 Matthew J. Korn
Matthew J. Korn Quinton J. Mandle
Quinton J. Mandle Jack M. Parent
Jack M. Parent- 1Department of Neurology, University of Michigan Medical Center, Ann Arbor, MI, USA
- 2VA Ann Arbor Healthcare System, Ann Arbor, MI, USA
Many animal models of temporal lobe epilepsy (TLE) exhibit altered neurogenesis arising from progenitors within the dentate gyrus subgranular zone (SGZ). Aberrant integration of new neurons into the existing circuit is thought to contribute to epileptogenesis. In particular, adult-born neurons that exhibit ectopic migration and hilar basal dendrites (HBDs) are suggested to be pro-epileptogenic. Loss of reelin signaling may contribute to these morphological changes in patients with epilepsy. We previously demonstrated that conditional deletion of the reelin adaptor protein, disabled-1 (Dab1), from postnatal mouse SGZ progenitors generated dentate granule cells (DGCs) with abnormal dendritic development and ectopic placement. To determine whether the early postnatal loss of reelin signaling is epileptogenic, we conditionally deleted Dab1 in neural progenitors and their progeny on postnatal days 7–8 and performed chronic video-EEG recordings 8–10 weeks later. Dab1-deficient mice did not have spontaneous seizures but exhibited interictal epileptiform abnormalities and a significantly reduced latency to pilocarpine-induced status epilepticus. After chemoconvulsant treatment, over 90% of mice deficient for Dab1 developed generalized motor convulsions with tonic-clonic movements, rearing, and falling compared to <20% of wild-type mice. Recombination efficiency, measured by Cre reporter expression, inversely correlated with time to the first sustained seizure. These pro-epileptogenic changes were associated with decreased neurogenesis and increased numbers of hilar ectopic DGCs. Interestingly, neurons co-expressing the Cre reporter comprised a fraction of these hilar ectopic DGCs cells, suggesting a non-cell autonomous effect for the loss of reelin signaling. We also noted a dispersion of the CA1 pyramidal layer, likely due to hypomorphic effects of the conditional Dab1 allele, but this abnormality did not correlate with seizure susceptibility. These findings suggest that the misplacement or reduction of postnatally-generated DGCs contributes to aberrant circuit development and hyperexcitability, but aberrant neurogenesis after conditional Dab1 deletion alone is not sufficient to produce spontaneous seizures.
Introduction
Mesial temporal lobe epilepsy (mTLE) is a common and often intractable focal epilepsy (Bender et al., 2004; Dube et al., 2012). Structural changes in the hippocampal dentate gyrus (DG) are implicated in mTLE pathogenesis, though the mechanisms underlying these changes are poorly understood. Neurogenesis is markedly disrupted in rodent models of mTLE (Parent et al., 1997, 2006; Jessberger et al., 2005, 2007; Shapiro et al., 2005; Murphy et al., 2011, 2012), leading to the idea that aberrantly integrated adult-born neurons (namely dentate granule cells, DGCs) produce seizures (Parent and Kron, 2012; Pun et al., 2012; Bielefeld et al., 2014; Cho et al., 2015) and contribute to associated comorbidities such as cognitive dysfunction and depression (Groticke et al., 2007; Muller et al., 2009; Zhang et al., 2010; Lesting et al., 2011; Levin et al., 2012; Klein et al., 2015).
The role of adult DGC neurogenesis is complex and there are several competing hypotheses as to how continuous integration of new neurons might be implicated in seizure development. Intact adult neurogenesis may mitigate acute seizure susceptibility (Iyengar et al., 2015) and suppressing adult neurogenesis with brain irradiation can exacerbate kindling progression (Raedt et al., 2007; Pekcec et al., 2011). However, the development of epilepsy after an acute insult, such as status epilepticus (SE), is attenuated if the seizure-induced increase in adult neurogenesis is inhibited by antimitotic agents (Jung et al., 2006; Sugaya et al., 2010) or pharmacogenetic methods (Cho et al., 2015). Perhaps most compelling is the finding that disrupting the integration of a subset of postnatal-born DGCs by suppressing phosphatase and tensin homolog (PTEN) signaling is sufficient to produce spontaneous seizures in mice, and the affected DGCs exhibit many of the aberrant changes seen in animal models of mTLE (Pun et al., 2012). Thus, there is an interest in identifying signaling pathways that regulate DGC neurogenesis and whose disruption might produce mTLE.
Reelin-expressing interneurons persist throughout the adult DG (Alcantara et al., 1998; Pesold et al., 1998; Gong et al., 2007) and reelin expression is decreased in human and experimental mTLE (Gong et al., 2007; Haas and Frotscher, 2010). It is proposed that altered neurogenesis in mTLE may arise in part due to disrupted reelin signaling (Haas et al., 2002; Gong et al., 2007; Kobow et al., 2009; Haas and Frotscher, 2010; Freiman et al., 2011). Loss of the reelin adaptor protein, disabled-1 (Dab1), imparts a similar developmental phenotype to loss of reelin function (Howell et al., 1997a; Sheldon et al., 1997; Olson et al., 2006). In prior work, we conditionally deleted Dab1 from nestin-expressing neural stem cells (NSCs) in the postnatal mouse subgranular zone (SGZ) and found that NSCs deficient in Dab1 generate adult-born neurons that migrate ectopically and have abnormal dendritic development (Teixeira et al., 2012). Here we explore whether the disrupted integration of Dab1-deficient early postnatal- through adult-born neurons leads to altered seizure susceptibility or epilepsy.
Materials and Methods
Animals and Tamoxifen Administration
All procedures and experiments were approved by the University of Michigan Institutional Committee on the Use and Care of Animals and were performed in accordance with guidelines developed by the National Institutes of Health. NestinCreERT2/R26RYFP/Dab1Flox/Flox triple transgenic mice were derived and maintained as previously described (Teixeira et al., 2012). Briefly, mice harboring the Dab1-conditional allele (Pramatarova et al., 2008) were crossed with Nestin-Cre-ERT2 mice (Lagace et al., 2007; “Line K” in Sun et al., 2014) and a recombination reporter line, R26R-YFP, containing a floxed-stop followed by yellow fluorescent protein (YFP) in the ROSA26 gene locus (Zambrowicz et al., 1997). Thus, tamoxifen (TMX)-inducible, cre-mediated recombination results in the deletion of Dab1 along with expression of YFP in nestin-expressing progenitors and their subsequent progeny. Bigenic mice with wild-type Dab1 treated with TMX and mice with mutant Dab1 but not treated with TMX were used as controls. Heterozygous mice (NestinCreERT2/R26RYFP/Dab1Flox/+) served as a gene dose comparison to the homozygous null mutants. We refer to homozygous mutants as Dab1Flox/Flox, heterozygous as Dab1Flox/+, and controls as Dab1+/+. Experimenters were blind to genotype and pups were treated i.p. once daily with TMX at 100 mg/kg (dissolved in 10% EtOH/90% sunflower oil, 12.5 mg/ml) or vehicle on P7 and P8. Animals survived to P55 before any additional manipulations were performed (Supplemental Figure 1). Unless otherwise noted, mice were housed under a 12 h light/dark cycle and were given food and water ad libitum.
Epidural Electrode Placement and Video-EEG
To determine if Dab1-deficient mice exhibit spontaneous seizures or epileptiform activity, we monitored animals with continuous video-EEG. Mice were implanted with four epidural screw electrodes at 2 months after the last administration of TMX. Procedures for affixing electrodes were performed as previously described (Lee et al., 2012; Kehrl et al., 2014; Wagnon et al., 2015). Mice were anesthetized with ketamine and xylazine and placed in a stereotaxic mouse adaptor (Stoelting). Six burr holes were made in the skull using a 3.2 mm steel bit (Meisinger USA, Centennial, Colorado). Electrodes were positioned and fastened (left and right frontal, left and right parietal, one cerebellar, and one reference over the sinus cavity) using mounting screws (E363/20; PlasticsOne, Roanoke, VA). The sockets were fitted into a 6-pin electrode pedestal and the entire apparatus was secured with dental cement (Stoelting). Animals received buprenorphine (0.05 mg/kg) every 12 h for 3 days after surgery. Following recovery, animals were monitored continuously for 10 days by video/EEG recording (Ceegraph Vision; Natus, Middleton, WI). Recordings were sampled at 256 Hz and concurrent video was analyzed offline and synced with EEG data in Persyst 12 (Persyst, Prescott, AZ). Seizures and epileptiform activity were assessed manually in their entirety by an observer blind to the genotypes. Interictal epileptiform discharges (IEDs) were defined as paroxysmal waveforms lasting 20–200 ms having spike or sharp-wave and after-going slow wave morphology, identified visually to be independent of preceding behavioral activity. Spontaneous seizures were defined as the abrupt appearance of rhythmic waveforms or runs of spikes/sharp waves that persisted for a minimum of 10 s and displayed an unequivocal evolution in frequency and morphology, followed by postictal suppression and return to baseline. Detailed evaluation of EEG waveforms was performed by randomly selecting a 4-h epoch from each day during the 10 day recording period.
