplitude of sIPSCs (n = 9 neurons). (G) No effects for heat-inactivated Tat (50 nM) were noted on the normalized data of the mean frequency and mean amplitude of mIPSCs (n = 10 neurons). Data are mean ± SEM. Significance was assessed by paired Student t-tests. *p < 0.05 vs. Control,#p < 0.05 vs. Tat (10 nM), $p < 0.05 vs. Tat (5 nM). Crt, Control; hTat, heat-inactivated Tat; MSNs, medium spiny neurons.
Effects of Morphine and Damgo in Combination with Tat
It is known that acute bath application of opioids, such as morphine, decrease GABAergic neurotransmission in different brain regions (Bajo et al., 2014; Bobeck et al., 2014; Yousefpour et al., 2014), however, the combined effects of opioids and Tat on GABA release are less clear. Thus, we examined the effects of opioids and Tat on IPSCs by performing patch-clamp recordings on striatal MSNs with bath application of morphine (1 μM) or a highly selective μ-opioid agonist damgo (1 μM) followed by Tat (10 nM) administration (Figure 2). The representative traces of sIPSCs for control, morphine (1 μM), and morphine + Tat (10 nM) are shown in Figure 2A. As shown in Figure 2B (right Panel), the mean (±SEM) frequency of sIPSCs (Hz) were as follows - for control: 1.77 ± 0.69, for morphine (1 μM): 1.37 ± 0.64, for morphine + Tat (10 nM): 0.90 ± 0.29; n = 9. Morphine significantly reducing sIPSC frequency compared to control [t(8) = 3.5, p = 0.008] with no further decrease by Tat (10 nM). As depicted in Figure 2B (left Panel), the mean (±SEM) amplitude of sIPSCs (pA) were as follows - for control: 32.20 ± 2.90, for morphine (1 μM): 33.68 ± 3.43, for morphine + Tat (10 nM): 35.93 ± 4.07; n = 9. No effects were noted on the mean amplitude of sIPSCs (Figure 2B). As shown in Figure 2C (right Panel), the mean (±SEM) frequency of mIPSCs (Hz) were as follows - for control: 0.70 ± 0.11, for morphine (1 μM): 0.61 ± 0.11, for morphine + Tat (10 nM): 0.57 ± 0.09; n = 7. For mIPSC frequency a significant morphine-induced decrease of mIPSC frequency was note compared to control [t(6) = 3.3, p = 0.016, n = 7 neurons, Figure 2C], with being further downregulated by Tat [morphine (1 μM) vs. morphine + Tat (10 nM): t(6) = 2.7, p = 0.034]. As depicted in Figure 2C (left Panel), the mean (±SEM) amplitude of mIPSCs (pA) were as follows - for control: 36.53 ± 3.04, for morphine (1 μM): 35.09 ± 2.01, for morphine + Tat (10 nM): 37.14 ± 2.55; n = 7. No effects were noted for the mean amplitude of mIPSCs (Figure 2C). As morphine acts on multiple subtypes of opioid receptors, we tested the selective μ-opioid receptor agonist damgo (1 μM). Similar effects were noted for damgo as seen with morphine. The representative traces of mIPSCs for control, damgo (1 μM), and damgo + Tat (10 nM) are shown in Figure 2D. As shown in Figure 2E (right Panel), the mean (±SEM) frequency of sIPSCs (Hz) were as follows - for control: 1.14 ± 0.27, for damgo (1 μM): 0.67 ± 0.19, for damgo + Tat (10 nM): 0.66 ± 0.20; n = 9. Damgo alone and damgo + Tat significantly decreased the mean frequency of sIPSC compared to control [t(8) = 2.4, p = 0.045 and t(8) = 2.4, p = 0.045, respectively]. As depicted in Figure 2E (left Panel), the mean (±SEM) amplitude of sIPSCs (pA) were as follows - for control: 33.08 ± 4.54, for damgo (1 μM): 38.40 ± 4.45, for damgo + Tat (10 nM): 33.69 ± 4.84, n = 9. No further downregulation of sIPSC frequency by Tat (10 nM) administration following damgo bath application was noted. As shown in Figure 2F (right Panel), the mean (±SEM) frequency of mIPSCs (Hz) were as follows - for control: 0.81 ± 0.16, for damgo (1 μM): 0.37 ± 0.06, for damgo + Tat (10 nM): 0.35 ± 0.06; n = 11. Similarly, damgo alone and damgo + Tat reduced the mean frequency of mIPSCs compared to control [t(10) = 2.6, p = 0.027 and t(10) = 2.7 p = 0.022, respectively]. As depicted in Figure 2F (left Panel), the mean (±SEM) amplitude of mIPSCs (pA) were as follows - for control: 28.77 ± 1.87, for damgo (1 μM): 30.41 ± 1.71, for damgo + Tat (10 nM): 30.42 ± 1.77, n = 11. No effects were noted on the mean amplitude of sIPSCs (Figure 2E) and mIPSCs (Figure 2F). Thus, morphine and damgo decrease GABAergic neurotransmission probably via a pre-synaptic mechanism as the mean amplitude of IPSCs was not significantly affected by opioids. Additionally, combined treatment of morphine and Tat further decreased action potential-independent GABA release presynaptically, which was not noted with combined damgo and Tat treatment.
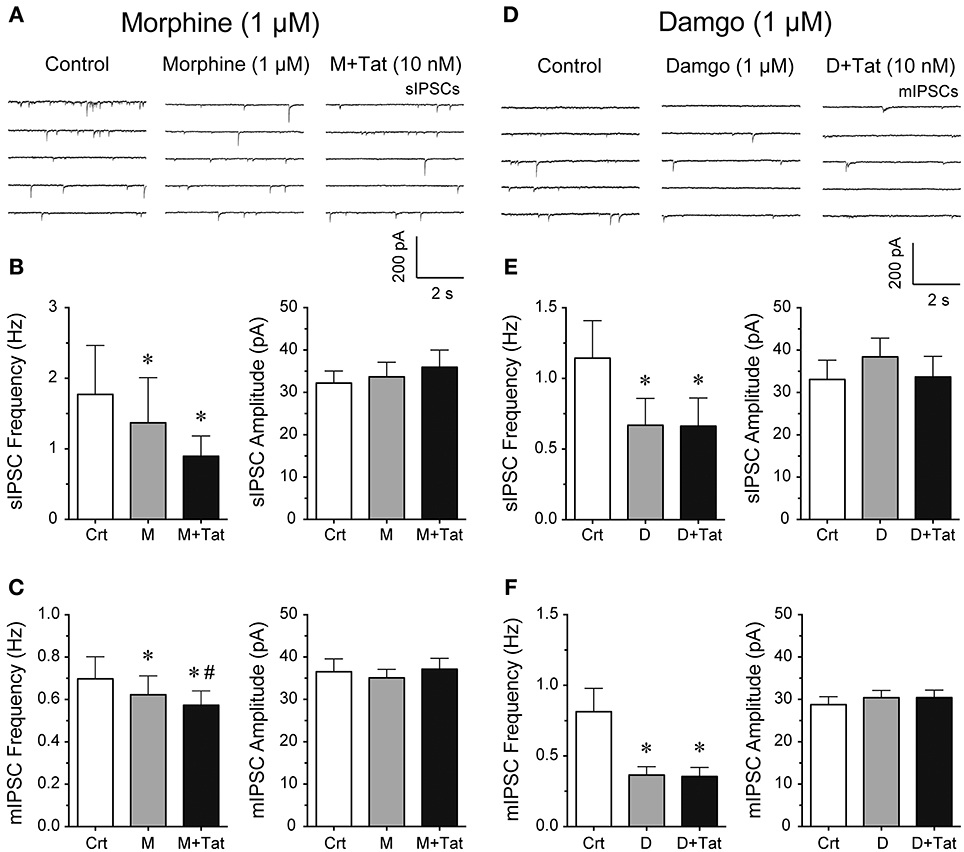
Figure 2. Morphine and damgo significantly decreased the frequency of IPSCs with Tat inducing a further depression of mIPSCs only in combination with morphine. (A–C) Morphine (1 μM) and Tat (10 nM) effects on IPSCs. (A) Representative traces show sIPSCs before morphine, after morphine, and after morphine + Tat application. (B) Morphine significantly decreased the mean frequency of sIPSCs, with morphine + Tat not changing the morphine-induced decrease in frequency of sIPSCs (n = 9 neurons). No effects were noted on the mean amplitude of sIPSCs (n = 9 neurons). (C) Morphine significantly decreased the mean frequency of mIPSCs with a further depression by Tat administration after morphine treatment (n = 7 neurons). No effects were noted on the mean amplitude of mIPSCs (n = 7 neurons). (D–F) Damgo (1 μM) and Tat (10 nM) effects on IPSCs. (D) Representative traces show mIPSCs before damgo, after damgo, and after damgo + Tat application. (E) Damgo significantly decreased the mean frequency of sIPSCs, with damgo + Tat not changing the damgo-induced decrease in frequency of sIPSCs (n = 9 neurons). No effects were noted on the mean amplitude of sIPSCs (n = 9 neurons). (F) Damgo significantly decreased the mean frequency of mIPSCs with no further depression of mIPSC frequency following Tat administration after damgo treatment (n = 11 neurons). No effects were noted on the mean amplitude of mIPSCs (n = 11 neurons). Data are mean ± SEM. Significance was assessed by paired Student t-tests. *p < 0.05 vs. Control, #p < 0.05 vs. M. Crt, Control; M, Morphine; D, Damgo.
