 Kathryn M. Lenz*
Kathryn M. Lenz* Bridget M. Nugent and Margaret M. McCarthy
Bridget M. Nugent and Margaret M. McCarthy
- Program in Neuroscience and Department of Physiology, University of Maryland School of Medicine, Baltimore, MD, USA
Steroid hormones of gonadal origin act on the neonatal brain to produce sex differences that underlie adult reproductive physiology and behavior. Neuronal sex differences occur on a variety of levels, including differences in regional volume and/or cell number, morphology, physiology, molecular signaling, and gene expression. In the rodent, many of these sex differences are determined by steroid hormones, particularly estradiol, and are established by diverse downstream effects. One brain region that is potently organized by estradiol is the preoptic area (POA), a region critically involved in many behaviors that show sex differences, including copulatory and maternal behaviors. This review focuses on the POA as a case study exemplifying the depth and breadth of our knowledge as well as the gaps in understanding the mechanisms through which gonadal hormones produce lasting neural and behavioral sex differences. In the POA, multiple cell types, including neurons, astrocytes, and microglia are masculinized by estradiol. Multiple downstream molecular mediators are involved, including prostaglandins, various glutamate receptors, protein kinase A, and several immune signaling molecules. Moreover, emerging evidence indicates epigenetic mechanisms maintain sex differences in the POA that are organized perinatally and thereby produce permanent behavioral changes. We also review emerging strategies to better elucidate the mechanisms through which genetics and epigenetics contribute to brain and behavioral sex differences.
Sex Differences in the Brain: Dogma and Beyond
Sex Determination and Sexual Differentiation
Early in normal development, genes on the sex chromosomes determine whether an animal will develop testes or ovaries, in a process deemed sex determination. In mammals, the primary sex determining signal is the SRY gene on the Y chromosome, which leads to the upregulation of testis determining factor in males and development of testes. In other words, animals with an SRY gene will develop testes and all associated male phenotypes, and in the absence of SRY, a default genetic program leads to ovarian development and all associated female phenotypes. Proceeding from this primary process of sex determination is sexual differentiation, whereby other tissues take on a male or female phenotype.
Sexual differentiation is largely controlled by the three major classes of steroid hormones that are produced by the gonads: androgens, estrogens, and progestins. In the traditional view of brain sexual differentiation, these gonadal hormones act on the brain during a perinatal sensitive period to organize a male or female phenotype. Subsequent to this differentiation, the same gonadal hormones act on the male or female brain across the lifespan to produce sex-specific behaviors. This classical view of hormonal effects on the brain and behavior was deemed the Organizational/Activational hypothesis following the groundbreaking work of Phoenix et al. (1959), and has been the paradigm used to study essentially every known sex difference in behavior. This predominant approach is used to first determine whether a sex difference in the brain or behavior is organized by early life gonadal hormones during a critical period, and then to see whether it is maintained or activated in adulthood by circulating gonadal hormone levels. Calling this approach “classical” suggests that it is not a perfect means to understand sex differences in the brain and behavior, however many prominent discoveries in the field have and continue to come from this basic approach. Nonetheless, there are multiple challenges to the dogma which now complicate our concepts of brain sexual differentiation, including the edicts that (1) brain feminization is a default process that occurs independently of estradiol, (2) the major mode of steroid hormone action is to bind nuclear steroid receptors and directly modulate gene transcription, (3) the gonad is the sole source of sex hormones, and (4) all sex differences in the brain and behavior result from the actions of sex hormones on the developing or adult brain. For each of these examples, data will be presented that now challenges the classic dogma.
Basic Mechanisms in Rodents
In rodents, the dominant organizing signal is the perinatal androgen surge from the testes that begins prenatally, peaks at the end of the embryonic period, and rapidly declines during the first postnatal day (Konkle and McCarthy, 2011). Androgens are converted to estrogens via the enzyme p450 aromatase, which is highly abundant and enzymatically active in the rodent brain during the testicular androgen surge (George and Ojeda, 1982; Roselli and Resko, 1993), especially in highly sexually dimorphic brain regions such as the hypothalamus (Konkle and McCarthy, 2011). Estrogens in the brain then proceed to initiate two parallel processes in males, masculinization, whereby the brain is made capable of producing adult male sex-specific behaviors, and defeminization, whereby the brain is stripped of its ability to produce female-specific behaviors (Figure 1). These processes occur across slightly different developmental windows (Wallen and Baum, 2002) and are controlled by differing mechanisms downstream of estradiol, even in the same brain region (Todd et al., 2005). In females, in the absence of the perinatal androgen surge, a program of feminization that has classically been viewed as “default” (Jost et al., 1973), yields a brain capable of producing adult female sex-specific behaviors. Additionally, the female brain is protected from estrogens of maternal origin prenatally by the binding protein, alpha-fetoprotein, which sequesters and prevents gonadal hormones from crossing the blood–brain-barrier (McEwen et al., 1975; Bakker et al., 2006). Both the dogmas that feminization is a default process and that alpha-fetoprotein protects the female brain from masculinization have been complicated by recent data. Behavioral analyses of female mice lacking functional aromatase indicates estrogens are necessary for the full feminization of female sexual behavior (Bakker and Baum, 2008), a notion also supported by the fact that alpha-fetoprotein levels drop precipitously as the ovaries begin to synthesize estrogens during the second postnatal week (Raynaud, 1973; Meijs-Roelofs and Kramer, 1979). Detection of alpha-fetoprotein within neurons suggests it is more appropriately characterized as a chaperone molecule, selectively transporting estradiol into the brain (Toran-Allerand, 1980, 1987; Bakker and Baum, 2008).
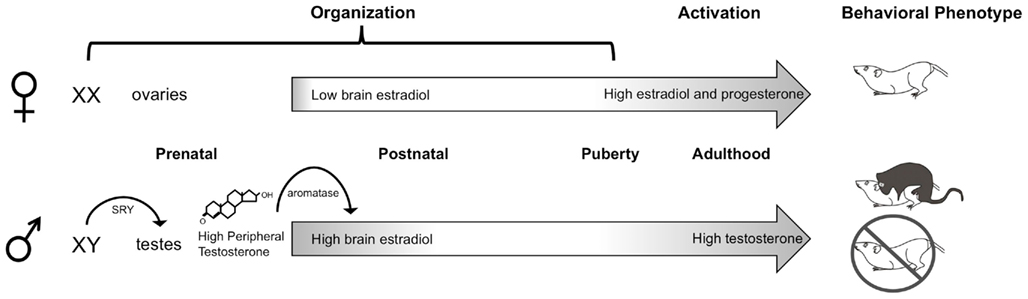
Figure 1. Neonatal organization of the brain and behavior. In male rodents, the SRY gene on the Y chromosome leads to the development of testes, which begin synthesizing androgens prenatally. Plasma testosterone levels rapidly increase late during the embryonic period and peak shortly after birth. Once in the brain, testosterone is converted by the p450 aromatase enzyme into estradiol, which acts to masculinize and defeminize the brain. A second organizational period occurs peripubertally, when steroid hormones begin to rise. In adulthood, the differentiated brain is activated by high levels of circulating gonadal hormones: androgens in males and estrogens and progestins in females, to produce sex differences in copulatory behaviors.
Hormone Receptors are Transcription Factors
The major mode of steroid hormone action is to bind to intracellular steroid hormone receptors, inducing them to dimerize, translocate to the nucleus, and bind to hormone response elements on DNA to regulate gene transcription (King and Greene, 1984; O’Malley and Tsai, 1992). When steroids bind to their cognate receptors, they decouple from a variety of molecules, including heat shock proteins (DeMarzo et al., 1991), as well as bind co-activators and co-repressors (O’Malley, 2006; Rosenfeld et al., 2006). Steroid hormone receptors can also regulate transcription independently of hormone response elements and direct binding onto DNA, for example, by complexing with other transcription factors, such as Fos (Uht et al., 1997). When steroid hormones recruit factors to the transcription complex, including co-activators and repressors, they modulate the transcriptional efficacy of both hormone receptors themselves and other transcription factors via epigenetic means such as methylation, acetylation, and phosphorylation (O’Malley, 2006; Rosenfeld et al., 2006). Interestingly, there are hormone dependent sex differences in expression of steroid receptor co-activators during the neonatal critical period for establishing sex differences in the brain (Auger et al., 2000).
More recent evidence implicates steroid hormone receptors, particularly estrogen receptors, in the regulation of a cell’s physiology and even of gene transcription that is independent of the classical mode of action via hormone response elements. There are multiple isoforms of the estrogen receptor: estrogen receptor alpha (ERα), beta (ERβ), and a third receptor, GPR-30, which can be located either in the membrane, cytosol, or nucleus. Steroid hormone receptors initiate a variety of signaling cascades via direct activation of kinases, proteases, and other molecules (Gu et al., 1996; Zhou et al., 1996; Watters et al., 1997; Bi et al., 2001; Abraham et al., 2004; Zadran et al., 2009). Many of these effects occur far more rapidly than classical gene transcription mediated effects could occur, changing cell physiology on the order of seconds (Kelly et al., 1976). Transmembrane steroid receptors mediate some of these rapid effects (Towle and Sze, 1983; Mermelstein and Micevych, 2008), and can be teased out from nuclear receptor effects by administering steroid hormones bound to membrane-impermeable molecules. These rapid membrane effects can also enhance or prime transcriptional effects (reviewed in Vasudevan and Pfaff, 2008).
De novo Synthesis of Steroid Hormones
The dogma that so-called gonadal hormones are only produced peripherally in the ovaries and testes has been challenged by strong evidence that the brain itself is steroidogenic. Select brain regions contain the enzymes necessary to synthesize estradiol from cholesterol and convergent evidence supports the hypothesis that the brain locally synthesizes estradiol (Schlinger and Arnold, 1992; Holloway and Clayton, 2001; Hojo et al., 2004). The amount of estradiol and testosterone measured in the neonatal brain varies widely from brain region to brain region and does not show a clear correlation with plasma hormone levels, suggesting region-specific synthesis (Konkle and McCarthy, 2011). The research on de novo steroidogenesis in the brain has been hampered by technical limitations in quantifying the low levels of hormones in brain tissue in a site-specific manner, especially in neonates. The question of how local synthesis of hormones contributes to brain sexual differentiation and adult brain physiology is an open question of much interest to the neuroendocrinology community because certain brain regions show more extensive hormonally mediated effects than others, and local differences in steroid hormone receptor expression have not entirely accounted for such effects.
