 Thanuja Gali Ramamoorthy
Thanuja Gali Ramamoorthy Ghazala Begum
Ghazala Begum Erika Harno
Erika Harno Anne White
Anne White- 1Faculty of Life Sciences, University of Manchester, Manchester, UK
- 2School of Clinical and Experimental Medicine, University of Birmingham, Birmingham, UK
- 3Faculty of Medical and Human Sciences, Centre for Endocrinology and Diabetes, University of Manchester, Manchester, UK
The prevalence of obesity in adults and children has increased globally at an alarming rate. Mounting evidence from both epidemiological studies and animal models indicates that adult obesity and associated metabolic disorders can be programmed by intrauterine and early postnatal environment- a phenomenon known as “fetal programming of adult disease.” Data from nutritional intervention studies in animals including maternal under- and over-nutrition support the developmental origins of obesity and metabolic syndrome. The hypothalamic neuronal circuits located in the arcuate nucleus controlling appetite and energy expenditure are set early in life and are perturbed by maternal nutritional insults. In this review, we focus on the effects of maternal nutrition in programming permanent changes in these hypothalamic circuits, with experimental evidence from animal models of maternal under- and over-nutrition. We discuss the epigenetic modifications which regulate hypothalamic gene expression as potential molecular mechanisms linking maternal diet during pregnancy to the offspring's risk of obesity at a later age. Understanding these mechanisms in key metabolic genes may provide insights into the development of preventative intervention strategies.
Introduction
The number of reported cases of obesity has risen dramatically in the developed world and is one of the greatest public health challenges we face due to the projected burden on future health care services. Current estimates suggest that more than a third of adults in the United States are obese and according to the Department of Health in the UK, more than half of the adult population is either overweight or obese (Department of Health, 2014). Obesity is also a risk factor for a number of other associated metabolic disorders such as type 2 diabetes, cardiovascular disease, hypertension, stroke, and osteoarthritis (Abbasi et al., 2002; Steinberger et al., 2003; Qatanani and Lazar, 2007; Chiarelli and Marcovecchio, 2008). It is undoubtedly multifactorial, occurring as a result of both complex genetic traits and environmental factors.
The gene-environment interaction represents a likely factor in the recent demographic shift toward a more obese phenotype. This comes as growing evidence shows that fetal and early postnatal environments influence the risk of developing obesity. Epidemiological and animal studies show that nutritional imbalances such as under- and over-nutrition during critical ontogenic periods induce persistent physiological alterations. These changes are strongly associated with metabolic disorders such as obesity, type 2 diabetes, and cardiovascular disease in adult offspring (Blackmore and Ozanne, 2013; Duque-Guimaraes and Ozanne, 2013; Hanson and Gluckman, 2014).
Programming of metabolic disorders in humans by intrauterine experiences was first explored by Barker et al. (1990). They observed a strong association between low birth weight and the risk of cardiovascular disease and type 2 diabetes in adulthood (Barker et al., 1990, 1993; Hales et al., 1991). Other retrospective analyses have also demonstrated that early life experiences can increase the susceptibility of developing metabolic disorders in adulthood (Ravelli et al., 1976, 1999). These observations led to the fetal programming hypothesis also known as “Developmental origins of health and disease (DOHaD)” or “thrifty phenotype” hypothesis. This theory suggests that the fetus will adapt to adverse changes in the intrauterine environment such as maternal undernutrition. However, increased nutritional availability in postnatal life can result in the development of metabolic syndrome (Hales and Barker, 2001). Emerging evidence confirms that maternal overnutrition during pregnancy and increased birth weight also increase the incidence of obesity in the offspring. Thus, the relationship between human birth weight and the potential of developing adult obesity and other metabolic disorders is a “U-shaped” curve with risk from both maternal under- and over-nutrition (Curhan et al., 1996; Pettitt and Jovanovic, 2001; Ong, 2006).
Importantly, fetal programming by both maternal under- and over-nutrition involves perturbations in the hypothalamic appetite regulatory systems which are established during early development (McMillen et al., 2005; Bouret, 2009; Stevens et al., 2011; Ross and Desai, 2014). The hypothalamus controls food intake and energy expenditure and is a prime target of developmental programming by maternal nutritional imbalance that predisposes the offspring to permanent risks of metabolic disorders. Epigenetic alterations of key metabolic genes in the hypothalamus are central in regulating this process. The purpose of this review is to discuss evidence from recent studies supporting hypothalamic programming of obesity and the role of epigenetic gene regulation in mediating phenotypic alterations in the offspring as a result of a maternal nutritional insult.
Hypothalamic Neuronal Circuits Controlling Energy Balance
Energy balance is controlled centrally, predominantly by the hypothalamus. The principal area containing these circuits is the arcuate nucleus (ARC), but within the hypothalamus the paraventricular nucleus (PVN) and the dorsomedial hypothalamus (DMH) also have a role in the regulation of energy balance. The ARC is known as the “feeding” center of the brain. It is a region close to the 3rd ventricle and the median eminence. This region of the brain has a “leaky” blood brain barrier, which allows the access of peripheral hormones to this part of the brain. The ARC contains both anorexigenic and orexigenic neurons. Together, the peptides released from these two sets of neurons counterbalance each other to control food intake and energy expenditure and ultimately have a major role in the control of body weight.
Anorexigenic ARC Neurons
The most studied anorexogenic neurons in the ARC are pro-opiomelanocortin (POMC) neurons as they synthesize POMC as well as cocaine and amphetamine regulated transcript peptides (CART), both of which decrease food intake. They are almost exclusively located in the ARC and project mainly to the PVN, lateral hypothalamus (LH) and the brainstem areas, which are all associated with control of energy balance (Figure 1).
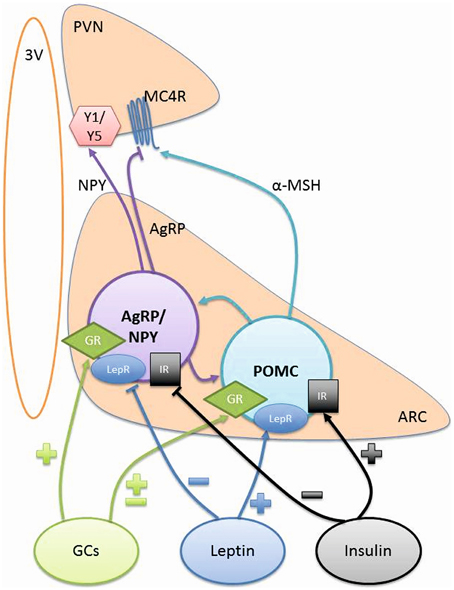
Figure 1. Hypothalamic signaling controlling energy balance. POMC and AgRP/NPY neurons project from the ARC to the PVN to control food intake and energy balance. The signaling from these neurons can be influenced by GCs, Leptin, and Insulin. ARC, arcuate nucleus; AgRP, agouti-related peptide; GCs, glucocorticoids; GR, glucocorticoid receptor; IR, insulin receptor; LepR, leptin receptor; MC4R, melanocortic 4 receptor; α-MSH, α melanocyte stimulating hormone; NPY, neuropeptide Y; POMC, pro-opiomelanocortin; PVN, paraventricular nucleus; Y1/Y5, Y1 and Y5 receptors; 3V, 3rd Ventricle.
POMC can be cleaved to numerous peptides including adrenocorticotrophic hormone (ACTH), β-endorphin and α-melanoctye stimulating hormone (α-MSH). The most widely studied anorexigenic neuropeptide in the ARC is α-MSH, which acts as an agonist at melanocortin 3 receptors (MC3R) and melanocortin 4 receptors (MC4R) in the hypothalamus. These receptors are highly expressed on second order neurons in the PVN and their activation leads to a reduction in food intake and an increase in energy utilization. Understanding the function of POMC has been enhanced by the use of knock-out models. The global POMC knock-out mouse has hyperphagia, leading to excessive body weight gain (Coll et al., 2005). Additionally, deletion of either the MC3R (Butler et al., 2001) or MC4R (Huszar et al., 1997) also leads to an obese phenotype with increased fat mass. Furthermore, genetic mutations in the POMC gene have been shown to cause obesity in patients (Krude et al., 1998; Challis et al., 2002; Farooqi et al., 2006; Creemers et al., 2008). Therefore, POMC derived peptides clearly have roles in reducing food intake to control body weight.
The other major anorexigenic neuropeptide in the ARC is CART, however, it is less widely studied than POMC, due to a lack of understanding of the receptors for this peptide (Lau and Herzog, 2014). CART is released from the same ARC neurons that release POMC and has a similar effect to reduce food intake and increase energy expenditure. But in contrast to POMC, CART is more widely expressed throughout the brain and the peripheral tissues (Douglass et al., 1995; Couceyro et al., 1997; Ekblad, 2006; Vrang, 2006; Kasacka et al., 2012). When CART knock-out mice are fed a high caloric diet, they demonstrate the expected increases in food intake and body weight (Asnicar et al., 2001; Wierup et al., 2005; Moffett et al., 2006), giving an insight into the role of CART in energy balance.
Orexigenic ARC Neurons
The orexigenic neurons in the ARC primarily release neuropeptide Y (NPY), agouti-related peptide (AgRP), and γ-aminobutyric acid (GABA) to increase food intake. Their cell bodies are located in the ARC and the neurons project to other hypothalamic regions including the PVN, DMH, perifornical area (Karatsoreos et al., 2010), LH, and the medial preoptic area (MPO) (Chronwall et al., 1985; Morris, 1989).
The role of these neurons in the control of food intake has been highlighted in a recent study where each of the individual components were deleted, demonstrating that NPY, AgRP, and GABA have a synergistic role in the control of food intake (Krashes et al., 2013). However, ablation of the whole orexigenic ARC neuron postnatally leads to starvation of the animal (Gropp et al., 2005; Luquet et al., 2005), indicating the essential role of these neurons in regulating appetite.
AgRP is an antagonist at the MC4R, blocking the effects of α-MSH in the energy regulating pathway and thereby increasing food intake. Interestingly, deletion of AgRP from the neuron can result in an unchanged (Corander et al., 2011) or a very mild phenotype, which is only seen in older mice (Wortley et al., 2005). These mice have a decrease in body weight from 6 months of age, associated with an increase in energy expenditure. Although, knocking out the peptide has such a small effect, administration of AgRP by intracerebroventricular (icv) injection increases food intake, which is sustained for up to a week (Rossi et al., 1998; Hagan et al., 2000). More recently, using genetic manipulation it was noted that AgRP has a role in prolonged, rather than short-term feeding (Krashes et al., 2013).
NPY binds to Y1 and Y5 receptors in the hypothalamus to exert its effects on food intake and energy balance (Morton and Schwartz, 2001). Its role in these processes has been clearly demonstrated in a well-designed study, where an icv injection of NPY induced feeding in rodents and which was maintained for a number of hours (Clark et al., 1984). Furthermore, repeated injections of NPY over several days resulted in increased food intake, body weight and adiposity (Stanley et al., 1986). Conversely, injecting an antisense oligonucleotide or antiserum to NPY decreased food intake and body weight gain in rats (Shibasaki et al., 1993; Hulsey et al., 1995). Curiously, global knock-out of NPY results in normal body weight and food intake as well as a normal response to a 48 h fast (Erickson et al., 1996). This must be a consequence of other pathways compensating in the absence of NPY.
As well as the two neuropeptides, the neurotransmitter GABA is also released from orexigenic ARC neurons. From studies using designer receptors exclusively activated by designer drug (DREADD) technology, it appears that GABA has a similar role to NPY, such that it can influence the initial phase of feeding and can act as a substitute for NPY (Krashes et al., 2013). Together, the three factors released from orexigenic ARC neurons promote an increase in food intake.
Glucocorticoids and the Regulation of Food Intake
Glucocorticoids (GCs) are steroid hormones secreted by the adrenal gland under the control of hypothalamic-pituitary-adrenal (HPA) axis and are known to have a role in the regulation of food intake as well as energy balance. They exert their effects by binding and activating the glucocorticoid receptor (GR). This triggers a cascade of events resulting in GR acting as a transcription factor and binding to glucocorticoid response elements (GREs) such as those present in the promoter regions of AgRP (Lee et al., 2013) and NPY (Misaki et al., 1992).
Models of GC excess in mice have highlighted their ability to increase food intake and body weight. Both active corticosterone (equivalent to cortisol in humans) and inactive 11-dehydrocorticosterone (cortisone in humans) can mediate these effects (Karatsoreos et al., 2010; Harno et al., 2013; Morgan et al., 2014). Furthermore, these models mimicked metabolic syndrome and Cushing's syndrome, both of which are associated with glucocorticoid excess and are characterized by hyperphagia. Similar clinical symptoms can be seen as a result of the adverse effects seen with administration of chronic synthetic “steroid” (glucocorticoid) treatment. In addition, adrenalectomy and removal of endogenous GCs leads to a reduction in body weight and food intake in genetically obese rodents (Marchington et al., 1983; Makimura et al., 2000).
