 Michela Deleidi
Michela Deleidi Madeline Jäggle
Madeline Jäggle Graziella Rubino
Graziella Rubino
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 03 June 2015
Sec. Neuropharmacology
Volume 9 - 2015 | https://doi.org/10.3389/fnins.2015.00172
This article is part of the Research Topic Dysmetabolism, obesity and inflammation: three prominent actors in the drama of major neuropsychiatric disorders View all 13 articles
As we age, the immune system undergoes a process of senescence accompanied by the increased production of proinflammatory cytokines, a chronic subclinical condition named as “inflammaging”. Emerging evidence from human and experimental models suggest that immune senescence also affects the central nervous system and promotes neuronal dysfunction, especially within susceptible neuronal populations. In this review we discuss the potential role of immune aging, inflammation and metabolic derangement in neurological diseases. The discovery of novel therapeutic strategies targeting age-linked inflammation may promote healthy brain aging and the treatment of neurodegenerative as well as neuropsychiatric disorders.
Aging is a complex phenomenon characterized by the progressive decline of physiological function and tissue homeostasis leading to increased vulnerability to degeneration and death. One of the most recognized effects of aging is the dysregulation of the immune system as a result of defects in both initiation and resolution of immune responses (immunosenescence) and chronic low-grade inflammation (inflammaging) (Franceschi et al., 2007; Montecino-Rodriguez et al., 2013). This chronic subclinical condition has been linked to an increased incidence of metabolic syndrome, atherosclerosis, cancer, and neurodegenerative diseases (Franceschi et al., 2007). Similarly, recent work suggests that microglia, the innate immune cells of the brain, undergo a process of senescence that may in turn contribute to the development of neurological diseases in elderly people (Wong, 2013). In this review we discuss recent advances on the link between immune aging and neurological disorders and we describe how the metabolic changes of the aging body, immunosenescence, and inflammation can exacerbate immunological contributions to neurodegenerative and neuropsychiatric diseases.
As we age, the immune system undergoes a process of senescence characterized by a progressive decline in immune function associated with an increased frequency of infections and chronic diseases. Aging affects both the adaptive and the innate immune system (Larbi et al., 2011; Goronzy et al., 2012; Shaw et al., 2013). The most relevant changes are observed in the adaptive immune system; immunosenescence is characterized by a decrease of naïve T cells and a concomitant increase in memory cells, a progressive reduction of the TCR repertoire and decreased proliferation in vitro (Arnold et al., 2011; Frasca et al., 2011; Nikolich-Zugich et al., 2012; Fulop et al., 2013). With regard to innate immunity, studies in animal models and humans have shown age-associated alterations including myeloid skewing (Dykstra et al., 2011), impairment of neutrophil chemotaxis and effector function (Wenisch et al., 2000; Butcher et al., 2001; Fulop et al., 2004), defects in NK cells (Le Garff-Tavernier et al., 2010) and monocyte dysregulation (Hearps et al., 2012). Immunosenescence is accompanied by a low-grade chronic proinflammatory environment in multiple tissues characterized by increased production of proinflammatory cytokines, such as interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α), acute-phase proteins, reactive oxygen species (ROS), and autoantibodies. This proinflammatory environment has been defined as “inflammaging” (Franceschi et al., 2000, 2007). Several mechanisms contribute to inflammaging, including the dysregulation of the adaptive vs. innate immunity (Franceschi et al., 2000) and cellular senescence. Senescent cells secrete a variety of proinflammatory cytokines, chemokines, growth factors and proteases collectively termed as senescence-associated secretory phenotype (SASP) (Freund et al., 2010; Chinta et al., 2014; Ovadya and Krizhanovsky, 2014). The SASP has beneficial effects, such as the reinforcement of the tumor-suppressing cell state (Acosta et al., 2008), prevention of fibrosis (Jun and Lau, 2010), and clearance of senescent and tumor cells (Xue et al., 2007). On the other hand, cellular senescence and the SASP drive the chronic inflammatory environment that is a major contributor to the development of aging-associated diseases. Chronic infections promote immunosenescence and inflammaging (Koch et al., 2007); cytomegalovirus (CMV) promotes age-like immune changes (Derhovanessian et al., 2011) and CMV reactivation has been associated with increased levels of IL-6 and TNF and premature mortality (Stowe et al., 2007; Roberts et al., 2010). However, further studies are needed to assess the link between CMV infection and inflammatory markers in the elderly (Bartlett et al., 2012). Other chronic infectious diseases such as HCV and HIV may also have a role in immunosenescence (Gruener et al., 2001; Zapata and Shaw, 2014). With effective antiretroviral therapy, the life expectancy of HIV patients has significantly improved and a link between chronic infection, immune dysregulation and age-related comorbidities has become evident (Deeks and Phillips, 2009; Lederman et al., 2013). Antigen-dependent clonal expansion of memory T cells as well as premature immune senescence have been shown in HIV patients (Appay et al., 2002, 2011). However, there is still debate as to whether HIV accelerates such immune aging (High et al., 2012). Interestingly, despite the increased life expectancy, the incidence of cognitive impairment in these patients remains high and mounting evidence suggests that persistent inflammation and immune dysregulation play a key role in HIV-associated cognitive disorders (Hong and Banks, 2015). Further corroborating evidence linking chronic infections, aging, and immunosenescence comes from experimental studies showing that lymphocytic choriomeningitis virus (LCMV) infection leads to a reduction of specific antiviral T cell responses in aged mice (Mekker et al., 2012).
Parasites such as Toxoplasma gondii (T. gondii) also contribute to immune dysregulation. Chronic infection with T. gondii is characterized by the presence of intraneuronal cysts that are controlled by the immune system (Suzuki et al., 2010). Growing evidence shows a link between chronic infection and CD8 T-cell dysfunction that in turn may promote the psychiatric disturbances often observed in these patients (Bhadra et al., 2011, 2013; Torrey et al., 2012). Genetic predisposition leading to an increased tendency toward uncontrolled inflammatory responses can also accelerate immunosenescence and inflammaging. With this respect, the Leiden Longevity Study (LLS) showed that individuals enriched for longevity genetic traits are less susceptible to CMV-associated immune alterations with aging (Derhovanessian et al., 2010). Hormonal changes such as the decreased production of estrogen or androgen also influence the secretion of cytokines (Maggio et al., 2006; Abu-Taha et al., 2009). Finally, alterations of mitochondrial function and metabolic changes in the adipose tissue contribute to immunosenescence and inflammaging.
Senescent and hyperactive microglia have been detected in the aged and diseased brain (Streit et al., 2004, 2009). Microglia are the innate immune cells of the central nervous system (CNS) and are involved in several physiological and pathological brain functions. They play essential roles in brain development and actively support neural circuitries and plasticity either through the removal of synaptic elements or the secretion of neurotransmitters and neurotrophic factors (Nimmerjahn et al., 2005; Wake et al., 2009; Kettenmann et al., 2011). During infections, brain injury or neurodegenerative diseases, resting microglia transform into an active state and release immune molecules including cytokines, ROS, and growth factors. Activated microglia exert beneficial functions such as the phagocytic clearance of pathogens and cellular debris. However, uncontrolled immune reactions contribute to neuronal dysfunction and death over time. During aging, rodent microglia undergo replicative senescence characterized by telomere shortening and loss of homogenous distribution throughout the brain (Flanary et al., 2007; Hefendehl et al., 2014). Experimental evidence suggest that microglial motility and paghocytic activity decrease with age leading to the formation of glial lipofuscin granules with subsequent accumulation of damaged mitochondria and oxidative stress (Brunk and Terman, 2002; Szweda et al., 2003; Xu et al., 2008; Damani et al., 2011; Hefendehl et al., 2014). In addition, histo-pathological studies show increased expression of interleukin-1α as well as CD11b/CR3 in humans suggesting that uncontrolled microglial activation occurs with normal brain aging (Sheng et al., 1998; Strohmeyer et al., 2002). Animal studies show increased microglial expression of MHC class II antigens and CD68 (Perry et al., 1993; Sheffield and Berman, 1998) as well as decreased arborization and abnormal cytoplasmic structures (Streit et al., 2004, 2009; Conde and Streit, 2006). Interestingly, these morphological changes precede the appearance of tau neuropathology in Alzheimer's disease (AD) patients suggesting that a loss of microglial function contributes to the onset of the disease (Streit et al., 2009).
The cellular and molecular mechanisms controlling brain immune senescence and neuro-inflammaging are still unknown. Several studies support the existence of a bidirectional communication between the peripheral immune system and the brain mainly through immune molecules, including cytokines, chemokines, acute phase proteins, and complement factors (Wrona, 2006). On the other hand, the aging brain is able to modulate the immune system and to promote the recruitment of immune cells from the periphery, especially lymphocytes, which contribute to immunosenescence and neuro-inflammaging (Gemechu and Bentivoglio, 2012). Thus, peripheral immunosenescence and inflammaging can modulate microglial phenotype and reactivity and drive low-grade brain inflammation (von Bernhardi et al., 2010). This hypothesis is supported by the observation that systemic low-grade inflammation is accompanied by an increase of cytokines in the brain and an imbalance between pro- and anti-inflammatory molecules (Ye and Johnson, 2001; Lukiw, 2004; Streit et al., 2004).
Systemic inflammation promotes the activation of brain immune cells in humans as well as in non-human primates (Brydon et al., 2008; Harrison et al., 2009; Hannestad et al., 2012) and can prime neurons and immune cells in the brain increasing the risk of developing neurological disorders (Deleidi and Isacson, 2012; Perry and Holmes, 2014). The molecular pathways that link low-grade systemic inflammation to brain aging and neuroinflammation remain unclear.
In summary, the aging of the peripheral immune system can promote or exacerbate microglial senescence and drive neuroinflammation (Figure 1). The individual genetic background as well as infections and environmental exposures over a lifetime could further exacerbate the dysregulation of immune responses, increasing the risk of neurological disorders.
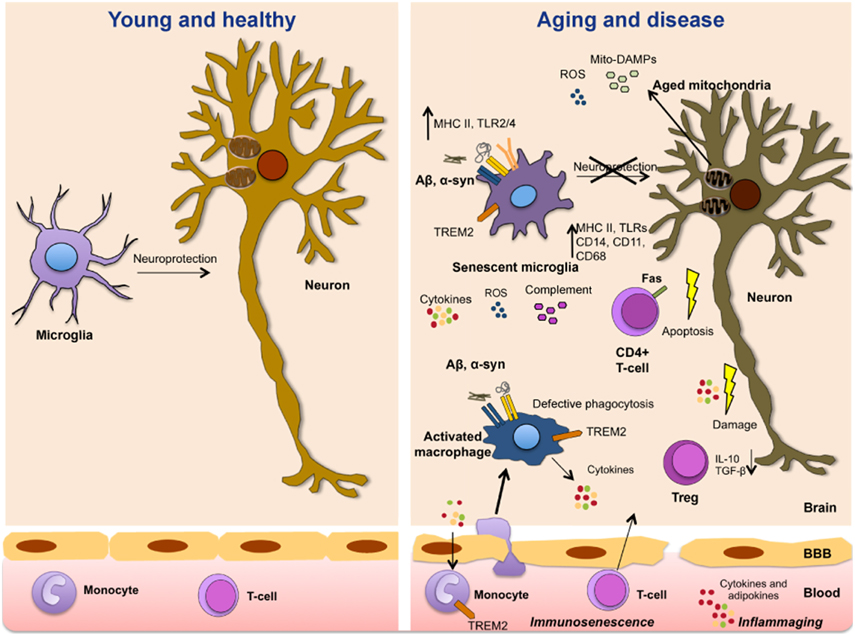
Figure 1. Immune aging, dysmetabolism, and inflammation in neurological diseases. The aging of the immune system is accompanied by a progressive senescence of immune cells (immunosenescence) and a chronic proinflammatory environment characterized by increased levels of cytokines and adipokines (inflammaging). These immune molecules modulate neuronal function and prime microglial cells and vulnerable neuronal populations. With aging, microglial cells also become senescent, lose their neuroprotective function and become more prone to abnormal inflammatory activation. Amyloid deposition and pathogenetic forms of α-synuclein activate microglia with the subsequent release of proinflammatory mediators. Aged dysfunctional mitochondria play a fundamental role in immunosenescence and age-related inflammation by the production of ROS and DAMPs that activate the NLRP3 inflammasome and immune cells triggering inflammatory reactions. In turn, inflammatory mediators aggravate mitochondrial dysfunction. High immune risk profile, infections and metabolic syndrome promote abnormal inflammatory reactions that contribute to disease onset and progression.
