 Sammy Kimoloi
Sammy Kimoloi Khalid Rashid
Khalid Rashid- 1Department of Medical Laboratory Sciences, Masinde Muliro University of Science and Technology, Kakamega, Kenya
- 2Biochemistry and Molecular Biology Department, Egerton University, Nakuru, Kenya
Cerebral malaria (CM) is the most severe complication associated with Plasmodium falciparum infection. The exact pathogenic mechanisms leading to the development of CM remains poorly understood while the mortality rates remain high. Several potential mechanisms including mechanical obstruction of brain microvasculature, inflammation, oxidative stress, cerebral energy defects, and hemostatic dysfunction have been suggested to play a role in CM pathogenesis. However, these proposed mechanisms, even when considered together, do not fully explain the pathogenesis and clinicopathological features of human CM. This necessitates consideration of alternative pathogenic mechanisms. P. falciparum generates substantial amounts of ammonia as a catabolic by-product, but lacks detoxification mechanisms. Whether this parasite-derived ammonia plays a pathogenic role in CM is presently unknown, despite its potential to cause localized brain ammonia elevation and subsequent neurotoxic effects. This article therefore, explores and proposes a potential role of parasite-derived ammonia in the pathogenesis and neuropathology of CM. A consideration of parasite-derived ammonia as a factor in CM pathogenesis provides plausible explanations of the various features observed in CM patients including how a largely intravascular parasite can cause neuronal dysfunction. It also provides a framework for rational development and testing of novel drugs targeting the parasite's ammonia handling.
Introduction
Cerebral malaria (CM) is one of the most common and fatal complications associated with Plasmodium falciparum infection (Newton et al., 2000). Despite the availability of effective anti-malarial drugs, CM mortality rates in malaria afflicted regions remain high, ranging between 15 and 20% (Mishra and Newton, 2009). This has partly been attributed to the fact that a number of patients succumb to CM before the onset of administered anti-malarial therapeutic effects (Mishra and Newton, 2009). Transient and persistent neurocognitive impairments are also common among cerebral malaria survivors, mostly children (van der Wal et al., 2005; Idro et al., 2010). Several mechanisms have been hypothesized to play a role in the pathogenesis of CM including; mechanical obstruction of brain microvasculature by sequestered parasitized red blood cells (PRBCs), inflammation, hemostatic dysfunction, excessive parasite-derived lactate, and oxidative stress (Berendt et al., 1994; Medana et al., 2001; van der Heyde et al., 2006; Mariga et al., 2014). However, the exact pathogenesis of CM remains poorly understood and the various clinicopahtological features of human CM cannot be fully explained by the existing hypotheses (van der Heyde et al., 2006). Additionally, adjunctive therapies targeting some of these earlier proposed pathogenic mechanisms have failed to improve CM treatment outcomes or have been found detrimental in human clinical studies (White et al., 2013). Exploration of other potential pathogenic factors is therefore needed in order to understand the underlying CM pathogenesis and develop adjunctive therapies that can possibly prolong anti-malarial therapeutic window and reduce neurocognitive sequelae among survivors.
Ammonia is a neurotoxic metabolic by-product whose blood concentration is tightly maintained at levels of less than 50 μmol/L mainly by the action of liver glutamine synthetase, which has a lower Km for ammonia than carbamoyl-phosphate synthetase (Arn et al., 1990; Cohn and Roth, 2004; Adeva et al., 2012). The brain astrocytic glutamine synthetase also plays a minor role, converting upto 25% of blood derived ammonia to glutamine (Eid and Lee, 2013). Several factors including, acute liver failure (ALF), urea cycle disorders, Reye's syndrome and hepatic drug toxity may however interfere with the normal ammonia metabolism leading to hyperammonemia (Cohn and Roth, 2004). Regardless of the cause, elevations of blood ammonia concentration above the normal physiological levels often lead to encephalopathies (Haberle, 2013). Interestingly, the clinicopathological features of acquired and congenital hyperammonemic encephalopathies such as coma, brain edema, and seizures (Summar et al., 2008; Haberle, 2013) are analogous to some of the features observed in CM patients (Idro et al., 2005). Moreover, experimental cerebral malaria (ECM) in murine models has also revealed metabolic changes that point toward parasite-induced perturbation of ammonia detoxification (Ghosh et al., 2012). These observations, coupled to the fact that P. falciparum generates large amounts of ammonia in the absence of intrinsic parasite detoxification mechanisms (Zeuthen et al., 2006) clearly suggests a potential role of parasite-derived ammonia in CM pathogenesis.
It was recently hypothesized that development of ALF in malaria patients may lead to accumulation of ammonia and other neurotoxins, which then enter the brain aided by blood brain barrier (BBB) breakdown and ultimately cause neurological damage and manifestation of CM (Martins and Daniel-Ribeiro, 2013). However, several studies indicates that parasite-induced liver dysfunction is not a common occurrence in P. falciparum malaria (Anand et al., 1992; Murthy et al., 1998; Nacher et al., 2001; Mazumder et al., 2002; Mohanty et al., 2004; Prommano et al., 2005; Kochar et al., 2010; Whitten et al., 2011). Therefore, it is conceivable that sequestered parasites in the brain vasculature known to produce large amounts of ammonia as a catabolic by-product, may disrupt normal brain ammonia metabolism leading to local accumulation of ammonia. This possibility has however not been considered as a pathogenic factor in CM. The aim of the current article is therefore to explore potential mechanisms by which P. falciparum may cause localized elevation of brain ammonia and subsequent neurotoxicity.
Human Brain Ammonia Uptake and Metabolism
The human brain usually takes up ammonia from cerebral blood vessels using different transport mechanisms. The non-protonated NH3 form is easily taken up by simple diffusion and there is usually a positive linear correlation between arterial ammonia concentration and brain uptake (Cooper and Plum, 1987). The ammonium ion NH+4 on the other hand does not easily cross the BBB but is transported by carrier-mediated processes that utilize an array of potassium channels and transporters or by substituting other cations with similar hydrated radius (Ott and Larsen, 2004). Once in the astrocytes, ammonia combines with glutamate in an ATP-dependent reaction, catalyzed by glutamine synthetase (l-glutamate:ammonia ligase (ADP-forming; E.C.6.3.1.2) (GS) to form glutamine, which is then released into the blood stream (Cooper and Plum, 1987; Adeva et al., 2012) (Figure 1). Since the brain lacks effective urea cycle, this astrocyte GS catalyzed conversion of ammonia to glutamine and release into the blood stream is almost the only means to limit brain ammonia concentration (Cooper and Plum, 1987; Adeva et al., 2012). The astrocyte GS reaction also plays a role during acute systemic hyperammonemic conditions, where excess blood ammonia (NH3/ NH+4) is avidly taken up by the astrocytes and converted to glutamine, leading to increased brain and blood glutamine levels (Olde Damink et al., 2002; Brusilow et al., 2010).
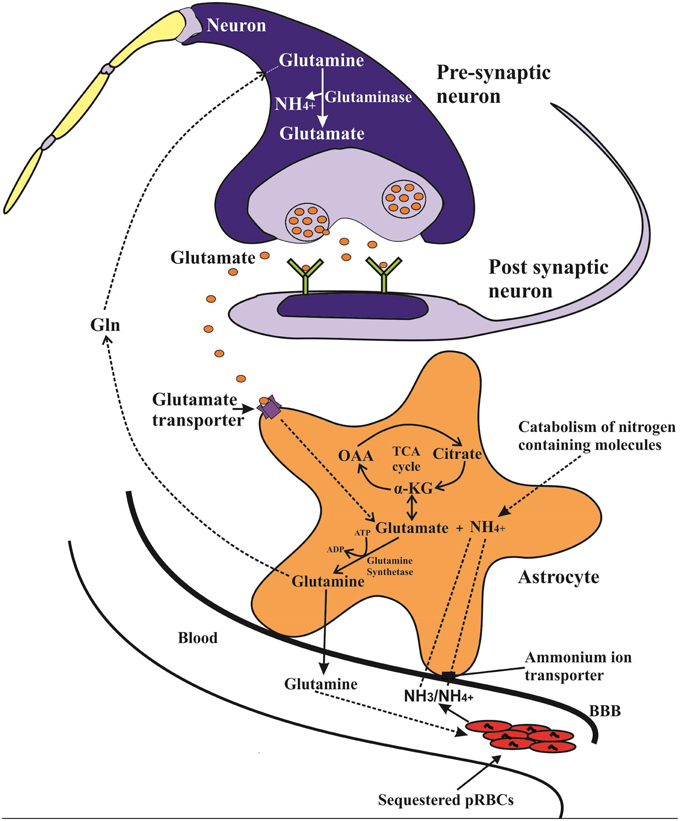
Figure 1. Potential ammonia-glutamine vicious cycle in which intravascular malaria parasites catabolize glutamine, releasing ammonia, which is taken up into the astrocyte via transporters including NCCK1 and conjugated with glutamate (derived from α-ketoglutarate) to form glutamine in a glutamine synthetase (GS) catalyzed reaction. The generated glutamine may then be released back to blood thus resupplying the parasite with the necessary glutamine. The GS catalyzed reaction also plays a key role in the glutamine-glutamate cycle, in which released synaptic glutamate is recycled back to the glutamatergic neurons in form of glutamine and converted by glutaminase to form glutamate neurotransmitter.
