 Cyrus Vahdatpour
Cyrus Vahdatpour Adam H. Dyer
Adam H. Dyer Daniela Tropea
Daniela Tropea- 1School of Medicine, Trinity College Dublin, Dublin, Ireland
- 2Department of Psychiatry, Trinity College Dublin, Dublin, Ireland
Insulin-Like Growth Factor 1 (IGF-1) is a neurotrophic polypeptide with crucial roles to play in Central Nervous System (CNS) growth, development and maturation. Following interrogation of the neurobiology underlying several neurodevelopmental disorders and Autism Spectrum Disorders (ASD), both recombinant IGF-1 (mecasermin) and related derivatives, such as (1-3)IGF-1, have emerged as potential therapeutic approaches. Clinical pilot studies and early reports have supported the safety/preliminary efficacy of IGF-1 and related compounds in the treatment of Rett Syndrome, with evidence mounting for its use in Phelan McDermid Syndrome and Fragile X Syndrome. In ASD, clinical trials are ongoing. Here, we review the role of IGF-1 in the molecular etiologies of these conditions in addition to the accumulating evidence from early clinical studies highlighting the possibility of IGF-1 and related compounds as potential treatments for these childhood-onset neurodevelopmental disorders.
Introduction
Insulin-Like Growth Factor 1 (IGF1) is a polypeptide hormone and a member of a superfamily of related insulin like hormones termed Insulin Like Peptides (ILPs) (Fernandez and Torres-Alemán, 2012). IGF-1 is primarily released by hepatocytes in response to Growth Hormone, but is also produced in the Central Nervous System (CNS) where it has pleiotrophic effects on all major CNS cell types (Bach et al., 1991; O'Kusky and Ye, 2012). IGF-1 has crucial roles to play in the development, growth and maturation of the CNS and its synapses, roles which have been reviewed extensively elsewhere (Bach et al., 1991; Popken et al., 2004; Aberg et al., 2006; Llorens-Martín et al., 2009; Corvin et al., 2012; O'Kusky and Ye, 2012; Supeno et al., 2013; Huat et al., 2014; Dyer et al., 2016).
IGF-1 and IGF1 receptor (IGF1R) expression levels have a definite spatio-temporal patterns (Bach et al., 1991; Bartlett et al., 1992; Bondy and Lee, 1993; Zhang et al., 2007). Of note, the abundance of IGF-1R expression over IGF-1 hints at the importance of peripherally produced IGF-1 in mediating the effects of IGF-1 on the CNS (Fernandez and Torres-Alemán, 2012). IGF-1 acts on its glycoprotein receptor (IGF1R), a tyrosine kinase receptor, to activate canonical signaling pathways, including: (i) the PI3K (phosphatidylinositol-3 kinase)—AKT1 (serine-threonine-specific protein kinase)—FOXO (forkhead box protein O) and (ii) the RAS—MAPK (mitogen-activated protein kinases)—ERK (extracellular signal regulated kinases) pathway. Both of these pathways have important roles to play including cell cycle regulation, gene expression, protein synthesis, autophagy, apoptosis and remodeling of the cytoskeleton (Fernandez and Torres-Alemán, 2012; Costales and Kolevzon, 2015).
Once released in the serum, IGF-1 can be cleaved to yield an amino terminal glycine-proline-glutamate (GPE tripeptide) and a truncated IGF-1 form called des-N-(1-3)-IGF-1, lacking the N-terminal GPE tripeptide, which has greatly reduced affinity for IGFBPs and is therefore, is more potent than IGF-1. (1-3)IGF-1 is an active metabolite with neuroprotective effects as well as effects on excitatory synaptic markers such as synapsin 1 and post-synaptic density 95 (PSD-95), recapitulating many of the effects of IGF-1 on synaptic maturation and plasticity (Guan et al., 1999; Corvin et al., 2012). Of note, this effect of (1-3) IGF-1 may be different in neuronal and non-neuronal cell populations. In one report studying the effects of IGF-1 acting on its canonical pathways in neuronal and non-neuronal cells, IGF-1 was found to increase pAkt statining in neurons but not glial cells, whilst (1-3) IGF-1 increased staining in glial cells but not neurons (Corvin et al., 2012). Thus, signaling by IGF-1 and its active tripeptide may demonstrate different effects in different CNS cell populations, adding further to the biological complexity of IGF-1 signaling. One mechanism of action of (1-3)IGF-1 may be to indirectly activate the IGF-1R via an increase in endogenous IGF-1 release (Corvin et al., 2012). Another important aspect of IGF-1 physiology is its binding to IGF Binding Proteins (IGFBP), which regulate its bioavailability, localization and activity (Ocrant et al., 1990; Clemmons, 1998; Hwa et al., 1999; Firth and Baxter, 2002).
Disruption of IGF-1 function has profound phenotypic consequences both in murine models and in humans, underscoring the important role of IGF-1 in CNS development and maturation (Beck et al., 1995; Woods et al., 1996; Netchine et al., 2011). In the present Mini-Review, we address the role of perturbed IGF-1 signaling and the therapeutic potential of IGF-1 and (1-3)IGF-1 in neurodevelopmental disorders such as Rett Syndrome (RTT), Fragile X Syndrome (FXS), Phelan McDermid Syndrome (PMDS) as well as broader Autism Spectrum Disorder (ASD).
Rett Syndrome
Rett Syndrome (RTT) is a pervasive X-Linked neurodevelopmental disorder affecting 1:10,000 female (Percy and Lane, 2004). RTT is characterized by an apparently normal development, followed by a subsequent regression in psychomotor, social and cognitive abilities, deficits in social interaction and a loss of acquired fine motor skills and purposive hand movements. In the CNS, RTT is characterized by microcephaly, neuronal atrophy and leads to cardiorespiratory problems (Julu et al., 2001; Hagberg, 2002). At present, the treatment of RTT represents an unmet therapeutic need.