Aging is characterized by important changes in the distribution of total and regional adipose tissue (Kuk et al., 2009). Overnutrition and obesity are associated with an increased risk of metabolic syndrome, a complex disorder characterized by hypertension, insulin resistance, glucose intolerance, and hyperlipidemia. Both hypertrophic fat cells and immune cells infiltrate the adipose tissue and secrete inflammatory molecules that can promote systemic low-grade inflammation. Adipocytes secrete more than 600 bioactive molecules, which are collectively termed as adipokines and include proinflammatory mediators, such as TNF-α, monocyte chemoattractant protein (MCP)-1, and IL-6, as well as anti-inflammatory molecules (Trayhurn and Wood, 2004). A link between obesity and inflammation has been initially hypothesized based on the observation of increased levels of IL-6 and C-reactive protein (CRP) in obese animals and humans (Yudkin et al., 1999; Festa et al., 2001; Moschen et al., 2011). Interestingly, concentrations of these cytokines decrease after weight loss (Ryan and Nicklas, 2004; Rao, 2012). Circulating adipokines have immune as well as neuronal functions and can influence brain inflammation. For example, leptin, one of the first adipokines identified, is a proinflammatory cytokine sharing structural similarities with IL-2, IL-6, IL-12, and granulocyte colony stimulating factor (Zhang et al., 1997). Leptin has important immune functions, especially related to T lymphocyte proliferation (Lord et al., 1998; Carbone et al., 2012). In the CNS, it also regulates appetite and energy balance (Friedman and Halaas, 1998) and contributes to development of sickness-type behavior during experimental inflammation (Pohl et al., 2014). Thus, excess adiposity drives low-grade systemic inflammation that increases the risk of developing insulin resistance, type 2 diabetes, cardiovascular disease, stroke, cancer, and neurodegeneration (Tchernof and Despres, 2013).
The link between obesity-induced inflammation and type 2 diabetes is further supported by the observation that the development of insulin resistance correlates with immune cell infiltration (Hotamisligil, 2006). Interestingly, obesity is also linked to mitochondrial dysfunction and endoplasmic reticulum (ER) stress (Arruda et al., 2014), which in turn leads to impaired glucose metabolism. In fact, the earliest hallmark of insulin resistance is the decreased expression of PPARγ coactivator 1-α (PGC1α), a positive regulator of mitochondrial biogenesis and respiration (Patti et al., 2003). Decreased expression of PGC1α has been linked to aging and chronic inflammation in humans and mice (Ghosh et al., 2011; Sczelecki et al., 2014).
In summary, fat accumulation and changes in fat redistribution with age lead to the dysregulation of adipokine secretion and metabolic changes linked to systemic and local inflammatory responses that, in turn, contribute to accelerated aging as well as cardiovascular and neurological disorders.
Because of their function in oxidative stress processes, mitochondria have a fundamental role in aging (Green et al., 2011). Altered mitochondrial function is central to several acute and chronic inflammatory diseases and increasing evidence show that mitochondria may contribute to immunosenescence and inflammaging via ROS production, calcium exchange, ATP and NF-κB activation. Mitochondrial dysfunction can be both the cause and the consequence of inflammation. Aged dysfunctional mitochondria are able to modulate innate immunity through redox-sensitive inflammatory signaling pathways or the direct activation of the NLRP3 inflammasome (through ROS, mtDNA, and ATP) (Kepp et al., 2011; Shimada et al., 2012). Mitochondria are an important source of damage-associated molecular patterns (mito-DAMPs) that activate Toll-like membrane receptors (TLRs) and cytoplasmic nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) (Krysko et al., 2011). Interestingly, DAMPs and mito-DAMPs directly activate antigen-presenting cells and other non-immune cells such as astrocytes (Mathew et al., 2012). On the other hand, inflammatory mediators, including inflammatory cytokines, ROS, and nitric oxide (NO), impair mitochondrial function by inducing the accumulation of mtDNA mutations and inhibiting mitochondrial respiratory chain and energy production (Lopez-Armada et al., 2013).
In recent years, the role of the immune system in the development of neuropsychiatric disorders has become more apparent. The immune system communicates with the brain and regulates behavior and many other critical neurological functions (Wilson et al., 2002). Thus, age-related immune dysregulation may be relevant to the pathophysiology of neuropsychiatric conditions. This hypothesis is further supported by the observation that psychiatric manifestations are common in autoimmune diseases including systemic lupus erythematosus, CNS vasculitis, Whipple's disease, Sjögren's syndrome, and Behçet's disease [reviewed in (Kayser and Dalmau, 2011)]. In addition, patients with acute isolated psychosis have been diagnosed with synaptic autoimmune encephalitis and antineuronal antibodies targeting synaptic proteins have been demonstrated (Kayser and Dalmau, 2011). Finally, it is well established that infections and inflammation during the perinatal period increase the risk of developing neurological and neuropsychiatric diseases in the adulthood (Hagberg et al., 2015). Infectious diseases can cause direct neuronal damage; in other circumstances, such as after LCMV infection or chronic T. gondii infection, neuronal dysfunction may also be linked to immune reactions. Chronic immune activation is a potential key contributor to disease pathology, even in the absence of overt cell death. It is also well established that viruses can alter neurotransmission and activate the kanyurine pathway causing chronic fatigue, cognitive impairment and mood disorders (Weissenborn et al., 2006). In particular, the alteration of the kanyurine pathway is linked to an increased risk for neurological and neuropsychiatric disorders (Schwarcz et al., 2012). It has also been hypothesized that viruses such as LCMV alter inhibitory circuits causing unbalanced excitatory neurotransmission and neuronal death via excitotoxicity (“Virus induced disinhibition and excitotoxicity”) (Pearce et al., 2000). Finally, long-term alterations in neurogenesis have been described in mice after LCMV infection (Sharma et al., 2002).
Several immune changes are associated with neuropsychiatric disorders; increased levels of inflammatory mediators have been reported in schizophrenia (Soderlund et al., 2009; Beumer et al., 2012a), depression (Dowlati et al., 2010) and autism spectrum disorders (ASD) (Vargas et al., 2005; Li et al., 2009). Altered monocytic as well as T- and B-cell function has been demonstrated in patients with schizophrenia and ASD (Ashwood et al., 2011; Beumer et al., 2012b; Braunschweig and van de Water, 2012; Jyonouchi et al., 2012; Muller et al., 2012). Further corroborating evidence comes from the observation that infections and chronic inflammatory diseases are linked to depression, anxiety as well as schizophrenia (Eaton et al., 2006; Loftis et al., 2008; Yolken and Torrey, 2008; Margaretten et al., 2011; Watkins and Treisman, 2012; Bhadra et al., 2013). Interestingly, many genes involved in the predisposition to schizophrenia are related to immune defense against invading pathogens, suggesting an involvement of immune dysregulation and infections in the pathogenesis of this disease (Carter, 2009; International Schizophrenia et al., 2009; Irish Schizophrenia Genomics C and the Wellcome Trust Case Control C, 2012). Neuropsychiatric disorders are also characterized by the activation of the brain innate immune system and increased density of activated microglia has been found in brains of schizophrenic and ASD patients (Vargas et al., 2005; Doorduin et al., 2009).
Aging, metabolic changes, obesity, and chronic stress modulate the communication between the immune system and the brain and may promote neuropsychiatric diseases. Neuropsychiatric disorders are indeed common in people with metabolic dysfunctions (Skilton et al., 2007). In these patients, inflammation is one major determinant of depressive symptoms (Capuron et al., 2008). Obesity is also a contributing factor; in obese women adiposity is associated with increased concentrations of inflammatory markers (IL-6, CRP, and adipokines) that correlate with depression and anxiety (Capuron et al., 2011). Interestingly, after surgery, weight loss correlates with reduced markers of inflammation and is associated with improvement of psychiatric symptoms (Capuron et al., 2011).
The brain, in turn, is able to regulate the immune system via neurotransmitter signals (Felten et al., 1987). As a consequence, psychological stress and depression are often associated with impaired immune function and increased risk of acute respiratory infections (Cohen et al., 1991, 2012). In addition, premature shortening of telomeres in leukocytes has been reported in subjects with chronic stress and depression as well as in mice exposed to stressful conditions (Epel et al., 2004; Simon et al., 2006; Kotrschal et al., 2007; Karabatsiakis et al., 2014). Stress responses pathways including the hypothalamic–pituitary–adrenal (HPA) axis and chronic immune stimulation, which are often associated with mood disorders, could be the cause of telomere shortening and accelerated aging observed in these patients (Glaser and Kiecolt-Glaser, 2005). The role of telomere shortening in other psychiatric disorders such as schizophrenia is still controversial and further studies are therefore warranted (Nieratschker et al., 2013; Kota et al., 2014).
AD is the most common form of dementia in the elderly affecting more than 35 million people worldwide. In AD, the progressive neuronal loss in the medial temporal lobe and many other brain regions causes cognitive decline. The pathological hallmarks of AD are extracellular β-amyloid (Aβ) plaques, intracellular neurofibrillary tangles and gliosis consisting of activated microglia and astrocytes surrounding β-amyloid plaques (Joachim and Selkoe, 1992). The pathogenesis of the common sporadic forms of AD is still unknown. Neuroinflammation and microglial activation have been proposed as key players in the pathogenesis of the disease (Wyss-Coray, 2006). Inflammation is an early event in the amyloid pathology and precedes plaque deposition in experimental models of AD (Ferretti et al., 2012; Wright et al., 2013). Amyloid deposition leads to microglial activation (Meyer-Luehmann et al., 2008) and the production of proinflammatory mediators that contribute to disease pathogenesis. However, microglia also play a beneficial role in restricting senile plaque formation by clearing Aβ deposits (Simard et al., 2006) and secreting neuroprotective factors. It has been proposed that the loss of such neuroprotective function rather than microglial inflammatory activation contributes to AD. Microglial senescence and dysfunction have been described in AD (Streit et al., 2009). Oxidative stress-related mechanisms that precedes Aβ oligomerization can trigger microglia senescence and dystrophy (Sutherland et al., 2013). Senescent microglia upregulate immune receptors such as MHC II, CD68, CD14, CD11, and TLRs (Sheffield and Berman, 1998; Landreth and Reed-Geaghan, 2009; Hart et al., 2012) and become more prone to trigger inflammatory response to Aβ (Reed-Geaghan et al., 2009). Thus, loss and gain of microglial function are not mutually exclusive and can both play a role in disease pathogenesis and progression. Loss of function in peripheral monocytes and macrophages can also contribute to disease pathogenesis. It has been shown that, upon stimulation, monocytes from AD patients secrete more proinflammatory cytokines compared to monocytes from healthy subjects (Bonotis et al., 2008). In addition, phagocytosis of Aβ is defective in peripheral macrophages from AD patients and this may elicit a compensatory response of the adaptive immune system (Fiala et al., 2005). AD-linked genetic mutations and metabolic risk factors such as obesity, lipid dysmetabolism, and chronic inflammation can trigger immune cell dysfunction and promote cognitive decline. Interestingly, two recent GWAS studies have identified mutations in Trem2 (triggering receptor expressed on myeloid cells 2) as risk factors for AD (Guerreiro et al., 2013; Jonsson et al., 2013). TREM2 is an innate immune receptor expressed on the cell surface of microglia, macrophages, osteoclasts and immature dendritic cells (Colonna, 2003). It regulates the phagocytic ability of myeloid cells and has anti-inflammatory properties by inhibiting cytokine secretion (Takahashi et al., 2005; Turnbull et al., 2006). Apolipoprotein E allele ε4 (APOE*ε4), the best-established genetic risk factor for sporadic AD, can also influence the degree of microglial activation in response to Aβ deposition (Barger and Harmon, 1997; Rodriguez et al., 2014). Associations have been reported between APOE*ε4 and clinical outcomes in infectious diseases further supporting a link between the patient genetic profile and the degree of inflammatory responses (Gerard et al., 2005; Itzhaki and Wozniak, 2006).
