
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 25 May 2016
Sec. Neuropharmacology
Volume 10 - 2016 | https://doi.org/10.3389/fnins.2016.00205
This article is part of the Research Topic Structure-based drug design for diagnosis and treatment of neurological diseases View all 11 articles
 Mercedes Unzeta1*
Mercedes Unzeta1* Gerard Esteban2
Gerard Esteban2 Irene Bolea1
Irene Bolea1 Wieslawa A. Fogel3
Wieslawa A. Fogel3 Rona R. Ramsay4
Rona R. Ramsay4 Moussa B. H. Youdim5
Moussa B. H. Youdim5 Keith F. Tipton2
Keith F. Tipton2 José Marco-Contelles6
José Marco-Contelles6HIGHLIGHTS
• ASS234 is a MTDL compound containing a moiety from Donepezil and the propargyl group from the PF 9601N, a potent and selective MAO B inhibitor. This compound is the most advanced anti-Alzheimer agent for preclinical studies identified in our laboratory.
• Derived from ASS234 both multipotent donepezil-indolyl (MTDL-1) and donepezil-pyridyl hybrids (MTDL-2) were designed and evaluated as inhibitors of AChE/BuChE and both MAO isoforms. MTDL-2 showed more high affinity toward the four enzymes than MTDL-1.
• MTDL-3 and MTDL-4, were designed containing the N-benzylpiperidinium moiety from Donepezil, a metal- chelating 8-hydroxyquinoline group and linked to a N-propargyl core and they were pharmacologically evaluated.
• The presence of the cyano group in MTDL-3, enhanced binding to AChE, BuChE and MAO A. It showed antioxidant behavior and it was able to strongly complex Cu(II), Zn(II) and Fe(III).
• MTDL-4 showed higher affinity toward AChE, BuChE.
• MTDL-3 exhibited good brain penetration capacity (ADMET) and less toxicity than Donepezil. Memory deficits in scopolamine-lesioned animals were restored by MTDL-3.
• MTDL-3 particularly emerged as a ligand showing remarkable potential benefits for its use in AD therapy.
Alzheimer's disease (AD), the most common form of adult onset dementia, is an age-related neurodegenerative disorder characterized by progressive memory loss, decline in language skills, and other cognitive impairments. Although its etiology is not completely known, several factors including deficits of acetylcholine, β-amyloid deposits, τ-protein phosphorylation, oxidative stress, and neuroinflammation are considered to play significant roles in the pathophysiology of this disease. For a long time, AD patients have been treated with acetylcholinesterase inhibitors such as donepezil (Aricept®) but with limited therapeutic success. This might be due to the complex multifactorial nature of AD, a fact that has prompted the design of new Multi-Target-Directed Ligands (MTDL) based on the “one molecule, multiple targets” paradigm. Thus, in this context, different series of novel multifunctional molecules with antioxidant, anti-amyloid, anti-inflammatory, and metal-chelating properties able to interact with multiple enzymes of therapeutic interest in AD pathology including acetylcholinesterase, butyrylcholinesterase, and monoamine oxidases A and B have been designed and assessed biologically. This review describes the multiple targets, the design rationale and an in-house MTDL library, bearing the N-benzylpiperidine motif present in donepezil, linked to different heterocyclic ring systems (indole, pyridine, or 8-hydroxyquinoline) with special emphasis on compound ASS234, an N-propargylindole derivative. The description of the in vitro biological properties of the compounds and discussion of the corresponding structure-activity-relationships allows us to highlight new issues for the identification of more efficient MTDL for use in AD therapy.
Alzheimer's disease (AD) is one of the most common neurodegenerative diseases accounting for more than 80% of total dementia cases in elderly people. It is estimated that currently 47 million victims of AD exist worldwide and that number is expected to grow up to more than 130 million by 2050 as a result of life expectancy increase over the next decades (Thies and Bleiler, 2012, 2013). In 2015, the World Alzheimer Report estimated that the current annual societal and economic cost of dementia was US $818 billion worldwide and that amount is expected to rise up to 1 trillion by 2018. This study also reported that the cost associated with dementia has increased by 35% since 2010.
The clinical manifestations of AD are characterized by misfunctioning and gradual neuronal death resulting in a progressive memory deterioration and cognitive decline, related with the loss of cholinergic dysfunction. The anatomopathology of AD has been described by progressive loss of synaptic neurons triggering atrophy in the hippocampus and frontal and tempo-parietal cortex. Two distinctive hallmarks of AD include the presence of accumulated amyloid beta (Aβ) plaques around neurons (Glenner and Murphy, 1989) and hyperphosphorylated microtubules associated with tau protein in the form of intracellular neurofibrillary tangles (NFT) (Goedert et al., 2006).
The pathogenesis of this neurodegenerative disorder is not yet fully understood, but the scientific consensus is quite firm in describing it as a multifactorial disease caused by several elements. These include loss of cholinergic transmission, excessive protein misfolding and Aβ aggregation (Terry et al., 1964; Grundke-Iqbal et al., 1986), oxidative stress and free radical formation (Coyle and Puttfarcken, 1993), metal dyshomeostasis (Huang et al., 2004), excitotoxic, and neuroinflammatory processes (Coyle and Puttfarcken, 1993). Although a large number of genes has been associated with the AD late-onset condition, these appear to affect susceptibility or rate of progression rather than being directly causative (Bertram and Tanzi, 2008; Chouraki and Seshadri, 2014; Karch and Goate, 2015).
Cholinergic neurotransmission modulates cognitive function and cortical plasticity (Arendt and Bigl, 1986) and also plays key roles in the control of cerebral blood flow (Biesold et al., 1989), cortical activity (Détári et al., 1999), and the sleep-wake cycle (Lee et al., 2005). In 1971, Deutsch postulated the involvement of the cholinergic system in learning and memory (Deutsch, 1971), which was later corroborated in studies with animal models and humans (Fibiger, 1991; Schliebs, 2005).
The first physiological evidence for the involvement of the cholinergic system in AD pathology was a reduction in pre-synaptic acetylcholine (ACh) and a reduced expression of choline acetyltransferase (ChAT), the enzyme responsible for ACh synthesis. In addition, decreases in the binding parameters of both muscarinic acetylcholine receptors (mAChR) and nicotinic acetylcholine receptors (nAChR) have been reported. These findings suggest a direct link between cholinergic neurotransmission and AD, and constitute the basis for the so-called “cholinergic hypothesis of AD” (Deutsch, 1971), which the main therapeutic approach used to date to address the cognitive loss associated with AD is based on.
Cholinesterases (ChE) catalyse the hydrolysis of ACh into choline and acetic acid, an essential process in the cholinergic neurotransmission. There are two types of ChEs: acetylcholinesterase (AChE, EC 3.1.1.7) and butyrylcholinesterase (BuChE, EC 3.1.1.8). Both enzymes are type α/β hydrolases folded with an α-helix bound with β-sheet containing a catalytic domain (Ollis et al., 1992).
AChE is expressed in cholinergic neurons and neuromuscular junctions where its primary function is the rapid breakdown of the neurotransmitter ACh released during cholinergic neurotransmission. Although AChE and BuChE are structurally similar, resembling each other by more than 50%, both their significance and location are substantially different.
The structure of AChE has been widely studied since the 1990s. The active site of the enzyme lies on a bottom of a long and narrow cavity of 20 Å deep and contains a catalytic triad with amino acid residues Ser200, His440 and Glu327 that catalyses the hydrolysis of the ester bond. The active site also includes an anionic site or α-anionic site that interacts with the quaternary ammonium atom of ACh, ensuring its correct orientation. A peripheral anionic site (PAS) or β-anionic site, located on the enzyme surface around the cavity entrance (Figure 1) has been described to play an important role in AD. This site was first recognized in the 1960s as a target for AChE activity modulators (see Sussman et al., 1991) such as toxins and promising drugs. Furthermore, interaction of the Aβ peptide with the PAS contributes to the formation of amyloid plaques by accelerating the aggregation process. Propidium iodide is an example of an inhibitor binding to this site (Inestrosa et al., 1996).
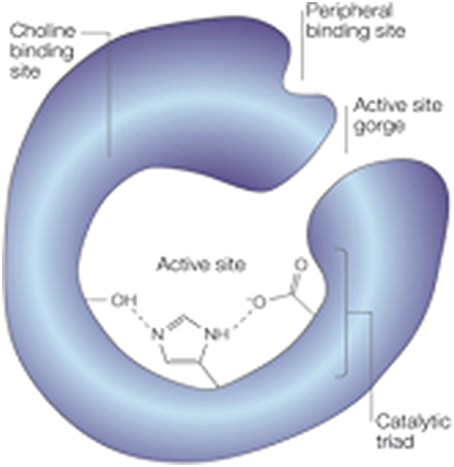
Figure 1. Structure of AChE displaying the active site at the bottom of a narrow gorge, lined with hydrophobic amino-acid side chains within the catalytic triad, the choline binding site, and the peripheral binding site (Reproduced, with permission from Soreq and Seidman, 2001).
One of the most consistent changes associated with AD has been the degeneration of neurons from the cholinergic nuclei in the basal forebrain region and their terminals in the hippocampus (Struble et al., 1982). The neuronal atrophy has been associated with a loss of cholinergic markers, occurring specifically in the nucleus basalis of Meynert (Vogels et al., 1990). The cholinergic system function, responsible for the storage and retrieval of items in memory, is therefore highly impaired in AD pathology, which has been corroborated by some neuroimaging data (Jack et al., 2010).
BuChE is expressed in the hippocampus and temporal neocortex but at lower levels than AChE, and associated with glial cells (Mesulam et al., 2002), suggesting that these enzymes may have complementary functions. In AD brain, neuritic plaques, and NFTs contain large amounts of AChE and BuChE, the latter being present in the neuroglia (Wright et al., 1993), Moreover, these enzymes have been reported to play a role in the processing of APP (Wright et al., 1993), although it is unclear whether their inhibition could influence the pathogenic course of AD. While AChE is expressed in nerve and blood cells, hydrolysing ACh, the biological significance of BuChE still remains poorly understood, although it has been reported to partially modulate or compensate for the diminished AChE activity in deficient animals (Xie et al., 2000).
Likewise AChE, glial BuChE hydrolyses ACh into choline and acetate (Daikhin and Yudkoff, 2000; Mesulam et al., 2002) but with a different kinetic behavior. While AChE predominates in neurons and exhibits high affinity for ACh, with low KM values, BuChE is mainly present in endothelia, glia and neuronal cells with low affinity (high KM value) for ACh (Soreq and Seidman, 2001).
In addition to its role in the hydrolysis of ACh, non-enzymatic functions have also been attributed to BuChE. Whereas, AChE may accelerate amyloid deposition in the brain of AD patients, as previously mentioned, BuChE can associate with Aβ protein possibly delaying the onset and rate of neurotoxic Aβ fibril formation as observed in vitro (Diamant et al., 2006).
Activity of BuChE has been found either unaltered or increased in certain AD brain regions (Perry et al., 1978; Ciro et al., 2012). The increase has been associated with amyloid plaques and NFTs (Geula and Mesulam, 1989; Guillozet et al., 1997). In addition to changes in activity, changes in AChE and BuChE protein expression also occur during the progression of AD. An increase in the levels of glial-derived BuChE and decrease in synaptic AChE have been observed, triggering a dramatic increase in the BuChE: AChE ratio in cortical regions from 0.6, in healthy conditions, to 11 in AD pathology (Giacobini, 2003). The observed changes in BuChE activity and expression throughout the course of AD, and its relationship with cognitive function, emphasize the potential value of BuChE and AChE inhibition as therapeutic targets in AD condition.
The amyloid cascade hypothesis postulates that neurodegeneration in AD is caused by abnormal accumulation of Aβ plaques in various areas of the brain (Hardy and Higgins, 1992; Evin and Weidemann, 2002). This accumulation acts as a pathological trigger for a cascade that includes neuritic injury, formation of NFTs via tau protein to neuronal dysfunction and cell death (Hardy and Higgins, 1992; Selkoe, 1994). Genetic, biochemical, and pathological evidences support this hypothesis as the primary cause of AD (Kayed et al., 2003). The Aβ senile plaques are composed by Aβ peptides, which consist of 39–43 amino acid residues proteolytically derived from the sequential enzymatic action of β- and γ-secretases on transmembrane APP (Coulson et al., 2000). The length of Aβ peptides varies at the C-terminal according to the cleavage pattern of APP, with Aβ1−40 being the most prevalent form, followed by the hydrophobic form Aβ1−42 that aggregates faster (Perl, 2010). Within plaques, Aβ peptides and β-sheet conformation assemble and polymerise into structurally distinct forms such as fibrils, protofibers, and polymorphic oligomers (Selkoe, 1994).
The kinetics of the aggregation process of Aβ peptide follows a sigmoidal curve owing to the presence of β-sheets in its structure (LeVine, 1993), and it can be monitored in vitro by the use of dying molecules. The aggregating process comprises two main phases: lag phase and elongation phase. Over the lag or nucleation phase, soluble monomers or dimers with random-coil structures form a nucleus that proceeds rapidly to the formation of fibrils. At the end of this phase, low-weight soluble oligomeric species are formed, which are spherical, globular, and described as micelles or amorphous aggregates. The molecular mass of these species varies between 25 and 50 KDa (Lambert et al., 1998). Throughout the elongation phase, the oligomeric species link together to form high-molecular weight oligomers of up to 1000 KDa approximately, (Huang et al., 2000). These prefibrillar species transform into protofilaments or protofibrils, which are short, flexible, and fibrillar. The protofibrils are the precursors of full-length fibrils that are formed by simple lateral association and structural organization. Mature fibrils are straight, unbranched, twisted and can reach up to 10 μm in length (Dobson and Batich, 2004).
The Aβ aggregation process is highly susceptible to many factors, including pH, ionic strength of the solvent, purification process or temperature. These are responsible for complications in reproducibility when experimentally assayed in vitro. Distinct oligomerisation and assembly processes between Aβ1−40 and Aβ1−42 have been described (Bitan et al., 2003). While Aβ1−42 exhibits higher neurotoxicity and is able to form hexamers, heptamers, or octamers, Aβ1−40 reaches equilibrium from monomers to tetramers. These variances are related to differences in the Ile-41-Ala-42 dipeptide at the C-terminus of Aβ. Until recently, it was assumed that fibrillary Aβ deposits were the responsible agents for the neuronal injury and neurodegenerative process of AD (Hardy and Higgins, 1992; Selkoe, 1994). However, later findings showed that soluble oligomeric species were able to disrupt synaptic function (Lambert et al., 1998) and recent data support the belief that soluble dimeric species are highly toxic (Jin et al., 2011). The mechanism by which Aβ induces cell damage has been reported to be caused by reactive oxygen species (ROS) production (Schubert et al., 1995), altered signaling pathways (Mattson, 1997), mitochondrial dysfunction (Shoffner, 1997), and interaction with biometals (Jin et al., 2011).
Both Aβ deposition and plaque formation also lead to local microglia activation, cytokine release, reactive astrocytosis, and multi-protein inflammatory responses (Heppner et al., 2015; Figure 2). The multifaceted biochemical and structural changes in surrounding axons, dendrites, and neuronal cell bodies also induce synapse loss as well as a remarkable cerebral atrophy in AD (Braak and Braak, 1994).
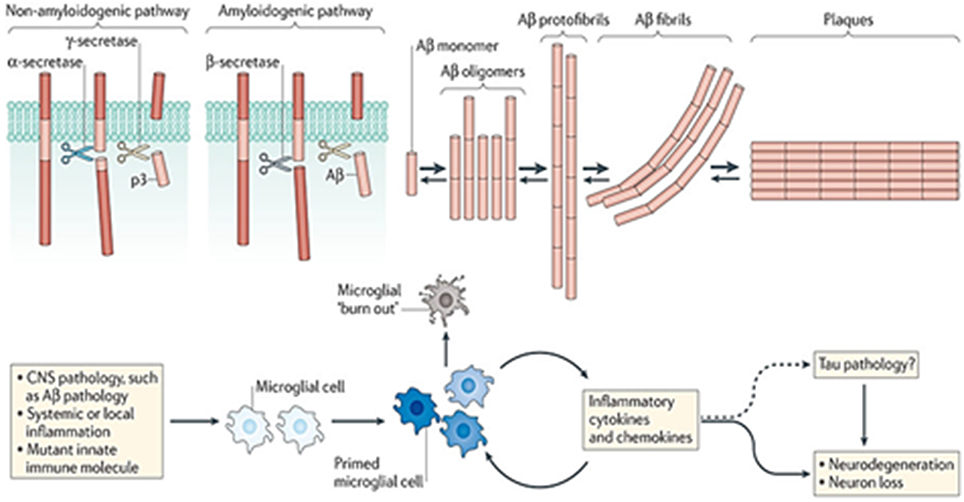
Figure 2. Role of APP cleavage processing in AD and microglial activation (Reproduced, with permission from Heppner et al., 2015).
Nonetheless, to date no correlation between Aβ accumulation with extent of neuronal loss and cognitive dysfunction has been reported. In addition, direct Aβ-peptide neurotoxicity has been difficult to be identified in animal models, suggesting the existence of key intermediates between amyloidosis and neurodegeneration (Nelson et al., 2012; Serrano-Pozo et al., 2013). In addition to these studies, genetic research has suggested that neurodegenerative processes occurring in AD are the consequence of an imbalance between Aβ peptide production and clearance. Since the postulation of the amyloid hypothesis of AD, extensive but, so far unsuccessful, efforts have been undertaken in clinical research in order to develop novel therapeutics, this hypothesis has continued to gain support over the last two decades, particularly from genetic studies.
Within any functional aerobic cell, the process involved in respiration inevitably generates ROS (Petersen et al., 2007). In particular, redox reactions are necessary for the generation of ATP and free radical intermediates are produced via the establishment of a proton gradient in oxidative phosphorylation. Multiple damaging mechanisms coexist in AD pathology, affecting each other at multiple levels (Von Bernhardi and Eugenín, 2012). In this respect, oxidative stress, which could be secondary to several other pathophysiological events, appears to be central in the progression of AD pathogenesis. Experimental evidence indicates that disturbances of the tissue redox state are important factors in early-stage AD, including the activation of multiple cell signaling pathways that contribute to the initial progression of the neurodegenerative process (Feng and Wang, 2012).
Evidence of ROS and reactive nitrogen species (RNS)-mediated injury has been reported in AD (Praticò, 2008). Increased levels of oxidative damage to biomolecules including proteins, lipids, carbohydrates, and nucleic acids have been detected in a number of studies (Moreira et al., 2008; Fukuda et al., 2009; Sultana et al., 2011). In addition, levels of antioxidant enzymes were found to be altered in specific AD brain regions (Sultana et al., 2011). Consequently, the “oxidative stress hypothesis of AD” emerged as a key factor in both the onset and progression of the disease. Oxidative stress is a widespread cellular process that currently lacks a specific treatment target such as a receptor or a single major metabolic pathway (Galasko et al., 2012). The wide variety of sources and sites of oxidative stress production paralleled by an even higher heterogeneity in the antioxidant responses. Specifically, the activity of cytochrome oxidase and pyruvate and α-ketoglutarate dehydrogenase complexes (key enzymes for the ATP supply) have been reported to be decreased as a result of oxidative damage (Aliev et al., 2011). In AD, there are established connexions between oxidative stress and other key AD events that amplify its complexity (Figure 3).