More than 85% of RTT is caused by a mutation in methyl CpG-binding protein 2 (MeCP2), encoding the protein MECP2, which has roles both inside and outside the nucleus (Kaufmann et al., 2005; Chahrour et al., 2008). Atypical cases of RTT may also be caused, in less than 10% cases, by mutations in cyclin-dependent kinase-like 5 (CDK-L5) and in the Forkead Box G1 (FOXG1) in 1% cases (Cahrour and Zoghbi, 2007).
The deletion of the MeCP2 gene in mouse models recapitulates many of the autonomic, motor and cognitive features of the human RTT phenotype (Banerjee et al., 2012; Castro et al., 2014; Katz et al., 2016), together with reduced connectivity (Armstrong, 2005; Chapleau et al., 2009) and defects in neurotransmitter and receptor expression. MeCP2 loss in mice results in an alteration in excitatory-inhibitory balance with reduced excitation/increased inhibition in cortical samples/tissue (Dani et al., 2005; Durand et al., 2012).
MeCP2 deficient brains demonstrate large numbers of modest transcriptional changes, both positive and negative (Katz et al., 2016). One well characterized target of MECP2 function is Brain Derived Neurotrophic Factor (BDNF), an important modulator of CNS growth, which shows synergy with IGF1 in the CNS (Ding et al., 2006). BDNF is down-regulated in murine models of RTT as well as patients with RTT (Chang et al., 2006; Zhou et al., 2006). Although studies in rats have shown that there is clear exchange of BDNF between the brain and the periphery (Pan et al., 1998) and viceversa (Poduslo and Curran, 1996), other studies have failed to raise BDNF in the brain to therapeutic levels, suggesting crossing through the blood-brain barrier (BBB) of this neurotrophin to be insufficient for clinical purposes (Pardridge et al., 1994). IGF-1 is a potential alternative, which on crossing the BBB acts on the same pathways as BDNF (such as the PI3K-Akt and MAP-ERK pathways) and appears important for BNDF effects on activity dependent plasticity (Pardridge et al., 1994; Ding et al., 2006).
In Mecp2 mutant mice, administration of both IGF-1 and (1-3)IGF-1 reverses many of the features of the RTT phenotype (Chen and Russo-Neustadt, 2007; Castro et al., 2014). Castro et al. (2014) demonstrated reduced IGF-1 levels in MeCP2 mutant mice, with subsequent daily administration of IGF-1 resulting in an improved lifespan, weight and autonomic parameters in the knockout mice. IGF-1 significantly improved abnormalities in activity dependent plasticity in MeCP2 mutant mice (using a monocular deprivation paradigm). Similar effects are observed with (1-3) IGF-1 administration (Tropea et al., 2009), together with improved spine density, synaptic amplitude and increased excitatory synaptic markers (Tropea et al., 2009). Furthermore, in a mouse model of atypical RTT with mutations in CDLK5, IGF-1 was demonstrated to rescue deficits in dendritic spine instability and expression of PSD-95 adding further support to IGF-1 as a potential treatment in RTT (Della Sala et al., 2016).
One mechanism by which IGF-1 exerts its effects in RTT may be by acting on its canonical signaling pathways (such as PI3K and MAPK pathways as above). Interestingly, a recent study demonstrates that IGF-1 application may actually increase nuclear MeCP2 transcript and protein (Tropea et al., 2016). Activity dependent plasticity was also shown to modulate MeCP2 expression and this additionally demonstrates the profound effects of IGF-1 on cellular neuroplasticity in the CNS. Restoration of these abnormalities in activity-dependent plasticity may be one important way in which IGF-1 may exert its effects in RTT. Interestingly, MeCP2 may affect IGF-1 levels by regulation of IGFBPs which has been demonstrated for IGFBP3 in both murine models and humans (Itoh et al., 2007).
IGF-1 is already indicated in the pediatric population for severe growth failure and IGF-1 deficiency. In RTT patients, two early studies have been performed demonstrating the tolerability and safety of IGF-1 as a potential treatment (Pini et al., 2012; Khwaja et al., 2014). Khwaja et al. (2014) demonstrated the safety of IGF-1 in 12 patients with MeCP2 mutations (9 with RTT) with a 4 week multiple ascending dose (40–120 ug/kg bd) followed by a 20 week open label extension, without any serious adverse events or hypoglycamia. A previous clinical study had demonstrated safety in 6 patients with RTT receiving twice daily injections of IGF-1 for a 6 month period (Pini et al., 2012). This was followed by a single case study of one of the patients reporting the safety of repeated treatment with IGF-1 for a second 6 month cycle (Pini et al., 2014).
Preliminary efficacy analysis by Khwaja et al. (2014) demonstrated an improvement in cardiorespiratory parameters, some neurobehavioral parameters and EEG measures of mood and anxiety (reversed frontal alpha band asymmetry). Pini et al. analyzed 10 patients whom had received IGF-1 treatment in a clinical study and compared various parameters to age and disease severity matched controls (Pini et al., 2016). They reported a significant improvement in disease severity as assessed by clinicians as well as two independent and blinded observers using a novel video based scoring system. Whilst this evidence is preliminary, this early data demonstrating efficacy of IGF-1 in RTT is encouraging. The results of ongoing Phase 2 clinical trials using IGF-1 in RTT are eagerly awaited (NCT01777542). Furthermore, an analog of (1-3)IGF-1, NNZ-2566 (administered orally) has demonstrated efficacy in both clinician and caregiver assessments in an industry-led Phase 2 trial on patients with RTT aged 15-45 (NCT01703533), with a Phase 2 trial on younger patients in progress (NCT02715115). Taken together, there is encouraging evidence for the use of IGF-1 and (1-3) IGF-1 in the treatment of RTT (see Tables 1, 2).