Metabolic syndrome has been associated with impaired cognitive function and increased risk of AD (Razay et al., 2007). Patients with diabetes mellitus have an increased risk of developing AD (Arvanitakis et al., 2004). Several studies show a negative correlation between body mass index (BMI) and cognitive performance (Elias et al., 2005; Jeong et al., 2005; Hassing et al., 2010). The mechanisms by which obesity influences risk of AD are still unclear. Obesity may contribute to disease onset by promoting low-grade systemic inflammation (Odegaard and Chawla, 2013; Erion et al., 2014), the disruption of the integrity of the blood brain barrier (Gustafson et al., 2007), and altered lipid metabolism (Hottman et al., 2014). Associations between systemic inflammatory markers and cognitive decline have been described with conflicting results and further studies are warranted (Engelhart et al., 2004; Tan et al., 2007; Sundelof et al., 2009; Singh-Manoux et al., 2014).
In summary, aged-related metabolic changes, immunosenescence and inflammaging can induce loss of microglial function and promote neuroinflammation and Aβ accumulation over time (Figure 1).
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease that affects motor neurons in the brain and spinal cord leading to progressive paralysis and death within 2 to 5 years after onset of symptoms. The etiology of ALS remains largely unknown. Most cases are sporadic, whereas approximately 5–10% of the cases are caused by an autosomal dominant mutation. The pathological hallmarks of ALS are the presence of cytoplasmic ubiquitinated protein inclusions in degenerating motor neurons and intense inflammatory reactions characterized by microglial/astroglial activation and infiltration of peripheral T cells (Pasinelli and Brown, 2006). Thus, both innate and adaptive immune responses are involved in the pathogenesis of ALS (McGeer and McGeer, 2002; Henkel et al., 2004). Experimental and clinical evidence support the hypothesis that microglia activation occurs at early stages of disease (Turner et al., 2004). In experimental ALS, the microglial phenotype changes with disease progression, with a shift from the M2 phenotype in the early disease phase to M1 phenotype at later stages, suggesting that the decreased function of neuroprotective microglia correlates with disease progression (Beers et al., 2011; Liao et al., 2012). Moreover, the degeneration of peripheral motor axons is preceded by the recruitment and activation of macrophages (Chiu et al., 2009). T cells also play a role in ALS as suggested by the presence of CD4+ and CD8+ T lymphocyte infiltrates surrounding degenerating motor neurons (Troost et al., 1989; Engelhardt et al., 1993). Recent studies have shown that regulatory T lymphocytes (Tregs) and cytotoxic T lymphocytes (Teffs) have distinct roles in the pathogenesis and progression of ALS (Beers et al., 2011). Increased numbers of endogenous Tregs and M2 microglia predominate at early stages of disease, whereas Th1 lymphocytes and M1 microglia are more abundant during the rapidly progressing phase (Beers et al., 2011; Liao et al., 2012). These data suggest that inflammation plays a neuroprotective role during the initial stages, whereas immune dysfunction correlates with the progression of the disease at later stages. With regard to ALS, it is still unclear whether immunosenescence, inflammaging, and dysmetabolism affect the disease risk. ALS is associated with metabolic alterations, including include weight loss, hypermetabolism, and hyperlipidemia (Dupuis et al., 2011), and dyslipidemia has been described as a protective factor in ALS (Dupuis et al., 2008; Sutedja et al., 2011). However, further research is needed to clarify the relationship between aged-associated metabolic derangements, inflammation and sporadic ALS.
PD is the second most common neurodegenerative disorder affecting over 4 million people worldwide. In PD, the interaction between aging, individual genetic vulnerability, and environmental factors leads to the death of dopaminergic (DA) neurons in the substantia nigra (SN) pars compacta and other vulnerable neuronal populations. Interestingly, the dysfunction of the autonomic nervous system (ANS) and brainstem pathology precedes midbrain DA cell loss and motor symptoms (Braak et al., 2006). Even though inflammation has been associated with the pathogenesis of PD, it is still under debate whether this is a cause or consequence to neuronal death. Several lines of evidence support a role for both the innate and the adaptive immune system in disease pathogenesis. These include the increased levels of proinflammatory cytokines observed in PD patients (Dobbs et al., 1999) and the association of the disease risk with certain HLA genetic variants (Hamza et al., 2010). In PD, both native and pathogenic forms of α-synuclein activate microglial TLRs (TLR2 and TLR4) triggering the release of proinflammatory cytokines and ROS (Beraud et al., 2011; Stefanova et al., 2011; Codolo et al., 2013; Fellner et al., 2013). Interestingly, nitrated forms of α-synuclein exacerbate peripheral adaptive immune responses (Benner et al., 2008).
Metabolic syndrome has been proposed as a risk factor for PD. Increased BMI and high cholesterol are potential risk factors for PD (Hu et al., 2006, 2008). However, studies have yielded inconsistent findings and further investigations are needed to elucidate this association (Palacios et al., 2011). Diabetes increases PD risk in prospective studies and has been associated with severity of disease (Xu et al., 2011; Cereda et al., 2012). It has been suggested that common dysregulated pathways converging on mitochondrial dysfunction, ER stress, and inflammation as well as glucose and lipid metabolism, link diabetes, and PD (Santiago and Potashkin, 2013). Interestingly, high calorie and high fat diet exacerbates autonomic abnormalities in α-synuclein mutant mice suggesting a link between metabolism and disease pathology (Griffioen et al., 2013; Rotermund et al., 2014). It has been proposed that energy intake influences the vulnerability of neurons during aging by modulating the production of neurotrophic factors and inflammation (Maswood et al., 2004; Youm et al., 2015).
The role of the systemic immune system in PD is still unclear. It has been shown that peripheral monocytes from PD patients have a proinflammatory phenotype and impaired phagocytic function compared to controls (Grozdanov et al., 2014). In addition, T-lymphocytes, but not B-lymphocytes, are present in the SN of MPTP-intoxicated mice and PD patients (Brochard et al., 2009). Abnormalities in peripheral T cells, including decrease in the number of CD4(+) T cell subsets and Treg dysfunction have also been demonstrated in PD patients (Calopa et al., 2010; Saunders et al., 2012). These data support the existence of a peripheral immune dysfunction in PD and are complemented by the association of viral infections with disease onset. Viral infections and inflammatory reactions are a possible environmental trigger for PD (Deleidi and Isacson, 2012). A viral etiology for PD is based on epidemiological studies showing a high incidence of severe progressive parkinsonism in people developing encephalopathy after the 1918–1919 influenza outbreak (Ravenholt and Foege, 1982). In addition, parkinsonism has also been described in patients infected with other viruses such as Japanese encephalitis virus, Epstein-Barr virus, Coxsackie, St. Louis, West Nile and HIV viruses (Jang et al., 2009). Even though viral parkinsonism is extremely rare, an emerging hypothesis is that viral infections cause inflammatory reactions that prime vulnerable neurons to degenerate in response to other cellular insults over time (Deleidi et al., 2010). Finally, as many PD-linked genetic mutations play a role in the regulation of the immune system (Deleidi and Gasser, 2013), it is likely that genetic vulnerability predisposes to the development of midbrain DA neurodegeneration via inflammatory mechanisms.
In summary, more work is required to determine the role of age-linked inflammatory changes and dysmetabolism in the pathogenesis of PD. Nevertheless, emerging evidence suggest that the interplay between aging and genetic vulnerability results in increased susceptibility to infections or abnormal inflammatory reactions that overtime predispose to PD. A fundamental question to address is whether metabolic and inflammatory changes are strong predictors of neurodegeneration in PD patients. Understanding the molecular mechanisms regulating such responses and their link with aging and metabolism may provide potential opportunities for therapeutic targeting in PD.
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the CNS in which the interaction between environmental exposures and genetic predisposition leads to persistent inflammation, oligodendrocyte death, and loss of the myelin sheath. Even though the sequence of events that initiate the disease remains largely unknown, MS is generally believed to be an immune-mediated disorder that occurs in genetically susceptible people. Diverse disease processes, including autoimmunity, viral infections, and metabolic changes may lead to the formation of inflammatory demyelinated plaques (Hedstrom et al., 2014; Belbasis et al., 2015). An accelerated aging could also play a role in the development of autoimmune diseases such as MS. Premature immune aging has been described in MS patients (Thewissen et al., 2005, 2007). It has been hypothesized that both genetic and environmental factors such as viral infections accelerate immunosenescence in these patients and contribute to disease pathogenesis.
Potential therapeutic strategies targeting the age-associated inflammatory immune phenotype include anti-inflammatory drugs and lifestyle interventions such as diet, exercise and nutritional modifications. Common anti-inflammatory drugs, vaccines against CMV and HIV and novel strategies, either small molecules or monoclonal antibodies, targeting inflammatory pathways (e.g., IL1-β, TNFα, IL-6, mTOR), could be used in mono- or combination therapy. The association between nonsteroidal anti-inflammatory drug (NSAID) treatment and lower incidence of AD and PD is still controversial (in t' Veld et al., 2001; Chen et al., 2003; Wahner et al., 2007; Gao et al., 2011; Rees et al., 2011; Cote et al., 2012; Alzheimer's Disease Anti-inflammatory Prevention Trial Research, 2013). Epidemiological studies suggest that the risk reduction with NSAIDs decreases with age (Zandi et al., 2002) and may be affected by individual genetic traits such as the APOE genotype (Szekely et al., 2008). Further studies are therefore warranted to assess the role of anti-inflammatory drugs and identify patient subgroups that could benefit from such interventions. With respect to neuropsychiatric disorders, it is noteworthy that many antipsychotics and antidepressants decrease the levels of pro-inflammatory cytokines and inhibit immune-inflammatory pathways in humans and experimental models of inflammation (Miller et al., 2011; Walker, 2013). Interestingly, Tyring and colleagues showed that Etanercept, an anti-TNF-α molecule, improves fatigue and depression symptoms in patients with psoriasis (Tyring et al., 2006). Therapeutic strategies targeting cellular senescence and SASP-induced inflammation may be beneficial to dampen inflammaging and delay the onset and progression of neurological diseases (Wang et al., 2010; Tchkonia et al., 2013). Ideally, these drugs should delay the accumulation of senescent cells or promote their clearance, without interfering with anti-oncogenic pathways. With this respect, inducers of heat shock response (e.g., heat shock, physical exercise, and diet) can efficiently inhibit the SASP and reduce obesity-dependent chronic inflammation (Newsholme and de Bittencourt, 2014).
The NLRP3 inflammasome is another potential therapeutic target. The NLRP3 inflammasome is an immune sensor that could link systemic and brain aged-related inflammation (Youm et al., 2013). Interestingly, Youm et al. showed that caloric restriction in animal models reduces chronic inflammation with a mechanism involving the inhibition of NLRP3 inflammasome and reduced levels of IL-1β and IL-18 in human monocytes by the ketone metabolite β-hydroxybutyrate (Youm et al., 2015). Thus, dietary or pharmacological approaches should be exploited to treat NLRP3-mediated inflammatory diseases.
Calorie restriction improves health and survival in several species including non-human primates (Colman et al., 2014; Fontana and Partridge, 2015). Even though it is still an open question whether it extends lifespan in humans, several beneficial effects of calorie restriction have been shown including the improvement of cognitive function in elderly people (Witte et al., 2009) and a shift of the transcriptional profile toward a younger state in muscle cells (Mercken et al., 2013). Further understanding of the molecular mechanisms underlying the beneficial effects of calorie restriction in humans will help devise novel drugs and dietary supplements that can prevent or delay aging-related diseases.