Combined Tat and Morphine Effects Are Blocked by Naloxone and Partially by CTAP
To examine more in detail through which receptor the combined Tat and morphine-induced decrease in GABAergic neurotransmission is mediated, striatal slices were pretreated with naloxone (1 μM), a high affinity antagonist for the μ-opioid receptor but also some affinity for the κ-, and δ-opioid receptors, or CTAP (1 μM), a selective μ-opioid receptor antagonist (Figure 3). The representative traces of sIPSCs for naloxone (1 μM) followed by Tat (10 nM) and then morphine (1 μM) administration are shown in Figure 3A. As shown in Figure 3B, the mean (±SEM) frequency of sIPSCs (Hz) were as follows - for control: 0.48 ± 0.06, for naloxone (1 μM): 0.49 ± 0.07, for naloxone + Tat (10 nM): 0.49 ± 0.06, for naloxone + Tat + morphine (1 μM): 0.52 ± 0.07. Pretreating striatal slices with the opioid antagonist naloxone (1 μM) revealed no significant effects for Tat (10 nM) or combined Tat and morphine (1 μM) treatment on the mean frequency of sIPSCs (n = 12 neurons, Figure 3B) and the mean frequency of mIPSCs (n = 10 neurons, Figure 3C). As shown in Figure 3C, the mean (±SEM) frequency of mIPSCs (Hz) were as follows - for control: 0.45 ± 0.09, for naloxone (1 μM): 0.44 ± 0.08, for naloxone + Tat (10 nM): 0.44 ± 0.10, for naloxone + Tat + morphine (1 μM): 0.45 ± 0.09. No significant effects were noted on the mean amplitude of sIPSCs and mIPSCs (data not shown). To test if the combined Tat (10 nM) and morphine (1 μM) effects were μ-opioid receptor specific, CTAP (1 μM) was used as a pretreatment (Figures 3D–F). The representative traces of mIPSCs for CTAP (1 μM) followed by Tat (10 nM) and then morphine (1 μM) administration are shown in Figure 3D. As shown in Figure 3E, the mean (±SEM) frequency of sIPSCs (Hz) were as follows - for control: 0.70 ± 0.17, for CTAP (1 μM): 0.66 ± 0.18, for CTAP + Tat (10 nM): 0.63 ± 0.14, for CTAP + Tat + morphine (1 μM): 0.65 ± 0.20. Similar to naloxone, CTAP (1 μM) revealed no significant effects for Tat (10 nM) or combined Tat and morphine (1 μM) treatment on the mean frequency of sIPSCs (n = 11 neurons, Figure 3E). As shown in Figure 3F, the mean (±SEM) frequency of mIPSCs (Hz) were as follows - for control: 0.51 ± 0.07, for CTAP (1 μM): 0.50 ± 0.07, for CTAP + Tat (10 nM): 0.48 ± 0.07, for CTAP + Tat + morphine (1 μM): 0.44 ± 0.06. Interestingly, whereas CTAP (1 μM) blocked the significant effects of Tat (10 nM) on the mean frequency of sIPSCs, the significant effect of combined Tat and morphine (1 μM) treatment on mIPSC frequency was not blocked [CTAP (1 μM) + Tat (10 nM) vs. CTAP + Tat + morphine (1 μM) t(9) = 3.0, p = 0.016, n = 10 neurons, Figure 3F]. No significant effects were noted on the mean amplitude of sIPSCs and mIPSCs (data not shown). Thus, the significant decrease of GABA release with combined Tat and morphine treatment appears to involve predominantly μ-opioid receptor-related mechanisms.
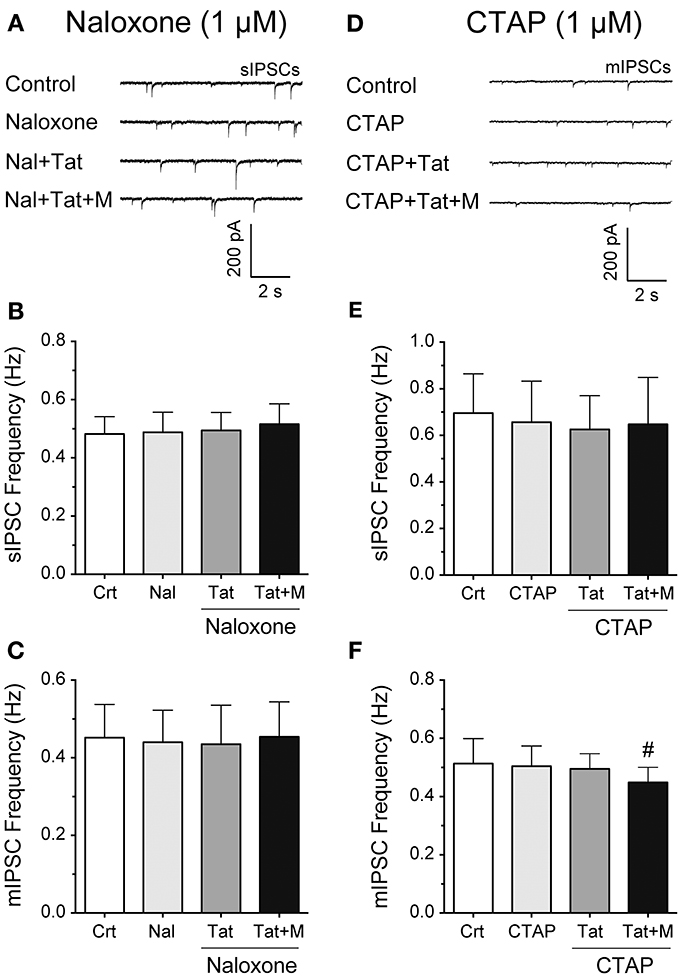
Figure 3. Naloxone and partially CTAP blocked the effects of Tat and/or morphine on the frequency of IPSCs. (A–C) Naloxone (1 μM), Tat (10 nM), and morphine (1 μM) effects on the mean frequency of IPSCs. (A) Representative traces show sIPSCs before and after naloxone treatment, followed by Tat, and morphine application. (B) No significant effects were noted on the mean frequency of sIPSCs for Tat or combined Tat and morphine treatment in the presence of naloxone (n = 12 neurons). (C) Similarly, in the presence of naloxone, no significant effects were noted on the mean frequency of mIPSCs for Tat (10 nM) or combined Tat and morphine treatment (n = 10 neurons). (D–F) CTAP (1 μM), Tat (10 nM) and morphine (1 μM) effects on the mean frequency of IPSCs. (D) Representative traces show mIPSCs before and after CTAP treatment, followed by Tat, and morphine (1 μM) application. (E) No significant effects were noted on the mean frequency of sIPSCs for Tat or combined Tat and morphine treatment in the presence of CTAP (n = 11 neurons). (F) In the presence of CTAP, combined Tat and morphine treatment significantly downregulated the mean frequency of mIPSCs compared to Tat alone (n = 10 neurons). Data are mean ± SEM. Significance was assessed by paired Student t-tests. #p < 0.05 vs. CTAP + Tat. Crt, Control; M, Morphine.