Moreover, the astrocytic GS reaction is involved in glutamine-glutamate (GABA) cycle, which serves an important role in glutamatergic and GABAergic neurotransmission. In this cycle glutamate released from the glutamatergic neurons is taken up by the astrocytes and converted by GS reaction to glutamine, which is then transported back to neurons and converted to glutamate by neuronal phosphate-activated glutaminase with a concomitant release of ammonia (Zwingmann and Butterworth, 2005; Hertz, 2013).
Can P. falciparum Cause Local Elevation of Brain Ammonia Levels?
The blood stage of P. falciparum degrades host hemoglobin to obtain free amino acids and imports some amino acids, particularly glutamine from the host serum (Elford et al., 1985; Goldberg, 2005). Although the parasite utilizes most of these free amino acids for protein biosynthesis, glutamine is usually deamidated and further deaminated and oxidized to 2-oxogluterate in order to maintain parasite's cellular levels of reduced NADPH + H+ (Werner et al., 2005). This deamidation and subsequent deamination reactions generates two molecules of ammonia per glutamine molecule. Thus, the malaria parasites require large amounts of glutamine and can produce substantial amounts of ammonia during the developmental life stages that require NADPH + H+. Indeed, an ammonia production rate of 0.8 fmol/h per parasite or 9 mmol/h per litre of packed infected erythrocytes has been demonstrated in the early trophozoite stage of P. falciparum (Zeuthen et al., 2006). Production of such amounts of ammonia may present a huge toxicity challenge to a parasite that lack ammonia-detoxifying machinery. However, studies suggest that the parasite may circumvent the toxic effects of ammonia by exporting it to the erythrocyte cytoplasm via P. falciparum aquaglyceroporin (PfAQP) and eventually into the extra-erythrocytic fluid (Zeuthen et al., 2006). Therefore, sequestration of a large number of metabolically active trophozoites and schizonts in a particular tissue vasculature may lead to release of massive amounts of ammonia into the blood stream.
In the human host, sequestration of PRBCs occur mainly in the brain microvasculature, with up to 10–20% of the total parasite biomass being estimated to be sequestered in the brain of some fatal CM cases (White et al., 2013). The sequestered PRBCs usually contain the mature trophozoite and schizonts (Silamut et al., 1999), which as already noted, takes up large amounts of glutamine from the extracellular fluid with concomitant increase in ammonia production (Elford et al., 1985; Zeuthen et al., 2006). The release of this parasite-derived ammonia in the human host most likely increases brain ammonia uptake, owing to reduced blood flow in congested microvessels and the fact that the brain ammonia uptake usually increases proportionally to the prevailing blood ammonia levels (Cooper and Plum, 1987). Subsequent detoxification of the increased brain ammonia via astrocytic GS reaction would in turn generate glutamine, which would then be released into the blood stream, as is the case during acute hyperammonemic conditions (Olde Damink et al., 2002). Interestingly, this would set up a vicious cycle whereby the sequestered parasites get rid of its toxic ammonia and at the same time sustains a continuous supply of glutamine by exploiting the host's astrocytic ammonia metabolic pathway (Figure 1). Such a cycle would enhance survival and rapid expansion of metabolically active sequestered parasite mass in the brain, thus sustaining and escalating ammonia production. Indeed malaria parasites are known to exploit or dysregulate other host's metabolic pathways to favor their survival and proliferation (Olszewski et al., 2009). However, the brain ammonia detoxification pathway capacity is limited and might be saturated by a sustained high rate of parasite-derived ammonia, thus leading to its accumulation and neurotoxicty. Moreover, but to a minor extent, an increased astrocytic glutamine output may also lead to increased glutamine uptake and subsequent release of ammonia by glutaminase-catalyzed reaction in the neurons. Therefore, P. falciparum particularly the sequestered parasites in the brain can create foci of increased ammonia production that may directly elevate brain ammonia, without the involvement of liver dysfunction.
Secondarily, other factors associated with P. falciparum infection may further contribute toward elevation of brain ammonia levels. Sequestered PRBCs, rosettes and reduced RBC deformability for example, may transiently reduce local microvascular perfusion leading to local tissue hypoxia. In such tissue regions, ammonia accumulation may be accelerated due to hypoxia-driven increase in amino acid catabolism (Cooper and Plum, 1987). Sequestered PRBCs in the brain may also create foci of enhanced immune cells activation and inflammatory responses (Porta et al., 1993; Patnaik et al., 1994; Medana et al., 2001). Activation of immune cells has been associated with increased uptake and utilization of glutamine to meet high energy demands required for effective immune functions (Curi et al., 1999). Glutamine catabolism by the activated immune cells may thus lead to enhanced ammonia production in the affected brain areas. Moreover, elevated reactive oxygen species (ROS) levels during their respiratory burst may compromise astrocytic ammonia detoxification events via inhibition of the the ROS sensitive GS (Brand et al., 1999), further aggravating the ammonia burden in the affected brain regions.
Hypoglycemia known to occur in up to one third of CM patients on admission (Idro et al., 2005) results in impaired energy metabolism and may also reduce ATP production. This would in turn exacerbate GS inhibition as a consequence of its reliance on ATP for enzymatic activity further contributing to ammonia accumulation. Fever and seizures, have also been shown to increase the rate of ammonia production in the vertebrate body (Cooper and Plum, 1987). Therefore, malaria induced fever and initial seizure episodes (mainly febrile) can contribute to heightened production of ammonia in the brain.
It is also possible that an initial slight to moderate increase in brain ammonia concentration by parasite's glutamine catabolic processes impairs the Krebs cycle, probably via inhibition of key metabolic enzymes like α-ketoglutarate (αKG) dehydrogenase (Cooper and Plum, 1987) and electron transport chain enzymes (Qureshi et al., 1998). Furthermore, the carbon needed to detoxify the excess parasite-derived ammonia depletes α-KG from cells, thereby additionally affecting the Krebs cycle (Ott et al., 2005). Pathophysiological concentrations of ammonia have also been shown to increase ROS production with resultant induction of mitochondrial permeability transition (MPT) in the astrocyte (Rama Rao et al., 2005). All these may ultimately reduce astrocyte ATP production thus setting up a vicious cycle in which ammonia impairs its own detoxification, leading to marked accumulation of ammonia and a further impairment of energy metabolism. Similarly, an initial parasite driven increase of ammonia can also decrease astrocyte expression of glutamate transporters (Chan et al., 2000) or increase ROS production (Rama Rao et al., 2005), further impairing ammonia brain metabolism.
In summary therefore, presence of a large number of metabolically active parasites in the brain can directly generate substantial amount of ammonia, as a metabolic byproduct, which may in turn overwhelm brain ammonia metabolism, leading to localized elevation of brain ammonia. The sequestration of PRBCs in the brain microvasculature is most likely the key factor that creates the large pool of parasites necessary to overwhelm the brain's ammonia detoxification capacity.
Elevated Brain Ammonia Hypothesis
In the human body ammonia exist at equilibrium as NH3(a weak base) and NH+4 (weak acid), with the latter having electrolytic conductance and ionic properties very similar to those of K+ ion (Bosoi and Rose, 2009). NH3 readily permeates membrane while the protonated NH+4 form crosses membranes via channels and substitution of K+ ion at its transporters (Nagaraja and Brookes, 1998). Elevated levels of ammonia alters brain function via direct as well as indirect mechanisms and has been considered to be a key factor in the pathogenesis of various metabolic encephalopathies (Butterworth et al., 1987; Butterworth, 1998; Bosoi and Rose, 2009; Haberle, 2013). We hypothesize that acute localized elevation of brain ammonia primarily by sequestered malaria parasites initiates a cascade of toxic effects that ultimately lead to the various CM clinical manifestations including brain edema, seizures, coma and neurological sequlae (Figure 2).
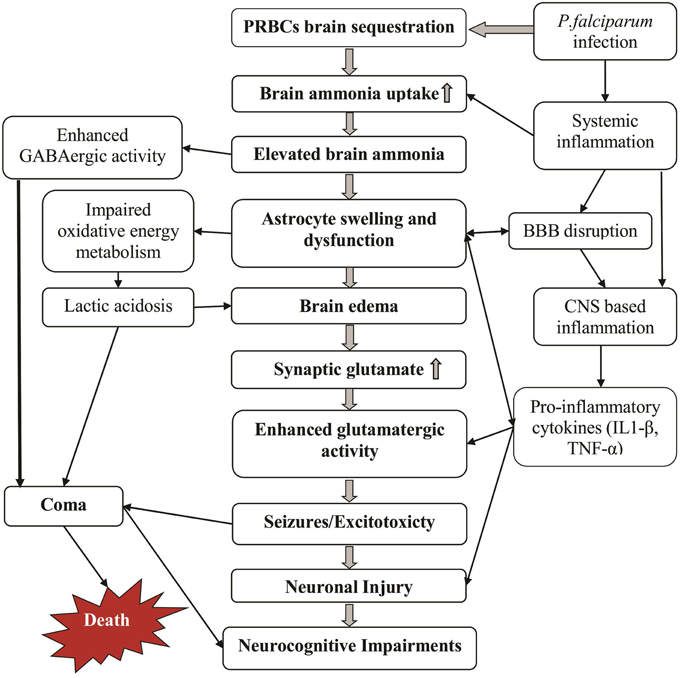
Figure 2. Potential mechanisms by which cerebral malaria manifestations can develop. Massive sequestration of parasites in brain microvasculature increases brain ammonia levels and thereby causing ammonia toxicity (middle and left columns). Some additional effects of the malaria infection including inflammation and disrupted BBB further enhance this toxicity (right column). The cause of death may be multifactorial including deep coma, edema and prolonged repetitive seizures.