Nutritional supplements such as natural anti-inflammatory molecules can dampen the inflammatory environment and promote healthy aging. For example, omega-3 essential fatty acids decrease the levels of IL-1, IL-6, TNF, and CRP (Simopoulos, 2002) and improve cognitive function in aged mice (Cutuli et al., 2014).
Regular and moderate physical activity is the most effective and affordable prevention strategy that reduces aged- and obesity-linked inflammation through the increased release of immunomodulant molecules (Cotman et al., 2007; Handschin and Spiegelman, 2008). Exercise improves cognitive, cardiac and immune function and reduces risk factors such as diabetes, hypertension, and cardiovascular diseases (Phillips et al., 2014). Interestingly, exercise also improves mitochondrial biogenesis and respiration by increasing the expression of PGC1α that, in turn, may have anti-inflammatory properties (Handschin and Spiegelman, 2008). Drugs or nutritional supplements that enhance mitochondrial function represent a potential therapeutic strategy for diseases associated with chronic low-grade inflammation.
Finally, it is important to remark that in current drug trials, inclusion criteria are usually based on clinical symptoms, therefore failing to take into account the underlying heterogeneity of the molecular pathogenesis and genetic risk patterns of complex neurological diseases. Thus, identifying the correlation between genomic risk profiles and the dysregulation of specific molecular inflammatory pathways will allow the identification of novel targets for therapeutic intervention and the stratification of patients toward personalized medicine.
Aging predisposes to disease via several mechanisms that in part converge on inflammatory pathways. As we age, the immune system undergoes a process of senescence accompanied by the increased production of inflammatory cytokines. Given the bidirectional communication between the peripheral immune system and the brain, it is not surprising that systemic inflammation and metabolic changes interfere with brain immunological processes and neuronal function. Immune challenges such as systemic infections, age-related immunological and metabolic changes can alter this communication and disrupt brain plasticity. Under these circumstances, immune molecules in the periphery signal to neurons and glial cells via humoral routes or the sympathetic nervous system. Numerous studies have now provided evidence that increased levels of inflammatory molecules impair behavioral and cognitive performances (Lynch, 2010). Hence, this strengthens the hypothesis that the age-associated dysregulation of immune function and systemic as well as brain inflammaging contribute to increased risk of cognitive dysfunction and neurodegenerative diseases.
In summary, the interplay between aging, genetic predisposition, and environmental exposures initiate systemic and local metabolic changes as well as inflammatory reactions that predispose to neuropsychiatric and neurodegenerative diseases (Figure 1). We would further argue that targeting age-associated inflammation, especially in selected patients with high immune risk profiles, can be an advantageous strategy to prevent or delay the onset of these diseases. Further understanding the cellular and molecular mechanisms that control immunosenescence and inflammaging both in the periphery and in the brain will help unravel how the immune and nervous system communicate and devise novel drugs to promote healthy aging.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Marie Curie Career Integration Grant MC CIG304108 (MD), Fritz Thyssen Foundation grant 10.13.1.155 (MD). We apologize to authors whose original papers could not be cited due to space constraints.
Abu-Taha, M., Rius, C., Hermenegildo, C., Noguera, I., Cerda-Nicolas, J. M., Issekutz, A. C., et al. (2009). Menopause and ovariectomy cause a low grade of systemic inflammation that may be prevented by chronic treatment with low doses of estrogen or losartan. J. Immunol. 183, 1393–1402. doi: 10.4049/jimmunol.0803157
Acosta, J. C., O'Loghlen, A., Banito, A., Guijarro, M. V., Augert, A., Raguz, S., et al. (2008). Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018. doi: 10.1016/j.cell.2008.03.038
Alzheimer's Disease Anti-inflammatory Prevention Trial Research, G. (2013). Results of a follow-up study to the randomized Alzheimer's Disease Anti-inflammatory Prevention Trial (ADAPT). Alzheimers Dement. 9, 714–723. doi: 10.1016/j.jalz.2012.11.012
Appay, V., Dunbar, P. R., Callan, M., Klenerman, P., Gillespie, G. M., Papagno, L., et al. (2002). Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 8, 379–385. doi: 10.1038/nm0402-379
Appay, V., Fastenackels, S., Katlama, C., Ait-Mohand, H., Schneider, L., Guihot, A., et al. (2011). Old age and anti-cytomegalovirus immunity are associated with altered T-cell reconstitution in HIV-1-infected patients. AIDS 25, 1813–1822. doi: 10.1097/QAD.0b013e32834640e6
Arnold, C. R., Wolf, J., Brunner, S., Herndler-Brandstetter, D., and Grubeck-Loebenstein, B. (2011). Gain and loss of T cell subsets in old age–age-related reshaping of the T cell repertoire. J. Clin. Immunol. 31, 137–146. doi: 10.1007/s10875-010-9499-x
Arruda, A. P., Pers, B. M., Parlakgul, G., Guney, E., Inouye, K., and Hotamisligil, G. S. (2014). Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 20, 1427–1435. doi: 10.1038/nm.3735
Arvanitakis, Z., Wilson, R. S., Bienias, J. L., Evans, D. A., and Bennett, D. A. (2004). Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol. 61, 661–666. doi: 10.1001/archneur.61.5.661
Ashwood, P., Krakowiak, P., Hertz-Picciotto, I., Hansen, R., Pessah, I. N., and Van de Water, J. (2011). Altered T cell responses in children with autism. Brain Behav. Immun. 25, 840–849. doi: 10.1016/j.bbi.2010.09.002
Barger, S. W., and Harmon, A. D. (1997). Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 388, 878–881. doi: 10.1038/42257
Bartlett, D. B., Firth, C. M., Phillips, A. C., Moss, P., Baylis, D., Syddall, H., et al. (2012). The age-related increase in low-grade systemic inflammation (Inflammaging) is not driven by cytomegalovirus infection. Aging Cell 11, 912–915. doi: 10.1111/j.1474-9726.2012.00849.x
Beers, D. R., Henkel, J. S., Zhao, W., Wang, J., Huang, A., Wen, S., et al. (2011). Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 134, 1293–1314. doi: 10.1093/brain/awr074
Belbasis, L., Bellou, V., Evangelou, E., Ioannidis, J. P., and Tzoulaki, I. (2015). Environmental risk factors and multiple sclerosis: an umbrella review of systematic reviews and meta-analyses. Lancet Neurol. 14, 263–273. doi: 10.1016/S1474-4422(14)70267-4
Benner, E. J., Banerjee, R., Reynolds, A. D., Sherman, S., Pisarev, V. M., Tsiperson, V., et al. (2008). Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE 3:e1376. doi: 10.1371/journal.pone.0001376
Beraud, D., Twomey, M., Bloom, B., Mittereder, A., Ton, V., Neitzke, K., et al. (2011). alpha-Synuclein Alters Toll-Like Receptor Expression. Front. Neurosci. 5:80. doi: 10.3389/fnins.2011.00080
Beumer, W., Drexhage, R. C., De Wit, H., Versnel, M. A., Drexhage, H. A., and Cohen, D. (2012a). Increased level of serum cytokines, chemokines and adipokines in patients with schizophrenia is associated with disease and metabolic syndrome. Psychoneuroendocrinology 37, 1901–1911. doi: 10.1016/j.psyneuen.2012.04.001
Beumer, W., Gibney, S. M., Drexhage, R. C., Pont-Lezica, L., Doorduin, J., Klein, H. C., et al. (2012b). The immune theory of psychiatric diseases: a key role for activated microglia and circulating monocytes. J. Leukoc. Biol. 92, 959–975. doi: 10.1189/jlb.0212100
Bhadra, R., Cobb, D. A., Weiss, L. M., and Khan, I. A. (2013). Psychiatric disorders in toxoplasma seropositive patients–the CD8 connection. Schizophr. Bull. 39, 485–489. doi: 10.1093/schbul/sbt006
Bhadra, R., Gigley, J. P., Weiss, L. M., and Khan, I. A. (2011). Control of Toxoplasma reactivation by rescue of dysfunctional CD8+ T-cell response via PD-1-PDL-1 blockade. Proc. Natl. Acad. Sci. U.S.A. 108, 9196–9201. doi: 10.1073/pnas.1015298108
Bonotis, K., Krikki, E., Holeva, V., Aggouridaki, C., Costa, V., and Baloyannis, S. (2008). Systemic immune aberrations in Alzheimer's disease patients. J. Neuroimmunol. 193, 183–187. doi: 10.1016/j.jneuroim.2007.10.020
Braak, H., de Vos, R. A., Bohl, J., and Del Tredici, K. (2006). Gastric alpha-synuclein immunoreactive inclusions in Meissner's and Auerbach's plexuses in cases staged for Parkinson's disease-related brain pathology. Neurosci. Lett. 396, 67–72. doi: 10.1016/j.neulet.2005.11.012
Braunschweig, D., and van de Water, J. (2012). Maternal autoantibodies in autism. Arch. Neurol. 69, 693–699. doi: 10.1001/archneurol.2011.2506
Brochard, V., Combadiere, B., Prigent, A., Laouar, Y., Perrin, A., Beray-Berthat, V., et al. (2009). Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Invest. 119, 182–192. doi: 10.1172/JCI36470
Brunk, U. T., and Terman, A. (2002). The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur. J. Biochem. 269, 1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x
Brydon, L., Harrison, N. A., Walker, C., Steptoe, A., and Critchley, H. D. (2008). Peripheral inflammation is associated with altered substantia nigra activity and psychomotor slowing in humans. Biol. Psychiatry 63, 1022–1029. doi: 10.1016/j.biopsych.2007.12.007
Butcher, S. K., Chahal, H., Nayak, L., Sinclair, A., Henriquez, N. V., Sapey, E., et al. (2001). Senescence in innate immune responses: reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J. Leukoc. Biol. 70, 881–886.
Calopa, M., Bas, J., Callen, A., and Mestre, M. (2010). Apoptosis of peripheral blood lymphocytes in Parkinson patients. Neurobiol. Dis. 38, 1–7. doi: 10.1016/j.nbd.2009.12.017
Capuron, L., Poitou, C., Machaux-Tholliez, D., Frochot, V., Bouillot, J. L., Basdevant, A., et al. (2011). Relationship between adiposity, emotional status and eating behaviour in obese women: role of inflammation. Psychol. Med. 41, 1517–1528. doi: 10.1017/S0033291710001984
Capuron, L., Su, S., Miller, A. H., Bremner, J. D., Goldberg, J., Vogt, G. J., et al. (2008). Depressive symptoms and metabolic syndrome: is inflammation the underlying link? Biol. Psychiatry 64, 896–900. doi: 10.1016/j.biopsych.2008.05.019
Carbone, F., La Rocca, C., and Matarese, G. (2012). Immunological functions of leptin and adiponectin. Biochimie 94, 2082–2088. doi: 10.1016/j.biochi.2012.05.018
Carter, C. J. (2009). Schizophrenia susceptibility genes directly implicated in the life cycles of pathogens: cytomegalovirus, influenza, herpes simplex, rubella, and Toxoplasma gondii. Schizophr. Bull. 35, 1163–1182. doi: 10.1093/schbul/sbn054
Cereda, E., Barichella, M., Cassani, E., Caccialanza, R., and Pezzoli, G. (2012). Clinical features of Parkinson disease when onset of diabetes came first: A case-control study. Neurology 78, 1507–1511. doi: 10.1212/WNL.0b013e3182553cc9
Chen, H., Zhang, S. M., Hernan, M. A., Schwarzschild, M. A., Willett, W. C., Colditz, G. A., et al. (2003). Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch. Neurol. 60, 1059–1064. doi: 10.1001/archneur.60.8.1059
Chinta, S. J., Woods, G., Rane, A., Demaria, M., Campisi, J., and Andersen, J. K. (2014). Cellular senescence and the aging brain. Exp. Gerontol. doi: 10.1016/j.exger.2014.09.018. [Epub ahead of print].