Direct Genetic Effects on Sexual Differentiation
The dogma that steroid hormones are the sole inducer of sex differences in phenotype has been strongly and effectively challenged by data indicating that genetic effects, which may be independent of gonadal hormones or interact with gonadal hormones, also contribute to sexual differentiation. A new model that has been particularly illuminating is the four core genotypes (FCG) mouse model in which the SRY gene is deleted from the Y chromosome and inserted on an autosome, thereby making a two-by-two matrix possible wherein sex chromosome complement can be dissociated from gonadal sex and the corresponding hormonal milieu (De Vries et al., 2002). In this way it is possible to determine whether a sex difference is primarily programmed by sex chromosomes, gonadal hormones, or an interaction between the two. Thus far, results indicate gonadal hormones are the major inducer of sex differences in reproductive brain areas and behaviors (De Vries et al., 2002; Wagner et al., 2004), but that other sex differences, including differences in aggression, nociception, habit formation, and certain autoimmune disorders, are shaped by sex chromosome complement more strongly than hormones (reviewed in Arnold and Chen, 2009). Additionally, certain sex differences, such as that in vasopressin innervation of the lateral septum, are controlled by both hormonal and sex chromosome effects (De Vries et al., 2002). Another model in which a transcription factor necessary for gonadal development, steroidogenic factor 1 (SF-1), is genetically deleted, results in an agonadal animal from embryogenesis (Ingraham et al., 1994) illustrates a similar principle. This model shows that while many brain sex differences are hormonally regulated during development, including those in the preoptic area (POA) discussed below, certain developmental events in sexual differentiation occur independent from the gonad (reviewed in Budefeld et al., 2012).
The Preoptic Area as a Case Study
While many brain regions are studied in the context of sex differences, the POA, lends itself well to the study of sex differences in the brain from the genomic/epigenomic to the behavioral because it (1) shows prominent sexual dimorphism on a variety of levels, (2) is particularly sensitive and responsive to gonadal hormones both developmentally and in adulthood, and (3) is implicated in behaviors which show robust sex differences. The POA also illustrates the wide variety of mechanisms that can lead to sex differences in morphology and behavior (Figure 2).
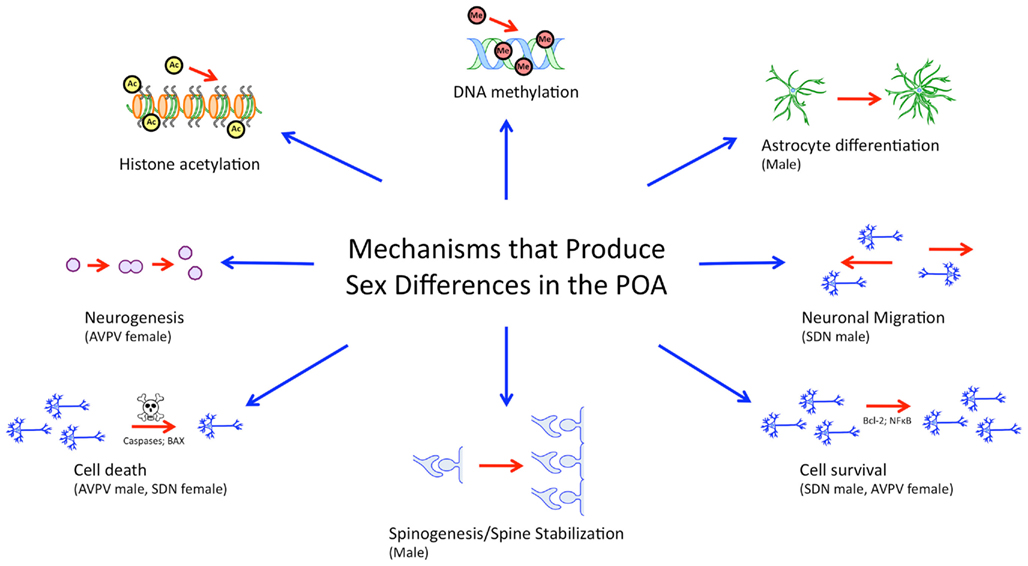
Figure 2. Mechanisms governing sexual differentiation of the preoptic area. Sexual differentiation of the preoptic area results from the complex interplay of diverse hormonally regulated mechanisms, including neurogenesis, cell survival and death, morphological differentiation of individual cells, and epigenetic activity. These mechanisms are subregion specific, cell-type specific, and occur during temporally limited critical periods during development.
Sexual Dimorphism and Hormone Dependence in the POA
There are several prominent sex differences in the POA, from the morphological to the molecular. Two neighboring subnuclei in the POA, the sexually dimorphic nucleus (SDN), and the anteroventral periventricular nucleus (AVPV), show opposite morphological sex differences in volume and cell number. Males have a five- to sevenfold larger SDN and consequently more cells in the SDN than females (Gorski, 1978). The perinatal androgen surge, followed by androgen aromatization to estradiol, protects SDN cells in males from programmed cell death (Davis et al., 1996). Estradiol leads to increased expression of the NMDA receptor subunit NR1 and antagonizing NMDA-type glutamate receptors in males increases cell death in the SDN (Hsu et al., 2001, 2005). NMDA receptor activation leads to a down regulation of the pro-apoptotic protein, Bax, and upregulation of the anti-apoptotic protein Bcl-2, thereby diminishing pro-apoptotic activity of caspase-3 (Tsukahara et al., 2006, 2008). Transcription of the gene for neural epidermal growth factor-like like-2 (NELL-2) is also regulated by estradiol (Choi et al., 2010) and further protects cells in the SDN from apoptosis (Jeong et al., 2008). Throughout the POA, the rate and frequency of neuronal migration during perinatal development is regulated by estradiol (Knoll et al., 2007). In the SDN specifically, radial spreading of neurons later during the perinatal period (postnatal days 7–15) in males amplifies the sex difference in SDN volume (Wolfe et al., 2005; Orikasa et al., 2010). Thus the phenotypic endpoint of a larger SDN in males is the result of a complex interplay of hormonal modulation of gene expression and neuronal responsiveness.
In contrast to the SDN, the female AVPV is larger than the male’s, and regulates the luteinizing hormone (LH) surge that produces ovulation in females. The AVPV contains multiple cell types, including GABAergic/glutamatergic neurons, dopamine neurons, and kisspeptin-expressing neurons, each of which is found in different amounts in males versus females. Opposite to the SDN–POA, the perinatal androgen surge drives cell death in the AVPV; treating a neonatal female with androgens reduces the number of dopaminergic neurons to that of males (Simerly et al., 1985). Estradiol upregulates caspase activity in dopaminergic neurons to induce apoptosis, and administering a caspase inhibitor prevents estradiol-induced cell death in males (Waters and Simerly, 2009). Another cell type, GABAergic/glutamatergic expressing neurons, are twice as numerous in females (Ottem et al., 2004), and this sex difference, unlike that in dopaminergic neurons, is regulated by the proinflammatory molecule, tumor necrosis factor alpha (TNFα). TNFα is higher in females and suppressed in males by the associated molecule TNF receptor-associated factor 2 inhibiting protein (Trip). In males Trip prevents NFKβ activation, Bcl-2 gene expression, and the resultant cell survival that occurs in females (Krishnan et al., 2009). The pro-apoptotic protein Bax is also higher in males and regulates the death of GABAergic neurons (Forger et al., 2004), but this effect on Bax is independent of Trip and, in addition, does not impact the cell death of dopamine neurons. Females also have significantly more cells positive for the neuropeptide, kisspeptin, than males, a sex difference that is regulated by estradiol (Kauffman et al., 2007; Homma et al., 2009) but not by Bax (Semaan and Kauffman, 2010). Currently the downstream mechanisms involved in this effect on kisspeptin neurons remain unelucidated. The overall sex difference in AVPV cell number is further enhanced by hormonally regulated neurogenesis that occurs in females during puberty (Ahmed et al., 2008).
The fact that the SDN and AVPV show opposing sex differences which are shaped by estradiol exposure precludes the temptingly convenient conclusion that estradiol acts via one conserved mechanism across the brain to produce sexual dimorphism. And not only do different signaling events govern sexual differentiation in the SDN versus AVPV, but combinations of different developmental processes, including cell death and cell migration in the SDN, and cell death and neurogenesis in the AVPV, converge to produce these sex differences. The example of the AVPV also illustrates that gonadal hormones can co-opt a variety of signaling pathways to produce the same effect (cell death) in different cell types within the same nucleus.
There are additional morphological sex differences in the POA. Projections to and from the POA differ. For example, there are sex differences in the density of peptidergic inputs from the bed nucleus of the stria terminalis to the POA, including galanin, substance P, and cholecystokinin (CCK), all of which are organized developmentally and not changed by adult hormonal treatment (Polston and Simerly, 2003). Along similar lines, females show a 20-fold denser projection of calcitonin gene-related peptide containing fibers to posterior regions of the hypothalamus (Herbison and Dye, 1993; Edelmann et al., 2007) and males have more fibers positive for nitrous oxide synthase in the AVPV than females on the day of birth (Edelmann et al., 2007). In some cases, the number or distribution of cells positive for such peptides also show a sex difference with, for example, males having more CCK-positive cells in the medial preoptic nucleus, and females having more CCK-positive cells in the periventricular preoptic nucleus (Micevych et al., 1987).
Morphological sex differences in the POA also exist at the level of individual cells. There are sex differences in astrocytes, with male astrocytes having more complex morphology and longer process length than females (Amateau and McCarthy, 2002b). There are notable sex differences in neurons as well, with male POA neurons having two- to threefold more dendritic spines per length of dendrite than females (Amateau and McCarthy, 2002a, 2004). As with the AVPV and SDN, this sex difference in dendritic spine density is organized by estradiol (Amateau and McCarthy, 2002a, 2004). For each of these sex differences, their induction depends on the presence of estradiol, can be produced by administering estradiol to female pups neonatally, and the effects are stable into adulthood regardless of the adult hormonal milieu. The case of sex differences in dendritic spines in the POA will be discussed in detail below. We have elucidated much of the downstream genetic and molecular signaling pathway through which estradiol organizes the sex difference in dendritic spine density, and this offers insight into the diverse means through which hormones can shape the developing brain. Moreover, recent work suggests microglial cells, which are the resident immune cells of the brain, also show sex differences in both number and morphology during the neonatal organizational period, which may also contribute to sexual differentiation.