Tat Effects on IPSCs Depend on Extracellular and Intracellular Calcium
To understand the mechanisms by which Tat decreases GABAergic synaptic neurotransmission, we examined the involvement of extracellular calcium, voltage-gated calcium channels, and intracellular calcium (Figure 4). Manipulating extracellular and intracellular calcium decreased the frequency of sIPSCs by 33.3% when removing extracellular calcium from the aCSF [control vs. 0 Ca2+ control: t(12) = 4.1, p < 0.001, n = 13 neurons], by 43.9% when blocking the voltage-gated calcium channels with CdCl2 [200 μM; control vs. CdCl2 control: t(9) = 3.5, p = 0.006, n = 10 neurons], and by 26.9% when depleting intracellular calcium stores with the endoplasmic reticulum calcium pump inhibitor thapsigargin [control vs. thapsigargin control: t(9) = 2.3, p = 0.050, n = 10 neurons; Figure 4A]. Importantly, whereas Tat (10 nM; 0.91 ± 0.22) significantly reduced the frequency of sIPSCs in the presence of normal aCSF (1.34 ± 0.39) [t(12) = 2.5, p = 0.027, n = 13 neurons], no Tat effects were noted when extracellular calcium was removed from the aCSF [n = 13 neurons; control: 0.61 ± 0.09, Tat (10 nM): 0.56 ± 0.07], voltage-gated calcium channels were blocked with CdCl2 [200 μM; n = 10 neurons; control: 0.67 ± 0.17, Tat (10 nM): 0.61 ± 0.16], or intracellular calcium stores were depleted from calcium with thapsigargin [1 μM; n = 9 neurons; control: 0.42 ± 0.10, Tat (10 nM): 0.46 ± 0.10; Figure 4A], indicating that extracellular and intracellular calcium are necessary for the Tat-induced decrease in sIPSC frequency. No significant overall treatment effects were noted for the amplitude of sIPSCs for any condition (Figure 4B). As shown in Figure 4B, the mean (±SEM) amplitude of sIPSCs (pA) for the different conditions were as follows—control condition: normal aCSF: 37.40 ± 4.09, Tat (10 nM): 43.82 ± 4.00; zero extracellular Ca2+: aCSF: 30.24 ± 1.55, Tat (10 nM): 29.99 ± 1.85; CdCl2: aCSF: 27.69 ± 1.97, Tat (10 nM): 24.16 ± 2.10; thapsigargin: aCSF: 30.24 ± 2.05, Tat (10 nM): 28.35 ± 1.55. For mIPSCs, the representative traces before and after Tat application for each condition are shown in Figure 4C. Similar to effects on sIPSCs, Tat (10 nM; 0.92 ± 0.32) significantly downregulated the frequency of mIPSCs in normal aCSF (1.10 ± 0.36) [t(14) = 2.7, p = 0.018, n = 15 neurons], but no significant Tat effects were noted in the absence of extracellular calcium [n = 8 neurons; control: 0.55 ± 0.09, Tat (10 nM): 0.49 ± 0.07], in the presence of CdCl2 [200 μM; n = 9 neurons; control: 0.50 ± 0.31, Tat (10 nM): 0.54 ± 0.31], and in the presence of thapsigargin [1 μM; n = 10 neurons; control: 0.32 ± 0.06, Tat (10 nM): 0.30 ± 0.06; Figure 4D]. Further, no significant Tat (10 nM) effects were noted for the amplitude of mIPSCs (Figure 4E). As shown in Figure 4E, the mean (±SEM) amplitude of mIPSCs (pA) for the different conditions were as follows—control condition: normal aCSF: 34.18 ± 2.46, Tat (10 nM): 30.72 ± 3.37; zero extracellular Ca2+: aCSF: 28.36 ± 1.46, Tat (10 nM): 27.69 ± 1.39; CdCl2: aCSF: 22.83 ± 1.37, Tat (10 nM): 21.66 ± 1.39; thapsigargin: aCSF: 25.90 ± 1.76, Tat (10 nM): 24.47 ± 2.32. Thus, the significant Tat (10 nM) effects on the frequency of IPSCs were abolished when removing extracellular and/or intracellular calcium, indicating that extracellular and intracellular calcium are necessary for the Tat-induced decrease in GABA release.
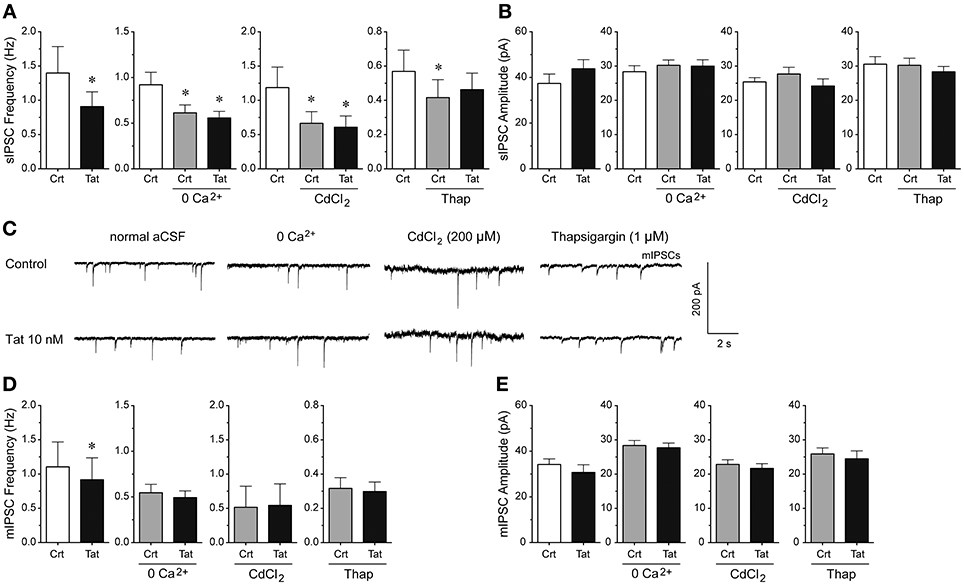
Figure 4. Extracellular and intracellular calcium contribute to the significant Tat (10 nM) effects on the mean frequency of IPSCs. (A) The significant Tat (10 nM) effect on the mean frequency of sIPSCs (n = 13 neurons) was blocked by zero extracellular Ca2+ (n = 13 neurons), CdCl2 (200 μM, n = 10 neurons), and thapsigargin (1 μM, n = 10 neurons). (B) No Tat effects were noted on the mean amplitude of sIPSCs. (C) Representative traces show sIPSCs before and after application of Tat (10 nM) in the presence of normal aCSF, zero extracellular Ca2+, CdCl2 (200 μM), and thapsigargin (1 μM). (D) The significant Tat (10 nM) effect on the mean frequency of mIPSCs (n = 15 neurons) was blocked by zero extracellular Ca2+ (n = 8 neurons), CdCl2 (200 μM, n = 9 neurons), and thapsigargin (1 μM, n = 10 neurons). (E) No Tat effects were noted on the mean amplitude of mIPSCs in any condition. Data are mean ± SEM. Significance was assessed by paired Student t-tests. *p < 0.05 vs. Control of corresponding condition. aCSF, artificial cerebrospinal fluid; Crt, Control; Thap, Thapsigargin.
Discussion
In the present study, we investigated (1) the HIV-1 Tat effects on the GABAergic neurotransmitter system, (2) the combined effects of Tat and opioids on the GABAergic system, and (3) the underlying mechanisms by which Tat affects GABAergic neurotransmission.
Tat Concentration-Dependently Decreases GABAergic Neurotransmission
It is well known that Tat is excitotoxic by activating glutamatergic NMDA receptors (Magnuson et al., 1995; Haughey et al., 2001; Pérez et al., 2001; Longordo et al., 2006; Li et al., 2008; Aksenov et al., 2012). Further, Tat has been shown to directly affect glutamatergic neurotransmission by increasing the frequency of miniature excitatory postsynaptic currents (mEPSCs) (Brailoiu et al., 2008) and neuronal excitability (Ngwainmbi et al., 2014; Fitting et al., 2015). However, as glutamate and GABA are the most abundant neurotransmitters in the brain, neuronal excitability is critically dependent on the level of inhibition, and accordingly changes of inhibitory synaptic efficacy has great impact on neuronal function and neuronal network processing (Petroff, 2002).
The present study provides direct evidence that Tat concentration-dependently decreases inhibitory transmitter release in the dorsolateral striatum. The observation that Tat alters sIPSCs and mIPSCs in medium spiny GABAergic projection neurons (MSNs) indicates that Tat has effects on intrinsic interneuron excitability and modifies the GABA release machinery. Specifically, our data suggest that action potential (AP)-independent IPSCs (mIPSCs) contribute mostly to overall IPSCs in our MSN recordings. It is known that AP-independent spontaneous vesicular release of GABA in the CNS mediates mIPSCs (Axmacher et al., 2004). Another source of mIPSCs is known to be the reversal of GABA transporter (GAT), which may result in additional GABA release under certain physiological conditions (Allen et al., 2004; Wu et al., 2007; Jin et al., 2011). Further, as in the present study glutamate receptor blockers were present in the bath, the GABA component of the AP-dependent synaptic potential, which arises largely from collateral branches of the MSNs themselves, was inhibited, thus contributing to smaller AP-dependent GABA release (sIPSCs). The marked reduction of the mean frequency of IPSCs by Tat (10 nM), without any change in the mean amplitude of IPSCs, suggests that Tat acts at presynaptic GABAergic terminals and suppresses inhibitory synaptic input to MSNs. As a decrease in inhibitory synaptic inputs would facilitate an increase in postsynaptic activity, the presynaptic inhibition of IPSCs may play a role in the excitatory effects of Tat on the activities of MSNs, besides direct excitation through postsynaptic actions (Kim et al., 2008).
Interestingly, in the present study, the highest concentration of Tat (50 nM) altered the amplitude of IPSCs, suggesting that Tat may modulate IPSCs also through postsynaptic mechanisms, such as alterations in number of postsynaptic receptors, postsynaptic sensitivity or conductance. The discrepancy of Tat (50 nM) increasing the amplitude of sIPSCs but decreasing the amplitude of mIPSCs is not well understood. For the increased Tat effects noted on the sIPSC amplitude, AP-driven events may play a role due to intrinsic properties of the presynaptic cell or network activity. It is suggested that tonically active GABAergic interneurons might contribute to the Tat-induced increase in sIPSC amplitude as fast-spiking parvalbumin interneurons in the pyramidal layer have been demonstrated to be selectively vulnerable to Tat (Marks et al., 2016). Additionally, other types of GABAergic interneurons make connections with the spiny neurons, including interneurons that express tyrosine hydroxylase (Xenias et al., 2015) and neuropeptide Y (Beatty et al., 2012). As we did notice a decrease in sIPSC frequency, it should be noted that the probability of opening also depends upon how much GABA is released (presynaptic), which influences the sIPSC frequency. In contrast, mIPSC amplitude is AP-independent, thus Tat effects on postsynaptic receptors, such as single channel conductance through an open GABA pore, or a change in the number of channels could contribute to a postsynaptic decrease in mIPSC amplitude. Further, the decrease in mIPSC amplitude could be due to the internalization of specific receptors. TNF-α is known to causes internalization of GABAA receptors, resulting in fewer surface GABAA receptors and a decrease in inhibitory synaptic strength (Stellwagen et al., 2005). TNF-α has been shown to play a central role in initiating inflammatory cascades in Tat-exposed astroglia and microglia (Benveniste and Benos, 1995; Luo et al., 2003; El-Hage et al., 2008) as well as in HIV-infected patients (Zhao et al., 2014). It is important to note however, that the effect on amplitude was only noticed for the high Tat (50 nM) concentration, in contrast to Tat (10 nM) that did not produce significant effects on ISPC amplitude.