Brain edema and the associated raised intracranial pressure (ICP) and intracranial hypertension commonly observed in CM (Idro et al., 2005; Medana et al., 2011) may occur primarily due to ammonia-induced astrocyte swelling. This is because acute elevation of brain ammonia by the parasite and subsequent uptake into the brain would stimulate astrocytic GS catalyzed glutamine synthesis, leading to a rapid accumulation of glutamine. Intracellular accumulation of glutamine is known to cause astrocyte swelling via various mechanisms including increased osmotic stress (Norenberg et al., 1991; Butterworth, 1998), or enhanced oxidative/nitrative stress (ONS) and induction of MPT following uptake into the mitochondria (Albrecht and Norenberg, 2006). The physico-chemical similarities between NH+4 and K+ may also play a role in excess ammonia mediated atrocyte swelling as suggested by studies showing that ammonia activates Na-K-Cl cotransporter-1 (NKCC1) and oubain-activated signaling pathway (Rama Rao et al., 2005, 2014a; Jayakumar et al., 2011; Song and Du, 2014). Moreover, excess ammonia increases the levels of aquaporin 4 water channel (AQP4) in the brain (Pan et al., 2010; Bodega et al., 2012), which may play a role in astrocyte swelling and brain edema (Eefsen et al., 2010; Rama Rao et al., 2010b, 2014b). While these mechanisms are yet to be investigated in the pathogenesis of CM edema, a few recent studies points toward a potential role. Brain autopsy studies of CM patients with cereberal edema, for example found a trend toward increased AQP4 channels in the brain stem (Medana et al., 2011). Increased brain and blood glutamine levels have also been reported in ECM (Ghosh et al., 2012), lending support to our postulated ammonia-mediated genesis of cytotoxic edema in CM.
The brain endothelial cells also expresses NKCC1, whose activation increases Na+ and Cl− transport into brain and has been implicated in the genesis of edema in stroke and experimental ischemic models (O'Donnell et al., 2004). Recent studies also indicate that ammonia activates the nuclear factor kappa B (NF-κB) pathway in brain endothelial cells, thereby contributing to astrocyte swelling and edema in ALF (Jayakumar et al., 2012, 2014). Therefore, it is possible that the widespread activation of brain endothelial cells (Combes et al., 2004; Idro et al., 2010) and up-regulation of NF-κB pathway by PRBCs (Tripathi et al., 2006, 2009) may partly be driven by the parasite-derived ammonia. The endothelial cell activation coupled with ammonia-mediated stimulation of BBB endothelial cell NKCC1 co-transporter activity, may in turn further contribute to the development and progression of brain swelling during CM.
Seizures and altered consciousness including deep coma are also commonly observed among CM patients, particularly children (Molyneux et al., 1989; Idro et al., 2010). We postulate that these manifestations are due to unique chemical properties of ammonia and its effects on key excitatory and inhibitory neurotransmitter systems at pathophysiological levels. A rapid increase in ammonia would not only result in intracellular alkalinization (increased intracellular pH) of astrocytes but also impairs extracellular K+ buffering capacity of the astrocytes (Marcaggi and Coles, 2001; Bosoi and Rose, 2009; Rangroo Thrane et al., 2013). The consequent rise in extracellular [K+] and overactivation of the Na+-K+-2Cl−cotransporter isoform 1 (NKCC1) in neurons in turn leads to selective impairment of cortical inhibitory GABAergic networks, neuronal disinhibition and seizures if ammonia concentration becomes very high (Rangroo Thrane et al., 2013).
Moreover, excess ammonia enhances excitatory glutamatergic neurotransmission via different mechanisms. Elevated ammonia for, example reduces astrocytic glutamate uptake and increases neuronal glutamate release, leading to elevated extracellular glutamate levels and enhanced excitatory glutamatergic activity (Raabe, 1987; Rose, 2002; Chan and Butterworth, 2003). Acute exposure of astrocytes to ammonia and consequent intracellular alkalinization also results in a calcium-dependent glutamate release (Rose et al., 2005). Collectively, these would increase extracellular glutamate thereby enhancing glutamatergic-mediated excitatory neurotransmission. Enhanced glutamatergic activity predispose the brain to seizures and excitotoxity (Meldrum, 1994; Stafstrom and Sasaki-Adams, 2003). Recent studies also suggest that prolonged exposure of astrocytes to extracellular glutamate down-regulates astrocytic inward rectifying potassium (Kir4.1) channels (Obara-Michlewska et al., 2011, 2014), a phenomenon which has been shown to be epileptogenic (Jansen et al., 2005).
In line with these observations, the seizures observed in CM may thus arise due ammonia-induced neuronal disinhibition and enhancement of glutamatergic activity. In support of these postulated mechanisms, biochemical changes indicative of increased glutamatergic activity have previously been reported in a murine CM model (Rae et al., 2004). The enhanced glutamatergic activity may however be transient since prolonged presence of increased glutamate in the synapse may cause downregulation of postsynaptic glutamate receptors, as an intrinsic protective response. Both N-methyl-D-aspartate (NMDA) and non-NMDA glutamate receptors for example are reduced in animal models of ALF, and this has been suggested to act as a counter mechanism against the detrimental effect of prolonged elevated extracellular levels of glutamate (Chan and Butterworth, 2003). Downregulation of glutamate mediated excitatory synaptic transmission, coupled with direct and indirect enhancement of GABAergic system by ammonia would depress brain activity, leading to coma (Basile, 2002). Indeed, an elevation of CNS tissue ammonia to 0.5 μmol/g, has been suggested to sufficiently disturb excitatory and inhibitory neurotransmission, leading to initiation of acute encephalopathy (Raabe, 1987).
Notably and of particular interest is that the coma in CM usually occurs rapidly often succeeding seizure onset and most patients recover quickly from the coma without much neurological impairments upon effective anti-malarial therapy (Molyneux et al., 1989; Idro et al., 2010). These observations fits well with our proposed patho-mechanisms and sequence of events i.e., elevated ammonia transiently enhances excitatory neurotransmission followed by compensatory measures that depresses brain activity ultimately leading to coma. Similarly, the rapid recovery from coma by most CM patients without much neurological impairment can be plausibly explained by our postulated mechanisms. A timely and effective anti-malarial therapy would basically reduce the quantity of sequestered ammonia-producing parasites, which in turn would promote restoration of normal brain ammonia metabolism, and return to normal neurotransmission.
Hypoglycaemia and lactic acidosis are also commonly observed among CM patients (Idro et al., 2005). While several factors such as reduced food intake, vomiting, hypoxia, heavy parasitemia, and systemic organ failures may lead to hypoglycemia and lactic acidosis, we hypothesize that ammonia-driven alteration of brain energy metabolism may play a major role. Ammonia disrupts the Krebs cycle via inhibition of alpha-ketoglutarate dehydrogenase (KGDH), leading to reduced mitochondrial production of ATP and reducing equivalents such as nicotinamide adenine dinucleotide (NADH) (Ott et al., 2005). Elevated ammonia also causes a metabolic shift toward glycolytic metabolism with concomitant increase in lactate formation (Kala and Hertz, 2005; Andersson et al., 2009). Increased lactate concentrations inhibit astrocytic glutamate uptake capacity (Andersson et al., 2009), which may further cripple the Krebs cycle through diminished anaplerotic replenishment of KGDH. The resultant reliance of the glycolytic pathway ATP production by the astrocytes in the wake of an increased need for ATP-dependent detoxification of excess ammonia would not only increase lactate and reduce pyruvate/lactate ratio but will also lead to rapid depletion of blood glucose. The lactic acidosis observed in CM may thus be largely due to ammonia-induced crippling of Krebs cycle and switch to the lactate producing glycolytic pathway. The observed hypoglycemia may be partly due to rapid depletion of glucose by the energetically inefficient glycolytic pathway, which generates only two molecules of ATP per glucose molecule. Such a consideration, may explain why restoration of normoglycemia in hypoglycemic CM patients, is not often associated with a change in the level of consciousness (Newton et al., 2000).
Increased CSF quinolinic acid and picolinic acid has been observed in children with CM but the exact cause is not well-understood (Dobbie et al., 2000; Medana et al., 2003). We hypothesize that the effects of elevated brain ammonia on tryptophan uptake and metabolism play a key role. Generally, free tryptophan is metabolized via the kynurenine pathway into a series of bioactive metabolites including kynurenine, kynurenic acid, 3-Hydroxykynurenine, 3-Hydroxyanthranilic acid, picolinic acid, anthranilic acid, xanthurenic acid, and quinolinic acid (Stone and Darlington, 2002; Stone et al., 2013). Among these metabolites, quinolinic acid is an excitotoxic N-methyl-D-aspartate (NMDA) glutamate receptor agonist, while kynurenic acid is a neuroprotective glutamate receptor antagonist (Stone and Perkins, 1981; Stone, 2001; Schwarcz and Pellicciari, 2002; Guillemin, 2012). Quinolinic acid is also a pro-oxidant, an immunomodulator and promotes the formation of hyperphosphorylated tau proteins in the nervous system (Guillemin et al., 2003; Rahman et al., 2009). Notably, the synthesis of the neuroprotectant kynurenic acid in the brain occur mainly within the astrocytes, while the synthesis of 3-hydroxykynurenine and further downstream metabolites including quinolinic acid occur in microglia (Stone et al., 2013). Excess ammonia is known to increase brain tryptophan uptake even in absence of any liver function derangement (Bachmann and Colombo, 1983). It also inhibits kynurenic acid synthesis from kynurenine in a dose-dependent manner (Saran et al., 1998) and increases cerebral blood flow (Aggarwal et al., 1994; Jalan et al., 2004b). By increasing cerebral blood flow as CM progresses, elevated ammonia may thus increase tryptophan uptake from the blood. However, subsequent metabolism will be skewed in favor of increased level of microglia-derived quinolinic acid and less of neuroprotectant kynurenic acid since elevated ammonia also reduces kynurenic acid synthesis (Saran et al., 1998). The increased quinolinic acid would in turn contribute to altered NMDA receptor activity and excitotoxity (Stone and Perkins, 1981; Guillemin, 2012) as well as increased CNS proinflammatory cytokine production and oxidative stress (Guillemin et al., 2003).