Pilocarpine Administration and Seizure Threshold
Mice were injected intraperitoneally with scopolamine methyl nitrate (5 mg/kg) to block peripheral cholinergic effects, followed 20 min later with pilocarpine hydrochloride (289 mg/kg), a muscarinic cholinergic agonist, to produce SE. Animals were monitored for 45 min and were given additional half-doses (not exceeding twice the original dose) of pilocarpine if no seizure activity was detected.
The clinical manifestation of SE was scored using a modified Racine scale (Racine, 1972). The scoring was: 0 for mice displaying no abnormal behavior; 1 for periods of freezing, chewing or nodding; 2–3 if tremors and shaking developed, along with straub tail; 4 for mice with seizures that included rearing or clonus, followed by a clear recovery; 5 was used for mice with rearing and clonus, along with running and falling; 6 for severe and repeated stage 5 seizures where animals either did not recover between events or expired.
Seizure threshold was defined as the latency (in minutes) from the administration of pilocarpine until the animal experienced the electrographic onset of SE, consisting of either a prolonged seizure lasting at least 3 min or 3 contiguous seizure events less than 2-min apart from one another without intervening complete behavioral recovery. Electrographic onset was defined by a distinctive shift in background activity whereby low-amplitude high frequency waves evolved into large, rhythmic epileptiform discharges, at least one standard deviation greater in amplitude compared to background activity.
Tissue Processing and Immunohistochemistry
Immediately after a 90-min SE episode, mice were injected with an overdose of sodium pentobarbital and perfused intracardially with 0.9% saline followed by 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS), pH 7.4. Brains were fixed overnight, cryoprotected in 30% sucrose at 4°C, and cyrosectioned at 40 μm in the coronal plane using a Leica CM1850 cyrostat (Leica Microsystems, St. Louis, MO). Sections were collected into cyropreservative (sucrose and ethylene glycol in 50 mM phosphate buffer) as series of every 12th section through the entire extent of the septotemporal axis and stored at −20°C until processed for immunohistoschemistry (IHC). Tissues were rinsed thoroughly in Tris buffered saline (TBS, pH 7.6) and incubated for 90 min at room temperature in a blocking solution containing 10% normal goat serum, triton x-100, glycine, and bovine serum albumin in TBS. Floating sections were incubated overnight at 4°C with a cell type-specific antibody along with a chicken anti-GFP (1:2000, Aves Labs - GFP-1020; Cho et al., 2015) to enhance the endogenous YFP fluorescence and confirm colocalization. Cell-type specific antibodies included rabbit anti-doublecortin (DCX, 1:1000, AbCam - ab18723; Lee and Umemori, 2013), mouse anti-NeuN (1:2000, Chemicon - MAB377; Miltiadous et al., 2013), and rabbit anti-Prox1 (1:2000, Millipore - AB5475; Farrar et al., 2005; Parent et al., 2006). We verified deletion of Dab1 by using a rabbit anti-Dab1 antibody (1:1000, Sigma − ab-232; Howell et al., 1997b; Teixeira et al., 2012 and Figures 3A,B'). Rabbit anti-MAP2 (2a+2b) antibody (1:1000, Sigma-M2320; Quadrato et al., 2014) was used to visualize hippocampal dendritic organization. Rabbit Ki67 antibody was used to label actively dividing cells (1:1000, Vector Labs–VP-K451; Singer et al., 2009). Alexa secondary antibodies (1:600; ThermoFisher) were used for single or double immunofluorescence. In some instances tissue was briefly rinsed with Hoescht 33342 (1:5000) for a bisbenzimide nuclear counterstain. Low-magnification images were acquired on a Leica DMI 6000 inverted scope and higher magnification images for co-localization were collected as thin optical section (1–2 μm) z-stacks on a Nikon A-1 laser scanning confocal microscope.
Data Quantification
For colocalization with each cell type-specific antigen, we quantified the left or right hemisphere of 4–5 sections per animal. Three-dimensional stacks were viewed as a maximum intensity projection and a grid comprised of 100 × 100 μm squares was placed over the image in ImageJ 1.48 (NIH, Bethesda, MD). Squares containing the GCL and SGZ were numbered and at least three were selected at random for quantification. Cells expressing the reporter were identified in the z-stack and colocalization of the Alexa secondary was confirmed in x and y dimension using Nikon Element Viewer (Melville, NY). No fewer than 5 cells were counted per section and a minimum of 30 cells were counted per animal. Values are reported as cells that express YFP and cell type–specific antigen over total number of YFP-positive cells. For recombination efficiency, we estimated the number of YFP-labeled cells per 100 μm2 by averaging the number of cells over three tissue sections for each animal. We verified localization of each cell type-specific antigen with bisbenzimide. Density of DCX+ cells was estimated with the aid of MicroBrightField Neurolucida (MBF, Williston, VT). For each animal, we selected a 500 μm linear region along the GCL and included cells within the SGZ and GCL. Only whole round or spherical cell bodies were counted and fragments of DCX labeling less than 5 μm were excluded. Cells were tagged with a digital marker through each optical section to prevent double-counting. Clusters of cells where one cell body merged into another were counted as one, thus our quantification underestimates the total number of DCX-positive cells. This number is reported as average number of DCX-expressing neurons per 500 μm. The density of hilar ectopic granule cells (HEGCs) was determined by counting all Prox1+ cells within a defined region of interest restricted to the hilar region of the DG, delineated as 20 μm from the bottom of the GCL. Representative images of labeling were exported as TIFF files and edited using Corel PaintShop Pro X7 (Corel, Ottawa, Ontario, Canada). Images were cropped to fit the dimensions of each figure.
Statistical Analyses
All statistical analyses were made using JMP Pro 11 software (SAS, Cary, NC). Data sets were checked for normal distributions. ANOVA with post-hoc test was performed to assess differences across multiple comparisons. Individual comparisons were made using Tukey's HSD test to correct for sample number. We performed a non-parametric comparison using Wilcoxon's t-test for comparison between groups for latency and Spearman's ρ or Fisher's Exact Test (small sample sizes) for multivariate correlations. Values are reported as mean ± SEM, for all statistical comparisons: *p < 0.05, **p < 0.02.
Results
Dab1–Deficient Adult Mice Exhibit Infrequent Interictal EEG Abnormalities and Rare Seizure-Like Events
We reviewed 10 day video-EEG records and found that no vehicle treated Dab1Flox/Flox (n = 10), vehicle or TMX treated Dab1Flox/+ (n = 5) or TMX treated Dab1+/+ (n = 9), exhibited seizures or IEDs (Figure 1A). A total of 3 of the 17 Dab1Flox/Flox mice that received TMX exhibited IEDs. One mouse exhibited brief myoclonic spasms, with preceding spike-wave discharges (black arrowheads, Figure 1B). We identified a second animal (Figure 1C) with recurrent epileptiform discharges at regular intervals with background suppression and slowing, coinciding with prolonged periods of immobility, tremors, and labored movement. A third mouse (Figure 1D) displayed intermittent 2–4 s discharges of large amplitude theta waves that were occasionally followed by brief spasms, not associated with an obvious epileptiform EEG discharge. Lastly, a single mouse exhibited intermittent spike-wave discharges (black arrowheads, Figure 1E) throughout the 10-day recording period but experienced no behavioral events. These observations indicate that deletion of Dab1 in postnatal NSCs does not cause overt spontaneous convulsive seizures, but leads to hyperexcitable network changes that can generate spike discharges and sporadic EEG patterns with behavioral events on an interictal-to-ictal continuum.
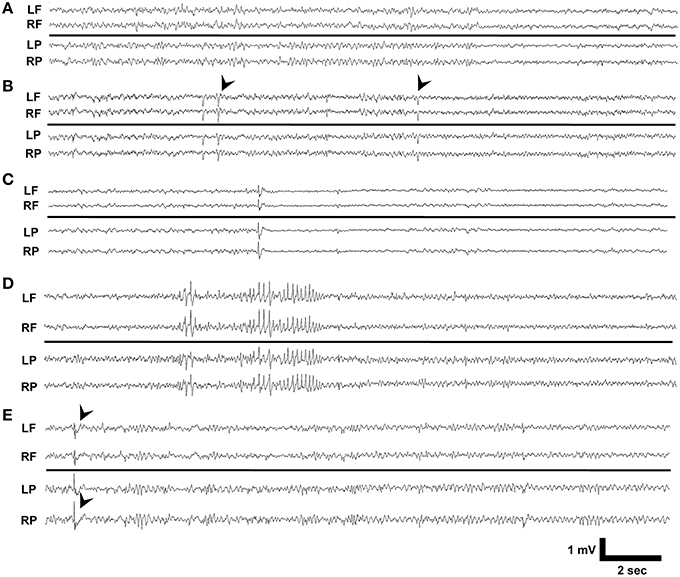
Figure 1. EEG phenotypes of Dab1Flox/Flox mutants. (A) Typical interictal EEG which exhibits no abnormalities, interictal spikes, or epileptiform discharges. (B) Intermittent sharp waves and background slowing which occasionally was associated with a myoclonic spasm (black arrowheads). This patterned activity persisted for 3 min. (C) Prolonged periods of low amplitude beta with intermittent spikes coinciding with behavioral inactivity. (D) High amplitude, sharply contoured theta waveforms that preceded myoclonus. (E) Interictal spike discharges (black arrowheads) appeared throughout the 10 day recording period as the sole abnormality in this mouse. LF, left frontal; RF, right frontal; LP, left parietal; RP, right parietal.