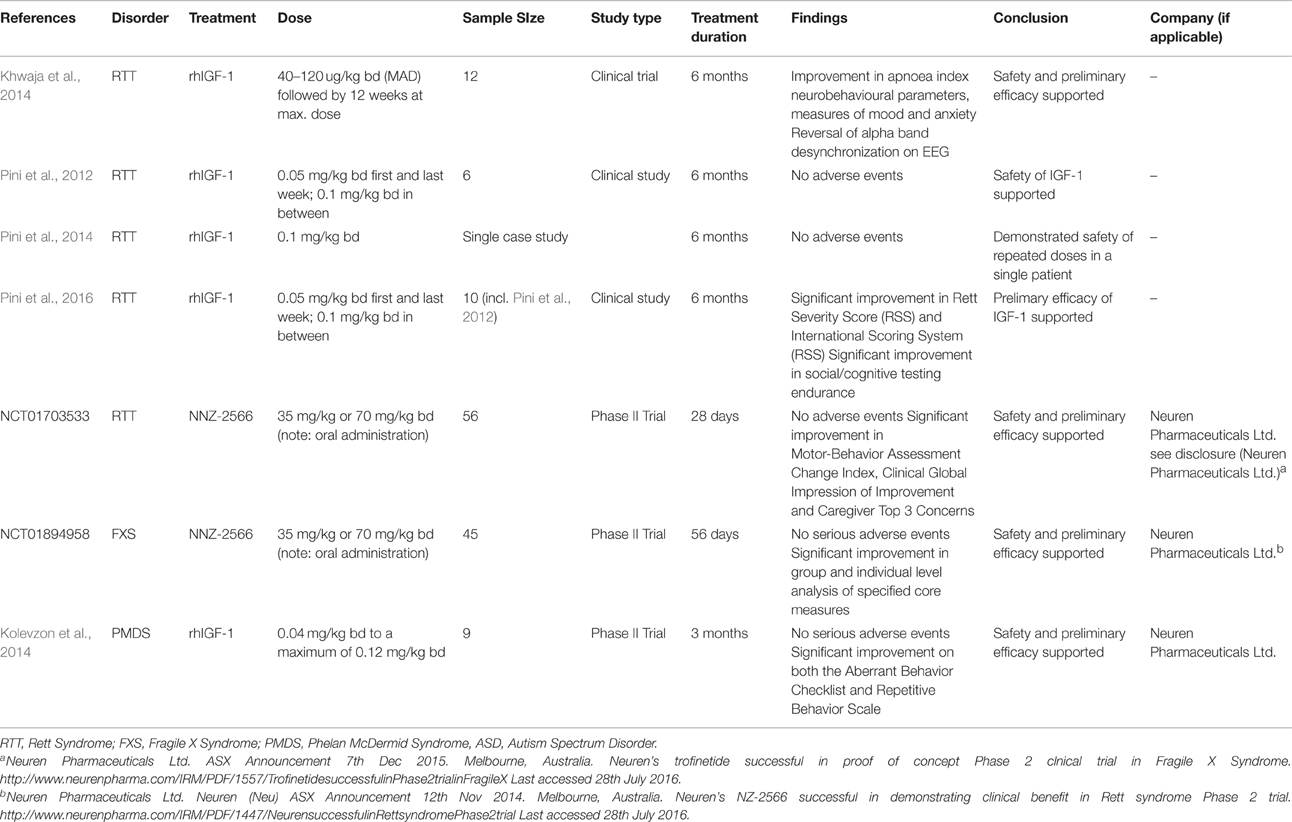
Table 1. Previous studies and trials examining IGF-1 as a potential treatment in several childhood-onset neurodevelopmental disorders.

Table 2. Ongoing trials examining IGF-1 as a potential treatment in several childhood-onset neurodevelopmental disorders.
Fragile X Syndrome
Fragile X Syndrome (FXS) results from a mutation in the Fmr1 gene, encoding the protein Fragile X Mental Retardation Protein (FMRP1). The disorder is characterized by learning disability, social anxiety and attention deficit disorder, impaired social interactions and seizures as well as an abnormal physical phenotype with macro-orchidism and facial dysmorphism (Hagerman, 1997; Garber et al., 2008). The treatment of FXS represents a largely unmet clinical need.
FMRP, an mRNA binding protein, expressed in neuronal cell bodies and dendrites acts to regulate protein translation at the synapse, with important roles to play in activity-dependent plasticity (e.g., inhibition of translation triggered by mGluR1/5 in response to neuronal stimulation) (Bhakar et al., 2012). FMRP may also have important presynaptic effects on neuronal transmission, mediated via its effects on large conductance Ca2+ activated K+ channels (Deng et al., 2013).
Several studies have reported disruptions in MAPK/ERK signaling in FXS. Weng et al. (2008) have demonstrated delayed early-phase phosphorylation of ERK in mice deficient in FMRP whilst Curia et al. (2013) demonstrate a resistance to seizures with dephosphorylation of p-ERK in Fmr1 mice (Weng et al., 2008; Curia et al., 2013). In the PI3K pathway, the p110beta catalytic subunit can be regulated by FMRP, with p110beta and PI3K activity elevated in Fmr1 knockout neurons. This suggests that dysregulation of PI3K signaling may be involved in the synaptic deficits seen in FXS. Indeed, inhibition of PI3K activity may correct dysregulated synaptic protein synthesis, AMPA internalization and spine density defects in knockout neurons (Gross et al., 2010). Thus, alterations in the same canonical pathways stimulated by IGF-1 and other neurotrophins may underlie a large part of the synaptic pathology seen in FXS.