Systemic inflammatory response is common during P. falciparum malaria infections, leading to increased blood concentration of both pro-inflammatory and anti-inflammatory cytokines. Among some patients who develop CM, various blood pro-inflammatory cytokines have been reported to increase including interleukin 1 beta (IL-1β), IL-6, and tumor necrosis factor alpha (TNF-α) (Kwiatkowski et al., 1990; Ringwald et al., 1991; Brown et al., 1999; Wenisch et al., 1999; Akanmori et al., 2000; Lyke et al., 2004). Therefore, a key role of inflammation in CM pathogenesis and pathology cannot be overlooked. We hypothesize that parasite-elicited systemic inflammation potentiates the proposed elevation of brain ammonia and subsequent neurotoxicity. This may include potentiation of brain ammonia uptake (Duchini et al., 1996) as well as the untoward effects of excess ammonia on neurocognitive functions (Shawcross et al., 2004; Marini and Broussard, 2006), astrocyte osmotic balance (Oh et al., 1992; Rama Rao et al., 2010a) and synaptic neurotransmission (Casamenti et al., 1999; Hu et al., 2000; Medana et al., 2001). The systemic inflammation may also synergize ammonia-induced pathogenic alteration of cerebral blood flow (CBF) and increased ICP as previously observed both in human and animal studies of hyperammonemia (Jalan et al., 2004b; Pedersen et al., 2007). Moreover, inflammatory cytokines particularly TNF-α promotes sequestration of PRBCs (Idro et al., 2010), the primary event that leads to elevation of brain ammonia according to our hypothesis.
Potentially also, elevated brain ammonia may elicit CNS based inflammatory response via direct activation of microglia and astrocytes. This is because CNS based cells particularly astrocytes, microglia, and oligodendrocytes are known to initiate inflammation in response to local noxious stimuli (Lucas et al., 2006) and high concentrations of ammonia has been shown to be a potent inflammatory stimuli. In one in vitro study, for example, ammonia was reported to directly stimulate secretion of TNF-α and IL-8 by immortalized human microglia (CHME-5) and astroglioma (GL-15) cells even in the absence of any other stimulus (Atanassov et al., 1995). Such a possibility of ammonia induced CNS inflammatory response, may explain the observed widespread activation of microglial throughout the brain in white and gray matter and far beyond areas of petechial bleedings or sequestered PRBCs foci during CM (Schluesener et al., 1998). It may also explain the induction and CNS production of the pro-inflammatory cytokines that has been observed in children with CM (Brown et al., 1999; John et al., 2008). Moreover, it may explain the therapeutic failure of drugs such as pentoxifylline (Idro et al., 2010), since such drugs may not cross the BBB to inhibit activated microglia.
Apoptosis of neurons, endothelial cell and astrocytes has been suggested to play a role in the genesis of symptoms and signs of experimental animal CM (Wiese et al., 2006; Lackner et al., 2007). Evidence also suggest that P. falciparum induces apoptosis of neurons, brain endothelial cell and astrocytes and might play a role in the pathogenesis of human CM pathology (Pino et al., 2003; Toure et al., 2008; Idro et al., 2010; N'dilimabaka et al., 2014). However, the cause and exact mechanisms underlying induction of apoptosis in these cells is not fully understood. Elevated brain ammonia might play a role. In one study, exposure of C6 glioma cells to 5 or 10 mM ammonia for 96 h induced apoptosis in 50% of the cells, via activation of Protein kinase C (PKC) and nuclear transcription factor kappa B (NFκB) pathways (Buzanska et al., 2000). In another study, ammonia was shown to induce apoptosis of rat hippocampal neurons via calcineurin-mediated BAD Ser155 dephosphorylation (Yang et al., 2004). Ammonia was also reported to induce apoptosis in primary cortical neurons, possibly via reduced phosphorylation and activation of protein kinase B (PKB or Akt), a key component in the activation of cellular pro-survival pathways (Klejman et al., 2005). Over activation of NMDA receptors which can be caused by elevated ammonia, has also been shown to induce cleavage and activation of pro-apoptotic caspases (Tenneti et al., 1998).
Interestingly, similar mechanisms seem to mediate the apoptosis in experimental animal CM and P. falciparum in vitro models. Activation of pro-apototic caspase 3, for example, was found to play a key role in neuronal and astrocytic apoptosis in experimental murine CM (Pino et al., 2003; Potter et al., 2006; Lackner et al., 2007). Recently also, reduced activation of Akt and decreased inhibition of the glycogen kinase synthase beta (GSK3β) was demonstrated in the brains of mice infected with Plasmodium berghei ANKA (Dai et al., 2012). Our postulated parasite-driven elevated brain ammonia might thus activate pro-apoptotic pathways and impair of pro-survival pathways, leading to apoptosis of neurons, astrocytes, and endothelial cells, thereby contributing to acute and long-term neuropathology.
Elevated brain ammonia can lead to long-term neurocognitive sequlae observed among CM survivors via various pathomechanisms. First, the widespread activation of microglia by elevated ammonia might predispose neurons to oxidative burst and lysosomal damage (Schluesener et al., 1998). Secondly, the overactivation of NMDA receptors by increased extracellular glutamate acting in synergy with high brain quinolinic acid, IL-1ß, and TNF-α concentration can lead to excitotoxic damage of NMDA receptor containing neurons and/or “kindling.” Such a mechanism may be particularly important in the development of long-term learning impairment and increased susceptibility to epilepsy later on in life among peadiatric CM survivors, as indicated by studies in young rats (Stafstrom and Sasaki-Adams, 2003). Thirdly, the derangement of energy metabolism within the brain as a result of elevated ammonia-mediated Krebs cycle inhibition can cause brain cellular damage or death, possibly via enhanced cerebral proteolysis as proposed by Ott et al. (2005). Fourth, the ammonia induced astrocyte swelling and ROS production, α-KDH inhibition and pro-oxidant kynurenines production may predispose the neurons to enhanced damaging effects of oxidative stress. Additionally and perhaps most important is that neurcognitive sequlae may develop due to ammonia induced apoptotic death of brain cells (Buzanska et al., 2000; Yang et al., 2003, 2004). Any of these mechanisms acting singly or cooperatively could lead to neuronal damage, death or persistent disturbed synaptic processes, which may in turn manifest as long-term neurocognitive impairments, following a CM episode.
Consideration of parasite-driven brain ammonia elevation as a central pathogenic factor can therefore account for the genesis of the various manifestations observed in CM patients. It also allows for the integration of various patho-mechanisms into a clear sequence of events during the genesis and progression of CM. Although there are several ethical and practical challenges to testing of the hypothesized mechanisms in humans with CM, a number of approaches are possible. CSF ammonia, glutamine, aspartate, and alanine levels can be measured in well-defined groups of CM patients. Post mortem analysis of ammonia metabolizing enzymes and level of ammonia metabolism related amino acids like glutamate, glutamine, and aspartate could also be done in brain tissues obtained from fatal cases of cerebral malaria. The outcome of clinical interventions that limits either blood, brain ammonia levels, or prevent ammonia-induced brain dysfunctions may also provide an indirect additional approach.
Murine models of CM can also provide an alternative approach to test the current hypothesis and its predictions with the added advantage of allowing experimental manipulations. Blood, CSF, and brain ammonia levels can be studied in both the fatal and resolving mouse models of CM. Similarly, pre-mortem and post-mortem studies of key enzymes involved in ammonia metabolism including glutamine synthetase, glutamate dehydrogenase, and glutaminase can provide data to evaluate this hypothesis.
Therapeutic Implications
If elevated brain ammonia plays a central role in CM pathogenesis, then interventions that reduces ammonia formation or toxicity could potential improve CM treatment outcome. Protein intake restriction during cerebral malaria and promotion of anabolism by rapid administration of intravenous dextrose solution could therefore reduce ammonia formation and toxicity. Similarly, aggressive control of factors such as fever and seizures, which enhances ammonia formation, may be beneficial.