Adult Mice Deficient in Dab1 Exhibit More Severe Seizures and a Reduced Latency to the Onset of Pilocarpine-Induced SE
To probe whether postnatal Dab1 deletion in DGCs alters susceptibility to developing SE after pilocarpine administration, we injected drug while animals were still being monitored by video-EEG. In addition to scoring the severity of the seizures, we measured the latency to onset of the first seizure and to the beginning of SE.
All Dab1Flox/Flox mice, regardless of whether or not they were treated with TMX, developed SE (Figure 2A, n = 20). This was in contrast to only 50% of Dab1+/+ (n = 4) and 67% of Dab1Flox/+ (n = 5) mice, indicating that mutant mice are significantly more likely to develop SE (Fisher's Exact Test; p < 0.02). Mutants typically required less pilocarpine to develop SE, but we determined that this variability was not significant (Fisher's Exact Test; p = 0.27). In some cases, mice that failed to develop SE exhibited brief seizures. Of the animals that did develop behavioral and electrographic SE, only mutants that received TMX (n = 12) were likely to develop severe, stage 5 seizures (Figure 2B, Fisher's Exact Test; p < 0.02). Only three mice expired during the course of the experiments (1 each in Dab1+/+ + TMX, Dab1Flox/Flox + vehicle, and Dab1Flox/Flox + TMX groups), and mortality did not differ between groups (Fisher's Exact Test; p = 0.86).
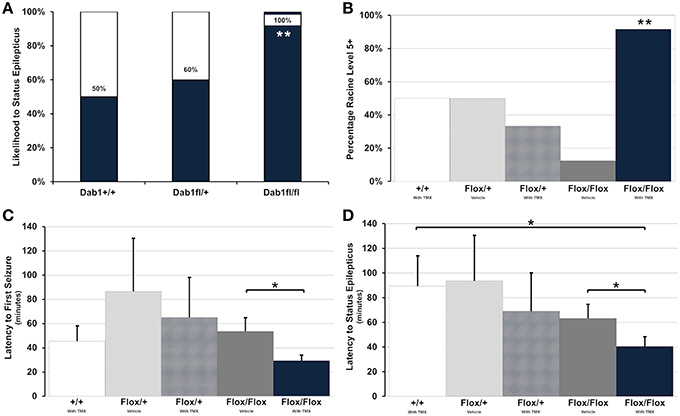
Figure 2. Conditional deletion of Dab1 reduces SE latency and increases seizure severity. (A) Dab1Flox/Flox mice were significantly more likely to develop SE compared to Dab1Flox/+ or Dab1+/+ mice. (B) Compared to all other treatment groups, TMX treated Dab1Flox/Flox mice were significantly more likely to develop severe, Racine stage 5 or 6 seizures following the administration of pilocarpine. (C) TMX-treated Dab1Flox/Flox mice exhibited the shortest latency to the first seizure and (D) a significantly shorter latency to SE compared to vehicle treated Dab1Flox/Flox or Dab1+/+mice. For all statistical comparisons: *p < 0.05, **p < 0.02.
Dab1Flox/Flox mice treated with TMX consistently had the earliest onset to the first seizure (Figure 2C), which was significantly shorter compared to Dab1Flox/Flox given vehicle (Dab1Flox/Flox + TMX: n = 12, 29.4 ± 4.1; Dab1Flox/Flox + vehicle: n = 8, 53.6 ± 10.7; p < 0.05). Dab1Flox/Flox mice had a significantly reduced latency to SE (Figure 2D, 40.5 ± 7.4) compared to Dab1Flox/Flox animals treated with vehicle (63.3 ± 10.7; p < 0.05) and mice with WT Dab1 (89.3 ± 24.2; p < 0.05), but were not different than Dab1Flox/+ mice given TMX (69.0 ± 30.6; p = 0.17) or vehicle (93.5 ± 36.5; p = 0.08). From these data we conclude that mice with conditional Dab1 deletion in postnatal DGCs develop seizures that evolve into SE more quickly than controls, and exhibit a greater seizure severity after pilocarpine treatment.
Recombination Efficiency and Deletion of Dab1 is Negatively Correlated with Latency to SE
Dab1 protein is predominately expressed throughout the SGZ of adult mice (white arrowheads, Figure 3A), and at lower levels in the GCL (white arrow, Figure 3A). Sporadic Dab1-positive neurons were also seen in the hilus and inner molecular layer. Treatment with TMX resulted in the expression of YFP (Figure 3B) and loss in the expression of virtually all Dab1 protein (Figure 3B′). YFP expression was limited to regions that exhibit postnatal neurogenesis.
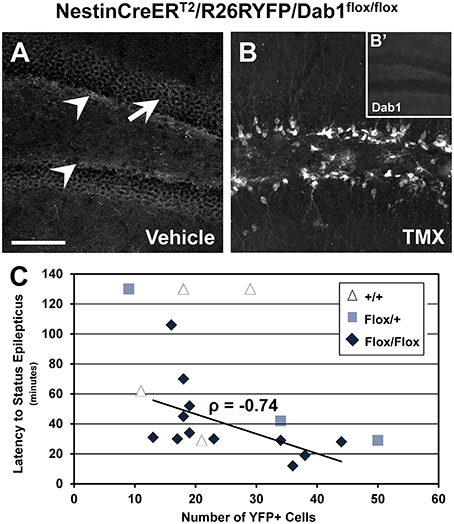
Figure 3. Recombination efficiency and latency to status epilepticus in NestinCreERT2/R26RYFP/Dab1Flox/Flox mice. (A–B′) Confocal images of sections through the DG immunostained for Dab1 (A,B′) or YFP (B). (A) Dab1 expression is distributed through the SGZ (arrowheads) of the DG in untreated Dab1Flox/Flox mice. Scattered labeling is present in the GCL (arrow) and to a lesser extent within the molecular layer and hilus. (B) Two days of TMX treatment induces Cre activation and recombination in Dab1Flox/Flox mice, resulting in substantial YFP reporter expression 2 months later. (B′) Note that virtually no Dab1 expression remains in the DG after TMX treatment. (C) There is a significant inverse correlation between recombination efficiency and latency to SE in TMX treated Dab1Flox/Flox mice, indicating that the latency to SE is influenced by reelin signaling in postnatal- or adult-born DGCs. No correlation exists for Dab1+/+ mice. Scale bar = 100 μm (A,B), 250 μm in (B′).
Using reporter activity as a proxy for recombination efficiency, we sought to determine if latency to SE varied as a function of the loss of Dab1 protein (Figure 3C). We found a significant inverse correlation between recombination efficiency after TMX treatment and SE onset latency for Dab1Flox/Flox (n = 12; ρ = −0.74, p < 0.02), an inverse correlation for Dab1Flox/+ (n = 3; ρ = −1), but not Dab1+/+ (n = 4; ρ = 0.41) mice. This finding suggests that progressively greater loss of Dab1 results in a more pronounced susceptibility to pilocarpine-induced SE. Thus, despite the absence of definite spontaneous seizures, altered circuitry associated with the conditional deletion of Dab1 appears to be pro-epileptic.
Dab1Flox/Flox Mice Exhibit a Dispersed CA1 Pyramidal Cell Layer which does not Correlate with Seizure Phenotype
We next sought to examine structural alternations potentially underlying the increased seizure susceptibility caused by conditional Dab1 deletion in postnatal NSCs and their progeny. Mice homozygous for floxed Dab1 have reduced Dab1 protein levels from P1-7, even without cre driven recombination (Pramatarova et al., 2008; Teixeira et al., 2014). This coincides with the period of development when pyramidal neurons migrate away from the ventricular zone and the hippocampal primordium to stratify in the outer lamina of CA1 (Stanfield and Cowan, 1979; Nakahira and Yuasa, 2005). It is not surprising then that the CA1 pyramidal layer is disrupted in mutants whether treated with vehicle or TMX (Figures 4A–C). MAP2ab labeling, which allows for visualization of CA1 apical dendrites, revealed that the ectopic layer of pyramidal neurons maintain their apical arbors (Figures 4D–F). In contrast to the highly organized dendritic fields of the inner layer, dendrites emanating from ectopic pyramidal neurons were notably tortuous (white arrow in Figure 4F′). This phenotype did not appear to contribute to the reduced seizure latency as we found no correlation between the mean width of the CA1 layer and latency to SE (Figure 4G) in vehicle (ρ = −0.36, p = 0.43) or TMX treated (ρ = −0.14, p = 0.66) mice, regardless of genotype (data not shown).