Sex Differences in Behavior and the POA
The POA has been implicated as a key regulator of several behaviors that show prominent sex differences, including female sexual behavior (Leedy, 1984; Bast et al., 1987), maternal behavior (Numan, 1986), and male sexual behavior (Larsson and Heimer, 1964). The POA receives input from the olfactory bulb via the amygdala and BNST and the central tegmental field; in turn the POA projects to several brain regions that are also critical for motivation and expression of sex, including the hypothalamus, ventral tegmental area, and brainstem (Simerly and Swanson, 1986, 1988). Mating increases expression of Fos, an immediate early gene marker of activation, in the POA, as do mating-related olfactory cues (Baum and Everitt, 1992; Bressler and Baum, 1996; Veening and Coolen, 1998). Male sexual behavior is comprised of a motivational component, the drive to seek sex, and a consummatory component, the physical performance of mounts, intromissions, and ejaculations; the POA contributes to both of these components. Ablation of the POA in adulthood diminishes the display of male sexual behavior (Larsson and Heimer, 1964; Christensen et al., 1977); conversely, electrical stimulation of the POA facilitates copulation (Malsbury, 1971). To date, it has not been possible to directly tie changes in behavior to underlying changes in morphology or synaptic physiology, however strong correlations exist between the two with regard to sexual behavior and the POA. Electrophysiological recording of freely behaving rats during sexual behavior shows increased firing rates of POA neurons during copulatory thrusting and intromission (Shimura et al., 1994) and latencies and numbers of mounts and intromissions correlate with the number of dendritic spines on POA neurons, which are a major site of excitatory input to these cells (Amateau and McCarthy, 2002a, 2004; Wright and McCarthy, 2009). In addition to these consummatory aspects of sexual behavior, certain motivational aspects of sexual behavior, such as the active pursuit of a female prior to mounting, are also associated with an increase in POA neuronal firing rate (Shimura et al., 1994). This motivation for sex and its rewarding properties are regulated by projections from the POA to the mesolimbic dopamine system (reviewed in Stolzenberg and Numan, 2011). Interestingly, this crosstalk with the mesolimbic dopamine system is not only important for male sexual behavior, but also for maternal behavior and female sexual behavior, suggesting the POA is more broadly involved in appetitive aspects of reproductive behavior.
Cellular Mechanisms Mediating Sexual Differentiation of the POA
Once the neonatal sex difference and hormone dependence of POA dendritic spine density was established, the question turned to downstream mechanism. Again thwarting expectation, estradiol’s actions on dendritic spines in the POA are mediated by an unlikely candidate, the proinflammatory lipid molecule, prostaglandin E2 (PGE2). In the neonatal POA, estradiol leads to a two-fold increase in the gene products and protein levels of the major synthesizing enzymes for prostaglandins, cyclooxygenase 1 and 2 (COX-1 and COX-2), and a resultant sevenfold increase in PGE2 levels (Amateau and McCarthy, 2002a). Estradiol’s effects are both specific to PGE2, not changing levels of similar prostanoid molecules, such as PGF2, and also the effects on PGE2 on spines are specific to the POA, not occurring in the nearby hippocampus or medial basal hypothalamus (Amateau and McCarthy, 2002a). PGE2 is downstream of estradiol, with co-administration of estradiol and the COX inhibitor indomethacin failing to masculinize spine density in females (Amateau and McCarthy, 2002a, 2004). PGE2 is also sufficient to masculinize adult copulatory behavior, as evidenced by females masculinized with PGE2 and given adult androgens showing masculine patterns of mounting and intromitting when paired with a receptive female (Amateau and McCarthy, 2004). That the behavioral effects of PGE2 are specific to masculinization of sexual behavior is confirmed by a lack of effect on female sexual behavior and maternal behavior, both of which are also mediated by or influenced by the POA (Todd et al., 2005).
There are four main receptors for PGE2, referred to as EP1–EP4, all of which are present in the developing POA (Burks et al., 2007), but only EP2 and EP4 are necessary for the full masculinization of POA morphology and behavior (Wright et al., 2008), and both signal via the cAMP-protein kinase A (PKA) cascade. The effects of PGE2 on POA spine density are also mimicked by glutamate (Amateau and McCarthy, 2002a), and PKA is a known modulator of glutamate receptor dynamics, particularly the AMPA-type receptor, throughout the brain. Both PKA and AMPA receptor activation are necessary for the effect of PGE2 on dendritic spine induction and masculinization of sex behavior (Wright and McCarthy, 2009). Activated by EP2 and EP4, PKA phosphorylates AMPA receptors in the POA and induces increased membrane insertion of AMPA receptor into the cell membrane (Wright et al., 2008; Lenz et al., 2011). Thus the effects of PGE2 on increasing AMPA receptor phosphorylation and membrane insertion converge with glutamate release to masculinize spine density and sexual behavior.
While the signaling machinery controlling sexual differentiation of spine density in the POA is initiated by a classical endocrine means, the downstream signaling is shared by many brain regions and systems to induce morphological plasticity. For example, the same signaling pathway that leads to the sex difference in POA spine density, namely cAMP to PKA to phosphorylated AMPA receptor subunits to increased AMPA membrane insertion, glutamatergic signaling, and spine stabilization, exactly parallels the cascade that contributes to LTP and dendritic spine stabilization in the hippocampus or cortex (Esteban et al., 2003; Wang et al., 2005). This example underscores the versatility of hormones to co-opt already existing mechanisms in the brain to produce morphological sex differences.
Neuroendocrinology Meets Neuroimmunology
In studying sex differences in the brain, we routinely investigate the crosstalk between two highly interactive physiological systems, the nervous, and endocrine systems. Recent and emerging evidence has also begun to highlight a dynamic crosstalk between the immune system and both nervous and endocrine systems in the context of sex differences in the brain. In addition to the effects of PGE2 on masculinizing the POA developmentally, prostaglandins in the POA and median eminence are downstream of estradiol and facilitate the generation of the adult LH surge in females that leads to ovulation (Ojeda et al., 1979; Rage et al., 1997). Specifically, prostaglandins induce rapid retraction of astroglial processes that allow GnRH to be released from neurosecretory terminals at the median eminence (de Seranno et al., 2010). The source of theses prostaglandins are astrocytes (Clasadonte et al., 2011) and yet another cell type: microglia, which are the brain’s resident immune cells (Adachi et al., 2009).
Microglia are the primary immunocompetent cells of the brain, responding to injury and inflammation with the production of proinflammatory molecules, including prostaglandins, nitric oxide, cytokines, and chemokines (Gehrmann et al., 1995; Kim and de Vellis, 2005). Concomitant with this production of proinflammatory molecules, microglial cells undergo a dramatic change from a ramified, quiescent morphology to an activated, ameboid morphology. The activated microglial profile is observed in the brain following viral or bacterial infection, traumatic injury, stroke, or in the context of a neurodegenerative disease (Licinio et al., 1993; Ling and Wong, 1993; Katila et al., 1994; Mittleman et al., 1997; Munn, 2000; Vargas et al., 2005; Fabene et al., 2010) and can play both damaging and neuroprotective roles (Marchetti et al., 2005; Streit, 2005). Microglia are also primarily in the activated state in the normal neonatal brain through the first postnatal week (Ling et al., 1990; Wu et al., 1992; Ling and Wong, 1993; Dalmau et al., 1997) and contribute to normal neurodevelopmental processes, including axon guidance, neurite growth, pruning, and apoptosis (Mallat and Chamak, 1994; Kingham et al., 1999; Polazzi and Contestabile, 2002; Marin-Teva et al., 2004; Tremblay et al., 2011). Given that microglia are active neonatally and contribute to neurodevelopmental processes known to be involved in sexual differentiation, the natural next question is whether microglia and their signaling are involved in producing sex differences in the brain (Figure 3).
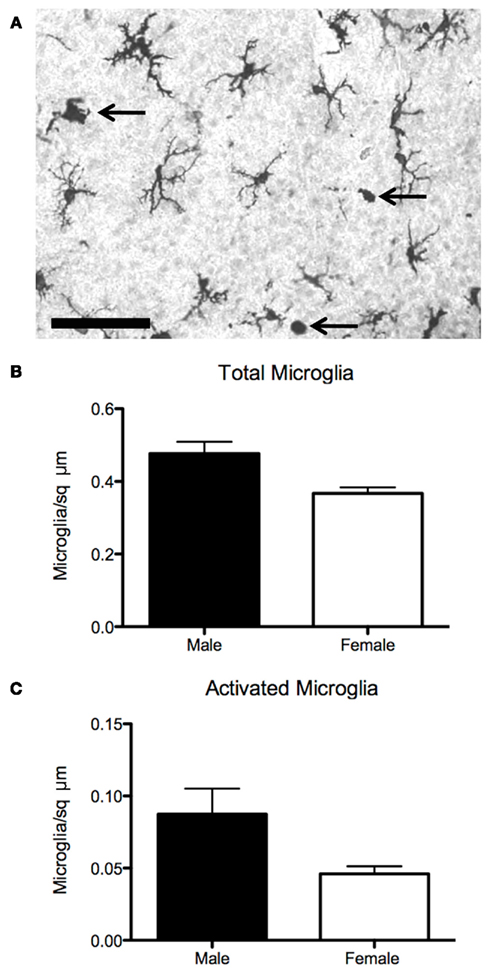
Figure 3. Sex differences in microglia in the preoptic area. (A) Established sex differences in number and morphology of neurons and astrocytes in the preoptic are being joined by sex differences in microglia, the brain’s resident immune cell. (B) Males have more microglia than females on postnatal day 2, when stained for the microglial marker Iba1. (C) Males also have more activated, ameboid microglia (arrows in 3A), which are associated with secretion of prostanoids, cytokines, chemokines, as well as phagocytosis. Scale bar = 50 μm.
Microglia express both isoforms of the estrogen receptor (Bruce-Keller et al., 2000) and respond to estradiol with both up and down regulation of various cytokines (Dimayuga et al., 2005). Estradiol is proposed as a key regulator of glial function in the context of neurological disease and injury by dictating which downstream cytokines, chemokines, and other signaling molecules that glia will produce, which in turn determines whether microglia and astrocytes have neuroprotective or neurotoxic effects (Morale et al., 2006). Estrogens also inhibit microglial apoptotic signaling (Dimayuga et al., 2005) suggesting steroid hormones may regulate microglial number in the developing brain.
Microglia and cytokines both contribute to the apoptotic signaling cascades that produce sex differences in POA cell number. For example, media from primary microglial cultures potentiates estrogen-induced pro-apoptotic signaling of Fas/Fas ligand, a molecule of the TNFα family, possibly through the signaling of interleukins 1 and 6 (Nilsen et al., 2000). As previously discussed, TNFα signaling in the AVPV also mediates the striking sex difference in GABAergic/glutamatergic cell number (Krishnan et al., 2009). With regard to sex behavior, amyloid beta precursor protein (APP), a molecule known to induce microglial activation in the context of neurodegenerative disease, is upregulated in the POA of mice that show persistent mating following castration (Park et al., 2010), suggesting that the production of normal male sex behavior can be modulated by this inflammatory molecule. Since microglia are both responsive to and capable of producing prostaglandins and are estrogen-sensitive, a potential role in establishing sex differences in dendritic spines in the POA and adult male sex behavior warrants investigation. Our recent data suggests that there may in fact be sex differences in microglia in the developing POA (Figure 3).
Many neuropsychiatric disorders have been linked to the immune system or early life infection, including autism, schizophrenia, and depression (Boksa, 2010; Derecki et al., 2010; Kelley and Dantzer, 2011), and all of these disorders are gender biased in either frequency or severity. Therefore, understanding how hormones modulate the immune system and vice versa provides explanatory power in searching for illness susceptibility genes in genome wide association studies of neuropsychiatric and neurological disorders. Moreover, understanding the basic genetic and non-genetic mechanisms of how sex differences in the brain develop can provide clues into which individuals may be at risk for developing these disorders, and how brain development goes awry to increase this susceptibility.
