

- 1 School of Veterinary Medicine and Science, University of Nottingham, Leicestershire, UK
- 2 Wellcome Trust Sanger Institute, Cambridge, UK
The animal nervous system processes information from the environment and mediates learning and memory using molecular signaling pathways in the postsynaptic terminal of synapses. Postsynaptic neurotransmitter receptors assemble to form multiprotein complexes that drive signal transduction pathways to downstream cell biological processes. Studies of mouse and Drosophila postsynaptic proteins have identified key roles in synaptic physiology and behavior for a wide range of proteins including receptors, scaffolds, enzymes, structural, translational, and transcriptional regulators. Comparative proteomic and genomic studies identified components of the postsynaptic proteome conserved in eukaryotes and early metazoans. We extend these studies, and examine the conservation of genes and domains found in the human postsynaptic density with those across the three superkingdoms, archaeal, bacteria, and eukaryota. A conserved set of proteins essential for basic cellular functions were conserved across the three superkingdoms, whereas synaptic structural and many signaling molecules were specific to the eukaryote lineage. Genes involved with metabolism and environmental signaling in Escherichia coli including the chemotactic and ArcAB Two-Component signal transduction systems shared homologous genes in the mammalian postsynaptic proteome. These data suggest conservation between prokaryotes and mammalian synapses of signaling mechanisms from receptors to transcriptional responses, a process essential to learning and memory in vertebrates. A number of human postsynaptic proteins with homologs in prokaryotes are mutated in human genetic diseases with nervous system pathology. These data also indicate that structural and signaling proteins characteristic of postsynaptic complexes arose in the eukaryotic lineage and rapidly expanded following the emergence of the metazoa, and provide an insight into the early evolution of synaptic mechanisms and conserved mechanisms of learning and memory.
Introduction
The postsynaptic terminal is important in learning and memory in invertebrates and vertebrates and the molecular signaling events within it regulate forms of neuronal plasticity. While it has been known for many decades that activation of postsynaptic neurotransmitter receptors is essential for transmitting information between neurons, the molecular mechanisms of plasticity, which underlie adaptive behaviors, remain incompletely understood. Proteomic methods in combination with other approaches have identified a large number of proteins in the postsynaptic terminal of excitatory synapses in Drosophila, mice, and rats, with over 1000 different proteins reported in mammalian synapses ( Husi et al., 2000; Walikonis et al., 2000; Satoh et al., 2002; Jordan et al., 2004; Li et al., 2004; Peng et al., 2004; Yoshimura et al., 2004; Trinidad et al., 2005, 2008; Cheng et al., 2006; Collins et al., 2006; Dosemeci et al., 2006, 2007; Coba et al., 2009; Fernandez et al., 2009; Hahn et al., 2009; Selimi et al., 2009). The postsynaptic density (PSD) is enriched in particular protein classes and protein domains and with the use of protein–protein interaction data and molecular network investigation the functional organization and signaling properties of this complex set of proteins have been outlined (Pocklington et al., 2006a,b; Emes et al., 2008b; Bayes et al., 2011). Biochemical evidence for the physiological role of these signaling networks in the PSD was found when activation of the N-methyl-D-aspartate (NMDA) subtype of glutamate receptor resulted in changes in phosphorylation in over 130 PSD proteins of the many functional classes within the networks (Coba et al., 2009). Further support for the physiological importance of these complex sets of proteins comes from mouse knockout studies: in the literature there are over 130 mouse mutants reported with synaptic physiological phenotypes affecting PSD proteins. Moreover, these mutations fall into the wide range of classes of proteins found at the synapse providing compelling evidence for their function.
Within the PSD are multiprotein complexes of structural and signaling proteins linking membrane bound receptors to downstream signaling pathways. The first proteomic characterization of a neurotransmitter receptor complex isolated from brain was the NMDA receptor complex which revealed 77 proteins (Husi et al., 2000). More recently, the same complexes were isolated using genetically targeted Tandem Affinity Purification tags in PSD-95, providing a stringent two-step affinity purification system, which revealed over 100 proteins (Fernandez et al., 2009). For a description of the technical issues relevant to biochemical purification of synaptic complexes and issues of contamination see (Collins et al., 2005; Dosemeci et al., 2007; Fernandez et al., 2009, Delint-Ramirez et al., 2010). Together with data from many other classes of receptors, and data obtained by a variety of complementary methods, it is now clear that neurotransmitter receptors are components of large signaling complexes embedded in the postsynaptic terminal.
A recent study of the proteome of the human PSD (hPSD) from the neocortex identified 1461 proteins (Bayes et al., 2011). This set of proteins was found to be involved in over 133 genetic diseases affecting the nervous system. Using genetic data linked to phenotype data in both mouse and humans, it was found that the phenotypes that were enriched in the hPSD included cognitive, affective, and motor functions (Bayes et al., 2011). This study also compared the conservation of protein sequencing in rodent and primate lineages and found a high degree of sequence constraint on hPSD proteins. This constraint was highest in the proteins forming the PSD-95 complexes and highly connected hub proteins supporting the view that the protein interactions in the postsynaptic complexes are functionally important.
Given the importance of postsynaptic proteins in animal behavior and human disease, it is of interest to understand how the molecular complexity of the postsynaptic proteome evolved. Comparative genomic methods have broadly focused on two complementary strategies: studies of specific proteins or classes of synaptic proteins (e.g., glutamate receptors or scaffold proteins; Kosik, 2009; Ryan and Grant, 2009) and comprehensive studies of the overall postsynaptic proteome (Pocklington et al., 2006b; Emes et al., 2008b; Ryan et al., 2008; Kosik, 2009; Ryan and Grant, 2009; Bayes et al., 2011). The evolutionary studies of the postsynaptic proteome reported a high degree of conservation of genes since the divergence of multicellular eukaryotes with a core set termed the protosynapse (Emes et al., 2008b; Ryan and Grant, 2009). The functional classes of proteins in the protosynapse set include those necessary for assembling neurotransmitter receptor complexes (Pocklington et al., 2006b; Fernandez et al., 2009). These classes of proteins are required for adaptive behaviors in unicellular eukaryotes, marine invertebrates, insects, and mammals. This suggests that the origins of adaptive behavior in complex nervous systems involved molecular mechanisms originating in ancestral unicellular organisms.
The relationship of the postsynaptic proteome with prokaryote pathways has yet to be explored. Whilst genome-wide studies have been conducted to identify elements conserved across the superkingdoms of life (for example see Koonin, 2003; Tatusov et al., 2003; Makarova et al., 2007). These studies did not specifically address the postsynaptic mechanisms, which we now report. In this study we have used the new proteomic data from the human neocortex PSD proteome (Bayes et al., 2011), and thus provides the first comparative genomic study based on the human synapse. Our study also extends previous phylogenetic studies to a range of bacteria and archaea and hence predating the last eukaryotic common ancestor (LECA, see Figure 1). These approaches allow us to identify those parts of the postsynaptic proteome conserved in the superkingdoms of eukaryotes, bacteria, and archaea and hence examine the most ancient limits of the origin of the sets of proteins found at human synapses.