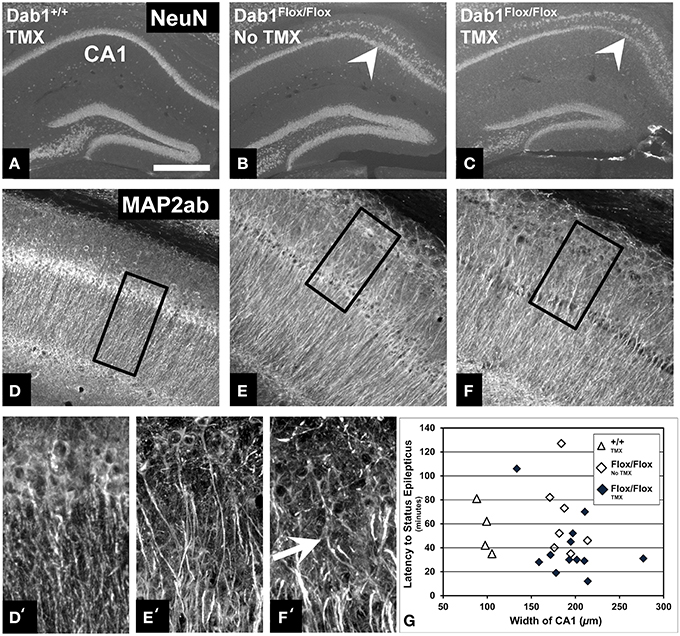
Figure 4. Abnormalities of the CA1 pyramidal layer do not correlate with latency to SE. (A) Labeling for NeuN demonstrates that the CA1 pyramidal cell layer of mice with WT Dab1 is single layered compared to the ectopic layer (above the white arrowheads, which point to the “normal” layer) found in hypomorphic mice harboring floxed-Dab1, regardless if they are treated with vehicle (B) or TMX (C). Similar to mice with WT Dab1 (D), the primary pyramidal layer of Dab1Flox/Flox mice treated with vehicle (E) or TMX (F), exhibit intact apical dendrites. (D′) Dendrites from pyramidal neurons of WT mice extend perpendicular to the long access of the cell layer. In contrast, dendrites of the ectopic pyramidal layer in mice with Floxed-Dab1, treated with vehicle (E′) or TMX (F′) have curved apical arbors (white arrow) with overlapping domains. (D′–F′) are the boxed regions in (D–F), respectively. (G) Despite this pronounced phenotype, there was no correlation between overall width of the CA1 pyramidal layer and latency to SE. Scale bar = 500 μm (A–C), 300 μm (D–F), and 50 μm (D′–F').
Dab1-Deficient NSCs Produce Fewer Immature Neurons but More HEGCs, the Degree of which Correlates with Reduced Seizure Latency
Previous work suggests that loss of Dab1 in postnatal NSCs and their progeny alters DGC development and maturation (Teixeira et al., 2012). To assess the degree of neurogenesis and location of DGCs after conditional Dab1 deletion, we performed IHC to identify proliferating cells (Ki67), immature neurons (DCX), and post-mitotic granule cells (Prox1).
We found significantly fewer actively proliferating cells in Dab1Flox/Flox mice (Figure 5A, n = 6; 10.3 ± 1.0) compared to controls (Figures 5B,C; n = 5; 25.5 ± 5.5; p < 0.02). Consistent with the decreased cell proliferation, there were also less DCX-labeled immature neurons observed in TMX treated Dab1Flox/Flox mice compared to wild-type vehicle treated controls (Figures 5D,E), suggesting decreased neurogenesis. As with our previous findings (Teixeira et al., 2012), immature neurons exhibited stunted dendrites with atrophic apical branches (white arrowheads, Figure 5E). We found fewer DCX-positive immature neurons in TMX treated Dab1Flox/Flox mice (Figure 5F; n = 14; 19.6 ± 2.2 per 500 linear μm) compared to TMX treated Dab1+/+ mice (n = 8; 32.6 ± 3.4; p < 0.02), but not Dab1Flox/+ mice (n = 3; 22.2 ± 6.7; p = 0.10).
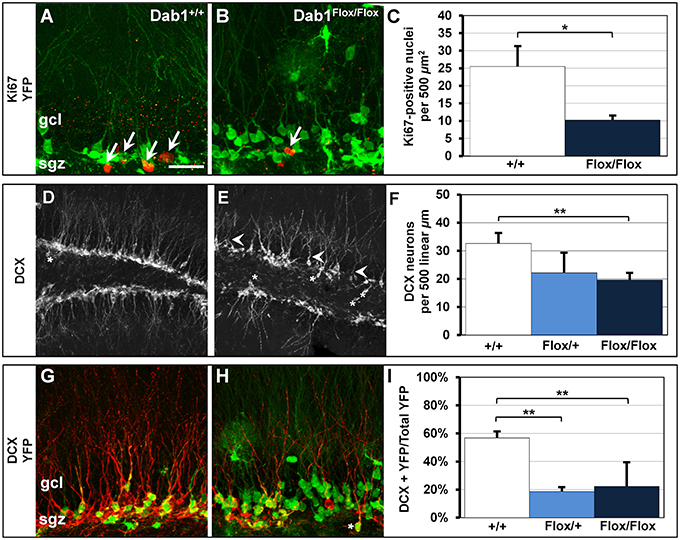
Figure 5. Cell proliferation and DCX-positive immature neurons are reduced in Dab1-deficient mice. (A) Ki67-labeled nuclei are distributed throughout the SGZ of Dab1+/+ mice treated with TMX and are often seen in clusters (white arrows). (B) In contrast, sparsely distributed Ki67-labeled nuclei (white arrow) are observed in Dab1Flox/Flox mice treated with TMX. (C) There are significantly fewer Ki67-labeled nuclei in Dab1Flox/Flox mice treated with TMX compared to controls treated with TMX. (D) DCX-expressing immature neurons are distributed throughout the SGZ and inner GCL of WT mice. Most have distinct apical dendrites (white arrowheads) and very few are found in the hilus (asterisk). (E) In contrast, we observe a sparse distribution of DCX-positive immature neurons in Dab1-deificient mice. Immature neurons exhibit fewer, atrophic, apical dendrites (white arrowheads) and several are located ectopically (asterisks). (F) There are significantly fewer DCX-expressing immature neurons throughout the SGZ and GCL of Dab1Flox/Flox mice treated with TMX compared to controls, but not Dab1Flox/+ mice, treated with TMX. (G) DCX/YFP co-expressing immature neurons are distributed throughout the SGZ and inner third of the granule cell layer. (H) Despite the appearance of more YFP labeling, fewer YFP+ cells co-localized with DCX throughout the SGZ, and some appeared ectopic (asterisks). (I) Significantly fewer DCX/YFP double-labeled cells appeared in Dab1Flox/+ and Dab1Flox/Flox mice compared to controls. Scale bar (in A) = 20 μm for (A,B,G,H) and 100 μm for (D,E).
We then quantified the percentage of DCX-positive cells that co-expressed YFP to determine how loss of Dab1 directly affects neuronal differentiation (Figures 5G,H). We observed significantly fewer DCX+/YFP+ immature neurons in TMX-treated Dab1Flox/Flox (Figure 5I; n = 17; 22.3% ± 0.03) and Dab1Flox/+ mice (n = 3; 18.6% ± 0.04) compared to TMX-treated controls (n = 9; 56.8% ± 0.05, p < 0.02). From these data we conclude that the loss of Dab1 reduces the proliferation of DG neural progenitors, which contributes to the decrease in the number of immature neurons.
We previously demonstrated that adult-born neurons generated from Dab1-deficient, nestin-expressing NSCs and their progeny migrate ectopically (Teixeira et al., 2012). Here we confirm the observation that mice deficient for Dab1 exhibit significantly more Prox1+ HEGCs (Figures 6A–C; n = 12, 6.48 ± 0.75 per 100 μm2) than TMX-treated controls (n = 6, 1.20 ± 0.36 per 100 μm2, p < 0.02) or Dab1Flox/+ mice (n = 2, 1.83 ± 0.12 per 100 μm2, p < 0.05). Notably, the number of HEGCs inversely correlated with the latency to onset of SE (Figure 6D; n = 13, ρ = −0.56, p < 0.05). Initially, we reasoned that it was the cell autonomous deletion of Dab1 from nestin-expressing NSCs that was responsible for causing Dab1-deficient neurons to migrate ectopically away from the GCL. In examining YFP+/Prox1+ granule cells (data not shown), we noted that the majority of HEGCs were negative for YFP reporter expression (Figure 6E, 83.95% ± 0.03). Taken together, the conditional deletion of Dab1 from postnatal nestin-expressing NSCs results in a non-cell autonomous ectopic migration of granule cells into the hilus, and their presence correlates with a reduced latency to SE onset.