GCs also have a role in altering the expression of both ARC orexigenic and anorexigenic neuropeptides. This has been explored by either depleting the levels of GCs or significantly increasing the amounts of GC exposure. AgRP is positively regulated by GCs in multiple studies (Savontaus et al., 2002; Coll et al., 2005; Kim et al., 2013; Lee et al., 2013). The results are less clear cut for NPY with studies demonstrating that dexamethasone, a synthetic GC, increases NPY expression (Goto et al., 2006; Yi et al., 2012), but corticosterone, the endogenous GC in rodents, has no effect (Coll et al., 2005). The control of POMC by GCs is also controversial with studies showing both positive (Savontaus et al., 2002; Uchoa et al., 2012) and negative (Beaulieu et al., 1988; Makimura et al., 2000; Gyengesi et al., 2010) regulation by GCs.
Other Peripheral Hormones Acting on Neural Networks
Other peripheral hormones, principally leptin and insulin, can cross the blood brain barrier to act in the hypothalamus on ARC orexigenic and anorexigenic neurons. Both of these peripheral hormones have receptors on both classes of neurons, allowing these factors to affect firing and signaling of the neuron and in turn have an effect on food intake and energy balance.
Leptin, released by adipocytes, circulates in direct proportion to the amount of adipose tissue (Considine et al., 1996). It is anorexigenic and some of its effects are mediated by POMC neurons as a result of increasing α-MSH secretion (Cowley et al., 2001; Munzberg et al., 2003; Guo et al., 2004). In addition, orexigenic AgRP/NPY neurons also express leptin receptors (Elmquist et al., 1998) and leptin reduces the expression of both AgRP and NPY neuropeptides (Stephens et al., 1995; Mizuno and Mobbs, 1999). Evidence for the functional role of leptin has come from studies where leptin has been administered centrally in rodents and decreases food intake and body weight (Campfield et al., 1995; Weigle et al., 1995). There are also studies in obese children with congenital leptin deficiency, where leptin replacement has been able to reduce body weight and hyperphagia (Farooqi et al., 1999). However, leptin is not a suitable treatment for most patients with obesity as they already have high circulating levels of leptin because they have developed leptin resistance, a condition where leptin is unable to induce its effect of lowering of food intake and therefore body weight (Schneeberger et al., 2014).
Insulin can act in the ARC to produce an anorexigenic effect, reducing food intake and body weight (Woods et al., 1979). Insulin receptors are widely expressed in the brain and co-localize with both POMC and AgRP neurons (Marks et al., 1990; Benoit et al., 2002). Activation of the insulin signaling pathway in the ARC decreases NPY and AgRP expression as well as increasing the expression of POMC to give the anorexigenic effect (Schwartz et al., 1992; Sipols et al., 1995; Benoit et al., 2002; Plum et al., 2006). As with leptin, there is evidence for central insulin resistance, decreasing the ability of insulin to reduce food intake in obese patients (Hallschmid et al., 2008).
Another peripheral hormone that plays an important role in the regulation of energy balance is the gut-derived orexigenic peptide hormone, ghrelin. It acts via the growth hormone secretagogue receptor (GHS-R) which is present at a high density in the hypothalamus, specifically in the nuclei involved in body weight and food intake regulation such as the ARC, ventromedial hypothalamus, DMH, and PVN (Mitchell et al., 2001; Zigman et al., 2006). The ghrelin-induced increase in food intake is mediated by the NPY/AgRP pathway as genetic ablation or pharmacological inactivation of AgRP and NPY blocks the orexigenic effects of ghrelin (Nakazato et al., 2001; Shintani et al., 2001; Chen et al., 2004). Moreover, central administration of ghrelin to rats leads to an increase in the mRNA expression of both NPY and AgRP (Kamegai et al., 2001; Nakazato et al., 2001; Shintani et al., 2001).
Experimental Evidence for the Programming of Appetite and Obesity
There are many examples within the literature demonstrating a link between maternal nutritional insults and detrimental effects on the fetus. Both maternal under- and over-nutrition have been extensively described with additional factors such as the timing of the insult, the gender of the offspring and importantly, the severity of the nutritional change leading to complex outcomes. As a result, deciphering the pathways and the key genes affected is challenging. The hypothalamic appetite signaling pathway is imperative in these processes and is altered following perturbations in the maternal nutritional environment in fetal and postnatal offspring.
Maternal Undernutrition and its Effects on the Offspring
Maternal undernutrition is recognized as being a cause of fetal programming leading to the offspring having an increased propensity to develop hypertension, type 2 diabetes and cardiovascular disease (Barker, 2005). The Dutch famine (1944–1945) has been the basis for initial pioneering epidemiological studies demonstrating the negative associations between maternal environment and disease outcomes in the offspring (Ravelli et al., 1976). One of the main characteristics of maternally programmed offspring is altered birth weight, which was a marker that these studies initially utilized to identify programmed offspring. It was found that mothers exposed to famine during early gestation had offspring with normal birth weight but with an increased possibility of becoming obese, whereas mothers exposed to famine later in pregnancy had offspring with lower birth weights (Ravelli et al., 1976; Sharkey et al., 2009). These findings clearly show that exposing the fetus to maternal undernutrition during different times of fetal development can induce differential outcomes. Longitudinal analysis of this cohort revealed that at approximately 50 years of age the maternally-programmed offspring had compromised glucose tolerance and higher insulin concentrations (Ravelli et al., 1999; Mostyn and Symonds, 2009). Extensive analysis of these individuals has led to the understanding that these changes may be a consequence of impaired insulin secretion (de Rooij et al., 2006). However, the precise mechanisms governing these changes are difficult to elucidate due to the restrictions of using the human model. This is particularly true when trying to assess the hypothalamic regulation of energy balance. As a result, several animal models have been established to characterize the potential pathways involved.
Caloric Restriction, Intrauterine Artery Ligation, and Twinning as Models of Adverse Events in Pregnancy
A range of programming models have been developed to mimic the human maternal undernutrition paradigm. These animal models are critical in defining the systems and underlying mechanisms that may be involved in the increased susceptibility of the children to develop disease in adulthood, following exposure to maternal nutritional programming. These models have included caloric restriction, intrauterine artery ligation, and twinning. Caloric restriction has been one of the most common paradigms, with studies reducing the calorie intake by 50–70% in the earlier studies (Delahaye et al., 2008; Breton et al., 2009) and then to more moderate levels in more recent studies (Stevens et al., 2010). Initially the high levels of caloric restriction were essential in defining changes that may be occurring in mothers exposed to famine. However, using moderate restriction could be more applicable to mimicking the effects of women dieting during the initial periods of conception to increase their chances of conceiving or because of social pressures to be thin. These studies have been vital in highlighting the effects of maternal undernutrition on hypothalamic mechanisms in the fetus. Nevertheless, the inconsistencies between the investigations about the severity and timing of the insult should be addressed.
One of the proposed mechanisms for maternal programming of the fetus is altered placental blood flow leading to the birth of small for gestational age babies (Sanders et al., 2004). Bilateral uterine ligation techniques have been developed as a tool for investigating the effects of placental insufficiency (Gagnon, 2003). Glucose is a necessary component of the maternal and fetal system, which the fetus will utilize to aid many developmental processes. Accordingly, any induced or natural decreases in blood flow can dramatically reduce the levels of glucose that the fetus is exposed to, leading to hypoglycaemia. This in turn can impact on insulin levels and pancreatic β-cell function. Consequently, the fetus must adapt to this situation by altering other metabolic parameters with expected adverse outcomes such as impaired glucose tolerance. However, there is evidence to suggest that this model of maternal programming and its outputs are difficult to reproduce (Neitzke et al., 2008). Therefore, models of bilateral uterine artery ligation require further development before significant conclusions can be made about any changes that are observed.
Over the years the number of mothers giving birth to twins has risen due to artificial reproductive technology and multiple ovulations, because mothers are having children later in life (Blondel et al., 2002; Ombelet et al., 2006; Muhlhausler et al., 2011). More recently, twins have been found to have some similarities with programmed offspring, having reduced birth weight, potentially due to the twins being exposed to a maternal environment where the fetuses must compete with each other for nutrition (Rumball et al., 2008). Additionally, twins have a reduced placenta size, as well as being in a restricted space. This may alter their developmental trajectory to provide compensatory mechanisms to ensure their survival. Studies have already found that these possible compensatory mechanisms can lead to altered glucose regulation and higher fat mass in adulthood (Poulsen et al., 1997; Ribel-Madsen et al., 2012). Despite these findings there are inconsistencies in the development of diseases in adult twins (Phillips et al., 2001; Muhlhausler et al., 2011). This may be because these investigations have omitted to use singletons as their control and therefore, it is difficult to ascertain if any changes are because of the inherited genotype of the animal or if they are caused by the maternal environment (Muhlhausler et al., 2011). Nevertheless, some studies have compared their data to effects seen in singletons. They have found changes similar to maternally undernourished animals with impaired glucose and insulin signaling and altered hypothalamic pathways (Rumball et al., 2008; Begum et al., 2012) indicating that these mechanisms are important in programming.
The Effects of Maternal Undernutrition on the Hypothalamic Appetite Signaling Pathway
The development of a functional hypothalamus occurs in two phases, which involves neurogenesis and the formation of functional circuits (axon growth and synaptogenesis). In precocial species such as humans, sheep and non-human primates, the development of hypothalamic neural projections occurs at the fetal stage. However, in altricial rodents the hypothalamus is immature at birth and development continues during the first 2 weeks of postnatal life when neurons send axonal projections to their target sites and functional synapses are formed (Bouret, 2010). As a result the rodent hypothalamus is not fully formed until after weaning. Thus, any changes in the maternal nutritional environment during these critical periods will severely impact on this system potentially altering the offspring's developmental trajectory. Several studies on maternal perinatal undernutrition in rodent models have produced key evidence that these programmed offspring have low birth weight, exhibited hyperphagia in postnatal life and develop obesity, insulin resistance in adulthood (Vickers et al., 2000; Yura et al., 2005). This hyperphagia is exacerbated when the offsprings are fed high caloric diet in the post-weaning period (Vickers et al., 2000; Yura et al., 2005). Moreover, alterations in hypothalamic cell proliferation and impaired axonal elongation have been observed in the perinatally undernourished offspring (Delahaye et al., 2008; Breton et al., 2009; Garcia et al., 2010). These changes modify the density of the NPY and POMC neurons. This could have direct consequences on the animal's food intake and energy expenditure and depending on which neurons are affected it could increase food consumption and body weight. Supporting evidence for this was provided by a study which showed a greater level of food intake as a direct result of the maternal insult (Garcia et al., 2010).
In addition to structural variations in the hypothalamus, these models have also presented modifications in the expression of ARC neuropeptides that have a central role in the energy regulating pathway. This includes the anorexigenic POMC, and the orexigenic neuropeptides, AgRP and NPY. Despite clear differences in the severity and timing of the nutritional insult, the outcome of maternal undernourishment is consistent, with increased hypothalamic NPY and AgRP and decreased POMC gene expression in postnatal rodent offspring (Lopez et al., 2005; Delahaye et al., 2008; Breton et al., 2009; Cripps et al., 2009; Shin et al., 2012).
As the hypothalamus develops postnatally in rodents, animal models that specifically target the postnatal period have been developed. This has been done by adjusting the litter size in rats and mice. It has been found that pups raised in large litters (therefore less maternal milk) have attenuated pre-weaning growth. This was associated with an increased number of NPY neurons and higher NPY concentrations in the ARC (Plagemann et al., 1999c). These results show that postnatal undernutrition in rats leads to alterations in hypothalamic neural circuits regulating food intake and body weight.
Whilst the rodent model has been extremely useful for characterizing maternal programming paradigms, the length of gestation and the level of hypothalamic development during this time is not comparable to humans (Symonds and Budge, 2009; Bouret, 2010). As a result findings from species that are more developmentally similar to humans, such as sheep, are vital in enhancing our further understanding. These studies have shown that moderate levels of maternal undernutrition during early gestation led to increases in GR expression levels but no changes in POMC or NPY mRNA expression at the fetal stage (Stevens et al., 2010; Begum et al., 2013). However, evidence from higher mammals such as baboons have shown a decrease in POMC expression and increased NPY expression in the baboon fetus (Li et al., 2013). Therefore, changes may become apparent depending on the insult and the species investigated.
There is a general lack of studies analyzing adult offspring. This is problematic because the impact of programming on the susceptibility of the adult offspring to develop diseases is the key issue. A few longitudinal studies have been reported (Stevens et al., 2010; Begum et al., 2013). In these, it has been shown that maternal undernutrition leads to increases in GR expression levels which are sustained from fetal to adult life. There were also associated decreases in POMC expression in adulthood which may be a consequence of the change in GR (Stevens et al., 2010; Begum et al., 2013).
The Effects of Maternal Overnutrition on the Hypothalamic Energy Regulating Pathway
Over recent years the focus of research in the programming field has moved from studying the effects of maternal undernutrition to maternal overnutrition. Investigating this programming paradigm has become increasingly vital due to the rise in the levels of obesity within developed countries such as the USA and UK. Partly to blame for this surge in obesity is the increased consumption of high caloric foods by the mothers, combined with reduced levels of exercise. The number of epidemiological studies establishing a clear link between increased maternal body mass index and the offsprings' susceptibility to disease in adulthood are extensive (Oken et al., 2008; Drake and Reynolds, 2010; Reynolds et al., 2010). An interesting study by Patti (2013), found that mothers who underwent bariatric surgery had children with a decreased propensity to develop obesity compared to offspring born before the mother was subject to surgery. This further highlights the negative implications of maternal overnutrition for the offspring (Patti, 2013).