Chiu, I. M., Phatnani, H., Kuligowski, M., Tapia, J. C., Carrasco, M. A., Zhang, M., et al. (2009). Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 106, 20960–20965. doi: 10.1073/pnas.0911405106
Codolo, G., Plotegher, N., Pozzobon, T., Brucale, M., Tessari, I., Bubacco, L., et al. (2013). Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 8:e55375. doi: 10.1371/journal.pone.0055375
Cohen, S., Janicki-Deverts, D., Doyle, W. J., Miller, G. E., Frank, E., Rabin, B. S., et al. (2012). Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc. Natl. Acad. Sci. U.S.A. 109, 5995–5999. doi: 10.1073/pnas.1118355109
Cohen, S., Tyrrell, D. A., and Smith, A. P. (1991). Psychological stress and susceptibility to the common cold. N. Engl. J. Med. 325, 606–612. doi: 10.1056/NEJM199108293250903
Colman, R. J., Beasley, T. M., Kemnitz, J. W., Johnson, S. C., Weindruch, R., and Anderson, R. M. (2014). Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 5:3557. doi: 10.1038/ncomms4557
Colonna, M. (2003). TREMs in the immune system and beyond. Nat. Rev. Immunol. 3, 445–453. doi: 10.1038/nri1106
Conde Jr., and Streit, W. J. (2006). Microglia in the aging brain. J. Neuropathol. Exp. Neurol. 65, 199–203. doi: 10.1097/01.jnen.0000202887.22082.63
Cote, S., Carmichael, P. H., Verreault, R., Lindsay, J., Lefebvre, J., and Laurin, D. (2012). Nonsteroidal anti-inflammatory drug use and the risk of cognitive impairment and Alzheimer's disease. Alzheimers Dement. 8, 219–226. doi: 10.1016/j.jalz.2011.03.012
Cotman, C. W., Berchtold, N. C., and Christie, L. A. (2007). Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci. 30, 464–472. doi: 10.1016/j.tins.2007.06.011
Cutuli, D., De Bartolo, P., Caporali, P., Laricchiuta, D., Foti, F., Ronci, M., et al. (2014). n-3 polyunsaturated fatty acids supplementation enhances hippocampal functionality in aged mice. Front. Aging Neurosci. 6:220. doi: 10.3389/fnagi.2014.00220
Damani, M. R., Zhao, L., Fontainhas, A. M., Amaral, J., Fariss, R. N., and Wong, W. T. (2011). Age-related alterations in the dynamic behavior of microglia. Aging Cell 10, 263–276. doi: 10.1111/j.1474-9726.2010.00660.x
Deeks, S. G., and Phillips, A. N. (2009). HIV infection, antiretroviral treatment, ageing, and non-AIDS related morbidity. BMJ 338:a3172. doi: 10.1136/bmj.a3172
Deleidi, M., and Gasser, T. (2013). The role of inflammation in sporadic and familial Parkinson's disease. Cell Mol. Life Sci. 70, 4259–4273. doi: 10.1007/s00018-013-1352-y
Deleidi, M., and Isacson, O. (2012). Viral and inflammatory triggers of neurodegenerative diseases. Sci. Trans. Med. 4:121ps123. doi: 10.1126/scitranslmed.3003492
Deleidi, M., Hallett, P. J., Koprich, J. B., Chung, C. Y., and Isacson, O. (2010). The Toll-like receptor-3 agonist polyinosinic:polycytidylic acid triggers nigrostriatal dopaminergic degeneration. J. Neurosci. 30, 16091–16101. doi: 10.1523/JNEUROSCI.2400-10.2010
Derhovanessian, E., Maier, A. B., Beck, R., Jahn, G., Hahnel, K., Slagboom, P. E., et al. (2010). Hallmark features of immunosenescence are absent in familial longevity. J. Immunol. 185, 4618–4624. doi: 10.4049/jimmunol.1001629
Derhovanessian, E., Maier, A. B., Hahnel, K., Beck, R., de Craen, A. J., Slagboom, E. P., et al. (2011). Infection with cytomegalovirus but not herpes simplex virus induces the accumulation of late-differentiated CD4+ and CD8+ T-cells in humans. J. Gen. Virol. 92, 2746–2756. doi: 10.1099/vir.0.036004-0
Dobbs, R. J., Charlett, A., Purkiss, A. G., Dobbs, S. M., Weller, C., and Peterson, D. W. (1999). Association of circulating TNF-alpha and IL-6 with ageing and parkinsonism. Acta Neurol. Scand. 100, 34–41. doi: 10.1111/j.1600-0404.1999.tb00721.x
Doorduin, J., de Vries, E. F., Willemsen, A. T., de Groot, J. C., Dierckx, R. A., and Klein, H. C. (2009). Neuroinflammation in schizophrenia-related psychosis: a PET study. J. Nucl. Med. 50, 1801–1807. doi: 10.2967/jnumed.109.066647
Dowlati, Y., Herrmann, N., Swardfager, W., Liu, H., Sham, L., Reim, E. K., et al. (2010). A meta-analysis of cytokines in major depression. Biol. Psychiatry 67, 446–457. doi: 10.1016/j.biopsych.2009.09.033
Dupuis, L., Corcia, P., Fergani, A., Gonzalez De Aguilar, J. L., Bonnefont-Rousselot, D., Bittar, R., et al. (2008). Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 70, 1004–1009. doi: 10.1212/01.wnl.0000285080.70324.27
Dupuis, L., Pradat, P. F., Ludolph, A. C., and Loeffler, J. P. (2011). Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 10, 75–82. doi: 10.1016/S1474-4422(10)70224-6
Dykstra, B., Olthof, S., Schreuder, J., Ritsema, M., and de Haan, G. (2011). Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 208, 2691–2703. doi: 10.1084/jem.20111490
Eaton, W. W., Byrne, M., Ewald, H., Mors, O., Chen, C. Y., Agerbo, E., et al. (2006). Association of schizophrenia and autoimmune diseases: linkage of Danish national registers. Am. J. Psychiatry 163, 521–528. doi: 10.1176/appi.ajp.163.3.521
Elias, M. F., Elias, P. K., Sullivan, L. M., Wolf, P. A., and D'Agostino, R. B. (2005). Obesity, diabetes and cognitive deficit: the framingham heart study. Neurobiol. Aging 26(Suppl. 1), 11–16. doi: 10.1016/j.neurobiolaging.2005.08.019
Engelhardt, J. I., Tajti, J., and Appel, S. H. (1993). Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch. Neurol. 50, 30–36. doi: 10.1001/archneur.1993.00540010026013
Engelhart, M. J., Geerlings, M. I., Meijer, J., Kiliaan, A., Ruitenberg, A., van Swieten, J. C., et al. (2004). Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Arch. Neurol. 61, 668–672. doi: 10.1001/archneur.61.5.668
Epel, E. S., Blackburn, E. H., Lin, J., Dhabhar, F. S., Adler, N. E., Morrow, J. D., et al. (2004). Accelerated telomere shortening in response to life stress. Proc. Natl. Acad. Sci. U.S.A. 101, 17312–17315. doi: 10.1073/pnas.0407162101
Erion Jr., Wosiski-Kuhn, M., Dey, A., Hao, S., Davis, C. L., Pollock, N. K., et al. (2014). Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J. Neurosci. 34, 2618–2631. doi: 10.1523/JNEUROSCI.4200-13.2014
Fellner, L., Irschick, R., Schanda, K., Reindl, M., Klimaschewski, L., Poewe, W., et al. (2013). Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 61, 349–360. doi: 10.1002/glia.22437
Felten, D. L., Felten, S. Y., Bellinger, D. L., Carlson, S. L., Ackerman, K. D., Madden, K. S., et al. (1987). Noradrenergic sympathetic neural interactions with the immune system: structure and function. Immunol. Rev. 100, 225–260. doi: 10.1111/j.1600-065X.1987.tb00534.x
Ferretti, M. T., Bruno, M. A., Ducatenzeiler, A., Klein, W. L., and Cuello, A. C. (2012). Intracellular Abeta-oligomers and early inflammation in a model of Alzheimer's disease. Neurobiol. Aging 33, 1329–1342. doi: 10.1016/j.neurobiolaging.2011.01.007
Festa, A., D'Agostino, R. Jr., Williams, K., Karter, A. J., Mayer-Davis, E. J., Tracy, R. P., et al. (2001). The relation of body fat mass and distribution to markers of chronic inflammation. Int. J. Obes. Relat. Metab. Disord. 25, 1407–1415. doi: 10.1038/sj.ijo.0801792
Fiala, M., Lin, J., Ringman, J., Kermani-Arab, V., Tsao, G., Patel, A., et al. (2005). Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer's disease patients. J. Alzheimers Dis. 7, 221–232; discussion:255–262.
Flanary, B. E., Sammons, N. W., Nguyen, C., Walker, D., and Streit, W. J. (2007). Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res. 10, 61–74. doi: 10.1089/rej.2006.9096
Fontana, L., and Partridge, L. (2015). Promoting health and longevity through diet: from model organisms to humans. Cell 161, 106–118. doi: 10.1016/j.cell.2015.02.020
Franceschi, C., Bonafe, M., Valensin, S., Olivieri, F., De Luca, M., Ottaviani, E., et al. (2000). Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N.Y. Acad. Sci. 908, 244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x
Franceschi, C., Capri, M., Monti, D., Giunta, S., Olivieri, F., Sevini, F., et al. (2007). Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 128, 92–105. doi: 10.1016/j.mad.2006.11.016
Frasca, D., Diaz, A., Romero, M., Landin, A. M., and Blomberg, B. B. (2011). Age effects on B cells and humoral immunity in humans. Ageing Res. Rev. 10, 330–335. doi: 10.1016/j.arr.2010.08.004
Freund, A., Orjalo, A. V., Desprez, P. Y., and Campisi, J. (2010). Inflammatory networks during cellular senescence: causes and consequences. Trends Mol. Med. 16, 238–246. doi: 10.1016/j.molmed.2010.03.003
Friedman, J. M., and Halaas, J. L. (1998). Leptin and the regulation of body weight in mammals. Nature 395, 763–770. doi: 10.1038/27376
Fulop, T., Larbi, A., and Pawelec, G. (2013). Human T cell aging and the impact of persistent viral infections. Front. Immunol. 4:271. doi: 10.3389/fimmu.2013.00271
Fulop, T., Larbi, A., Douziech, N., Fortin, C., Guerard, K. P., Lesur, O., et al. (2004). Signal transduction and functional changes in neutrophils with aging. Aging cell 3, 217–226. doi: 10.1111/j.1474-9728.2004.00110.x
Gao, X., Chen, H., Schwarzschild, M. A., and Ascherio, A. (2011). Use of ibuprofen and risk of Parkinson disease. Neurology 76, 863–869. doi: 10.1212/WNL.0b013e31820f2d79
Gemechu, J. M., and Bentivoglio, M. (2012). T Cell Recruitment in the Brain during Normal Aging. Front. Cell. Neurosci. 6:38. doi: 10.3389/fncel.2012.00038
Gerard, H. C., Wildt, K. L., Whittum-Hudson, J. A., Lai, Z., Ager, J., and Hudson, A. P. (2005). The load of Chlamydia pneumoniae in the Alzheimer's brain varies with APOE genotype. Microb. Pathog. 39, 19–26. doi: 10.1016/j.micpath.2005.05.002