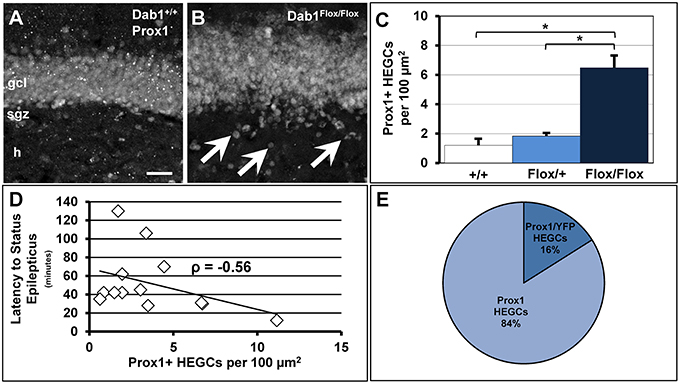
Figure 6. Conditional deletion of Dab1 results in non-cell autonomous ectopic migration of Prox1+ granule cells that inversely correlates with latency to SE. (A) The GCL of Dab1+/+ mice given TMX is compact with very few Prox1+ cells found outside the SGZ or GCL. (B) The overall structure of the GCL is disrupted in TMX-treated Dab1Flox/Flox mice, with many Prox1+ HEGCs evident (arrowheads). (C) The density of HEGCs in Dab1Flox/Flox mice was significantly greater compared to controls and Dab1Flox/+ mice after TMX treatment. *p < 0.05. (D) The density of Prox1 + HEGCs inversely correlated with latency to SE. (E) The majority of Prox1 + HEGCs were negative for YFP, supporting a non-cell autonomous effect of the loss of reelin signaling. Scale bar = 50 μm in (A,B).
Discussion
We sought to determine if changes that arise following conditional deletion of Dab1 from postnatal NSCs and their progeny cause spontaneous seizures. We observed no definite behavioral or electrographic seizures, but some mice experienced single myoclonic spasms associated with EEG discharges, and others showed three different patterns of epileptiform or paroxysmal discharges among 4 of 17 TMX-treated mutant mice. These findings suggest that the conditional loss of reelin signaling in postnatally-generated DGCs does not result in a robust epilepsy phenotype. However, we did find that TMX-treated Dab1Flox/Flox mice are more likely to develop severe SE after administration of the chemoconvulstant pilocarpine, with a shorter latency than TMX-treated heterozygous or wild-type mice, as well as vehicle-treated homozygous littermates. Histological analyses confirmed that the postnatal loss of Dab1 in NSCs induces the ectopic migration of DGCs (Teixeira et al., 2012) and reduces ongoing adult neurogenesis. Possible mechanisms for this latter finding may involve promoting NSC quiescence, changing the phenotype of NSC progeny from neuronal to glial (Teixeira et al., 2012), or decreasing the proliferation or survival of transit amplifying progenitors or immature neurons.
The Development of HEGCs in Dab1Flox/Flox Mice is not Sufficient to Generate Spontaneous Seizures
Among the more notable changes in Dab1Flox/Flox mice is the ectopic placement of adult-born granule cells into the hilus and along the border between the GCL and the inner molecular layer of the dentate gyrus (Figure 6; Teixeira et al., 2012). Dispersion of the GCL and HEGCs are hallmark features in humans with mTLE (Lurton et al., 1998; Parent et al., 2006; Blumcke et al., 2009; Bae et al., 2010) and can be reproduced in models that induce SE with kainic acid, pilocarpine, or limbic kindling (Bouilleret et al., 1999; McCloskey et al., 2006; Botterill et al., 2015). Several studies have confirmed that the loss of reelin correlates with aberrant migration (Haas et al., 2002; Heinrich et al., 2006; Gong et al., 2007; Haas and Frotscher, 2010), but our study is unique in several ways. In contrast to the overt developmental phenotype of the reeler mutant mouse, conditional deletion of Dab1 from postnatal nestin-expressing NSCs allows us to explore whether the aberrant development of neonatal- and adult-born neurons disrupt an already established circuit. Secondly, our approach evaluates the contribution of displaced neonatal- or adult-born DGCs to a potential seizure phenotype, separate from the ectopic dispersion of mature DGCs in the DG (Heinrich et al., 2006). Another striking observation is the large percentage of ectopic Prox1+/YFP- DGCs, presumably those expressing normal levels of Dab1. Based upon this finding, we postulate that although the loss of Dab1 in the progeny of NSCs may contribute to a migration deficit, the majority of HEGCs migrate into the hilus due to a non-cell autonomous effect of postnatal Dab1 deletion.
A prevailing hypothesis is that HEGCs contribute to the development of spontaneous seizures and epilepsy (Scharfman and Pierce, 2012), but our observation that mice with ectopically located granule cells are largely seizure-free suggests that there are unaccounted for factors that promote seizure development. Our findings are similar to the study by Koyama et al. (2012) where they demonstrated that generating aberrantly located neurons without inducing seizures was not sufficient to produce rats with epilepsy. Deletion of the pro-apoptotic gene BAX generates mice with HEGCs, and though the mice are deficient in a pattern separation task, they also remain seizure-free (Myers et al., 2013). These observations, along with the current study, challenge the hypothesis that HEGCs are sufficient for driving spontaneous seizures.
This does not eliminate the possibility that HEGCs can act as “hub” cells within a seizure network (Morgan and Soltesz, 2008; Cameron et al., 2011). There is clear electrophysiological evidence that they are hyperexcitable and make aberrant connections (Scharfman et al., 2000; Zhan and Nadler, 2009; Althaus et al., 2015). There is also strong correlative evidence between the presence of HEGCs and seizure severity (McCloskey et al., 2006; Hester and Danzer, 2013). It is possible that our deletion is not sufficient enough to generate enough aberrantly placed GCs. In support of this idea, the mouse with the highest recombination efficiency and that also had the greatest density of HEGCs did not show abnormalities on video/EEG monitoring. However, this mouse did exhibit the shortest latency to SE. We also considered the possibility that we did not allow enough time for the aberrant neurons to integrate. We addressed this by extending the period after TMX to 4 months, but these mice were also seizure-free. Though we cannot definitively exclude the possibility that there are subcortical seizures with a limited focal onset, HEGCs are not sufficient to cause epilepsy with clinical manifestations via cortical EEG, including generalized tonic-clonic seizures.
Notably, the study showing spontaneous seizure development after postnatal deletion of PTEN in murine DGCs also demonstrated substantial structural abnormalities in PTEN-deficient HEGCs and those in the GCL. These changes included increased soma size and an excess of dendritic spines, features that are likely important for the development of spontaneous seizures (Pun et al., 2012). We did not observe such structural abnormalities after postnatal Dab1 deletion. In summary, our findings suggest that HEGCs produce network instability, but additional aberrant structural or functional features are necessary to cause epilepsy.
CA1 Pyramidal Cell Layer Disruption in Dab1 Hypomorphs does not Contribute to Hyperexcitability in the Hippocampus
Mice expressing floxed dab1 alleles, whether they are treated with vehicle or TMX, have been shown to express less protein during early postnatal development than the wild-type, and this hypomorphic effect causes abnormalities in the cerebellum and CA1, but not the dentate gyrus (Herrick and Cooper, 2002; Matsuki et al., 2008; Pramatarova et al., 2008). More recently, this structural phenotype was implicated in causing deficits in maternal behavior and spatial learning (Teixeira et al., 2014). Other genetic models that exhibit a similar heterotopia of CA1 exhibit deficits in long-term potentiation (Petrone et al., 2003). Occasionally, a bilayered CA1 pyramidal cell layer is even present in human mTLE hippocampal specimens (Sloviter et al., 2004). All of these observations are consistent with a role of this morphological abnormality in mTLE or its associated comorbidities. We noted that the CA1 phenotype was particularly severe in our Dab1Flox/Flox mice (Figures 4A–C), raising concerns that the ectopic CA1 may contribute to overall hyperexcitability. The strongest evidence to the contrary is the fact that Dab1Flox/Flox mice that exhibit a severe CA1 dispersion in the absence of TMX have a seizure latency which is significantly longer compared to Dab1Flox/Flox mice treated with TMX. This is particularly remarkable considering the dendritic disorganization found among the ectopically located CA1 pyramidal neurons (Figures 4D–F).
Recent work examining the onset of spontaneous seizure in the pilocarpine model of mTLE in the rat indicates that the firing rate of CA1 increases preictally and may indicate a focal ictal onset zone (Fujita et al., 2014). Despite the dramatic dispersion of CA1, the lack of hyperexcitability leads us to conclude that aberrant migration of CA1 pyramidal cells is not sufficient to drive spontaneous seizures. This further supports the role of the DG in seizure susceptibility and underscores the implications for aberrantly integrated adult-born neurons (Krook-Magnuson et al., 2015).