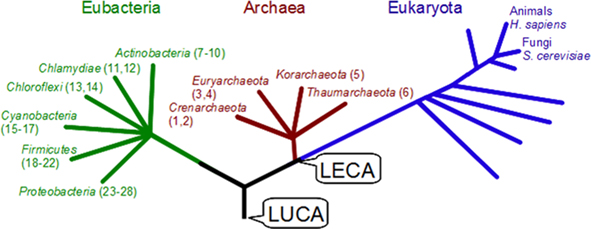
Figure 1. Phylogeny of compared species. The generalized phylogeny of the three superkingdoms studied, adapted from (Woese et al., 1990). Numbers in parentheses relate to full species names and taxonomy given in Supplemental Table S1. LECA, last eukaryotic common ancestor. LUCA, last universal common ancestor.
Materials and Methods
When inferring homology at these large evolutionary distances, many phylogenetic and evolutionary studies often rely on serial gene comparison utilizing the robust BLAST tool (Altschul et al., 1997). However, to avoid the potential errors associated with an automated unsupervised approach, or the problem of directionality when using a single reference as the start point of similarity searches, we applied a novel approach using a multiple sequence alignment of available vertebrate species as the start point of our similarity searches. The whole genome alignments of 44 vertebrate genomes obtained from the UCSC genome browser (see Table S1 in Supplementary Material) were used to generate a hidden Markov model (HMM) for each transcript. These were then used to search the predicted peptide sequences of 28 bacterial and archaeal species (see Table S2 in Supplementary Material). In addition, shared protein domains were identified by comparison to Pfam (Finn et al., 2010) and the position of predicted domains within homologous regions was determined. Using this approach we have robustly identified those parts of the hPSD conserved across the three superkingdoms Eubacteria, Arcahea, and Eukaryota.
Proteins of the hPSD (Bayes et al., 2011) were mapped to a total of 1431 unique human genes with known HUGO identifiers. In turn these HUGO identifiers could be matched to a total of 4721 transcripts of the March 2006 hg18 human genome build available via the UCSC genome browser. Exon level alignments obtained from the UCSC genome browser http://hgdownload.cse.ucsc.edu/goldenPath/hg18/multiz44way/ were parsed to generate multiple sequence files (see Table S1 in Supplementary Material for species aligned). These files were translated and screened to remove sequences with in frame stop codons suggesting a species specific pseudogenization event.
Protein alignments were then parsed to remove poorly aligned regions using the Gblocks algorithm (Castresana, 2000) with the following criteria; maximum number of contiguous non-conserved positions = 8, minimum length of a block = 5, gap positions allowed in all sequences. 4457 transcript alignments (representing a total of 1429 genes) were parsed in this way and were analyzed further. These alignments were used to generate a HMM using hmmbuild of the HMMer package (HMMer version 3.0b2 with default settings; Eddy, 1998). In turn hmmsearch (HMMer version 3.0b2 with maximum E-value cutoff of E < 0.001; Eddy, 1998) was used to compare each HMM to the predicted protein coding sequences of 28 prokaryote species consisting of six archaea (Aeropyrum pernix K1, Pyrobaculum aerophilum str. IM2, Archaeoglobus fulgidus DSM4304, Methanococcus jannaschii DSM2661, Candidatus Korarchaeum cryptofilum OPF8, and Cenarchaeum symbiosum A) and 22 eubacteria (Mycobacterium avium 104, Mycobacterium tuberculosis H37Rv.EB1, Rubrobacter xylanophilus DSM 9941, Cryptobacterium curtum DSM 15641, Lentisphaera araneosa HTCC2155, Chlamydia pneumoniae AR39, Chloroflexus aggregans DSM 9485, Herpetosiphon aurantiacus ATCC 23779, Synechococcus sp. CC9311, Prochlorococcus marinus str. AS9601, Microcystis aeruginosa NIES-843, Bacillus anthracis A0039, Bacillus cereus 10987, Bacillus subtilis, Staphylococcus aureus subsp. aureus COL, Streptococcus agalactiae 515, Aeromonas hydrophila ATCC7966, Campylobacter jejuni RM1221, E. coli K12.EB1.55, Helicobacter pylori 26695, Pseudomonas fluorescens Pf-5, and Wolbachia pipientis wMel). Additionally a single species of single celled eukaryote Saccharomyces cerevisiae was included (see Table S2 in Supplementary Material for a complete taxonomy of species tested). A cut off of E-value ≤ 0.001 was used in the HMM search to identify a region of high sequence similarity, from this homology was inferred. For perfectly conserved genes a representative phylogeny was determined from the alignment of all vertebrate transcripts and prokaryote sequences. Maximum likelihood trees were generated using PhyML (Guindon and Gascuel, 2003) under the WAG model (Whelan and Goldman, 2001; see Figure S1 in Supplementary Material).
Protein domains were identified in each of the predicted proteomes by comparison of each protein in turn to the Pfam-A HMM library (build September 20091) using the hmmscan tool of HMMer (Eddy, 19981) an E-value cutoff of E ≤ 0.01 was used to determine a significant hit. Gene ontology (GO; Harris et al., 2004) terms were associated to predicted protein domains using Pfam2GO (build October 2009).
For each biochemical pathway significant conservation was detected by comparing the number of pathway members with a homolog detected by the HMM search process to the expected number of genes conserved in a pathway of the same size using the binomial test. To avoid the potential inclusion of false positives the Bonferroni correction was applied to the binomial test.
Results
Conservation of the hPSD with Prokaryote Genomes
Conservation of hPSD genes
The HMM search and alignment identified a large number of genes with regions of significant similarity (Figure 2A): 1117 (78.2%) of genes show significant similarity to any one of the species tested (archaeal, bacterial, or yeast Figure 2Bi), and 407 genes (28.5%) were conserved in eukaryotes and a single member of both the archaeal and bacterial superkingdoms. Homologs of 804 (56.3%) genes were detected in either a single archaeal or bacterial gene (Figure 2Bii). 754 (52.8%) had homologs with at least a single bacterial species (Figure 2Biii) and 517 (36.2%) were conserved in at least a single archaeal species (Figure 2Biv). (For details of genes with identified homologs see Supplemental Table S3). These homologous genes represent 65 family types including enzymes, ribosomal proteins, and kinases, representing a conserved core set of the hPSD genes predating the origins of multicellular eukaryotic life.