Once again rodent models have been essential in detailing the effects of maternal high fat diet on the offspring. These studies have not only described changes that occur during pregnancy, but also those that may take place during lactation (Guo and Jen, 1995; Samuelsson et al., 2008; Tamashiro et al., 2009). As expected, the outcome of these studies has been the development of obesity in the offspring accompanied by impaired insulin and glucose homeostasis, as well as many other outcomes such as cardiovascular diseases (Williams et al., 2014). However, unlike maternal undernutrition, the findings from the maternal overnutrition studies have been conflicting and therefore, more difficult to interpret. Whilst some studies are in agreement that there are impaired anorexigenic and orexigenic nerve fiber projections from the ARC to the PVN (Bouret et al., 2008; Chen et al., 2008; Kirk et al., 2009), the levels of neuropeptide expression are extremely diverse. For example, when the mothers were fed a high fat diet throughout gestation and lactation there was increased POMC mRNA and lower amounts of NPY expression in the programmed offspring (Chen et al., 2008). The rise in POMC may have occurred to provide some form of compensatory mechanism within these animals. In contrast, some studies have shown decreases in POMC and NPY (Morris and Chen, 2009), whilst another found increases in CART, NPY, and AgRP (Lopez et al., 2005). Although the standard animal model of maternal overnutrition during gestation and lactation, gives rise to adverse effects in the offspring, it does not explain which effects are caused by gestational and which by lactational exposure to maternal high fat feeding. Indeed, Vogt et al. (2014), have demonstrated the importance in the timing of the maternal insult and the effects that this can have on fetal outcome. In this study it was found that giving mothers a high fat diet during lactation induced changes in the POMC and AgRP nerve fiber projections in the hypothalamus (Vogt et al., 2014).
In the aforementioned study, one possible underlying mechanism suggested to induce modified fiber projections was altered insulin signaling (Vogt et al., 2014). Indeed, when the insulin receptor on POMC neurons was ablated in offspring from mothers fed a high fat diet, this led to a rescue in the previously distorted nerve projections. As a result, this study is the first to demonstrate a clear link between altered insulin signaling in the hypothalamus and perturbations in the hypothalamic energy regulating pathways in a maternally programmed paradigm. Since in humans, ARC neuronal projections arise during the third trimester, these results indicate that changes in maternal glucose metabolism during this critical window may have metabolic effects on the offspring throughout life. This investigation did not rule out the possibility of other mechanisms affecting the structure of the hypothalamus and other studies have suggested that changes in hypothalamic connections may also be associated with altered leptin resistance (Kirk et al., 2009). These findings have been essential in determining potential underlying reasons for the changes that are being observed in maternal programming paradigms. Nevertheless, there are still large gaps in our knowledge of how nutritional insults impact on the maternal and fetal interface such that they are affecting peripheral regulators to modify central pathways in a negative manner.
Another approach to studying postnatal overfeeding is by reducing the litter size. Raising pups in small litters post birth can potentially decrease competition for the mother's milk and therefore increase nutritional availability. As a result, there is a greater chance of the pups developing obesity, hyperphagia, and impaired glucose tolerance. Indeed, one study found that this led to increased numbers of neurons producing the orexigenic neuropeptide galanin in pups from reduced litters (Plagemann et al., 1999b). Taken together, these results demonstrate that exposure to prenatal or postnatal overnutrition in rodents results in substantial changes in the development of the hypothalamic architecture.
Maternal Gestational Diabetes Programs the Hypothalamic Neural Network Controlling Appetite in Offspring
Gestational diabetes is a common complication predisposing offspring to obesity and diabetes. The effect of maternal insulin deficiency on the offspring's hypothalamic appetite regulating system has been studied using rodents injected with the pancreatic islet toxin streptozotocin (STZ) during early pregnancy to develop a model of diabetes. The pups of diabetic mothers presented with impaired metabolic regulation including increased glucose and insulin levels, hyperphagia and increased body weight. In addition, they exhibited structural alterations affecting the density of AgRP and anorexigenic αMSH containing fibers in the PVN with altered leptin sensitivity (Steculorum and Bouret, 2011b). In a similar animal model, Plagemann et al. (1999a) has shown that offspring of diabetic dams have an increased number of galanin and NPY containing neurons (Plagemann et al., 1999a). Taken together, these results highlight that maternal gestational diabetes leads to malprogramming of hypothalamic orexigenic and anorexigenic systems in the offspring, contributing to metabolic disorders later in life. Perhaps more importantly, these acquired alterations in hypothalamic appetite related circuits can be prevented by normalizing gestational hyperglycaemia (Franke et al., 2005).
Programming Affecting Leptin, Insulin, Ghrelin and Glucocorticoids, and the Impact on their Role in the Hypothalamus
As mentioned above, leptin can bind to ARC anorexigenic and orexigenic neurons influencing transcriptional activity. Increases in leptin levels are critical for the developing brain in assisting with increased fiber density and ensuring correct axonal projections from the ARC to other areas of the hypothalamus. Additionally, leptin is crucial for neuronal differentiation and migration (Bouret et al., 2004; Udagawa et al., 2007). As a result, it is clearly evident that this hypothalamic energy regulating pathway is susceptible to changes in leptin levels which could be impaired in offspring of programmed mothers. Indeed, studies outlined above found altered leptin levels contributing to increased food intake in the offspring (Delahaye et al., 2008; Garcia et al., 2010). However, the exact mechanisms leading to these changes are yet to be elucidated.
Many studies have shown impaired insulin regulation in fetuses from nutritionally programmed mothers (Snoeck et al., 1990; Chamson-Reig et al., 2006; Theys et al., 2009; Todd et al., 2009). It is understood that these changes are occurring because of modifications in the pancreas, such that there is deficient insulin production and secretion. Further evidence suggests that a maternal low protein diet can lead to pancreatic islets with deficient responses to glucose stimulation (Theys et al., 2009). There is also significantly reduced β-cell proliferation found in postnatal offspring (Chamson-Reig et al., 2006). In part these changes in the pancreas could be a compensatory mechanism developed by the fetus in response to hypoglycaemia experienced as a consequence of insufficient blood flow across the placenta. It is important to note that only one investigation has clearly described a direct link between peripheral insulin exerting its action on the insulin receptor in the hypothalamus and impaired POMC nerve fiber projections in the hypothalamus (Vogt et al., 2014).
Recently, ghrelin has been shown to play an important role in the normal development of ARC neural projections in neonatal mice and perturbations in its action during early life can affect leptin sensitivity and cause lifelong metabolic disturbances (Steculorum et al., 2015). Furthermore, acute ghrelin injection to mouse pups during second week of postnatal life lead to altered levels POMC and NPY mRNA (Steculorum and Bouret, 2011a). These results highlight the importance of the effect of ghrelin in influencing normal hypothalamic development. Moreover, ghrelin levels are altered by early life nutrition decreasing its ability to activate ARC neurons (Collden et al., 2015).
Additionally, although key regulators of the hypothalamic pathway such as leptin have been investigated in relation to maternal programming paradigms, very few studies have examined GCs. In particular, changes in the GR have been shown to influence the expression of neuropeptides and therefore, GR presents itself as an ideal candidate for further study. Indeed Li et al. (2013) and Begum et al. (2013) have demonstrated clear increases in hypothalamic GR expression associated with adverse outcomes within the offspring of maternally undernourished baboons and sheep, respectively. These studies highlight the need for more research to be carried out on the role of GR and other hypothalamic regulators, which the field currently lacks.
Gender Specific Effects of Maternal Programming within the Offspring
Whilst there has been much work carried out to determine the effects of maternal programming, some investigations have failed to take into account the possibility of gender specific differences in the offspring. The possibility of sexually dimorphic outcomes has been clearly highlighted in several recent studies (Todd et al., 2009; Matthews and Phillips, 2012; Begum et al., 2013). For example, in maternally undernourished offspring it was found that males had an increased propensity to reduced insulin levels and were more sensitive to changes in insulin (Todd et al., 2009). Evidence from the literature suggests that this may be caused by the male brain being more sensitive to the nutritional alterations that are associated with leptin and insulin resistance (Clegg et al., 2003). Furthermore, it was found that male offspring from undernourished ewes developed an increase in fat mass which was not observed in the females (Begum et al., 2013). It could be hypothesized that these changes may have occurred as a consequence of decreased central actions of insulin, leading to lower levels of POMC expression and reduced inhibition of food intake. Investigations detailing the effects of maternal programming on the hypothalamic energy regulating pathway have found associated changes in leptin and insulin levels (Delahaye et al., 2008; Vogt et al., 2014). These peptides can be regulated by the levels of estrogen, which could provide a compensatory mechanism in females to circumvent the effects of under- or over- nourishment. However, more work needs to be carried out in order to fully understand sexually dimorphic changes as a consequence of maternal programming.
Molecular Mechanisms Underlying Developmental Programming of Obesity
The literature clearly demonstrates that environmental insults during critical periods of fetal development and early postnatal life can influence energy expenditure pathways, with permanent consequences for obesity risk. One plausible explanation is that nutritional insults during early life may interfere with organogenesis and thus altering organ structure and function (Langley-Evans, 2009). Another mechanism to explain developmental programming is epigenetic modifications, which regulate gene expression patterns in a spatial and temporal manner. Epigenetic marks are established during fetal development and early postnatal life (Jaenisch and Bird, 2003). These marks exhibit developmental plasticity in response to the environment and are maintained throughout life. Therefore, perturbations to such processes represent a principal candidate mechanism by which early life nutritional insults can induce persistent phenotypic alterations in adulthood. Accordingly, there has been recent growing interest in the role of epigenetic dysregulation of appetite regulatory genes in metabolic disorders such as obesity (Gluckman et al., 2009; Stevens et al., 2011; Waterland, 2014).
Epigenetic Mechanisms
Epigenetics is the study of stable alterations in gene activity that occur without changes in the underlying DNA sequence. Epigenetic modifications play a significant role in regulating chromatin assembly, DNA replication and repair as well as gene expression during cell differentiation and development (Delage and Dashwood, 2008). The candidate epigenetic mechanisms include methylation of cytosines in CpG dinucleotides, posttranslational modifications of histone proteins and micro-RNA mediated gene regulation. Most importantly, these epigenetic modifications function synergistically to maintain chromatin conformations in a cell type specific manner to influence transcriptional activity. Up to now, most of the studies of early life influences on epigenetic regulation of genes have focused on DNA methylation and a few on histone modifications. Therefore, the following section will focus on describing these two mechanisms and the potential influence they may have on central genes in the metabolic pathways of the fetus that may be altered as a consequence of maternal programming.
Influence of DNA Methylation
DNA methylation is the most prominent, well-understood, and highly stable epigenetic modification. It is involved in controlling gene transcription, parental imprinting, X-chromosome inactivation, and also in preventing retrotransposon activity and genomic instability (Jones, 2012). The majority of DNA methylation occurs at the C5 position of cytosine residues that are adjacent to a guanine residue, known as a CpG dinucleotide. DNA methylation is a covalent modification characterized by the addition of a methyl group to the cytosine residue of DNA. This is catalyzed by DNA methyltransferases (DNMTs) which use S-adenosylmethionine (SAM) as a cofactor and methyl donor. DNMT3a and DNMT3b are de novo methyltransferases which methylate cytosine residues at the previously unmethylated CpG sites during germ cell development and embryogenesis. DNMT1 is a maintenance methyltransferase which predominantly methylates hemimethylated CpG dinucleotides and copies the methylation pattern in the newly synthesized DNA during replication (Hermann et al., 2004).
DNA methylation can affect gene expression by two mechanisms: (1) by directly preventing the binding of transcription factors to their recognition sites on the DNA (Watt and Molloy, 1988), and (2) it can silence gene expression by promoting the binding of methyl-CpG-binding proteins (MBPs) which directly recognize methylated DNA and recruit transcriptional co-repressor complexes containing histone deacetylases (HDACs) and histone methyltransferases (Boyes and Bird, 1991; Jones et al., 1998; Nan et al., 1998). As a result, DNA methylation can effectively carry out its regulation of transcriptional activity. In mammals, 70–80% of CpG dinucleotides are methylated and the remaining unmethylated regions of the genome that have higher levels of guanine and cytosine content, are clustered together in CpG islands (Illingworth and Bird, 2009). There are CpG islands in promoter regions of 70% of mammalian genes and promoter CpG methylation is generally associated with gene silencing (Bird, 2002).