Reelin Signaling Modulates Adult Neurogenesis
Previous reports have demonstrated a role for reelin signaling in regulating embryonic (Won et al., 2006; Lakoma et al., 2011) and postnatal neurogenesis (Pujadas et al., 2010). We observed a significant decrease in DCX-expressing immature neurons in the adult DG of Dab1-deficient mice, which is novel in this context because the deletion of Dab1 targets NSCs that primarily contribute to ongoing adult neurogenesis. Interestingly, we found no difference in the number of NeuN+/YFP+ cells (data not shown), suggesting that Dab1-deficient NSCs differentiate into neurons at the same rate or higher than intact NSCs. One possible interpretation of these findings is that the percentage of neurons surviving after differentiation increases to compensate for the overall decrease in levels of cell proliferation (Tanapat et al., 2001). Another explanation, not necessary mutually exclusive, is that deletion of Dab1 causes newly-generated neurons to mature faster, resulting in a greater proportion of NeuN+/YFP+ neurons. Prolonged seizures have been shown to have this effect in a mouse model of mTLE (Overstreet-Wadiche et al., 2006), and together with our findings these data further implicate a role for the loss of reelin in structural DG abnormalities associated with mTLE. Future work should be directed at determining how Dab1 deletion influences the survival of newly generated neurons.
Recent work suggests that mice with reduced adult neurogenesis (by x-irradiation or pharmacogenetic deletion) exhibit a potentiated response to chemoconvulsants (Iyengar et al., 2015). Moreover, the pharmacogenetic suppression of adult neurogenesis leads to a potentially compensatory plasticity within the DG involving disinhibition of pre-existing DGCs (Singer et al., 2011). These observations are consistent with our findings that Dab1-deficent mice have dramatically fewer immature neurons and a reduced latency to pilocarpine-induced SE. Our results therefore highlight the importance of adult neurogenesis in maintaining network stability and suggest that this circuit is a potential target for anti-epileptogenic interventions.
Author Contributions
MK and JP all contributed to the design and interpretation of the results. MK and QM collected, processed and quantified data included in this manuscript. MK and JP drafted and revised the contents of this manuscript; MK, QM, and JP approved the final version and its conclusions. MK, QM, and JP agreed to be accountable for the contents of this report.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like thank Corey Powell and the Consulting for Statistics, Computing, and Analytics Research office at the University of Michigan for their assistance. These studies were supported by a NIH Grant to JP (NS058585) and Epilepsy Foundation Postdoctoral Fellowship to MK.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnins.2016.00063
Supplementary Figure 1. Flowchart of procedures and experimental time points. Animals were treated with TMX, 2 months later they were fitted for EEG and monitored for 10 days. At the end of the recording period, we challenged the seizure sensitivity with pilocarpine. Once in SE, mice were monitored for 90 min, at which time they were euthanized.
References
Alcantara, S., Ruiz, M., D'arcangelo, G., Ezan, F., De Lecea, L., Curran, T., et al. (1998). Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J. Neurosci. 18, 7779–7799.
Althaus, A. L., Sagher, O., Parent, J. M., and Murphy, G. G. (2015). Intrinsic neurophysiological properties of hilar ectopic and normotopic dentate granule cells in human temporal lobe epilepsy and a rat model. J. Neurophysiol. 113, 1184–1194. doi: 10.1152/jn.00835.2014
Bae, E. K., Jung, K. H., Chu, K., Lee, S. T., Kim, J. H., Park, K. I., et al. (2010). Neuropathologic and clinical features of human medial temporal lobe epilepsy. J. Clin. Neurol. 6, 73–80. doi: 10.3988/jcn.2010.6.2.73
Bender, R. A., Dube, C., and Baram, T. Z. (2004). Febrile seizures and mechanisms of epileptogenesis: insights from an animal model. Adv. Exp. Med. Biol. 548, 213–225. doi: 10.1007/978-1-4757-6376-8_15
Bielefeld, P., Van Vliet, E. A., Gorter, J. A., Lucassen, P. J., and Fitzsimons, C. P. (2014). Different subsets of newborn granule cells: a possible role in epileptogenesis? Eur. J. Neurosci. 39, 1–11. doi: 10.1111/ejn.12387
Blumcke, I., Kistner, I., Clusmann, H., Schramm, J., Becker, A. J., Elger, C. E., et al. (2009). Towards a clinico-pathological classification of granule cell dispersion in human mesial temporal lobe epilepsies. Acta Neuropathol. 117, 535–544. doi: 10.1007/s00401-009-0512-5
Botterill, J. J., Guskjolen, A. J., Marks, W. N., Caruncho, H. J., and Kalynchuk, L. E. (2015). Limbic but not non-limbic kindling impairs conditioned fear and promotes plasticity of NPY and its Y2 receptor. Brain Struct. Funct. 220, 3641–3655. doi: 10.1007/s00429-014-0880-z
Bouilleret, V., Ridoux, V., Depaulis, A., Marescaux, C., Nehlig, A., and Le Gal La Salle, G. (1999). Recurrent seizures and hippocampal sclerosis following intrahippocampal kainate injection in adult mice: electroencephalography, histopathology and synaptic reorganization similar to mesial temporal lobe epilepsy. Neuroscience 89, 717–729. doi: 10.1016/S0306-4522(98)00401-1
Cameron, M. C., Zhan, R. Z., and Nadler, J. V. (2011). Morphologic integration of hilar ectopic granule cells into dentate gyrus circuitry in the pilocarpine model of temporal lobe epilepsy. J. Comp. Neurol. 519, 2175–2192. doi: 10.1002/cne.22623
Cho, K. O., Lybrand, Z. R., Ito, N., Brulet, R., Tafacory, F., Zhang, L., et al. (2015). Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat. Commun. 6:6606. doi: 10.1038/ncomms7606
Dube, C. M., Mcclelland, S., Choy, M. K., Brewster, A. L., Noam, Y., and Baram, T. Z. (2012). “Fever, febrile seizures and epileptogenesis,” in Jasper's Basic Mechanisms of the Epilepsies, 4th Edn., eds. J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen, and A. V. Delgado-Escueta (Bethesda, MD: National Center for Biotechnology Information).
Farrar, C. E., Huang, C. S., Clarke, S. G., and Houser, C. R. (2005). Increased cell proliferation and granule cell number in the dentate gyrus of protein repair-deficient mice. J. Comp. Neurol. 493, 524–537. doi: 10.1002/cne.20780
Freiman, T. M., Eismann-Schweimler, J., and Frotscher, M. (2011). Granule cell dispersion in temporal lobe epilepsy is associated with changes in dendritic orientation and spine distribution. Exp. Neurol. 229, 332–338. doi: 10.1016/j.expneurol.2011.02.017
Fujita, S., Toyoda, I., Thamattoor, A. K., and Buckmaster, P. S. (2014). Preictal activity of subicular, CA1, and dentate gyrus principal neurons in the dorsal hippocampus before spontaneous seizures in a rat model of temporal lobe epilepsy. J. Neurosci. 34, 16671–16687. doi: 10.1523/JNEUROSCI.0584-14.2014
Gong, C., Wang, T. W., Huang, H. S., and Parent, J. M. (2007). Reelin regulates neuronal progenitor migration in intact and epileptic hippocampus. J. Neurosci. 27, 1803–1811. doi: 10.1523/JNEUROSCI.3111-06.2007
Groticke, I., Hoffmann, K., and Loscher, W. (2007). Behavioral alterations in the pilocarpine model of temporal lobe epilepsy in mice. Exp. Neurol. 207, 329–349. doi: 10.1016/j.expneurol.2007.06.021
Haas, C. A., Dudeck, O., Kirsch, M., Huszka, C., Kann, G., Pollak, S., et al. (2002). Role for reelin in the development of granule cell dispersion in temporal lobe epilepsy. J. Neurosci. 22, 5797–5802.
Haas, C. A., and Frotscher, M. (2010). Reelin deficiency causes granule cell dispersion in epilepsy. Exp. Brain Res. 200, 141–149. doi: 10.1007/s00221-009-1948-5
Heinrich, C., Nitta, N., Flubacher, A., Muller, M., Fahrner, A., Kirsch, M., et al. (2006). Reelin deficiency and displacement of mature neurons, but not neurogenesis, underlie the formation of granule cell dispersion in the epileptic hippocampus. J. Neurosci. 26, 4701–4713. doi: 10.1523/JNEUROSCI.5516-05.2006
Herrick, T. M., and Cooper, J. A. (2002). A hypomorphic allele of dab1 reveals regional differences in reelin-Dab1 signaling during brain development. Development 129, 787–796.