Genetics of Sex Differences in the POA
Though much attention has been paid to morphological and physiological sex differences in the POA and how they relate to behavioral sex differences, there is little knowledge of the broad gene networks regulating sexual differentiation in the POA, or any other brain region that shows sex differences in morphology, physiology, or mediates a sex difference in behavior. For each specific sex difference discussed above, some upstream genetic contributors have been determined. In the case of the AVPV, the cell survival and cell death related genes Bax and Bcl-2 are differentially expressed between males and females, as is the gene for the TNFalpha related molecule, Trip (Forger et al., 2004; Krishnan et al., 2009). The genes for ER alpha, beta, and KISS1, the gene for kisspeptin, show sex differences in expression in the neonatal AVPV at the time of sexual differentiation (Cao and Patisaul, 2011). In the case of the SDN, several genes such as NELL-2 (Jeong et al., 2008), RNA binding motif protein 3 (RBM3), and alpha-tubulin (Hsu et al., 2005; neurotrophic related genes), as well as Bcl-2, cytochrome oxidase subunits II and III (Hsu et al., 2005; apoptosis related genes) show changes in expression. In the case of sex differences in dendritic spines, the critical effect of estradiol is to upregulate the COX genes responsible for prostaglandin synthesis. Occasional other instances of sex differences or hormonal effects on gene expression in the developing POA can be found. For example, estradiol upregulates expression of the progesterone receptor in the POA (Wagner et al., 2001; Quadros et al., 2002), but this effect has not been related to a specific developmental process. Yet, while we have some knowledge of candidate genes that relate to sex differences in the POA, we do not have an integrated concept of whether and/or how these genes interact.
Mice with null mutations, so-called knockout mice, provide some insight into the role of hormones in producing behavioral sex differences. Mice with dysfunctional estrogen receptors, androgen receptors, progesterone receptors, or the aromatase enzyme have all been analyzed for reproductive-related behaviors, and abnormalities reported for sexual behavior, aggression, and olfactory investigation (Bakker et al., 2004; Schneider et al., 2005; Bodo et al., 2006; Kudwa et al., 2006; Raskin et al., 2009; Juntti et al., 2010). To date, models have not distinguished organizational from activational effects of hormones, and as of yet, few studies have looked for genetic changes downstream of the mutated receptor. Another model is the steroidogenic factor 1 knockout mouse (SF-1-KO), in which animals develop without gonads or adrenal glands (Ingraham et al., 1994). As SF-1 is a crucial regulator of steroidogenic enzymes (Lala et al., 1992), brain-specific SF-1 knockouts have been used to determine whether a sex difference in the brain is hormonally mediated or potentially controlled by other genetic sex differences (reviewed in Majdic and Tobet, 2011). As with any knockout model, a notable caveat is that compensatory mechanisms could mask developmental effects that normally rely on gonadal hormone action.
It is self-evident that a set of genes during the neonatal critical period must be differentially expressed or repressed in males and females due to the androgen surge from the testes. Gonadal hormone receptors are known transcription factors, and there are additional neonatal sex differences in transcription factors such as CREB (Auger et al., 2002), yet the downstream changes in gene expression are still largely unknown. Despite the increase in gene microarray studies in biomedical science, large-scale experiments have yet to be published that definitively elucidate what genes are differentially expressed in the POA between (1) males and females, (2) hormonally untreated and treated animals, or (3) wild-type and steroid receptor knockout mice during the critical period for sexual differentiation, let alone any experimental inquiry into whether manipulation of such candidate genes significantly influences sexual differentiation. This type of discovery-based research is much needed to push the study of sex differences into the future.
Epigenetics in the POA
Epigenetic processes are critical to normal development and may underlie the development of sex differences in the brain and behavior, both before and after hormonally induced sexual differentiation of the brain. Processes such as imprinting and X inactivation occur in diploid animals during early embryonic differentiation to ensure that monoallelic gene expression is achieved (Okamoto et al., 2004). Imprinting is a means of controlling gene expression that results in preferential maternal or paternal allelic inheritance of specific genes (Reik and Walter, 2001), and offspring sex influences parental allelic inheritance within the POA (Gregg et al., 2010). Interestingly, in light of the emerging effects of the immune system on hormones and brain sex differences, interleukin-18, a cytokine involved in neuroinflammation linked to sex differences in disease, has a strong maternal origin in the female POA (Ramagopalan et al., 2009; Alboni et al., 2010). X inactivation silences one copy of the X chromosome in females to ensure the expression of a single set of X-linked genes, although several genes “escape” X inactivation and are thereby expressed at twice the level in females, or have homologs on the Y chromosome. Many genes associated with higher cognitive function appear to be X-linked and when mutated often result in mental impairment, implicating their importance in neural development (Zechner et al., 2001).
There is emerging interest in the epigenetic mechanisms contributing to the maintenance of permanent changes to the neural substrate established during hormonally induced sexual differentiation of the brain (McCarthy et al., 2009). Early environmental inputs alter DNA methylation and histone modification patterns, and can result in lifelong changes in gene expression, which in some cases can be transgenerational (Dunn et al., 2010; Roth and Sweatt, 2010). These epigenetic marks are therefore considered a potential mechanism by which hormone exposure during the critical period for sexual differentiation of the brain maintains long-term sex differences in gene expression, and may be the basis for permanent, hormonally organized structural and behavioral differences. The process of sexual differentiation of the brain is an ideal model process to study epigenetics and the POA in particular offers several advantages. Epigenetic changes induced by masculinizing exogenous factors such as maternal behavior and gonadal hormone exposure impact both DNA methylation and histone modifications within the POA (Nugent et al., 2010; Auger and Auger, 2011; Auger et al., 2011; Bale, 2011; Nugent and McCarthy, 2011).
DNA methylation is the process whereby DNA methyltransferase enzymes (DNMTs) covalently bind methyl groups to the C5 position of cytosine residues adjacent to guanine residues (referred to as CpG sites). This modification is traditionally thought to cause long-lasting decreases in gene expression by either, (1) recruitment of methyl-binding and chromatin remodeling proteins, causing chromatin to condense, or (2) by blocking transcriptional machinery from accessing start sites on a gene’s promoter (Tate and Bird, 1993). The methylation status of DNA is not only reliant on the activity of DNMTs, but also on the complex of proteins bound to methylated cytosines because members of these corepressor/coactivator complexes often contain histone-modifying enzymes (Klose and Bird, 2006). In fact, DNA methylation and histone modifications are so tightly linked that inhibiting one can alter the other (He et al., 2005). Histone acetylation has been the most widely studied chromatin modification studied in regards to sexual differentiation of the brain. The impact of acetylation on chromatin rigidity depends upon the specific loci where the modification is placed and which histone tail is modified (Jenuwein and Allis, 2001). The existence of histone deacetylase enzymes (HDACS), which remove acetyl groups reversing the work of histone acetyl-transferases (HATs), reveals the transient nature of these post-translational modifications. DNA methylation is also dynamic and can change in response to cellular signaling, altering methylation patterns in a matter of minutes to hours (Métivier et al., 2008; Kim et al., 2009). There is also growing support for the idea that active demethylation occurs in post-mitotic cells (Bhutani et al., 2011). Despite the dynamic nature of the epigenetic marks, much of the data on sexual differentiation of the brain consists of snapshots in time of DNA methylation status and histone modifications, most frequently during the critical period.
Gonadal steroid hormone receptors are known to recruit coactivating and corepressing proteins, which have intrinsic histone-modifying capabilities, to transcription start sites (Bonthuis et al., 2011; Itkonen and Mills, 2011). Thus activation of steroid receptors by high levels of circulating hormones during the critical period for sexual differentiation of the brain may influence histone modifications. Sex differences and hormonally induced changes in lysine 9/14 acetylation (associated with gene expression) and lysine 9 trimethylation (associated with gene silencing) of histone H3 have been reported in the developing mouse cortex and hippocampus; however, the same study found no influence of sex or hormones on these histone modifications in the POA (Tsai et al., 2009). A different study found that sex differences in histone H3 and H4 acetylation at the ERα and aromatase promoters within the POA depends on which developmental time point is examined (Matsuda et al., 2011). Pharmacological inhibition of HDAC enzyme activity or antisense oligonucleotide-mediated knockdown of HDAC2 and HDAC4 during the critical period for sexual differentiation of the brain significantly impairs male sexual behavior in adulthood, suggesting the importance of histone acetylation in organization of the male brain. Pharmacological techniques have successfully been used to investigate the importance of histone acetylation in the sexual differentiation of other regions of the brain as well (Murray et al., 2009). A variety of inhibitors of DNMT activity are available and may prove valuable to future investigations of the role of DNA methylation in sexual differentiation.
Most studies on sex and hormonally induced differences in DNA methylation have focused on differences in methylation levels at particular CpG sites on specific promoters, most notably on genes for steroid hormone receptors. In particular many researchers have focused on the methylation status of ERα within the POA during the critical period for sexual differentiation of the brain, since activation of ERα is known to be essential for masculinization (Kudwa et al., 2006). A major experience in the life of the neonate, maternal licking, and grooming, influences methylation patterns in the promoter region of the gene for ERα. Male and female rodents receive differing amounts of anogenital licking and grooming during early development, with males receiving significantly more maternal licking in the first two postnatal weeks (Moore and Morelli, 1979). The high levels of anogenital licking typically received by males are associated with masculinization of ERα expression levels in the POA (Champagne et al., 2006). Reduced maternal care increases methylation on the ERα 1b promoter, and decreases ERα gene expression within the POA, but not in other regions (Champagne et al., 2006; Cameron et al., 2008). Conversely, simulated maternal grooming increases ERα 1b promoter methylation and decreases ERα expression in females to male-like levels (Kurian et al., 2010). The contradictory findings that natural maternal care versus simulated maternal care results in opposite methylation patterns on the same promoter region exemplifies the complexity and at times contradictory results regarding hormone receptor promoter methylation in the brain during sexual differentiation.
The impact of hormones on DNA methylation in the developing POA is also debated, perhaps due to the slightly different developmental time points and experimental methods used between laboratories. For instance, females have higher levels of ERα gene expression in the POA at postnatal day 10 (Kurian et al., 2010), a time point at the end of the critical period for sexual differentiation of the brain, when hormone levels within the brain are relatively low (Konkle and McCarthy, 2011). Administration of estradiol on the first 2 days of life increases ERα promoter methylation and decreases ERα gene expression by postnatal day 8 in the female, although comparisons of promoter methylation between females, hormonally masculinized females, and males were not made (Kurian et al., 2010). A later study found that males have lower levels of methylation than females on the ERα promoter and that estradiol treatment administered to females on the first 2 days of life decreases, or masculinizes methylation patterns on the ERα promoter by postnatal day 2 (Schwarz et al., 2010). Importantly, methylation of the ERα promoter is dynamic across the animal’s lifespan. At postnatal day 20, before the onset of puberty, there are no differences in methylation on the ERα promoter between males, females, and females treated neonatally with a masculinizing dose of estradiol. However, in adulthood, following the onset of adult gonadal steroid circulation, males and females treated neonatally with estradiol have lower levels of methylation at one CpG site on the ERα promoter compared to control females. Thus DNA methylation is (1) responsive to hormones, (2) transitory, (3) potentially organized by developmental hormone exposure, and (4) subsequently influenced by activational hormones in the adult.