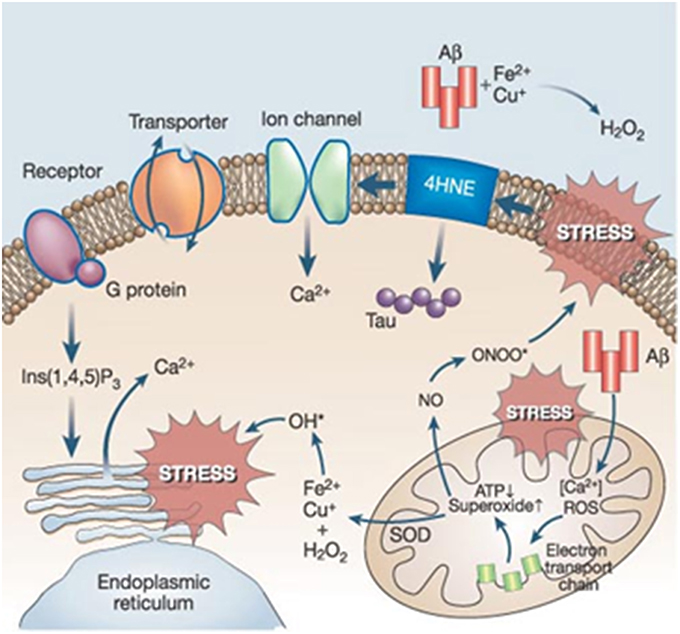
Figure 3. Prevailing connections between oxidative stress and other key players in AD (Reproduced, with permission from Mattson, 2004).
The presence of oxidation markers in the early stages of AD indicates that oxidative stress precedes the appearance of some other hallmarks (Rosini et al., 2014). Moreover, the accumulation of oxidative active modified products, such as 8-hydroxyguanosine (8-OHG) and nitrotyrosine in the cytoplasm of cerebral neurons from Down's syndrome patients has been reported to precede amyloid deposition (Odetti et al., 1998; Nunomura et al., 2000). This finding was also demonstrated in an APP transgenic mice model of AD (Smith et al., 1998). In post-mortem brains of myocardial infarction (MCI) patients, CSF, plasma, and urine increased levels of lipid peroxidation, nuclear acid oxidation, and diminished levels of antioxidant enzymes were found (Keller et al., 2005; Butterfield et al., 2006). Furthermore, heme oxygenase-1 (HO-1) levels were also reported to be increased in both AD and MCI post-mortem brain tissues (Schipper et al., 2006). Such findings might indicate increased oxidative stress as a general response to tissue damage in the brain, which may therefore contribute to its further progression.
Although Aβ and its aggregated senile plaques are undoubtedly involved in the neurodegenerative process of AD, the chronology of their presence has been now deemed secondary by many studies (Castellani et al., 2006; Zhu et al., 2007). Evidence suggests that secretion and deposition of Aβ within the neurons are compensatory measures taken by cells in effort to protect themselves against damage triggered by oxidative stress (Hayashi et al., 2007; Nakamura et al., 2007). Mitochondrial abnormalities, initially caused by gradual oxidative disturbances are major contributors of ROS to the cell (Bonda et al., 2010). Oxidative perturbances in mitochondrial operations result in impaired metabolic capacity (Wang et al., 2009) that would prevent the effective transfer of electrons along the respiratory chain during oxidative phosphorylation and the further generation of excessive ROS. This abnormality creates a vicious positive-feedback cycle where ROS produce oxidative stress that eventually yields more ROS along with other cellular detriments. ROS gradually accumulate in the cell and once a threshold is reached, it is no longer able to control the debilitating cycle propagation and a compensatory “steady state” is unavoidably initiated in order to regain control of its environment (Zhu et al., 2007). This hyper-defensive state, intended to prolong cell life, increases vulnerability to additional insults such as Aβ peptides. The cell therefore becomes subject to oxidative damage produced by oligomerisation and aggregation (Wang et al., 2009), that in turn, leads to neuroinflammation, mitochondrial damage, and further ROS generation.
In addition, cellular oxidative damage has also been linked to tau hyperphosphorylation and formation of NFTs (Lee et al., 2004). As a consequence of this aberrant cycle, cells succumb to neurodegeneration exhibiting the distinctive cognitive decline and dementia descriptive of AD (Zhu et al., 2007). Altogether, the primary role of oxidative stress in both AD onset and progression open the possibility of developing specific disease-modifying antioxidant approaches to confronting the disease.
Numerous studies in AD and other neurodegenerative disorders have described an increase in the levels of oxidative stress reflected by a deregulated content of metals iron, copper, and zinc in the brain of patients. Recent findings have strongly pointed to brain oxidative stress as one of the earliest changes in AD pathogenesis that might play a central role in the disease progression (Guglielmotto et al., 2010; Lee et al., 2010). Redox-active metals, especially Fe2+ and Cu2+ are capable of stimulating free radical formation via the Fenton reaction, thereby increasing protein and DNA oxidation and enhancing lipid peroxidation (Khalil et al., 2011).
Metal ions have also been reported to mediate Aβ toxicity in AD (Duce et al., 2010). The Aβ peptide itself has been shown to be a strong redox-active catalyst able to produce hydrogen peroxide and OH− in presence of copper or iron, which, in turn, are enriched in the amyloid cores of senile plaques (Huang et al., 1999, 2004). Metal ions can also interact with Aβ peptide enhancing its self-aggregation and oligomerisation at low physiological concentrations or under mildly acidic conditions (Huang et al., 1999). Moreover, metals can promote tau hyperphosphorylation and subsequent formation of NFTs by inducing aggregation upon tau interaction with Aβ (Yamamoto et al., 2002).
The presence of an iron-responsive element (IRE) in the 5′ untranslated regions (UTR) of AβPP mRNA was revealed as another link between iron metabolism and AD (Rogers et al., 2002; Silvestri and Camaschella, 2008). Iron closely upregulates intracellular levels of APP holo-protein by a mechanism that is similar to the translational control of ferritin L- and H- mRNAs through an IRE in their 5′ UTR (Rogers et al., 2002; Silvestri and Camaschella, 2008).
Copper is an essential trace element for all living animals. Copper levels are controlled by homeostatic mechanisms to prevent an excess, or a deficiency, that may trigger a unique set of adverse health effects. Copper can undergo redox cycling between Cu1+ and Cu2+ and the activities of some copper-containing enzymes, including superoxide dismutase (SOD), cytochrome-c oxidase, ceruloplasmin, and tyrosinase, have important biological functions. However, copper is also involved in the formation of free radicals by the Fenton reaction, in which free copper catalyses the formation of toxic hydroxyl radicals from physiologically available hydrogen peroxide (Barnham et al., 2004). Two classic disorders related to copper metabolism are Menkes and Wilson diseases, in which neurodegeneration is a common complication (Kodama et al., 1999; Merle et al., 2007).
In AD pathology, copper is mislocalized in brains, where decreased levels of this metal have been reported in affected regions (Deibel et al., 1996; Magaki et al., 2007) with enrichment in amyloid plaques and tangles (Lovell et al., 1998). Copper is released into the glutamatergic synaptic cleft facilitated by the copper-transporting P-type ATPase ATP7A at concentrations of ~15 μM (Hartter and Barnea, 1988; Hopt et al., 2003). There, it causes S-nitrosylation of NMDA receptors, inhibiting their activation (Schlief et al., 2005, 2006).
Aβ toxicity has been linked to the presence of redox metals, especially the binding to copper as well as the non-redox zinc ions (Hou and Zagorski, 2006; Tougu et al., 2008), although the precise mechanisms involved are still under investigation (Syme et al., 2004; Ma et al., 2006; Minicozzi et al., 2008; Alies et al., 2011). The copper binding site to Aβ1−42 has high affinity (Kd = 7.03 10−18 M; logKapp = 17.2), as measured by competitive metal-capture analysis, whereas that of Aβ1−40 is 10.3 (Atwood et al., 2000). The binding to copper has also been shown to modify and accelerate Aβ aggregation.
Copper also promotes dityrosine cross-linking of Aβ, which further accelerates Aβ aggregation (Atwood et al., 2000; Ali et al., 2005). The copper-induced Aβ oligomer contains a membrane-penetrating structure with histidine bridging (Curtain et al., 2001; Smith et al., 2006). This fact underlines the importance of histidine in the copper-Aβ interaction, which results in oligomers rather than the fibrils that are formed in the presence of zinc (Jiao and Yang, 2007). The copper-Aβ complex has been reported to exhibit cytotoxic properties (You et al., 2012) as this binding increases Aβ-induced cell death in cell culture (Wu et al., 2008; Perrone et al., 2010). Some hypotheses for this finding include the inhibition of cytochrome c (Crouch et al., 2005) and the increase of oxidative stress, triggered by the generation of hydrogen peroxide by Aβ and copper via a catalytic cycle (Huang et al., 1999). APP also interacts with copper by binding it between residues 142 and 166 (White et al., 1999; Barnham et al., 2003; Cappai et al., 2004) and catalytically oxidizing Cu1+ to Cu2+ (Multhaup et al., 1996). Furthermore, copper promotes APP internalization, whereas copper deficiency promotes Aβ secretion but not APP cleavage (Acevedo et al., 2011). Therefore, APP may not directly influence copper homeostasis, but inappropriate interaction with copper may be neurotoxic.
Tau phosphorylation and aggregation may also be induced by copper. Certain fragments in the four-repeat microtubule-binding domain of tau were shown to aggregate in the presence of copper in vitro (Ma et al., 2006; Zhou et al., 2007). Copper binding to tau induces hydrogen peroxide production in vitro (Su et al., 2007) and NFTs have also been shown to bind to copper in a redox-dependent manner as a source of ROS within the neuron (Sayre et al., 2000). In addition, copper exposure has been reported to induce tau hyperphosphorylation promoting tau pathology in a mouse model of AD (Kitazawa et al., 2009).
The brain is the main pool of zinc within the body (Frederickson, 1989). Zinc is an essential component for many enzymes and transcription factors. In healthy conditions, most of the zinc content is located in membrane-bound metalloproteins (MP I, II, and III), loosely bound to zinc within the cytoplasm, and vesicular zinc, enriched in synapses. It has several functional roles in signal transmission. Synaptic transmission releases high concentrations of zinc into the synaptic cleft (Frederickson, 1989; Vogt et al., 2000; Qian and Noebels, 2005), where it acts as an antagonist of GABA (Molnar and Nadler, 2001; Ruiz et al., 2004) and NMDA receptors (Paoletti et al., 1997; Traynelis et al., 1998; Vogt et al., 2000) and activates the G-protein-coupled receptor GPR39 (Besser et al., 2009).
Inconsistent reports of zinc levels in AD have been published. Early surveys of brain tissue found no difference in zinc levels between AD and controls. Yet, later studies showed a decrease in zinc levels in neo-cortex (Danscher et al., 1997), superior frontal and parietal gyri, medial temporal gyrus and thalamus and hippocampus (Corrigan et al., 1993; Panayi et al., 2002). However, in other reports elevated zinc levels in AD-affected amygdala, hippocampus and cerebellum, (Deibel et al., 1996; Danscher et al., 1997), olfactory areas (Samudralwar et al., 1995), and superior temporal gyrus (Religa et al., 2006) were described.
The cause of this apparent zinc dysregulation remains unknown, yet it may be involved in alterations of proteins such as metallothionein III, which is found in neurons and reduced in AD brains (Uchida et al., 1991; Yu et al., 2001). In contrast, metallothionein I/II was found increased in astrocytes of post-mortem and preclinical AD brains (Adlard et al., 1998). In AD pathogenesis, enriched zinc levels have been found in amyloid plaques (Bush et al., 1994c). Studies using microparticle-induced X-ray emission tomography demonstrated a three-fold in zinc concentrations-surrounding neuropils in the amigdala (Lovell et al., 1998). These findings were later corroborated by histochemistry (Suh et al., 2000), autometallographic tracing (Stoltenberg et al., 2005) and synchrotron-based infrared and X-ray imaging (Miller et al., 2006).
Aβ binds to zinc at residues 6–28 (Bush et al., 1993, 1994a,b,c), with up to three zinc ions bound to histidines 6, 13, and 14 (Damante et al., 2009), inducing the aggregation of Aβ into soluble precipitates (Bush et al., 1994c). Therefore, zinc, like copper, accelerates Aβ-induced toxicity and zinc sequestration into amyloid deposits (Bush et al., 1994c) (Faller et al., 2013), inducing loss of functional zinc in the synapses. This loss may contribute to cognitive decline in AD due to ZnT-3 depletion (Adlard et al., 2010).
Zinc may also interfere with APP processing and function as these processes are coordinated by secretases under the zinc regulation. This metal increases the synthesis of PS-1 (Park et al., 2001); however, the γ-secretase activity is inhibited by Zn2+ (Hoke et al., 2007). Zinc binds directly to Aβ, hiding its proteolytic cleavage site (Bush et al., 1994b) and therefore inhibiting its degradation by matrix metalloproteases (Crouch et al., 2009). Secreted Aβ is normally degraded by proteases such as neprilysin within a short period of time. However, zinc and other metals enhance the oligomerisation and accumulation of the amyloid protein.
After being incorporated into the membrane, the conformation of Aβ changes and it aggregates on the membranes. These aggregates are able to form channels, which unlike endogenous Ca2+ channels, are not regulated by standard channel blockers. Thus, a continuous flow of Ca2+ is initiated and disruption of calcium homeostasis triggers several apoptotic pathways, including free radical formation and tau phosphorylation leading to cell death. Conversely, zinc secreted into synaptic clefts, inhibits Aβ-induced Ca2+ entry, and thus confers a protective function in AD (Kawahara et al., 2014).
In agreement with these findings, in vivo studies revealed that high intake of dietary zinc triggered elevated expression of APP and enhanced amyloidogenic APP cleavage and Aβ deposition in APP/PS1 transgenic mice (Borchardt et al., 2000; Cuajuangco and Faget, 2003; Wang et al., 2010). Finally, zinc may also be involved in tau pathology as it is enriched in tangle-containing neurons (Suh et al., 2000). Zinc moderates translation of tau and modulates its phosphorylation by affecting the activities of GSK-3β, protein kinase B, ERK1/2, and c-Jun N-terminal kinase (An et al., 2005; Pei et al., 2006; Lei et al., 2011). Zinc can also bind to tau monomers, altering their conformation (Boom et al., 2009) and inducing both aggregation and fibrillation of this protein (Mo et al., 2009).
Iron is a fundamental element in biology for oxygen transport and energy metabolism. It is a transition element that exists in oxidative states from −2 to +8, although in biological systems only ferrous (Fe2+) and ferric (Fe3+) states normally exist. The cycling between ferric to ferrous states is used in biology for various redox reactions essential to life. However, deleterious reactions with oxygen, such as Fenton reaction, which is catalyzed by the free ferrous iron and may involve bound Fe(IV) (Freinbichler et al., 2009) are a source of oxidative stress (Zigler et al., 2015).
In the brain, iron is involved in development (Beard, 2003), metabolic and neurotransmitter systems (Agarwal, 2001). It is therefore regulated by multiple proteins, reflecting its involvement in different cellular functions. In AD brains, iron is enriched in both NFTs (Smith et al., 1998) and senile plaques (Goodman, 1953) with an estimated concentration of three-fold higher that of the normal neuropil levels (Lovell et al., 1998). Iron accumulation occurs in the cortex but not in the cerebellum (Andrasi et al., 1995; Duce et al., 2010). The iron storage protein ferritin binds to most iron within the brain and its levels increase with age and in AD (Bartzokis and Tishler, 2000). Transferrin (Tf) is an extracellular iron-transporting protein expressed in the brain that exchanges iron between cells. The complex Fe-Tf is endocytosed (Eckenroth et al., 2011) and iron is reduced to its ferrous state by an unknown ferrirreductase.
Recently, AD-associated APP was identified as a neuronal ferroxidase (Duce et al., 2010). APP-knockout mice exhibit iron accumulation in the brain and peripheral tissues and loss of APP ferroxidase activity in AD brains is coincident with iron retention in the tissues (Duce et al., 2010). Recent studies reported that iron-export capability of APP requires tau protein (Lei et al., 2012), which is involved in axonal transport (Lei et al., 2010) and binds to APP (Islam and Levy, 1997). Moreover, the loss of tau in mice is reported to cause age-related iron accumulation (Lei et al., 2012). The iron-regulatory system is disturbed in AD brains and ferritin has been reported to be elevated and co-localized with senile plaques expressed in astrocytes (Connor et al., 1992) and increased in frontal cortex of AD brains (Loeffler et al., 1995). However, the amount of APP was not significantly reduced in cortex (Duce et al., 2010) but both ferroxidase activity and soluble tau protein levels were observed to be decreased (Shin et al., 1992; Khatoon et al., 1994; Zhukareva et al., 2001; Van Eersel et al., 2009; Duce et al., 2010). Genetic factors have also been linked to higher susceptibility to iron burden in AD (Blázquez et al., 2007; Bertram and Tanzi, 2008; Percy et al., 2008).
Iron burden enriches around the senile plaque region (Meadowcroft et al., 2009) and promotes Aβ aggregation in vitro (Mantyh et al., 1993). Furthermore, iron-aggregated Aβ toxicity in cell culture (Schubert and Chevion, 1995) is mediated by ROS production (Rottkamp et al., 2001) or by the activation of Bcl-2/Bax-related apoptosis pathway (Kuperstein and Yavin, 2003). As mentioned above, iron also binds to tau protein, but only Fe3+ has reported to induce aggregation of hyperphosphorylated tau protein, which can be reversed by reducing Fe3+ to Fe2+ (Yamamoto et al., 2002) or by the use of iron-chelators (Amit et al., 2008). In AD brain, NFTs bind iron, which acts as a source of ROS within the neurons (Sayre et al., 2000). A decrease of tau phosphorylation in hippocampal neurons treated with Fe3+ (Egaña et al., 2003), corresponding with a decrease in CDK5 activity. Conversely, treatment with Fe2+ also resulted in hyperphosphorylation (Lovell et al., 2004) without inducing aggregation, possibly resulting from upstream activation of the ERK1/2, MAPK pathway (Muñoz et al., 2006; Huang et al., 2007). Recently, oxygen tension-dependent transcriptional factor, hypoxia inducible factor-1α (HIF-1α) has emerged as a potential target in neurodegenerative diseases. This protein regulates intracellular iron by binding to HIF-responsive elements (HREs) that are located within the genes of iron-related proteins such as Tf receptor and heme oxygenase-1 (Petousi and Robbins, 2014).