Chemical compounds that act as metabolic “sinks” for ammonia can also be explored as adjuncts in the management of CM. These include compounds such as benzoate, sodium phenylacetate, and calcium phenylbutyrate. These agents are currently used in the management of various hyperammonemic disorders. Hypothermia and other interventions aimed at mitigating ammonia induced brain dysfunction and damage may also be tested as potential adjunct therapies in the management of CM. Hypothermia has been found to reduce ICP in patients with ALF (Jalan et al., 1999, 2004a). It would be therefore interesting to test the efficacy of mild hypothermia in reducing CM-induced brain edema and raised ICP. More recently, several experimental agents including NMDA receptor antagonists, drugs antagonizing endogenous ammonia-activated ouabain-like compounds, Na-K-Cl cotransporter-1 (NKCC1) inhibitors, and L-histidine have shown therapeutic potential in ALF animal models (Rama Rao et al., 2010c; Jayakumar et al., 2011; Cauli et al., 2013; Song and Du, 2014). These agents may also represent novel and potentially relevant experimental adjunctive therapies in CM.
Conclusion
The similar clinicopathological features of CM and hyperammonemic encephalopathies as well as the available evidence suggesting that P. falciparum can produce substantial amount of ammonia warrants the consideration of parasite-derived ammonia as a potential key factor in the pathogenesis of cerebral malaria. Predictable consequences of P. falciparum-induced elevation of brain ammonia can account for most of clinicopathology of CM and therefore provide hints of potential adjunctive therapies. Targeting of the parasite's ammonia production and transport via PfAQP blockade could also be a novel therapeutic strategy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledges, Professor Ayub Ofulla for helpful critique of the earlier drafts.
References
Adeva, M. M., Souto, G., Blanco, N., and Donapetry, C. (2012). Ammonium metabolism in humans. Metab. Clin. Exp. 61, 1495–1511. doi: 10.1016/j.metabol.2012.07.007
Aggarwal, S., Kramer, D., Yonas, H., Obrist, W., Kang, Y., Martin, M., et al. (1994). Cerebral hemodynamic and metabolic changes in fulminant hepatic failure: a retrospective study. Hepatology 19, 80–87. doi: 10.1002/hep.1840190114
Akanmori, B. D., Kurtzhals, J. A., Goka, B. Q., Adabayeri, V., Ofori, M. F., Nkrumah, F. K., et al. (2000). Distinct patterns of cytokine regulation in discrete clinical forms of Plasmodium falciparum malaria. Eur. Cytokine Netw. 11, 113–118.
Albrecht, J., and Norenberg, M. D. (2006). Glutamine: a Trojan horse in ammonia neurotoxicity. Hepatology 44, 788–794. doi: 10.1002/hep.21357
Anand, A. C., Ramji, C., Narula, A. S., and Singh, W. (1992). Malarial hepatitis: a heterogeneous syndrome? Natl. Med. J. India 5, 59–62.
Andersson, A. K., Adermark, L., Persson, M., Westerlund, A., Olsson, T., and Hansson, E. (2009). Lactate contributes to ammonia-mediated astroglial dysfunction during hyperammonemia. Neurochem. Res. 34, 556–565. doi: 10.1007/s11064-008-9819-1
Arn, P. H., Hauser, E. R., Thomas, G. H., Herman, G., Hess, D., and Brusilow, S. W. (1990). Hyperammonemia in women with a mutation at the ornithine carbamoyltransferase locus. A cause of postpartum coma. N. Engl. J. Med. 322, 1652–1655. doi: 10.1056/NEJM199006073222307
Atanassov, C. L., Muller, C. D., Dumont, S., Rebel, G., Poindron, P., and Seiler, N. (1995). Effect of ammonia on endocytosis and cytokine production by immortalized human microglia and astroglia cells. Neurochem. Int. 27, 417–424. doi: 10.1016/0197-0186(95)00023-2
Bachmann, C., and Colombo, J. P. (1983). Increased tryptophan uptake into the brain in hyperammonemia. Life Sci. 33, 2417–2424. doi: 10.1016/0024-3205(83)90635-5
Basile, A. S. (2002). Direct and indirect enhancement of GABAergic neurotransmission by ammonia: implications for the pathogenesis of hyperammonemic syndromes. Neurochem. Int. 41, 115–122. doi: 10.1016/S0197-0186(02)00032-3
Berendt, A. R., Tumer, G. D., and Newbold, C. I. (1994). Cerebral malaria: the sequestration hypothesis. Parasitol. Today 10, 412–414. doi: 10.1016/0169-4758(94)90238-0
Bodega, G., Suarez, I., Lopez-Fernandez, L. A., Garcia, M. I., Kober, M., Penedo, M., et al. (2012). Ammonia induces aquaporin-4 rearrangement in the plasma membrane of cultured astrocytes. Neurochem. Int. 61, 1314–1324. doi: 10.1016/j.neuint.2012.09.008
Bosoi, C. R., and Rose, C. F. (2009). Identifying the direct effects of ammonia on the brain. Metab. Brain Dis. 24, 95–102. doi: 10.1007/s11011-008-9112-7
Brand, A., Leibfritz, D., and Richter-Landsberg, C. (1999). Oxidative stress-induced metabolic alterations in rat brain astrocytes studied by multinuclear NMR spectroscopy. J. Neurosci. Res. 58, 576–585.
Brown, H., Turner, G., Rogerson, S., Tembo, M., Mwenechanya, J., Molyneux, M., et al. (1999). Cytokine expression in the brain in human cerebral malaria. J. Infect. Dis. 180, 1742–1746. doi: 10.1086/315078
Brusilow, S. W., Koehler, R. C., Traystman, R. J., and Cooper, A. J. (2010). Astrocyte glutamine synthetase: importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics 7, 452–470. doi: 10.1016/j.nurt.2010.05.015
Butterworth, R. F., Giguere, J. F., Michaud, J., Lavoie, J., and Layrargues, G. P. (1987). Ammonia: key factor in the pathogenesis of hepatic encephalopathy. Neurochem. Pathol. 6, 1–12. doi: 10.1007/BF02833598
Butterworth, R. F. (1998). Effects of hyperammonaemia on brain function. J. Inherit. Metab. Dis. 21(Suppl. 1), 6–20. doi: 10.1023/A:1005393104494
Buzanska, L., Zablocka, B., Dybel, A., Domanska-Janik, K., and Albrecht, J. (2000). Delayed induction of apoptosis by ammonia in C6 glioma cells. Neurochem. Int. 37, 287–297. doi: 10.1016/S0197-0186(00)00030-9
Casamenti, F., Prosperi, C., Scali, C., Giovannelli, L., Colivicchi, M. A., Faussone-Pellegrini, M. S., et al. (1999). Interleukin-1beta activates forebrain glial cells and increases nitric oxide production and cortical glutamate and GABA release in vivo: implications for Alzheimer's disease. Neuroscience 91, 831–842. doi: 10.1016/S0306-4522(98)00680-0
Cauli, O., Gonzalez-Usano, A., Cabrera-Pastor, A., Gimenez-Garzo, C., Lopez-Larrubia, P., Ruiz-Sauri, A., et al. (2013). Blocking NMDA receptors delays death in rats with acute liver failure by dual protective mechanisms in kidney and brain. Neuromolecular Med. 16, 360–375. doi: 10.1007/s12017-013-8283-5
Chan, H., and Butterworth, R. F. (2003). Cell-selective effects of ammonia on glutamate transporter and receptor function in the mammalian brain. Neurochem. Int. 43, 525–532. doi: 10.1016/S0197-0186(03)00043-3
Chan, H., Hazell, A. S., Desjardins, P., and Butterworth, R. F. (2000). Effects of ammonia on glutamate transporter (GLAST) protein and mRNA in cultured rat cortical astrocytes. Neurochem. Int. 37, 243–248. doi: 10.1016/S0197-0186(00)00026-7
Cohn, R. M., and Roth, K. S. (2004). Hyperammonemia, bane of the brain. Clin. Pediatr. (Phila) 43, 683–689. doi: 10.1177/000992280404300801
Combes, V., Taylor, T. E., Juhan-Vague, I., Mege, J. L., Mwenechanya, J., Tembo, M., et al. (2004). Circulating endothelial microparticles in malawian children with severe falciparum malaria complicated with coma. JAMA 291, 2542–2544. doi: 10.1001/jama.291.21.2542-b
Cooper, A. J., and Plum, F. (1987). Biochemistry and physiology of brain ammonia. Physiol. Rev. 67, 440–519.
Curi, R., Newsholme, P., Pithon-Curi, T. C., Pires-de-Melo, M., Garcia, C., Homem-de-Bittencourt Junior, P. I., et al. (1999). Metabolic fate of glutamine in lymphocytes, macrophages and neutrophils. Braz. J. Med. Biol. Res. 32, 15–21. doi: 10.1590/S0100-879X1999000100002
Dai, M., Freeman, B., Shikani, H. J., Bruno, F. P., Collado, J. E., Macias, R., et al. (2012). Altered regulation of Akt signaling with murine cerebral malaria, effects on long-term neuro-cognitive function, restoration with lithium treatment. PLoS ONE 7:e44117. doi: 10.1371/journal.pone.0044117
Dobbie, M., Crawley, J., Waruiru, C., Marsh, K., and Surtees, R. (2000). Cerebrospinal fluid studies in children with cerebral malaria: an excitotoxic mechanism? Am. J. Trop. Med. Hyg. 62, 284–290.
Duchini, A., Govindarajan, S., Santucci, M., Zampi, G., and Hofman, F. M. (1996). Effects of tumor necrosis factor-alpha and interleukin-6 on fluid-phase permeability and ammonia diffusion in CNS-derived endothelial cells. J. Investig. Med. 44, 474–482.
