
- Department of Anatomy and Neurobiology, Virginia Commonwealth University Medical Center, Richmond, VA, USA
Neuronal plasticity is an essential property of the brain that is impaired in different neurological conditions. Phosphodiesterase type 1 (PDE1) inhibitors can enhance levels of the second messengers cAMP/cGMP leading to the expression of neuronal plasticity-related genes, neurotrophic factors, and neuroprotective molecules. These neuronal plasticity enhancement properties make PDE1 inhibitors good candidates as therapeutic agents in many neurological conditions. However, the lack of specificity of the drugs currently available poses a challenge to the systematic evaluation of the beneficial effect of these agents. The development of more specific drugs may pave the way for the use of PDE1 inhibitors as therapeutic agents in cases of neurodevelopmental conditions such as fetal alcohol spectrum disorders and in degenerative disorders such as Alzheimer’s and Parkinson’s.
During the last decades several groups have been actively pursuing the development of pharmacological agents that can improve neuronal plasticity in normal subjects or restore this function in different neurological conditions. Among these agents, phosphodiesterase type 1 (PDE1) inhibitors have been tested in animal models and humans showing promising therapeutic use (Dyke and Montana, 1999; Josselyn and Nguyen, 2005; Rose et al., 2005).
Neuronal plasticity is the lifelong ability of the brain to reorganize neural pathways. This property is essential during the development when refinement of connections lead to the precise wiring of the brain; in recovery after lesions, and in learning and memory processes. Therefore, the disruption of this crucial neuronal feature can result in alterations in normal brain development and cognitive problems (Nithianantharajah and Hannan, 2006). In fact, neuronal plasticity is disrupted in disorders of brain development such as Fetal alcohol spectrum disorders (Medina and Krahe, 2008), Down syndrome (Siarey et al., 1997, 2005), and Angelman syndrome (Yashiro et al., 2009) and in Neurodegenerative disorders such as Alzheimer’s (Nalbantoglu et al., 1997; Marcello et al., 2008) and Parkinson’s disease (Morgante et al., 2006; Bagetta et al., 2010). Therefore, because of PDE1 inhibitors are good candidates as plasticity enhancers, there is potential for its use as a therapeutic approach in this conditions. In addition, PDE1 inhibitors have also been shown potential to exert a neuroprotective role (Chen et al., 2007), which could be particularly interesting for treatment of neurodegenerative conditions.
Phosphodiesterase are enzymes that catalyze the hydrolysis of the 3′ cyclic phosphate bonds of adenosine and/or guanosine 3′, 5′ cyclic monophosphate (Beavo, 1995). These enzymes have been grouped into several families based on their regulation and substrate specificity. Some PDEs such as type 4 are specific for cAMP, while others such PDE5, specific for cGMP (Beavo, 1995). The PDE1 family is activated by Ca++/calmodulin and is capable to act on both cAMP and cGMP. Calmodulin is activated by calcium and can bind and activate adenyl cyclase leading to conversion of AMP into cAMP (Brostom et al., 1975; Ferguson and Storm, 2004). However, once in the cytoplasm, calmodulin can activate PDE1 causing the conversion of cAMP into AMP and cGMP to GMP (Goraya and Cooper, 2005). PDE1 presents several isoforms; PDE1A1 is present in the lung and heart, whereas PDE1A2 and PDE1B1 in the brain (Kakkar et al., 1999). While PDE1A2 is highly expressed in cerebral cortex and hippocampus, PDE1B1 is expressed mainly in dopaminergic regions such as the striatum and nucleus accumbens (Beavo, 1995; Kakkar et al., 1999). PDE1C is highly expressed in the heart, however it can also be found in the CNS, such as in olfactory neuroepithelium and, in small quantities, substantia nigra and thalamus (Menniti et al., 2006; Lakics et al., 2010).
Here we will provide a brief overview of the mechanisms of action of PDE1 inhibitors and discuss the potential for the therapeutic use of these drugs in neurological disorders, focusing on neuronal plasticity improvement.
How PDE Inhibition can Improve Neuronal Plasticity?
Glutamatergic transmission leads to an increase of intracellular Ca++, and cAMP/cGMP levels triggering cascades that will ultimately lead to the phosphorylation and activation of the transcription factors cAMP responsive element binding protein (CREB) and serum response factor (SRF; Figure 1). Activation of CREB and/or SRF can lead to the expression of plasticity-related genes, which in turn promote the functional and morphological changes necessary for neuronal plasticity to occur (Frank and Greenberg, 1994; Atkins et al., 1998; Silva et al., 1998; Josselyn and Nguyen, 2005; Etkin et al., 2006; Pintchovski et al., 2009) such as alteration of cytoskeleton protein complexes (Lavaur et al., 2008) and remodeling of dendritic spines (Suzuki et al., 2007). CREB is one of the most important molecules in neuronal plasticity processes and it is crucial for several paradigms such as long-term potentiation (LTP; Barco et al., 2002; Bourtchouladze et al., 2003), learning and memory (Frank and Greenberg, 1994; Silva et al., 1998; Lamprecht, 2005), ocular dominance plasticity (Mower et al., 2002), and barrel cortex plasticity (Glazewski et al., 1999). Recent studies have shown that SRF is also implicated in neuronal plasticity and is related to LTP (Ramanan et al., 2005), LTD (Etkin et al., 2006), and spatial memory formation (Tyan et al., 2008).