Recent research and clinical trials have confirmed that HIF-1α activation may be a potent strategy for postponing the pathogenesis and ameliorating the outcomes of AD. In this context, the use of iron chelators such as M30 has been reported to increase the levels of this protein by inducing the expression of its target genes VEGF and EPO. In addition, overexpression of HIF-1α has been shown to protect cultured cortical neurons from Aβ-induced neurotoxicity, through activating glycolytic and hexose monophosphate shunt-related enzymes. Therefore, an additional use of iron-chelators to overexpress HIF-1α may contribute to preventing neuron death and ameliorating the symptomatology of AD by inducing the expression of neuroprotection-related genes (Benkler et al., 2010).
Thus, in the context of AD pathology, the involvement of both oxidative stress and metal ions in AD (Ayton et al., 2013), indicates the use of antioxidant-chelators as potential beneficial agents in the prevention and treatment of the disease, by scavenging free radicals and, more importantly, by blocking their production, as well as restoring normal metal balance.
Although abnormal accumulation of metals has been reported in AD for many years, it has been only recently considered as a key early factor in AD, closely related to increased levels of both oxidative stress and amyloid beta-protein toxicity in Barone (2016). The development of metal-chelators has emerged, concurrently, as a novel pharmacological approach against this neurological disorder. In this regard Youdim and co-workers have reported a novel multifunctional molecule, M30, which combines iron-chelation with and monoamine oxidase (MAO) inhibitory properties as a possible drug for the treatment of AD (Zheng et al., 2005; Avramovich-Tirosh et al., 2007).
Inflammation is a defensive mechanism of the body against multiple threats such as infections and injury. It is a complex event that involves both soluble factors and specialized cells (Brown et al., 2007). Similar inflammatory processes occur in the brain and peripheral tissues. In the brain, glial cells, including astrocytes and microglia, undergo activation under pro-inflammatory conditions by the increase in the production of inflammatory cytokines in the CNS, which become deleterious and leads to progressive tissue damage in degenerative diseases (Morales et al., 2014). Infiltration by peripheral macrophages may also occur (Rogers et al., 1996).
In chronic disorders such as AD pathology, inflammation plays a critical role. It has been reported that insoluble fibrillar Aβ surrounding microglia, reactive astrocytes and dystrophic neurites contributes to the process of neuronal degeneration by initiating a series of cellular events which are able to elicit an immune response. Moreover, Aβ deposition in parenchyma and blood vessels has been described to trigger microglial migration and mediation of acute and chronic inflammatory response against the aggregates, thus inducing the production of nitric oxide (NO), ROS, pro-inflammatory cytokines such as tumor necrotic factor α (TNFα) or inteleukins-1β and -6 (IL-1β, IL-6) and prostaglandin E2 (PGE2), which eventually may promote neuronal death (Kitazawa et al., 2009).
One of the major observations in late AD is the loss of neuronal cells and brain shrinkage. Specific loss of dopaminergic cells has led to the use of deprenyl or selegiline as adjunctive therapy in PD to prevent removal of dopamine by metabolism (Birkmayer et al., 1977). The same principle applies in AD, where the decrease in all neurotransmitters is due mainly to a loss of neuronal connections.
Monoamine oxidases (MAO, EC 1.4.3.4) are flavin adenine dinucleotide (FAD)-containing enzymes that catalyse the oxidative deamination of primary, secondary, or tertiary amines, producing the corresponding aldehyde, hydrogen peroxide and ammonia. There are two isoforms of MAO present in mammals: MAO A and MAO B, which share ~70% sequence identity and are encoded by separate genes located on the X chromosome. The C-terminal regions of MAOs are transmembrane α-helices that anchor the enzymes to the mitochondrial outer membrane leaving the rest of the protein exposed to the cytoplasm. The entry of a substrate or inhibitor into the active site of MAOs is predicted to occur near the intersection of the enzyme with the surface of the membrane (De Colibus et al., 2005; Edmondson et al., 2007).
In selective MAO-knockout mice, the two MAO isoforms resulted in significantly different effects on both metabolism and behavior. Whereas, the lack of MAO A triggered aggressive phenotypes as a result of elevated levels of 5-HT and NA and lesser of DA (Chester et al., 2015), only the levels of 2-phenylethylamine were increased in MAO B-knockout mice, which exhibited traits such as sensation seeking, impulsiveness and extraversion. MAO A and MAO B were originally distinguished by their sensitivity to nanomolar concentrations of the acetylenic inhibitors clorgyline and l-deprenyl, respectively, as well as by their substrate specificities. The content of MAO varies over lifetime as MAO A appears before MAO B, the brain level of which increases dramatically after birth (Tsang et al., 1986; Strolin-Benedetti et al., 1992; Nicotra et al., 2004). MAO B levels continue to increase throughout the lifetime and in AD progression. In rat peripheral nervous system, MAO is localized in the endothelial cells of the endoneurial vessels, Schwann cells and unmyelinated axons of some neurons (Matsubayashi et al., 1986).
In human brain, MAO activity differs among regions. While the highest levels are found in basal ganglia and hippocampus, the lowest are observed in the cerebellum and neocortex (O'Carroll et al., 1983). The anatomical distribution of MAO isoforms in human brains were confirmed by positron-emission topography (PET) using intravenous 11C-labeled irreversible inhibitors (Fowler et al., 1987; Saura et al., 1992). Immunohistochemical studies have revealed that serotonergic neurons and astrocytes are rich in MAO B whereas catecholaminergic neurons mainly contain MAO A (Westlund et al., 1988). Caudate dopaminergic nerve endings contain only MAO A and small amounts of this isoform are also found in the serotonergic nerve terminals (O'Carroll et al., 1987). Some differences in the dopamine metabolism have been observed between species. For instance, only MAO A isoform is involved in rat brain, whereas MAO B is primary responsible for dopamine metabolism in human brain (Fornai et al., 1999).
Inhibitors of MAO, reversible and irreversible, equipotent on both isoforms or highly selective, have been used for more than 60 years for the treatment of depression (Mendelewicz and Youdim, 1983; Youdim et al., 2006). The most successful approach has been the use of selective irreversible inhibitors as exemplified by deprenyl, now used in PD therapy. The chemical moiety of this compound includes the propargyl group, which is readily incorporated into different scaffolds to cause MAO inhibition in compounds that are designed for other targets. This finding has been identified as one of the mechanisms of various MTDL to delay the progression of neurodegeneration as it will be discussed below.
At present, only five drugs have been approved by the U.S. Food and Drug Administration (FDA, USA) for use in AD. These compounds are mainly based on the “cholinergic hypothesis” and aimed at re-establishing functional cholinergic neurotransmission. Four of these: tacrine (1), rivastigmine (2), donepezil (3), and galantamine (4) are cholinesterase inhibitors, whereas memantine (5) is a NMDA receptor antagonist (Figure 4).
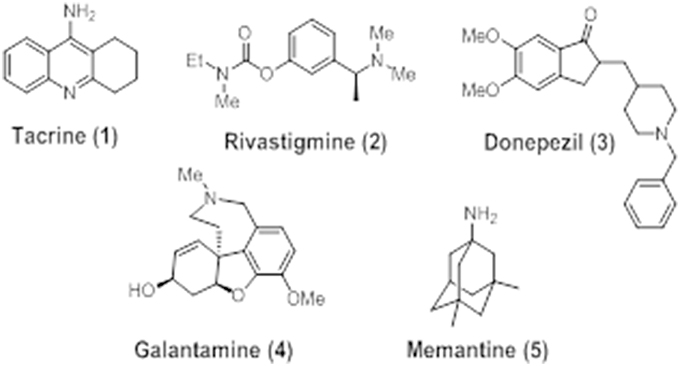
Figure 4. Chemical structures of FDA-approved agents for use in AD: tacrine (1), donepezil (2), rivastigmine (3), galantamine (4), and memantine (5).
Tacrine or tetrahydroaminoacridine (THA), marketed as Cognex®, was the first drug to be approved for use in AD by the FDA in 1993. It is a competitive AChE inhibitor with high lipid solubility (Nielsen et al., 1989) also able to interact with muscarinic receptors (Adem, 1993) and MAO A and B (Adem et al., 1989). Although it had some relatively small benefits on cognition, tacrine was withdrawn from the market in 2013 due to the high incidence of side effects, mostly related to hepatotoxicity (Qizilbash et al., 1998).
Rivastigmine, marketed under the trade name of Exelon® since 1998, is a non-selective pseudoreversible ChE inhibitor reported to produce enhanced benefits over AChE inhibition alone (Touchon et al., 2006; Bullock and Lane, 2007). A recent study using a transdermal patch reported a reduction in the prevalence of side effects as well as positive results after administering rivastigmine to mild-to-moderate AD patients. These studies reported better outcomes, in comparison with placebo group, for rate of cognitive function decline and daily living activities (Birks et al., 2015).
Galantamine (Razadyne®) is a well-studied drug approved in 2001 for the treatment of AD, possessing a dual mechanism of action to attenuate the symptoms of cognitive decline in AD. It is a reversible selective competitive AChE inhibitor (Greenblatt et al., 1999) that is able to simultaneously modulate nAChERs (Dajas-Bailador et al., 2003). The main mechanism of action of this molecule is the ability to increase the content of ACh, enhancing cholinergic neurotransmission and therefore improving cognition in AD patients by delay of ACh catabolism at synapse. Moreover, galantamine has also demonstrated to directly interact with the nAChER as low-affinity agonist, allosterically binding to a distinct binding site from where nicotinic agonists, such as ACh, choline, or carbachol, bind (Akk and Steinbach, 2005).Moriguchi et al. (2003) showed that galantamine also potentiates the activity of NMDARs, so that the dual mechanism of action on both cholinergic and glutamatergic systems in AD patients may elucidate the improvement in cognition, memory, and learning that have been found in clinical trials with this drug (Wilkinson and Murray, 2001).
Memantine, marketed as Namenda® or Ebixa®, is a glutamatergic agent and the first and only NMDAR antagonist approved in 2003 for the treatment of moderate-to-severe AD and dementia. In clinical trials, memantine has shown significant benefits on cognition, function, and global status of AD patients. Moreover, it appears to be harmless and well-tolerated, with a safety profile compared to placebo (Doody et al., 2007). Memantine binds to NMDARs with a low-micromolar IC50 value. Furthermore, it exhibits poor selectivity among NMDARs subtypes, with under micromolar IC50 values for NR2A, NR2B, NR2C, and NR2D receptors expressed in Xenopus oocytes (Parsons et al., 1999). Although memantine was initially regarded as a poor drug candidate for AD therapy, the mechanisms by which exerts clinical benefits and its safe profile have attracted a great considerable interest in the field of medicinal chemistry (Lipton, 2006). In addition, memantine has also been reported to exhibit neuroprotective activities against Aβ toxicity (Tremblay et al., 2000; Miguel-Hidalgo et al., 2002; Hu et al., 2007), tau phosphorylation (Song et al., 2008), neuroinflammation (Willard et al., 2000), and oxidative stress (Figueiredo et al., 2013; Liu et al., 2013).
Donepezil, marketed under the trade name of Aricept® in 1996, is a brain-permeable reversible non-competitive ChE inhibitor approved for use in AD (Birks and Harvey, 2006) and currently the most widely prescribed drug for the treatment of this disease. Donepezil is highly selective for AChE over BuChE activity (405:1) (Nochi et al., 1995). Compared to other approved AChE inhibitors, donepezil is similarly effective in ameliorating cognitive and functional decline in AD with comparable safety and tolerability. Moreover, beneficial effects have been observed at lower doses (5 mg/day), which is highly valuable at minimizing adverse reactions. Although clinical trials with donepezil have been reported, modest but reproducible, improvements in cognition and global functioning in treated patients, as compared to placebo. These effects were not permanent as patients, continued to exhibit a decline in cognitive functioning over time (Doody et al., 2007).
In recent years, the consistent failure of pharmacological approaches targeting traditional AD targets such as Aβ and tau protein together with the existing number of drugs to be tested in pre-dementia stages of the disease has left no effective disease-modifying treatment of AD in sight. Thus, additional benefits of donepezil might be revealed with the use of this molecule in combination with other active moieties.
The lack of therapeutic effectiveness of the current FDA-approved drugs based on the single-target paradigm for the treatment of cognition and memory decline, prompted to the rational design and development of a novel and improved pharmacological approach against AD: the Multi-Target-Directed Ligands (MTDL) or “dirty drugs” (Buccafusco and Terry, 2000; Youdim et al., 2005; León et al., 2013). These molecules are conceived to directly interact weakly with multiple targets associated with AD by the molecular hybridisation of different pharmacophore moieties from identified bioactive molecules. The development of MTDLs obviates the challenge of simultaneously administering multiple drugs with potentially different degrees of bioavailability, pharmacokinetics, and metabolism. Furthermore, this pharmacological approach also provides AD patients with a simplification of the therapeutic regimen (Figure 5).
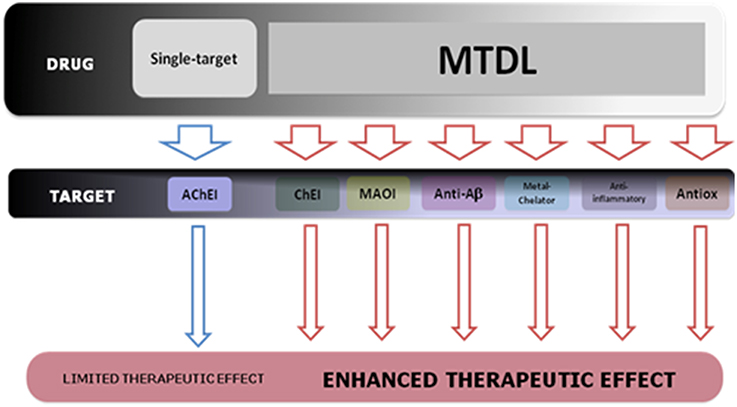
Figure 5. Scheme of the therapeutic design strategy of MTDL for the treatment of the multifaceted nature of AD pathology.
To date, the therapeutic potential of MTDLs for the treatment of complex neurodegenerative diseases and age-related cognitive impairment has not been realized. Multifaceted diseases such as AD, mainly caused by perturbations of the complex intracellular network that links to tissue and organs systems, needs a more comprehensive pharmacological approach. Therefore, approaches relying on targeting single proteins/mechanisms are not likely to have effective outcomes. Because of the existence of feedback mechanisms in biological systems, enzyme inhibition may not be able to decrease activity in long term, but instead, may result in increased activity, as reported from the use of rapid-reversible AChE inhibitors that significantly increased both protein activity and expression levels in the CSF of AD patients after long-term treatment (Darreh-Shori and Soininen, 2010).
Since 2005, literature has shown several promising results from applying this innovative approach to drug design. Well-known drugs such as donepezil, tacrine, or rivastigmine (Bolognesi et al., 2007; Samadi et al., 2011) as well as bioactive natural products such as curcumin (Malar and Devi, 2014), berberine (Jiang et al., 2011; Shan et al., 2011), or 8-hydroxyquinoline (Gomes et al., 2014) have been used as structural scaffolds for the development and search of new chemical entities with multiple properties or MTDLs for the treatment of AD. These new entities may be considered as simplified versions or lead drugs possessing great potential as real alternatives to the current unsuccessful pharmacological therapies for effectively fighting against AD and other complex diseases.
The next sections include an overview on the outcomes obtained from the biological assessment of several MTDLs molecules that have been lately reported by some of us. All the compounds described bear the N-benzylpiperidine group present in donepezil and the N-propargylamine motif present in PF9601N, a potent and selective MAO B inhibitor with neuroprotective effects in vitro and in vivo using different experimental models of neurodegenerative diseases (Cutillas et al., 2002; Pérez and Unzeta, 2003; Pérez et al., 2003; Battaglia et al., 2006; Sanz et al., 2009). Both scaffolds were linked by different heterocyclic ring systems, such as pyridine, indole or 8-hydroxyquinoline, allowing the facile synthesis of different MTDL molecules as promising drugs to be used in AD therapy (Figure 6). The inhibitory profile of these new MTDL molecules inhibiting ChE and MAO, their antioxidant, anti-β-aggregating, anti-inflammatory, and anti-apoptotic behavior together with their metal-chelating properties were determined and results from comparative studies were assessed. Taken together, the outcomes reported with these derivatives reveal a potential improvement of the current pharmacological therapy of AD.
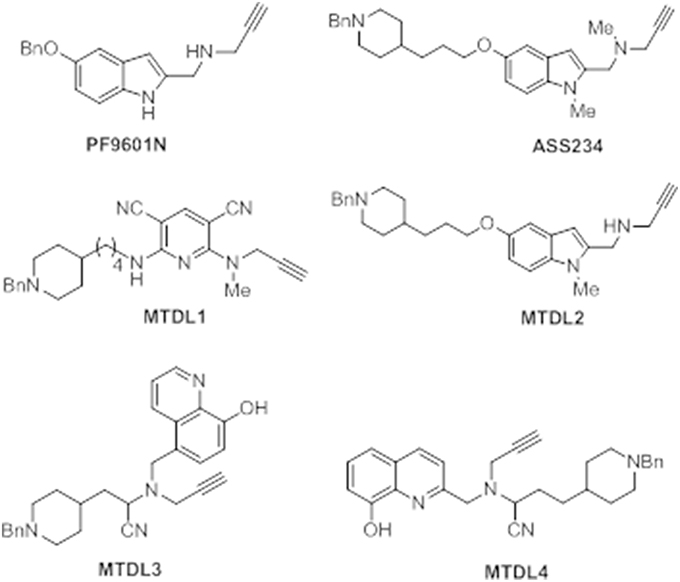
Figure 6. Structure of compounds PF9601N, ASS234, and the MTDL1-4 described in this review.
ASS234 {N-[5-(3-(1-benzylpiperidin-4-yl)propoxy)-1-methyl-1H-indol-2-ylmethyl]-N-methylprop-2-yn-1-amine} (Figure 7) was first identified by some of us as a suitable candidate for use in AD as a result of extensive screening of several series of novel derivatives. This multipotent molecule was specifically developed to combine the anti-cholinergic activity of donepezil, as an AChE inhibitor widely used in AD therapy, with a propargylamine moiety derived from selective MAO B inhibitor, PF9601N (Figure 7) with neuroprotective properties (Bolea et al., 2011).
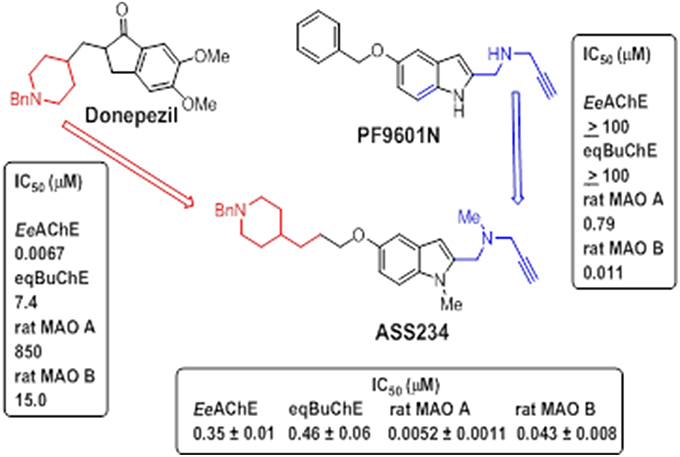
Figure 7. Design strategy of ASS234. IC50 values (in μM) of donepezil, PF9601N and ASS234 inhibiting both ChEs and MAO activities are shown Bolea et al. (2011).