Eefsen, M., Jelnes, P., Schmidt, L. E., Vainer, B., Bisgaard, H. C., and Larsen, F. S. (2010). Brain expression of the water channels aquaporin-1 and -4 in mice with acute liver injury, hyperammonemia and brain edema. Metab. Brain Dis. 25, 315–323. doi: 10.1007/s11011-010-9213-y
Eid, T., and Lee, T. S. (2013). Reassessing the role of astrocytes in ammonia neurotoxicity. Nat. Med. 19, 1572–1574. doi: 10.1038/nm.3420
Elford, B. C., Haynes, J. D., Chulay, J. D., and Wilson, R. J. (1985). Selective stage-specific changes in the permeability to small hydrophilic solutes of human erythrocytes infected with Plasmodium falciparum. Mol. Biochem. Parasitol. 16, 43–60. doi: 10.1016/0166-6851(85)90048-9
Ghosh, S., Sengupta, A., Sharma, S., and Sonawat, H. M. (2012). Metabolic fingerprints of serum, brain, and liver are distinct for mice with cerebral and noncerebral malaria: a (1)H NMR spectroscopy-based metabonomic study. J. Proteome Res. 11, 4992–5004. doi: 10.1021/pr300562m
Goldberg, D. E. (2005). Hemoglobin degradation. Curr. Top. Microbiol. Immunol. 295, 275–291. doi: 10.1007/3-540-29088-5_11
Guillemin, G. J., Croitoru-Lamoury, J., Dormont, D., Armati, P. J., and Brew, B. J. (2003). Quinolinic acid upregulates chemokine production and chemokine receptor expression in astrocytes. Glia 41, 371–381. doi: 10.1002/glia.10175
Guillemin, G. J. (2012). Quinolinic acid, the inescapable neurotoxin. FEBS J. 279, 1356–1365. doi: 10.1111/j.1742-4658.2012.08485.x
Haberle, J. (2013). Clinical and biochemical aspects of primary and secondary hyperammonemic disorders. Arch. Biochem. Biophys. 536, 101–108. doi: 10.1016/j.abb.2013.04.009
Hertz, L. (2013). The Glutamate-Glutamine (GABA) Cycle: importance of late postnatal development and potential reciprocal interactions between biosynthesis and degradation. Front. Endocrinol. (Lausanne) 4:59. doi: 10.3389/fendo.2013.00059
Hu, S., Sheng, W. S., Ehrlich, L. C., Peterson, P. K., and Chao, C. C. (2000). Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 7, 153–159. doi: 10.1159/000026433
Idro, R., Jenkins, N. E., and Newton, C. R. (2005). Pathogenesis, clinical features, and neurological outcome of cerebral malaria. Lancet Neurol. 4, 827–840. doi: 10.1016/S1474-4422(05)70247-7
Idro, R., Marsh, K., John, C. C., and Newton, C. R. (2010). Cerebral malaria: mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatr. Res. 68, 267–274. doi: 10.1203/PDR.0b013e3181eee738
Jalan, R., Olde Damink, S. W., Deutz, N. E., Hayes, P. C., and Lee, A. (2004a). Moderate hypothermia in patients with acute liver failure and uncontrolled intracranial hypertension. Gastroenterology 127, 1338–1346. doi: 10.1053/j.gastro.2004.08.005
Jalan, R., Olde Damink, S. W., Hayes, P. C., Deutz, N. E., and Lee, A. (2004b). Pathogenesis of intracranial hypertension in acute liver failure: inflammation, ammonia and cerebral blood flow. J. Hepatol. 41, 613–620. doi: 10.1016/j.jhep.2004.06.011
Jalan, R., Sw, O. D., Deutz, N. E., Lee, A., and Hayes, P. C. (1999). Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 354, 1164–1168. doi: 10.1016/S0140-6736(98)12440-6
Jansen, L. A., Uhlmann, E. J., Crino, P. B., Gutmann, D. H., and Wong, M. (2005). Epileptogenesis and reduced inward rectifier potassium current in tuberous sclerosis complex-1-deficient astrocytes. Epilepsia 46, 1871–1880. doi: 10.1111/j.1528-1167.2005.00289.x
Jayakumar, A. R., Tong, X. Y., Curtis, K. M., Ruiz-Cordero, R., Abreu, M. T., and Norenberg, M. D. (2014). Increased toll-like receptor 4 in cerebral endothelial cells contributes to the astrocyte swelling and brain edema in acute hepatic encephalopathy. J. Neurochem. 128, 890–903. doi: 10.1111/jnc.12516
Jayakumar, A. R., Tong, X. Y., Ospel, J., and Norenberg, M. D. (2012). Role of cerebral endothelial cells in the astrocyte swelling and brain edema associated with acute hepatic encephalopathy. Neuroscience 218, 305–316. doi: 10.1016/j.neuroscience.2012.05.006
Jayakumar, A. R., Valdes, V., and Norenberg, M. D. (2011). The Na-K-Cl cotransporter in the brain edema of acute liver failure. J. Hepatol. 54, 272–278. doi: 10.1016/j.jhep.2010.06.041
John, C. C., Panoskaltsis-Mortari, A., Opoka, R. O., Park, G. S., Orchard, P. J., Jurek, A. M., et al. (2008). Cerebrospinal fluid cytokine levels and cognitive impairment in cerebral malaria. Am. J. Trop. Med. Hyg. 78, 198–205.
Kala, G., and Hertz, L. (2005). Ammonia effects on pyruvate/lactate production in astrocytes–interaction with glutamate. Neurochem. Int. 47, 4–12. doi: 10.1016/j.neuint.2005.04.001
Klejman, A., Wegrzynowicz, M., Szatmari, E. M., Mioduszewska, B., Hetman, M., and Albrecht, J. (2005). Mechanisms of ammonia-induced cell death in rat cortical neurons: roles of NMDA receptors and glutathione. Neurochem. Int. 47, 51–57. doi: 10.1016/j.neuint.2005.04.006
Kochar, D. K., Tanwar, G. S., Khatri, P. C., Kochar, S. K., Sengar, G. S., Gupta, A., et al. (2010). Clinical features of children hospitalized with malaria–a study from Bikaner, northwest India. Am. J. Trop. Med. Hyg. 83, 981–989. doi: 10.4269/ajtmh.2010.09-0633
Kwiatkowski, D., Hill, A. V., Sambou, I., Twumasi, P., Castracane, J., Manogue, K. R., et al. (1990). TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet 336, 1201–1204. doi: 10.1016/0140-6736(90)92827-5
Lackner, P., Burger, C., Pfaller, K., Heussler, V., Helbok, R., Morandell, M., et al. (2007). Apoptosis in experimental cerebral malaria: spatial profile of cleaved caspase-3 and ultrastructural alterations in different disease stages. Neuropathol. Appl. Neurobiol. 33, 560–571. doi: 10.1111/j.1365-2990.2007.00833.x
Lucas, S. M., Rothwell, N. J., and Gibson, R. M. (2006). The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 147(Suppl. 1), S232–S240. doi: 10.1038/sj.bjp.0706400
Lyke, K. E., Burges, R., Cissoko, Y., Sangare, L., Dao, M., Diarra, I., et al. (2004). Serum levels of the proinflammatory cytokines interleukin-1 beta (IL-1beta), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect. Immun. 72, 5630–5637. doi: 10.1128/IAI.72.10.5630-5637.2004
Marcaggi, P., and Coles, J. A. (2001). Ammonium in nervous tissue: transport across cell membranes, fluxes from neurons to glial cells, and role in signalling. Prog. Neurobiol. 64, 157–183. doi: 10.1016/S0301-0082(00)00043-5
Mariga, S. T., Kolko, M., Gjedde, A., and Bergersen, L. H. (2014). Lactate transport and receptor actions in cerebral malaria. Front. Neurosci. 8:125. doi: 10.3389/fnins.2014.00125
Marini, J. C., and Broussard, S. R. (2006). Hyperammonemia increases sensitivity to LPS. Mol. Genet. Metab. 88, 131–137. doi: 10.1016/j.ymgme.2005.12.013
Martins, Y. C., and Daniel-Ribeiro, C. T. (2013). A new hypothesis on the manifestation of cerebral malaria: the secret is in the liver. Med. Hypotheses 81, 777–783. doi: 10.1016/j.mehy.2013.08.005
Mazumder, R., Mishra, R. K., Mazumder, H., and Mukherjee, P. (2002). Jaundice in falciparum malaria–some prospective observations. J. Indian Med. Assoc. 100, 312–314.
Medana, I. M., Chaudhri, G., Chan-Ling, T., and Hunt, N. H. (2001). Central nervous system in cerebral malaria: “Innocent bystander” or active participant in the induction of immunopathology? Immunol. Cell Biol. 79, 101–120. doi: 10.1046/j.1440-1711.2001.00995.x
Medana, I. M., Day, N. P., Sachanonta, N., Mai, N. T., Dondorp, A. M., Pongponratn, E., et al. (2011). Coma in fatal adult human malaria is not caused by cerebral oedema. Malar. J. 10:267. doi: 10.1186/1475-2875-10-267
Medana, I. M., Day, N. P., Salahifar-Sabet, H., Stocker, R., Smythe, G., Bwanaisa, L., et al. (2003). Metabolites of the kynurenine pathway of tryptophan metabolism in the cerebrospinal fluid of Malawian children with malaria. J. Infect. Dis. 188, 844–849. doi: 10.1086/377583
Meldrum, B. S. (1994). The role of glutamate in epilepsy and other CNS disorders. Neurology 44, S14–23.