Although theses studies clearly demonstrate a role for DNA methylation in sexual differentiation of the POA, the discrepancies in the directionality of ERα methylation in this region make it difficult to draw concrete conclusions. Quantifying methylation at individual CpG sites across a promoter raises questions as to the meaning and functional significance of small changes in methylation at a given site. It appears that some sites, perhaps sites within a transcription factor’s response element, are more important than others for determining gene expression. But, if a sex difference and hormonally induced change in methylation at a specific CpG site does not correlate with a sex difference in gene expression, is it functionally meaningful? Moreover, how large does a difference in methylation at a given site need to be to impact gene expression? Could DNA methylation have additional roles besides just controlling gene expression? Effects of environmental endocrine disrupting chemicals on methylation of ERα in the POA indicate that in addition to altering gene expression, methylation can dictate the use of alternative promoters, which may influence the selective expression of ERα transcript variants (Monje et al., 2007). The possibility that hormonally induced changes in methylation dictate promoter usage and transcript variant expression is an understudied aspect of epigenetics but may provide a better understanding of how hormones organize the brain.
Alternative approaches to studying the impact of epigenetic modifications on the maintenance of sexual differentiated traits are needed. DNA methylation and histone modifications have been thoroughly studied on hormone receptor promoters, but these largely descriptive studies have done little to push the field of developmental neuroendocrinology forward. Since less is known about them, two other epigenetic contributors have received little to no attention in sexual differentiation research: (1) microRNAs (miRNAs), and (2) hydroxymethylation. miRNAs are short, naturally occurring, non-coding segments of RNA involved in the posttranscriptional silencing of genes. Steroid hormones have been shown to regulate miRNA expression, which can in turn result in silencing of steroid hormone receptor gene expression (reviewed in Cochrane et al., 2010). Sex differences in these epigenetic regulators have been found in whole brain homogenates, and testosterone and prenatal stress alters their expression suggesting they may play an important role in sexual differentiation (Morgan and Bale, 2011). There have been no categorizations of miRNAs in the POA, or other sexually dimorphic regions of the brain, as of yet. A variant of methylated DNA, 5-hydroxymethylcytosine, was recently discovered to be highly abundant in the brain (Kriaucionis and Heintz, 2009) but because of technical limitations in the study of hydroxymethylation, little is known about its function (Jin et al., 2010). 5-hydroxymethylcytosine residues appear to be generated by oxidation of 5-methylcytosine and are associated with active transcription during early development (Tahiliani et al., 2009; Ficz et al., 2011). These marks may be a means for DNA demethylation or may be necessary for cellular pluripotency prior to phenotypic commitment (Ficz et al., 2011; Guo et al., 2011).
Large-scale investigations of the transcriptome and epigenome during and after sexual differentiation would allow for the identification of candidate genes involved in permanent steroid-induced organization associated with masculinized versus defeminized versus feminized brain phenotypes. New technologies such as RNAseq and MethylC-seq can enable high throughput sequencing of the transcriptome and epigenome across sexes and developmental time points, enabling the study of sex differences in copy number variation and single nucleotide polymorphisms of specific genes. Techniques such as these have the ability to revolutionize the study of sex differences in the brain.
Conclusion and Future Directions
Sex differences in the brain result from a complex interaction between steroid hormones synthesized by both the gonads and the brain, sex chromosome effects, and the environment that occurs early in ontogeny and continues throughout the lifespan. The POA exhibits sex differences in a variety of morphological and physiological parameters that make it an ideal brain region to study the mechanisms that govern sexual differentiation of brain and behavior. Although we know much about how sex differences in the POA and other sexually differentiated brain regions are established in isolation, we still lack a circuit-level understanding of how multiple sexually differentiated brain regions and/or cell types influence each other’s development. Many sex differences in brain morphology, physiology, and behavior have been characterized and intensely studied, yet research on sex differences in the brain is still in early days relative to the rest of neuroscience because many research programs fail to even include female animals as subjects. Moreover, it is not currently possible to directly link a change in brain morphology, physiology, or gene expression to a resulting change in behavior in a complex mammalian model. The future of sex differences research will be enriched by the molecular and epigenetic techniques that allow for making direct connections, and to ask more complex questions about the interplay between hormones, genes and environment in establishing and maintaining sexual dimorphism in the brain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abraham, I. M., Todman, M. G., Korach, K. S., and Herbison, A. E. (2004). Critical in vivo roles for classical estrogen receptors in rapid estrogen actions on intracellular signaling in mouse brain. Endocrinology 145, 3055–3061.
Adachi, S., Fujioka, H., Kakehashi, C., Matsuwaki, T., Nishihara, M., and Akema, T. (2009). Possible involvement of microglia containing cyclooxygenase-1 in the accumulation of gonadotrophin-releasing hormone in the preoptic area in female rats. J. Neuroendocrinol. 21, 1029–1037.
Ahmed, E. I., Zehr, J. L., Schulz, K. M., Lorenz, B. H., DonCarlos, L. L., and Sisk, C. L. (2008). Pubertal hormones modulate the addition of new cells to sexually dimorphic brain regions. Nat. Neurosci. 11, 995–997.
Alboni, S., Cervia, D., Sugama, S., and Conti, B. (2010). Interleukin 18 in the CNS. J. Neuroinflammation 7. doi:10.1186/1742-2094-7-9
Amateau, S. K., and McCarthy, M. M. (2002a). A novel mechanism of dendritic spine plasticity involving estradiol induction of prostaglandin-E2. J. Neurosci. 22, 8586–8596.
Amateau, S. K., and McCarthy, M. M. (2002b). Sexual differentiation of astrocyte morphology in the developing rat preoptic area. J. Neuroendocrinol. 14, 904–910.
Amateau, S. K., and McCarthy, M. M. (2004). Induction of PGE2 by estradiol mediates developmental masculinization of sex behavior. Nat. Neurosci. 7, 643–650.
Arnold, A. P., and Chen, X. (2009). What does the “four core genotypes” mouse model tell us about sex differences in the brain and other tissues?. Front. Neuroendocrinol. 30, 1–9.
Auger, A. P., and Auger, C. J. (2011). Epigenetic turn ons and turn offs: Chromatin reorganization and brain differentiation. Endocrinology 152, 349–353.
Auger, A. P., Jessen, H. M., and Edelmann, M. N. (2011). Epigenetic organization of brain sex differences and juvenile social play behavior. Horm. Behav. 59, 358–363.
Auger, A. P., Perrot-Sinal, T. S., Auger, C. J., Ekas, L. A., Tetel, M. J., and McCarthy, M. M. (2002). Expression of the nuclear receptor coactivator, cAMP response element-binding protein, is sexually dimorphic and modulates sexual differentiation of neonatal rat brain. Endocrinology 143, 3009–3016.
Auger, A. P., Tetel, M. J., and McCarthy, M. M. (2000). Steroid receptor coactivator-1 (SRC-1) mediates the development of sex-specific brain morphology and behavior. Proc. Natl. Acad. Sci. U.S.A. 97, 7551–7555.
Bakker, J., and Baum, M. J. (2008). Role for estradiol in female-typical brain and behavioral sexual differentiation. Front. Neuroendocrinol. 29, 1–16.
Bakker, J., De Mees, C., Douhard, Q., Balthazart, J., Gabant, P., Szpirer, J., and Szpirer, C. (2006). Alpha-fetoprotein protects the developing female mouse brain from masculinization and defeminization by estrogens. Nat. Neurosci. 9, 220–226.
Bakker, J., Honda, S., Harada, N., and Balthazart, J. (2004). Restoration of male sexual behavior by adult exogenous estrogens in male aromatase knockout mice. Horm. Behav. 46, 1–10.
Bale, T. L. (2011). Sex differences in prenatal epigenetic programming of stress pathways. Stress 14, 348–356.
Bast, J. D., Hunts, C., Renner, K. J., Morris, R. K., and Quadagno, D. M. (1987). Lesions in the preoptic area suppressed sexual receptivity in ovariectomized rats with estrogen implants in the ventromedial hypothalamus. Brain Res. Bull. 18, 153–158.
Baum, M. J., and Everitt, B. J. (1992). Increased expression of c-fos in the medial preoptic area after mating in male rats: role of afferent inputs from the medial amygdala and midbrain central tegmental field. Neuroscience 50, 627–646.
Bi, R., Foy, M. R., Vouimba, R. M., Thompson, R. F., and Baudry, M. (2001). Cyclic changes in estradiol regulate synaptic plasticity through the MAP kinase pathway. Proc. Natl. Acad. Sci. U.S.A. 98, 13391–13395.
Bodo, C., Kudwa, A. E., and Rissman, E. F. (2006). Both estrogen receptor-alpha and -beta are required for sexual differentiation of the anteroventral periventricular area in mice. Endocrinology 147, 415–420.
Boksa, P. (2010). Effects of prenatal infection on brain development and behavior: a review of findings from animal models. Brain Behav. Immun. 24, 881–897.
Bonthuis, P. J., Patteson, J. K., and Rissman, E. F. (2011). Acquisition of sexual receptivity: roles of chromatin acetylation, estrogen receptor-α, and ovarian hormones. Endocrinology 152, 3172–3181.
Bressler, S. C., and Baum, M. J. (1996). Sex comparison of neuronal Fos immunoreactivity in the rat vomeronasal projection circuit after chemosensory stimulation. Neuroscience 71, 1063–1072.
Bruce-Keller, A. J., Keeling, J. L., Keller, J. N., Huang, F. F., Camondola, S., and Mattson, M. P. (2000). Antiinflammatory effects of estrogen on microglial activation. Endocrinology 141, 3646–3656.
Budefeld, T., Tobet, S. A., and Majdic, G. (2012). Steroidogenic factor 1 and the central nervous system. J. Neuroendocrinol. 24, 225–235.
Burks, S. R., Wright, C. L., and McCarthy, M. M. (2007). Exploration of prostanoid receptor subtype regulating estradiol and prostaglandin E2 induction of spinophilin in developing preoptic area neurons. Neuroscience 146, 1117–1127.
Cameron, N., Shahrokh, D., Del Corpo, A., Dhir, S., Szyf, M., Champagne, F., and Meaney, M. (2008). Epigenetic programming of phenotypic variations in reproductive strategies in the rat through maternal care. J. Neuroendocrinol. 20, 795–801.