ASS234 was revealed as a potent inhibitor of both MAO A and MAO B, with an IC50 values of 5.2 ± 1.1 nM and 43.1 ± 7.9 nM, respectively. It also inhibited both ChEs exhibiting IC50 values of 0.35 ± 0.001 and 0.46 ± 0.06 μM toward AChE and BuChE, respectively. In comparison with donepezil and PF9601N, analyzed under the same experimental conditions, donepezil was ineffective at inhibiting MAO activities whereas PF9601N potently and selectively inhibited MAO B isoform but displayed no interaction with both ChEs (Bolea et al., 2011). These data indicate that ASS234 combines the desirable properties of donepezil and PF9601N being capable of simultaneously enhancing both cholinergic and monoaminergic transmission as well as possessing neuroprotective effects (Bolea et al., 2013).
Kinetic studies showed that ASS234 is a-reversible inhibitor of both ChEs, with micromolar affinity, and a highly potent irreversible MAO A inhibitor with similar behavior to clorgyline (Esteban et al., 2014). The parent propargylamine, PF9601N, behaves similarly to l-deprenyl as an effective selective MAO B inhibitor. Both PF9601N and ASS234 display as mechanism-based MAO inhibitors. The selectivity of ASS234 as a MAO A inhibitor, indicated by the IC50 values, for both purified and membrane-bound protein samples (Pérez et al., 1999; Bolea et al., 2011), might be attributed to the shared propargylamine structure, which is larger and more hydrophobic in ASS234. The initial reversible binding parameter (Ki value of 0.2 μM) indicated that ASS234 has slightly lower affinity for MAO A than clorgyline (Ki value of 0.04 μM), and this was also reflected in higher rate constant (k1) for the irreversible reaction.
UV-VIS spectral analyses showed an irreversible modification of the MAO flavin group by ASS234 similar to that found with other propargyl MAO inhibitors. The crystal structure of human MAO B in complex with ASS234 revealed the formation of a covalent adduct with the N5 atom of the flavin cofactor. The N-benzylpiperidine moiety of the compound was not able to fully bind to the intact molecule to the MAO B active site, which ruled out the possibility that the inhibitor may have undergone degradation. Thus, these outcomes demonstrated the efficacy of ASS234 as inhibitor of both the “classic” AD-targeted ChE enzymes and the MAOs (Esteban et al., 2014).
Following the search for additional activities with multi-target ASS234, the potential modulation of the monoaminergic transmission and metabolism by this molecule were also investigated both in vitro and in vivo (Van Schoors et al., unpublished results). Since some of the behavioral alterations occurring in AD such as depression are possibly caused by monoaminergic dysfunction, the therapeutic use of antidepressant drugs based on the selective inhibition of MAO A, an enzyme possessing an indispensable role in the metabolic regulation of neurotransmitters 5-HT, NA, DA, and/or on the blockage of the corresponding reuptake systems at the pre-synaptic nerve endings, may also be considered.
In this context, a significant increase in the levels of 5-HT associated with a reduction in the levels of its metabolite 5-HIAA was observed as a result of irreversible inhibition of MAO A in SH-SY5Y cells treated with ASS234. Comparatively, similar though less pronounced effects were found with highly-selective MAO A inhibitor clorgyline under the same experimental conditions, thus revealing an active effect of this multi-target drug to in enhancing 5-HT levels. In contrast, DA levels were significantly decreased in PC12 cells that had been treated with ASS234 for 24 h. This finding may be due to a reduction of both activity and expression of the enzymes responsible for DA synthesis, tyrosine hydroxylase and aromatic L-amino acid decarboxylase as a response to initial rapid elevation of DA levels following the blockage of MAO activity. Both MAO A inhibitors ASS234 and clorgyline were able to modulate the levels of DA metabolites DOPAC and HVA, which were decreased after incubations. In vivo microdialysis studies of the effects upon administration of ASS234 in freely-moving rats also revealed alterations in the extracellular monoamines levels in both hippocampus and prefrontal cortex, two brain areas that are highly-impaired in AD. Interestingly, the effect of ASS234 differed from brain areas and dosage. In hippocampus, levels of 5-HT and NA were increased whereas those of DA and NA markedly augmented in prefrontal cortex.
These differences might be attributed to the presence of metabolizing enzymes in both brain areas as well as to the complexity of the monoaminergic neurochemical interactions existing in the brain. In addition, ASS234, as a multi-target compound, might be able to modulate other neurobiological systems and therefore producing a complex pharmacological outcome. The results from the in vivo studies revealed a time-dependent effect on extracellular levels of monoaminergic neurotransmitters after administration of a single, subcutaneous, dose of ASS234. This results could be used in further studies to assess both the pharmacokinetics and bioavailability of the compound in reaching its therapeutic target. Many factors, including route of administration and the existence of active metabolites may contribute to the observed delay in the in vivo effects observed with ASS234.
In AD, levels of DA and NA are diminished in cortex and hippocampus while those of 5-HT are decreased in hippocampus (Reinikainen et al., 1988), producing some distinctive behavioral symptoms including depressive-like disorder, psychosis, or memory impairment related to alterations in serotonergic, catecholaminergic, and cholinergic neurotransmissions (Vermeiren et al., 2015). Therefore, the outcomes observed in vivo upon the administration of ASS234 in addition with those previously found, confirm the potential value of this compound to be used as a modulator of the monoaminergic transmission in AD, both in terms of therapy and for further elucidation of the mechanisms involved.
The therapeutic potential of ASS234 has been also evaluated following its administration to a rat model of vascular dementia (Stasiak et al., 2014). These experiments involved the permanent bilateral occlusion of the common carotid arteries (BCCAO) with experimental vascular dementia. In this rat model, the administration of ASS234 for five consecutive days resulted in a potent and selective inhibition of brain MAO A activity as well as a concurrent increase in the concentrations of serotonin and catecholamines dopamine and noradrenaline. BCCAO resulted in impaired time parameters and memory functions as measured by hole-board memory tests in rats. Treatment of BCCAO rats with ASS234 showed less negative effects on cognitive parameters and exerted a significant positive effect on working memory.
Aggregation and deposition of Aβ is a key pathological hallmark of AD and there is increasing evidence suggesting that the neurotoxicity of this peptide is related to the formation of toxic oligomeric aggregates. In this regard, ASS234 has demonstrated to possess anti-Aβ1−42 aggregating capacity and also an ability to reduce the presence of oligomeric forms of β-amyloid, as the most toxic species in AD (Bolea et al., 2013). Moreover, ASS234 was able to inhibit AChE-catalyzed Aβ aggregation by binding to the PAS of the enzyme. ASS234 also showed a protective effects on the Aβ1−42-mediated cytotoxicity induced in human neuroblastoma cells by preventing the activation of the intrinsic mitochondrial pathway of apoptosis. Moreover, ASS234 displayed significant antioxidant properties, arising from the increase in the expression of catalase and SOD-1 in human neuroblastoma SH-SY5Y cells. This compound also has the capacity to capture free radicals in vitro, as measured by the well-established ORAC-FL (Oxygen radical Absorbance Capacity) fluorescent method. It is noteworthy that in a recent in vivo approach using transgenic APP/PS1AE9 mice given ASS234, both Aβ plaques in cortex and activated glia were found decreased (Serrano et al., 2016).
These findings, summarized in Figure 8, allow us to conclude that ASS234 is able to interact with multiple targets relevant to AD. They also indicate that this is an interesting MTDL molecule that should be considered for therapeutic development against AD.
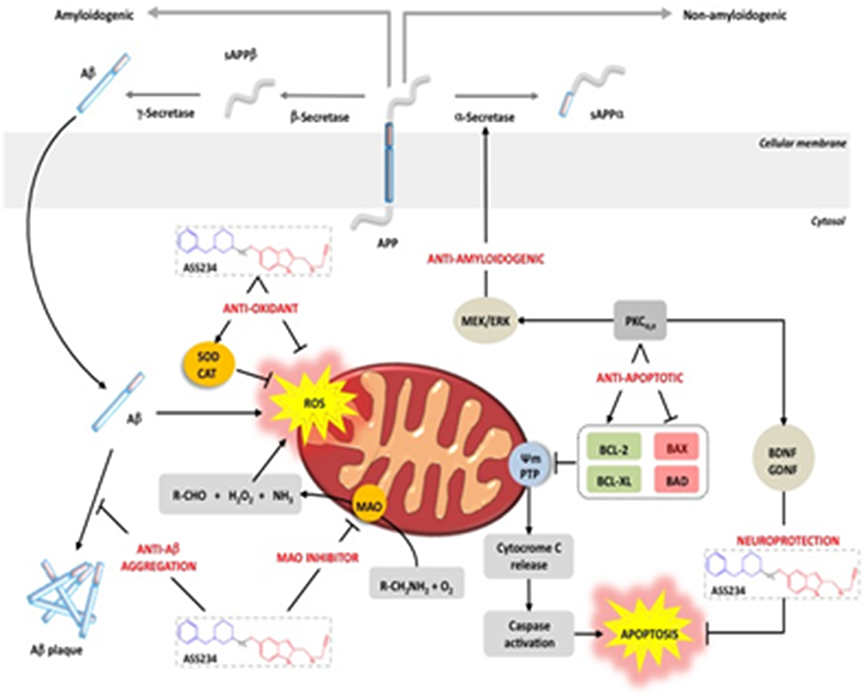
Figure 8. Schematic representation of ASS234 targets involved in AD pathogenesis. ASS234 forms a N5 flavin adduct (like clorgyline) with MAO A and it is able to block AChE-induced Aβ aggregation. ASS234 shows antioxidant and anti-apoptotic properties and it is able to induce neuroprotection through the Wnt pathway. ASS234 also shows less toxicity than donepezil in HepG2 cells (With permission of Bolea et al., 2013b).
With the aim of searching for improved MTDLs, two series of novel structurally-derived compounds, derived from ASS234 as multipotent donepezil-indolyl and donepezil-pyridyl hybrids were designed and pharmacologically evaluated for their potential use in AD. The synthetic strategy that was used to design these novel donepezil-pyridyl hybrids is outlined in Figures 9, 10.
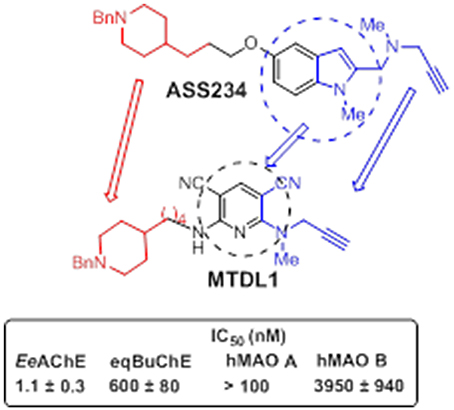
Figure 9. Designed structure of hybrid MTDL1 and IC50 values for the inhibition of ChE and MAO enzymes (Bautista-Aguilera et al., 2014a).
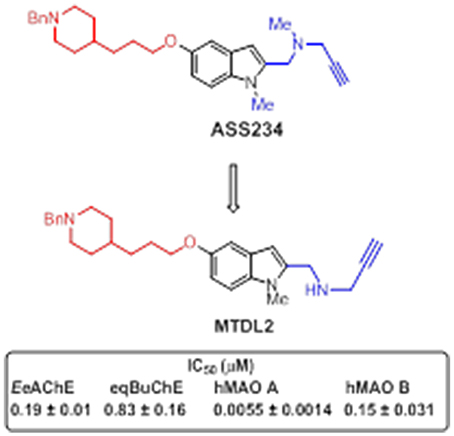
Figure 10. Structure of hybrid MTDL2 and IC50 values for the inhibition of ChEs and MAO enzymes (Bautista-Aguilera et al., 2014b).
Among all the compounds tested, the donepezil-pyridyl hybrid MTDL-1 (Figure 9) was identified as a potent AChE inhibitor (IC50 = 1.1 nM) and a moderate BuChE inhibitor (IC50 = 0.6 μM) with total selectivity toward human MAO B (Bautista-Aguilera et al., 2014a). Interestingly, the design and development of non-selective ChE inhibitors for use in AD appears of valuable pharmacological concern, as the hydrolysis of neurotransmitter ACh may largely occur via BuChE catalysis in aged AD brains, the activity levels of which have been found elevated on late-stages of the disease as it is also the case with MAO B. Since BuChE is also found in glial cells that are recruited and activated around the plaques and tangles, the inhibition of this enzyme might provide additional benefits at reducing neuroinflammation. Suitable drug-likeness profile and ADMET properties of these two novel MTDLs were also confirmed by 3D-QSAR studies.
The donepezil-indolyl hybrid molecule MTDL-2 (Figure 10), exhibited an interesting profile as potent MAO A inhibitor (IC50 = 5.5 nM) that was moderately able to inhibit MAO B (IC50 = 150 nM), AChE (IC50 = 190 nM), and BuChE (IC50 = 830 nM) (Bautista-Aguilera et al., 2014b). Moreover, this compound appeared to be a mixed-type AChE inhibitor able to span both the CAS and PAS of this enzyme, as found by molecular modeling studies. Interestingly, docking simulations revealed that the selective inhibition of MAO isoforms by propargyl-containing compounds accounts for the orientation on their propargylamine and phenyl moieties in MAO, which energetically affects the interaction with the active site.
Newly-designed MTDL-3 and MTDL-4 derivatives containing the N-benzylpiperidine moiety from donepezil and a metal-chelating 8-hydroxyquinoline group (Figures 11, 12), were linked to a central N-propargylamine core and pharmacologically evaluated (Wang et al., 2014; Wu et al., 2015). This molecule contains a moiety also present in the compound M30, a brain-permeable and iron-chelating compound with antioxidant activity displaying neuroprotective activity in animal models (Salkovic-Petrisic et al., 2015). M30 modulates HIF-α-related glycolytic genes in the frontal cortex of APP/PS1 mice used as AD model (Mechlovich et al., 2014). These studies also showed M30 to have beneficial effects on several major hallmarks of AD (Kupershmidt et al., 2012), indicating the potential value of incorporating this moiety into MTDL-3 (Figure 11).
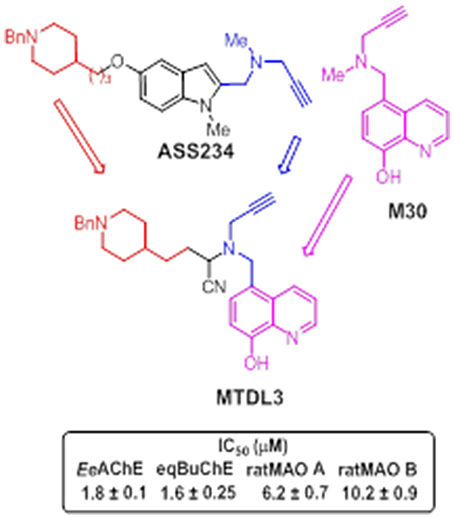
Figure 11. Structure of hybrid MTDL3 and IC50 values for the inhibition of ChEs and MAO enzymes (Wang et al., 2014).
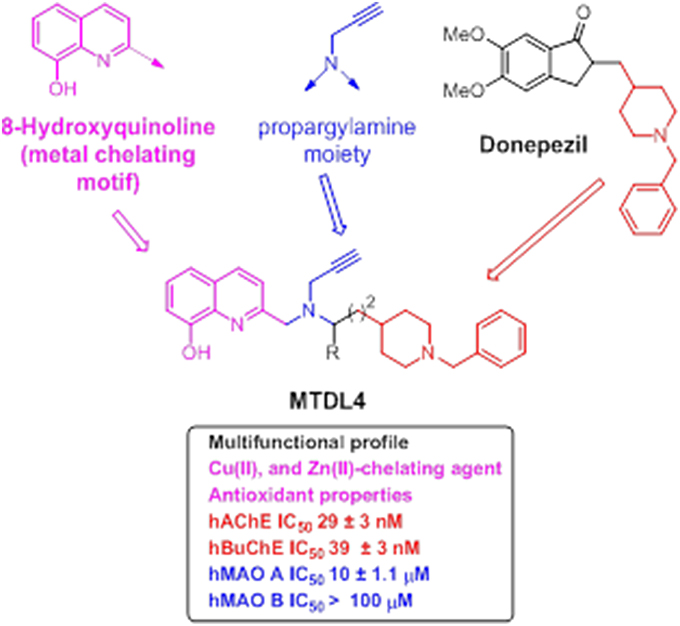
Figure 12. Structure of hybrid MTDL-4 and IC50 values for the inhibition of ChEs and MAO enzymes (Wu et al., 2015).
MTDL-3 was revealed as an irreversible MAO inhibitor and mixed-type ChE inhibitor, in low micromolar range, able to strongly complex Cu (II), Zn (II), and Fe (III) (Wang et al., 2014). In addition, the cyano group, which was only present in some derivatives, was found to enhance the binding to AChE, BuChE, and MAO A (Wang et al., 2014). Although α-aminonitriles have seldom been investigated as ChE inhibitors, some previous studies described nitriles as MAO inhibitors, and reported that cyanide potentiates irreversible MAO inhibition (Ramadan et al., 2007). Although cyanide is known to be a poor reversible inhibitor of the oxidation of benzylamine by MAO (Houslay and Tipton, 1973), previous studies have indicated that the inhibitory activity of several compounds against MAO is potentiated by cyanide (Davison, 1957; Ramadan and Tipton, 1998; Ramadan et al., 1999, 2007; Juárez-Jiménez et al., 2014). An earlier study by Davison (1957) on the inhibition of mitochondrial MAO by the irreversible inhibitor iproniazid revealed that, although at low concentrations, this compound alone had little effect on enzyme activity, while the inhibitory activity was significantly enhanced in the presence of cyanide ions. A similar potentiating effect was also reported for phenelzine and pheniprazine (Ramadan et al., 1999). Such studies demonstrated that, in case of pheniprazine, cyanide potentiates the inhibition by increasing the apparent binding affinity to MAO without producing significant changes in the rate of the reaction that yields the irreversibly inhibited species (Ramadan et al., 2007). This finding suggests that the potentiating effect of cyanide can be ascribed to an activation mechanism whereby cyanide assists the binding of the inhibitor to the enzyme.
Theoretical ADMET analyses, showed MTDL-3 to exhibit good drug-likeness properties and brain penetration capacity for CNS activity. Moreover, less toxicity than donepezil in HepG2 cells was also revealed with these derivatives. These findings are important, of noteworthy relevance since the simultaneous administration of multiple therapeutic agents (polypharmacology) may lead to potentially lethal side effects triggered by drug-to-drug interactions. Therefore, such undesirable outcomes might be significantly reduced by the use of multi-target derivatives such as MTDL-3. Furthermore, these molecules also displayed good performance in terms of translational science since scopolamine-induced memory deficits were partially restored by MTDL-3, confirming its effective pharmacokinetics in vivo.