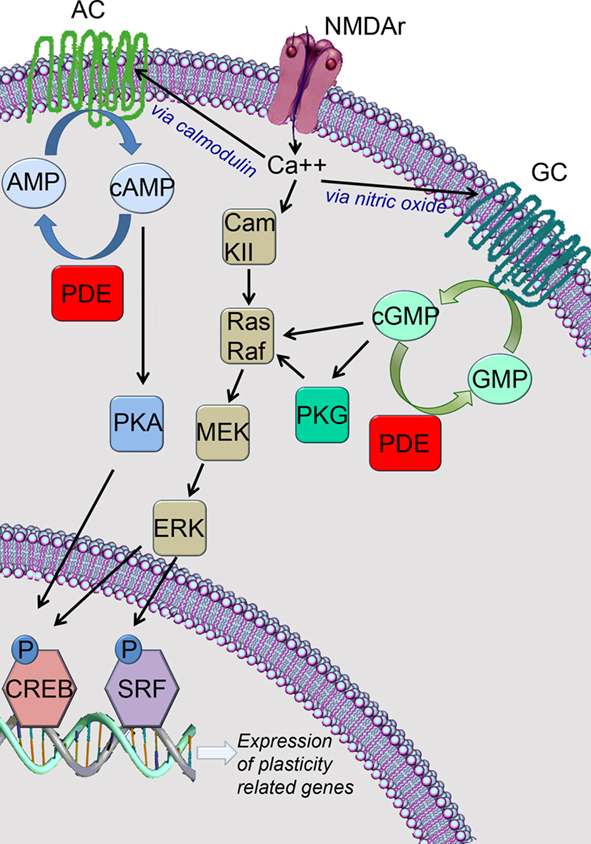
Figure 1. Inhibition of phosphodiesterase type 1 (PDE1) lead to the increase of cAMP and cGMP and ultimately to the expression of plasticity-related genes. AC, adenyl cyclase; GC, guanylate cyclase; CREB, cAMP responding element binding protein; SRF, serum response factor.
Another way that PDE inhibition can improve neuronal plasticity is trough potentiation of glutamatergic transmission (Figure 2). The increase in cAMP/cGMP levels caused by PDE1 inhibition, can lead to the phosphorylation of AMPA receptors promoting its incorporation into the synapse (Serulle et al., 2007) and leading to facilitation of the glutamatergic transmission. AMPA receptors can move in and out the synaptic membrane affecting glutamatergic transmission by making NMDA receptors functional (Malinow and Malenka, 2002). At the resting membrane potential, the NMDA receptor is blocked by Mg++ (Mayer et al., 1984; Nowak et al., 1984). However, activation of AMPA receptors can depolarize the postsynaptic membrane, releasing the Mg++ blockade and making the NMDA receptor functional (Mayer et al., 1984; Nowak et al., 1984). Therefore, NMDA receptor activation can be facilitated or reduced respectively by insertion/removal of AMPA receptors in/from the postsynaptic membrane (Malinow and Malenka, 2002; Malenka and Bear, 2004).
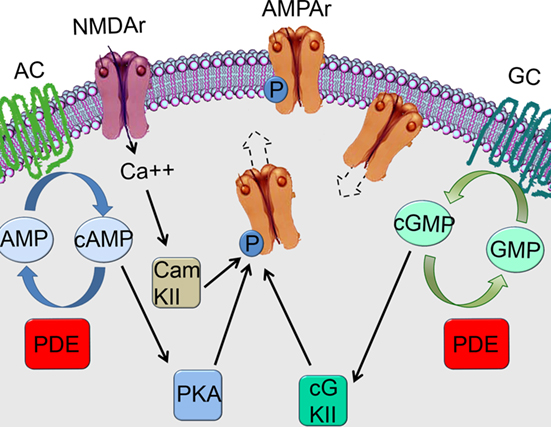
Figure 2. Inhibition of phosphodiesterase type 1 may lead to phosphorylation of AMPA receptors and its incorporation to the synapse. AC, adenyl cyclase; GC, guanylate cyclase.