Cao, J., and Patisaul, H. B. (2011). Sexually dimorphic expression of hypothalamic estrogen receptors alpha and beta and Kiss1 in neonatal male and female rats. J. Comp. Neurol. 519, 2954–2977.
Champagne, F. A., Weaver, I. C. G., Diorio, J., Dymov, S., Szyf, M., and Meaney, M. J. (2006). Maternal care associated with methylation of the estrogen receptor-{alpha} 1b promoter and estrogen receptor-{alpha} expression in the medial preoptic area of female offspring. Endocrinology 147, 2909–2915.
Choi, E. J., Kim, D. H., Kim, J. G., Kim, D. Y., Kim, J. D., Seol, O. J., Jeong, C. S., Park, J. W., Choi, M. Y., Kang, S. G., Costa, M. E., Ojeda, S. R., and Lee, B. J. (2010). Estrogen-dependent transcription of the NEL-like 2 (NELL2) gene and its role in protection from cell death. J. Biol. Chem. 285, 25074–25084.
Christensen, L. W., Nance, D. M., and Gorski, R. A. (1977). Effects of hypothalamic and preoptic lesions on reproductive behavior in male rats. Brain Res. Bull. 2, 137–141.
Clasadonte, J., Poulain, P., Hanchate, N. K., Corfas, G., Ojeda, S. R., and Prevot, V. (2011). Prostaglandin E2 release from astrocytes triggers gonadotropin-releasing hormone (GnRH) neuron firing via EP2 receptor activation. Proc. Natl. Acad. Sci. U.S.A. 108, 16104–16109.
Cochrane, D. R., Cittelly, D. M., and Richer, J. K. (2010). Steroid receptors and microRNAs: relationships revealed. Steroids 76, 1–10.
Dalmau, I., Finsen, B., Tonder, N., Zimmer, J., Gonzalez, B., and Castellano, B. (1997). Development of microglia in the prenatal rat hippocampus. J. Comp. Neurol. 377, 70–84.
Davis, A. M., Grattan, D. R., Selmanoff, M., and McCarthy, M. M. (1996). Sex differences in glutamic acid decarboxylase mRNA in neonatal rat brain: implications for sexual differentiation. Horm. Behav. 30, 538–552.
de Seranno, S., d’Anglemont de Tassigny, X., Estrella, C., Loyens, A., Kasparov, S., Leroy, D., Ojeda, S. R., Beauvillain, J. C., and Prevot, V. (2010). Role of estradiol in the dynamic control of tanycyte plasticity mediated by vascular endothelial cells in the median eminence. Endocrinology 151, 1760–1772.
De Vries, G. J., Rissman, E. F., Simerly, R. B., Yang, L. Y., Scordalakes, E. M., Auger, C. J., Swain, A., Lovell-Badge, R., Burgoyne, P. S., and Arnold, A. P. (2002). A model system for study of sex chromosome effects on sexually dimorphic neural and behavioral traits. J. Neurosci. 22, 9005–9014.
DeMarzo, A. M., Beck, C. A., Onate, S. A., and Edwards, D. P. (1991). Dimerization of mammalian progesterone receptors occurs in the absence of DNA and is related to the release of the 90-kDa heat shock protein. Proc. Natl. Acad. Sci. U.S.A. 88, 72–76.
Derecki, N. C., Privman, E., and Kipnis, J. (2010). Rett syndrome and other autism spectrum disorders – brain diseases of immune malfunction?. Mol. Psychiatry 15, 355–363.
Dimayuga, F. O., Reed, J. L., Carnero, G. A., Wang, C., Dimayuga, E. R., Dimayuga, V. M., Perger, A., Wilson, M. E., Keller, J. N., and Bruce-Keller, A. J. (2005). Estrogen and brain inflammation: effects on microglial expression of MHC, costimulatory molecules and cytokines. J. Neuroimmunol. 161, 123–136.
Dunn, G. A., Morgan, C. P., and Bale, T. L. (2010). Sex-specificity in transgenerational epigenetic programming. Horm. Behav. 59, 290–295.
Edelmann, M., Wolfe, C., Scordalakes, E. M., Rissman, E. F., and Tobet, S. (2007). Neuronal nitric oxide synthase and calbindin delineate sex differences in the developing hypothalamus and preoptic area. Dev. Neurobiol. 67, 1371–1381.
Esteban, J. A., Shi, S. H., Wilson, C., Nuriya, M., Huganir, R. L., and Malinow, R. (2003). PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci. 6, 136–143.
Fabene, P. F., Bramanti, P., and Constantin, G. (2010). The emerging role for chemokines in epilepsy. J. Neuroimmunol. 224, 22–27.
Ficz, G., Branco, M. R., Seisenberger, S., Santos, F., Krueger, F., Hore, T. A., Marques, C. J., Andrews, S., and Reik, W. (2011). Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 473, 398–402.
Forger, N. G., Rosen, G. J., Waters, E. M., Jacob, D., Simerly, R. B., and de Vries, G. J. (2004). Deletion of Bax eliminates sex differences in the mouse forebrain. Proc. Natl. Acad. Sci. U.S.A. 101, 13666–13671.
Gehrmann, J., Matsumoto, Y., and Kreutzberg, G. W. (1995). Microglia: intrinsic immuneffector cell of the brain. Brain Res. Brain Res. Rev. 20, 269–287.
George, F. W., and Ojeda, S. R. (1982). Changes in aromatase activity in the rat brain during embryonic, neonatal, and infantile development. Endocrinology 111, 522–529.
Gregg, C., Zhang, J., Butler, J. E., Haig, D., and Dulac, C. (2010). Sex-specific parent-of-origin allelic expression in the mouse brain. Science 329, 682–685.
Gu, G., Rojo, A. A., Zee, M. C., Yu, J., and Simerly, R. B. (1996). Hormonal regulation of CREB phosphorylation in the anteroventral periventricular nucleus. J. Neurosci. 16, 3035–3044.
Guo, J. U., Su, Y., Zhong, C., Ming, G., and Song, H. (2011). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434.
He, F., Ge, W., Martinowich, K., Becker-Catania, S., Coskun, V., Zhu, W., Wu, H., Castro, D., Guillemot, F., and Fan, G. (2005). A positive autoregulatory loop of jak-STAT signaling controls the onset of astrogliogenesis. Nat. Neurosci. 8, 616–625.
Herbison, A. E., and Dye, S. (1993). Perinatal and adult factors responsible for the sexually dimorphic calcitonin gene-related peptide-containing cell population in the rat preoptic area. Neuroscience 54, 991–999.
Hojo, Y., Hattori, T. A., Enami, T., Furukawa, A., Suzuki, K., Ishii, H. T., Mukai, H., Morrison, J. H., Janssen, W. G., Kominami, S., Harada, N., Kimoto, T., and Kawato, S. (2004). Adult male rat hippocampus synthesizes estradiol from pregnenolone by cytochromes P45017alpha and P450 aromatase localized in neurons. Proc. Natl. Acad. Sci. U.S.A. 101, 865–870.
Holloway, C. C., and Clayton, D. F. (2001). Estrogen synthesis in the male brain triggers development of the avian song control pathway in vitro. Nat. Neurosci. 4, 170–175.
Homma, T., Sakakibara, M., Yamada, S., Kinoshita, M., Iwata, K., Tomikawa, J., Kanazawa, T., Matsui, H., Takatsu, Y., Ohtaki, T., Matsumoto, H., Uenoyama, Y., Maeda, K., and Tsukamura, H. (2009). Significance of neonatal testicular sex steroids to defeminize anteroventral periventricular kisspeptin neurons and the GnRH/LH surge system in male rats. Biol. Reprod. 81, 1216–1225.
Hsu, H. K., Shao, P. L., Tsai, K. L., Shih, H. C., Lee, T. Y., and Hsu, C. (2005). Gene regulation by NMDA receptor activation in the SDN-POA neurons of male rats during sexual development. J. Mol. Endocrinol. 34, 433–445.
Hsu, H. K., Yang, R. C., Shih, H. C., Hsieh, Y. L., Chen, U. Y., and Hsu, C. (2001). Prenatal exposure of testosterone prevents SDN-POA neurons of postnatal male rats from apoptosis through NMDA receptor. J. Neurophysiol. 86, 2374–2380.
Ingraham, H. A., Lala, D. S., Ikeda, Y., Luo, X., Shen, W. H., Nachtigal, M. W., Abbud, R., Nilson, J. H., and Parker, K. L. (1994). The nuclear receptor steroidogenic factor 1 acts at multiple levels of the reproductive axis. Genes Dev. 8, 2302–2312.
Itkonen, H., and Mills, I. G. (2011). Chromatin binding of the androgen receptor in prostate cancer. Mol. Cell. Endocrinol. PMID: 21989426. [Epub ahead of print].
Jeong, J. K., Ryu, B. J., Choi, J., Kim, D. H., Choi, E. J., Park, J. W., Park, J. J., and Lee, B. J. (2008). NELL2 participates in formation of the sexually dimorphic nucleus of the pre-optic area in rats. J. Neurochem. 106, 1604–1613.
Jin, S. G., Kadam, S., and Pfeifer, G. P. (2010). Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res. 38. doi:10.1093/nar/gkq223
Jost, A., Vigier, B., Prepin, J., and Perchellet, J. P. (1973). Studies on sex differentiation in mammals. Recent Prog. Horm. Res. 29, 1–41.
Juntti, S. A., Tollkuhn, J., Wu, M. V., Fraser, E. J., Soderborg, T., Tan, S., Honda, S., Harada, N., and Shah, N. M. (2010). The androgen receptor governs the execution, but not programming, of male sexual and territorial behaviors. Neuron 66, 260–272.
Katila, H., Hurme, M., Wahlbeck, K., Appelberg, B., and Rimon, R. (1994). Plasma and cerebrospinal fluid interleukin-1 beta and interleukin-6 in hospitalized schizophrenic patients. Neuropsychobiology 30, 20–23.
Kauffman, A. S., Gottsch, M. L., Roa, J., Byquist, A. C., Crown, A., Clifton, D. K., Hoffman, G. E., Steiner, R. A., and Tena-Sempere, M. (2007). Sexual differentiation of Kiss1 gene expression in the brain of the rat. Endocrinology 148, 1774–1783.
Kelley, K. W., and Dantzer, R. (2011). Alcoholism and inflammation: neuroimmunology of behavioral and mood disorders. Brain Behav. Immun. 25(Suppl. 1), S13–S20.
Kelly, M. J., Moss, R. L., and Dudley, C. A. (1976). Differential sensitivity of preoptic-septal neurons to microelectrophoresed estrogen during the estrous cycle. Brain Res. 114, 152–157.
Kim, M. S., Kondo, T., Takada, I., Youn, M. Y., Yamamoto, Y., Takahashi, S., Matsumoto, T., Fujiyama, S., Shirode, Y., and Yamaoka, I. (2009). DNA demethylation in hormone-induced transcriptional derepression. Nature 461, 1007–1012.