Impressive preclinical evidence comes from a study by Deacon et al. (2015) using an analog of (1-3)IGF-1, NNZ-2566. In Fmr1 knockout mice, NNZ-2566 demonstrated a significantly reduced brain phospho-ERK and phospho-Akt. Similarly, NNZ-2566 resulted in a significant reduction in spine numbers, which are increased in the Fmr1 mice in comparison to controls. Improvement in hyperactivity and anxiety, learning and memory deficits was also observed (Deacon et al., 2015). In patients with FXS, a Phase 2 industry-led clinical trial has been completed using NNZ-2566, with clinical improvement in many of the core symptoms of FXS as disclosed by Neuren Pharmaceuticals (NCT01894958). The exact efficacy of IGF-1 related compounds such as NNZ-2566 awaits further clarification, but this early clinical evidence is encouraging.
Phelan-McDermid Syndrome
Phelan-McDermid Syndrome (PMDS) is another monogenic neurodevelopmental disorder and results from deletions in the SHANK3 gene on chromosome 22q13.3. Affected patients demonstrate global developmental delay, severe impairments in speech and intellectual disability (Phelan and McDermid, 2012). The protein product of SHANK3 is a key scaffolding protein present in the post-synaptic density of excitatory synapses, with key roles in activity-dependent plasticity and the functional maintenance of these synapses.
In 3-week old hippocampal neurons treated with siRNA to inhibit SHANK3 synthesis, there was a decreased number and an increased length of dendritic spines (Roussignol et al., 2005). Similarly, application of SHANK3 resulted in the formation of spine-like protrusions containing SHANK3 in aspiny neurons supporting the role of SHANK3 in spine formation and synaptic plasticity. Further, application of SHANK3 increased immunoreactivity for AMPAR subunits in cell body and dendritic spines (but not for GABA subunits), supporting its role in the function and maintenance of excitatory synapses (Roussignol et al., 2005). Using mice deficient in Shank3, Bozdagi et al. found reduced amplitude of miniature excitatory post-synaptic currents (mEPSCs) in the hippocampus and impaired Long Term Potentiation (LTP), with only transient spine expansion present (Bozdagi et al., 2010).
In Shank3 deficient mice, intraperitoneal injection of IGF-1, administered daily for a 2 week period reversed deficits in AMPAR receptors and LTP described above. Similar results were also seen for the active tripeptide (1-3) IGF-1 (Bozdagi et al., 2013). Recently, similar effects for IGF-1 on Shank3 deficient human neurons has been demonstrated. By using induced pluripotent stem cells (iPSCs) from patients with PMDS and using them to produce functional neurons, Shcheglovitov et al. demonstrated that these neurons had a reduced expression of SHANK3 with accompanying defects in excitatory synaptic transmission as seen in the murine models above (Shcheglovitov et al., 2013). Treatment of the SHANK3 deficient neurons with IGF-1 increased the amplitude and frequency of EPSCs, restored the amplitude of evoked AMPA and NMDAR EPSCs and restored NMDA receptor currents on application of NMDA (Shcheglovitov et al., 2013). IGF-1 also caused a 340% increase in the fraction of puncta expressing PSD-95 in PMDS neurons (Shcheglovitov et al., 2013).
In a double blind, placebo controlled Phase 2 trial reported by Kolevzon et al., the safety and preliminary efficacy of IGF-1 treatment in PMDS were reported on 9 patients with PMDS aged 5–15 (Kolevzon et al., 2014). IGF-1 treatment was associated with significant improvements in social impairment and restrictive behaviors (Aberrant Behavior Checklist and Repetitive Behavior Scale). No serious adverse events occurred with the main side effects including sleep disturbance, hypoglycemia (<50 mg/dL), constipation, increased appetite and mood changes/irritability. This encouraging evidence awaits further confirmation and exploration in further clinical trials in the PMDS population.
Autism Spectrum Disorder
Autism Spectrum Disorder is a heterogenous neurodevelopmental disorders characterized by deficits in social interaction and in speech and language with narrowed interests and repetitive behaviors (Wang and Doering, 2015). Autism affects about 3–6 per 1000 of the population, although recent estimates place its prevalence higher at 1 in 68 (CDC, 2012). The most substantial clue to ASD etiology is its substantial heritability (−90%) (Freitag, 2007). ASD demonstrates complex genetics, and whilst it is a genetically heterogenous disorder, evidence repeatedly implicates genes involved in synaptic development, function and activity-dependent plasticity by both common and rare variation (Wang et al., 2009; Hussman et al., 2011; De Rubeis et al., 2014). The potent effects of IGF-1 on synaptic function, maintenance and plasticity make it a potentially attractive target for the treatment of ASDs.
Vanhala et al. reported low levels of IGF-1 in children with autism, however sample sizes were small (n = 11) (Vanhala et al., 2006). In 25 young children with a diagnosis of autism, it was subsequently shown that IGF-1 levels were significantly reduced (Riikonen et al., 2001), and in those with a diagnosis of autism, Cerebrospinal Fluid (CSF) IGF-1 was correlated with head size (Mills et al., 2007). On measuring urinary IGF-1 excretion, Anlar et al. (2007) demonstrated that IGF-1 level was significantly lower in autistic children than in age matched controls. In contrast, a larger study has demonstrated significantly higher levels of IGF-1 in children with autism (Marchetto et al., 2016). The exact role of the IGF-1 axis in ASD awaits further clarification.