Mishra, S. K., and Newton, C. R. (2009). Diagnosis and management of the neurological complications of falciparum malaria. Nat. Rev. Neurol. 5, 189–198. doi: 10.1038/nrneurol.2009.23
Mohanty, N., Satpathy, S. K., and Nanda, P. (2004). Hepatopathy in complicated falciparum malaria: report from eastern India. Trans. R. Soc. Trop. Med. Hyg. 98, 753–754. doi: 10.1016/S0035-9203(03)00023-3
Molyneux, M. E., Taylor, T. E., Wirima, J. J., and Borgstein, A. (1989). Clinical features and prognostic indicators in paediatric cerebral malaria: a study of 131 comatose Malawian children. Q. J. Med. 71, 441–459.
Murthy, G. L., Sahay, R. K., Sreenivas, D. V., Sundaram, C., and Shantaram, V. (1998). Hepatitis in falciparum malaria. Trop. Gastroenterol. 19, 152–154.
Nacher, M., Treeprasertsuk, S., Singhasivanon, P., Silachamroon, U., Vannaphan, S., Gay, F., et al. (2001). Association of hepatomegaly and jaundice with acute renal failure but not with cerebral malaria in severe falciparum malaria in Thailand. Am. J. Trop. Med. Hyg. 65, 828–833.
Nagaraja, T. N., and Brookes, N. (1998). Intracellular acidification induced by passive and active transport of ammonium ions in astrocytes. Am. J. Physiol. 274, C883–C891.
N'dilimabaka, N., Taoufiq, Z., Zougbede, S., Bonnefoy, S., Lorthiois, A., Couraud, P. O., et al. (2014). P. falciparum isolate-specific distinct patterns of induced apoptosis in pulmonary and brain endothelial cells. PLoS ONE 9:e90692. doi: 10.1371/journal.pone.0090692
Newton, C. R., Hien, T. T., and White, N. (2000). Cerebral malaria. J. Neurol. Neurosurg. Psychiatr. 69, 433–441. doi: 10.1136/jnnp.69.4.433
Norenberg, M. D., Baker, L., Norenberg, L. O., Blicharska, J., Bruce-Gregorios, J. H., and Neary, J. T. (1991). Ammonia-induced astrocyte swelling in primary culture. Neurochem. Res. 16, 833–836. doi: 10.1007/BF00965694
Obara-Michlewska, M., Pannicke, T., Karl, A., Bringmann, A., Reichenbach, A., Szeliga, M., et al. (2011). Down-regulation of Kir4.1 in the cerebral cortex of rats with liver failure and in cultured astrocytes treated with glutamine: implications for astrocytic dysfunction in hepatic encephalopathy. J. Neurosci. Res. 89, 2018–2027. doi: 10.1002/jnr.22656
Obara-Michlewska, M., Ruszkiewicz, J., Zielinska, M., Verkhratsky, A., and Albrecht, J. (2014). Astroglial NMDA receptors inhibit expression of K4.1 channels in glutamate-overexposed astrocytes in vitro and in the brain of rats with acute liver failure. Neurochem. Int. doi: 10.1016/j.neuint.2014.10.006. [Epub ahead of print].
O'Donnell, M. E., Lam, T. I., Tran, L., and Anderson, S. E. (2004). The role of the blood-brain barrier Na-K-2Cl cotransporter in stroke. Adv. Exp. Med. Biol. 559, 67–75.
Oh, Y. J., Francis, J. W., Markelonis, G. J., and Oh, T. H. (1992). Interleukin-1-beta and tumor necrosis factor-alpha increase peripheral-type benzodiazepine binding sites in cultured polygonal astrocytes. J. Neurochem. 58, 2131–2138. doi: 10.1111/j.1471-4159.1992.tb10955.x
Olde Damink, S. W., Deutz, N. E., Dejong, C. H., Soeters, P. B., and Jalan, R. (2002). Interorgan ammonia metabolism in liver failure. Neurochem. Int. 41, 177–188. doi: 10.1016/S0197-0186(02)00040-2
Olszewski, K. L., Morrisey, J. M., Wilinski, D., Burns, J. M., Vaidya, A. B., Rabinowitz, J. D., et al. (2009). Host-parasite interactions revealed by Plasmodium falciparum metabolomics. Cell Host Microbe 5, 191–199. doi: 10.1016/j.chom.2009.01.004
Ott, P., Clemmesen, O., and Larsen, F. S. (2005). Cerebral metabolic disturbances in the brain during acute liver failure: from hyperammonemia to energy failure and proteolysis. Neurochem. Int. 47, 13–18. doi: 10.1016/j.neuint.2005.04.002
Ott, P., and Larsen, F. S. (2004). Blood-brain barrier permeability to ammonia in liver failure: a critical reappraisal. Neurochem. Int. 44, 185–198. doi: 10.1016/S0197-0186(03)00153-0
Pan, C. F., Zhu, S. M., and Zheng, Y. Y. (2010). Ammonia induces upregulation of aquaporin-4 in neocortical astrocytes of rats through the p38 mitogen-activated protein kinase pathway. Chin. Med. J. 123, 1888–1892.
Patnaik, J. K., Das, B. S., Mishra, S. K., Mohanty, S., Satpathy, S. K., and Mohanty, D. (1994). Vascular clogging, mononuclear cell margination, and enhanced vascular permeability in the pathogenesis of human cerebral malaria. Am. J. Trop. Med. Hyg. 51, 642–647.
Pedersen, H. R., Ring-Larsen, H., Olsen, N. V., and Larsen, F. S. (2007). Hyperammonemia acts synergistically with lipopolysaccharide in inducing changes in cerebral hemodynamics in rats anaesthetised with pentobarbital. J. Hepatol. 47, 245–252. doi: 10.1016/j.jhep.2007.03.026
Pino, P., Vouldoukis, I., Kolb, J. P., Mahmoudi, N., Desportes-Livage, I., Bricaire, F., et al. (2003). Plasmodium falciparum–infected erythrocyte adhesion induces caspase activation and apoptosis in human endothelial cells. J. Infect. Dis. 187, 1283–1290. doi: 10.1086/373992
Porta, J., Carota, A., Pizzolato, G. P., Wildi, E., Widmer, M. C., Margairaz, C., et al. (1993). Immunopathological changes in human cerebral malaria. Clin. Neuropathol. 12, 142–146.
Potter, S. M., Chan-Ling, T., Rosinova, E., Ball, H. J., Mitchell, A. J., and Hunt, N. H. (2006). A role for Fas-Fas ligand interactions during the late-stage neuropathological processes of experimental cerebral malaria. J. Neuroimmunol. 173, 96–107. doi: 10.1016/j.jneuroim.2005.12.004
Prommano, O., Chaisri, U., Turner, G. D., Wilairatana, P., Ferguson, D. J., Viriyavejakul, P., et al. (2005). A quantitative ultrastructural study of the liver and the spleen in fatal falciparum malaria. Southeast Asian J. Trop. Med. Public Health 36, 1359–1370.
Qureshi, K., Rao, K. V., and Qureshi, I. A. (1998). Differential inhibition by hyperammonemia of the electron transport chain enzymes in synaptosomes and non-synaptic mitochondria in ornithine transcarbamylase-deficient spf-mice: restoration by acetyl-L-carnitine. Neurochem. Res. 23, 855–861. doi: 10.1023/A:1022406911604
Raabe, W. (1987). Synaptic transmission in ammonia intoxication. Neurochem. Pathol. 6, 145–166. doi: 10.1007/BF02833604
Rae, C., Mcquillan, J. A., Parekh, S. B., Bubb, W. A., Weiser, S., Balcar, V. J., et al. (2004). Brain gene expression, metabolism, and bioenergetics: interrelationships in murine models of cerebral and noncerebral malaria. FASEB J. 18, 499–510. doi: 10.1096/fj.03-0543com
Rahman, A., Ting, K., Cullen, K. M., Braidy, N., Brew, B. J., and Guillemin, G. J. (2009). The excitotoxin quinolinic acid induces tau phosphorylation in human neurons. PLoS ONE 4:e6344. doi: 10.1371/journal.pone.0006344
Rama Rao, K. V., Jayakumar, A. R., and Norenberg, M. D. (2005). Role of oxidative stress in the ammonia-induced mitochondrial permeability transition in cultured astrocytes. Neurochem. Int. 47, 31–38. doi: 10.1016/j.neuint.2005.04.004
Rama Rao, K. V., Jayakumar, A. R., and Norenberg, M. D. (2014a). Brain edema in acute liver failure: mechanisms and concepts. Metab. Brain Dis. 29, 927–936. doi: 10.1007/s11011-014-9502-y
Rama Rao, K. V., Jayakumar, A. R., Tong, X., Alvarez, V. M., and Norenberg, M. D. (2010a). Marked potentiation of cell swelling by cytokines in ammonia-sensitized cultured astrocytes. J. Neuroinflammation 7:66. doi: 10.1186/1742-2094-7-66
Rama Rao, K. V., Jayakumar, A. R., Tong, X., Curtis, K. M., and Norenberg, M. D. (2010b). Brain aquaporin-4 in experimental acute liver failure. J. Neuropathol. Exp. Neurol. 69, 869–879. doi: 10.1097/NEN.0b013e3181ebe581
Rama Rao, K. V., Reddy, P. V., Tong, X., and Norenberg, M. D. (2010c). Brain edema in acute liver failure: inhibition by L-histidine. Am. J. Pathol. 176, 1400–1408. doi: 10.2353/ajpath.2010.090756
Rama Rao, K. V., Verkman, A. S., Curtis, K. M., and Norenberg, M. D. (2014b). Aquaporin-4 deletion in mice reduces encephalopathy and brain edema in experimental acute liver failure. Neurobiol. Dis. 63, 222–228. doi: 10.1016/j.nbd.2013.11.018
Rangroo Thrane, V., Thrane, A. S., Wang, F., Cotrina, M. L., Smith, N. A., Chen, M., et al. (2013). Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat. Med. 19, 1643–1648. doi: 10.1038/nm.3400
Ringwald, P., Peyron, F., Vuillez, J. P., Touze, J. E., Le Bras, J., and Deloron, P. (1991). Levels of cytokines in plasma during Plasmodium falciparum malaria attacks. J. Clin. Microbiol. 29, 2076–2078.