Kim, S. U., and de Vellis, J. (2005). Microglia in health and disease. J. Neurosci. Res. 81, 302–313.
King, W. J., and Greene, G. L. (1984). Monoclonal antibodies localize oestrogen receptor in the nuclei of target cells. Nature 307, 745–747.
Kingham, P. J., Cuzner, M. L., and Pocock, J. M. (1999). Apoptotic pathways mobilized in microglia and neurones as a consequence of chromogranin A-induced microglial activation. J. Neurochem. 73, 538–547.
Klose, R. J., and Bird, A. P. (2006). Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31, 89–97.
Knoll, J. G., Wolfe, C. A., and Tobet, S. A. (2007). Estrogen modulates neuronal movements within the developing preoptic area–anterior hypothalamus. Eur. J. Neurosci. 26, 1091–1099.
Konkle, A. T., and McCarthy, M. M. (2011). Developmental time course of estradiol, testosterone, and dihydrotestosterone levels in discrete regions of male and female rat brain. Endocrinology 152, 223–235.
Kriaucionis, S., and Heintz, N. (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in purkinje neurons and the brain. Science 324, 929–930.
Krishnan, S., Intlekofer, K. A., Aggison, L. K., and Petersen, S. L. (2009). Central role of TRAF-interacting protein in a new model of brain sexual differentiation. Proc. Natl. Acad. Sci. U.S.A. 106, 16692–16697.
Kudwa, A. E., Michopoulos, V., Gatewood, J. D., and Rissman, E. F. (2006). Roles of estrogen receptors alpha and beta in differentiation of mouse sexual behavior. Neuroscience 138, 921–928.
Kurian, J. R., Olesen, K. M., and Auger, A. P. (2010). Sex differences in epigenetic regulation of the estrogen receptor-{alpha} promoter within the developing preoptic area. Endocrinology 151, 2297–2305.
Lala, D. S., Rice, D. A., and Parker, K. L. (1992). Steroidogenic factor I, a key regulator of steroidogenic enzyme expression, is the mouse homolog of fushi tarazu-factor I. Mol. Endocrinol. 6, 1249–1258.
Larsson, K., and Heimer, L. (1964). Mating behaviour of male rats after lesions in the preoptic area. Nature 202, 413–414.
Leedy, M. G. (1984). Effects of small medial preoptic lesions on estrous cycles and receptivity in female rats. Psychoneuroendocrinology 9, 189–196.
Lenz, K. M., Wright, C. L., Martin, R. C., and McCarthy, M. M. (2011). Prostaglandin E2 regulates AMPA receptor phosphorylation and promotes membrane insertion in preoptic area neurons and glia during sexual differentiation. PLoS ONE 6, e18500. doi:10.1371/journal.pone.0018500
Licinio, J., Seibyl, J. P., Altemus, M., Charney, D. S., and Krystal, J. H. (1993). Elevated CSF levels of interleukin-2 in neuroleptic-free schizophrenic patients. Am. J. Psychiatry 150, 1408–1410.
Ling, E. A., Kaur, L. C., Yick, T. Y., and Wong, W. C. (1990). Immunocytochemical localization of CR3 complement receptors with OX-42 in amoeboid microglia in postnatal rats. Anat. Embryol. 182, 481–486.
Ling, E. A., and Wong, W. C. (1993). The origin and nature of ramified and amoeboid microglia: a historical review and current concepts. Glia 7, 9–18.
Majdic, G., and Tobet, S. (2011). Cooperation of sex chromosomal genes and endocrine influences for hypothalamic sexual differentiation. Front. Neuroendocrinol. 32, 137–145.
Mallat, M., and Chamak, B. (1994). Brain macrophages: neurotoxic or neurotrophic effector cells?. J. Leukoc. Biol. 56, 416–422.
Malsbury, C. W. (1971). Facilitation of male rat copulatory behavior by electrical stimulation of the medial preoptic area. Physiol. Behav. 7, 797–805.
Marchetti, B., Serra, P. A., L’Episcopo, F., Tirolo, C., Caniglia, S., Testa, N., Cioni, S., Gennuso, F., Rocchitta, G., Desole, M. S., Mazzarino, M. C., Miele, E., and Morale, M. C. (2005). Hormones are key actors in gene x environment interactions programming the vulnerability to Parkinson’s disease: glia as a common final pathway. Ann. N. Y. Acad. Sci. 1057, 296–318.
Marin-Teva, J. L., Dusart, I., Colin, C., Gervais, A., van Rooijen, N., and Mallat, M. (2004). Microglia promote the death of developing Purkinje cells. Neuron 41, 535–547.
Matsuda, K. I., Mori, H., Nugent, B. M., Pfaff, D. W., McCarthy, M. M., and Kawata, M. (2011). Histone deacetylation during brain development is essential for permanent masculinization of sexual behavior. Endocrinology 152, 2760–2767.
McCarthy, M. M., Auger, A. P., Bale, T. L., De Vries, G. J., Dunn, G. A., Forger, N. G., Murray, E. K., Nugent, B. M., Schwarz, J. M., and Wilson, M. E. (2009). The epigenetics of sex differences in the brain. J. Neurosci. 29, 12815.
McEwen, B. S., Plapinger, L., Chaptal, C., Gerlach, J., and Wallach, G. (1975). Role of fetoneonatal estrogen binding proteins in the associations of estrogen with neonatal brain cell nuclear receptors. Brain Res. 96, 400–406.
Meijs-Roelofs, H. M., and Kramer, P. (1979). Maturation of the inhibitory feedback action of oestrogen on follicle-stimulating hormone secretion in the immature female rat: a role for alpha-foetoprotein. J. Endocrinol. 81, 199–208.
Mermelstein, P. G., and Micevych, P. E. (2008). Nervous system physiology regulated by membrane estrogen receptors. Rev. Neurosci. 19, 413–424.
Métivier, R., Gallais, R., Tiffoche, C., Le Péron, C., Jurkowska, R. Z., Carmouche, R. P., Ibberson, D., Barath, P., Demay, F., and Reid, G. (2008). Cyclical DNA methylation of a transcriptionally active promoter. Nature 452, 45–50.
Micevych, P. E., Park, S. S., Akesson, T. R., and Elde, R. (1987). Distribution of cholecystokinin-immunoreactive cell bodies in the male and female rat: I. Hypothalamus. J. Comp. Neurol. 255, 124–136.
Mittleman, B. B., Castellanos, F. X., Jacobsen, L. K., Rapoport, J. L., Swedo, S. E., and Shearer, G. M. (1997). Cerebrospinal fluid cytokines in pediatric neuropsychiatric disease. J. Immunol. 159, 2994–2999.
Monje, L., Varayoud, J., Luque, E. H., and Ramos, J. G. (2007). Neonatal exposure to bisphenol A modifies the abundance of estrogen receptor {alpha} transcripts with alternative 5’-untranslated regions in the female rat preoptic area. J. Endocrinol. 194, 201–212.
Moore, C. L., and Morelli, G. A. (1979). Mother rats interact differently with male and female offspring. J. Comp. Physiol. Psychol. 93, 677–684.
Morale, M. C., Serra, P. A., L’Episcopo, F., Tirolo, C., Caniglia, S., Testa, N., Gennuso, F., Giaquinta, G., Rocchitta, G., Desole, M. S., Miele, E., and Marchetti, B. (2006). Estrogen, neuroinflammation and neuroprotection in Parkinson’s disease: glia dictates resistance versus vulnerability to neurodegeneration. Neuroscience 138, 869–878.
Morgan, C. P., and Bale, T. L. (2011). Early prenatal stress epigenetically programs dysmasculinization in second-generation offspring via the paternal lineage. J. Neurosci. 31, 11748–11755.
Munn, N. A. (2000). Microglia dysfunction in schizophrenia: an integrative theory. Med. Hypotheses 54, 198–202.
Murray, E. K., Hien, A., de Vries, G. J., and Forger, N. G. (2009). Epigenetic control of sexual differentiation of the bed nucleus of the stria terminalis. Endocrinology 150, 4241–4247.
Nilsen, J., Mor, G., and Naftolin, F. (2000). Estrogen-regulated developmental neuronal apoptosis is determined by estrogen receptor subtype and the Fas/Fas ligand system. J. Neurobiol. 43, 64–78.
Nugent, B. M., and McCarthy, M. M. (2011). Epigenetic underpinnings of developmental sex differences in the brain. Neuroendocrinology 93, 150–158.
Nugent, B. M., Schwarz, J. M., and McCarthy, M. M. (2010). Hormonally-mediated epigenetic changes to steroid receptors in the developing brain: implications for sexual differentiation. Horm. Behav. 59, 338–344.
Numan, M. (1986). The role of the medial preoptic area in the regulation of maternal behavior in the rat. Ann. N. Y. Acad. Sci. 474, 226–233.
Ojeda, S. R., Negro-Vilar, A., and McCann, S. M. (1979). Release of prostaglandin Es by hypothalamic tissue: evidence for their involvement in catecholamine-induced luteinizing hormone-releasing hormone release. Endocrinology 104, 617–624.
Okamoto, I., Otte, A. P., Allis, C. D., Reinberg, D., and Heard, E. (2004). Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 303, 644–649.
O’Malley, B. W., and Tsai, M. J. (1992). Molecular pathways of steroid receptor action. Biol. Reprod. 46, 163–167.
Orikasa, C., Kondo, Y., Usui, S., and Sakuma, Y. (2010). Similar numbers of neurons are generated in the male and female rat preoptic area in utero. Neurosci. Res. 68, 9–14.
Ottem, E. N., Godwin, J. G., Krishnan, S., and Petersen, S. L. (2004). Dual-phenotype GABA/glutamate neurons in adult preoptic area: sexual dimorphism and function. J. Neurosci. 24, 8097–8105.
Park, J. H., Bonthius, P. J., Tsai, H. W., Bekiranov, S., and Rissman, E. F. (2010). Amyloid beta precursor protein regulates male sexual behavior. J. Neurosci. 30, 9967–9972.
Phoenix, C. H., Goy, R. W., Gerall, A. A., and Young, W. C. (1959). Organizing action of prenatally administered testosterone propionate on the tissues mediating mating behavior in the female guinea pig. Endocrinology 65, 369–382.
Polazzi, E., and Contestabile, A. (2002). Reciprocal interactions between microglia and neurons: from survival to neuropathology. Rev. Neurosci. 13, 221–242.
Polston, E. K., and Simerly, R. B. (2003). Sex-specific patterns of galanin, cholecystokinin, and substance P expression in neurons of the principal bed nucleus of the stria terminalis are differentially reflected within three efferent preoptic pathways in the juvenile rat. J. Comp. Neurol. 465, 551–559.
Quadros, P. S., Pfau, J. L., Goldstein, A. Y., De Vries, G. J., and Wagner, C. K. (2002). Sex differences in progesterone receptor expression: a potential mechanism for estradiol-mediated sexual differentiation. Endocrinology 143, 3727–3739.