Transcriptional Regulation by Histone Modifications
Histones are targets for epigenetic modifications which have profound effects on transcriptional activity, because of their role in packaging DNA into nuclear chromatin. The N-terminal region of core histones designated as H2A, H2B, H3, and H4 are subjected to numerous covalent modifications which influence both chromatin organization and gene expression. The posttranslational modifications of histones include acetylation, methylation, phosphorylation, ubiquitination, and sumoylation (Kouzarides, 2007). Acetylation of histones at specific lysine residues is a highly dynamic process regulated by two families of enzymes with opposing actions, histone acetyltransferases (HATs) and HDACs. HATs catalyze the transfer of acetyl groups to the lysine residues utilizing acetyl-CoA as cofactor whereas HDACs function to reverse acetylation (Sterner and Berger, 2000). Histone acetylation induces open chromatin conformation and correlates with transcriptional competence. Specifically, acetylation at histone 3 lysine 9 and 14 (H3K9/14ac) is associated with transcriptional initiation and H3K36ac is associated with transcriptional elongation. Histone methylation can be targeted at either lysine or arginine residues by lysine (HKMT) or arginine (PRMT) methyltransferases respectively. Histone arginine methylation is involved in transcriptional activity whereas lysine methylation has been associated with either activation or repression depending on the residue modified. For example, methylation of H3K4 is associated with transcriptional activation whereas methylation of H3K9 and K27 correlates with gene silencing (Kouzarides, 2007). Thus, combinations of different histone modifications can influence the regulation of the opening and closing of the chromatin resulting in activation or repression of a given gene. Many studies have used these patterns of histone modifications, termed the “histone code,” as a marker for epigenetic analysis within their models.
Epigenetic Programming of Obesity: Candidate Gene Studies Vs. Genome Wide Association Studies
Given that epigenetic modifications are influenced by environmental perturbations, several groups have investigated the effect of maternal nutrition on the epigenetic changes of key hypothalamic genes regulating energy balance and thereby impacting on obesity in the offspring. These are discussed in the following sections of the review. A candidate gene approach has several strengths, narrowing the focus on the role of specific genes enabling direct phenotypic correlations to be made with increased statistical power. However, this approach does not further the understanding of the role of possible alterations in novel genes or pathways in the offspring, which ultimately shape developmental trajectories. This highlights the need for the genome-wide association study (GWAS) to unravel novel pathways involved in fetal programming of obesity. Accordingly, a few studies have focused on the impact of in utero environmental insults on the offspring's genome-wide epigenetic alterations (Altobelli et al., 2013; Crudo et al., 2013; Suter et al., 2014). Although these studies are instrumental in identifying novel genes and pathways associated with fetal programming, they have limitations such as low statistical power with multiple testing problems (Amos et al., 2011). Thus, future studies should combine genome-wide analysis with candidate-gene approaches, which are key to identifying the mechanisms involved in the increased risk of metabolic disorders in the offspring.
Epigenetic Effects on Hypothalamic POMC as a Consequence of Maternal Programming
In addition to genetic polymorphisms in POMC (Krude et al., 1998; Challis et al., 2002; Farooqi et al., 2006; Creemers et al., 2008), epigenetic variations could contribute to an individual's risk of obesity (Kuehnen et al., 2012). The anorexigenic POMC is a likely target for epigenetic modifications by programming because there is good evidence for its regulation by promoter methylation (Newell-Price et al., 2001). The POMC gene has two CpG islands, one identified at the 5′-promoter region and one downstream around the intron 2 and exon 3 boundary (Newell-Price, 2003; Kuehnen et al., 2012). However, the POMC promoter regions necessary for the regulation of the POMC gene in the pituitary, and the hypothalamus, are different. In the hypothalamus its expression is regulated by two highly conserved distal neuronal enhancers, nPE1 and nPE2 located 10–12 kb upstream of the transcriptional start site (de Souza et al., 2005). Several studies have illustrated that an altered nutritional milieu perinatally can program epigenetic changes in the key regulator elements of the POMC promoter thereby affecting the appetite signaling pathway and inducing an obese phenotype in the offspring. For instance, maternal undernourishment during the periconception period in sheep leads to fetal hypothalamic POMC hypomethylation in a region that is closely related to the nPE1 and nPE2 POMC enhancer regions (Stevens et al., 2010, 2011). Another investigation by Plagemann et al. (2009) demonstrated that neonatal overnutrition in rats was associated with hypermethylation of CpG sites within the Sp1 binding site, which is required for the leptin and insulin mediated expression of POMC in the hypothalamus. These acquired methylation patterns were accompanied by a metabolic phenotype including obesity and increased leptin, insulin and glucose levels (Plagemann et al., 2009). This study clearly demonstrates epigenetic programming of POMC by early life nutrition and its effect on the risk of developing metabolic disorders. Additional investigations have shown hypermethylation of the POMC proximal promoter region in 28 day old postnatal mice reared by dams with dietary supplementation of conjugated linoleic acids (Zhang et al., 2014). This led to suppression of POMC expression and increased food intake contributing to catch-up growth and metabolic changes such as hyperglycaemia and insulin resistance (Zhang et al., 2014). These results show that changes in early life nutrition can elicit epigenetic and associated expression changes in hypothalamic POMC in the offspring.
In a clinical study, Kuehnen et al. (2012) have shown that hypermethylation of a few distinct CpG sites at the intron 2-exon 3 border (CpG island 2) of the POMC gene from peripheral blood cell (PBC) DNA was found in obese compared to normal weight children. This increased methylation was associated with reduced binding of the p300 histone acetyltransferase and decreased POMC gene expression in human PBCs. However, it is difficult to understand how epigenetic changes in the POMC gene in PBCs relates to changes in the gene in the hypothalamus as many epigenetic modifications are tissue specific and related to the function of the gene in one specific tissue. Although, it could be that the CpG island 2 of the human POMC gene is a metastable epiallele (Waterland, 2014).
All of these studies indicate that POMC is an emerging candidate for epigenetic modifications as a consequence of maternal programming. Despite this, more focused investigations need to be carried out delineating the full extent of any epigenetic modifications and the potential mechanisms that may be causing them. Some of these underlying mechanisms could be associated with altered dietary factors which act as cofactors of many epigenetic modifier enzymes such as DNMTs, HATs, and histone methyltransferases which are discussed in detail in the later section of the review. However, this hypothesis still requires further investigation.
Programming Epigenetic Changes in the Hypothalamic GR
GR is a nuclear receptor that is important in many developmental and metabolic pathways. It is also involved in the hypothalamic control of energy homeostasis by regulating the expression of neuropeptides such as POMC, NPY, and AgRP as discussed above (Beaulieu et al., 1988; Misaki et al., 1992; Drouin et al., 1993; Wardlaw et al., 1998; Gyengesi et al., 2010; Lee et al., 2013). Thus, hypothalamic GR represents a prime candidate for fetal programming which induces alterations in energy balance, predisposing the offspring to metabolic disorders.
Evidence shows a striking link between effects of maternal care on the stress response and the epigenetic status of the GR gene in the offspring (reviewed in Zhang et al., 2013) indicating that the GR is a candidate for programming. In a well-established sheep model of undernutrition, it was shown that maternal undernourishment resulted in decreased GR promoter methylation, and alterations in histone modifications associated with increased GR mRNA expression in the fetal hypothalamic regions involved in energy balance (Stevens et al., 2010; Begum et al., 2012). Fascinatingly, these epigenetic alterations of GR persisted up to 5 years of age in adult offspring and were accompanied by decreased POMC mRNA expression. These results show the enduring nature of the impact on expression changes in the neuronal networks regulating food intake and energy expenditure (Begum et al., 2013). Interestingly, epigenetic changes in the GR gene as a result of altered maternal nutrition have been observed in other tissues such as liver and hippocampus. These results indicate programming effects on the methylation patterns of tissue-specific alternate first exons or promoters of the GR gene in the offspring (Lillycrop et al., 2005, 2007; Ke et al., 2011). Lillycrop et al. (2005) demonstrated that folic acid supplementation to a protein-restricted maternal diet prevented the epigenetic changes observed in the GR gene in the liver of the offspring. These results suggest the role of one-carbon metabolism in the epigenetic phenotype of the offspring (Lillycrop et al., 2005). Therefore, dietary alterations of these nutrients could provide a link between maternal diet and changes in the offspring's epigenetic signature.
Epigenetic Alterations in Other Hypothalamic Genes as a Result of Programming
Altered nutritional experiences during fetal or postnatal life impact on the epigenetic status of the other key hypothalamic genes involved in energy balance. For example, Mahmood et al. (2013) have shown that feeding new born rats with high-carbohydrate formula milk resulted in increased mRNA expression of NPY. This correlated with epigenetic alterations such as hypomethylation of specific CpG sites and increased H3K9 acetylation in the proximal promoter region of NPY and was associated with adult-onset obesity (Mahmood et al., 2013). Interestingly, overfeeding during neonatal life leads to hypermethylation in the promoter of the hypothalamic insulin receptor involved in the regulation of food intake and metabolism (Plagemann et al., 2010). Another study has shown that feeding mice with high fat diet during pregnancy and lactation resulted in global and promoter hypomethylation of hypothalamic dopamine and opioid-related genes in the offspring, leading to increased expression of these genes. These offspring showed more preference for palatable food rich in sucrose and fat (Vucetic et al., 2010). This suggests that dietary modulation during critical stages of the growth period can also affect epigenetic regulation of genes involved in food intake, body weight, and energy homeostasis thereby subsequent risk of developing obesity and related disorders.
Mechanisms of Changes Induced in the Epigenome as a Result of Programming
The precise mechanisms causing epigenetic changes induced by developmental programming is largely unknown. Altered levels of different nutrients, hormones or stress are likely to be the mediators of downstream signals of environmental insults. Micronutrients such as folate, choline, methionine, and vitamins are methyl group donors that contribute to the production of SAM through one-carbon metabolism. Since SAM is the universal methyl donor for both DNA and protein methyltransferases, variation in its levels can affect methylation patterns. For example, methyl donor supplementation to a high fat maternal diet prevented adverse effects such as body weight gain, behavioural changes and changes in DNA methylation and gene expression in the brain that were observed in the offspring of dams fed high fat diet (Carlin et al., 2013). Another way by which dietary factors can modify the epigenome is by modulating the expression or activity of epigenetic modifiers such as DNMTs. Accordingly, folic acid and other methyl group donors have also been shown to alter the expression levels of DNMT1, DNMT3a, and methyl CpG binding proteins involved in epigenetic gene silencing (Ghoshal et al., 2006; Lillycrop et al., 2007). The activities of other epigenetic enzymes such as HATs, HDACs, and demethylases are also susceptible to changes in the levels of cofactors such as acetyl-CoA, NAD, FAD, α-ketoglutarate and thus respond to altered nutrient intake (reviewed in Kaelin and McKnight, 2013). Additionally, evidence shows that both glucose and glucose metabolism induce genome-wide epigenetic alterations (Pirola et al., 2011). Therefore, it is conceivable that dietary factors can influence DNA methylation and histone modifications and thus affect signaling mechanisms regulating energy balance. However, despite this evidence, it is still not clear how fetal programming targets gene and tissue specific epigenetic modifications associated with metabolic disorders. It could be that the intensity and timing of the insult during early embryogenesis may impact the epigenetic alterations in the key metabolic genes in developing tissues. However, future studies are needed to understand the mechanisms by which the offspring's epigenomic configuration could be altered by maternal diet.
Transgenerational Effects on Epigenetic Programming of Obesity
Evidence from human and animal studies indicate that developmental programming of obesity and other metabolic disorders is a transgenerational phenomenon and the transmission of effects to subsequent generations may occur even in the absence of exposure to adverse conditions (Aiken and Ozanne, 2014). Although mechanisms underlying transgenerational transmission of programming are not clear, evidence shows that it could be due to epigenetic inheritance via both maternal and paternal lineages (Vickers, 2014). The involvement of epigenetic mechanisms in transgenerational amplification of obesity was further confirmed when methyl donor supplementation (e.g., folic acid) during development prevented the increase in body weight in subsequent generations caused by maternal obesity (Waterland et al., 2008). Several studies using animal models have shown transgenerational effects of maternal under- or over-nutrition on glucose metabolism (Martin et al., 2000; Benyshek et al., 2004; Gniuli et al., 2008; Pinheiro et al., 2008; Jimenez-Chillaron et al., 2009) and cardiovascular and HPA axis function in F2 offspring (Bertram et al., 2008). However, the effect on appetite regulation in subsequent generations has not been widely studied and therefore requires further investigation. A complete review of transgenerational effects on epigenetic programming of obesity is outside the scope of this review. However, understanding the mechanisms of transgenerational inheritance is important for the development of intervention strategies to modulate the effects of developmental programming of offspring, grandoffspring and beyond.
Conclusion
The compelling evidence reviewed here clearly shows that early life nutritional imbalance can induce persistent alterations, determining the permanent risk of obesity and other metabolic disorders in later life. Furthermore, epigenetic mechanisms might play an important role in these processes. Maternal under- and over-nutrition cause altered epigenetic regulation of genes that control food intake and energy expenditure in the hypothalamic neural network resulting in lifelong metabolic changes (Figure 2). This clearly suggests that the rise in the obesity epidemic could be at least in part explained by early life nutritional imbalances. The timing and the intensity of such events also seem to have a significant role to play in this process. This has led to the hypothesis that “we are what we eat and also what our mothers ate.” Our understanding of how environmental stimuli such as nutrition can induce epigenetic alterations is far from complete and further work is required in this area. However, given that there is clear evidence that these epigenetic modifications are reversible, there are possibilities that intervention strategies could combat the rise in obesity.