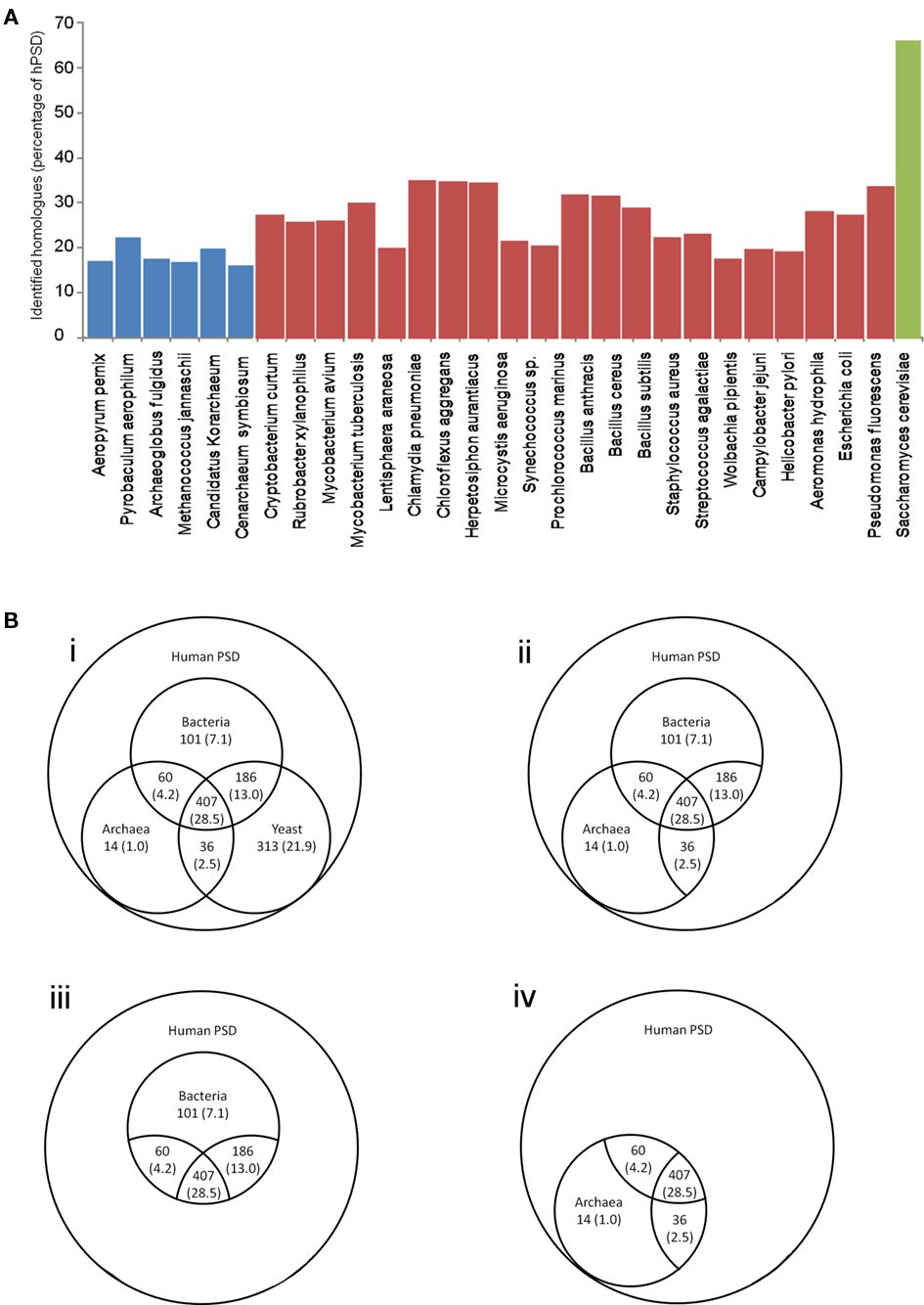
Figure 2. Numbers of hPSD homologs in phylogenetic groups. (A) Histogram of homologs identified in each species tested as a percent of the human PSD dataset (blue archaea, red bacteria, green S. cerevisiae). (B) i Comparison of number of identified hPSD proteome homologs across three superkingdoms, ii subset of i highlighting number of identified hPSD proteome homologs identified in at least a single bacterial and archaeal species, iii subset of i highlighting number of identified hPSD proteome homologs in bacteria, iv subset of i highlighting number of identified hPSD proteome homologs in archaea. In all sections, percentage of 1429 hPSD genes tested are shown in parentheses.
Within this set were 61 genes conserved across all species, which raises the possibility that their presence in these species resulted from horizontal gene transfer. Consistent with this, we noted that synthetase enzymes were the most abundant (including alanyl-tRNA, aspartyl-tRNA, glutamyl-prolyl tRNA, phenylalanyl-tRNA, histidyl-tRNA lysyl-tRNA synthetases, and IARS protein) and tRNA synthetases have been described as having undergone almost exclusive horizontal gene transfer in their evolutionary history (Wolf et al., 1999; Koonin et al., 2001). We investigated the phylogenetic relationship of the conserved genes and of 10 perfectly conserved tRNA synthetases (AARS, DARS, FARSA, HARS2, IARS, KARS, MARS, SARS, TARSL2, and VARS), only AARS and FARSA show an apparent monophylogeny consistent with the expected three superkingdoms (see Figure S1 in Supplementary Material). Additionally, six ribosomal proteins (L8, S14, S15A, S16, S18, and S5) were also conserved in all species tested, and as previously reported for other translation machinery (Koonin et al., 2001), these genes appear to be monophylogenetic without obvious evidence of horizontal gene transfer (see Figure S1 in Supplementary Material). Additional genes for which the analysis of phylogeny gives little evidence of horizontal gene transfer include four translation elongation factors (EEF1a1, EEF1A2, EEF2, and TUFM), three lyases (ENO1, ENO2, and ENO3) two chaperon proteins (CCT4 and HSPD1), and the G protein OLA1. The remaining 37 genes, of the group of genes conserved across all species do not show clear monophylogeny consistent with the superkingdoms and hence are candidates for evidence of horizontal gene transfer during their evolution (Figure S1 in Supplementary Material).
We next classified the homologs into protein types based on Panther domain composition and available mammalian gene annotations. The hPSD was categorized into a total of 22 protein supercategories. A total of 20 of these 22 have detectable homologs in both archaeal and bacterial species (Figure 3). Categories of proteins (shown in italics) with high conservation across all kingdoms include Enzymes, Protein folding/Chaperones, Ribosomal proteins, and Kinases. Within the Enzymes categories were sub-categories dehydrogenases, ligases, hydrolases, transketolases, synthases, RNA helicases, lyases, and nucleases, which have a homolog in greater than 50% of archaeal or bacterial species tested. The synthetases are very well conserved [archaea having on average homologs of 26.2 (77%) of the 34 human genes and bacteria 28.5 (84%)], as are dehydrogenases [archaea have on average homologs of 35.5 (54%) of the 66 human genes, bacteria 48.0 (73%)]. Additionally, translation factors, nucleotide kinases, chromatin remodeling, and cation transporters were also well conserved.