Hester, M. S., and Danzer, S. C. (2013). Accumulation of abnormal adult-generated hippocampal granule cells predicts seizure frequency and severity. J. Neurosci. 33, 8926–8936. doi: 10.1523/JNEUROSCI.5161-12.2013
Howell, B. W., Gertler, F. B., and Cooper, J. A. (1997a). Mouse disabled (mDab1): a Src binding protein implicated in neuronal development. EMBO J. 16, 121–132. doi: 10.1093/emboj/16.1.121
Howell, B. W., Hawkes, R., Soriano, P., and Cooper, J. A. (1997b). Neuronal position in the developing brain is regulated by mouse disabled-1. Nature 389, 733–737. doi: 10.1038/39607
Iyengar, S. S., Lafrancois, J. J., Friedman, D., Drew, L. J., Denny, C. A., Burghardt, N. S., et al. (2015). Suppression of adult neurogenesis increases the acute effects of kainic acid. Exp. Neurol. 264, 135–149. doi: 10.1016/j.expneurol.2014.11.009
Jessberger, S., Romer, B., Babu, H., and Kempermann, G. (2005). Seizures induce proliferation and dispersion of doublecortin-positive hippocampal progenitor cells. Exp. Neurol. 196, 342–351. doi: 10.1016/j.expneurol.2005.08.010
Jessberger, S., Zhao, C., Toni, N., Clemenson, G. D. Jr., Li, Y., and Gage, F. H. (2007). Seizure-associated, aberrant neurogenesis in adult rats characterized with retrovirus-mediated cell labeling. J. Neurosci. 27, 9400–9407. doi: 10.1523/JNEUROSCI.2002-07.2007
Jung, K. H., Chu, K., Lee, S. T., Kim, J., Sinn, D. I., Kim, J. M., et al. (2006). Cyclooxygenase-2 inhibitor, celecoxib, inhibits the altered hippocampal neurogenesis with attenuation of spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neurobiol. Dis. 23, 237–246. doi: 10.1016/j.nbd.2006.02.016
Kehrl, J. M., Sahaya, K., Dalton, H. M., Charbeneau, R. A., Kohut, K. T., Gilbert, K., et al. (2014). Gain-of-function mutation in Gnao1: a murine model of epileptiform encephalopathy (EIEE17)? Mamm. Genome 25, 202–210. doi: 10.1007/s00335-014-9509-z
Klein, S., Bankstahl, J. P., Loscher, W., and Bankstahl, M. (2015). Sucrose consumption test reveals pharmacoresistant depression-associated behavior in two mouse models of temporal lobe epilepsy. Exp. Neurol. 263, 263–271. doi: 10.1016/j.expneurol.2014.09.004
Kobow, K., Jeske, I., Hildebrandt, M., Hauke, J., Hahnen, E., Buslei, R., et al. (2009). Increased reelin promoter methylation is associated with granule cell dispersion in human temporal lobe epilepsy. J. Neuropathol. Exp. Neurol. 68, 356–364. doi: 10.1097/NEN.0b013e31819ba737
Koyama, R., Tao, K., Sasaki, T., Ichikawa, J., Miyamoto, D., Muramatsu, R., et al. (2012). GABAergic excitation after febrile seizures induces ectopic granule cells and adult epilepsy. Nat. Med. 18, 1271–1278. doi: 10.1038/nm.2850
Krook-Magnuson, E., Armstrong, C., Bui, A., Lew, S., Oijala, M., and Soltesz, I. (2015). In vivo evaluation of the dentate gate theory in epilepsy. J. Physiol. 593, 2379–2388. doi: 10.1113/JP270056
Lagace, D. C., Whitman, M. C., Noonan, M. A., Ables, J. L., Decarolis, N. A., Arguello, A. A., et al. (2007). Dynamic contribution of nestin-expressing stem cells to adult neurogenesis. J. Neurosci. 27, 12623–12629. doi: 10.1523/JNEUROSCI.3812-07.2007
Lakoma, J., Garcia-Alonso, L., and Luque, J. M. (2011). Reelin sets the pace of neocortical neurogenesiss. Development 138, 5223–5234. doi: 10.1242/dev.063776
Lee, C. H., Javed, D., Althaus, A. L., Parent, J. M., and Umemori, H. (2012). Neurogenesis is enhanced and mossy fiber sprouting arises in FGF7-deficient mice during development. Mol. Cell. Neurosci. 51, 61–67. doi: 10.1016/j.mcn.2012.07.010
Lee, C. H., and Umemori, H. (2013). Suppression of epileptogenesis-associated changes in response to seizures in FGF22-deficient mice. Front. Cell. Neurosci. 7:43. doi: 10.3389/fncel.2013.00043
Lesting, J., Geiger, M., Narayanan, R. T., Pape, H. C., and Seidenbecher, T. (2011). Impaired extinction of fear and maintained amygdala-hippocampal theta synchrony in a mouse model of temporal lobe epilepsy. Epilepsia 52, 337–346. doi: 10.1111/j.1528-1167.2010.02758.x
Levin, J. R., Serrano, G., and Dingledine, R. (2012). Reduction in delayed mortality and subtle improvement in retrograde memory performance in pilocarpine-treated mice with conditional neuronal deletion of cyclooxygenase-2 gene. Epilepsia 53, 1411–1420. doi: 10.1111/j.1528-1167.2012.03584.x
Lurton, D., El Bahh, B., Sundstrom, L., and Rougier, A. (1998). Granule cell dispersion is correlated with early epileptic events in human temporal lobe epilepsy. J. Neurol. Sci. 154, 133–136. doi: 10.1016/S0022-510X(97)00220-7
Matsuki, T., Pramatarova, A., and Howell, B. W. (2008). Reduction of Crk and CrkL expression blocks reelin-induced dendritogenesis. J. Cell Sci. 121, 1869–1875. doi: 10.1242/jcs.027334
McCloskey, D. P., Hintz, T. M., Pierce, J. P., and Scharfman, H. E. (2006). Stereological methods reveal the robust size and stability of ectopic hilar granule cells after pilocarpine-induced status epilepticus in the adult rat. Eur. J. Neurosci. 24, 2203–2210. doi: 10.1111/j.1460-9568.2006.05101.x
Miltiadous, P., Kouroupi, G., Stamatakis, A., Koutsoudaki, P. N., Matsas, R., and Stylianopoulou, F. (2013). Subventricular zone-derived neural stem cell grafts protect against hippocampal degeneration and restore cognitive function in the mouse following intrahippocampal kainic acid administration. Stem Cells Transl. Med. 2, 185–198. doi: 10.5966/sctm.2012-0074
Morgan, R. J., and Soltesz, I. (2008). Nonrandom connectivity of the epileptic dentate gyrus predicts a major role for neuronal hubs in seizures. Proc. Natl. Acad. Sci. U.S.A. 105, 6179–6184. doi: 10.1073/pnas.0801372105
Muller, C. J., Groticke, I., Bankstahl, M., and Loscher, W. (2009). Behavioral and cognitive alterations, spontaneous seizures, and neuropathology developing after a pilocarpine-induced status epilepticus in C57BL/6 mice. Exp. Neurol. 219, 284–297. doi: 10.1016/j.expneurol.2009.05.035
Murphy, B. L., Hofacer, R. D., Faulkner, C. N., Loepke, A. W., and Danzer, S. C. (2012). Abnormalities of granule cell dendritic structure are a prominent feature of the intrahippocampal kainic acid model of epilepsy despite reduced postinjury neurogenesis. Epilepsia 53, 908–921. doi: 10.1111/j.1528-1167.2012.03463.x
Murphy, B. L., Pun, R. Y., Yin, H., Faulkner, C. R., Loepke, A. W., and Danzer, S. C. (2011). Heterogeneous integration of adult-generated granule cells into the epileptic brain. J. Neurosci. 31, 105–117. doi: 10.1523/JNEUROSCI.2728-10.2011
Myers, C. E., Bermudez-Hernandez, K., and Scharfman, H. E. (2013). The influence of ectopic migration of granule cells into the hilus on dentate gyrus-CA3 function. PLoS ONE 8:e68208. doi: 10.1371/journal.pone.0068208
Nakahira, E., and Yuasa, S. (2005). Neuronal generation, migration, and differentiation in the mouse hippocampal primoridium as revealed by enhanced green fluorescent protein gene transfer by means of in utero electroporation. J. Comp. Neurol. 483, 329–340. doi: 10.1002/cne.20441
Olson, E. C., Kim, S., and Walsh, C. A. (2006). Impaired neuronal positioning and dendritogenesis in the neocortex after cell-autonomous Dab1 suppression. J. Neurosci. 26, 1767–1775. doi: 10.1523/JNEUROSCI.3000-05.2006
Overstreet-Wadiche, L. S., Bromberg, D. A., Bensen, A. L., and Westbrook, G. L. (2006). Seizures accelerate functional integration of adult-generated granule cells. J. Neurosci. 26, 4095–4103. doi: 10.1523/JNEUROSCI.5508-05.2006
Parent, J. M., Elliott, R. C., Pleasure, S. J., Barbaro, N. M., and Lowenstein, D. H. (2006). Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Ann. Neurol. 59, 81–91. doi: 10.1002/ana.20699
Parent, J. M., and Kron, M. M. (2012). “Neurogenesis and Epilepsy,” in Jasper's Basic Mechanisms of the Epilepsies, 4th Edn., eds. J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen, and A. V. Delgado-Escueta (Bethesda, MD: National Center for Biotechnology Information).