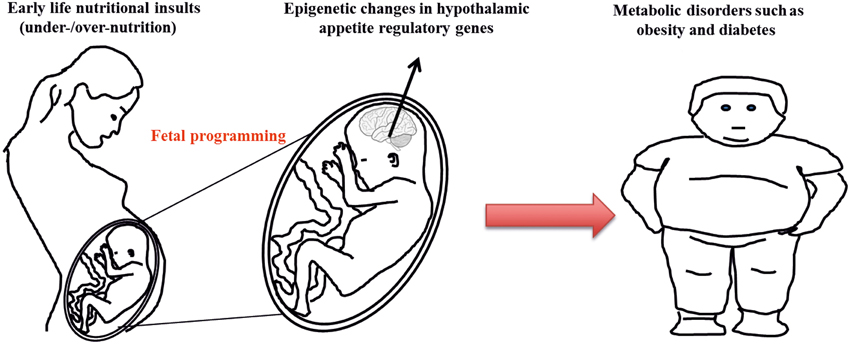
Figure 2. Epigenetic programming of obesity. Altered nutritional milieu at critical developmental period can program alterations in the epigenetic signature of hypothalamic appetite regulatory genes leading to changes in their gene expression pattern and increased susceptibility to metabolic disorders such as obesity and diabetes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Barbara Mawer Endowment fund and UK National Institute of Health Research Manchester Biomedical Research Center.
References
Abbasi, F., Brown, B. W. Jr., Lamendola, C., McLaughlin, T., and Reaven, G. M. (2002). Relationship between obesity, insulin resistance, and coronary heart disease risk. J. Am. Coll. Cardiol. 40, 937–943. doi: 10.1016/S0735-1097(02)02051-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aiken, C. E., and Ozanne, S. E. (2014). Transgenerational developmental programming. Hum. Reprod. Update 20, 63–75. doi: 10.1093/humupd/dmt043
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Altobelli, G., Bogdarina, I. G., Stupka, E., Clark, A. J., and Langley-Evans, S. (2013). Genome-wide methylation and gene expression changes in newborn rats following maternal protein restriction and reversal by folic acid. PLoS ONE 8:e82989. doi: 10.1371/journal.pone.0082989
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Amos, W., Driscoll, E., and Hoffman, J. I. (2011). Candidate genes versus genome-wide associations: which are better for detecting genetic susceptibility to infectious disease? Proc. Biol. Sci. 278, 1183–1188. doi: 10.1098/rspb.2010.1920
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Asnicar, M. A., Smith, D. P., Yang, D. D., Heiman, M. L., Fox, N., Chen, Y. F., et al. (2001). Absence of cocaine- and amphetamine-regulated transcript results in obesity in mice fed a high caloric diet. Endocrinology 142, 4394–4400. doi: 10.1210/endo.142.10.8416
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barker, D. J. (2005). The developmental origins of insulin resistance. Horm. Res. 64(Suppl. 3), 2–7. doi: 10.1159/000089311
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barker, D. J., Bull, A. R., Osmond, C., and Simmonds, S. J. (1990). Fetal and placental size and risk of hypertension in adult life. BMJ 301, 259–262. doi: 10.1136/bmj.301.6746.259
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barker, D. J., Gluckman, P. D., Godfrey, K. M., Harding, J. E., Owens, J. A., and Robinson, J. S. (1993). Fetal nutrition and cardiovascular disease in adult life. Lancet 341, 938–941. doi: 10.1016/0140-6736(93)91224-A
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Beaulieu, S., Gagne, B., and Barden, N. (1988). Glucocorticoid regulation of proopiomelanocortin messenger ribonucleic acid content of rat hypothalamus. Mol. Endocrinol. 2, 727–731. doi: 10.1210/mend-2-8-727
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Begum, G., Davies, A., Stevens, A., Oliver, M., Jaquiery, A., Challis, J., et al. (2013). Maternal undernutrition programs tissue-specific epigenetic changes in the glucocorticoid receptor in adult offspring. Endocrinology 154, 4560–4569. doi: 10.1210/en.2013-1693
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Begum, G., Stevens, A., Smith, E. B., Connor, K., Challis, J. R., Bloomfield, F., et al. (2012). Epigenetic changes in fetal hypothalamic energy regulating pathways are associated with maternal undernutrition and twinning. FASEB J. 26, 1694–1703. doi: 10.1096/fj.11-198762
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Benoit, S. C., Air, E. L., Coolen, L. M., Strauss, R., Jackman, A., Clegg, D. J., et al. (2002). The catabolic action of insulin in the brain is mediated by melanocortins. J. Neurosci. 22, 9048–9052.
Benyshek, D. C., Johnston, C. S., and Martin, J. F. (2004). Post-natal diet determines insulin resistance in fetally malnourished, low birthweight rats (F1) but diet does not modify the insulin resistance of their offspring (F2). Life Sci. 74, 3033–3041. doi: 10.1016/j.lfs.2003.11.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bertram, C., Khan, O., Ohri, S., Phillips, D. I., Matthews, S. G., and Hanson, M. A. (2008). Transgenerational effects of prenatal nutrient restriction on cardiovascular and hypothalamic-pituitary-adrenal function. J. Physiol. 586, 2217–2229. doi: 10.1113/jphysiol.2007.147967
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bird, A. (2002). DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21. doi: 10.1101/gad.947102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Blackmore, H. L., and Ozanne, S. E. (2013). Maternal diet-induced obesity and offspring cardiovascular health. J. Dev. Orig. Health Dis. 4, 338–347. doi: 10.1017/S2040174412000761
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Blondel, B., Kogan, M. D., Alexander, G. R., Dattani, N., Kramer, M. S., Macfarlane, A., et al. (2002). The impact of the increasing number of multiple births on the rates of preterm birth and low birthweight: an international study. Am. J. Public Health 92, 1323–1330. doi: 10.2105/AJPH.92.8.1323
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bouret, S. G. (2009). Early life origins of obesity: role of hypothalamic programming. J. Pediatr. Gastroenterol. Nutr. 48(Suppl. 1), S31–S38. doi: 10.1097/MPG.0b013e3181977375
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bouret, S. G. (2010). Role of early hormonal and nutritional experiences in shaping feeding behavior and hypothalamic development. J. Nutr. 140, 653–657. doi: 10.3945/jn.109.112433
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bouret, S. G., Draper, S. J., and Simerly, R. B. (2004). Trophic action of leptin on hypothalamic neurons that regulate feeding. Science 304, 108–110. doi: 10.1126/science.1095004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bouret, S. G., Gorski, J. N., Patterson, C. M., Chen, S., Levin, B. E., and Simerly, R. B. (2008). Hypothalamic neural projections are permanently disrupted in diet-induced obese rats. Cell Metab. 7, 179–185. doi: 10.1016/j.cmet.2007.12.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Boyes, J., and Bird, A. (1991). DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell 64, 1123–1134. doi: 10.1016/0092-8674(91)90267-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Breton, C., Lukaszewski, M. A., Risold, P. Y., Enache, M., Guillemot, J., Riviere, G., et al. (2009). Maternal prenatal undernutrition alters the response of POMC neurons to energy status variation in adult male rat offspring. Am. J. Physiol. Endocrinol. Metab. 296, E462–472. doi: 10.1152/ajpendo.90740.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Butler, A. A., Marks, D. L., Fan, W., Kuhn, C. M., Bartolome, M., and Cone, R. D. (2001). Melanocortin-4 receptor is required for acute homeostatic responses to increased dietary fat. Nat. Neurosci. 4, 605–611. doi: 10.1038/88423
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Campfield, L. A., Smith, F. J., Guisez, Y., Devos, R., and Burn, P. (1995). Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science 269, 546–549. doi: 10.1126/science.7624778
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Carlin, J., George, R., and Reyes, T. M. (2013). Methyl donor supplementation blocks the adverse effects of maternal high fat diet on offspring physiology. PLoS ONE 8:e63549. doi: 10.1371/journal.pone.0063549
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Challis, B. G., Pritchard, L. E., Creemers, J. W., Delplanque, J., Keogh, J. M., Luan, J., et al. (2002). A missense mutation disrupting a dibasic prohormone processing site in pro-opiomelanocortin (POMC) increases susceptibility to early-onset obesity through a novel molecular mechanism. Hum. Mol. Genet. 11, 1997–2004. doi: 10.1093/hmg/11.17.1997
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chamson-Reig, A., Thyssen, S. M., Arany, E., and Hill, D. J. (2006). Altered pancreatic morphology in the offspring of pregnant rats given reduced dietary protein is time and gender specific. J. Endocrinol. 191, 83–92. doi: 10.1677/joe.1.06754
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, H., Simar, D., Lambert, K., Mercier, J., and Morris, M. J. (2008). Maternal and postnatal overnutrition differentially impact appetite regulators and fuel metabolism. Endocrinology 149, 5348–5356. doi: 10.1210/en.2008-0582
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, H. Y., Trumbauer, M. E., Chen, A. S., Weingarth, D. T., Adams, J. R., Frazier, E. G., et al. (2004). Orexigenic action of peripheral ghrelin is mediated by neuropeptide Y and agouti-related protein. Endocrinology 145, 2607–2612. doi: 10.1210/en.2003-1596
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chiarelli, F., and Marcovecchio, M. L. (2008). Insulin resistance and obesity in childhood. Eur. J. Endocrinol. 159(Suppl. 1), S67–S74. doi: 10.1530/EJE-08-0245
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chronwall, B. M., Dimaggio, D. A., Massari, V. J., Pickel, V. M., Ruggiero, D. A., and O'Donohue, T. L. (1985). The anatomy of neuropeptide-Y-containing neurons in rat brain. Neuroscience 15, 1159–1181. doi: 10.1016/0306-4522(85)90260-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Clark, J. T., Kalra, P. S., Crowley, W. R., and Kalra, S. P. (1984). Neuropeptide Y and human pancreatic polypeptide stimulate feeding behavior in rats. Endocrinology 115, 427–429. doi: 10.1210/endo-115-1-427
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Clegg, D. J., Riedy, C. A., Smith, K. A., Benoit, S. C., and Woods, S. C. (2003). Differential sensitivity to central leptin and insulin in male and female rats. Diabetes 52, 682–687. doi: 10.2337/diabetes.52.3.682
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Coll, A. P., Challis, B. G., Lopez, M., Piper, S., Yeo, G. S., and O'Rahilly, S. (2005). Proopiomelanocortin-deficient mice are hypersensitive to the adverse metabolic effects of glucocorticoids. Diabetes 54, 2269–2276. doi: 10.2337/diabetes.54.8.2269
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Collden, G., Balland, E., Parkash, J., Caron, E., Langlet, F., Prevot, V., et al. (2015). Neonatal overnutrition causes early alterations in the central response to peripheral ghrelin. Mol. Metab. 4, 15–24. doi: 10.1016/j.molmet.2014.10.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Considine, R. V., Sinha, M. K., Heiman, M. L., Kriauciunas, A., Stephens, T. W., Nyce, M. R., et al. (1996). Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 334, 292–295. doi: 10.1056/NEJM199602013340503
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Corander, M. P., Rimmington, D., Challis, B. G., O'Rahilly, S., and Coll, A. P. (2011). Loss of agouti-related peptide does not significantly impact the phenotype of murine POMC deficiency. Endocrinology 152, 1819–1828. doi: 10.1210/en.2010-1450
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Couceyro, P. R., Koylu, E. O., and Kuhar, M. J. (1997). Further studies on the anatomical distribution of CART by in situ hybridization. J. Chem. Neuroanat. 12, 229–241. doi: 10.1016/S0891-0618(97)00212-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cowley, M. A., Smart, J. L., Rubinstein, M., Cerdan, M. G., Diano, S., Horvath, T. L., et al. (2001). Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411, 480–484. doi: 10.1038/35078085
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Creemers, J. W., Lee, Y. S., Oliver, R. L., Bahceci, M., Tuzcu, A., Gokalp, D., et al. (2008). Mutations in the amino-terminal region of proopiomelanocortin (POMC) in patients with early-onset obesity impair POMC sorting to the regulated secretory pathway. J. Clin. Endocrinol. Metab. 93, 4494–4499. doi: 10.1210/jc.2008-0954
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cripps, R. L., Martin-Gronert, M. S., Archer, Z. A., Hales, C. N., Mercer, J. G., and Ozanne, S. E. (2009). Programming of hypothalamic neuropeptide gene expression in rats by maternal dietary protein content during pregnancy and lactation. Clin. Sci. 117, 85–93. doi: 10.1042/CS20080393
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Crudo, A., Suderman, M., Moisiadis, V. G., Petropoulos, S., Kostaki, A., Hallett, M., et al. (2013). Glucocorticoid programming of the fetal male hippocampal epigenome. Endocrinology 154, 1168–1180. doi: 10.1210/en.2012-1980
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Curhan, G. C., Chertow, G. M., Willett, W. C., Spiegelman, D., Colditz, G. A., Manson, J. E., et al. (1996). Birth weight and adult hypertension and obesity in women. Circulation 94, 1310–1315. doi: 10.1161/01.CIR.94.6.1310
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Delage, B., and Dashwood, R. H. (2008). Dietary manipulation of histone structure and function. Annu. Rev. Nutr. 28, 347–366. doi: 10.1146/annurev.nutr.28.061807.155354
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Delahaye, F., Breton, C., Risold, P. Y., Enache, M., Dutriez-Casteloot, I., Laborie, C., et al. (2008). Maternal perinatal undernutrition drastically reduces postnatal leptin surge and affects the development of arcuate nucleus proopiomelanocortin neurons in neonatal male rat pups. Endocrinology 149, 470–475. doi: 10.1210/en.2007-1263
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Department of Health. (2014). Adult Obesity and Type 2 Diabetes [Online]. Available online at: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/338934/Adult_obesity_and_type_2_diabetes_.pdf (Accessed).
de Rooij, S. R., Painter, R. C., Phillips, D. I., Osmond, C., Michels, R. P., Godsland, I. F., et al. (2006). Impaired insulin secretion after prenatal exposure to the Dutch famine. Diabetes Care 29, 1897–1901. doi: 10.2337/dc06-0460
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
de Souza, F. S., Santangelo, A. M., Bumaschny, V., Avale, M. E., Smart, J. L., Low, M. J., et al. (2005). Identification of neuronal enhancers of the proopiomelanocortin gene by transgenic mouse analysis and phylogenetic footprinting. Mol. Cell. Biol. 25, 3076–3086. doi: 10.1128/MCB.25.8.3076-3086.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Douglass, J., McKinzie, A. A., and Couceyro, P. (1995). PCR differential display identifies a rat brain mRNA that is transcriptionally regulated by cocaine and amphetamine. J. Neurosci. 15, 2471–2481.