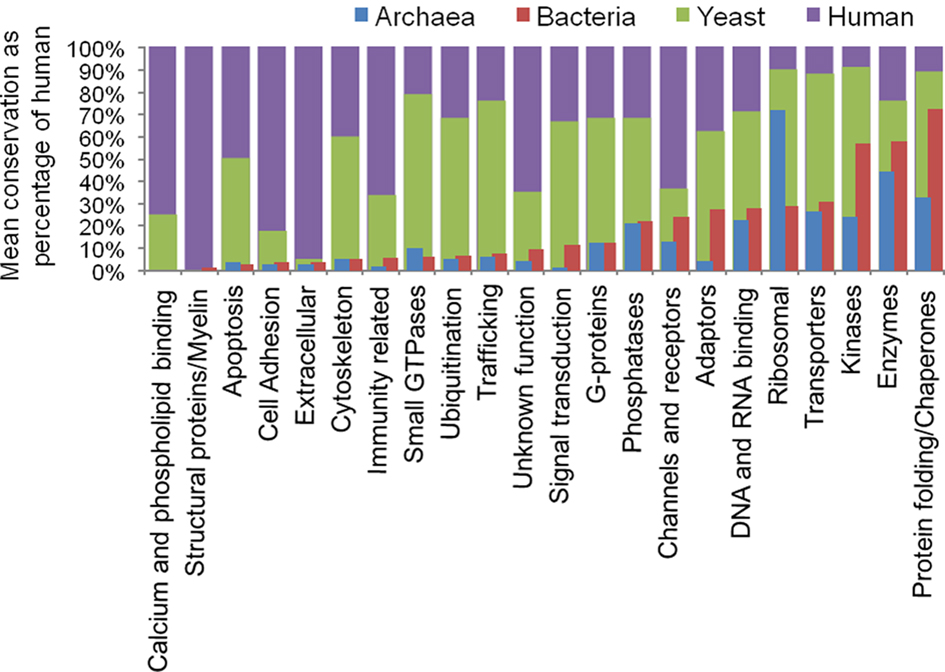
Figure 3. Conservation of hPSD protein types. Relative composition of identified protosynapse compared to human postsynaptic density. Mean bacterial or archaeal percentage of identified homologs were compared to the percent of human genes in a protein supercategory.
Conservation of protein domains
Protein domains are considered as building blocks of the overall function of a protein and the evolutionary constraint on these regions of proteins is reflected in high conservation between taxonomic groups (Emes, 2008a). To determine the extent that aligned regions between homologs overlapped with conserved protein domains, the co-occurrence of protein domains in aligned regions was determined. It was found that whilst the number of hPSD transcripts which could be inferred as homologous between prokaryotic and eukaryotic species ranged from 13.4 (Methanococcus jannaschii) to 29.8% (Herpetosiphon aurantiacus), an average of 96.5% of these aligned regions share a conserved domain between humans and the prokaryote species tested (minimum of 93.0%, Aeromonas hydrophila; maximum of 99.0%, Cenarchaeum symbiosum). The functional roles of these conserved domains were categorized by mapping to GO terms2 and then compared with domains conserved in eukaryotes (domains and GO terms conserved at LECA) and those conserved in all three superkingdoms (domains and GO terms conserved at LUCA; see Figure 4). At the LUCA we observed the conservation of basic biological processes, where the largest conserved groups were domains classified as involved in translation (25 conserved domains, including 10 ribosomal protein and 12 tRNA synthase domains), carbohydrate metabolic process (6 conserved domains), glycolysis (6 conserved domains), and tRNA aminoacylation for protein translation (5 tRNA synthase domains). Since LECA we observed the conservation of domains involved in signal transduction (6 conserved domains), vesicle mediated and intracellular protein transport (16 domains including clathrins, Sec complex domains, and adaptor protein complex domains), and ATP synthesis coupled proton transport (5 conserved ATP synthase domains, see Figure 4).
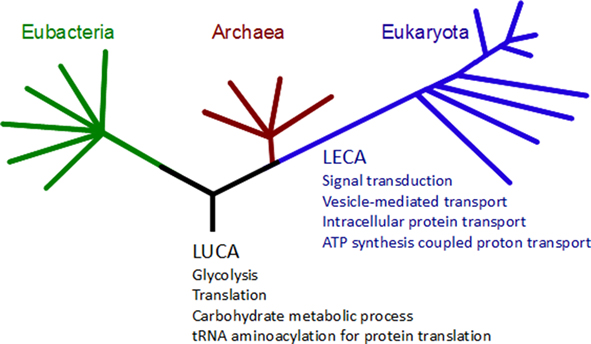
Figure 4. Gene ontology (GO) expansions and innovations associated with conserved protein domains. Protein domains conserved between human and tested species were used to determine associated GO terms (see Materials and Methods). GO terms shown in black are associated with protein domains conserved in eukaryote, archaeal, and bacterial species. GO terms shown in blue are associated with Pfam domains not shared between humans and any archaeal or bacterial species tested.
Conservation of functional pathways
To further dissect any functional conservation across the three superkingdoms we utilized the collection of 309 pathways from E. coli (K-12 substrain MG16553) to identify conservation of interacting sets of proteins. A single bacteria species was selected for this analysis as the interaction data for E. coli is well curated and thought to be a more complete interactome than that available for other prokaryote species. Analysis resulted in a total of 10 pathways identified as having a significantly greater number of homologs than expected by chance (see Table S4 in Supplementary Material). Five significant pathways (“glycine biosynthesis I”; “glutamine biosynthesis I”; “aspartate biosynthesis”; “proline degradation I”; and “tRNA charging pathway”) contain the synthases and transaminases discussed above which are involved in amino acid biosynthesis. Three significant pathways (“octaprenyl diphosphate biosynthesis”; “superpathway of glycolysis, pyruvate dehydrogenase, TCA, and glyoxylate bypass”; and “TCA cycle”) can be grouped as involved in energy generation and a final two conserved pathways are involved in Fatty acid biosysnthesis (“cis-vaccenate biosynthesis” and “PRPP biosynthesis I”). These pathways comprise basic processes consistent across all cellular life. This supports the conclusion that the majority of the homologs of the hPSD comprise basic cellular machinery and not the structural or signal transduction proteins associated with the chemical synapse.