Rage, F., Lee, B. J., Ma, Y. J., and Ojeda, S. R. (1997). Estradiol enhances prostaglandin E2 receptor gene expression in luteinizing hormone-releasing hormone (LHRH) neurons and facilitates the LHRH response to PGE2 by activating a glia-to-neuron signaling pathway. J. Neurosci. 17, 9145–9156.
Ramagopalan, S., Yee, I., Dyment, D., Orton, S. M., Marrie, R., Sadovnick, A., and Ebers, G. (2009). Parent-of-origin effect in multiple sclerosis. Neurology 73, 602–605.
Raskin, K., de Gendt, K., Duittoz, A., Liere, P., Verhoeven, G., Tronche, F., and Mhaouty-Kodja, S. (2009). Conditional inactivation of androgen receptor gene in the nervous system: effects on male behavioral and neuroendocrine responses. J. Neurosci. 29, 4461–4470.
Raynaud, J. P. (1973). Influence of rat estradiol binding plasma protein (EBP) on uterotrophic activity. Steroids 21, 249–258.
Reik, W., and Walter, J. (2001). Genomic imprinting: parental influence on the genome. Nat. Rev. Genet. 2, 21–32.
Roselli, C. E., and Resko, J. A. (1993). Aromatase activity in the rat brain: hormonal regulation and sex differences. J. Steroid Biochem. Mol. Biol. 44, 499–508.
Rosenfeld, M. G., Lunyak, V. V., and Glass, C. K. (2006). Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 20, 1405–1428.
Roth, T. L., and Sweatt, J. D. (2010). Annual research review: epigenetic mechanisms and environmental shaping of the brain during sensitive periods of development. J. Child Psychol. Psychiatry 52, 398–408.
Schlinger, B. A., and Arnold, A. P. (1992). Circulating estrogens in a male songbird originate in the brain. Proc. Natl. Acad. Sci. U.S.A. 89, 7650–7653.
Schneider, J. S., Burgess, C., Sleiter, N. C., DonCarlos, L. L., Lydon, J. P., O’Malley, B., and Levine, J. E. (2005). Enhanced sexual behaviors and androgen receptor immunoreactivity in the male progesterone receptor knockout mouse. Endocrinology 146, 4340–4348.
Schwarz, J. M., Nugent, B. M., and McCarthy, M. M. (2010). Developmental and hormone-induced epigenetic changes to estrogen and progesterone receptor genes in brain are dynamic across the life span. Endocrinology 151, 4871–4881.
Semaan, S. J., and Kauffman, A. S. (2010). Sexual differentiation and development of forebrain reproductive circuits. Curr. Opin. Neurobiol. 20, 424–431.
Shimura, T., Yamamoto, T., and Shimokochi, M. (1994). The medial preoptic area is involved in both sexual arousal and performance in male rats: re-evaluation of neuron activity in freely moving animals. Brain Res. 640, 215–222.
Simerly, R. B., and Swanson, L. W. (1986). The organization of neural inputs to the medial preoptic nucleus of the rat. J. Comp. Neurol. 246, 312–342.
Simerly, R. B., and Swanson, L. W. (1988). Projections of the medial preoptic nucleus: a Phaseolus vulgaris leucoagglutinin anterograde tract-tracing study in the rat. J. Comp. Neurol. 270, 209–242.
Simerly, R. B., Swanson, L. W., Handa, R. J., and Gorski, R. A. (1985). Influence of perinatal androgen on the sexually dimorphic distribution of tyrosine hydroxylase-immunoreactive cells and fibers in the anteroventral periventricular nucleus of the rat. Neuroendocrinology 40, 501–510.
Stolzenberg, D. S., and Numan, M. (2011). Hypothalamic interaction with the mesolimbic DA system in the control of the maternal and sexual behaviors in rats. Neurosci. Biobehav. Rev. 35, 826–847.
Streit, W. J. (2005). Microglia and neuroprotection: implications for Alzheimer’s disease. Brain Res. Brain Res. Rev. 48, 234–239.
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., Agarwal, S., Iyer, L. M., Liu, D. R., and Aravind, L. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935.
Tate, P. H., and Bird, A. P. (1993). Effects of DNA methylation on DNA-binding proteins and gene expression. Curr. Opin. Genet. Dev. 3, 226–231.
Todd, B. J., Schwarz, J. M., and McCarthy, M. M. (2005). Prostaglandin-E2: a point of divergence in estradiol-mediated sexual differentiation. Horm. Behav. 48, 512–521.
Toran-Allerand, C. D. (1980). Coexistence of alpha-fetoprotein, albumin and transferrin immunoreactivity in neurones of the developing mouse brain. Nature 286, 733–735.
Toran-Allerand, C. D. (1987). Neuronal uptake of alpha-fetoprotein (AFP) synthesized and secreted by hepatocytes in liver/brain co-cultures. Neurosci. Lett. 83, 35–40.
Towle, A. C., and Sze, P. Y. (1983). Steroid binding to synaptic plasma membrane: differential binding of glucocorticoids and gonadal steroids. J. Steroid Biochem. 18, 135–143.
Tremblay, M. E., Stevens, B., Sierra, A., Wake, H., Bessis, A., and Nimmerjahn, A. (2011). The role of microglia in the healthy brain. J. Neurosci. 31, 16064–16069.
Tsai, H. W., Grant, P. A., and Rissman, E. F. (2009). Sex differences in histone modifications in the neonatal mouse brain. Epigenetics 4, 47–53.
Tsukahara, S., Hojo, R., Kuroda, Y., and Fujimaki, H. (2008). Estrogen modulates Bcl-2 family protein expression in the sexually dimorphic nucleus of the preoptic area of postnatal rats. Neurosci. Lett. 432, 58–63.
Tsukahara, S., Kakeyama, M., and Toyofuku, Y. (2006). Sex differences in the level of Bcl-2 family proteins and caspase-3 activation in the sexually dimorphic nuclei of the preoptic area in postnatal rats. J. Neurobiol. 66, 1411–1419.
Uht, R. M., Anderson, C. M., Webb, P., and Kushner, P. J. (1997). Transcriptional activities of estrogen and glucocorticoid receptors are functionally integrated at the AP-1 response element. Endocrinology 138, 2900–2908.
Vargas, D. L., Nascimbene, C., Krishnan, C., Zimmerman, A. W., and Pardo, C. A. (2005). Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 57, 67–81.
Vasudevan, N., and Pfaff, D. W. (2008). Non-genomic actions of estrogens and their interaction with genomic actions in the brain. Front. Neuroendocrinol. 29, 238–257.
Veening, J. G., and Coolen, L. M. (1998). Neural activation following sexual behavior in the male and female rat brain. Behav. Brain Res. 92, 181–193.
Wagner, C. K., Pfau, J. L., De Vries, G. J., and Merchenthaler, I. J. (2001). Sex differences in progesterone receptor immunoreactivity in neonatal mouse brain depend on estrogen receptor alpha expression. J. Neurobiol. 47, 176–182.
Wagner, C. K., Xu, J., Pfau, J. L., Quadros, P. S., De Vries, G. J., and Arnold, A. P. (2004). Neonatal mice possessing an Sry transgene show a masculinized pattern of progesterone receptor expression in the brain independent of sex chromosome status. Endocrinology 145, 1046–1049.
Wallen, K., and Baum, M. J. (2002). “Masculinization and defeminization in altricial and precocial mammals: Comparative aspects of steroid hormone action,” in Hormones, Brain, and Behavior, ed. D. Pfaff (New York: Academic Press), 385–424.
Wang, J. Q., Arora, A., Yang, L., Parelkar, N. K., Zhang, G., Liu, X., Choe, E. S., and Mao, L. (2005). Phosphorylation of AMPA receptors: mechanisms and synaptic plasticity. Mol. Neurobiol. 32, 237–249.
Waters, E. M., and Simerly, R. B. (2009). Estrogen induces caspase-dependent cell death during hypothalamic development. J. Neurosci. 29, 9714–9718.
Watters, J. J., Campbell, J. S., Cunningham, M. J., Krebs, E. G., and Dorsa, D. M. (1997). Rapid membrane effects of steroids in neuroblastoma cells: effects of estrogen on mitogen activated protein kinase signalling cascade and c-fos immediate early gene transcription. Endocrinology 138, 4030–4033.
Wolfe, C. A., Van Doren, M., Walker, H. J., Seney, M. L., McClellan, K. M., and Tobet, S. A. (2005). Sex differences in the location of immunochemically defined cell populations in the mouse preoptic area/anterior hypothalamus. Brain Res. Dev. Brain Res. 157, 34–41.
Wright, C. L., Burks, S. R., and McCarthy, M. M. (2008). Identification of prostaglandin E2 receptors mediating perinatal masculinization of adult sex behavior and neuroanatomical correlates. Dev. Neurobiol. 68, 1406–1419.
Wright, C. L., and McCarthy, M. M. (2009). Prostaglandin E2-induced masculinization of brain and behavior requires protein kinase A, AMPA/kainate, and metabotropic glutamate receptor signaling. J. Neurosci. 29, 13274–13282.
Wu, C. H., Wen, C. Y., Shieh, J. Y., and Ling, E. A. (1992). A quantitative and morphometric study of the transformation of amoeboid microglia into ramified microglia in the developing corpus callosum in rats. J. Anat. 181(Pt 3), 423–430.
Zadran, S., Qin, Q., Bi, X., Zadran, H., Kim, Y., Foy, M. R., Thompson, R., and Baudry, M. (2009). 17-Beta-estradiol increases neuronal excitability through MAP kinase-induced calpain activation. Proc. Natl. Acad. Sci. U.S.A. 106, 21936–21941.
Zechner, U., Wilda, M., Kehrer-Sawatzki, H., Vogel, W., Fundele, R., and Hameister, H. (2001). A high density of X-linked genes for general cognitive ability: a run-away process shaping human evolution?. Trends Genet. 17, 697–701.
Keywords: sex difference, development, preoptic area, estradiol, hormone, epigenetics
Citation: Lenz KM, Nugent BM and McCarthy MM (2012) Sexual differentiation of the rodent brain: dogma and beyond. Front. Neurosci. 6:26. doi: 10.3389/fnins.2012.00026
Received: 29 November 2011;
Paper pending published: 04 January 2012;
Accepted: 04 February 2012;
Published online: 21 February 2012.
Edited by:
Elena Jazin, Uppsala University, SwedenReviewed by:
Susan E. Bergeson, Texas Tech University Health Sciences Center, USAXi Huang, University of California San Francisco, USA
Copyright: © 2012 Lenz, Nugent and McCarthy. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Kathryn M. Lenz, Department of Physiology, University of Maryland School of Medicine, 655 West Baltimore Street, BRB 5-014, Baltimore, 21201 MD, USA. e-mail:a21sZW56QHVtYXJ5bGFuZC5lZHU=