Interestingly, in a recent report using neurons derived from patients with ASD, Marchetto et al. found a partial rescue of deficits in neuronal networks (neuronal spike number and activity) on application of IGF-1 (Marchetto et al., 2016). At present, clinical trials of IGF-1 in Autism Spectrum Disorder are ongoing, with a phase 2 trial currently recruiting (NCT01970345). The trial aims to pilot the use of IGF-1 as a novel treatment for the core symptoms of ASD. The results of this trial are eagerly awaited. If successful, further trials will be needed in the ASD population to determine the exact efficacy of IGF-1 as a potential treatment, as well as studies to investigate the specific groups of patients with ASD which benefit the most from treatment with recombinant IGF-1.
Potential Molecular Mechanisms of IGF1 and Derivates in Different Disorders
There are three potential mechanisms by which IGF-1 may exert its effects in these neurodevelopmental disorders. The first is increased glutamatergic transmission, as seen in various preclinical studies (Tropea et al., 2009; Corvin et al., 2012; Castro et al., 2014; Marchetto et al., 2016). A potential consequence of this includes effects related to increased synaptic potentiation and plasticity (Bozdagi et al., 2013). A second potential action of IGF-1 in these neurodevelopmental disorders includes activation of molecular pathways involved in growth and connectivity (PI3K and MAPK). In RTT models, where these pathways are down-regulated, IGF1 induces an increase in the relative markers of neuronal function (Tropea et al., 2009; Castro et al., 2014), whilst in FXS models, where they are up-regulated, an (1-3)IGF1 analog induces a decrease in the cellular pathways (Deacon et al., 2015). This may be the result of a homeostatic action of IGF-1, in re-establishing basal levels of activity in these canonical signaling pathways. The third potential mechanism relates to effects on transcription. This has been demonstrated in recent studies of IGF-1 on mecp2 transcript (Tropea et al., 2016). More work is needed to clarify the direct and indirect effects of IGF1 and derivates, and their action in different cell types.
Conclusion
Preliminary evidence is beginning to emerge from well validated murine models and early clinical studies, that treatment with recombinant human IGF-1 (rhIGF-1/mecasermin) and derived compounds may be of benefit in several childhood onset neurodevelopmental disorders. Whilst the evidence base is preliminary, further clinical trials and studies are needed in order to quantify the effects of IGF-1 on patients with these disorders, as well as identifying particular patients which may derive maximum benefit from treatment with IGF-1 and related compounds. The results of ongoing clinical trials are eagerly awaited.
Author Contributions
CV, AD, and DT had substantial roles in the drafting, writing and editing of the review and final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aberg, N. D., Brywe, K. G., and Isgaard, J. (2006). Aspects of growth hormone and insulin-like growth factor-I related to neuroprotection, regeneration, and functional plasticity in the adult brain. ScientificWorldJournal 6, 53–80. doi: 10.1100/tsw.2006.22
Anlar, B., Oktem, F., Bakkaloglu, B., Haliloglu, M., Oguz, H., Unal, F., et al. (2007). Urinary epidermal and insulin-like growth factor excretion in autistic children. Neuropediatrics 38, 151–153. doi: 10.1055/s-2007-990282
Armstrong, D. D. (2005). Neuropathology of Rett syndrome. J. Child Neurol. 20, 747–753. doi: 10.1177/08830738050200082401
Bach, M. A., Shen-Orr, Z., Lowe, W. L. Jr., Roberts, C. T. Jr., and LeRoith, D. (1991). Insulin-like growth factor I mRNA levels are developmentally regulated in specific regions of the rat brain. Brain Res. Mol. Brain Res. 10, 43–48. doi: 10.1016/0169-328X(91)90054-2
Banerjee, A., Castro, J., and Sur, M. (2012). Rett syndrome: genes, synapses, circuits, and therapeutics. Front. Psychiatry 3:34. doi: 10.3389/fpsyt.2012.00034
Bartlett, W. P., Li, X. S., and Williams, M. (1992). Expression of IGF-1 mRNA in the murine subventricular zone during postnatal development. Brain Res. Mol. Brain Res. 12, 285–291. doi: 10.1016/0169-328X(92)90131-T
Beck, K. D., Powell-Braxton, L., Widmer, H. R., Valverde, J., and Hefti, F. (1995). Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron 14, 717–730. doi: 10.1016/0896-6273(95)90216-3
Bhakar, A. L., Dölen, G., and Bear, M. F. (2012). The pathophysiology of fragile X (and what it teaches us about synapses). Annu. Rev. Neurosci. 35, 417–443. doi: 10.1146/annurev-neuro-060909-153138
Bondy, C. A., and Lee, W. H. (1993). Patterns of insulin-like growth factor and IGF receptor gene expression in the brain. Functional implications. Ann. N.Y. Acad. Sci. 692, 33–43.
Bozdagi, O., Sakurai, T., Papapetrou, D., Wang, X., Dickstein, D. L., Takahashi, N., et al. (2010). Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 1:15. doi: 10.1186/2040-2392-1-15
Bozdagi, O., Tavassoli, T., and Buxbaum, J. D. (2013). Insulin-like growth factor-1 rescues synaptic and motor deficits in a mouse model of autism and developmental delay. Mol. Autism 4:9. doi: 10.1186/2040-2392-4-9
Cahrour, M., and Zoghbi, H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437. doi: 10.1016/j.neuron.2007.10.001
Castro, J., Garcia, R. I., Kwok, S., Banerjee, A., Petravicz, J., Woodson, J., et al. (2014). Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett Syndrome. Proc. Natl. Acad. Sci. U.S.A. 111, 9941–9946. doi: 10.1073/pnas.1311685111
Centers for Disease Control Prevention (2012). Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. Available online at: http://www.cdc.gov/mmwr/volumes/65/ss/ss6503a1.htm (Accessed on: September 26, 2016).