Ghosh, S., Lertwattanarak, R., Lefort, N., Molina-Carrion, M., Joya-Galeana, J., Bowen, B. P., et al. (2011). Reduction in reactive oxygen species production by mitochondria from elderly subjects with normal and impaired glucose tolerance. Diabetes 60, 2051–2060. doi: 10.2337/db11-0121
Glaser, R., and Kiecolt-Glaser, J. K. (2005). Stress-induced immune dysfunction: implications for health. Nat. Rev. Immunol. 5, 243–251. doi: 10.1038/nri1571
Goronzy, J. J., Li, G., Yu, M., and Weyand, C. M. (2012). Signaling pathways in aged T cells - a reflection of T cell differentiation, cell senescence and host environment. Semin. Immunol. 24, 365–372. doi: 10.1016/j.smim.2012.04.003
Green, D. R., Galluzzi, L., and Kroemer, G. (2011). Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333, 1109–1112. doi: 10.1126/science.1201940
Griffioen, K. J., Rothman, S. M., Ladenheim, B., Wan, R., Vranis, N., Hutchison, E., et al. (2013). Dietary energy intake modifies brainstem autonomic dysfunction caused by mutant alpha-synuclein. Neurobiol. Aging 34, 928–935. doi: 10.1016/j.neurobiolaging.2012.07.008
Grozdanov, V., Bliederhaeuser, C., Ruf, W. P., Roth, V., Fundel-Clemens, K., Zondler, L., et al. (2014). Inflammatory dysregulation of blood monocytes in Parkinson's disease patients. Acta Neuropathol. 128, 651–663. doi: 10.1007/s00401-014-1345-4
Gruener, N. H., Lechner, F., Jung, M. C., Diepolder, H., Gerlach, T., Lauer, G., et al. (2001). Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J. Virol. 75, 5550–5558. doi: 10.1128/JVI.75.12.5550-5558.2001
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Gustafson, D. R., Karlsson, C., Skoog, I., Rosengren, L., Lissner, L., and Blennow, K. (2007). Mid-life adiposity factors relate to blood-brain barrier integrity in late life. J. Int. Med. 262, 643–650. doi: 10.1111/j.1365-2796.2007.01869.x
Hagberg, H., Mallard, C., Ferriero, D. M., Vannucci, S. J., Levison, S. W., Vexler, Z. S., et al. (2015). The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 11, 192–208. doi: 10.1038/nrneurol.2015.13
Hamza, T. H., Zabetian, C. P., Tenesa, A., Laederach, A., Montimurro, J., Yearout, D., et al. (2010). Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat. Genet. 42, 781–785. doi: 10.1038/ng.642
Handschin, C., and Spiegelman, B. M. (2008). The role of exercise and PGC1alpha in inflammation and chronic disease. Nature 454, 463–469. doi: 10.1038/nature07206
Hannestad, J., Gallezot, J. D., Schafbauer, T., Lim, K., Kloczynski, T., Morris, E. D., et al. (2012). Endotoxin-induced systemic inflammation activates microglia: [(1)(1)C]PBR28 positron emission tomography in nonhuman primates. Neuroimage 63, 232–239. doi: 10.1016/j.neuroimage.2012.06.055
Harrison, N. A., Brydon, L., Walker, C., Gray, M. A., Steptoe, A., Dolan, R. J., et al. (2009). Neural origins of human sickness in interoceptive responses to inflammation. Biol. Psychiatry 66, 415–422. doi: 10.1016/j.biopsych.2009.03.007
Hart, A. D., Wyttenbach, A., Perry, V. H., and Teeling, J. L. (2012). Age related changes in microglial phenotype vary between CNS regions: grey versus white matter differences. Brain Behav. Immun. 26, 754–765. doi: 10.1016/j.bbi.2011.11.006
Hassing, L. B., Dahl, A. K., Pedersen, N. L., and Johansson, B. (2010). Overweight in midlife is related to lower cognitive function 30 years later: a prospective study with longitudinal assessments. Dement. Geriatr. Cogn. Disord. 29, 543–552. doi: 10.1159/000314874
Hearps, A. C., Martin, G. E., Angelovich, T. A., Cheng, W. J., Maisa, A., Landay, A. L., et al. (2012). Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell 11, 867–875. doi: 10.1111/j.1474-9726.2012.00851.x
Hedstrom, A. K., Lima Bomfim, I., Barcellos, L., Gianfrancesco, M., Schaefer, C., Kockum, I., et al. (2014). Interaction between adolescent obesity and HLA risk genes in the etiology of multiple sclerosis. Neurology 82, 865–872. doi: 10.1212/WNL.0000000000000203
Hefendehl, J. K., Neher, J. J., Suhs, R. B., Kohsaka, S., Skodras, A., and Jucker, M. (2014). Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 13, 60–69. doi: 10.1111/acel.12149
Henkel, J. S., Engelhardt, J. I., Siklos, L., Simpson, E. P., Kim, S. H., Pan, T., et al. (2004). Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann. Neurol. 55, 221–235. doi: 10.1002/ana.10805
High, K. P., Brennan-Ing, M., Clifford, D. B., Cohen, M. H., Currier, J., Deeks, S. G., et al. OARWGo Aging (2012). HIV and aging: state of knowledge and areas of critical need for research. A report to the NIH Office of AIDS Research by the HIV and Aging Working Group. J. Acquir. Immune Defic. Syndr. 60(Suppl. 1), S1–S18. doi: 10.1097/QAI.0b013e31825a3668
Hong, S., and Banks, W. A. (2015). Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain Behav. Immun. 45, 1–12. doi: 10.1016/j.bbi.2014.10.008
Hotamisligil, G. S. (2006). Inflammation and metabolic disorders. Nature 444, 860–867. doi: 10.1038/nature05485
Hottman, D. A., Chernick, D., Cheng, S., Wang, Z., and Li, L. (2014). HDL and cognition in neurodegenerative disorders. Neurobiol Dis 72(Pt A), 22–36. doi: 10.1016/j.nbd.2014.07.015
Hu, G., Antikainen, R., Jousilahti, P., Kivipelto, M., and Tuomilehto, J. (2008). Total cholesterol and the risk of Parkinson disease. Neurology 70, 1972–1979. doi: 10.1212/01.wnl.0000312511.62699.a8
Hu, G., Jousilahti, P., Nissinen, A., Antikainen, R., Kivipelto, M., and Tuomilehto, J. (2006). Body mass index and the risk of Parkinson disease. Neurology 67, 1955–1959. doi: 10.1212/01.wnl.0000247052.18422.e5
in t' Veld, B. A., Ruitenberg, A., Hofman, A., Launer, L. J., van Duijn, C. M., Stijnen, T., et al. (2001). Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N. Engl. J. Med. 345, 1515–1521. doi: 10.1056/NEJMoa010178
International Schizophrenia, C., Purcell, S. M., Wray, N. R., Stone, J. L., Visscher, P. M., O'Donovan, M. C., et al. (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752. doi: 10.1038/nature08185
Irish Schizophrenia Genomics C the Wellcome Trust Case Control C. (2012). Genome-wide association study implicates HLA-C*01:02 as a risk factor at the major histocompatibility complex locus in schizophrenia. Biol. Psychiatry 72, 620–628. doi: 10.1016/j.biopsych.2012.05.035
Itzhaki, R. F., and Wozniak, M. A. (2006). Herpes simplex virus type 1, apolipoprotein E, and cholesterol: a dangerous liaison in Alzheimer's disease and other disorders. Prog. Lipid Res. 45, 73–90. doi: 10.1016/j.plipres.2005.11.003
Jang, H., Boltz, D. A., Webster, R. G., and Smeyne, R. J. (2009). Viral parkinsonism. Biochim. Biophys. Acta 1792, 714–721. doi: 10.1016/j.bbadis.2008.08.001
Jeong, S. K., Nam, H. S., Son, M. H., Son, E. J., and Cho, K. H. (2005). Interactive effect of obesity indexes on cognition. Dement. Geriatr. Cogn. Disord. 19, 91–96. doi: 10.1159/000082659
Joachim, C. L., and Selkoe, D. J. (1992). The seminal role of beta-amyloid in the pathogenesis of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 6, 7–34. doi: 10.1097/00002093-199205000-00003
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Jun, J. I., and Lau, L. F. (2010). The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 12, 676–685. doi: 10.1038/ncb2070
Jyonouchi, H., Geng, L., Streck, D. L., and Toruner, G. A. (2012). Immunological characterization and transcription profiling of peripheral blood (PB) monocytes in children with autism spectrum disorders (ASD) and specific polysaccharide antibody deficiency (SPAD): case study. J. Neuroinflammation 9:4. doi: 10.1186/1742-2094-9-4
Karabatsiakis, A., Kolassa, I. T., Kolassa, S., Rudolph, K. L., and Dietrich, D. E. (2014). Telomere shortening in leukocyte subpopulations in depression. BMC psychiatry 14:192. doi: 10.1186/1471-244X-14-192
Kayser, M. S., and Dalmau, J. (2011). The emerging link between autoimmune disorders and neuropsychiatric disease. J. Neuropsychiatry Clin. Neurosci. 23, 90–97. doi: 10.1176/appi.neuropsych.23.1.90
Kepp, O., Galluzzi, L., and Kroemer, G. (2011). Mitochondrial control of the NLRP3 inflammasome. Nat. Immunol. 12, 199–200. doi: 10.1038/ni0311-199
Kettenmann, H., Hanisch, U. K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Koch, S., Larbi, A., Ozcelik, D., Solana, R., Gouttefangeas, C., Attig, S., et al. (2007). Cytomegalovirus infection: a driving force in human T cell immunosenescence. Ann. N.Y. Acad. Sci. 1114, 23–35. doi: 10.1196/annals.1396.043
Kota, L. N., Purushottam, M., Moily, N. S., and Jain, S. (2014). Shortened telomere in unremitted schizophrenia. Psychiatry Clin. Neurosci. 69, 292–297. doi: 10.1111/pcn.12260
Kotrschal, A., Ilmonen, P., and Penn, D. J. (2007). Stress impacts telomere dynamics. Biol. Lett. 3, 128–130. doi: 10.1098/rsbl.2006.0594
Krysko, D. V., Agostinis, P., Krysko, O., Garg, A. D., Bachert, C., Lambrecht, B. N., et al. (2011). Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 32, 157–164. doi: 10.1016/j.it.2011.01.005
Kuk, J. L., Saunders, T. J., Davidson, L. E., and Ross, R. (2009). Age-related changes in total and regional fat distribution. Ageing Res. Rev. 8, 339–348. doi: 10.1016/j.arr.2009.06.001
Landreth, G. E., and Reed-Geaghan, E. G. (2009). Toll-like receptors in Alzheimer's disease. Curr. Top. Microbiol. Immunol. 336, 137–153. doi: 10.1007/978-3-642-00549-7_8
Larbi, A., Pawelec, G., Wong, S. C., Goldeck, D., Tai, J. J., and Fulop, T. (2011). Impact of age on T cell signaling: a general defect or specific alterations? Ageing Res. Rev. 10, 370–378.