Drake, A. J., and Reynolds, R. M. (2010). Impact of maternal obesity on offspring obesity and cardiometabolic disease risk. Reproduction 140, 387–398. doi: 10.1530/REP-10-0077
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Drouin, J., Sun, Y. L., Chamberland, M., Gauthier, Y., De Lean, A., Nemer, M., et al. (1993). Novel glucocorticoid receptor complex with DNA element of the hormone-repressed POMC gene. EMBO J. 12, 145–156.
Duque-Guimaraes, D. E., and Ozanne, S. E. (2013). Nutritional programming of insulin resistance: causes and consequences. Trends Endocrinol. Metab. 24, 525–535. doi: 10.1016/j.tem.2013.05.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ekblad, E. (2006). CART in the enteric nervous system. Peptides 27, 2024–2030. doi: 10.1016/j.peptides.2005.12.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Elmquist, J. K., Bjorbaek, C., Ahima, R. S., Flier, J. S., and Saper, C. B. (1998). Distributions of leptin receptor mRNA isoforms in the rat brain. J. Comp. Neurol. 395, 535–547.
Erickson, J. C., Hollopeter, G., and Palmiter, R. D. (1996). Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science 274, 1704–1707. doi: 10.1126/science.274.5293.1704
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Farooqi, I. S., Drop, S., Clements, A., Keogh, J. M., Biernacka, J., Lowenbein, S., et al. (2006). Heterozygosity for a POMC-null mutation and increased obesity risk in humans. Diabetes 55, 2549–2553. doi: 10.2337/db06-0214
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Farooqi, I. S., Jebb, S. A., Langmack, G., Lawrence, E., Cheetham, C. H., Prentice, A. M., et al. (1999). Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N. Engl. J. Med. 341, 879–884. doi: 10.1056/NEJM199909163411204
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Franke, K., Harder, T., Aerts, L., Melchior, K., Fahrenkrog, S., Rodekamp, E., et al. (2005). “Programming” of orexigenic and anorexigenic hypothalamic neurons in offspring of treated and untreated diabetic mother rats. Brain Res. 1031, 276–283. doi: 10.1016/j.brainres.2004.11.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gagnon, R. (2003). Placental insufficiency and its consequences. Eur. J. Obstet. Gynecol. Reprod. Biol. 110(Suppl. 1), S99–S107. doi: 10.1016/S0301-2115(03)00179-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Garcia, A. P., Palou, M., Priego, T., Sanchez, J., Palou, A., and Pico, C. (2010). Moderate caloric restriction during gestation results in lower arcuate nucleus NPY- and alphaMSH-neurons and impairs hypothalamic response to fed/fasting conditions in weaned rats. Diabetes Obes. Metab. 12, 403–413. doi: 10.1111/j.1463-1326.2009.01174.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ghoshal, K., Li, X., Datta, J., Bai, S., Pogribny, I., Pogribny, M., et al. (2006). A folate- and methyl-deficient diet alters the expression of DNA methyltransferases and methyl CpG binding proteins involved in epigenetic gene silencing in livers of F344 rats. J. Nutr. 136, 1522–1527.
Gluckman, P. D., Hanson, M. A., Buklijas, T., Low, F. M., and Beedle, A. S. (2009). Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat. Rev. Endocrinol. 5, 401–408. doi: 10.1038/nrendo.2009.102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gniuli, D., Calcagno, A., Caristo, M. E., Mancuso, A., Macchi, V., Mingrone, G., et al. (2008). Effects of high-fat diet exposure during fetal life on type 2 diabetes development in the progeny. J. Lipid Res. 49, 1936–1945. doi: 10.1194/jlr.M800033-JLR200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goto, M., Arima, H., Watanabe, M., Hayashi, M., Banno, R., Sato, I., et al. (2006). Ghrelin increases neuropeptide Y and agouti-related peptide gene expression in the arcuate nucleus in rat hypothalamic organotypic cultures. Endocrinology 147, 5102–5109. doi: 10.1210/en.2006-0104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gropp, E., Shanabrough, M., Borok, E., Xu, A. W., Janoschek, R., Buch, T., et al. (2005). Agouti-related peptide-expressing neurons are mandatory for feeding. Nat. Neurosci. 8, 1289–1291. doi: 10.1038/nn1548
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guo, F., and Jen, K. L. (1995). High-fat feeding during pregnancy and lactation affects offspring metabolism in rats. Physiol. Behav. 57, 681–686. doi: 10.1016/0031-9384(94)00342-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guo, L., Munzberg, H., Stuart, R. C., Nillni, E. A., and Bjorbaek, C. (2004). N-acetylation of hypothalamic alpha-melanocyte-stimulating hormone and regulation by leptin. Proc. Natl. Acad. Sci. U.S.A. 101, 11797–11802. doi: 10.1073/pnas.0403165101
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gyengesi, E., Liu, Z. W., D'Agostino, G., Gan, G., Horvath, T. L., Gao, X. B., et al. (2010). Corticosterone regulates synaptic input organization of POMC and NPY/AgRP neurons in adult mice. Endocrinology 151, 5395–5402. doi: 10.1210/en.2010-0681
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hagan, M. M., Rushing, P. A., Pritchard, L. M., Schwartz, M. W., Strack, A. M., Van Der Ploeg, L. H., et al. (2000). Long-term orexigenic effects of AgRP-(83—132) involve mechanisms other than melanocortin receptor blockade. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R47–R52.
Hales, C. N., and Barker, D. J. (2001). The thrifty phenotype hypothesis. Br. Med. Bull. 60, 5–20. doi: 10.1093/bmb/60.1.5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hales, C. N., Barker, D. J., Clark, P. M., Cox, L. J., Fall, C., Osmond, C., et al. (1991). Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 303, 1019–1022. doi: 10.1136/bmj.303.6809.1019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hallschmid, M., Benedict, C., Schultes, B., Born, J., and Kern, W. (2008). Obese men respond to cognitive but not to catabolic brain insulin signaling. Int. J. Obes. (Lond). 32, 275–282. doi: 10.1038/sj.ijo.0803722
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hanson, M. A., and Gluckman, P. D. (2014). Early developmental conditioning of later health and disease: physiology or pathophysiology? Physiol. Rev. 94, 1027–1076. doi: 10.1152/physrev.00029.2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Harno, E., Cottrell, E. C., Keevil, B. G., Deschoolmeester, J., Bohlooly, Y. M., Andersen, H., et al. (2013). 11-Dehydrocorticosterone causes metabolic syndrome, which is prevented when 11beta-HSD1 is knocked out in livers of male mice. Endocrinology 154, 3599–3609. doi: 10.1210/en.2013-1362
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hermann, A., Gowher, H., and Jeltsch, A. (2004). Biochemistry and biology of mammalian DNA methyltransferases. Cell. Mol. Life Sci. 61, 2571–2587. doi: 10.1007/s00018-004-4201-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hulsey, M. G., Pless, C. M., White, B. D., and Martin, R. J. (1995). ICV administration of anti-NPY antisense oligonucleotide: effects on feeding behavior, body weight, peptide content and peptide release. Regul. Pept. 59, 207–214. doi: 10.1016/0167-0115(95)00110-W
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huszar, D., Lynch, C. A., Fairchild-Huntress, V., Dunmore, J. H., Fang, Q., Berkemeier, L. R., et al. (1997). Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88, 131–141. doi: 10.1016/S0092-8674(00)81865-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Illingworth, R. S., and Bird, A. P. (2009). CpG islands–“a rough guide”. FEBS Lett. 583, 1713–1720. doi: 10.1016/j.febslet.2009.04.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jaenisch, R., and Bird, A. (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33(Suppl.), 245–254. doi: 10.1038/ng1089
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jimenez-Chillaron, J. C., Isganaitis, E., Charalambous, M., Gesta, S., Pentinat-Pelegrin, T., Faucette, R. R., et al. (2009). Intergenerational transmission of glucose intolerance and obesity by in utero undernutrition in mice. Diabetes 58, 460–468. doi: 10.2337/db08-0490
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jones, P. A. (2012). Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492. doi: 10.1038/nrg3230
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jones, P. L., Veenstra, G. J., Wade, P. A., Vermaak, D., Kass, S. U., Landsberger, N., et al. (1998). Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19, 187–191. doi: 10.1038/561
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kaelin, W. G. Jr., and McKnight, S. L. (2013). Influence of metabolism on epigenetics and disease. Cell 153, 56–69. doi: 10.1016/j.cell.2013.03.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kamegai, J., Tamura, H., Shimizu, T., Ishii, S., Sugihara, H., and Wakabayashi, I. (2001). Chronic central infusion of ghrelin increases hypothalamic neuropeptide Y and Agouti-related protein mRNA levels and body weight in rats. Diabetes 50, 2438–2443. doi: 10.2337/diabetes.50.11.2438
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Karatsoreos, I. N., Bhagat, S. M., Bowles, N. P., Weil, Z. M., Pfaff, D. W., and McEwen, B. S. (2010). Endocrine and physiological changes in response to chronic corticosterone: a potential model of the metabolic syndrome in mouse. Endocrinology 151, 2117–2127. doi: 10.1210/en.2009-1436
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kasacka, I., Janiuk, I., Lewandowska, A., Bekisz, A., and Lebkowski, W. (2012). Distribution pattern of CART-containing neurons and cells in the human pancreas. Acta Histochem. 114, 695–699. doi: 10.1016/j.acthis.2011.12.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ke, X., McKnight, R. A., Caprau, D., O'Grady, S., Fu, Q., Yu, X., et al. (2011). Intrauterine growth restriction affects hippocampal dual specificity phosphatase 5 gene expression and epigenetic characteristics. Physiol. Genomics 43, 1160–1169. doi: 10.1152/physiolgenomics.00242.2010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, S. G., Lee, B., Kim, D. H., Kim, J., Lee, S., Lee, S. K., et al. (2013). Control of energy balance by hypothalamic gene circuitry involving two nuclear receptors, neuron-derived orphan receptor 1 and glucocorticoid receptor. Mol. Cell. Biol. 33, 3826–3834. doi: 10.1128/MCB.00385-13
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kirk, S. L., Samuelsson, A. M., Argenton, M., Dhonye, H., Kalamatianos, T., Poston, L., et al. (2009). Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS ONE 4:e5870. doi: 10.1371/journal.pone.0005870
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128, 693–705. doi: 10.1016/j.cell.2007.02.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Krashes, M. J., Shah, B. P., Koda, S., and Lowell, B. B. (2013). Rapid versus delayed stimulation of feeding by the endogenously released AgRP neuron mediators GABA, NPY, and AgRP. Cell Metab. 18, 588–595. doi: 10.1016/j.cmet.2013.09.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Krude, H., Biebermann, H., Luck, W., Horn, R., Brabant, G., and Gruters, A. (1998). Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 19, 155–157. doi: 10.1038/509
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kuehnen, P., Mischke, M., Wiegand, S., Sers, C., Horsthemke, B., Lau, S., et al. (2012). An Alu element-associated hypermethylation variant of the POMC gene is associated with childhood obesity. PLoS Genet. 8:e1002543. doi: 10.1371/journal.pgen.1002543
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Langley-Evans, S. C. (2009). Nutritional programming of disease: unravelling the mechanism. J. Anat. 215, 36–51. doi: 10.1111/j.1469-7580.2008.00977.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lau, J., and Herzog, H. (2014). CART in the regulation of appetite and energy homeostasis. Front. Neurosci. 8:313. doi: 10.3389/fnins.2014.00313
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, B., Kim, S. G., Kim, J., Choi, K. Y., Lee, S., Lee, S. K., et al. (2013). Brain-specific homeobox factor as a target selector for glucocorticoid receptor in energy balance. Mol. Cell. Biol. 33, 2650–2658. doi: 10.1128/MCB.00094-13
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, C., McDonald, T. J., Wu, G., Nijland, M. J., and Nathanielsz, P. W. (2013). Intrauterine growth restriction alters term fetal baboon hypothalamic appetitive peptide balance. J. Endocrinol. 217, 275–282. doi: 10.1530/JOE-13-0012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lillycrop, K. A., Phillips, E. S., Jackson, A. A., Hanson, M. A., and Burdge, G. C. (2005). Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J. Nutr. 135, 1382–1386.