Further, the effects of Tat on the inhibitory GABAergic system indicate to be variable with multiple studies reporting different results (Brailoiu et al., 2008; Fitting et al., 2013; Hargus and Thayer, 2013; Zucchini et al., 2013; Marks et al., 2016). A recent study demonstrated that Tat applied in vitro as well as expressed in the brain in vivo induced a decrease in GABA exocytosis in the cortex, while GABA exocytosis was unchanged in the hippocampus (Zucchini et al., 2013). Further, it appears that Tat has various effects on different inhibitory synaptic proteins, with dowregulating synaptotagmin 2 (Syt2) in the hippocampus of Tat transgenic mice (Fitting et al., 2013), whereas upregulating gephyrin in the hippocampus in vitro and in vivo (Fitting et al., 2013; Hargus and Thayer, 2013). Thus, depending on the brain region and the inhibitory synaptic input the GABAergic system might be differently affected by Tat.
Overall the present study demonstrates that Tat modulates inhibitory GABAergic influence on MSNs in the dorsolateral striatum through both presynaptic and postsynaptic mechanisms, depending on Tat concentration.
Differential Effects of Tat in Combination with Morphine or Damgo on the GABAergic System
Multiple studies have investigated the effects of opioids on GABAergic synaptic transmission with demonstrating that opioids inhibit GABA-mediated neurotransmission (Vaughan and Christie, 1997; Vaughan et al., 1997; Zhang et al., 2015). As reported in the present study it has been shown previously that opioids decrease the frequency of IPSCs but do not change the amplitude of IPSCs, indicating that neurotransmission is inhibited presynaptically (Vaughan and Christie, 1997; Vaughan et al., 1997). More interestingly, whereas a combined effect was noted for morphine and Tat on the frequency of mIPSCs, the selective μ-opioid receptor agonist damgo did not further downregulate mIPSC frequency in the presence of Tat. It is known that morphine and damgo, which primarily act on the μ-opioid receptors, have high efficacy in inhibiting calcium channels, whereas damgo but not morphine has high efficacy for p38 mitogen-activated protein kinase activation that regulates inflammation and is activated by cytokine production (Tibbles and Woodgett, 1999; Tan et al., 2009). As Tat has been shown to activate p38 mitogen-activated protein kinase (Li et al., 2005; Li and Lau, 2007; Gupta et al., 2010), differential caspase-dependent pathways could be used by Tat and morphine to inhibit GABA release and cause a downregulation when applied in combination.
Additionally, in contrast to damgo, morphine is not selective to μ-opioid receptors but also has effects on other opioid receptors. That other subtypes of opioid receptors besides the μ-opioid receptor seem to be involved in the combined opioid and Tat effects on GABAergic neurotransmission is supported by the finding that naloxone but not CTAP was able to block the combined opioid and Tat effects on GABAergic neurotransmission, specifically for mIPSC frequency. It should be noted that the μ-opioid receptor inhibition with CTAP blocked combined Tat and morphine-induced decrease in sIPSC frequency, but not in mIPSC frequency. The involvement of δ-opioid receptors has been shown in a previous study, indicating that a selective δ-opioid receptor agonist (D-Pen(2,5)-enkephalin, DPDPE) but not a selective κ-opioid receptors agonist [(+)-(5 alpha,7 alpha,8 beta)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide; U-69593] decreased IPSCs with a co-localization of δ-opioid receptors and dopamine 1 (D1)-type receptors in the rat dorsolateral striatum (Ambrose et al., 2006). Naloxone is a high affinity antagonist for the μ-opioid receptor but also has some affinity on the κ-, and δ-opioid receptors, whereas CTAP is a selective μ-opioid receptor antagonist. Previous studies have shown that κ-, and δ-opioid receptors are involved in the regulation of inhibitory transmission (Ford et al., 2006; Bosse et al., 2014; Gilpin et al., 2014). Specifically, δ-opioid receptors inhibit GABA release via a presynaptic site of action in the striatum (Jiang and North, 1992) and are localized mainly to presynaptic terminals of inhibitory synapses (Svingos et al., 1998). In addition to a regulating function, δ-opioid receptors have also been shown to downregulate GABAA receptor expression in the cortex of δ-opioid receptor transgenic mice (Feng et al., 2011). Besides morphine's effects on μ- opioid receptors but also on κ-, and δ-opioid receptors, it additionally has been shown that morphine can activate nociceptin/orphanin FQ peptide receptors (Ueda et al., 2000; Kest et al., 2001). The specific receptor that might contribute to the further downregulation of GABA release by combined Tat and morphine treatment can not be conclusively determined in the present study and needs further investigation.
Additionally, it needs to be pointed out that certain subpopulation of MSNs might show differential responsiveness to treatments, such as Tat and opioids. μ-opioid receptors are generally thought to reside in striosome (not matrix) MSNs (Cui et al., 2014). Even though μ-opioid receptors are expressed in both the D1-type MSNs of the direct pathway and D2-type MSNs of the indirect pathway, it appears that at least in some striosomes there is an overabundance of D1-type MSNs (Fujiyama et al., 2011; Watabe-Uchida et al., 2012; Cui et al., 2014), and further μ-opioid receptors might be specifically located in D1-MSNs of the direct pathway (Gerfen, 1985; Crittenden and Graybiel, 2011; Cui et al., 2014). Thus, the differential contribution of the D1-type MSNs and D2-type MSNs to the effects of morphine and damgo in the context of Tat exposure needs to be assessed in more detail.
Decrease of GABAergic Neurotransmission by Tat is Dependent on Extracellular and Intracellular Calcium
Studies have demonstrated that Tat alters calcium with inducing abnormal and excessive calcium influx and increasing intracellular calcium release that consequentially elevates cytosolic free calcium levels and leads to neurotoxicity (Haughey and Mattson, 2002; Hu, 2016). However, little research has been done to examine the involvement of calcium in the context of Tat on GABAergic synaptic neurotransmission. GABA has been shown to be released via calcium-dependent mechanisms that involve different subtypes of voltage-gated calcium channels (Alamilla and Gillespie, 2013; Nelson et al., 2014). The present study demonstrates that the observed Tat-induced decrease in GABAergic neurotransmission is dependent on extracellular and intracellular calcium. It is known that Tat increases calcium via inositol 1,4,5-trisphosphate (IP3)-regulated and/or ryanodine-regulated pools (Lipton, 1994; Nath and Geiger, 1998; Haughey et al., 1999; Fitting et al., 2014) with subsequent increases in calcium that enters the cell extracellularly via voltage-gated calcium channels (Bonavia et al., 2001). An increase in intracellular calcium could thus, account for the reduction in the amplitude of mIPSCs seen in the present study, as increases in intracellular calcium can lead to a suppression of the mean amplitude of IPSCs (Brussaard et al., 1996). Interestingly, there was an increase noted for the mean amplitude of sIPSCs by Tat (50 nM), indicating that the AP-dependent release of GABA from transmitter vesicles was decreased for frequency but increased for amplitude. As the increase in amplitude was only seen for sIPSCs and not mIPSCs the amplitude change could be related to a selective effect on intrinsic interneuron excitability and needs to be further studied.
The present study further suggests that the effects of Tat on GABA release appear to be mediated via μ-opioid receptors, as CTAP was able to block the Tat-induced decrease of GABA release. Opioid receptors are widely expressed in the striatum and the GABA modulation by μ-opioid receptors is consistent with anatomical data showing that neurons are immunopositive for μ-opioid receptors on pre- (axonal) and postsynaptic (dendritic) locations in different brain regions (Arvidsson et al., 1995; Drake and Milner, 1999). Additionally, it is known that μ-opioid receptors are extensively co-localized with paravalbumin-positive neurons (Drake and Milner, 2006), with a recent study demonstrating that neurons expressing parvalbumin in the pyramidal layer and neurons expressing somatostatin in stratum oriens are selectively vulnerable to Tat (Marks et al., 2016). Overall, the present study demonstrates that the Tat-induced decrease in GABAergic neurotransmission is dependent on extracellular and intracellular calcium, potentially involving a μ-opioid receptor-related pathway.