Le Garff-Tavernier, M., Beziat, V., Decocq, J., Siguret, V., Gandjbakhch, F., Pautas, E., et al. (2010). Human NK cells display major phenotypic and functional changes over the life span. Aging Cell 9, 527–535. doi: 10.1111/j.1474-9726.2010.00584.x
Lederman, M. M., Funderburg, N. T., Sekaly, R. P., Klatt, N. R., and Hunt, P. W. (2013). Residual immune dysregulation syndrome in treated HIV infection. Adv. Immunol. 119, 51–83. doi: 10.1016/B978-0-12-407707-2.00002-3
Li, X., Chauhan, A., Sheikh, A. M., Patil, S., Chauhan, V., Li, X. M., et al. (2009). Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 207, 111–116. doi: 10.1016/j.jneuroim.2008.12.002
Liao, B., Zhao, W., Beers, D. R., Henkel, J. S., and Appel, S. H. (2012). Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 237, 147–152. doi: 10.1016/j.expneurol.2012.06.011
Loftis, J. M., Huckans, M., Ruimy, S., Hinrichs, D. J., and Hauser, P. (2008). Depressive symptoms in patients with chronic hepatitis C are correlated with elevated plasma levels of interleukin-1beta and tumor necrosis factor-alpha. Neurosci. Lett. 430, 264–268. doi: 10.1016/j.neulet.2007.11.001
Lopez-Armada, M. J., Riveiro-Naveira, R. R., Vaamonde-Garcia, C., and Valcarcel-Ares, M. N. (2013). Mitochondrial dysfunction and the inflammatory response. Mitochondrion 13, 106–118. doi: 10.1016/j.mito.2013.01.003
Lord, G. M., Matarese, G., Howard, J. K., Baker, R. J., Bloom, S. R., and Lechler, R. I. (1998). Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 394, 897–901. doi: 10.1038/29795
Lukiw, W. J. (2004). Gene expression profiling in fetal, aged, and Alzheimer hippocampus: a continuum of stress-related signaling. Neurochem. Res. 29, 1287–1297. doi: 10.1023/B:NERE.0000023615.89699.63
Lynch, M. A. (2010). Age-related neuroinflammatory changes negatively impact on neuronal function. Front. Aging Neurosci. 1:6. doi: 10.3389/neuro.24.006.2009
Maggio, M., Basaria, S., Ble, A., Lauretani, F., Bandinelli, S., Ceda, G. P., et al. (2006). Correlation between testosterone and the inflammatory marker soluble interleukin-6 receptor in older men. J. Clin. Endocrinol. Metab. 91, 345–347. doi: 10.1210/jc.2005-1097
Margaretten, M., Julian, L., Katz, P., and Yelin, E. (2011). Depression in patients with rheumatoid arthritis: description, causes and mechanisms. Int. J. Clin. Rheumtol. 6, 617–623. doi: 10.2217/ijr.11.62
Maswood, N., Young, J., Tilmont, E., Zhang, Z., Gash, D. M., Gerhardt, G. A., et al. (2004). Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 101, 18171–18176. doi: 10.1073/pnas.0405831102
Mathew, A., Lindsley, T. A., Sheridan, A., Bhoiwala, D. L., Hushmendy, S. F., Yager, E. J., et al. (2012). Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. J. Alzheimers Dis. 30, 617–627. doi: 10.3233/JAD-2012-120145
McGeer, P. L., and McGeer, E. G. (2002). Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 26, 459–470. doi: 10.1002/mus.10191
Mekker, A., Tchang, V. S., Haeberli, L., Oxenius, A., Trkola, A., and Karrer, U. (2012). Immune senescence: relative contributions of age and cytomegalovirus infection. PLoS Pathog. 8:e1002850. doi: 10.1371/journal.ppat.1002850
Mercken, E. M., Crosby, S. D., Lamming, D. W., JeBailey, L., Krzysik-Walker, S., Villareal, D. T., et al. (2013). Calorie restriction in humans inhibits the PI3K/AKT pathway and induces a younger transcription profile. Aging Cell 12, 645–651. doi: 10.1111/acel.12088
Meyer-Luehmann, M., Spires-Jones, T. L., Prada, C., Garcia-Alloza, M., de Calignon, A., Rozkalne, A., et al. (2008). Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature 451, 720–724. doi: 10.1038/nature06616
Miller, B. J., Buckley, P., Seabolt, W., Mellor, A., and Kirkpatrick, B. (2011). Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol. Psychiatry 70, 663–671. doi: 10.1016/j.biopsych.2011.04.013
Montecino-Rodriguez, E., Berent-Maoz, B., and Dorshkind, K. (2013). Causes, consequences, and reversal of immune system aging. J. Clin. Invest. 123, 958–965. doi: 10.1172/JCI64096
Moschen, A. R., Molnar, C., Enrich, B., Geiger, S., Ebenbichler, C. F., and Tilg, H. (2011). Adipose and liver expression of interleukin (IL)-1 family members in morbid obesity and effects of weight loss. Mol. Med. 17, 840–845. doi: 10.2119/molmed.2010.00108
Muller, N., Wagner, J. K., Krause, D., Weidinger, E., Wildenauer, A., Obermeier, M., et al. (2012). Impaired monocyte activation in schizophrenia. Psychiatry Res. 198, 341–346. doi: 10.1016/j.psychres.2011.12.049
Newsholme, P., and de Bittencourt, P. I. Jr. (2014). The fat cell senescence hypothesis: a mechanism responsible for abrogating the resolution of inflammation in chronic disease. Curr. Opin. Clin. Nutr. Metab. Care 17, 295–305. doi: 10.1097/MCO.0000000000000077
Nieratschker, V., Lahtinen, J., Meier, S., Strohmaier, J., Frank, J., Heinrich, A., et al. (2013). Longer telomere length in patients with schizophrenia. Schizophr. Res. 149, 116–120. doi: 10.1016/j.schres.2013.06.043
Nikolich-Zugich, J., Li, G., Uhrlaub, J. L., Renkema, K. R., and Smithey, M. J. (2012). Age-related changes in CD8 T cell homeostasis and immunity to infection. Semin. Immunol. 24, 356–364. doi: 10.1016/j.smim.2012.04.009
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Odegaard, J. I., and Chawla, A. (2013). Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science 339, 172–177. doi: 10.1126/science.1230721
Ovadya, Y., and Krizhanovsky, V. (2014). Senescent cells: SASPected drivers of age-related pathologies. Biogerontology 15, 627–642. doi: 10.1007/s10522-014-9529-9
Palacios, N., Gao, X., McCullough, M. L., Jacobs, E. J., Patel, A. V., Mayo, T., et al. (2011). Obesity, diabetes, and risk of Parkinson's disease. Mov. Disord. 26, 2253–2259. doi: 10.1002/mds.23855
Pasinelli, P., and Brown, R. H. (2006). Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 7, 710–723. doi: 10.1038/nrn1971
Patti, M. E., Butte, A. J., Crunkhorn, S., Cusi, K., Berria, R., Kashyap, S., et al. (2003). Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. U.S.A. 100, 8466–8471. doi: 10.1073/pnas.1032913100
Pearce, B. D., Valadi, N. M., Po, C. L., and Miller, A. H. (2000). Viral infection of developing GABAergic neurons in a model of hippocampal disinhibition. Neuroreport 11, 2433–2438. doi: 10.1097/00001756-200008030-00019
Perry, V. H., and Holmes, C. (2014). Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 10, 217–224. doi: 10.1038/nrneurol.2014.38
Perry, V. H., Matyszak, M. K., and Fearn, S. (1993). Altered antigen expression of microglia in the aged rodent CNS. Glia 7, 60–67. doi: 10.1002/glia.440070111
Phillips, C., Baktir, M. A., Srivatsan, M., and Salehi, A. (2014). Neuroprotective effects of physical activity on the brain: a closer look at trophic factor signaling. Front. Cell. Neurosci. 8:170. doi: 10.3389/fncel.2014.00170
Pohl, J., Woodside, B., and Luheshi, G. N. (2014). Leptin modulates the late fever response to LPS in diet-induced obese animals. Brain Behav. Immun. 42, 41–47. doi: 10.1016/j.bbi.2014.07.017
Rao, S. R. (2012). Inflammatory markers and bariatric surgery: a meta-analysis. Inflamm. Res. 61, 789–807. doi: 10.1007/s00011-012-0473-3
Ravenholt, R. T., and Foege, W. H. (1982). 1918 influenza, encephalitis lethargica, parkinsonism. Lancet 2, 860–864. doi: 10.1016/S0140-6736(82)90820-0
Razay, G., Vreugdenhil, A., and Wilcock, G. (2007). The metabolic syndrome and Alzheimer disease. Arch. Neurol. 64, 93–96. doi: 10.1001/archneur.64.1.93
Reed-Geaghan, E. G., Savage, J. C., Hise, A. G., and Landreth, G. E. (2009). CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J. Neurosci. 29, 11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009
Rees, K., Stowe, R., Patel, S., Ives, N., Breen, K., Clarke, C. E., et al. (2011). Non-steroidal anti-inflammatory drugs as disease-modifying agents for Parkinson's disease: evidence from observational studies. Cochrane Database Syst. Rev. CD008454. doi: 10.1002/14651858.CD008454.pub2
Roberts, E. T., Haan, M. N., Dowd, J. B., and Aiello, A. E. (2010). Cytomegalovirus antibody levels, inflammation, and mortality among elderly Latinos over 9 years of follow-up. Am. J. Epidemiol. 172, 363–371. doi: 10.1093/aje/kwq177
Rodriguez, G. A., Tai, L. M., LaDu, M. J., and Rebeck, G. W. (2014). Human APOE4 increases microglia reactivity at Abeta plaques in a mouse model of Abeta deposition. J. Neuroinflammation 11:111. doi: 10.1186/1742-2094-11-111
Rotermund, C., Truckenmuller, F. M., Schell, H., and Kahle, P. J. (2014). Diet-induced obesity accelerates the onset of terminal phenotypes in alpha-synuclein transgenic mice. J. Neurochem. 131, 848–858. doi: 10.1111/jnc.12813
Ryan, A. S., and Nicklas, B. J. (2004). Reductions in plasma cytokine levels with weight loss improve insulin sensitivity in overweight and obese postmenopausal women. Diabetes Care 27, 1699–1705. doi: 10.2337/diacare.27.7.1699
Santiago, J. A., and Potashkin, J. A. (2013). Shared dysregulated pathways lead to Parkinson's disease and diabetes. Trends Mol. Med. 19, 176–186. doi: 10.1016/j.molmed.2013.01.002
Saunders, J. A., Estes, K. A., Kosloski, L. M., Allen, H. E., Dempsey, K. M., Torres-Russotto, D. R., et al. (2012). CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson's disease. J. Neuroimmune Pharmacol. 7, 927–938. doi: 10.1007/s11481-012-9402-z
Schwarcz, R., Bruno, J. P., Muchowski, P. J., and Wu, H. Q. (2012). Kynurenines in the mammalian brain: when physiology meets pathology. Nat. Rev. Neurosci. 13, 465–477. doi: 10.1038/nrn3257
Sczelecki, S., Besse-Patin, A., Abboud, A., Kleiner, S., Laznik-Bogoslavski, D., Wrann, C. D., et al. (2014). Loss of Pgc-1alpha expression in aging mouse muscle potentiates glucose intolerance and systemic inflammation. Am. J. Physiol. Endocrinol. Metab. 306, E157–E167. doi: 10.1152/ajpendo.00578.2013
Sharma, A., Valadi, N., Miller, A. H., and Pearce, B. D. (2002). Neonatal viral infection decreases neuronal progenitors and impairs adult neurogenesis in the hippocampus. Neurobiol. Dis. 11, 246–256. doi: 10.1006/nbdi.2002.0531
Shaw, A. C., Goldstein, D. R., and Montgomery, R. R. (2013). Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 13, 875–887. doi: 10.1038/nri3547
Sheffield, L. G., and Berman, N. E. (1998). Microglial expression of MHC class II increases in normal aging of nonhuman primates. Neurobiol. Aging 19, 47–55. doi: 10.1016/S0197-4580(97)00168-1
Sheng, J. G., Mrak, R. E., and Griffin, W. S. (1998). Enlarged and phagocytic, but not primed, interleukin-1 alpha-immunoreactive microglia increase with age in normal human brain. Acta Neuropathol. 95, 229–234. doi: 10.1007/s004010050792
Shimada, K., Crother, T. R., Karlin, J., Dagvadorj, J., Chiba, N., Chen, S., et al. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414. doi: 10.1016/j.immuni.2012.01.009
Simard, A. R., Soulet, D., Gowing, G., Julien, J. P., and Rivest, S. (2006). Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron 49, 489–502. doi: 10.1016/j.neuron.2006.01.022
Simon, N. M., Smoller, J. W., McNamara, K. L., Maser, R. S., Zalta, A. K., Pollack, M. H., et al. (2006). Telomere shortening and mood disorders: preliminary support for a chronic stress model of accelerated aging. Biol. Psychiatry 60, 432–435. doi: 10.1016/j.biopsych.2006.02.004
Simopoulos, A. P. (2002). The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother. 56, 365–379. doi: 10.1016/S0753-3322(02)00253-6