Parent, J. M., Yu, T. W., Leibowitz, R. T., Geschwind, D. H., Sloviter, R. S., and Lowenstein, D. H. (1997). Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J. Neurosci. 17, 3727–3738.
Pekcec, A., Lupke, M., Baumann, R., Seifert, H., and Potschka, H. (2011). Modulation of neurogenesis by targeted hippocampal irradiation fails to affect kindling progression. Hippocampus 21, 866–876. doi: 10.1002/hipo.20802
Pesold, C., Impagnatiello, F., Pisu, M. G., Uzunov, D. P., Costa, E., Guidotti, A., et al. (1998). Reelin is preferentially expressed in neurons synthesizing gamma-aminobutyric acid in cortex and hippocampus of adult rats. Proc. Natl. Acad. Sci. U.S.A. 95, 3221–3226. doi: 10.1073/pnas.95.6.3221
Petrone, A., Battaglia, F., Wang, C., Dusa, A., Su, J., Zagzag, D., et al. (2003). Receptor protein tyrosine phosphatase alpha is essential for hippocampal neuronal migration and long-term potentiation. EMBO J. 22, 4121–4131. doi: 10.1093/emboj/cdg399
Pramatarova, A., Chen, K., and Howell, B. W. (2008). A genetic interaction between the APP and Dab1 genes influences brain development. Mol. Cell. Neurosci. 37, 178–186. doi: 10.1016/j.mcn.2007.09.008
Pujadas, L., Gruart, A., Bosch, C., Delgado, L., Teixeira, C. M., Rossi, D., et al. (2010). Reelin regulates postnatal neurogenesis and enhances spine hypertrophy and long-term potentiation. J. Neurosci. 30, 4636–4649. doi: 10.1523/JNEUROSCI.5284-09.2010
Pun, R. Y., Rolle, I. J., Lasarge, C. L., Hosford, B. E., Rosen, J. M., Uhl, J. D., et al. (2012). Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron 75, 1022–1034. doi: 10.1016/j.neuron.2012.08.002
Quadrato, G., Elnaggar, M. Y., Duman, C., Sabino, A., Forsberg, K., and Di Giovanni, S. (2014). Modulation of GABAA receptor signaling increases neurogenesis and suppresses anxiety through NFATc4. J. Neurosci. 34, 8630–8645. doi: 10.1523/JNEUROSCI.0047-14.2014
Racine, R. J. (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32, 281–294. doi: 10.1016/0013-4694(72)90177-0
Raedt, R., Boon, P., Persson, A., Alborn, A. M., Boterberg, T., Van Dycke, A., et al. (2007). Radiation of the rat brain suppresses seizure-induced neurogenesis and transiently enhances excitability during kindling acquisition. Epilepsia 48, 1952–1963. doi: 10.1111/j.1528-1167.2007.01146.x
Scharfman, H. E., Goodman, J. H., and Sollas, A. L. (2000). Granule-like neurons at the hilar/CA3 border after status epilepticus and their synchrony with area CA3 pyramidal cells: functional implications of seizure-induced neurogenesis. J. Neurosci. 20, 6144–6158.
Scharfman, H. E., and Pierce, J. P. (2012). New insights into the role of hilar ectopic granule cells in the dentate gyrus based on quantitative anatomic analysis and three-dimensional reconstruction. Epilepsia 53(Suppl. 1), 109–115. doi: 10.1111/j.1528-1167.2012.03480.x
Shapiro, L. A., Korn, M. J., and Ribak, C. E. (2005). Newly generated dentate granule cells from epileptic rats exhibit elongated hilar basal dendrites that align along GFAP-immunolabeled processes. Neuroscience 136, 823–831. doi: 10.1016/j.neuroscience.2005.03.059
Sheldon, M., Rice, D. S., D'arcangelo, G., Yoneshima, H., Nakajima, K., Mikoshiba, K., et al. (1997). Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature 389, 730–733. doi: 10.1038/39601
Singer, B. H., Gamelli, A. E., Fuller, C. L., Temme, S. J., Parent, J. M., and Murphy, G. G. (2011). Compensatory network changes in the dentate gyrus restore long-term potentiation following ablation of neurogenesis in young-adult mice. Proc. Natl. Acad. Sci. U.S.A. 108, 5437–5442. doi: 10.1073/pnas.1015425108
Singer, B. H., Jutkiewicz, E. M., Fuller, C. L., Lichtenwalner, R. J., Zhang, H., Velander, A. J., et al. (2009). Conditional ablation and recovery of forebrain neurogenesis in the mouse. J. Comp. Neurol. 514, 567–582. doi: 10.1002/cne.22052
Sloviter, R. S., Kudrimoti, H. S., Laxer, K. D., Barbaro, N. M., Chan, S., Hirsch, L. J., et al. (2004). “Tectonic” hippocampal malformations in patients with temporal lobe epilepsy. Epilepsy Res. 59, 123–153. doi: 10.1016/j.eplepsyres.2004.04.001
Stanfield, B. B., and Cowan, W. M. (1979). The development of the hippocampus and dentate gyrus in normal and reeler mice. J. Comp. Neurol. 185, 423–459. doi: 10.1002/cne.901850303
Sugaya, Y., Maru, E., Kudo, K., Shibasaki, T., and Kato, N. (2010). Levetiracetam suppresses development of spontaneous EEG seizures and aberrant neurogenesis following kainate-induced status epilepticus. Brain Res. 1352, 187–199. doi: 10.1016/j.brainres.2010.06.061
Sun, M. Y., Yetman, M. J., Lee, T. C., Chen, Y., and Jankowsky, J. L. (2014). Specificity and efficiency of reporter expression in adult neural progenitors vary substantially among nestin-CreER(T2) lines. J. Comp. Neurol. 522, 1191–1208. doi: 10.1002/cne.23497
Tanapat, P., Hastings, N. B., Rydel, T. A., Galea, L. A., and Gould, E. (2001). Exposure to fox odor inhibits cell proliferation in the hippocampus of adult rats via an adrenal hormone-dependent mechanism. J. Comp. Neurol. 437, 496–504. doi: 10.1002/cne.1297
Teixeira, C. M., Kron, M. M., Masachs, N., Zhang, H., Lagace, D. C., Martinez, A., et al. (2012). Cell-Autonomous Inactivation of the Reelin Pathway Impairs Adult Neurogenesis in the Hippocampus. J. Neurosci. 32, 12051–12065. doi: 10.1523/JNEUROSCI.1857-12.2012
Teixeira, C. M., Masachs, N., Muhaisen, A., Bosch, C., Perez-Martinez, J., Howell, B., et al. (2014). Transient downregulation of Dab1 protein levels during development leads to behavioral and structural deficits: relevance for psychiatric disorders. Neuropsychopharmacology 39, 556–568. doi: 10.1038/npp.2013.226
Wagnon, J. L., Korn, M. J., Parent, R., Tarpey, T. A., Jones, J. M., Hammer, M. F., et al. (2015). Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Hum. Mol. Genet. 24, 506–515. doi: 10.1093/hmg/ddu470
Won, S. J., Kim, S. H., Xie, L., Wang, Y., Mao, X. O., Jin, K., et al. (2006). Reelin-deficient mice show impaired neurogenesis and increased stroke size. Exp. Neurol. 198, 250–259. doi: 10.1016/j.expneurol.2005.12.008
Zambrowicz, B. P., Imamoto, A., Fiering, S., Herzenberg, L. A., Kerr, W. G., and Soriano, P. (1997). Disruption of overlapping transcripts in the ROSA beta geo 26 gene trap strain leads to widespread expression of beta-galactosidase in mouse embryos and hematopoietic cells. Proc. Natl. Acad. Sci. U.S.A. 94, 3789–3794. doi: 10.1073/pnas.94.8.3789
Zhan, R. Z., and Nadler, J. V. (2009). Enhanced tonic GABA current in normotopic and hilar ectopic dentate granule cells after pilocarpine-induced status epilepticus. J. Neurophysiol. 102, 670–681. doi: 10.1152/jn.00147.2009
Keywords: neural stem cells, adult neurogenesis, temporal lobe epilepsy, pilocarpine
Citation: Korn MJ, Mandle QJ and Parent JM (2016) Conditional Disabled-1 Deletion in Mice Alters Hippocampal Neurogenesis and Reduces Seizure Threshold. Front. Neurosci. 10:63. doi: 10.3389/fnins.2016.00063
Received: 15 December 2015; Accepted: 10 February 2016;
Published: 25 February 2016.
Edited by:
Laura López-Mascaraque, Instituto Cajal-Consejo Superior de Investigaciones Científicas, SpainReviewed by:
Helen E. Scharfman, New York State Department of Health, USAAshok K. Shetty, Texas A&M Health Science Center College of Medicine at Scott & White, USA
Copyright © 2016 Korn, Mandle and Parent. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jack M. Parent, cGFyZW50QHVtaWNoLmVkdQ==