Previously we reported that a significant proportion of genes encoding PSD proteins were members of known signal transduction pathways mediating environmental responses (15% of yeast orthologs of genes encoding mouse PSD proteins (Emes et al., 2008b). Of the annotated E. coli signal transduction pathways the ArcAB two-component signal transduction system involved in response to anaerobic environments showed a significant (p < 0.05 binomial test) conservation of homologs between E. coli and humans. However, this significance is lost if a Bonferroni multiple hypothesis correction is imposed. The ArcAB system comprises ArcB (a membrane associated sensor kinase and phosphatase) and ArcA (a cytosolic transcription factor). An additional 19 E. coli pathways can be described as involved in the sensing of extracellular environment including 18 members of the “two-component signal transduction systems” and the “chemotactic signal transduction system.” These pathways contained a total of 46 E. coli genes, of these only the chemotaxis protein CheA of the chemotactic signal transduction system [homologous to human 3-methyl-2-oxobutanoate dehydrogenase kinase, branched-chain ketoacid dehydrogenase kinase (BCKDK)] and the sensor kinase arcB (homologous to human pyruvate dehydrogenase kinase, PDK3), which is a membrane associated protein responsive to anaerobic conditions and part of the ArcAB two-component signal transduction system mentioned above, have homologs detectable by our approach. This suggests that a proportion of prokaryotic signaling systems capable of transmitting signals from the external environment to gene expression are also found in the in human postsynaptic proteins.
Conservation of Vertebrate hPSD Genes and Evidence for Eukaryote Innovation and Expansion
Conservation of genes in the eukaryote lineage
It is of interest to identify hPSD genes lacking identifiable homologs in the bacterial or archaeal superkingdoms, since they likely represent eukaryote innovations or expansions since LECA (although the possibility of gene loss or rapid sequence divergence since the common ancestor, making detection of homologs not possible cannot be excluded). Toward this objective we identified genes conserved between yeast and vertebrates, which were not detected in prokaryotes. They included proteins involved in Ubiquitination, Trafficking, and the cytoskeleton and G-proteins. Genes that were largely vertebrate specific with poor conservation in yeast, bacteria, and archaea included Calcium and phospholipid binding, cell adhesion, and structural proteins (Figure 3). These expansions include the eight calcium and phospholipid binding proteins (Annexins and GLUT4) and the single lipase gene, which do not have homologs in any of the bacterial or archaeal species. Additionally, on average only 5% of genes encoding human synapse cytoskeleton proteins have an identified homolog in the bacterial and archaeal species whilst 60% are conserved in yeast, representing rapid expansion at the base of the eukaryotes. When these categories are investigated in more detail it can be seen that of the cytoskeletal proteins certain categories are particularly underrepresented in the prokaryotes, for example the actin-related genes [archaea have a mean number of 3.8 detectable homologs (4%) of the 96 human genes, bacteria 4.6 (5%)] and cytoskeleton motor subgroups [archaea have a mean number of 1.8 (4%) detectable homologs of the 42 human genes, bacteria 1.4 (3%)].
Conservation of protein domains
One hundred ninety nine domains in hPSD proteins, which were absent in prokaryotes, were shared in aligned regions between vertebrates and yeast proteins. These domains mapped to a total of 344 GO terms. Within the biological process ontology the most abundant were “intracellular protein transport” (18 domains, including 5 adapter protein complex and 4 clathrin protein domains), “vesicle mediated transport” (14 domains, including 5 adapter protein complex and 4 clathrin protein domains), “signal transduction” [6 conserved domains Protein phosphatase 2A regulatory B subunit (Pfam B56), 3′5′-cyclic nucleotide phosphodiesterase (Pfam PDEase_I), Phosphatidylinositol-specific phospholipase (Pfam PI-PLC-Y), RhoGap, Regulatory subunit of type II PKA R-subunit (Pfam RIIa), Secreted protein acidic and rich in cysteine Ca2+ binding region (Pfam SPARC_Ca_bdg)], and “ATP synthesis coupled proton transport” (5 conserved domains). See Table S5 in Supplementary Material for counts of conserved domains in all species tested. Again this supports the assumption that the signaling functions associated with the hPSD are conserved in the eukaryotes alone. To further investigate the conservation of functional entities between prokaryotes and eukaryotes, biochemical pathway composition was analyzed.
Conservation of functional pathways in eukaryote homologs of the hPSD
Analysis of yeast biochemical pathways using data from the Saccharomyces genome database4 identifies only seven pathways with a significant abundance of conserved synapse proteome genes (see Table S6 in Supplementary Material). As with the E. coli analysis these pathways are broadly related to amino acid synthesis and utilization (“glutamine biosynthesis” and “proline utilization”) and energy generation (“glycerol degradation,” “2-ketoglutarate dehydrogenase complex,” “pyruvate dehydrogenase complex,” “glycolysis and TCA cycle, aerobic respiration”).
In light of recent research highlighting the importance of members of the hPSD in human nervous system disease (Bayes et al., 2011), we investigated if any of the 61 genes conserved across the three superkingdoms are associated with disease. By comparison to this previous publication (see Bayes et al., 2011; Table S5 in Supplementary Material), nine genes were associated with human disease in the OMIM database (ABCD1, ABCD3, DLD, ENO3, FH, HSPD1, PGK1, TUFM, and VCP5). Of these ABCD1 (adrenoleukodytrophy), ABCD3 (Zellweger syndrome), DLD (Leigh syndrome and Maple syrup urine disease, MSUD), FH (fumarase deficiency), HSPD1 (Spastic paraplegia 13 and hypomyelinating leukodystrophy), TUFM (combined oxidative phosphorylation deficiency 4), and VCP (Inclusion body myopathy with early onset Paget disease and frontotemporal dementia) are associated with disease of the CNS.
Discussion
The postsynaptic proteome of metazoan organisms is highly complex and here we use comparative genomic methods to explore those components that are conserved in prokaryotes. Through the identification of conserved proteins, domains, and pathways, we identified potential building blocks in bacteria and archaea, which predate the divergence of the eukaryotes. The 407 genes (28% of the hPSD) identified as homologs of vertebrates and at least one bacteria, archaea, and S. cerevisiae highlighted the breadth of protein types conserved across the three superkingdoms. Whilst some of these homologs are only identified in a single bacterial or archaeal species, and could potentially reflect an artifact, of the genes that were conserved across all species tested (and lacking evidence of horizontal gene transfer) there were synthetases, dehydrogenases, and lyases which contribute to a wide range of reactions, and elongation factor and ribosomal proteins key to the highly conserved process of translation (Nakamoto, 2009). These genes conserved across the three superkingdoms are ancestral to the formation of the neuronal synapse.