Singh-Manoux, A., Dugravot, A., Brunner, E., Kumari, M., Shipley, M., Elbaz, A., et al. (2014). Interleukin-6 and C-reactive protein as predictors of cognitive decline in late midlife. Neurology 83, 486–493. doi: 10.1212/WNL.0000000000000665
Skilton, M. R., Moulin, P., Terra, J. L., and Bonnet, F. (2007). Associations between anxiety, depression, and the metabolic syndrome. Biol. Psychiatry 62, 1251–1257. doi: 10.1016/j.biopsych.2007.01.012
Soderlund, J., Schroder, J., Nordin, C., Samuelsson, M., Walther-Jallow, L., Karlsson, H., et al. (2009). Activation of brain interleukin-1beta in schizophrenia. Mol. Psychiatry 14, 1069–1071. doi: 10.1038/mp.2009.52
Stefanova, N., Fellner, L., Reindl, M., Masliah, E., Poewe, W., and Wenning, G. K. (2011). Toll-like receptor 4 promotes alpha-synuclein clearance and survival of nigral dopaminergic neurons. Am. J. Pathol. 179, 954–963. doi: 10.1016/j.ajpath.2011.04.013
Stowe, R. P., Kozlova, E. V., Yetman, D. L., Walling, D. M., Goodwin, J. S., and Glaser, R. (2007). Chronic herpesvirus reactivation occurs in aging. Exp. Gerontol. 42, 563–570. doi: 10.1016/j.exger.2007.01.005
Streit, W. J., Braak, H., Xue, Q. S., and Bechmann, I. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 118, 475–485. doi: 10.1007/s00401-009-0556-6
Streit, W. J., Sammons, N. W., Kuhns, A. J., and Sparks, D. L. (2004). Dystrophic microglia in the aging human brain. Glia 45, 208–212. doi: 10.1002/glia.10319
Strohmeyer, R., Ramirez, M., Cole, G. J., Mueller, K., and Rogers, J. (2002). Association of factor H of the alternative pathway of complement with agrin and complement receptor 3 in the Alzheimer's disease brain. J. Neuroimmunol. 131, 135–146. doi: 10.1016/S0165-5728(02)00272-2
Sundelof, J., Kilander, L., Helmersson, J., Larsson, A., Ronnemaa, E., Degerman-Gunnarsson, M., et al. (2009). Systemic inflammation and the risk of Alzheimer's disease and dementia: a prospective population-based study. J. Alzheimers Dis. 18, 79–87. doi: 10.3233/JAD-2009-1126
Sutedja, N. A., van der Schouw, Y. T., Fischer, K., Sizoo, E. M., Huisman, M. H., Veldink, J. H., et al. (2011). Beneficial vascular risk profile is associated with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatr. 82, 638–642. doi: 10.1136/jnnp.2010.236752
Sutherland, G. T., Chami, B., Youssef, P., and Witting, P. K. (2013). Oxidative stress in Alzheimer's disease: Primary villain or physiological by-product? Redox Rep. 18, 134–141. doi: 10.1179/1351000213Y.0000000052
Suzuki, Y., Wang, X., Jortner, B. S., Payne, L., Ni, Y., Michie, S. A., et al. (2010). Removal of Toxoplasma gondii cysts from the brain by perforin-mediated activity of CD8+ T cells. Am. J. Pathol. 176, 1607–1613. doi: 10.2353/ajpath.2010.090825
Szekely, C. A., Breitner, J. C., Fitzpatrick, A. L., Rea, T. D., Psaty, B. M., Kuller, L. H., et al. (2008). NSAID use and dementia risk in the Cardiovascular Health Study: role of APOE and NSAID type. Neurology 70, 17–24. doi: 10.1212/01.wnl.0000284596.95156.48
Szweda, P. A., Camouse, M., Lundberg, K. C., Oberley, T. D., and Szweda, L. I. (2003). Aging, lipofuscin formation, and free radical-mediated inhibition of cellular proteolytic systems. Ageing Res. Rev. 2, 383–405. doi: 10.1016/S1568-1637(03)00028-X
Takahashi, K., Rochford, C. D., and Neumann, H. (2005). Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647–657. doi: 10.1084/jem.20041611
Tan, Z. S., Beiser, A. S., Vasan, R. S., Roubenoff, R., Dinarello, C. A., Harris, T. B., et al. (2007). Inflammatory markers and the risk of Alzheimer disease: the Framingham Study. N/eurology 68, 1902–1908. doi: 10.1212/01.wnl.0000263217.36439.da
Tchernof, A., and Despres, J. P. (2013). Pathophysiology of human visceral obesity: an update. Physiol. Rev. 93, 359–404. doi: 10.1152/physrev.00033.2011
Tchkonia, T., Zhu, Y., van Deursen, J., Campisi, J., and Kirkland, J. L. (2013). Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest. 123, 966–972. doi: 10.1172/JCI64098
Thewissen, M., Linsen, L., Somers, V., Geusens, P., Raus, J., and Stinissen, P. (2005). Premature immunosenescence in rheumatoid arthritis and multiple sclerosis patients. Ann. N. Y. Acad. Sci. 1051, 255–262. doi: 10.1196/annals.1361.066
Thewissen, M., Somers, V., Venken, K., Linsen, L., van Paassen, P., Geusens, P., et al. (2007). Analyses of immunosenescent markers in patients with autoimmune disease. Clin. Immunol. 123, 209–218. doi: 10.1016/j.clim.2007.01.005
Torrey, E. F., Bartko, J. J., and Yolken, R. H. (2012). Toxoplasma gondii and other risk factors for schizophrenia: an update. Schizophr. Bull. 38, 642–647. doi: 10.1093/schbul/sbs043
Trayhurn, P., and Wood, I. S. (2004). Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br. J. Nutr. 92, 347–355. doi: 10.1079/BJN20041213
Troost, D., van den Oord, J. J., de Jong, J. M., and Swaab, D. F. (1989). Lymphocytic infiltration in the spinal cord of patients with amyotrophic lateral sclerosis. Clin. Neuropathol. 8, 289–294.
Turnbull, I. R., Gilfillan, S., Cella, M., Aoshi, T., Miller, M., Piccio, L., et al. (2006). Cutting edge: TREM-2 attenuates macrophage activation. J. Immunol. 177, 3520–3524. doi: 10.4049/jimmunol.177.6.3520
Turner, M. R., Cagnin, A., Turkheimer, F. E., Miller, C. C., Shaw, C. E., Brooks, D. J., et al. (2004). Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 15, 601–609. doi: 10.1016/j.nbd.2003.12.012
Tyring, S., Gottlieb, A., Papp, K., Gordon, K., Leonardi, C., Wang, A., et al. (2006). Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet 367, 29–35. doi: 10.1016/S0140-6736(05)67763-X
Vargas, D. L., Nascimbene, C., Krishnan, C., Zimmerman, A. W., and Pardo, C. A. (2005). Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 57, 67–81. doi: 10.1002/ana.20315
von Bernhardi, R., Tichauer, J. E., and Eugenin, J. (2010). Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J. Neurochem. 112, 1099–1114. doi: 10.1111/j.1471-4159.2009.06537.x
Wahner, A. D., Bronstein, J. M., Bordelon, Y. M., and Ritz, B. (2007). Nonsteroidal anti-inflammatory drugs may protect against Parkinson disease. Neurology 69, 1836–1842. doi: 10.1212/01.wnl.0000279519.99344.ad
Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S., and Nabekura, J. (2009). Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 29, 3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009
Walker, F. R. (2013). A critical review of the mechanism of action for the selective serotonin reuptake inhibitors: do these drugs possess anti-inflammatory properties and how relevant is this in the treatment of depression? Neuropharmacology 67, 304–317. doi: 10.1016/j.neuropharm.2012.10.002
Wang, C., Maddick, M., Miwa, S., Jurk, D., Czapiewski, R., Saretzki, G., et al. (2010). Adult-onset, short-term dietary restriction reduces cell senescence in mice. Aging 2, 555–566.
Watkins, C. C., and Treisman, G. J. (2012). Neuropsychiatric complications of aging with HIV. J. Neurovirol. 18, 277–290. doi: 10.1007/s13365-012-0108-z
Weissenborn, K., Ennen, J. C., Bokemeyer, M., Ahl, B., Wurster, U., Tillmann, H., et al. (2006). Monoaminergic neurotransmission is altered in hepatitis C virus infected patients with chronic fatigue and cognitive impairment. Gut 55, 1624–1630. doi: 10.1136/gut.2005.080267
Wenisch, C., Patruta, S., Daxbock, F., Krause, R., and Horl, W. (2000). Effect of age on human neutrophil function. J. Leukoc. Biol. 67, 40–45.
Wilson, C. J., Finch, C. E., and Cohen, H. J. (2002). Cytokines and cognition–the case for a head-to-toe inflammatory paradigm. J. Am. Geriatr. Soc. 50, 2041–2056. doi: 10.1046/j.1532-5415.2002.50619.x
Witte, A. V., Fobker, M., Gellner, R., Knecht, S., and Floel, A. (2009). Caloric restriction improves memory in elderly humans. Proc. Natl. Acad. Sci. U.S.A. 106, 1255–1260. doi: 10.1073/pnas.0808587106
Wong, W. T. (2013). Microglial aging in the healthy CNS: phenotypes, drivers, and rejuvenation. Front. Cell. Neurosci. 7:22. doi: 10.3389/fncel.2013.00022
Wright, A. L., Zinn, R., Hohensinn, B., Konen, L. M., Beynon, S. B., Tan, R. P., et al. (2013). Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP-J20 mouse model of Alzheimer's disease. PLoS ONE 8:e59586. doi: 10.1371/journal.pone.0059586
Wrona, D. (2006). Neural-immune interactions: an integrative view of the bidirectional relationship between the brain and immune systems. J. Neuroimmunol. 172, 38–58. doi: 10.1016/j.jneuroim.2005.10.017
Wyss-Coray, T. (2006). Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat. Med. 12, 1005–1015. doi: 10.1038/nm1484
Xu, H., Chen, M., Manivannan, A., Lois, N., and Forrester, J. V. (2008). Age-dependent accumulation of lipofuscin in perivascular and subretinal microglia in experimental mice. Aging cell 7, 58–68. doi: 10.1111/j.1474-9726.2007.00351.x
Xu, Q., Park, Y., Huang, X., Hollenbeck, A., Blair, A., Schatzkin, A., et al. (2011). Diabetes and risk of Parkinson's disease. Diabetes care 34, 910–915. doi: 10.2337/dc10-1922
Xue, W., Zender, L., Miething, C., Dickins, R. A., Hernando, E., Krizhanovsky, V., et al. (2007). Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660. doi: 10.1038/nature05529
Ye, S. M., and Johnson, R. W. (2001). An age-related decline in interleukin-10 may contribute to the increased expression of interleukin-6 in brain of aged mice. Neuroimmunomodulation 9, 183–192. doi: 10.1159/000049025
Yolken, R. H., and Torrey, E. F. (2008). Are some cases of psychosis caused by microbial agents? A review of the evidence. Mol. Psychiatry 13, 470–479. doi: 10.1038/mp.2008.5
Youm, Y. H., Grant, R. W., McCabe, L. R., Albarado, D. C., Nguyen, K. Y., Ravussin, A., et al. (2013). Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 18, 519–532. doi: 10.1016/j.cmet.2013.09.010
Youm, Y. H., Nguyen, K. Y., Grant, R. W., Goldberg, E. L., Bodogai, M., Kim, D., et al. (2015). The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 21, 263–269. doi: 10.1038/nm.3804
Yudkin, J. S., Stehouwer, C. D., Emeis, J. J., and Coppack, S. W. (1999). C-reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arterioscler. Thromb. Vasc. Biol. 19, 972–978.
Zandi, P. P., Anthony, J. C., Hayden, K. M., Mehta, K., Mayer, L., Breitner, J. C., et al. (2002). Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology 59, 880–886. doi: 10.1212/WNL.59.6.880
Zapata, H. J., and Shaw, A. C. (2014). Aging of the human innate immune system in HIV infection. Curr. Opin. Immunol. 29, 127–136. doi: 10.1016/j.coi.2014.06.007
Keywords: brain-immune interactions, aging, immunosenescence, inflammation, metabolism, neurodegenerative diseases, neuropsychiatric diseases
Citation: Deleidi M, Jäggle M and Rubino G (2015) Immune aging, dysmetabolism, and inflammation in neurological diseases. Front. Neurosci. 9:172. doi: 10.3389/fnins.2015.00172
Received: 28 February 2015; Accepted: 28 April 2015;
Published: 03 June 2015.
Edited by:
Tommaso Cassano, University of Foggia, ItalyReviewed by:
Bradley D. Pearce, Emory University, USACopyright © 2015 Deleidi, Jäggle and Rubino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michela Deleidi, German Center for Neurodegenerative Diseases (DZNE), Hertie Institute for Clinical Brain Research, University of Tübingen, Otfried-Müller. Str 27, 72076 Tübingen, Germany,bWljaGVsYS5kZWxlaWRpQGR6bmUuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.