Another related molecule, MTDL-4 (Figure 12), showed similar behavior as dual ChE/MAO inhibitor (Wu et al., 2015), which was identified from an initial pharmacological screening as an appropriate lead compound for further investigation. Subsequent investigations revealed further potentially valuable properties of this molecule.
Overall, the findings presented in this review robustly reinforce the suitability of MTDLs as an appropriate pharmacological approach to target disease progression in AD therapy. Amongst all the compounds tested, multi-target MTDL-3 particularly emerged as a ligand possessing a number of remarkable potential benefits for use in this neurological disorder including well-balanced dual AChE/MAO inhibition, strong metal-chelating activity, neuroprotective and anti-apoptotic properties, potent antioxidant capacities and anti-inflammatory action. Furthermore, MTDL-3 has effects as enhancer of cognitive functions. This prompts us to propose this molecule as the first racemic α-aminonitrile identified so far as a multifunctional chelator for biometals able to interact in two key enzymatic systems implicated in AD. We consider it as an important new lead compound that merits further investigation for the potential treatment of this disease.
MU, GE, JM, wrote the manuscript. RR, KT, and WF, corrected and revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
MU, GE, JM thank to MIneco (Spain) for support Grant SAF 2012-33304, SAF 2015-65586R. All authors thank EU COST Action CM1103 for its support.
AD, Alzheimer's disease; Aβ, amyloid beta; NFT, neurofibrillary tangles; AChE, acetylcholinesterase; BuChE, butyrylcholinesterase; MAO, monoamine oxidase; APP, amyloid precursor protein; ROS, reactive oxygen species; MTDL, multi-target-directed ligand.
Acevedo, K. M., Hung, Y. H., Dalziel, A. H., Li, Q. X., Laughton, K., Wikhe, K., et al. (2011). Copper promotes the trafficking of the amyloid precursor protein. J. Biol. Chem. 286, 8252–8262. doi: 10.1074/jbc.M110.128512
Adem, A. (1993). The next generation of cholinesterase inhibitors. Acta Neurol. Scand. Suppl. 149, 10–12. doi: 10.1111/j.1600-0404.1993.tb04246.x
Adem, A., Jossan, S. S., and Oreland, L. (1989). Tetrahydroaminoacridine inhibits human and rat brain monoamine oxidase. Neurosci. Lett. 107, 313–317. doi: 10.1016/0304-3940(89)90837-9
Adlard, P. A., Parncutt, J. M., Finkelstein, D. I., and Bush, A. I. (2010). Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer's disease? J. Neurosci. 30, 1631–1636. doi: 10.1523/jneurosci.5255-09.2010
Adlard, P. A., West, A. K., and Vickers, J. C. (1998). Increased density of metallothionein I/II-immunopositive cortical glial cells in the early stages of Alzheimer's disease. Neurobiol. Dis. 5, 349–356. doi: 10.1006/nbdi.1998.0203
Agarwal, K. N. (2001). Iron and the brain: neurotransmitter receptors and magnetic resonance spectroscopy. Br. J. Nutr. 85, S147–S150. doi: 10.1079/bjn2000307
Akk, G., and Steinbach, J. H. (2005). Galantamine activates muscle-type nicotinic acetylcholine receptors without binding to the acetylcholine-binding site. J. Neurosci. 25, 1992–2001. doi: 10.1523/JNEUROSCI.4985-04.2005
Ali, F. E., Separovic, F., Barrow, C. J., Cherny, R. A., Fraser, F., Bush, A. I., et al. (2005). Methionine regulates copper/hydrogen peroxide oxidation products of Abeta. J. Pept. Sci. 11, 353–360. doi: 10.1002/psc.626
Alies, B., Eury, H., Bijani, C., Rechignat, L., Faller, P., and Hureau, C. (2011). pH-dependent, Cu(II) coordination to amyloid-beta peptide: impact of sequence alterations, including the H6R and D7N familial mutations. Inorg. Chem. 50, 11192–11201. doi: 10.1021/ic201739n
Aliev, G., Li, Y., Palacios, H. H., and Obrenovich, M. E. (2011). Oxidative stress induced mitochondrial DNA deletion as a hallmark for the drug development in the context of the cerebrovascular diseases. Recent Pat. Cardiovasc. Drug Discov. 6, 222–241. doi: 10.2174/157489011797376942
Amit, T., Avramovich-Tirosh, Y., Youdim, M. B. H., and Mandel, S. (2008). Targeting multiple Alzheimer's disease etiologies with multimodal neuroprotective and neurorestorative iron chelators. FASEB J. 22, 1296–1305. doi: 10.1096/fj.07-8627rev
An, W. L., Bjorkdahl, C., Liu, R., Cowburn, R. F., Winblad, B., and Pei, J. J. (2005). Mechanism of zinc-induced phosphorylation of p70 S6 kinase and glycogen synthase kinase 3beta in SH-SY5Y neuroblastoma cells. J. Neurochem. 92, 1104–1115. doi: 10.1111/j.1471-4159.2004.02948.x
Andrasi, E., Farkas, E., Scheibler, H., Reffy, A., and Bezur, L. A. (1995). 'Zn, Cu, Mn and Fe levels in brain in Alzheimer's disease. Arch. Gerontol. Geriatr. 21, 89–97. doi: 10.1016/0167-4943(95)00643-Y
Arendt, T., and Bigl, V. (1986). Alzheimer plaques and cortical cholinergic innervation. Neuroscience 17, 277–279. doi: 10.1016/0306-4522(86)90243-5
Atwood, C. S., Scarpa, R. C., Huang, X., Moir, R. D., Jones, W. D., Fairlie, D. P., et al. (2000). Characterization of copper interactions with Alzheimer amyloid beta peptides: identification of an attomolar-affinity copper binding site on amyloid beta1-42. J. Neurochem. 75, 1219–1233. doi: 10.1046/j.1471-4159.2000.0751219.x
Avramovich-Tirosh, Y., Amit, T., Bar-Am, O., Zheng, H., Fridkin, M., and Youdim, B. (2007). Therapeutic targets and potential of the novel brain- permeable multifunctional iron chelator-monoamine oxidase inhibitor drug, M-30, for the treatment of Alzheimer's disease. J. Neurochem. 100, 490–502. doi: 10.1111/j.1471-4159.2006.04258.x
Ayton, S., Lei, P., and Bush, A. I. (2013). Metallostasis in Alzheimer's disease. Free Radic. Biol. Med. 62, 76–89. doi: 10.1016/j.freeradbiomed.2012.10.558
Barnham, K. J., Masters, C. L., and Bush, A. I. (2004). Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 3, 205–214. doi: 10.1038/nrd1330
Barnham, K. J., McKinstry, W. J., Multhaup, G., Galatis, D., Morton, C. J., Curtain, C. C., et al. (2003). Structure of the Alzheimer's disease amyloid precursor protein copper binding domain: a regulator of neuronal copper homeostasis. J. Biol. Chem. 278, 17401–17407. doi: 10.1074/jbc.M300629200
Barone, E. (2016). Oxidative stress and Alzheimer's Disease: Where do we stand? Curr. Alzheimer Res. 13, 108–111. doi: 10.2174/156720501302160101123849
Bartzokis, G., and Tishler, T. A. (2000). MRI evaluation of basal ganglia ferritin iron and neurotoxicity in Alzheimer's and Huntingon's disease. Cell. Mol. Biol. 46, 821–833.
Battaglia, V., Sanz, E., Salvi, M., Unzeta, M., and Toninello, A. (2006). Protective effect of PF 9601N on mitochondrial permeability transition pore. Cell. Mol. Life Sci. 63, 1440–1448. doi: 10.1007/s00018-006-6105-8
Bautista-Aguilera, O. M., Esteban, G., Bolea, I., Nikolic, K., Agbaba, D., Moraleda, I., et al. (2014b). Design, synthesis, pharmacological evaluation, QSAR analysis, molecular modeling and ADMET of novel donepezil-indolyl hybrids as multipotent cholinesterase/monoamine oxidase inhibitors for the potential treatment of Alzheimer's disease. Eur. J. Med. Chem. 75, 82–95. doi: 10.1016/j.ejmech.2013.12.028
Bautista-Aguilera, O. M., Esteban, G., Chioua, M., Nikolic, K., Agbaba, D., Moraleda, I., et al. (2014a). Multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer's disease: design, synthesis, biochemical evaluation, ADMET, molecular modeling, and QSAR analysis of novel donepezil-pyridyl hybrids. Drug Des. Devel. Ther. 8, 1893–18910. doi: 10.2147/DDDT.S69258
Beard, J. (2003). Iron deficiency alters brain development and functioning. J. Nutr. 133, 1468S–1472S.
Benkler, C., Offen, D., Melamed, E., Kupershmidt, L., Amit, T., Mandel, S., et al. (2010). Recent advances in amyotrophic lateral sclerosis research: Perspectives for personalized clinical application. EPMA J. 1, 343–361. doi: 10.1007/s13167-010-0026-1
Bertram, L., and Tanzi, R. E. (2008). Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat. Rev.Neurosci. 9, 768–778. doi: 10.1038/nrn2494
Besser, L., Chorin, E., Sekler, I., Silverman, W. F., Atkin, S., Russell, J. T., et al. (2009). Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J. Neurosci. 29, 2890–2901. doi: 10.1523/JNEUROSCI.5093-08.2009
Biesold, D., Inanami, O., Sato, A., and Sato, Y. (1989). Stimulation of the nucleus basalis of Meynert increases cerebral cortical blood flow in rats. Neurosci. Lett. 98, 39–44. doi: 10.1016/0304-3940(89)90370-4
Birkmayer, W., Riederer, P., Ambrozi, L., and Youdim, M. (1977). Implications of combined treatment with “Madopar” and l-Deprenyl in Prkinson's disease. Lancet 1, 439–443. doi: 10.1016/S0140-6736(77)91940-7
Birks, J., Grimley-Evans, J., Iakovidou, V., and Tsolaki, M. (2015). Rivastigmine for Alzheimer's disease. Cochrane Database Syst. Rev. 4:CD001191. doi: 10.1002/14651858.CD001191
Birks, J. S., and Harvey, R. J. (2006). Donepezil for dementia due to Alzheimer's disease. Cochrane Database Syst. Rev. 1:CD001190. doi: 10.1002/14651858.CD001190.pub2
Bitan, G., Kirkitadze, M. D., Lomakin, A., Vollers, S. S., Benedek, G. B., and Teplow, D. B. (2003). Amyloid beta -protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. U.S.A. 100, 330–335. doi: 10.1073/pnas.222681699
Blázquez, L., De Juan, D., Ruiz-Martinez, J., Emparanza, J. I., Saenz, A., Otaegui, D., et al. (2007). Genes related to iron metabolism and susceptibility to Alzheimer's disease in Basque population. Neurobiol. Aging 28, 1941–1943. doi: 10.1016/j.neurobiolaging.2006.08.009
Bolea, I., Gella, A., Monjas, L., Pérez, C., Rodríguez-Franco, M. I., Marco-Contelles, J., et al. (2013a). Multipotent, permeable drug ASS234 inhibits Aβ aggregation, possesses antioxidant properties and protects from Aβ-induced apoptosis in vitro. Curr. Alzheimer Res. 10, 797–808. doi: 10.2174/15672050113109990151
Bolea, I., Gella, A., and Unzeta, M. (2013b). Propargylamine-derived multitarget-ligands: fighting Alzheimer's disease with monoamine oxidase inhibitors. J. Neural. Trans. 120, 893–902. doi: 10.1007/s00702-012-0948-y
Bolea, I., Juárez-Jiménez, J., de Los Ríos, C., Chioua, M., Pouplana, R., Luque, F. J., et al. (2011). Synthesis, biological evaluation, and molecular modeling of donepezil and N-[(5-(benzyloxy)-1-methyl-1H-indol-2-yl)methyl]-N-methylprop-2-yn-1-amine hybrids as new multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer's disease. J. Med. Chem. 54, 8251–8270. doi: 10.1021/jm200853t
Bolognesi, M. L., Cavalli, A., Luca-Valgimigli, L., Bartolini, M., Rosini, M., Andrisano, V., et al. (2007). Multitarget-directed drug design strategy: from a dual binding site acetylcholinesterase inhibitor to a trifunctional compound against Alzheimer's disease. J. Med. Chem. 50, 6446–6449. doi: 10.1021/jm701225u
Bonda, D. J., Wang, X., Perry, G., Smith, M. A., and Zhu, X. (2010). Mitochondrial dynamics in Alzheimer disease: opportunities for future treatment strategies. Drugs Aging 27, 1–12. doi: 10.2165/11532140-000000000-00000
Boom, A., Authelet, M., Dedecker, R., Frederick, C., Van Heurck, R., Daubie, V., et al. (2009). Bimodal modulation of tau protein phosphorylation and conformation by extracellular Zn2+ in human-tau transfected cells. Biochim. Biophys. Acta 1793, 1058–1067. doi: 10.1016/j.bbamcr.2008.11.011
Borchardt, T., Schmidt, C., Camarkis, J., Cappai, R., Masters, C. L., Multhaup, G., et al. (2000). Differential effects of zinc on amyloid precursor protein (APP) processing in copper resistant variants of cultured Chinese hamster ovary cells. Cell. Mol. Biol. 46, 785–795.
Braak, H., and Braak, E. (1994). Morphological criteria for the recognition of Alzheimer's disease and the distribution pattern of cortical changes related to this disorder. Neurobiol. Aging 15, 355–356. doi: 10.1016/0197-4580(94)90032-9
Brown, K. L., Cosseau, C., Gardy, J. L., and Hancock, R. E. (2007). Complexities of targeting innate immunity to treat infection. Trends Immunol. 28, 260–266. doi: 10.1016/j.it.2007.04.005
Buccafusco, J. J., and Terry, A. V. Jr. (2000). Multiple central nervous system targets for elicing benefitial effects on memory and cognition. J. Pharmacol. Exp. Ther. 295, 438–446.
Bullock, R., and Lane, R. (2007). Executive dyscontrol in dementia, with emphasis on subcortical pathology and the role of butyrylcholinesterase. Curr. Alzheimer Res. 4, 277–293. doi: 10.2174/156720507781077313
Bush, A. I., Multhaup, G., Moir, R. D., Williamson, T. G., Small, D. H., Rumble, B., et al. (1993). A novel zinc (II) binding site modulates the function of the beta A4 amyloid protein precursor of Alzheimer's disease. J. Biol. Chem. 268, 16109–16112.
Bush, A. I., Pettingell, W. H. Jr. de Paradis, M. D., and Tanzi, R. E. (1994b). Modulation of A beta adhesiveness and secretase site cleavage by zinc. J. Biol. Chem. 269, 12152–12158.
Bush, A. I., Pettingell, W. H. Jr. de Paradis, M. D., Tanzi, R. E., and Wasco, W. (1994a). The amyloid beta-protein precursor and its mammalian homologues: evidence for a zinc-modulated heparin-binding superfamily. J. Biol. Chem. 269, 26618–26621.
Bush, A. I., Pettingell, W. H., Multhaup, G., de Paradis, M., Vonsattel, J. P., Gusella, J. F., et al. (1994c). Rapid induction of Alzheimer Abeta amyloid formation by zinc. Science 265, 1464–1467. doi: 10.1126/science.8073293
Butterfield, D. A., Reed, T., Perluigi, M., De Marco, C., Coccia, R., Cini, C., et al. (2006). Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci. Lett. 397, 170–173. doi: 10.1016/j.neulet.2005.12.017
Cappai, R., Kong, G., McKinstry, W., Galatis, D., Adams, J., Bellingham, S., et al. (2004). Structural and functional analysis of the Alzheimer's disease amyloid precursor protein copper binding domain. Neurobiol. Aging 25, s60. doi: 10.1016/s0197-4580(04)80203-3
Castellani, R. J., Lee, H. G., Zhu, X., Nunomura, A., Perry, G., and Smith, M. A. (2006). Neuropathology of Alzheimer disease: pathognomonic but not pathogenic. Acta Neuropathol. 111, 503–509. doi: 10.1007/s00401-006-0071-y
Chester, D. S., DeWall, C. N., Derefinko, K. J., Estus, S., Peters, J. R., Lynam, D. R., et al. (2015). Monoamine oxidase A (MAO A) genotype predicts greater aggression through impulsive reactivity to negative affect. Behav. Brain Res. 283, 97–101. doi: 10.1016/j.bbr.2015.01.034
Chouraki, V., and Seshadri, S. (2014). Genetics of Alzheimer's disease. Adv. Genet. 87, 245–294. doi: 10.1016/B978-0-12-800149-3.00005-6
Ciro, A., Park, J., Burkhard, G., Yan, N., and Geula, C. (2012). Biochemical differentiation of cholinesterases from normal and Alzheimer's disease cortex. Curr. Alzheimer Res. 9, 138–143. doi: 10.2174/156720512799015127
Connor, J. R., Menzies, S. L., St Martin, S. M., and Mufson, E. J. (1992). A histochemical study of iron, transferrin, and ferritin in Alzheimer's diseased brains. J. Neurosci. Res. 31, 75–83. doi: 10.1002/jnr.490310111
Corrigan, F. M., Reynolds, G. P., and Ward, N. I. (1993). Hippocampal tin, aluminum and zinc in Alzheimer's disease. Biometals 6, 149–154. doi: 10.1007/BF00205853
Coulson, E. J., Paliga, K., Beyreuther, K., and Masters, C. L. (2000). What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochemistry 36, 175–184. doi: 10.1016/S0197-0186(99)00125-4
Coyle, J. T., and Puttfarcken, P. (1993). Oxidative stress, glutamate and neurodegenerative dosorders. Science 262, 689–695. doi: 10.1126/science.7901908
Crouch, P. J., Blake, R., Duce, J. A., Ciccotosto, G. D., Li, Q. X., Barnham, K. J., et al. (2005). Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta (1-42). J. Neurosci. 25, 672–679. doi: 10.1523/JNEUROSCI.4276-04.2005
Crouch, P. J., Tew, D. J., Du, T., Nguyen, D. N., Caragounis, A., Filiz, G., et al. (2009). Restored degradation of the Alzheimer's amyloid-beta peptide by targeting amyloid formation. J. Neurochem. 108, 1198–1207. doi: 10.1111/j.1471-4159.2009.05870.x
Cuajuangco, M. P., and Faget, K. Y. (2003). Zinc takes the center stage: Its paradoxical role in Alzheimer's disease. Brain Res. Brain Res. Rev. 41, 44–56. doi: 10.1016/S0165-0173(02)00219-9
Curtain, C. C., Ali, F. E., Volitaskis, I., Cherny, R. A., Norton, R. S., Beyreuther, K., et al. (2001). Alzheimer's disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J. Biol. Chem. 276, 20466–22047. doi: 10.1074/jbc.M100175200
Cutillas, B., Ambrosio, S., and Unzeta, M. (2002). Neuroprotective effect of the monoamin oxidase inhibitor PF 9601N on rat nigral neurons after 6-OH-Dopamine striatal lesion. Neurosci. Lett. 329, 165–168. doi: 10.1016/S0304-3940(02)00614-6
Daikhin, Y., and Yudkoff, M. (2000). Compartmentation of brain glutamate metabolism in neurons and glia. J. Nutr. 130, 1026S–1031S.