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, Z., Wong, S. T., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi: 10.1126/science.1153252
Chang, Q., Khare, G., Dani, V., Nelson, S., and Jaenisch, R. (2006). The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 49, 341–348. doi: 10.1016/j.neuron.2005.12.027
Chapleau, C. A., Calfa, G. D., Lane, M. C., Albertson, A. J., Larimore, J. L., Kudo, S., et al. (2009). Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol. Dis. 35, 219–233. doi: 10.1016/j.nbd.2009.05.001
Chen, M. J., and Russo-Neustadt, A. A. (2007). Running exercise- and antidepressant-induced increases in growth and survival-associated signaling molecules are IGF-dependent. Growth Factors 25, 118–131. doi: 10.1080/08977190701602329
Clemmons, D. R. (1998). Role of insulin-like growth factor binding proteins in the control of IGF actions. Mol. Cell. Endocrinol. 140, 19–24.
Corvin, A. P., Molinos, I., Little, G., Donohoe, G., Gill, M., Morris, D. W., et al. (2012). Insulin-like growth factor 1 (IGF1) and its active peptide (1-3)IGF1 enhance the expression of synaptic markers in neuronal circuits through different cellular mechanisms. Neurosci. Lett. 520, 51–56. doi: 10.1016/j.neulet.2012.05.029
Costales, J. L., and Kolevzon, A. (2015). The therapeutic potential of insulin-like growth factor-1 in central nervous system disorders. Neurotherapeutics 12, 620–630. doi: 10.1007/s13311-015-0352-z
Curia, G., Gualtieri, F., Bartolomeo, R., Vezzalo, R., and Biagini, G. (2013). Resilience to audiogenic seizures is associated with p-ERK1/2 dephosphorylation in the subiculum of Fmr1 knockout mice. Front. Cell. Neurosci. 25:46. doi: 10.3389/fncel.2013.00046
Dani, V. S., Chang, Q., Maffei, A., Turrigiano, G. G., Jaenisch, R., Nelson, S. B., et al. (2005). Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 12560–12565. doi: 10.1073/pnas.0506071102
Deacon, R. M., Glass, L., Snape, M., Hurley, M. J., Altimiras, F. J., Biekofsky, R. R., et al. (2015). NNZ-2566, a novel analog of (1-3) IGF-1, as a potential therapeutic agent for fragile X syndrome. Neuromol. Med. 17, 71–82. doi: 10.1007/s12017-015-8341-2
Della Sala, G., Putignano, E., Chelini, G., Melani, R., Calcagno, E., Michele Ratto, G., et al. (2016). Dendritic spine instability in a mouse model of CDKL5 disorder is rescued by insulin-like growth factor 1. Mol. Psychiatry. 80, 302–311. doi: 10.1016/j.biopsych.2015.08.028
Deng, P. Y., Rotman, Z., Blundon, J. A., Cho, Y., Cui, J., Cavalli, V., et al. (2013). FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77, 696–711. doi: 10.1016/j.neuron.2012.12.018
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
Ding, Q., Vaynman, S., Akhavan, M., Ying, Z., and Gomez-Pinilla, F. (2006). Insulin-like growth factor I interfaces with brain-derived neurotrophic factor-mediated synaptic plasticity to modulate aspects of exercise-induced cognitive function. Neuroscience 140, 823–833. doi: 10.1016/j.neuroscience.2006.02.084
Durand, S., Patrizi, A., Quast, K. B., Hachingian, L., Pavlyuk, R., and Saxena, A. (2012). NMDA receptor regulation prevents regression of visual cortical function in the absence of Mecp2. Neuron 76, 1078–10790. doi: 10.1016/j.neuron.2012.12.004
Dyer, A. H., Vahdatpour, C., Sanfeliu, A., and Tropea, D. (2016). The role of Insulin-Like Growth Factor 1 (IGF-1) in brain development, maturation and neuroplasticity. Nueroscience 325, 89–99. doi: 10.1016/j.neuroscience.2016.03.056
Fernandez, A. M., and Torres-Alemán, I. (2012). The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 13, 225–239. doi: 10.1038/nrn3209
Firth, S. M., and Baxter, R. C. (2002). Cellular actions of the insulin-like growth factor binding proteins. Endocr. Rev. 23, 824–854. doi: 10.1210/er.2001-0033
Freitag, C. M. (2007). The genetics of autistic disorders and its clinical relevance: a review of the literature. Mol. Psychiatry 12, 2–22. doi: 10.1038/sj.mp.4001896
Garber, K. B., Visootsak, J., and Warren, S. T. (2008). Fragile X syndrome. Eur. J. Hum. Genet. 16, 666–6672. doi: 10.1586/14737159.2016.1135739
Gross, C., Nakamoto, M., Yao, X., Chan, C. B., Yim, S. Y., Ye, K., et al. (2010). Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J. Neurosci. 30, 10624–10638. doi: 10.1523/JNEUROSCI.0402-10.2010
Guan, J., Waldvogel, H. J., Faull, R. L., Gluckman, P. D., and Williams, C. E. (1999). The effects of the N-terminal tripeptide of insulin-like growth factor-1, glycine-proline-glutamate in different regions following hypoxic-ischemic brain injury in adult rats. Neuroscience 89, 649–659. doi: 10.1016/S0306-4522(98)00338-8
Hagberg, B. (2002). Clinical manifestations and stages of Rett syndrome. Ment. Retard. Disabil. Res. Rev. 8, 61–65. doi: 10.1002/mrdd.10020
Hagerman, R. J. (1997). Fragile X syndrome. molecular and clinical insights and treatment issues. West J. Med. 166, 129–137.