Lillycrop, K. A., Slater-Jefferies, J. L., Hanson, M. A., Godfrey, K. M., Jackson, A. A., and Burdge, G. C. (2007). Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br. J. Nutr. 97, 1064–1073. doi: 10.1017/S000711450769196X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lopez, M., Seoane, L. M., Tovar, S., Garcia, M. C., Nogueiras, R., Dieguez, C., et al. (2005). A possible role of neuropeptide Y, agouti-related protein and leptin receptor isoforms in hypothalamic programming by perinatal feeding in the rat. Diabetologia 48, 140–148. doi: 10.1007/s00125-004-1596-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Luquet, S., Perez, F. A., Hnasko, T. S., and Palmiter, R. D. (2005). NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science 310, 683–685. doi: 10.1126/science.1115524
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mahmood, S., Smiraglia, D. J., Srinivasan, M., and Patel, M. S. (2013). Epigenetic changes in hypothalamic appetite regulatory genes may underlie the developmental programming for obesity in rat neonates subjected to a high-carbohydrate dietary modification. J. Dev. Orig. Health Dis. 4, 479–490. doi: 10.1017/S2040174413000238
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Makimura, H., Mizuno, T. M., Roberts, J., Silverstein, J., Beasley, J., and Mobbs, C. V. (2000). Adrenalectomy reverses obese phenotype and restores hypothalamic melanocortin tone in leptin-deficient ob/ob mice. Diabetes 49, 1917–1923. doi: 10.2337/diabetes.49.11.1917
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marchington, D., Rothwell, N. J., Stock, M. J., and York, D. A. (1983). Energy balance, diet-induced thermogenesis and brown adipose tissue in lean and obese (fa/fa) Zucker rats after adrenalectomy. J. Nutr. 113, 1395–1402.
Marks, J. L., Porte, D. Jr., Stahl, W. L., and Baskin, D. G. (1990). Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology 127, 3234–3236. doi: 10.1210/endo-127-6-3234
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Martin, J. F., Johnston, C. S., Han, C. T., and Benyshek, D. C. (2000). Nutritional origins of insulin resistance: a rat model for diabetes-prone human populations. J. Nutr. 130, 741–744.
Matthews, S. G., and Phillips, D. I. (2012). Transgenerational inheritance of stress pathology. Exp. Neurol. 233, 95–101. doi: 10.1016/j.expneurol.2011.01.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McMillen, I. C., Adam, C. L., and Muhlhausler, B. S. (2005). Early origins of obesity: programming the appetite regulatory system. J. Physiol. 565, 9–17. doi: 10.1113/jphysiol.2004.081992
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Misaki, N., Higuchi, H., Yamagata, K., and Miki, N. (1992). Identification of glucocorticoid responsive elements (GREs) at far upstream of rat NPY gene. Neurochem. Int. 21, 185–189. doi: 10.1016/0197-0186(92)90145-H
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mitchell, V., Bouret, S., Beauvillain, J. C., Schilling, A., Perret, M., Kordon, C., et al. (2001). Comparative distribution of mRNA encoding the growth hormone secretagogue-receptor (GHS-R) in Microcebus murinus (Primate, lemurian) and rat forebrain and pituitary. J. Comp. Neurol. 429, 469–489.
Mizuno, T. M., and Mobbs, C. V. (1999). Hypothalamic agouti-related protein messenger ribonucleic acid is inhibited by leptin and stimulated by fasting. Endocrinology 140, 814–817. doi: 10.1210/en.140.2.814
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Moffett, M., Stanek, L., Harley, J., Rogge, G., Asnicar, M., Hsiung, H., et al. (2006). Studies of cocaine- and amphetamine-regulated transcript (CART) knockout mice. Peptides 27, 2037–2045. doi: 10.1016/j.peptides.2006.03.035
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Morgan, S. A., McCabe, E. L., Gathercole, L. L., Hassan-Smith, Z. K., Larner, D. P., Bujalska, I. J., et al. (2014). 11beta-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc. Natl. Acad. Sci. U.S.A. 111, E2482–E2491. doi: 10.1073/pnas.1323681111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Morris, B. J. (1989). Neuronal localisation of neuropeptide Y gene expression in rat brain. J. Comp. Neurol. 290, 358–368. doi: 10.1002/cne.902900305
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Morris, M. J., and Chen, H. (2009). Established maternal obesity in the rat reprograms hypothalamic appetite regulators and leptin signaling at birth. Int. J. Obes. (Lond). 33, 115–122. doi: 10.1038/ijo.2008.213
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Morton, G. J., and Schwartz, M. W. (2001). The NPY/AgRP neuron and energy homeostasis. Int. J. Obes. Relat. Metab. Disord 25(Suppl. 5), S56–S62. doi: 10.1038/sj.ijo.0801915
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mostyn, A., and Symonds, M. E. (2009). Early programming of adipose tissue function: a large-animal perspective. Proc. Nutr. Soc. 68, 393–400. doi: 10.1017/S002966510999022X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Muhlhausler, B. S., Hancock, S. N., Bloomfield, F. H., and Harding, R. (2011). Are twins growth restricted? Pediatr. Res. 70, 117–122. doi: 10.1038/pr.2011.342
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Munzberg, H., Huo, L., Nillni, E. A., Hollenberg, A. N., and Bjorbaek, C. (2003). Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology 144, 2121–2131. doi: 10.1210/en.2002-221037
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakazato, M., Murakami, N., Date, Y., Kojima, M., Matsuo, H., Kangawa, K., et al. (2001). A role for ghrelin in the central regulation of feeding. Nature 409, 194–198. doi: 10.1038/35051587
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nan, X., Ng, H. H., Johnson, C. A., Laherty, C. D., Turner, B. M., Eisenman, R. N., et al. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389. doi: 10.1038/30764
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Neitzke, U., Harder, T., Schellong, K., Melchior, K., Ziska, T., Rodekamp, E., et al. (2008). Intrauterine growth restriction in a rodent model and developmental programming of the metabolic syndrome: a critical appraisal of the experimental evidence. Placenta 29, 246–254. doi: 10.1016/j.placenta.2007.11.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Newell-Price, J. (2003). Proopiomelanocortin gene expression and DNA methylation: implications for Cushing's syndrome and beyond. J. Endocrinol. 177, 365–372. doi: 10.1677/joe.0.1770365
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Newell-Price, J., King, P., and Clark, A. J. (2001). The CpG island promoter of the human proopiomelanocortin gene is methylated in nonexpressing normal tissue and tumors and represses expression. Mol. Endocrinol. 15, 338–348. doi: 10.1210/mend.15.2.0599
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Oken, E., Rifas-Shiman, S. L., Field, A. E., Frazier, A. L., and Gillman, M. W. (2008). Maternal gestational weight gain and offspring weight in adolescence. Obstet. Gynecol. 112, 999–1006. doi: 10.1097/AOG.0b013e31818a5d50
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ombelet, W., Martens, G., De Sutter, P., Gerris, J., Bosmans, E., Ruyssinck, G., et al. (2006). Perinatal outcome of 12,021 singleton and 3108 twin births after non-IVF-assisted reproduction: a cohort study. Hum. Reprod. 21, 1025–1032. doi: 10.1093/humrep/dei419
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ong, K. K. (2006). Size at birth, postnatal growth and risk of obesity. Horm. Res. 65(Suppl. 3), 65–69. doi: 10.1159/000091508
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Patti, M. E. (2013). Intergenerational programming of metabolic disease: evidence from human populations and experimental animal models. Cell. Mol. Life Sci. 70, 1597–1608. doi: 10.1007/s00018-013-1298-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pettitt, D. J., and Jovanovic, L. (2001). Birth weight as a predictor of type 2 diabetes mellitus: the U-shaped curve. Curr. Diab. Rep. 1, 78–81. doi: 10.1007/s11892-001-0014-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Phillips, D. I., Davies, M. J., and Robinson, J. S. (2001). Fetal growth and the fetal origins hypothesis in twins—problems and perspectives. Twin Res. 4, 327–331. doi: 10.1375/1369052012669
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pinheiro, A. R., Salvucci, I. D., Aguila, M. B., and Mandarim-De-Lacerda, C. A. (2008). Protein restriction during gestation and/or lactation causes adverse transgenerational effects on biometry and glucose metabolism in F1 and F2 progenies of rats. Clin. Sci. 114, 381–392. doi: 10.1042/CS20070302
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pirola, L., Balcerczyk, A., Tothill, R. W., Haviv, I., Kaspi, A., Lunke, S., et al. (2011). Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res. 21, 1601–1615. doi: 10.1101/gr.116095.110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Plagemann, A., Harder, T., Brunn, M., Harder, A., Roepke, K., Wittrock-Staar, M., et al. (2009). Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome. J. Physiol. 587, 4963–4976. doi: 10.1113/jphysiol.2009.176156
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Plagemann, A., Harder, T., Melchior, K., Rake, A., Rohde, W., and Dorner, G. (1999a). Elevation of hypothalamic neuropeptide Y-neurons in adult offspring of diabetic mother rats. Neuroreport 10, 3211–3216.
Plagemann, A., Harder, T., Rake, A., Voits, M., Fink, H., Rohde, W., et al. (1999b). Perinatal elevation of hypothalamic insulin, acquired malformation of hypothalamic galaninergic neurons, and syndrome x-like alterations in adulthood of neonatally overfed rats. Brain Res. 836, 146–155.
Plagemann, A., Harder, T., Rake, A., Waas, T., Melchior, K., Ziska, T., et al. (1999c). Observations on the orexigenic hypothalamic neuropeptide Y-system in neonatally overfed weanling rats. J. Neuroendocrinol. 11, 541–546.
Plagemann, A., Roepke, K., Harder, T., Brunn, M., Harder, A., Wittrock-Staar, M., et al. (2010). Epigenetic malprogramming of the insulin receptor promoter due to developmental overfeeding. J. Perinat. Med. 38, 393–400. doi: 10.1515/jpm.2010.051
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Plum, L., Belgardt, B. F., and Bruning, J. C. (2006). Central insulin action in energy and glucose homeostasis. J. Clin. Invest. 116, 1761–1766. doi: 10.1172/JCI29063
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Poulsen, P., Vaag, A. A., Kyvik, K. O., Moller Jensen, D., and Beck-Nielsen, H. (1997). Low birth weight is associated with NIDDM in discordant monozygotic and dizygotic twin pairs. Diabetologia 40, 439–446. doi: 10.1007/s001250050698
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Qatanani, M., and Lazar, M. A. (2007). Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev. 21, 1443–1455. doi: 10.1101/gad.1550907
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ravelli, A. C., van Der Meulen, J. H., Osmond, C., Barker, D. J., and Bleker, O. P. (1999). Obesity at the age of 50 y in men and women exposed to famine prenatally. Am. J. Clin. Nutr. 70, 811–816.