Rose, C., Kresse, W., and Kettenmann, H. (2005). Acute insult of ammonia leads to calcium-dependent glutamate release from cultured astrocytes, an effect of pH. J. Biol. Chem. 280, 20937–20944. doi: 10.1074/jbc.M412448200
Rose, C. (2002). Increased extracellular brain glutamate in acute liver failure: decreased uptake or increased release? Metab. Brain Dis. 17, 251–261. doi: 10.1023/A:1021945515514
Saran, T., Hilgier, W., Kocki, T., Urbanska, E. M., Turski, W. A., and Albrecht, J. (1998). Acute ammonia treatment in vitro and in vivo inhibits the synthesis of a neuroprotectant kynurenic acid in rat cerebral cortical slices. Brain Res. 787, 348–350. doi: 10.1016/S0006-8993(98)00031-6
Schluesener, H. J., Kremsner, P. G., and Meyermann, R. (1998). Widespread expression of MRP8 and MRP14 in human cerebral malaria by microglial cells. Acta Neuropathol. 96, 575–580. doi: 10.1007/s004010050938
Schwarcz, R., and Pellicciari, R. (2002). Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J. Pharmacol. Exp. Ther. 303, 1–10. doi: 10.1124/jpet.102.034439
Shawcross, D. L., Davies, N. A., Williams, R., and Jalan, R. (2004). Systemic inflammatory response exacerbates the neuropsychological effects of induced hyperammonemia in cirrhosis. J. Hepatol. 40, 247–254. doi: 10.1016/j.jhep.2003.10.016
Silamut, K., Phu, N. H., Whitty, C., Turner, G. D., Louwrier, K., Mai, N. T., et al. (1999). A quantitative analysis of the microvascular sequestration of malaria parasites in the human brain. Am. J. Pathol. 155, 395–410. doi: 10.1016/S0002-9440(10)65136-X
Song, D., and Du, T. (2014). Ammonium activates ouabain-activated signalling pathway in astrocytes: therapeutic potential of ouabain antagonist. Curr. Neuropharmacol. 12, 334–341. doi: 10.2174/1570159X12666140828222115
Stafstrom, C. E., and Sasaki-Adams, D. M. (2003). NMDA-induced seizures in developing rats cause long-term learning impairment and increased seizure susceptibility. Epilepsy Res. 53, 129–137. doi: 10.1016/S0920-1211(02)00258-9
Stone, T. W., and Darlington, L. G. (2002). Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discov. 1, 609–620. doi: 10.1038/nrd870
Stone, T. W., and Perkins, M. N. (1981). Quinolinic acid: a potent endogenous excitant at amino acid receptors in CNS. Eur. J. Pharmacol. 72, 411–412. doi: 10.1016/0014-2999(81)90587-2
Stone, T. W., Stoy, N., and Darlington, L. G. (2013). An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol. Sci. 34, 136–143. doi: 10.1016/j.tips.2012.09.006
Stone, T. W. (2001). Kynurenines in the CNS: from endogenous obscurity to therapeutic importance. Prog. Neurobiol. 64, 185–218. doi: 10.1016/S0301-0082(00)00032-0
Summar, M. L., Dobbelaere, D., Brusilow, S., and Lee, B. (2008). Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr 97, 1420–1425. doi: 10.1111/j.1651-2227.2008.00952.x
Tenneti, L., D'emilia, D. M., Troy, C. M., and Lipton, S. A. (1998). Role of caspases in N-methyl-D-aspartate-induced apoptosis in cerebrocortical neurons. J. Neurochem. 71, 946–959. doi: 10.1046/j.1471-4159.1998.71030946.x
Toure, F. S., Ouwe-Missi-Oukem-Boyer, O., Bisvigou, U., Moussa, O., Rogier, C., Pino, P., et al. (2008). Apoptosis: a potential triggering mechanism of neurological manifestation in Plasmodium falciparum malaria. Parasite Immunol. 30, 47–51. doi: 10.1111/j.1365-3024.2007.00986.x
Tripathi, A. K., Sha, W., Shulaev, V., Stins, M. F., and Sullivan, D. J. Jr/ (2009). Plasmodium falciparum-infected erythrocytes induce NF-kappaB regulated inflammatory pathways in human cerebral endothelium. Blood 114, 4243–4252. doi: 10.1182/blood-2009-06-226415
Tripathi, A. K., Sullivan, D. J., and Stins, M. F. (2006). Plasmodium falciparum-infected erythrocytes increase intercellular adhesion molecule 1 expression on brain endothelium through NF-kappaB. Infect. Immun. 74, 3262–3270. doi: 10.1128/IAI.01625-05
van der Heyde, H. C., Nolan, J., Combes, V., Gramaglia, I., and Grau, G. E. (2006). A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 22, 503–508. doi: 10.1016/j.pt.2006.09.002
van der Wal, G., Verhagen, W. I., and Dofferhoff, A. S. (2005). Neurological complications following Plasmodium falciparum infection. Neth. J. Med. 63, 180–183.
Wenisch, C., Linnau, K. F., Looaresuwan, S., and Rumpold, H. (1999). Plasma levels of the interleukin-6 cytokine family in persons with severe Plasmodium falciparum malaria. J. Infect. Dis. 179, 747–750. doi: 10.1086/314630
Werner, C., Stubbs, M. T., Krauth-Siegel, R. L., and Klebe, G. (2005). The crystal structure of Plasmodium falciparum glutamate dehydrogenase, a putative target for novel antimalarial drugs. J. Mol. Biol. 349, 597–607. doi: 10.1016/j.jmb.2005.03.077
White, N. J., Turner, G. D., Day, N. P., and Dondorp, A. M. (2013). Lethal malaria: marchiafava and bignami were right. J. Infect. Dis. 208, 192–198. doi: 10.1093/infdis/jit116
Whitten, R., Milner, D. A. Jr. Yeh, M. M., Kamiza, S., Molyneux, M. E., and Taylor, T. E. (2011). Liver pathology in Malawian children with fatal encephalopathy. Hum. Pathol. 42, 1230–1239. doi: 10.1016/j.humpath.2010.11.019
Wiese, L., Kurtzhals, J. A., and Penkowa, M. (2006). Neuronal apoptosis, metallothionein expression and proinflammatory responses during cerebral malaria in mice. Exp. Neurol. 200, 216–226. doi: 10.1016/j.expneurol.2006.02.011
Yang, L., Omori, K., Otani, H., Suzukawa, J., and Inagaki, C. (2003). GABAC receptor agonist suppressed ammonia-induced apoptosis in cultured rat hippocampal neurons by restoring phosphorylated BAD level. J. Neurochem. 87, 791–800. doi: 10.1046/j.1471-4159.2003.02069.x
Yang, L., Omori, K., Suzukawa, J., and Inagaki, C. (2004). Calcineurin-mediated BAD Ser155 dephosphorylation in ammonia-induced apoptosis of cultured rat hippocampal neurons. Neurosci. Lett. 357, 73–75. doi: 10.1016/j.neulet.2003.12.032
Zeuthen, T., Wu, B., Pavlovic-Djuranovic, S., Holm, L. M., Uzcategui, N. L., Duszenko, M., et al. (2006). Ammonia permeability of the aquaglyceroporins from Plasmodium falciparum, Toxoplasma gondii and Trypansoma brucei. Mol. Microbiol. 61, 1598–1608. doi: 10.1111/j.1365-2958.2006.05325.x
Keywords: P. falciparum, cerebral malaria, parasite-derived ammonia, elevated ammonia, pathogenesis
Citation: Kimoloi S and Rashid K (2015) Potential role of Plasmodium falciparum-derived ammonia in the pathogenesis of cerebral malaria. Front. Neurosci. 9:234. doi: 10.3389/fnins.2015.00234
Received: 11 December 2014; Accepted: 18 June 2015;
Published: 03 July 2015.
Edited by:
Albert Gjedde, University of Copenhagen, DenmarkReviewed by:
Johannes Hirrlinger, University of Leipzig, GermanyLeif Hertz, College of Basic Medical Sciences, China Medical University, China
Copyright © 2015 Kimoloi and Rashid. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sammy Kimoloi, Department of Medical Laboratory Sciences, Masinde Muliro University of Science and Technology, Room 002, Science Park D-Block, PO BOX 190-50100, Kakamega, Kenya,a2ltb2xvaUB5YWhvby5jb20=