This study identifies the hPSD machinery conserved across the prokaryote/eukaryote boundary involved in amino acid biosynthesis and energy generation. In contrast, the majority of structural and signaling complexes of the PSD are conserved only in the eukaryotes. Within the eukaryotic lineage the postsynaptic proteome expanded to include small-GTPases, G-proteins, and proteins associated with signal transduction and trafficking. Following the divergence of the metazoans the postsynaptic proteome grew by addition/expansion of the protein classes; extracellular, cell adhesion, and structural proteins (see Figure 3). As expected, these results based on the hPSD are consistent with previous findings based on mouse and Drosophila proteomic data (Emes et al., 2008b). Within the extracellular proteins the signaling molecules fibrinogen β/γ, signal-regulatory protein alpha, versican, and growth factors FGF and VGF are notably lacking in the yeast and hence are examples of genes conserved within the metazoa.
The comparison of the shared domains between significantly similar proteins highlights the potential problems of unsupervised homology based approaches to infer the composition of ancient protein complexes. For example, the Shank proteins have been described as the central interacting protein of the backbone of the PSD (Alie and Manuel, 2010). We found both SHANK1 and SHANK3 to have significant similarity between the vertebrate alignment and multiple bacterial species (see Table S3). However by looking at the shared domains within the aligned region it can be seen that this is due to multiple shared ankyrin domains. This suggests that the function of these proteins will be different in prokaryote and vertebrate species. In addition, the bacterial sequences do not contain the SH3 or PDZ domains characteristic of the vertebrate Shank proteins. Thus, although it is useful to have this overview of numbers of genes with a significant match in the bacterial/archaeal groups, what does it tell us of the potential evolution and origin of the synapse proteome? For this we need to delve into an understanding of function of the conserved genes and ask what their potential roles are. By comparison of the numbers of observed and expected members of known biochemical pathways, we showed that while many individual genes exhibit significant sequence similarity, few pathways show significant conservation of members. Aside from those pathways with a single member, the significant E. coli pathways are the “tRNA charging pathway,” “superpathway of glycolysis, pyruvate dehydrogenase, TCA, and glyoxylate bypass,” and “TCA cycle.” This suggests that the processes of translation and energy generation are the only processes incorporated “wholesale” into the vertebrate postsynaptic proteome. Analysis of the conserved protein domains supports these findings: associated GO terms “translation,” “metabolic process,” “carbohydrate metabolic process,” and “glycolysis” were most abundant in the human:prokaryote shared domains. It is interesting to note that metabolic processes are critical for vertebrate synaptic function and emerging as a key mechanism involved with neurodegenerative disorders (Davey et al., 1998).
The presence of a system to sense environmental factors has been described in the eukaryote phylum Porifera (for review see Ryan and Grant, 2009). Although a “synapse-like” structure has not been observed in sponges, homologs of many of the PSD genes have been detected in the globular or flask cells predicted to have environmental sensing properties (Sakarya et al., 2007), and homologs of cell–cell signaling and adhesion genes have been detected (Nichols et al., 2006). Additionally the cnidaria have been shown to have homologs of a range of genes encoding proteins found in the synaptic proteome (Sakarya et al., 2007). To determine if these phenomena are associated with proteins of the protosynapse, we investigated if the conservation of genes involved in “environmental responses” seen previously in yeast (Emes et al., 2008b) is also conserved in an example prokaryote species. When complete pathways are analyzed, no significant evidence for conservation of the pathways was detected. However, two pathways involved in environmental signaling in E. coli “Chemotactic Signal Transduction System” and “ArcAB Two-Component Signal Transduction System” shared homologous genes found in the human synapse. Within the ArcAB system the conserved ArcB gene is a kinase and part of the anaerobic sensing machinery of E. coli and the human homolog PDK3 is a pyruvate dehydrogenase kinase which inhibits the pyruvate dehydrogenase complex involved in glucose metabolism. The conserved E. coli gene of the chemotactic signal system is the chemotaxis protein CheA a kinase involved in the control of transmission of sensory signals from chemoreceptors to flagellar motors (McNally and Matsumura, 1991). The homologous gene in humans, BCKDK regulates the activity of the branched-chain ketoacid dehydrogenase complex. The complex implicated in the branched-chain ketoaciduria or MSUD (Menkes et al., 1954) associated with neurological abnormalities (Hoffmann et al., 2006; Walsh and Scott, 2010). In mice loss of this gene is associated with growth deficiencies including brain development (Joshi et al., 2006) and in rats, behavior was altered by manipulation of branched-chain alpha-hydroxy acids which accumulate in MSUD (Vasques Vde et al., 2005).
The identification of homologs of genes relevant for human disease in the prokaryotes highlights the importance what are often dismissed as “house-keeping” genes in CNS function and human health. Whilst some of these may have been subject to horizontal gene transfer in their evolution, the phylogeny of two genes HSPD1 (Spastic paraplegia 13 and hypomyelinating leukodystrophy) and TUFM (combined oxidative phosphorylation deficiency 4) suggests that these genes have been conserved since LUCA without horizontal gene transfer and are required for CNS health in extant vertebrate animals. Whilst we have identified homologous genes encoding both the hPSD of vertebrates and proteins conserved in a range of prokaryote genomes, possession of homologs of genes which encode essential synapse proteins does not suggest that a macromolecular structure similar to the PSD is present in these organisms; simply that homologs of genes encoding these proteins are detectable. It is also important to recognize that these findings should not be misconstrued to imply there was a simple evolutionary trajectory from prokaryotes to human synapses. These conserved proteins and protein domains expands our understanding of the composition of the “genetic toolkit” (Rokas, 2008; Ruiz-Trillo et al., 2008) utilized in construction of the metazoan synapse. The lack of detectable structural and signaling hPSD molecules essential for synapse function in the prokaryote genomes fixes the upper limit for the origin of the protein interaction networks that comprise the PSD in its protosynaptic form to a point after the divergence of the prokaryote/eukaryote lineage.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a Royal Society UK Grant, RG080388 to Richard David Emes. Seth G. N. Grant was supported by the Wellcome Trust Genes to Cognition Programme, MRC, EU FP7 EUROSPIN, and SYNSYS programs. The authors would like to thank N. H. Komiyama and the anonymous reviewers for their helpful comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/neuroscience/neurogenomics/paper/10.3389/fnins.2011.00044
Footnotes
References
Alie, A., and Manuel, M. (2010). The backbone of the post-synaptic density originated in a unicellular ancestor of choanoflagellates and metazoans. BMC Evol. Biol. 10, 34. doi: 10.1186/1471-2148-10-34
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402.
Bayes, A., van de Lagemaat, L. N., Collins, M. O., Croning, M. D., Whittle, I. R., Choudhary, J. S., and Grant, S. G. (2011). Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 14, 19–21.
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552.