Conclusions
A summary of the findings of the present study is outlined in Figure 5. Our findings indicate that Tat as well as opioids, including morphine and damgo, separately, decreased AP-dependent and AP-independent GABA release in the striatum via a μ-opioid receptor-related mechanism, with Tat effects being dependent on extracellular and intracellular calcium. Combined Tat and opioid treatment revealed additive effects on GABA release only for Tat combined with morphine but not with damgo, which was blocked by naloxone but not CTAP. Based on the findings of the present study the decrease of GABA release by Tat and opioids is primarily mediated via μ-opioid receptors with additive effects on GABA release being potentially mediated via additional opioid receptors. Importantly, further studies are necessary to examine the effects of chronic Tat and opioid exposure on GABAergic neurotransmission to guide future research on HAND in the context of opiate drug use.
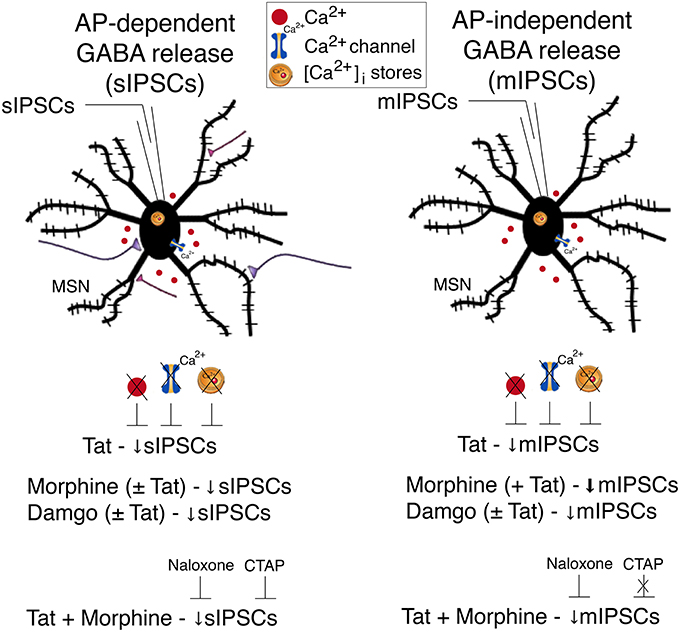
Figure 5. Summary of the effects on inhibitory GABAergic neurotransmission by HIV-1 Tat and opioids. Spontaneous inhibitory postsynaptic currents (sIPSCs) and miniature IPSCs (mIPSCs) were recorded from medium spiny neurons (MSNs) located in the dorsolateral striatum. A significant decrease in sIPSC frequency and mIPSC frequency was noted for Tat (10 nM) treatment. The significant Tat (10 nM) effect on the frequency of IPSCs was abolished when removing extracellular calcium (0 Ca2+), blocking voltage-dependent Ca2+ channels with cadmium chloride (CdCl2), and depleting intracellular endoplasmic reticulum calcium stores with thapsigargin, indicating that extracellular and intracellular calcium are necessary for the Tat-induced decrease in GABA release. For opioids, including morphine and damgo, separately and in combination with Tat, a significant decrease was noted for sIPSC frequency and mIPSC frequency, that was only potentiated for mIPSCs when combining Tat with morphine, but not damgo. Additionally, pretreatment with naloxone or CTAP prevented the combined Tat and morphine-induced decrease in sIPSCs frequency, whereas only naloxone, but not CTAP, prevented the combined Tat and morphine effect on mIPSCs frequency. It is hypothesized that the decrease of GABA release by Tat and opioids is primarily mediated via μ-opioid receptors with additive effects on GABA release being potentially mediated via additional opioid receptors. AP, action potential; MSN, medium spiny neuron; Ca2+, calcium; [Ca2+]i, intracellular calcium; sIPSC, spontaneous inhibitory postsynaptic currents; mIPSC, miniature inhibitory postsynaptic currents.
Ethics Statement
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Author Contributions
CX and SF designed the research; CX performed the electrophysiology experiments; CX and SF analyzed the data; CX and SF interpreted the data; SF wrote the paper; CX and SF discussed and edited the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge the support from the National Institute on Drug Abuse (NIDA R00 DA033878, R21 DA041903).
References
Aksenov, M. Y., Aksenova, M. V., Mactutus, C. F., and Booze, R. M. (2012). D1/NMDA receptors and concurrent methamphetamine + HIV-1 Tat neurotoxicity. J. Neuroimmune Pharmacol. 7, 599–608. doi: 10.1007/s11481-012-9362-3
Alamilla, J., and Gillespie, D. C. (2013). Maturation of calcium-dependent GABA, glycine, and glutamate release in the glycinergic MNTB-LSO pathway. PLoS ONE 8:e75688. doi: 10.1371/journal.pone.0075688
Albini, A., Ferrini, S., Benelli, R., Sforzini, S., Giunciuglio, D., Aluigi, M. G., et al. (1998). HIV-1 Tat protein mimicry of chemokines. Proc. Natl. Acad. Sci. U.S.A. 95, 13153–13158. doi: 10.1073/pnas.95.22.13153
Allen, N. J., Karadottir, R., and Attwell, D. (2004). Reversal or reduction of glutamate and GABA transport in CNS pathology and therapy. Pflugers Arch. 449, 132–142. doi: 10.1007/s00424-004-1318-x
Ambrose, L. M., Gallagher, S. M., Unterwald, E. M., and Van Bockstaele, E. J. (2006). Dopamine-D1 and delta-opioid receptors co-exist in rat striatal neurons. Neurosci. Lett. 399, 191–196. doi: 10.1016/j.neulet.2006.02.027
Anthony, I. C., Ramage, S. N., Carnie, F. W., Simmonds, P., and Bell, J. E. (2005). Does drug abuse alter microglial phenotype and cell turnover in the context of advancing HIV infection? Neuropathol. Appl. Neurobiol. 31, 325–338. doi: 10.1111/j.1365-2990.2005.00648.x
Antinori, A., Arendt, G., Becker, J. T., Brew, B. J., Byrd, D. A., Cherner, M., et al. (2007). Updated research nosology for HIV-associated neurocognitive disorders. Neurology 69, 1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b
Arango, J. C., Simmonds, P., Brettle, R. P., and Bell, J. E. (2004). Does drug abuse influence the microglial response in AIDS and HIV encephalitis? AIDS 18(Suppl. 1), S69–S74. doi: 10.1097/00002030-200401001-00010
Arvidsson, U., Riedl, M., Chakrabarti, S., Lee, J. H., Nakano, A. H., Dado, R. J., et al. (1995). Distribution and targeting of a mu-opioid receptor (MOR1) in brain and spinal cord. J. Neurosci. 15, 3328–3341.
Axmacher, N., Winterer, J., Stanton, P. K., Draguhn, A., and Müller, W. (2004). Two-photon imaging of spontaneous vesicular release in acute brain slices and its modulation by presynaptic GABAA receptors. Neuroimage 22, 1014–1021. doi: 10.1016/j.neuroimage.2004.02.009
Bajo, M., Madamba, S. G., Roberto, M., and Siggins, G. R. (2014). Acute morphine alters GABAergic transmission in the central amygdala during naloxone-precipitated morphine withdrawal: role of cyclic AMP. Front. Integr. Neurosci. 8:45. doi: 10.3389/fnint.2014.00045
Bauer, L. O., Ceballos, N. A., Shanley, J. D., and Wolfson, L. I. (2005). Sensorimotor dysfunction in HIV/AIDS: effects of antiretroviral treatment and comorbid psychiatric disorders. AIDS 19, 495–502. doi: 10.1097/01.aids.0000162338.66180.0b
Beatty, J. A., Sullivan, M. A., Morikawa, H., and Wilson, C. J. (2012). Complex autonomous firing patterns of striatal low-threshold spike interneurons. J. Neurophysiol. 108, 771–781. doi: 10.1152/jn.00283.2012
Behnisch, T., Francesconi, W., and Sanna, P. P. (2004). HIV secreted protein Tat prevents long-term potentiation in the hippocampal CA1 region. Brain Res. 1012, 187–189. doi: 10.1016/j.brainres.2004.03.037
Bell, J. E., Brettle, R. P., Chiswick, A., and Simmonds, P. (1998). HIV encephalitis, proviral load and dementia in drug users and homosexuals with AIDS. Effect of neocortical involvement. Brain 121(Pt 11), 2043–2052. doi: 10.1093/brain/121.11.2043
Benveniste, E. N., and Benos, D. J. (1995). TNF-alpha- and IFN-gamma-mediated signal transduction pathways: effects on glial cell gene expression and function. FASEB J. 9, 1577–1584.