Dajas-Bailador, F. A., Heimala, K., and Wonnacott, S. (2003). The allosteric potentiation of nicotinic acetylcholine receptors by galantamine is transduced into cellular responses in neurons: Ca2+ signals and neurotransmitter release. Mol. Pharmacol. 64, 1217–1226. doi: 10.1124/mol.64.5.1217
Damante, C. A., Osz, K., Nagy, Z., Pappalardo, G., Grasso, G., Impellizzeri, G., et al. (2009). Metal loading capacity of Abeta N-terminus: a combined potentiometric and spectroscopic study of zinc (II) complexes with Abeta (1-16), its short or mutated peptide fragments and its polyethylene glycolylated analogue. Inorg. Chem. 48, 10405–10415. doi: 10.1021/ic9012334
Danscher, G., Jensen, K. B., Frederickson, C. J., Kemp, K., Andreasen, A., Ravid, S., et al. (1997). Increased amount of zinc in the hippocampus and amygdala of Alzheimer's diseased brains: a proton-induced X-ray emission spectroscopic analysis of cryostat sections from autopsy material. J. Neurosci. Methods 76, 53–59. doi: 10.1016/S0165-0270(97)00079-4
Darreh-Shori, T., and Soininen, H. (2010). Effects of cholinesterase inhibitors on the activities and protein levels of cholinesterases in the cerebrospinal fluid of patients with Alzheimer's disease: A review of recent clinical studies. Curr. Alzheimer Res. 7, 67–73. doi: 10.2174/156720510790274455
Davison, A. N. (1957). The mechanism of the irreversible inhibition of rat-liver monomine oxidase by iproniazid. Biochem. J. 67, 316–322. doi: 10.1042/bj0670316
De Colibus, C. L., Li, M., Binda, C., Lustig, A., Edmondson, D. E., and Mattevi, A. (2005). Three-dimensional structure of human monoamine oxidase A (MAO A): relation to the structures of rat MAO A and human MAO B. Proc. Natl. Acad. Sci. U.S.A. 102, 12684–12689. doi: 10.1073/pnas.0505975102
Deibel, M. A., Ehmann, W. D., and Markesbery, W. R. (1996). Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer's disease: possible relation to oxidative stress. J. Neurol. Sci. 143, 137–142. doi: 10.1016/S0022-510X(96)00203-1
Détári, L., Rasmusson, D. D., and Semba, K. (1999). The role of basal forebrain neurons in tonic and phasic activation of the cerebral cortex. Prog. Neurobiol. 58, 249–277. doi: 10.1016/S0301-0082(98)00084-7
Deutsch, J. A. (1971). The cholinergic synapse and the site of memory. Science 174, 788–794. doi: 10.1126/science.174.4011.788
Diamant, S., Podoly, E., Friedler, A., Ligumsky, H., Livnah, O., and Soreq, H. (2006). Butyrylcholinesterase attenuates amyloid fibril formation in vitro. Proc. Natl. Acad. Sci. U.S.A. 103, 8628–8633. doi: 10.1073/pnas.0602922103
Dobson, J., and Batich, C. (2004). A potential iron-based mechanism for enhanced deposition of amyloid plaques due to cognitive stimulation in Alzheimer disease. J. Neuropathol. Exp. Neurol. 63, 674–675. doi: 10.1093/jnen/63.6.674
Doody, R. S., Tariot, P. N., Pfeiffer, E., Olin, J. T., and Graham, S. M. (2007). Meta-analysis of six-month memantine trials in Alzheimer's disease. Alzheimers Dement. 1, 7–17. doi: 10.1016/j.jalz.2006.10.004
Duce, J. A., Tsatsanis, A., Cater, M. A., James, S. A., Robb, E., Wikhe, K., et al. (2010). Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer's disease. Cell 142, 857–867. doi: 10.1016/j.cell.2010.08.014
Eckenroth, B. E., Steere, A. N., Chasteen, N. D., Everse, S. J., and Mason, A. B. (2011). How the binding of human transferrin primes the transferrin receptor potentiating iron release at endosomal pH. Proc. Natl. Acad. Sci. U.S.A. 108, 13089–13094. doi: 10.1073/pnas.1105786108
Edmondson, D. E., Binda, C., and Mattevi, A. (2007). Structural insights into the mechanism of amine oxidation by monoamine oxidases A and B. Arch. Biochem. Biophys. 464, 269–276. doi: 10.1016/j.abb.2007.05.006
Egaña, J. T., Zambrano, C., Núñez, M. T., González-Billault, C., and Maccioni, R. B. (2003). Iron-induced oxidative stress modifies tau phosphorylation patterns in hippocampal cell cultures. Biometals 16, 215–223.
Esteban, G., Allan, J., Samadi, A., Mattevi, A., Unzeta, M., Marco-Contelles, J., et al. (2014). Kinetic and structural analysis of irreversible inhibition of human monoamine oxidases by ASS234, a multi-target compound designed for use in Alzheimer's disease. Biochim. Biophys. Acta 1844, 1104–1110. doi: 10.1016/j.bbapap.2014.03.006
Evin, G., and Weidemann, A. (2002). Biogenesis and metabolism of Alzheimer's disease Abeta amyloid peptides. Peptides 23, 1285–1297. doi: 10.1016/S0196-9781(02)00063-3
Faller, P., Hureau, C., and Berthoumieu, O. (2013). Role of metal ions in the self-assembly of the Alzheimer's amyloid-beta peptide. Inorg. Chem. 52, 12193–12206. doi: 10.1021/ic4003059
Feng, Y., and Wang, X. (2012). Antioxidant therapies for Alzheimer's disease. Oxid. Med. Cell. Longev. 2012:472932. doi: 10.1155/2012/472932
Fibiger, H. C. (1991). Cholinergic mechanisms in learning, memory and dementia: a review of recent evidence. Trends Neurosci. 14, 220–223. doi: 10.1016/0166-2236(91)90117-D
Figueiredo, C. P., Clarke, J. R., Ledo, J. H., Ribeiro, F. C., Costa, C. V., Melo, H. M., et al. (2013). Memantine rescues transient cognitive impairment caused by high-molecular-weight aβ oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J. Neurosci. 33, 9626–9634. doi: 10.1523/JNEUROSCI.0482-13.2013
Fornai, F., Chen, K., Giorgi, F. S., Gesi, M., Alessandri, M. C., and Shih, J. C. (1999). Striatal dopamine metabolism in monoamine oxidase B-deficient mice: a brain dialysis study. J. Neurochem. 73, 2434–2440. doi: 10.1046/j.1471-4159.1999.0732434.x
Fowler, J. S., Mac Gregor, R. R., Wolf, A. P., Arnett, C. D., Devey, S. L., Schlyer, D., et al (1987). Mapping human brain monoamine oxidase A and B with 11C-labeled suicide inactivators and PET. Science 235, 481–485. doi: 10.1126/science.3099392
Frederickson, C. J. (1989). Neurobiology of zinc and zinc-containing neuron. Int. Rev. Neurobiol. 31, 145–238. doi: 10.1016/s0074-7742(08)60279-2
Freinbichler, W., Tipton, K. F., Della Corte, L., and Linert, W. (2009). Mechanistic aspects of the Fenton reaction under conditions approximated to the extracellular fluid. J. Inorg. Biochem. 103, 28–34. doi: 10.1016/j.jinorgbio.2008.08.014
Fukuda, M., Kanou, F., Shimada, N., Sawabe, M., Saito, Y., Murayama, S., et al. (2009). Elevated levels of 4-hydroxynonenal-histidine Michael adduct in the hippocampi of patients with Alzheimer's disease. Biomed. Res. 30, 229–233. doi: 10.2220/biomedres.30.227
Galasko, D. R., Peskind, E., Clark, C. M., Quinn, J. F., Ringman, J. M., Jicha, G. A., et al. (2012). Antioxidants for Alzheimer disease: A randomized clinical trial with cerebrospinal fluid biomarker measures. Arch. Neurol. 69, 836–841. doi: 10.1001/archneurol.2012.85
Geula, C., and Mesulam, M. (1989). Special properties of cholinesterases in the cerebral cortex of Alzheimer's disease. Brain Res. 498, 185–189. doi: 10.1016/0006-8993(89)90419-8
Giacobini, E. (2003). Cholinesterases: new roles in brain function and in Alzheimer's disease. Neurochem. Res. 28, 515–522. doi: 10.1023/A:1022869222652
Glenner, G. G., and Murphy, M. A. (1989). Amyloidosis of the nervous system. J. Neurol. Sci. 94, 1–28. doi: 10.1016/0022-510X(89)90214-1
Goedert, M., Klug, A., and Crowther, R. A. (2006). Tau protein, the paired helical filament and Alzheimer's disease. J. Alzheimers Dis. 9, 195–207.
Gomes, L. M., Vieira, R. P., Jones, M. R., Wang, M. C., Dyrager, C., Souza-Fagundes, E. M., et al. (2014). 8-Hydroxyquinoline Schiff-base compounds as antioxidants and modulators of copper-mediated Aβ peptide aggregation. J. Inorg. Biochem. 139, 106–116. doi: 10.1016/j.jinorgbio.2014.04.011
Goodman, L. (1953). Alzheimer's disease; a clinico-pathologic analysis of twenty three cases with a theory on pathogenesis. J. Nerv. Mental Dis. 118, 97–130. doi: 10.1097/00005053-195308000-00001
Greenblatt, H. M., Kryger, G., Lewis, T., Silman, I., and Sussman, J. L. (1999). Structure of acetylcholinesterase complexed with (-)-galanthamine at 2.3 A resolution. FEBS Lett. 463, 321–326. doi: 10.1016/S0014-5793(99)01637-3
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H., M., and Binder, L. I. (1986). Abnormal phosphorilation of the microtubule-associated protein tau in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 93, 4913–4917. doi: 10.1073/pnas.83.13.4913
Guglielmotto, M., Giliberto, L., Tamagno, E., and Tabaton, M. (2010). Oxidative stress mediates the pathogenic effect of different Alzheimer's disease risk factors. Front. Aging Neurosci. 2:3. doi: 10.3389/neuro.24.003.2010
Guillozet, A. L., Smiley, J. F., Mash, D. C., and Mesulam, M. M. (1997). Butyrylcholinesterase in the life cycle of amyloid plaques. Ann Neurol. 42, 909–918. doi: 10.1002/ana.410420613
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer's disease: The amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Hartter, D. E., and Barnea, A. (1988). Evidence for release of copper in the brain: Depolarization-induced release of newly taken-up 67 copper. Synapse 2, 412–415. doi: 10.1002/syn.890020408
Hayashi, T., Shishido, N., Nakayama, K., Nunomura, A., Smith, M. A., Perry, G., et al. (2007). Lipid peroxidation and 4-hydroxy-2-nonenal formation by copper ion bound to amyloid-beta peptide. Free Rad. Biol. Med. 43, 1552–1559. doi: 10.1016/j.freeradbiomed.2007.08.013
Heppner, F. L., Ransohoff, R. M., and Becher, B. (2015). Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372. doi: 10.1038/nrn3880
Hoke, D. E., Tan, J. L., Ilaya, N. T., Culvenor, J. G., Smith, S. J., White, A. R., et al. (2007). In vitro gamma-secretase cleavage of the Alzheimer's amyloid precursor protein correlates to a subset of presenilin complexes and is inhibited by zinc. FEBS J. 272, 5544–5557. doi: 10.1111/j.1742-4658.2005.04950.x
Hopt, A., Korte, S., Fink, H., Panne, U., Niessner, R., Jahn, R., et al. (2003). Methods for studying synaptosomal copper release. J. Neurosci. Methods 128, 159–172. doi: 10.1016/S0165-0270(03)00173-0
Hou, L., and Zagorski, M. G. (2006). NMR reveals anomalous copper (II) binding to the amyloid Abeta peptide of Alzheimer's disease. J. Am. Chem. Soc. 128, 9260–9261. doi: 10.1021/ja046032u
Houslay, M. D., and Tipton, K. F. (1973). The reaction pathway of rat liver monoamine oxidase. Biochem. J. 135, 173–186. doi: 10.1042/bj1350173
Hu, M., Schurdak, M. E., Puttfarcken, P. S., El Kouhen, R., Gopalakrishnan, M., and Li, J. (2007). High content screen microscopy analysis of amyloid beta 1-42-induced neurite outgrowth reduction in rat primary cortical neurons: neuroprotective effects of alpha 7 neuronal nicotinic acetylcholine receptor ligands. Brain Res. 1151, 227–235. doi: 10.1016/j.brainres.2007.03.051
Huang, T. H., Yang, D. S., Fraser, P. E., and Chakrabartty, A. (2000). Alternate aggregation pathways of the Alzheimer beta-amyloid peptide. An in vitro model of preamyloid. J. Biol. Chem. 275, 36436–36440. doi: 10.1074/jbc.M005698200
Huang, X., Cuajungco, M. P., Atwood, C. S., Hartshorn, M. A., Tyndall, G. R., Hanson, J. D., et al. (1999). Cu (II) potentiation of Alzheimer A-β neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J. Biol. Chem. 274, 37111–37116. doi: 10.1074/jbc.274.52.37111
Huang, X., Dai, J., Huang, C., Zhang, Q., Bhanot, O., and Pelle, E. (2007). Deferoxamine synergistically enhances iron-mediated AP-1 activation: a showcase of the interplay between extracellular-signal-regulated kinase and tyrosine phosphatase. Free Rad. Res. 41, 1135–1142. doi: 10.1080/10715760701609061
Huang, X., Moir, R. D., Tanzi, R. E., Bush, A. I., and Rogers, J. T. (2004). Redox-active metals, oxidative stress and Alzheimer's disease pathology. Ann. N.Y. Acad. Sci. 1012, 153–163. doi: 10.1196/annals.1306.012
Inestrosa, N. C., Alvarez, A., Perez, C. A., Moreno, R. D., Vicente, M., Linker, C., et al. (1996). Acetylcholinesterase accelerates assembly of amyloid-β-peptides into Alzheimer's Fibrils: possible role of the peripheral site of the enzyme. Neuron 16, 881–891.
Islam, K., and Levy, E. (1997). Carboxyl-terminal fragments of beta-amyloid precursor protein bind to microtubules and the associated protein tau. Am. J. Pathol. 151, 265–271.
Jack, C. R. Jr. Wiste, H. J., Vemuri, P., Weigand, S. D., Senjem, M. L., Zeng, G., et al. (2010). Brain beta-amyloid measures and magnetic resonance imaging atrophy both predict time-to-progression from mild cognitive impairment to Alzheimer's disease Neuroimaging Initiative. Brain 133, 3336–3348. doi: 10.1093/brain/awq277
Jiang, H., Wang, X., Huang, L., Luo, Z., Su, T., Ding, K., et al. (2011). Benzenediol-berberine hybrids: Multifunctional agents for Alzheimer's disease. Bioorg. Med. Chem. 19, 7228–7235. doi: 10.1016/j.bmc.2011.09.040
Jiao, Y., and Yang, P. (2007). Mechanism of copper (II) inhibiting Alzheimer's amyloid beta-peptide from aggregation: a molecular dynamics investigation. J. Phys. Chem. 111, 7646–7655. doi: 10.1021/jp0673359
Jin, M., Shepardson, N., Yang, T., Chen, G., Walsh, D., and Selkoe, D. J. (2011). Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 5819–5824. doi: 10.1073/pnas.1017033108
Juárez-Jiménez, J., Mendes, E., Galdeano, C., Martins, C., Silva, D. B., Marco-Contelles, J., et al. (2014). Exploring the structural basis of the selective inhibition of monoamine oxidase A by dicarbonitrile aminoheterocycles: Role of Asn181 and Ile335 validated by spectroscopic and computational studies. Biochim. Biophys. Acta 1844, 389–397. doi: 10.1016/j.bbapap.2013.11.003
Karch, C. M., and Goate, A. M. (2015). Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. doi: 10.1016/j.biopsych.2014.05.006
Kawahara, M., Mizuno, D., Koyama, H., Konora, K., Ohkawa, S., and Sadakane, Y. (2014). Disruption of Zinc homeostasis at the pathogenesis of senile dementia. Metallomics 6, 209–216. doi: 10.1039/C3MT00257H
Kayed, R., Head, E., Thompson, J. L., McIntire, T. M., Milton, S. C., Cotman, C. W., et al. (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489. doi: 10.1126/science.1079469
Keller, J. N., Schmitt, F. A., Scheff, S. W., Ding, Q., Chen, Q., Butterfield, D. A., et al. (2005). Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 64, 1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA
Khalil, M., Teunissen, C., and Langkammer, C. (2011). Iron and neurodegeneration in multiple sclerosis. Mult. Scler. Int. 2011:606807. doi: 10.1155/2011/606807
Khatoon, S., Grundke-Iqbal, I., and Iqbal, K. (1994). Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett. 351, 80–84. doi: 10.1016/0014-5793(94)00829-9
Kitazawa, M., Cheng, D., and Laferla, F. M. (2009). Chronic copper exposure exacerbates both amyloid and tau pathology and selectively deregulates CDK5 in a mouse model of AD. J. Neurochem. 108, 1550–1560. doi: 10.1111/j.1471-4159.2009.05901.x
Kodama, H., Murata, Y., and Kobayashi, M. (1999). Clinical manifestations and treatment of Menkes disease and its variants. Pediatr. Int. 41, 423–429. doi: 10.1046/j.1442-200x.1999.01095.x
Kupershmidt, L., Amit, T., Bar-Am, O., Youdim, M. B., and Weinreb, O. (2012). The novel multi-target iron chelating-radical scavenging compound M30 possesses beneficial effects on major hallmarks of Alzheimer's disease. Antioxid. Redox Signal. 17, 860–877. doi: 10.1089/ars.2011.4279
Kuperstein, F., and Yavin, E. (2003). Pro-apoptotic signaling in neuronal cells following iron and amyloid beta peptide neurotoxicity. J. Neurochem. 86, 114–125. doi: 10.1046/j.1471-4159.2003.01831.x
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
Lee, H. P., Zhu, X., Casadesus, G., Castellani, R. J., Nunomura, A., Smith, M. A., et al. (2010). Antioxidant approaches for the treatment of Alzheimer's disease. Expert Rev. Neurother. 10, 1201–1208. doi: 10.1586/ern.10.74
Lee, M. G., Hassani, O. K., Alonso, A., and Jones, B. E. (2005). Cholinergic basal forebrain neurons burst with theta during waking and paradoxical sleep. J. Neurosci. 25, 4365–4369. doi: 10.1523/JNEUROSCI.0178-05.2005
Lee, Y. J., Jeong, S. Y., Karbowski, M., Smith, C. L., and Youle, R. J. (2004). Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Bio. Cell. 15, 5001–5011. doi: 10.1091/mbc.E04-04-0294
Lei, P., Ayton, S., Bush, A. I., and Adlard, P. A. (2011). GSK-3 in neurodegenerative diseases. Int. J. Alzheimers Dis. 2011:189246. doi: 10.4061/2011/189246
Lei, P., Ayton, S., Finkelstein, D. I., Adlard, P. A., Masters, C. L., and Bush, A. I. (2010). Tau protein: Relevance to Parkinson's disease. Int. J. Biochem. Cell Biol. 42, 1775–1778. doi: 10.1016/j.biocel.2010.07.016
Lei, P., Ayton, S., Finkelstein, D. I., Spoerri, L., Ciccotosto, G. D., Wright, D. K., et al. (2012). Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 18, 291–295. doi: 10.1038/nm.2613
León, R., García, A. G., and Marco-Contelles, J. (2013). Recent advances in the multitaret-directed ligands approach for the treatment of Alzheimer's disease. Med. Res. Rev. 33, 139–189. doi: 10.1002/med.20248
LeVine, H. (1993). Thioflavine T interaction with synthetic Alzheimer's disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 2, 404–410. doi: 10.1002/pro.5560020312
Lipton, S. A. (2006). Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat. Rev. Drug Discov. 5, 160–170. doi: 10.1038/nrd1958
Liu, W., Xu, Z., Deng, Y., Xu, B., Wei, Y., and Yang, T. (2013). Protective effects of memantine against methylmercury-induced glutamate dyshomeostasis and oxidative stress in rat cerebral cortex. Neurotox. Res. 24, 320–337. doi: 10.1007/s12640-013-9386-3
Loeffler, D. A., Connor, J. R., Juneau, P. L., Snyder, B. S., Kanaley, L., DeMaggio, A. J., et al. (1995). Transferrin and iron in normal, Alzheimer's disease, and Parkinson's disease brain regions. J. Neurochem. 65, 710–724. doi: 10.1046/j.1471-4159.1995.65020710.x
Lovell, M. A., Robertson, J. D., Teesdale, W. J., Campbell, J. L., and Markesbery, W. R. (1998). Copper, iron and zinc in Alzheimer's disease senile plaques. J. Neurol. Sci. 58, 47–52. doi: 10.1016/S0022-510X(98)00092-6
Lovell, M. A., Xiong, S., Xie, C., Davies, P. L., and Markesbery, W. R. (2004). Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. J. Alzheimers Dis. 5, 659–671.