Cheng, D., Hoogenraad, C. C., Rush, J., Ramm, E., Schlager, M. A., Duong, D. M., Xu, P., Wijayawardana, S. R., Hanfelt, J., Nakagawa, T., Sheng, M., and Peng, J. (2006). Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell Proteomics 5, 1158–1170.
Coba, M. P., Pocklington, A. J., Collins, M. O., Kopanitsa, M. V., Uren, R. T., Swamy, S., Croning, M. D., Choudhary, J. S., and Grant, S. G. (2009). Neurotransmitters drive combinatorial multistate postsynaptic density networks. Sci. Signal. 2, ra19.
Collins, M. O., Husi, H., Yu, L., Brandon, J. M., Anderson, C. N., Blackstock, W. P., Choudhary, J. S., and Grant, S. G. (2006). Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J. Neurochem. 97(Suppl 1), 16–23.
Collins, M. O., Yu, L., Coba, M. P., Husi, H., Campuzano, I., Blackstock, W. P., Choudhary, J. S., and Grant, S. G. (2005). Proteomic analysis of in vivo phosphorylated synaptic proteins. J. Biol. Chem. 280, 5972–5982.
Davey, G. P., Peuchen, S., and Clark, J. B. (1998). Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J. Biol. Chem. 273, 12753–12757.
Delint-Ramirez, I., Fernandez, E., Bayes, A., Kicsi, E., Komiyama, N. H., and Grant, S. G. (2010). In vivo composition of NMDA receptor signaling complexes differs between membrane subdomains and is modulated by PSD-95 and PSD-93. J. Neurosci. 30, 8162–8170.
Dosemeci, A., Makusky, A. J., Jankowska-Stephens, E., Yang, X., Slotta, D. J., and Markey, S. P. (2007). Composition of the synaptic PSD-95 complex. Mol. Cell Proteomics 6, 1749–1760.
Dosemeci, A., Tao-Cheng, J. H., Vinade, L., and Jaffe, H. (2006). Preparation of postsynaptic density fraction from hippocampal slices and proteomic analysis. Biochem. Biophys. Res. Commun. 339, 687–694.
Emes, R. D., Pocklington, A. J., Anderson, C. N., Bayes, A., Collins, M. O., Vickers, C. A., Croning, M. D., Malik, B. R., Choudhary, J. S., Armstrong, J. D., and Grant, S. G. (2008b). Evolutionary expansion and anatomical specialization of synapse proteome complexity. Nat. Neurosci. 11, 799–806.
Fernandez, E., Collins, M. O., Uren, R. T., Kopanitsa, M. V., Komiyama, N. H., Croning, M. D., Zografos, L., Armstrong, J. D., Choudhary, J. S., and Grant, S. G. (2009). Targeted tandem affinity purification of PSD-95 recovers core postsynaptic complexes and schizophrenia susceptibility proteins. Mol. Syst. Biol. 5, 269.
Finn, R. D., Mistry, J., Tate, J., Coggill, P., Heger, A., Pollington, J. E., Gavin, O. L., Gunasekaran, P., Ceric, G., Forslund, K., Holm, L., Sonnhammer, E. L., Eddy, S. R., and Bateman, A. (2010). The Pfam protein families database. Nucleic Acids Res. 38, D211–D222.
Guindon, S., and Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704.
Hahn, C. G., Banerjee, A., Macdonald, M. L., Cho, D. S., Kamins, J., Nie, Z., Borgmann-Winter, K. E., Grosser, T., Pizarro, A., Ciccimaro, E., Arnold, S. E., Wang, H. Y., and Blair, I. A. (2009). The post-synaptic density of human postmortem brain tissues: an experimental study paradigm for neuropsychiatric illnesses. PLoS ONE 4, e5251. doi: 10.1371/journal.pone.0005251
Harris, M. A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., Eilbeck, K., Lewis, S., Marshall, B., Mungall, C., Richter, J., Rubin, G. M., Blake, J. A., Bult, C., Dolan, M., Drabkin, H., Eppig, J. T., Hill, D. P., Ni, L., Ringwald, M., Balakrishnan, R., Cherry, J. M., Christie, K. R., Costanzo, M. C., Dwight, S. S., Engel, S., Fisk, D. G., Hirschman, J. E., Hong, E. L., Nash, R. S., Sethuraman, A., Theesfeld, C. L., Botstein, D., Dolinski, K., Feierbach, B., Berardini, T., Mundodi, S., Rhee, S. Y., Apweiler, R., Barrell, D., Camon, E., Dimmer, E., Lee, V., Chisholm, R., Gaudet, P., Kibbe, W., Kishore, R., Schwarz, E. M., Sternberg, P., Gwinn, M., Hannick, L., Wortman, J., Berriman, M., Wood, V., de la Cruz, N., Tonellato, P., Jaiswal, P., Seigfried, T., and White, R. (2004). The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261.
Hoffmann, B., Helbling, C., Schadewaldt, P., and Wendel, U. (2006). Impact of longitudinal plasma leucine levels on the intellectual outcome in patients with classic MSUD. Pediatr. Res. 59, 17–20.
Husi, H., Ward, M. A., Choudhary, J. S., Blackstock, W. P., and Grant, S. G. (2000). Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 3, 661–669.
Jordan, B. A., Fernholz, B. D., Boussac, M., Xu, C., Grigorean, G., Ziff, E. B., and Neubert, T. A. (2004). Identification and verification of novel rodent postsynaptic density proteins. Mol. Cell Proteomics 3, 857–871.
Joshi, M. A., Jeoung, N. H., Obayashi, M., Hattab, E. M., Brocken, E. G., Liechty, E. A., Kubek, M. J., Vattem, K. M., Wek, R. C., and Harris, R. A. (2006). Impaired growth and neurological abnormalities in branched-chain alpha-keto acid dehydrogenase kinase-deficient mice. Biochem. J. 400, 153–162.
Koonin, E. V. (2003). Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat. Rev. Microbiol. 1, 127–136.
Koonin, E. V., Makarova, K. S., and Aravind, L. (2001). Horizontal gene transfer in prokaryotes: quantification and classification. Annu. Rev. Microbiol. 55, 709–742.
Kosik, K. S. (2009). Exploring the early origins of the synapse by comparative genomics. Biol. Lett. 5, 108–111.
Li, K. W., Hornshaw, M. P., Van Der Schors, R. C., Watson, R., Tate, S., Casetta, B., Jimenez, C. R., Gouwenberg, Y., Gundelfinger, E. D., Smalla, K. H., and Smit, A. B. (2004). Proteomics analysis of rat brain postsynaptic density. Implications of the diverse protein functional groups for the integration of synaptic physiology. J. Biol. Chem. 279, 987–1002.