Huat, T. J., Khan, A. A., Pati, S., Mustafa, Z., Abdullah, J. M., and Jaafar, H. (2014). IGF-1 enhances cell proliferation and survival during early differentiation of mesenchymal stem cells to neural progenitor-like cells. BMC Neurosci. 15:91. doi: 10.1186/1471-2202-15-91
Hussman, J. P., Chung, R. H., Griswold, A. J., Jaworski, J. M., Salyakina, D., Ma, D., et al. (2011). A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Mol. Autism 2:1. doi: 10.1186/2040-2392-2-1
Hwa, V., Oh, Y., and Rosenfeld, R. G. (1999). The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr. Rev. 20, 761–787. doi: 10.1210/er.20.6.761
Itoh, M., Ide, S., Takashima, S., Kudo, S., Nomura, Y., Segawa, M., et al. (2007). Methyl CpG-binding protein 2 (a mutation of which causes Rett syndrome) directly regulates insulin-like growth factor binding protein 3 in mouse and human brains. Neuropathol. Exp. Neurol. 66, 117–123. doi: 10.1097/nen.0b013e3180302078
Julu, P. O., Kerr, A. M., Apartopoulos, F., Al-Rawas, S., Engerstrom, I. W., Engerström, L., et al. (2001). Characterisation of breathing and associated central autonomic dysfunction in the Rett disorder. Arch. Dis. Child. 85, 29–37. doi: 10.1136/adc.85.1.29
Katz, D. M., Bird, A., Coenraads, M., Gray, S. J., Menon, D. U., Philpot, B. D., et al. (2016). Rett syndrome: crossing the threshold to clinical translation. Trends Neurosci. 39, 100–113. doi: 10.1016/j.tins.2015.12.008
Kaufmann, W. E., Johnston, M. V., and Blue, M. E. (2005). MeCP2 expression and function during brain development: implications for Rett syndrome's pathogenesis and clinical evolution. Brain Dev. 27(Suppl. 1), S77–S87. doi: 10.1016/j.braindev.2004.10.008
Khwaja, O. S., Ho, E., Barnes, K. V., O'Leary, H. M., Pereira, L. M., and Finkelstein, Y. (2014). Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 111, 4596–4601. doi: 10.1073/pnas.1311141111
Kolevzon, A., Bush, L., Wang, A. T., Halpern, D., Frank, Y., Grodberg, D., et al. (2014). A pilot controlled trial of insulin-like growth factor-1 in children with Phelan-McDermid syndrome. Mol. Autism 5:54. doi: 10.1186/2040-2392-5-54
Llorens-Martín, M., Torres-Alemán, I., and Trejo, J. L. (2009). Mechanisms mediating brain plasticity: IGF1 and adult hippocampal neurogenesis. Neuroscientist 15, 134–148. doi: 10.1177/1073858408331371
Marchetto, M. C., Belinson, H., Tian, Y., Freitas, B. C., Fu, C., Vadodaria, K. C., et al. (2016). Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol. Psychiatry. doi: 10.1038/mp.2016.95. [Epub ahead of print].
Mills, J. L., Hediger, M. L., Molloy, C. A., Chrousos, G. P., Manning-Courtney, P., Yu, K. F., et al. (2007). Elevated levels of growth-related hormones in autism and autism spectrum disorder. Clin. Endocrinol. 67, 230–237. doi: 10.1111/j.1365-2265.2007.02868.x
Netchine, I., Azzi, S., Le Bouc, Y., and Savage, M. O. (2011). IGF1 molecular anomalies demonstrate its critical role in fetal, postnatal growth and brain development. Best Pract. Res. Clin. Endocrinol. Metab. 25, 181–190. doi: 10.1016/j.beem.2010.08.005
Ocrant, I., Fay, C. T., and Parmelee, J. T. (1990). Characterization of insulin-like growth factor binding proteins produced in the rat central nervous system. Endocrinology 127, 1260–1267. doi: 10.1210/endo-127-3-1260
O'Kusky, J., and Ye, P. (2012). Neurodevelopmental effects of insulin-like growth factor signaling. Front. Neuroendocrinol. 33, 230–251. doi: 10.1016/j.yfrne.2012.06.002
Pan, W., Banks, W. A., Fasold, M. B., Bluth, J., and Kastin, A. J. (1998). Transport of brain-derived neurotrhophic factor across the blood brain barrier. Nueropharmacology 37, 1553–1561. doi: 10.1016/S0028-3908(98)00141-5
Pardridge, W. M., Kang, Y. S., and Buckiak, J. L. (1994). Transport of human recombinant brain-derived neurotrophic factor (BDNF) through the rat blood-brain barrier in vivo using vector-mediated peptide drug delivery. Pharm. Res. 11, 738–746. doi: 10.1023/A:1018940732550
Percy, A. K., and Lane, J. B. (2004). Rett syndrome: clinical and molecular update. Curr. Opin. Paediatr. 16, 670–677. doi: 10.1097/01.mop.0000143693.59408.ce
Phelan, K., and McDermid, H. E. (2012). The 22q13.3 deletion syndrome (Phelan-McDermid syndrome). Mol. Syndromol. 2, 186–201. doi: 10.1159/000334260
Pini, G., Congiu, L., Benicasa, A., DiMarco, P., Bigoni, S., Dyer, A. H., et al. (2016). Illness severity, social and cognitive ability, and EEG analysis of ten patients with rett syndrome treated with mecasermin (recombinant human IGF-1). Autism Res. Treat. 2016:5073078. doi: 10.1155/2016/5073078