Ma, Q. F., Hu, J., Wu, W. H., Liu, H. D., Du, J. T., Fu, Y., et al. (2006). Characterization of copper binding to the peptide amyloid-beta (1-16) associated with Alzheimer's disease. Biopolymers 83, 20–31. doi: 10.1002/bip.20523
Magaki, S., Raghavan, R., Mueller, C., Oberg, K. C., Vinters, H. V., and Kirsch, W. M. (2007). Iron, copper, and iron regulatory protein 2 in Alzheimer's disease and related dementias. Neurosci. Lett. 418, 72–76. doi: 10.1016/j.neulet.2007.02.077
Malar, D. S., and Devi, K. P. (2014). Dietary polyphenols for treatment of Alzheimer's disease: future research and development. Curr. Pharm. Biotechnol. 15, 330–342. doi: 10.2174/1389201015666140813122703
Mantyh, P. W., Ghilardi, J. R., Rogers, S., DeMaster, E., Allen, C. J., Stimson, E. R., et al. (1993). Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of beta-amyloid peptide. J. Neurochem. 61, 1171–1174. doi: 10.1111/j.1471-4159.1993.tb03639.x
Matsubayashi, K., Fukuyama, H., Akiguchi, I., Kameyama, M., Imai, H., and Maeda, T. (1986). Localization of monoamine oxidase (MAO) in the rat peripheral nervous system existence of MAO-containing unmyelinated axons. Brain Res. 368, 30–35. doi: 10.1016/0006-8993(86)91039-5
Mattson, M. P. (1997). Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol. Rev. 77, 1081–1132.
Mattson, M. P. (2004). Pathways towards and away from Alzheimer's disease. Nature 430, 631–639. doi: 10.1038/nature02621
Meadowcroft, M. D., Connor, J. R., Smith, M. B., and Yang, Q. X. (2009). MRI and histological analysis of beta-amyloid plaques in both human Alzheimer's disease and APP/PS1 transgenic mice. J. Magn. Reson. Imaging. 29, 997–1007. doi: 10.1002/jmri.21731
Mechlovich, D., Amit, T., Bar-Am, O., Mandel, S., Youdim, M. B., and Weinreb, O. (2014). The novel multi-target iron chelator, M30 modulates HIF-1α-related glycolytic genes and insulin signaling pathway in the frontal cortex of APP/PS1 Alzheimer's disease mice. Curr. Alzheimer Res. 2, 119–127. doi: 10.2174/1567205010666131212112529
Mendelewicz, J., and Youdim, M. B. H. (1983). L-Deprenyl: a selective monoamine oxidase type B inhibitor in the treatment of depression: a double-bind evaluation. Br. J. Psychiatry 142, 508–511. doi: 10.1192/bjp.142.5.508
Merle, U., Schaefer, M., Ferenci, P., and Stremmel, W. (2007). Clinical presentation, diagnosis and long-term outcome of Wilson's disease: A cohort study. Gut 56, 115–120. doi: 10.1136/gut.2005.087262
Mesulam, M., Guillozet, A., Shaw, P., and Quinn, B. (2002). Widely spread butyrylcholinesterase can hydrolyze acetylcholine in the normal and Alzheimer brain. Neurobiol. Dis. 9, 88–93. doi: 10.1006/nbdi.2001.0462
Miguel-Hidalgo, J. J., Álvarez, X. A., Cacabelos, R., and Quack, G. (2002). Neuroprotection by memantine against neurodegeneration induced by beta-amyloid (1-40). Brain Res. 958, 210–221. doi: 10.1016/S0006-8993(02)03731-9
Miller, L. M., Wang, Q., Telivala, T. P., Smith, R. J., Lanzirotti, A., and Miklossy, J. (2006). Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer's disease. J. Struct. Biol. 155, 30–37. doi: 10.1016/j.jsb.2005.09.004
Minicozzi, V., Stellato, F., Comai, M., Serra, M. D., Potrich, C., Meyer-Klaucke, W., et al. (2008). Identifying the minimal copper- and zinc-binding site sequence in amyloid-beta peptides. J. Biol. Chem. 283, 10784–10792. doi: 10.1074/jbc.M707109200
Mo, Z. Y., Zhu, Y. Z., Zhu, H. L., Fan, J. B., Chen, J., and Liang, Y. (2009). Low micromolar zinc accelerates the fibrillization of human tau via bridging of Cys-291 and Cys-322. J. Biol. Chem. 284, 34648–34657. doi: 10.1074/jbc.M109.058883
Molnar, P., and Nadler, J. V. (2001). Lack of effect of mossy fiber-released zinc on granule cell GABA (A) receptors in the pilocarpine model of epilepsy'. J. Neurophysiol. 85, 1932–1940.
Morales, I., Guzmán-Martínez, L., Cerda-Troncoso, C., Farias, G. A., and Maccioni, R. B. (2014). Neuroinflammation in the pathogenesis of Alzheimer's disease. A rational framework for the search of novel therapeutic approaches. Front. Cell Neurosci. 8:112. doi: 10.3389/fncel.2014.00112
Moreira, P. I., Nunomura, A., Nakamura, M., Takeda, A., Shenk, J. C., Aliev, G., et al. (2008). Nucleic acid oxidation in Alzheimer disease. Free Rad. Biol. Med. 44, 1493–1505. doi: 10.1016/j.freeradbiomed.2008.01.002
Moriguchi, S., Marszalec, W., Zhao, X., Yeh, J. Z., and Narahashi, T. (2003). Potentiation of N-methyl-D-aspartate-induced currents by the nootropic drug nefiracetam in rat cortical neurons. J. Pharmacol. Exp. Ther. 307, 160–167. doi: 10.1124/jpet.103.050823
Multhaup, G., Schlicksupp, A., Hesse, L., Beher, D., Ruppert, T., Masters, C. L., et al. (1996). The amyloid precursor protein of Alzheimer's disease in the reduction of copper (II) to copper (I). Science 271, 1406–1409. doi: 10.1126/science.271.5254.1406
Muñoz, P., Zavala, G., Castillo, K., Aguirre, P., Hidalgo, C., and Núñez, M. (2006). Effect of iron on the activation of the MAPK/ERK pathway in PC12 neuroblastoma cells. Biol. Res. 39, 189–190.
Nakamura, M., Shishido, N., Nunomura, A., Smith, M. A., Perry, G., Hayashi, Y., et al. (2007). Three histidine residues of amyloid-beta peptide control the redox activity of copper and iron. Biochemistry 46, 12737–12743. doi: 10.1021/bi701079z
Nelson, P. T., Alafuzoff, I., Bigio, E. H., Bouras, C., Braak, H., Cairns, N. J., et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. doi: 10.1097/NEN.0b013e31825018f7
Nicotra, A., Pierucci, F., Parvez, H., and Senatori, O. (2004). Monoamine oxidase expression during development and aging. Neurotoxicology 25, 155–165. doi: 10.1016/S0161-813X(03)00095-0
Nielsen, J. A., Mena, E. E., Williams, I. H., Nocerini, M. R., and Liston, D. (1989). Correlation of brain levels of 9-amino-1, 2, 3, 4-tetrahydroacridine (THA) with neurochemical and behavioral changes.' Eur. J. Pharmacol. 173, 53–64. doi: 10.1016/0014-2999(89)90008-3
Nochi, S., Asakawa, N., and Sato, T. (1995). Kinetic study on the inhibition of acetylcholinesterase by 1-benzyl-4-[(5,6-dimethoxy-1-indanon)-2-yl]methylpiperidine hydrochloride (E2020). Biol. Pharm. Bull. 18, 1145–1147. doi: 10.1248/bpb.18.1145
Nunomura, A., Perry, G., Pappolla, M. A., Friedland, R. P., Hirai, K., Chiba, S., et al. (2000). Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. J. Neuropathol. Exp. Neurol. 59, 1011–1017. doi: 10.1093/jnen/59.11.1011
O'Carroll, A. M., Fowler, C. J., Phillips, J. P., Tobbia, I., and Tipton, K. F. (1983). The deamination of dopamine by human brain monoamine oxidase. Specificity for the two enzyme forms in seven brain regions. Naunyn Schmiedebergs Arch. Pharmacol. 322, 198–202.
O'Carroll, A. M., Tipton, K. F., Sullivan, J. P., Fowler, C. J., and Ross, S. B. (1987). Intra- and extrasynaptosomal deamination of dopamine and noradrenaline by the two forms of human brain monoamine oxidase. Implications for the neurotoxicity of N-methyl-4-phenyl-1, 2, 3, 6- tetrahydropyridine in man. Biog. Amines 4, 165–178.
Odetti, P., Angelini, G., Dapino, D., Zaccheo, D., Garibaldi, S., Dagna-Bricarelli, F., et al. (1998). Early glycoxidation damage in brains from Down's syndrome. Biochem. Biophys. Res. Commun. 132, 849–851. doi: 10.1006/bbrc.1998.8186
Ollis, D. L., Cheah, E., Cygler, M., Dijkstra, B., Frolow, F., Franken, S. M., et al. (1992). The α/β hydrolase fold. Protein Eng. 5, 197–211. doi: 10.1093/protein/5.3.197
Panayi, A. E., Spyrou, N. M., Iversen, B. S., and White, M. A. (2002). Determination of cadmium and zinc in Alzheimer's brain tissue using inductively coupled plasma mass spectrometry. J. Neurol. Sci. 195, 1–10. doi: 10.1016/S0022-510X(01)00672-4
Paoletti, P., Ascher, P., and Neyton, J. (1997). High-affinity zinc inhibition of NMDA NR1–NR2A receptors. J. Neurosci. 17, 5711–5725.
Park, I. H., Jung, M. W., Mori, H., and Mook-Jung, I. (2001). Zinc enhances synthesis of presenilin 1 in mouse primary cortical culture. Biochem. Biophys. Res. Commun. 285, 680–688. doi: 10.1006/bbrc.2001.5243
Parsons, C. G., Danysz, W., Bartmann, A., Spielmanns, P., Frankiewicz, T., Hesselink, M., et al. (1999). Amino-alkyl-cyclohexanes are novel uncompetitive NMDA receptor antagonists with strong voltage-dependency and fast blocking kinetics: in vitro and in vivo characterization. Neuropharmacology 38, 85–108. doi: 10.1016/S0028-3908(98)00161-0
Pei, J. J., An, W. L., Zhou, X. W., Nishimura, T., Norberg, J., Benedikz, E., et al. (2006). P70 S6 kinase mediates tau phosphorylation and synthesis. FEBS Lett. 580, 107–114. doi: 10.1016/j.febslet.2005.11.059
Percy, M., Moalem, S., Garcia, A., Somerville, M. J., Hicks, M., Andrews, D., et al. (2008). Involvement of ApoE E4 and H63D in sporadic Alzheimer's disease in a folate-supplemented Ontario population. J. Alzheimers Dis. 14, 69–84.
Pérez, V., and Unzeta, M. (2003). PF9601N, anew MAO B inhibitor, attenuates MPTP-induced depletion of striatal dopamine levels in C57/BL6 mice. Nurochem. Int. 42, 221–239. doi: 10.1016/S0197-0186(02)00091-8
Pérez, V., Marco, J. L., Fernández-Álvarez, E., and Unzeta, M. (1999). Relevance of benzyloxy group in 2-indolyl methylamines in the selective MAO-B inhibition. Br. J. Pharmacol. 12, 869–876. doi: 10.1038/sj.bjp.0702600
Pérez, V., Romera, M., Lizcano, J. M., Marco, J. L., and Unzeta, M. (2003). Protective effect of PF 9601N, a novel MAO B inhibitor, on dopamine lesioned PC12 cells. J. Pharm. Pharmacol. 55, 713–716. doi: 10.1211/002235703765344649
Perl, D. P. (2010). Neuropathology of Alzheimer's disease. Mt Sinai J. Med. 77, 32–42. doi: 10.1002/msj.20157
Perrone, L., Mothes, E., Vignes, M., Mockel, A., Figueroa, C., Miquel, M. C., et al. (2010). Copper transfer from Cu-Abeta to human serum albumin inhibits aggregation, radical production and reduces Abeta toxicity. Chembiochem 11, 110–118. doi: 10.1002/cbic.200900474
Perry, E. K., Perry, R. H., Blessed, G., and Tomlinson, B. E. (1978). Changes in brain cholinesterases in senile dementia of Alzheimer type. Neuropathol. Appl. Neurobiol. 4, 273–277. doi: 10.1111/j.1365-2990.1978.tb00545.x
Petersen, R. B., Nunomura, A., Lee, H. G., Casadesus, G., Perry, G., Smith, M. A., et al. (2007). Signal transduction cascades associated with oxidative stress in Alzheimer's disease. J. Alzheimers Dis. 11, 143–152.
Petousi, N., and Robbins, P. A. (2014). Human adaptation to the hypoxia of high altitude: the Tibetan paradigm from the pregenomic to the postgenomic era. J. Appl. Physiol. 116, 875–884. doi: 10.1152/japplphysiol.00605.2013
Praticò, 2008###Praticò, D. (2008). Evidence of oxidative stress in Alzheimer's disease brain and antioxidant therapy: lights and shadows. Ann. N.Y. Acad. Sci. 1147, 70–78. doi: 10.1196/annals.1427.010
Qian, J., and Noebels, J. L. (2005). Visualization of transmitter release with zinc fluorescence detection at the mouse hippocampal mossy fibre synapse. J. Physiol. 566, 747–758. doi: 10.1113/jphysiol.2005.089276
Qizilbash, N., Whitehead, A., Higgins, J., Wilcock, G., Schneider, L., and Farlow, M. (1998). Cholinesterase inhibition for Alzheimer's disease: a meta-analysis of the tacrine trials. J. Am. Med. Assoc. 280, 1777–1782. doi: 10.1001/jama.280.20.1777
Ramadan, Z. B., Dostert, P., and Tipton, K. F. (1999). Some peculiar aspects of monoamine oxidase inhibition. Neurobiology 7, 159–174.
Ramadan, Z. B., and Tipton, K. F. (1998). Cyanide potentiates the inhibition of monoamine oxidase by hydrazine derivatives. J. Neurochem. 71, S36–S36.
Ramadan, Z. B., Wrang, M. L., and Tipton, K. F. (2007). Species differences in the selective inhibition of monoamine oxidase (1-methyl-2-phenylethyl) hydrazine and its potentiation by cyanide. Neurochem. Res. 32, 1783–1790. doi: 10.1007/s11064-007-9309-x
Reinikainen, K. J., Paljarvi, L., Huuskonen, M., Soininen, H., Laakso, M., and Riekkinen, P. J. (1988). A post-mortem study of noradrenergic, serotonergic and GABAergic neurons in Alzheimer's disease. J. Neurol. Sci. 84, 101–116. doi: 10.1016/0022-510X(88)90179-7
Religa, D., Strozyk, D., Cherny, R. A., Volitakis, I., Haroutunian, V., Winblad, B., et al. (2006). Elevated cortical zinc in Alzheimer disease. Neurology 67, 69–75. doi: 10.1212/01.wnl.0000223644.08653.b5
Rogers, J. T., Randall, J. D., Cahill, C. M., Eder, P. S., Huang, X., Gunshin, L., et al. (2002). An iron-responsive element type II in the 5-untranslated region of the Alzheimer's amyloid precursor protein transcript. J. Biol. Chem. 277, 45518–45528. doi: 10.1074/jbc.M207435200
Rogers, J., Webster, S., Lue, L. F., Brachova, L., Civin, W. H., Emmerling, M., et al. (1996). Inflammation and Alzheimer's disease pathogenesis. Neurobiol. Aging 17, 681–686. doi: 10.1016/0197-4580(96)00115-7
Rosini, M., Simoni, E., Milelli, A., Minarini, A., and Melchiorre, C. (2014). Oxidative stress in Alzheimer's disease: are we connecting the dots? J. Med. Chem. 57, 2821–2831. doi: 10.1021/jm400970m
Rottkamp, C. A., Raina, A. K., Zhu, X. W., Gaier, E., Bush, A. I., Atwood, C. S., et al. (2001). Redox-active iron mediates amyloid-b toxicity. Free Rad. Biol. Med. 30, 447–450. doi: 10.1016/S0891-5849(00)00494-9
Ruiz, A., Walker, M. C., Fabian-Fine, R., and Kullmann, D. M. (2004). Endogenous zinc inhibits GABA (A) receptors in a hippocampal pathway. J. Neurophysiol. 91, 1091–1096. doi: 10.1152/jn.00755.2003
Salkovic-Petrisic, M., Knezovic, A., Osmanovic-Barilar, J., Smailovic, U., Trkulja, V., Riederer, P., et al. (2015). Multi-target iron-chelators improve memory loss in a rat model of sporadic Alzheimer's disease. Life Sci. 136, 108–119. doi: 10.1016/j.lfs.2015.06.026
Samadi, A., Valderas, C., de los Ríos, C., Bastida, A., Chioua, M., González-Lafuente, L., et al. (2011). Cholinergic and neuroprotective drugs for the treatment of Alzheimer and neuronal vascular diseases. II. Synthesis, biological assessment, and molecular modelling of new tacrine analogues from highly substituted 2-aminopyridine-3-carbonitriles. Bioorg. Med. Chem. 19, 122–133. doi: 10.1016/j.bmc.2010.11.040
Samudralwar, D. L., Diprete, C. C., Ni, B. F., Ehmann, W. D., and Markesbery, W. (1995). Elemental imbalances in the olfactory pathway in Alzheimer's disease. J. Neurol. Sci. 130, 139–145. doi: 10.1016/0022-510X(95)00018-W
Sanz, E., Quintana, A., Hidalgo, J., Marco, J. L., and Unzeta, M. (2009). PF 9601N confers MAOB independent neuroprotection in ER-stress-induced cell death. Mol. Cell. Neurosci. 41, 19–31. doi: 10.1016/j.mcn.2009.01.005
Saura, J., Kettler, R., Da, P. M., and Richards, J. G. (1992). Quantitative enzyme radioautography with 3H-Ro 41-1049 and 3H-Ro 19-6327 in vitro: localization and abundance of MAO-A and MAO-B in rat CNS, peripheral organs, and human brain. J. Neurosci. 12, 1977–1999.