Berger, J. R., and Arendt, G. (2000). HIV dementia: the role of the basal ganglia and dopaminergic systems. J. Psychopharmacol. 14, 214–221. doi: 10.1177/026988110001400304
Berger, J. R., and Nath, A. (1997). HIV dementia and the basal ganglia. Intervirology 40, 122–131. doi: 10.1159/000150539
Bergevin, A., Girardot, D., Bourque, M. J., and Trudeau, L. E. (2002). Presynaptic mu-opioid receptors regulate a late step of the secretory process in rat ventral tegmental area GABAergic neurons. Neuropharmacology 42, 1065–1078. doi: 10.1016/S0028-3908(02)00061-8
Bertrand, S. J., Mactutus, C. F., Aksenova, M. V., Espensen-Sturges, T. D., and Booze, R. M. (2014). Synaptodendritic recovery following HIV Tat exposure: neurorestoration by phytoestrogens. J. Neurochem. 128, 140–151. doi: 10.1111/jnc.12375
Bobeck, E. N., Chen, Q., Morgan, M. M., and Ingram, S. L. (2014). Contribution of adenylyl cyclase modulation of pre- and postsynaptic GABA neurotransmission to morphine antinociception and tolerance. Neuropsychopharmacology 39, 2142–2152. doi: 10.1038/npp.2014.62
Bokhari, S. M., Hegde, R., Callen, S., Yao, H., Adany, I., Li, Q., et al. (2011). Morphine potentiates neuropathogenesis of SIV infection in rhesus macaques. J. Neuroimmune Pharmacol. 6, 626–639. doi: 10.1007/s11481-011-9272-9
Bonavia, R., Bajetto, A., Barbero, S., Albini, A., Noonan, D. M., and Schettini, G. (2001). HIV-1 Tat causes apoptotic death and calcium homeostasis alterations in rat neurons. Biochem. Biophys. Res. Commun. 288, 301–308. doi: 10.1006/bbrc.2001.5743
Bosse, K. E., Jutkiewicz, E. M., Schultz-Kuszak, K. N., Mabrouk, O. S., Kennedy, R. T., Gnegy, M. E., et al. (2014). Synergistic activity between the delta-opioid agonist SNC80 and amphetamine occurs via a glutamatergic NMDA-receptor dependent mechanism. Neuropharmacology 77, 19–27. doi: 10.1016/j.neuropharm.2013.08.027
Brailoiu, G. C., Brailoiu, E., Chang, J. K., and Dun, N. J. (2008). Excitatory effects of human immunodeficiency virus 1 Tat on cultured rat cerebral cortical neurons. Neuroscience 151, 701–710. doi: 10.1016/j.neuroscience.2007.11.031
Brussaard, A. B., Kits, K. S., and de Vlieger, T. A. (1996). Postsynaptic mechanism of depression of GABAergic synapses by oxytocin in the supraoptic nucleus of immature rat. J. Physiol. 497(Pt 2), 495–507. doi: 10.1113/jphysiol.1996.sp021783
Buzhdygan, T., Lisinicchia, J., Patel, V., Johnson, K., Neugebauer, V., Paessler, S., et al. (2016). Neuropsychological, neurovirological and neuroimmune aspects of abnormal GABAergic transmission in HIV infection. J. Neuroimmune Pharmacol. 11, 279–293. doi: 10.1007/s11481-016-9652-2
Crittenden, J. R., and Graybiel, A. M. (2011). Basal Ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front. Neuroanat. 5:59. doi: 10.3389/fnana.2011.00059
Cui, Y., Ostlund, S. B., James, A. S., Park, C. S., Ge, W., Roberts, K. W., et al. (2014). Targeted expression of mu-opioid receptors in a subset of striatal direct-pathway neurons restores opiate reward. Nat. Neurosci. 17, 254–261. doi: 10.1038/nn.3622
Dore, G. J., Correll, P. K., Li, Y., Kaldor, J. M., Cooper, D. A., and Brew, B. J. (1999). Changes to AIDS dementia complex in the era of highly active antiretroviral therapy. AIDS 13, 1249–1253. doi: 10.1097/00002030-199907090-00015
Drake, C. T., and Milner, T. A. (1999). Mu opioid receptors are in somatodendritic and axonal compartments of GABAergic neurons in rat hippocampal formation. Brain Res. 849, 203–215. doi: 10.1016/S0006-8993(99)01910-1
Drake, C. T., and Milner, T. A. (2006). Mu opioid receptors are extensively co-localized with parvalbumin, but not somatostatin, in the dentate gyrus. Neurosci. Lett. 403, 176–180. doi: 10.1016/j.neulet.2006.04.047
El-Hage, N., Bruce-Keller, A. J., Knapp, P. E., and Hauser, K. F. (2008). CCL5/RANTES gene deletion attenuates opioid-induced increases in glial CCL2/MCP-1 immunoreactivity and activation in HIV-1 Tat-exposed mice. J. Neuroimmune Pharmacol. 3, 275–285. doi: 10.1007/s11481-008-9127-1
Ellis, R., Langford, D., and Masliah, E. (2007). HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat. Rev. Neurosci. 8, 33–44. doi: 10.1038/nrn2040
Eugenin, E. A., D'Aversa, T. G., Lopez, L., Calderon, T. M., and Berman, J. W. (2003). MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J. Neurochem. 85, 1299–1311. doi: 10.1046/j.1471-4159.2003.01775.x
Feligioni, M., Raiteri, L., Pattarini, R., Grilli, M., Bruzzone, S., Cavazzani, P., et al. (2003). The human immunodeficiency virus-1 protein Tat and its discrete fragments evoke selective release of acetylcholine from human and rat cerebrocortical terminals through species-specific mechanisms. J. Neurosci. 23, 6810–6818.
Feng, Y., He, X., Yang, Y., Chen, J., Yin, K., and Xia, Y. (2011). Effect of delta-opioid receptor over-expression on cortical expression of GABAA receptor alpha1-subunit in hypoxia. Chin. J. Physiol. 54, 118–123. doi: 10.4077/CJP.2011.AMM047
Fitting, S., Ignatowska-Jankowska, B. M., Bull, C., Skoff, R. P., Lichtman, A. H., Wise, L. E., et al. (2013). Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 Tat transgenic mice. Biol. Psychiatry 73, 443–453. doi: 10.1016/j.biopsych.2012.09.026
Fitting, S., Knapp, P. E., Zou, S., Marks, W. D., Bowers, M. S., Akbarali, H. I., et al. (2014). Interactive HIV-1 Tat and morphine-induced synaptodendritic injury is triggered through focal disruptions in Na+ influx, mitochondrial instability, and Ca2+ overload. J. Neurosci. 34, 12850–12864. doi: 10.1523/JNEUROSCI.5351-13.2014
Fitting, S., Ngwainmbi, J., Kang, M., Khan, F. A., Stevens, D. L., Dewey, W. L., et al. (2015). Sensitization of enteric neurons to morphine by HIV-1 Tat protein. Neurogastroenterol. Motil. 27, 468–480. doi: 10.1111/nmo.12514
Fitting, S., Scoggins, K. L., Xu, R., Dever, S. M., Knapp, P. E., Dewey, W. L., et al. (2012). Morphine efficacy is altered in conditional HIV-1 Tat transgenic mice. Eur. J. Pharmacol. 689, 96–103. doi: 10.1016/j.ejphar.2012.05.029
Fitting, S., Xu, R., Bull, C., Buch, S. K., El-Hage, N., Nath, A., et al. (2010). Interactive comorbidity between opioid drug abuse and HIV-1 Tat: chronic exposure augments spine loss and sublethal dendritic pathology in striatal neurons. Am. J. Pathol. 177, 1397–1410. doi: 10.2353/ajpath.2010.090945
Ford, C. P., Mark, G. P., and Williams, J. T. (2006). Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J. Neurosci. 26, 2788–2797. doi: 10.1523/JNEUROSCI.4331-05.2006
Fujiyama, F., Sohn, J., Nakano, T., Furuta, T., Nakamura, K. C., Matsuda, W., et al. (2011). Exclusive and common targets of neostriatofugal projections of rat striosome neurons: a single neuron-tracing study using a viral vector. Eur. J. Neurosci. 33, 668–677. doi: 10.1111/j.1460-9568.2010.07564.x
Gelman, B. B., Chen, T., Lisinicchia, J. G., Soukup, V. M., Carmical, J. R., Starkey, J. M., et al. (2012). The National NeuroAIDS Tissue Consortium brain gene array: two types of HIV-associated neurocognitive impairment. PLoS ONE 7:e46178. doi: 10.1371/journal.pone.0046178
Gerfen, C. R. (1985). The neostriatal mosaic. I. Compartmental organization of projections from the striatum to the substantia nigra in the rat. J. Comp. Neurol. 236, 454–476. doi: 10.1002/cne.902360404
Gilpin, N. W., Roberto, M., Koob, G. F., and Schweitzer, P. (2014). Kappa opioid receptor activation decreases inhibitory transmission and antagonizes alcohol effects in rat central amygdala. Neuropharmacology 77, 294–302. doi: 10.1016/j.neuropharm.2013.10.005
Gupta, S., Knight, A. G., Gupta, S., Knapp, P. E., Hauser, K. F., Keller, J. N., et al. (2010). HIV-Tat elicits microglial glutamate release: role of NAPDH oxidase and the cystine-glutamate antiporter. Neurosci. Lett. 485, 233–236. doi: 10.1016/j.neulet.2010.09.019
Gurwell, J. A., Nath, A., Sun, Q., Zhang, J., Martin, K. M., Chen, Y., et al. (2001). Synergistic neurotoxicity of opioids and human immunodeficiency virus-1 Tat protein in striatal neurons in vitro. Neuroscience 102, 555–563. doi: 10.1016/S0306-4522(00)00461-9
Hargus, N. J., and Thayer, S. A. (2013). Human immunodeficiency virus-1 Tat protein increases the number of inhibitory synapses between hippocampal neurons in culture. J. Neurosci. 33, 17908–17920. doi: 10.1523/JNEUROSCI.1312-13.2013
Haughey, N. J., Holden, C. P., Nath, A., and Geiger, J. D. (1999). Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J. Neurochem. 73, 1363–1374. doi: 10.1046/j.1471-4159.1999.0731363.x
Haughey, N. J., and Mattson, M. P. (2002). Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J. Acquir. Immune Defic. Syndr. 31(Suppl. 2), S55–S61. doi: 10.1097/00126334-200210012-00005
Haughey, N. J., Nath, A., Mattson, M. P., Slevin, J. T., and Geiger, J. D. (2001). HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J. Neurochem. 78, 457–467. doi: 10.1046/j.1471-4159.2001.00396.x
Heaton, R. K., Franklin, D. R., Ellis, R. J., McCutchan, J. A., Letendre, S. L., Leblanc, S., et al. (2011). HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J. Neurovirol. 17, 3–16. doi: 10.1007/s13365-010-0006-1
Hu, G., Yao, H., Chaudhuri, A. D., Duan, M., Yelamanchili, S. V., Wen, H., et al. (2012). Exosome-mediated shuttling of microRNA-29 regulates HIV Tat and morphine-mediated neuronal dysfunction. Cell Death Dis. 3, e381. doi: 10.1038/cddis.2012.114
Hu, X. T. (2016). HIV-1 Tat-mediated calcium dysregulation and neuronal dysfunction in vulnerable brain regions. Curr. Drug Targets 17, 4–14. doi: 10.2174/1389450116666150531162212
Jiang, Z. G., and North, R. A. (1992). Pre- and postsynaptic inhibition by opioids in rat striatum. J. Neurosci. 12, 356–361.