Ravelli, G. P., Stein, Z. A., and Susser, M. W. (1976). Obesity in young men after famine exposure in utero and early infancy. N. Engl. J. Med. 295, 349–353. doi: 10.1056/NEJM197608122950701
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reynolds, R. M., Osmond, C., Phillips, D. I., and Godfrey, K. M. (2010). Maternal BMI, parity, and pregnancy weight gain: influences on offspring adiposity in young adulthood. J. Clin. Endocrinol. Metab. 95, 5365–5369. doi: 10.1210/jc.2010-0697
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ribel-Madsen, R., Fraga, M. F., Jacobsen, S., Bork-Jensen, J., Lara, E., Calvanese, V., et al. (2012). Genome-wide analysis of DNA methylation differences in muscle and fat from monozygotic twins discordant for type 2 diabetes. PLoS ONE 7:e51302. doi: 10.1371/journal.pone.0051302
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ross, M. G., and Desai, M. (2014). Developmental programming of appetite/satiety. Ann. Nutr. Metab. 64(Suppl. 1), 36–44. doi: 10.1159/000360508
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rossi, M., Kim, M. S., Morgan, D. G., Small, C. J., Edwards, C. M., Sunter, D., et al. (1998). A C-terminal fragment of Agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo. Endocrinology 139, 4428–4431. doi: 10.1210/endo.139.10.6332
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rumball, C. W., Oliver, M. H., Thorstensen, E. B., Jaquiery, A. L., Husted, S. M., Harding, J. E., et al. (2008). Effects of twinning and periconceptional undernutrition on late-gestation hypothalamic-pituitary-adrenal axis function in ovine pregnancy. Endocrinology 149, 1163–1172. doi: 10.1210/en.2007-1306
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Samuelsson, A. M., Matthews, P. A., Argenton, M., Christie, M. R., McConnell, J. M., Jansen, E. H., et al. (2008). Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension 51, 383–392. doi: 10.1161/HYPERTENSIONAHA.107.101477
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sanders, M., Fazzi, G., Janssen, G., Blanco, C., and De Mey, J. (2004). Prenatal stress changes rat arterial adrenergic reactivity in a regionally selective manner. Eur. J. Pharmacol. 488, 147–155. doi: 10.1016/j.ejphar.2004.02.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Savontaus, E., Conwell, I. M., and Wardlaw, S. L. (2002). Effects of adrenalectomy on AGRP, POMC, NPY and CART gene expression in the basal hypothalamus of fed and fasted rats. Brain Res. 958, 130–138. doi: 10.1016/S0006-8993(02)03674-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schneeberger, M., Gomis, R., and Claret, M. (2014). Hypothalamic and brainstem neuronal circuits controlling homeostatic energy balance. J. Endocrinol. 220, T25–T46. doi: 10.1530/JOE-13-0398
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schwartz, M. W., Sipols, A. J., Marks, J. L., Sanacora, G., White, J. D., Scheurink, A., et al. (1992). Inhibition of hypothalamic neuropeptide Y gene expression by insulin. Endocrinology 130, 3608–3616. doi: 10.1210/en.130.6.3608
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sharkey, D., Gardner, D. S., Symonds, M. E., and Budge, H. (2009). Maternal nutrient restriction during early fetal kidney development attenuates the renal innate inflammatory response in obese young adult offspring. Am. J. Physiol. Renal Physiol. 297, F1199–F1207. doi: 10.1152/ajprenal.00303.2009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shibasaki, T., Oda, T., Imaki, T., Ling, N., and Demura, H. (1993). Injection of anti-neuropeptide Y gamma-globulin into the hypothalamic paraventricular nucleus decreases food intake in rats. Brain Res. 601, 313–316. doi: 10.1016/0006-8993(93)91727-A
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shin, B. C., Dai, Y., Thamotharan, M., Gibson, L. C., and Devaskar, S. U. (2012). Pre- and postnatal calorie restriction perturbs early hypothalamic neuropeptide and energy balance. J. Neurosci. Res. 90, 1169–1182. doi: 10.1002/jnr.23013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shintani, M., Ogawa, Y., Ebihara, K., Aizawa-Abe, M., Miyanaga, F., Takaya, K., et al. (2001). Ghrelin, an endogenous growth hormone secretagogue, is a novel orexigenic peptide that antagonizes leptin action through the activation of hypothalamic neuropeptide Y/Y1 receptor pathway. Diabetes 50, 227–232. doi: 10.2337/diabetes.50.2.227
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sipols, A. J., Baskin, D. G., and Schwartz, M. W. (1995). Effect of intracerebroventricular insulin infusion on diabetic hyperphagia and hypothalamic neuropeptide gene expression. Diabetes 44, 147–151. doi: 10.2337/diab.44.2.147
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Snoeck, A., Remacle, C., Reusens, B., and Hoet, J. J. (1990). Effect of a low protein diet during pregnancy on the fetal rat endocrine pancreas. Biol. Neonate 57, 107–118. doi: 10.1159/000243170
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stanley, B. G., Kyrkouli, S. E., Lampert, S., and Leibowitz, S. F. (1986). Neuropeptide Y chronically injected into the hypothalamus: a powerful neurochemical inducer of hyperphagia and obesity. Peptides 7, 1189–1192. doi: 10.1016/0196-9781(86)90149-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Steculorum, S. M., and Bouret, S. G. (2011a). Developmental effects of ghrelin. Peptides 32, 2362–2366. doi: 10.1016/j.peptides.2011.06.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Steculorum, S. M., and Bouret, S. G. (2011b). Maternal diabetes compromises the organization of hypothalamic feeding circuits and impairs leptin sensitivity in offspring. Endocrinology 152, 4171–4179. doi: 10.1210/en.2011-1279
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Steculorum, S. M., Collden, G., Coupe, B., Croizier, S., Lockie, S., Andrews, Z. B., et al. (2015). Neonatal ghrelin programs development of hypothalamic feeding circuits. J. Clin. Invest. 125, 846–858. doi: 10.1172/JCI73688
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Steinberger, J., Daniels, S. R., American Heart Association Atherosclerosis, H., Obesity in the Young, C., and American Heart Association Diabetes, C. (2003). Obesity, insulin resistance, diabetes, and cardiovascular risk in children: an American Heart Association scientific statement from the Atherosclerosis, Hypertension, and Obesity in the Young Committee (Council on Cardiovascular Disease in the Young) and the Diabetes Committee (Council on Nutrition, Physical Activity, and Metabolism). Circulation 107, 1448–1453. doi: 10.1161/01.CIR.0000060923.07573.F2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stephens, T. W., Basinski, M., Bristow, P. K., Bue-Valleskey, J. M., Burgett, S. G., Craft, L., et al. (1995). The role of neuropeptide Y in the antiobesity action of the obese gene product. Nature 377, 530–532. doi: 10.1038/377530a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sterner, D. E., and Berger, S. L. (2000). Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64, 435–459. doi: 10.1128/MMBR.64.2.435-459.2000
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stevens, A., Begum, G., Cook, A., Connor, K., Rumball, C., Oliver, M., et al. (2010). Epigenetic changes in the hypothalamic proopiomelanocortin and glucocorticoid receptor genes in the ovine fetus after periconceptional undernutrition. Endocrinology 151, 3652–3664. doi: 10.1210/en.2010-0094
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stevens, A., Begum, G., and White, A. (2011). Epigenetic changes in the hypothalamic pro-opiomelanocortin gene: a mechanism linking maternal undernutrition to obesity in the offspring? Eur. J. Pharmacol. 660, 194–201. doi: 10.1016/j.ejphar.2010.10.111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Suter, M. A., Ma, J., Vuguin, P. M., Hartil, K., Fiallo, A., Harris, R. A., et al. (2014). In utero exposure to a maternal high-fat diet alters the epigenetic histone code in a murine model. Am. J. Obstet. Gynecol. 210, 463.e461–463.e411. doi: 10.1016/j.ajog.2013.10.097
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Symonds, M. E., and Budge, H. (2009). Nutritional models of the developmental programming of adult health and disease. Proc. Nutr. Soc. 68, 173–178. doi: 10.1017/S0029665109001049
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tamashiro, K. L., Terrillion, C. E., Hyun, J., Koenig, J. I., and Moran, T. H. (2009). Prenatal stress or high-fat diet increases susceptibility to diet-induced obesity in rat offspring. Diabetes 58, 1116–1125. doi: 10.2337/db08-1129
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Theys, N., Bouckenooghe, T., Ahn, M. T., Remacle, C., and Reusens, B. (2009). Maternal low-protein diet alters pancreatic islet mitochondrial function in a sex-specific manner in the adult rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1516–R1525. doi: 10.1152/ajpregu.00280.2009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Todd, S. E., Oliver, M. H., Jaquiery, A. L., Bloomfield, F. H., and Harding, J. E. (2009). Periconceptional undernutrition of ewes impairs glucose tolerance in their adult offspring. Pediatr. Res. 65, 409–413. doi: 10.1203/PDR.0b013e3181975efa
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Uchoa, E. T., Silva, L. E., de Castro, M., Antunes-Rodrigues, J., and Elias, L. L. (2012). Glucocorticoids are required for meal-induced changes in the expression of hypothalamic neuropeptides. Neuropeptides 46, 119–124. doi: 10.1016/j.npep.2012.02.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Udagawa, J., Hatta, T., Hashimoto, R., and Otani, H. (2007). Roles of leptin in prenatal and perinatal brain development. Congenit. Anom. (Kyoto). 47, 77–83. doi: 10.1111/j.1741-4520.2007.00150.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vickers, M. H. (2014). Developmental programming and transgenerational transmission of obesity. Ann. Nutr. Metab. 64(Suppl. 1), 26–34. doi: 10.1159/000360506
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vickers, M. H., Breier, B. H., Cutfield, W. S., Hofman, P. L., and Gluckman, P. D. (2000). Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am. J. Physiol. Endocrinol. Metab. 279, E83–E87.
Vogt, M. C., Paeger, L., Hess, S., Steculorum, S. M., Awazawa, M., Hampel, B., et al. (2014). Neonatal insulin action impairs hypothalamic neurocircuit formation in response to maternal high-fat feeding. Cell 156, 495–509. doi: 10.1016/j.cell.2014.01.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vrang, N. (2006). Anatomy of hypothalamic CART neurons. Peptides 27, 1970–1980. doi: 10.1016/j.peptides.2005.10.029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vucetic, Z., Kimmel, J., Totoki, K., Hollenbeck, E., and Reyes, T. M. (2010). Maternal high-fat diet alters methylation and gene expression of dopamine and opioid-related genes. Endocrinology 151, 4756–4764. doi: 10.1210/en.2010-0505
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wardlaw, S. L., McCarthy, K. C., and Conwell, I. M. (1998). Glucocorticoid regulation of hypothalamic proopiomelanocortin. Neuroendocrinology 67, 51–57. doi: 10.1159/000054298
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Waterland, R. A. (2014). Epigenetic mechanisms affecting regulation of energy balance: many questions, few answers. Annu. Rev. Nutr. 34, 337–355. doi: 10.1146/annurev-nutr-071813-105315
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Waterland, R. A., Travisano, M., Tahiliani, K. G., Rached, M. T., and Mirza, S. (2008). Methyl donor supplementation prevents transgenerational amplification of obesity. Int. J. Obes. (Lond). 32, 1373–1379. doi: 10.1038/ijo.2008.100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Watt, F., and Molloy, P. L. (1988). Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev. 2, 1136–1143. doi: 10.1101/gad.2.9.1136
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weigle, D. S., Bukowski, T. R., Foster, D. C., Holderman, S., Kramer, J. M., Lasser, G., et al. (1995). Recombinant ob protein reduces feeding and body weight in the ob/ob mouse. J. Clin. Invest. 96, 2065–2070. doi: 10.1172/JCI118254
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wierup, N., Richards, W. G., Bannon, A. W., Kuhar, M. J., Ahren, B., and Sundler, F. (2005). CART knock out mice have impaired insulin secretion and glucose intolerance, altered beta cell morphology and increased body weight. Regul. Pept. 129, 203–211. doi: 10.1016/j.regpep.2005.02.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Williams, L., Seki, Y., Vuguin, P. M., and Charron, M. J. (2014). Animal models of in utero exposure to a high fat diet: a review. Biochim. Biophys. Acta 1842, 507–519. doi: 10.1016/j.bbadis.2013.07.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Woods, S. C., Lotter, E. C., McKay, L. D., and Porte, D. Jr. (1979). Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 282, 503–505. doi: 10.1038/282503a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wortley, K. E., Anderson, K. D., Yasenchak, J., Murphy, A., Valenzuela, D., Diano, S., et al. (2005). Agouti-related protein-deficient mice display an age-related lean phenotype. Cell Metab. 2, 421–427. doi: 10.1016/j.cmet.2005.11.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yi, C. X., Foppen, E., Abplanalp, W., Gao, Y., Alkemade, A., La Fleur, S. E., et al. (2012). Glucocorticoid signaling in the arcuate nucleus modulates hepatic insulin sensitivity. Diabetes 61, 339–345. doi: 10.2337/db11-1239
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yura, S., Itoh, H., Sagawa, N., Yamamoto, H., Masuzaki, H., Nakao, K., et al. (2005). Role of premature leptin surge in obesity resulting from intrauterine undernutrition. Cell Metab. 1, 371–378. doi: 10.1016/j.cmet.2005.05.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, T. Y., Labonte, B., Wen, X. L., Turecki, G., and Meaney, M. J. (2013). Epigenetic mechanisms for the early environmental regulation of hippocampal glucocorticoid receptor gene expression in rodents and humans. Neuropsychopharmacology 38, 111–123. doi: 10.1038/npp.2012.149
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, X., Yang, R., Jia, Y., Cai, D., Zhou, B., Qu, X., et al. (2014). Hypermethylation of Sp1 binding site suppresses hypothalamic POMC in neonates and may contribute to metabolic disorders in adults: impact of maternal dietary CLAs. Diabetes 63, 1475–1487. doi: 10.2337/db13-1221
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zigman, J. M., Jones, J. E., Lee, C. E., Saper, C. B., and Elmquist, J. K. (2006). Expression of ghrelin receptor mRNA in the rat and the mouse brain. J. Comp. Neurol. 494, 528–548. doi: 10.1002/cne.20823
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: fetal programming, pro-opiomelanocortin, glucocorticoid receptor, epigenetics, hypothalamus
Citation: Gali Ramamoorthy T, Begum G, Harno E and White A (2015) Developmental programming of hypothalamic neuronal circuits: impact on energy balance control. Front. Neurosci. 9:126. doi: 10.3389/fnins.2015.00126
Received: 12 February 2015; Paper pending published: 03 March 2015;
Accepted: 26 March 2015; Published: 21 April 2015.
Edited by:
Marc Claret, Institut d'Investigacions Biomèdiques August Pi i Sunyer, SpainReviewed by:
Michiru Hirasawa, Memorial University, CanadaChristophe Breton, University of Lille, France
Copyright © 2015 Gali Ramamoorthy, Begum, Harno and White. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne White, Faculty of Life Sciences and Medical and Human Sciences, University of Manchester, AV Hill Building, Manchester M13 9PT, UKYW5uZS53aGl0ZUBtYW5jaGVzdGVyLmFjLnVr