Sayre, L. M., Perry, G., Harris, P. L., Liu, Y., Schubert, K. A., and Smith, M. A. (2000). In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer's disease: a central role for bound transition metals. J. Neurochem. 74, 270–279. doi: 10.1046/j.1471-4159.2000.0740270.x
Schipper, H. M., Bennett, D. A., Liberman, A., Bienias, J. L., Schneider, J. A., Kelly, J., et al. (2006). Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol. Aging 27, 252–261. doi: 10.1016/j.neurobiolaging.2005.01.016
Schliebs, R. (2005). Basal forebrain cholinergic dysfunction in Alzheimer's disease–interrelationship with beta-amyloid, inflammation and neurotrophin signaling. Neurochem. Res. 30, 895–908. doi: 10.1007/s11064-005-6962-9
Schlief, M. L., Craig, A. M., and Gitlin, J. D. (2005). NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J. Neurosci. 25, 239–246. doi: 10.1523/JNEUROSCI.3699-04.2005
Schlief, M. L., West, T., Craig, A. M., Holtzman, D. M., and Gitlin, J. D. (2006). Role of the Menkes copper-transporting ATPase in NMDA receptor-mediated neuronal toxicity. Proc. Natl. Acad. Sci. U.S.A. 103, 14919–14924. doi: 10.1073/pnas.0605390103
Schubert, D., Behl, C., Lesley, R., Brack, A., Dargusch, R., Sagara, Y., et al. (1995). Amyloid peptides are toxic via a common oxidative mechanism. Proc. Natl. Acad. Sci. U.S.A. 14, 1989–1993. doi: 10.1073/pnas.92.6.1989
Schubert, D., and Chevion, M. (1995). The role of iron in beta amyloid toxicity.' Biochem. Biophys. Res. Commun. 216, 702–707. doi: 10.1006/bbrc.1995.2678
Selkoe, D. J. (1994). Alzheimer's disease: a central role for amyloid. J. Neuropathol. Exp. Neurol. 53, 438–447. doi: 10.1097/00005072-199409000-00003
Serrano, M. P., Herrero-Labrador, R., Futch, H. S., Serrano, J., Romero, A., Fernandez, A. P., et al. (2016). The proof-of-concept of ASS234: Peripherally administered ASS234 enters the central nervous system and reduces pathology in a mouse model of Alzheimer's disease. J. Psychiatry Neurosci.
Serrano-Pozo, A., Qian, J., Monsell, S. E., Frosch, M. P., Betensky, R. A., and Hyman, B. (2013). Examination of the clinicopathologic continuum of Alzheimer disease in the autopsy cohort of the National Alzheimer Coordinating Center. J. Neuropathol. Exp. Neurol. 72, 1182–1192. doi: 10.1097/NEN.0000000000000016
Shan, W. J., Huang, L., Zhou, Q., Meng, F. C., and Li, X. S. (2011). Synthesis, biological evaluation of 9-N-substituted berberine derivatives as multi-functional agents of antioxidant, inhibitors of acetylcholinesterase, butyrylcholinesterase and amyloid-β aggregation. Eur. J. Med. Chem. 46, 5885–5893. doi: 10.1016/j.ejmech.2011.09.051
Shin, R. W., Iwaki, T., Kitamoto, T., Sato, Y., and Tateishi, J. (1992). Massive accumulation of modified tau and severe depletion of normal tau characterize the cerebral cortex and white matter of Alzheimer's disease: demonstration using the hydrated autoclaving method. Am. J. Pathol. 140, 937–945.
Shoffner, J. M. (1997). Oxidative phosphorylation defects and Alzheimer's disease. Neurogenetics 1, 13–19. doi: 10.1007/s100480050002
Silvestri, L., and Camaschella, C. (2008). A potential pathogenetic role of iron in Alzheimer's disease. J. Cell Mol. Med. 12, 1548–1550. doi: 10.1111/j.1582-4934.2008.00356.x
Smith, D. P., Smith, D. G., Curtain, C. C., Boas, J. F., Pilbrow, J. R., Ciccotosto, G. D., et al. (2006). Copper-mediated amyloid-beta toxicity is associated with an intermolecular histidine bridge. J. Biol. Chem. 281, 15145–15154. doi: 10.1074/jbc.M600417200
Smith, M. A., Hirai, K., Hsiao, K., Pappolla, M. A., Harris, P. L., Siedlak, S. L., et al. (1998). Amyloid-beta deposition in Alzheimer transgenic mice is associated with oxidative stress. J. Neurochem. 70, 2212–2215. doi: 10.1046/j.1471-4159.1998.70052212.x
Song, M. S., Rauw, G., Baker, G. B., and Kar, S. (2008). Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur. J. Neurosci. 28, 1989–2002. doi: 10.1111/j.1460-9568.2008.06498.x
Soreq, H., and Seidman, S. (2001). Acetylcholinesterase–new roles for an old actor. Nat. Rev. Neurosci. 2, 294–302. doi: 10.1038/35067589
Stasiak, A., Mussur, M., Unzeta, M., Samadi, A., Marco-Contelles, J. L., and Fogel, W. A. (2014). Effects of novel monoamine oxidases and cholinesterases targeting compounds on brain neurotransmitters and behavior in rat model of vascular dementia. Curr. Pharm. Des. 20, 161–171. doi: 10.2174/13816128113199990026
Stoltenberg, M., Bruhn, M., Sondergaard, C., Doering, P., West, M. J., Larsen, A., et al. (2005). Immersion autometallographic tracing of zinc ions in Alzheimer beta-amyloid plaques. Histochem. Cell Biol. 123, 605–611. doi: 10.1007/s00418-005-0787-0
Strolin-Benedetti, M., Dostert, P., and Tipton, K. F. (1992). Developmental aspects of the monoamine-degrading enzyme monoamine oxidase. Dev. Pharmacol. Ther. 18, 191–200.
Struble, R. G., Cork, L. C., Whitehouse, P. J., and Price, D. L. (1982). Cholinergic innervation in neuritic plaques. Science 216, 413–415. doi: 10.1126/science.6803359
Su, X. Y., Wu, W. H., Huang, Z. P., Hu, J., Lei, P., Yu, C. H., et al. (2007). Hydrogen peroxide can be generated by tau in the presence of Cu (II). Biochem. Biophys. Res. Commun. 358, 661–665. doi: 10.1016/j.bbrc.2007.04.191
Suh, S. W., Jensen, K. B., Jensen, M. S., Silva, D. S., Kesslak, P. J., Danscher, G., et al. (2000). Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer's diseased brains. Brain Res. 852, 274–278. doi: 10.1016/S0006-8993(99)02096-X
Sultana, R., Mecocci, P., Mangialasche, F., Cecchetti, R., Baglioni, M., and Butterfield, D. A. (2011). Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer's disease: insights into the role of oxidative stress in Alzheimer's disease and initial investigations into a potential biomarker for this dementing disorder. J. Alzheimers Dis. 24, 77–84. doi: 10.3233/JAD-2011-101425
Sussman, J. L., Harel, M., Frolow, F., Oefner, C., Goldman, A., Toker, L., et al. (1991). Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science 253, 872–879. doi: 10.1126/science.1678899
Syme, C. D., Nadal, R. C., Rigby, S. E. J., and Viles, J. H. (2004). Copper binding to the amyloid-beta (Abeta) peptide associated with Alzheimer's disease: folding, coordination geometry, pH dependence, stoichiometry, and affinity of Abeta-(1-28): insights from a range of complementary spectroscopic techniques. J. Biol. Chem. 279, 18169–18177. doi: 10.1074/jbc.M313572200
Terry, R. D., Gonatas, N. K., and Weiss, M. (1964). Ultrastructural studies in Alzheimer'spresenile dementia. Ann. J. Pathol. 44, 269–297.
Thies, W., and Bleiler, L. (2012). Alzheimer's association: Alzheimer's disease facts and figures. Alzheimers Dement. 8, 131–168.
Thies, W., and Bleiler, L. (2013). Alzheimer's disease: Facts and figures. Alzheimer Dement. 9, 208–215. doi: 10.1016/j.jalz.2013.02.003
Touchon, J., Bergman, H., Bullock, R., Rapatz, G., Nagel, J., and Lane, R. (2006). Response to rivastigmine or donepezil in Alzheimer's patients with symptoms suggestive of concomitant Lewy body pathology. Curr. Med. Res. Opin. 22, 49–59. doi: 10.1185/030079906X80279
Tougu, V., Karafin, A., and Palumaa, P. (2008). Binding of zinc (II) and copper (II) to the full-length Alzheimer's amyloid-beta peptide. J. Neurochem. 104, 1249–1259. doi: 10.1111/j.1471-4159.2007.05061.x
Traynelis, S. F., Burgess, M. F., Zheng, F., Lyuboslavsky, P., and Powers, J. L. (1998). Control of voltage-independent zinc inhibition of NMDA receptors by the NR1 subunit. J. Neurosci. 18, 6163–6175.
Tremblay, R., Chakravarthy, B., Hewitt, K., Tauskela, J., Morley, P., Atkinson, T., et al. (2000). Transient NMDA receptor inactivation provides long-term protection to cultured cortical neurons from a variety of death signals. J. Neurosci. 20, 7183–7192.
Tsang, D., Ho, K. P., and Wen, H. L. (1986). Ontogenesis of multiple forms of monoamine oxidase in rat brain regions and liver. Dev. Neurosci. 8, 243–250. doi: 10.1159/000112258
Uchida, Y., Takio, K., Titani, K., Ihara, Y., and Tomonaga, M. (1991). The growth inhibitory factor that is deficient in the Alzheimer's disease brain is a 68 amino acid metallothionein-like protein. Neuron 7, 337–347. doi: 10.1016/0896-6273(91)90272-2
Van Eersel, J., Bi, M., Ke, Y. D., Hodges, J. R., Xuereb, J. H., Gregory, G. C., et al. (2009). Phosphorylation of soluble tau differs in Pick's disease and Alzheimer's disease brains. J. Neural Trans. 116, 1243–1251. doi: 10.1007/s00702-009-0293-y
Vermeiren, Y., Van Dam, D., Aerts, T., Engelborghs, S., Martin, J. J., and De Deyn, P. P. (2015). The monoaminergic footprint of depression and psychosis in dementia with Lewy bodies compared to Alzheimer's disease. Alzheimers Res Ther. 7, 7. doi: 10.1186/s13195-014-0090-1
Vogels, O. J., Broere, C. A., ter Laak, H. J., ten Donkelaar, H. J., Nieuwenhuys, R., and Schulte, B. P. (1990). Cell loss and shrinkage in the nucleus basalis Meynert complex in Alzheimer's disease. Neurobiol. Aging 11, 3–13. doi: 10.1016/0197-4580(90)90056-6
Vogt, K., Mellor, J., Tong, G., and Nicoll, R. (2000). The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron 26, 187–196. doi: 10.1016/S0896-6273(00)81149-6
Von Bernhardi, R., and Eugenín, J. (2012). Alzheimer's disease: redox dysregulation as a common denominator for diverse pathogenic mechanisms. Antioxid. Redox Signal. 16, 974–1031. doi: 10.1089/ars.2011.4082
Wang, C. Y., Wang, T., Zheng, W., Zhao, B. L., Danscher, G., Chen, Y. H., et al. (2010). Zinc overload enhances APP cleavage and Abeta deposition in the Alzheimer mouse brain. PLoS ONE 5:e15349. doi: 10.1371/journal.pone.0015349
Wang, L., Esteban, G., Ojima, M., Bautista-Aguilera, O. M., Inokuchi, T., Moraleda, I., et al. (2014). Donepezil+propargylamine+8-hydroxyquinoline hybrids as new multifunctional metal-chelators, ChE and MAO inhibitors for the potential treatment of Alzheimer's disease. Eur. J. Med. Chem. 80, 543–561. doi: 10.1016/j.ejmech.2014.04.078
Wang, X., Su, B., Zheng, L., Perry, G., Smith, M. A., and Zhu, X. (2009). The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J. Neurochem. 109, 153–159. doi: 10.1111/j.1471-4159.2009.05867.x
Westlund, K. N., Denney, R. M., Kochersperger, L. M., Rose, R. M., and Abell, C. W. (1988). Distinct monoamine oxidase A and B populations in primate brain. Science 230, 181–183. doi: 10.1126/science.3875898
White, A. R., Multhaup, G., Maher, F., Bellingham, S., Camakaris, J., Zheng, H., et al. (1999). The Alzheimer's disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J. Neurosci. 19, 9170–9179.
Wilkinson, D., and Murray, J. (2001). Galantamine: a randomized, double-blind, dose comparison in patients with Alzheimer's disease. Int. J. Geriatr. Psychiatry 16, 852–857. doi: 10.1002/gps.409
Willard, L. B., Hauss-Wegrzyniak, B., Danysz, W., and Wenk, G. L. (2000). The cytotoxicity of chronic neuroinflammation upon basal forebrain cholinergic neurons of rats can be attenuated by glutamatergic antagonism or cyclooxygenase-2 inhibition. Exp. Brain Res. 134, 58–65. doi: 10.1007/s002210000446
Wright, C. I., Geula, C., and Mesulam, M. M. (1993). Neurological cholinesterases in the normal brain and in Alzheimer's disease: relationship to plaques, tangles, and patterns of selective vulnerability. Ann. Neurol. 34, 373–384. doi: 10.1002/ana.410340312
Wu, M. Y., Esteban, G., Brogi, S., Shionoya, M., Wang, L., Campiani, G., et al. (2015). Donepezil-like multifunctional agents: design, synthesis, molecular modelling and biological evaluation. Eur. J. Med. Chem. doi: 10.1016/j.ejmech.2015.10.001. [Epub ahead of print].
Wu, W., Lei, P., Liu, Q., Hu, J., Gunn, A. P., Chen, M., et al. (2008). Sequestration of copper from beta-amyloid promotes selective lysis by cyclen-hybrid cleavage agents. J. Biol. Chem. 283, 31657–31664. doi: 10.1074/jbc.M804722200
Xie, W., Stribley, J. A., Chatonnet, A., Wilder, P. J., Rizzino, A., McComb, R. D., et al. (2000). Postnatal developmental delay and supersensitivity to organophosphate in gene-targeted mice lacking acetylcholinesterase. J. Pharmacol. Exp. Ther. 293, 896–902.
Yamamoto, A., Shin, R. W., Hasegawa, K., Naiki, H., Sato, H., Kitamoto, T., et al. (2002). Iron (III) induces aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: Implications in the formation of neurofibrillary tangles of Alzheimer's disease. J. Neurochem. 82, 1137–1147. doi: 10.1046/j.1471-4159.2002.t01-1-01061.x
You, H., Tsutsui, S., Hameed, S., Kannanayakal, T. J., Chen, L., Xia, P., et al. (2012). Ab neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc. Natl. Acad. Sci. U.S.A. 109, 1737–1742. doi: 10.1073/pnas.1110789109
Youdim, M. B. H., Edmondson, D., and Tipton, K. F. (2006). The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 7, 295–309. doi: 10.1038/nrn1883
Youdim, M. B. H., Maruyama, W., and Naoi, M. (2005). Neuropharmacological, neuroprotective and amyloid precursor processing properties of selective MAO-B inhibitor antiparkinsonian drug, rasagiline. Drugs Today 41, 369–391. doi: 10.1358/dot.2005.41.6.893613
Yu, W. H., Lukiw, W. J., Bergeron, C., Niznik, H. B., and Fraser, P. E. (2001). Metallothionein III is reduced in Alzheimer's disease. Brain Res. 894, 37–45. doi: 10.1016/S0006-8993(00)03196-6
Zheng, H., Gal, S., Weiner, L. M., Bar-Am, O., Warshawsky, A., Fridkin, M., et al. (2005). Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases: in vitro studies on antioxidant activity, prevention of lipid peroxide formation and monoamine oxidase inhibition. J. Neurochem. 95, 68–78. doi: 10.1111/j.1471-4159.2005.03340.x
Zhou, L. X., Du, J. T., Zeng, Z. Y., Wu, W. H., Zhao, Y. F., Kanazawa, K., et al. (2007). Copper (II) modulates in vitro aggregation of tau peptide. Peptides 28, 2229–2234. doi: 10.1016/j.peptides.2007.08.022
Zhu, X., Lee, H. G., Perry, G., and Smith, M. A. (2007). Alzheimer disease, the two-hit hypothesis: an update. Biochim. Biophys. Acta 1772, 494–502. doi: 10.1016/j.bbadis.2006.10.014
Zhukareva, V., Vogelsberg-Ragaglia, V., Van Deerlin, V. M., Bruce, J., Shuck, T., Grossman, M., et al. (2001). Loss of brain tau defines novel sporadic and familial tauopathies with frontotemporal dementia. Ann. Neurol. 49, 165–175.
Keywords: multi-target-directed ligands, donepezil, oxidative stress, anti-β-amyloid aggregation, Alzheimer's disease
Citation: Unzeta M, Esteban G, Bolea I, Fogel WA, Ramsay RR, Youdim MBH, Tipton KF and Marco-Contelles J (2016) Multi-Target Directed Donepezil-Like Ligands for Alzheimer's Disease. Front. Neurosci. 10:205. doi: 10.3389/fnins.2016.00205
Received: 16 March 2016; Accepted: 25 April 2016;
Published: 25 May 2016.
Edited by:
Francisco Lopez-Munoz, University of Alcala, SpainReviewed by:
Massimo Grilli, University of Genova, ItalyCopyright © 2016 Unzeta, Esteban, Bolea, Fogel, Ramsay, Youdim, Tipton and Marco-Contelles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mercedes Unzeta, bWVyY2VkZXMudW56ZXRhQHVhYi5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.