Pini, G., Scusa, M. F., Benincasa, A., Bottliglioni, I., Congiu, L., Vahdatpour, C., et al. (2014). Repeated insulin-like growth factor 1 treatment in a patient with rett syndrome: a single case study. Front. Paediatry 2:52. doi: 10.3389/fped.2014.00052
Pini, G., Scusa, M. F., Congiu, L., Benincasa, A., Morescalchi, P., Bottliglioni, I., et al. (2012). IGF1 as a potential treatment for rett syndrome: safety assessment in six rett patients. Autism Res. Treat. 2012:679801. doi: 10.1155/2012/679801
Poduslo, J. F., and Curran, G. L. (1996). Permeability at the blood-brain and blood-nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3, BDNF. Brain Res. Mol. Brain Res. 36, 280–286. doi: 10.1016/0169-328X(95)00250-V
Popken, G. J., Hodge, R. F., Ye, P., Zhang, J., Ng, W., O'Kusky, J. R., et al. (2004). In vivo effects of insulin-like growth factor-I (IGF-I) on prenatal and early postnatal development of the central nervous system. Eur. J. Neurosci. 19, 2056–2068. doi: 10.1111/j.0953-816X.2004.03320.x
Riikonen, R., Turpeinen, U., and Riikonen, R. (2001). Cerebrospinal fluid insulin-like growth factors IGF-1 and IGF-2 in infantile autism. Dev. Med. Chlild Neurol. 43, 614–616. doi: 10.1017/S0012162201001116
Roussignol, G., Ango, F., Romorini, S., Tu, J. C., Sala, C., Worley, P. F., et al. (2005). Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J. Neurosci. 25, 3560–3570. doi: 10.1523/jneurosci.4354-04.2005
Shcheglovitov, A., Shcheglovitova, O., Yazawa, M., Portmann, T., Shu, R., Sebastino, V., et al. (2013). SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503, 267–271. doi: 10.1038/nature12618
Supeno, N. E., Pati, S., Hadi, R. A., Ghani, A. R., Mustafa, Z., Abdullah, J. M., et al. (2013). IGF-1 acts as controlling switch for long-term proliferation and maintenance of EGF/FGF-responsive striatal neural stem cells. Int. J. Med. Sci. 10, 522–531. doi: 10.7150/ijms.5325
Tropea, D., Diacometti, E., Wilson, N. R., Beard, C., McCurry, C., Fu, D. D., et al. (2009). Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. U.S.A. 106, 2029–2034. doi: 10.1073/pnas.0812394106
Tropea, D., Mortimer, N., Bellini, S., Molinos, I., Sanfeliu, A., Shovlin, S., et al. (2016). Expression of nuclear Methyl-CpG binding protein 2 (Mecp2) is dependent on neuronal stimulation and application of Insulin-like growth factor 1. Neurosci. Lett. 621, 111–116. doi: 10.1016/j.neulet.2016.04.024
Vanhala, R., Turpeinen, U., and Riikonen, R. (2006). Low levels of insulin-like growth factor-I in cerebrospinal fluid in children with autism. Dev. Med. Child Neurol. 48, 751–755. doi: 10.1017/S0012162206001605
Wang, H., and Doering, L. C. (2015). Autism spectrum disorders: emerging mechanisms and mechanism-based treatment. Front. Cell Neursci. 9:183. doi: 10.3389/fncel.2015.00183
Wang, K., Zhang, H., Ma, D., Bucan, M., Glessner, J. T., Abrahams, B. S., et al. (2009). Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459, 528–533. doi: 10.1038/nature07999
Weng, N., Weller, I. J., Sumis, A., Berry-Kravis, E., and Greenough, W. T. (2008). Early-phase ERK activation as a biomarker for metabolic status in fragile X syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 1253–1257. doi: 10.1002/ajmg.b.30765
Woods, K. A., Camacho-Hübner, C., Savage, M. O., and Clark, A. J. (1996). Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N. Engl. J. Med. 335, 1363–1367. doi: 10.1056/NEJM199610313351805
Zhang, J., Moats-Staats, B. M., Ye, P., and D'Ercole, A. J. (2007). Expression of insulin-like growth factor system genes during the early postnatal neurogenesis in the mouse hippocampus. J. Neurosci. Res. 85, 1618–1627. doi: 10.1002/jnr.21289
Keywords: IGF-1, autism spectrum disorders, Rett Syndrome, Fragile X Syndrome, Phelan-Mcdermid Syndrome
Citation: Vahdatpour C, Dyer AH and Tropea D (2016) Insulin-Like Growth Factor 1 and Related Compounds in the Treatment of Childhood-Onset Neurodevelopmental Disorders. Front. Neurosci. 10:450. doi: 10.3389/fnins.2016.00450
Received: 07 July 2016; Accepted: 20 September 2016;
Published: 30 September 2016.
Edited by:
Roberto Canitano, University Hospital of Siena, ItalyReviewed by:
Enrico Tongiorgi, University of Trieste, ItalyHansen Wang, University of Toronto, Canada
Om Sai Ramesh Vallamkonda, Lady Hardinge Medical College, India
Copyright © 2016 Vahdatpour, Dyer and Tropea. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Tropea, ZGFuaWVsYS50cm9wZWFAdGNkLmll