Makarova, K. S., Sorokin, A. V., Novichkov, P. S., Wolf, Y. I., and Koonin, E. V. (2007). Clusters of orthologous genes for 41 archaeal genomes and implications for evolutionary genomics of archaea. Biol. Direct 2, 33.
McNally, D. F., and Matsumura, P. (1991). Bacterial chemotaxis signaling complexes: formation of a CheA/CheW complex enhances autophosphorylation and affinity for CheY. Proc. Natl. Acad. Sci. U.S.A. 88, 6269–6273.
Menkes, J. H., Hurst, P. L., and Craig, J. M. (1954). A new syndrome: progressive familial infantile cerebral dysfunction associated with an unusual urinary substance. Pediatrics 14, 462–467.
Nakamoto, T. (2009). Evolution and the universality of the mechanism of initiation of protein synthesis. Gene 432, 1–6.
Nichols, S. A., Dirks, W., Pearse, J. S., and King, N. (2006). Early evolution of animal cell signaling and adhesion genes. Proc. Natl. Acad. Sci. U.S.A. 103, 12451–12456.
Peng, J., Kim, M. J., Cheng, D., Duong, D. M., Gygi, S. P., and Sheng, M. (2004). Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J. Biol. Chem. 279, 21003–21011.
Pocklington, A. J., Armstrong, J. D., and Grant, S. G. (2006a). Organization of brain complexity – synapse proteome form and function. Brief. Funct. Genomic. Proteomic. 5, 66–73.
Pocklington, A. J., Cumiskey, M., Armstrong, J. D., and Grant, S. G. N. (2006b). The proteomes of neurotransmitter receptor complexes form modular networks with distributed functionality underlying plasticity and behaviour. Mol. Syst. Biol. 2, E1–E14.
Rokas, A. (2008). The origins of multicellularity and the early history of the genetic toolkit for animal development. Annu. Rev. Genet. 42, 235–251.
Ruiz-Trillo, I., Roger, A. J., Burger, G., Gray, M. W., and Lang, B. F. (2008). A phylogenomic investigation into the origin of metazoa. Mol. Biol. Evol. 25, 664–672.
Ryan, T. J., Emes, R. D., Grant, S. G., and Komiyama, N. H. (2008). Evolution of NMDA receptor cytoplasmic interaction domains: implications for organisation of synaptic signalling complexes. BMC Neurosci. 9, 6. doi: 10.1186/1471-2202-9-6
Ryan, T. J., and Grant, S. G. (2009). The origin and evolution of synapses. Nat. Rev. Neurosci. 10, 701–712.
Sakarya, O., Armstrong, K. A., Adamska, M., Adamski, M., Wang, I. F., Tidor, B., Degnan, B. M., Oakley, T. H., and Kosik, K. S. (2007). A post-synaptic scaffold at the origin of the animal kingdom. PLoS ONE 2, e506. doi: 10.1371/journal.pone.0000506
Satoh, K., Takeuchi, M., Oda, Y., Deguchi-Tawarada, M., Sakamoto, Y., Matsubara, K., Nagasu, T., and Takai, Y. (2002). Identification of activity-regulated proteins in the postsynaptic density fraction. Genes Cells 7, 187–197.
Selimi, F., Cristea, I. M., Heller, E., Chait, B. T., and Heintz, N. (2009). Proteomic studies of a single CNS synapse type: the parallel fiber/purkinje cell synapse. PLoS Biol. 7, e83. doi: 10.1371/journal.pbio.1000083
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., Krylov, D. M., Mazumder, R., Mekhedov, S. L., Nikolskaya, A. N., Rao, B. S., Smirnov, S., Sverdlov, A. V., Vasudevan, S., Wolf, Y. I., Yin, J. J., and Natale, D. A. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 41. doi: 10.1186/1471-2105-4-41
Trinidad, J. C., Thalhammer, A., Specht, C. G., Lynn, A. J., Baker, P. R., Schoepfer, R., and Burlingame, A. L. (2008). Quantitative analysis of synaptic phosphorylation and protein expression. Mol. Cell Proteomics 7, 684–696.
Trinidad, J. C., Thalhammer, A., Specht, C. G., Schoepfer, R., and Burlingame, A. L. (2005). Phosphorylation state of postsynaptic density proteins. J. Neurochem. 92, 1306–1316.
Vasques Vde, C., Brinco, F., and Wajner, M. (2005). Intrahippocampal administration of the branched-chain alpha-hydroxy acids accumulating in maple syrup urine disease compromises rat performance in aversive and non-aversive behavioral tasks. J. Neurol. Sci. 232, 11–21.
Walikonis, R. S., Jensen, O. N., Mann, M., Provance, D. W. Jr., Mercer, J. A., and Kennedy, M. B. (2000). Identification of proteins in the postsynaptic density fraction by mass spectrometry. J. Neurosci. 20, 4069–4080.
Walsh, K. S., and Scott, M. N. (2010). Neurocognitive profile in a case of maple syrup urine disease. Clin. Neuropsychol. 24, 689–700.
Whelan, S., and Goldman, N. (2001). A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18, 691–699.
Woese, C. R., Kandler, O., and Wheelis, M. L. (1990). Towards a natural system of organisms: proposal for the domains archaea, bacteria, and eucarya. Proc. Natl. Acad. Sci. U.S.A. 87, 4576–4579.
Wolf, Y. I., Aravind, L., Grishin, N. V., and Koonin, E. V. (1999). Evolution of aminoacyl-tRNA synthetases – analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 9, 689–710.
Yoshimura, Y., Yamauchi, Y., Shinkawa, T., Taoka, M., Donai, H., Takahashi, N., Isobe, T., and Yamauchi, T. (2004). Molecular constituents of the postsynaptic density fraction revealed by proteomic analysis using multidimensional liquid chromatography-tandem mass spectrometry. J. Neurochem. 88, 759–768.
Keywords: synapse, brain, proteome, comparative genomics, phylogenetics
Citation: Emes RD and Grant SGN (2011) The human postsynaptic density shares conserved elements with proteomes of unicellular eukaryotes and prokaryotes. Front. Neurosci. 5:44. doi: 10.3389/fnins.2011.00044
Received: 08 November 2010;
Accepted: 16 March 2011;
Published online: 31 March 2011.
Edited by:
Jocelyne Caboche, Universite Pierre et Marie Curie, FranceReviewed by:
Ken Kosik, University of California at Santa Barbara, USAPhilippe Vernier, Centre National de la Recherche Scientifique, France
Copyright: © 2011 Emes and Grant. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Richard David Emes, School of Veterinary Medicine and Science, University of Nottingham, Sutton Bonington Campus, College Road, Sutton Bonington, Leicestershire LE12 5RD, UK. e-mail:cmljaGFyZC5lbWVzQG5vdHRpbmdoYW0uYWMudWs=