Jin, X. T., Galvan, A., Wichmann, T., and Smith, Y. (2011). Localization and function of GABA transporters GAT-1 and GAT-3 in the basal ganglia. Front. Syst. Neurosci. 5:63. doi: 10.3389/fnsys.2011.00063
Johnson, S. W., and North, R. A. (1992). Opioids excite dopamine neurons by hyperpolarization of local interneurons. J. Neurosci. 12, 483–488.
Kest, B., Hopkins, E., Palmese, C. A., Chen, Z. P., Mogil, J. S., and Pintar, J. E. (2001). Morphine tolerance and dependence in nociceptin/orphanin FQ transgenic knock-out mice. Neuroscience 104, 217–222. doi: 10.1016/S0306-4522(01)00037-9
Kim, H. J., Martemyanov, K. A., and Thayer, S. A. (2008). Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J. Neurosci. 28, 12604–12613. doi: 10.1523/JNEUROSCI.2958-08.2008
Klitenick, M. A., Dewitte, P., and Kalivas, P. W. (1992). Regulation of somatodendritic dopamine release in the ventral tegmental area by opioids and GABA: an in vivo microdialysis study. J. Neurosci. 12, 2623–2632.
Kumar, A. M., Borodowsky, I., Fernandez, B., Gonzalez, L., and Kumar, M. (2007). Human immunodeficiency virus type 1 RNA Levels in different regions of human brain: quantification using real-time reverse transcriptase-polymerase chain reaction. J. Neurovirol. 13, 210–224. doi: 10.1080/13550280701327038
Kure, K., Lyman, W. D., Weidenheim, K. M., and Dickson, D. W. (1990). Cellular localization of an HIV-1 antigen in subacute AIDS encephalitis using an improved double-labeling immunohistochemical method. Am. J. Pathol. 136, 1085–1092.
Lan, X., Rao, T. K., Chander, P. N., Skorecki, K., and Singhal, P. C. (2015). Apolipoprotein L1 (APOL1) Variants (Vs) a possible link between Heroin-associated Nephropathy (HAN) and HIV-associated Nephropathy (HIVAN). Front. Microbiol. 6:571. doi: 10.3389/fmicb.2015.00571
Li, J. C., and Lau, A. S. (2007). A role for mitogen-activated protein kinase and Ets-1 in the induction of interleukin-10 transcription by human immunodeficiency virus-1 Tat. Immunology 121, 337–348. doi: 10.1111/j.1365-2567.2007.02580.x
Li, J. C., Lee, D. C., Cheung, B. K., and Lau, A. S. (2005). Mechanisms for HIV Tat upregulation of IL-10 and other cytokine expression: kinase signaling and PKR-mediated immune response. FEBS Lett. 579, 3055–3062. doi: 10.1016/j.febslet.2005.04.060
Li, W., Huang, Y., Reid, R., Steiner, J., Malpica-Llanos, T., Darden, T. A., et al. (2008). NMDA receptor activation by HIV-Tat protein is clade dependent. J. Neurosci. 28, 12190–12198. doi: 10.1523/JNEUROSCI.3019-08.2008
Lipton, S. A. (1994). AIDS-related dementia and calcium homeostasis. Ann. N.Y. Acad. Sci. 747, 205–224. doi: 10.1111/j.1749-6632.1994.tb44411.x
Liu, B., Liu, X., and Tang, S. J. (2016). Interactions of opioids and HIV infection in the pathogenesis of chronic pain. Front. Microbiol. 7:103. doi: 10.3389/fmicb.2016.00103
Longordo, F., Feligioni, M., Chiaramonte, G., Sbaffi, P. F., Raiteri, M., and Pittaluga, A. (2006). The human immunodeficiency virus-1 protein transactivator of transcription up-regulates N-methyl-D-aspartate receptor function by acting at metabotropic glutamate receptor 1 receptors coexisting on human and rat brain noradrenergic neurones. J. Pharmacol. Exp. Ther. 317, 1097–1105. doi: 10.1124/jpet.105.099630
Luo, Y., Berman, M. A., Abromson-Leeman, S. R., and Dorf, M. E. (2003). Tumor necrosis factor is required for RANTES-induced astrocyte monocyte chemoattractant protein-1 production. Glia 43, 119–127. doi: 10.1002/glia.10231
Magnuson, D. S., Knudsen, B. E., Geiger, J. D., Brownstone, R. M., and Nath, A. (1995). Human immunodeficiency virus type 1 tat activates non-N-methyl-D-aspartate excitatory amino acid receptors and causes neurotoxicity. Ann. Neurol. 37, 373–380. doi: 10.1002/ana.410370314
Marks, W. D., Paris, J. J., Schier, C. J., Denton, M. D., Fitting, S., McQuiston, A. R., et al. (2016). HIV-1 Tat causes cognitive deficits and selective loss of parvalbumin, somatostatin, and neuronal nitric oxide synthase expressing hippocampal CA1 interneuron subpopulations. J. Neurovirol. doi: 10.1007/s13365-016-0447-2. [Epub ahead of print].
Masvekar, R. R., El-Hage, N., Hauser, K. F., and Knapp, P. E. (2015). GSK3beta-activation is a point of convergence for HIV-1 and opiate-mediated interactive neurotoxicity. Mol. Cell. Neurosci. 65, 11–20. doi: 10.1016/j.mcn.2015.01.001
Mayne, M., Holden, C. P., Nath, A., and Geiger, J. D. (2000). Release of calcium from inositol 1,4,5-trisphosphate receptor-regulated stores by HIV-1 Tat regulates TNF-alpha production in human macrophages. J. Immunol. 164, 6538–6542. doi: 10.4049/jimmunol.164.12.6538
McQuiston, A. R. (2007). Effects of μ-opioid receptor modulation on GABAB receptor synaptic function in hippocampal CA1. J. Neurophysiol. 97, 2301–2311. doi: 10.1152/jn.01179.2006
Musante, V., Summa, M., Neri, E., Puliti, A., Godowicz, T. T., Severi, P., et al. (2010). The HIV-1 viral protein Tat increases glutamate and decreases GABA exocytosis from human and mouse neocortical nerve endings. Cereb. Cortex 20, 1974–1984. doi: 10.1093/cercor/bhp274
Nath, A., and Geiger, J. (1998). Neurobiological aspects of human immunodeficiency virus infection: neurotoxic mechanisms. Prog. Neurobiol. 54, 19–33. doi: 10.1016/S0301-0082(97)00053-1
Nelson, A. B., Hammack, N., Yang, C. F., Shah, N. M., Seal, R. P., and Kreitzer, A. C. (2014). Striatal cholinergic interneurons Drive GABA release from dopamine terminals. Neuron 82, 63–70. doi: 10.1016/j.neuron.2014.01.023
Ngwainmbi, J., De, D. D., Smith, T. H., El-Hage, N., Fitting, S., Kang, M., et al. (2014). Effects of HIV-1 Tat on enteric neuropathogenesis. J. Neurosci. 34, 14243–14251. doi: 10.1523/JNEUROSCI.2283-14.2014
Noel, R. J. Jr., Rivera-Amill, V., Buch, S., and Kumar, A. (2008). Opiates, immune system, acquired immunodeficiency syndrome, and nonhuman primate model. J. Neurovirol. 14, 279–285. doi: 10.1080/13550280802078209
Pennartz, C. M., Berke, J. D., Graybiel, A. M., Ito, R., Lansink, C. S., van der Meer, M., et al. (2009). Corticostriatal interactions during learning, memory processing, and decision making. J. Neurosci. 29, 12831–12838. doi: 10.1523/JNEUROSCI.3177-09.2009
Pérez, A., Probert, A. W., Wang, K. K., and Sharmeen, L. (2001). Evaluation of HIV-1 Tat induced neurotoxicity in rat cortical cell culture. J. Neurovirol. 7, 1–10. doi: 10.1080/135502801300069575
Pérez-Casanova, A., Noel, R. J. Jr., Rivera-Amill, V., Husain, K., and Kumar, A. (2007). Morphine-mediated deterioration of oxidative stress leads to rapid disease progression in SIV/SHIV-infected macaques. AIDS Res. Hum. Retroviruses 23, 1004–1007. doi: 10.1089/aid.2006.0286
Petroff, O. A. (2002). GABA and glutamate in the human brain. Neuroscientist 8, 562–573. doi: 10.1177/1073858402238515
Prendergast, M. A., Rogers, D. T., Mulholland, P. J., Littleton, J. M., Wilkins, L. H. Jr., Self, R. L., et al. (2002). Neurotoxic effects of the human immunodeficiency virus type-1 transcription factor Tat require function of a polyamine sensitive-site on the N-methyl-D-aspartate receptor. Brain Res. 954, 300–307. doi: 10.1016/S0006-8993(02)03360-7
Rivera-Amill, V., Noel, R. J. Jr., García, Y., Rivera, I., Iszard, M., Buch, S., et al. (2010). Accelerated evolution of SIV env within the cerebral compartment in the setting of morphine-dependent rapid disease progression. Virology 398, 201–207. doi: 10.1016/j.virol.2009.11.046
Sabatier, J. M., Vives, E., Mabrouk, K., Benjouad, A., Rochat, H., Duval, A., et al. (1991). Evidence for neurotoxic activity of tat from human immunodeficiency virus type 1. J. Virol. 65, 961–967.
Sacktor, N., McDermott, M. P., Marder, K., Schifitto, G., Selnes, O. A., McArthur, J. C., et al. (2002). HIV-associated cognitive impairment before and after the advent of combination therapy. J. Neurovirol. 8, 136–142. doi: 10.1080/13550280290049615
Saylor, D., Dickens, A. M., Sacktor, N., Haughey, N., Slusher, B., Pletnikov, M., et al. (2016). HIV-associated neurocognitive disorder - pathogenesis and prospects for treatment. Nat. Rev. Neurol. 12, 234–248. doi: 10.1038/nrneurol.2016.27
Scott, J. C., Woods, S. P., Carey, C. L., Weber, E., Bondi, M. W., Grant, I., et al. (2011). Neurocognitive consequences of HIV infection in older adults: an evaluation of the “cortical” hypothesis. AIDS Behav. 15, 1187–1196. doi: 10.1007/s10461-010-9815-8
Shin, A. H., Kim, H. J., and Thayer, S. A. (2012). Subtype selective NMDA receptor antagonists induce recovery of synapses lost following exposure to HIV-1 Tat. Br. J. Pharmacol. 166, 1002–1017. doi: 10.1111/j.1476-5381.2011.01805.x
Singh, I. N., Goody, R. J., Dean, C., Ahmad, N. M., Lutz, S. E., Knapp, P. E., et al. (2004). Apoptotic death of striatal neurons induced by human immunodeficiency virus-1 Tat and gp120: differential involvement of caspase-3 and endonuclease G. J. Neurovirol. 10, 141–151. doi: 10.1080/13550280490441103
Speth, C., Schabetsberger, T., Mohsenipour, I., Stöckl, G., Würzner, R., Stoiber, H., et al. (2002). Mechanism of human immunodeficiency virus-induced complement expression in astrocytes and neurons. J. Virol. 76, 3179–3188. doi: 10.1128/JVI.76.7.3179-3188.2002
Stellwagen, D., Beattie, E. C., Seo, J. Y., and Malenka, R. C. (2005). Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J. Neurosci. 25, 3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005
Svingos, A. L., Clarke, C. L., and Pickel, V. M. (1998). Cellular sites for activation of delta-opioid receptors in the rat nucleus accumbens shell: relationship with Met5-enkephalin. J. Neurosci. 18, 1923–1933.
Tan, M., Walwyn, W. M., Evans, C. J., and Xie, C. W. (2009). p38 MAPK and beta-arrestin 2 mediate functional interactions between endogenous micro-opioid and alpha2A-adrenergic receptors in neurons. J. Biol. Chem. 284, 6270–6281. doi: 10.1074/jbc.M806742200
Tibbles, L. A., and Woodgett, J. R. (1999). The stress-activated protein kinase pathways. Cell. Mol. Life Sci. 55, 1230–1254. doi: 10.1007/s000180050369
Ueda, H., Inoue, M., Takeshima, H., and Iwasawa, Y. (2000). Enhanced spinal nociceptin receptor expression develops morphine tolerance and dependence. J. Neurosci. 20, 7640–7647.
Vaughan, C. W., and Christie, M. J. (1997). Presynaptic inhibitory action of opioids on synaptic transmission in the rat periaqueductal grey in vitro. J. Physiol. 498(Pt 2), 463–472. doi: 10.1113/jphysiol.1997.sp021872
Vaughan, C. W., Ingram, S. L., Connor, M. A., and Christie, M. J. (1997). How opioids inhibit GABA-mediated neurotransmission. Nature 390, 611–614. doi: 10.1038/37610
Wallace, D. R., Dodson, S. L., Nath, A., and Booze, R. M. (2006). δ opioid agonists attenuate TAT1-72-induced oxidative stress in SK-N-SH cells. Neurotoxicology 27, 101–107. doi: 10.1016/j.neuro.2005.07.008
Watabe-Uchida, M., Zhu, L., Ogawa, S. K., Vamanrao, A., and Uchida, N. (2012). Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron 74, 858–873. doi: 10.1016/j.neuron.2012.03.017
Westendorp, M. O., Frank, R., Ochsenbauer, C., Stricker, K., Dhein, J., Walczak, H., et al. (1995). Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 375, 497–500. doi: 10.1038/375497a0
Wu, Y., Wang, W., Díez-Sampedro, A., and Richerson, G. B. (2007). Nonvesicular inhibitory neurotransmission via reversal of the GABA transporter GAT-1. Neuron 56, 851–865. doi: 10.1016/j.neuron.2007.10.021
Xenias, H. S., Ibáñez-Sandoval, O., Koós, T., and Tepper, J. M. (2015). Are striatal tyrosine hydroxylase interneurons dopaminergic? J. Neurosci. 35, 6584–6599. doi: 10.1523/JNEUROSCI.0195-15.2015
Xu, C., Hermes, D. J., Mackie, K., Lichtman, A. H., Ignatowska-Jankowska, B. M., and Fitting, S. (2016). Cannabinoids occlude the HIV-1 tat-induced decrease in GABAergic neurotransmission in prefrontal cortex slices. J. Neuroimmune Pharmacol. 11, 316–331. doi: 10.1007/s11481-016-9664-y
Yousefpour, M., Naderi, N., Mansouri, Z., Janahmadi, M., Alizadeh, A. M., and Motamedi, F. (2014). The comparison of the effects of acute and repeated morphine administration on fast synaptic transmission in magnocellular neurons of supraoptic nucleus, plasma vasopressin levels, and urine volume of male rats. Iran J. Pharm. Res. 13, 975–985.
Zhang, W., Yang, H. L., Song, J. J., Chen, M., Dong, Y., Lai, B., et al. (2015). DAMGO depresses inhibitory synaptic transmission via different downstream pathways of mu opioid receptors in ventral tegmental area and periaqueductal gray. Neuroscience 301, 144–154. doi: 10.1016/j.neuroscience.2015.05.077
Zhao, L., Pu, S. S., Gao, W. H., Chi, Y. Y., Wen, H. L., Wang, Z. Y., et al. (2014). Effects of HIV-1 tat on secretion of TNF-alpha and IL-1beta by U87 cells in AIDS patients with or without AIDS dementia complex. Biomed. Environ. Sci. 27, 111–117. doi: 10.3967/bes2014.024
Zou, S., Fitting, S., Hahn, Y. K., Welch, S. P., El-Hage, N., Hauser, K. F., et al. (2011). Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at μ-opioid receptor-expressing glia. Brain 134, 3613–3628. doi: 10.1093/brain/awr281
Keywords: HIV-1 Tat, morphine, CTAP, μ-opioid receptor, GABA neurotransmission, striatum
Citation: Xu C and Fitting S (2016) Inhibition of GABAergic Neurotransmission by HIV-1 Tat and Opioid Treatment in the Striatum Involves μ-Opioid Receptors. Front. Neurosci. 10:497. doi: 10.3389/fnins.2016.00497
Received: 18 July 2016; Accepted: 17 October 2016;
Published: 08 November 2016.
Edited by:
Tod Edward Kippin, University of California, Santa Barbara, USACopyright © 2016 Xu and Fitting. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvia Fitting, sfitting@email.unc.edu