Neuroprotective Role of PDE1 Inhibitors
The cascade showed in Figure 1 is also related to neuroprotection. For instance, activation of CREB can lead to the expression of many neuroprotective and anti-apoptotic molecules. It is well-known that deprivation of neurotrophic factors can induce apoptosis and conversely, that the expression of these molecules can counteract the effect of pro-apoptotic factors (Raff et al., 1993). In this regard, CREB phosphorylation can lead to the expression of many protective neurotrophic factors such as BDNF, FGF, and TGF (Lonze and Ginty, 2002). In addition of its role in regulating neurotrophic factors expression, CREB activation can also play a role in neuroprotection trough more direct and effective mechanisms. One way is by the expression of the Bcl-2 anti-apoptotic protein (Kitagawa, 2007). In addition, it was recently demonstrated that CREB phosphorylation can lead to the expression of the peroxisome proliferator activated receptor gamma coactivator 1 alpha (PGC-1α), which acts as a reactive oxygen species scavenger (St-Pierre et al., 2006). CREB can also lead to neuroprotection by mediating the expression of NR4A orphan nuclear receptors (Volakakis et al., 2010). In fact, animals with deletion of NR4A gene present increased oxidative damage and increase of apoptosis after hippocampal insult (Volakakis et al., 2010).
Vinpocetine, a Classic PDE1 Inhibitor
The alkaloid vinpocetine (vinpocetine – ethyl apovincaminate) is a classic inhibitor of PDE1 activity (Vereczkey, 1985; Nicholson, 1990). Vinpocetine treatment has been shown to facilitate LTP (Molnar and Gaal, 1992; Molnar et al., 1994), enhance the structural dynamics of dendritical spines (Lendvai et al., 2003), improve memory retrieval (DeNoble, 1987), and enhance performance on cognitive tests in humans (Hindmarch et al., 1991). In a model of fetal alcohol spectrum disorders, vinpocetine was able to restore neuronal plasticity in visual cortex (Medina et al., 2006) as well as the functional organization of this area (Krahe et al., 2009). Furthermore, vinpocetine treatment was also shown to revert the effects of early alcohol exposure in learning performance in the water maze (Filgueiras et al., 2010). In rats treated with intracerebroventricular streptozocin, a paradigm that mimics some Alzheimer-like cognitive problems, a vinpocetine treatment was able to restore performance in the water maze and the passive avoidance test (Deshmukh et al., 2009).
In addition of its potential as a plasticity enhancer, it was recently demonstrated that vinpocetine has a strong anti-inflammatory effect (Jeon et al., 2010). Vinpocetine inhibits IKK, preventing IkB degradation and the consequent translocation of NF-κB to the nucleus. Surprisingly, this mechanism is independent of vinpocetine action on PDE1. This new action of vinpocetine, combined with its potential to enhance neuronal plasticity suggest that this drug may have beneficial effects in conditions such as Alzheimer’s and Parkinson’s where inflammation and poor neuronal plasticity are present (Medina, 2010).
The efficacy of the use of vinpocetine in clinical trials has been controversial. A comparison of multiple studies evaluating vinpocetine use in cases of dementia was recently conducted (Szatmari and Whitehouse, 2003). After excluding studies that had major flaws (such as lack of control groups or randomization), Szatmari and Whitehouse focused in three works (Fenzl et al., 1986; Blaha et al., 1989; Hindmarch et al., 1991). In these studies, a total of 583 patients were given vinpocetine (n = 377) or placebo (n = 206). Vinpocetine doses were between 15 and 60 mg/kg. Vinpocetine treatment (30 and 60 mg/kg doses) showed beneficial effects as accessed by significant changes in scores used to evaluate clinical improvement such as the Syndrom-Kurztest and the Clinical global impression scale (Szatmari and Whitehouse, 2003). Few adverse effects were reported. Despite of vinpocetine treated patients having presented statistically significant improvements in measured outcomes, the authors questioned the reliability of these studies due to lack of the details of the randomization process and the fact that the studies were performed before the current criteria for dementia were implemented (Szatmari and Whitehouse, 2003).
Another meta-analysis was done to review the effects of vinpocetine treatment on acute ischemic stroke (Bereczki and Fekete, 2009). However, the efficacy of vinpocetine was inconclusive due to the fact that most of the studies analyzed did not use double-blinded approaches and randomization. The only two studies that used more appropriate designs did not show significant differences between groups and were based in small samples (Bereczki and Fekete, 2009).
The use of vinpocetine in animal models suggests that vinpocetine has a role to play in restoring neuronal plasticity (learning and memory in particular) in different conditions. However, the findings from clinical trials are still controversial and more studies are needed. It is conceivable that one reason for this discrepancy is the great difference in the dosage used in basic science studies and the ones used in clinical trials. Most studies performed in animal models used doses between 10 and 20 mg/kg (DeNoble, 1987; Medina et al., 2006; Krahe et al., 2008; Deshmukh et al., 2009; Filgueiras et al., 2010). These doses translated to humans would result in approximately 1,000 mg, which would be roughly a 20-fold increase in the current doses used in clinical trials (Szatmari and Whitehouse, 2003; Bereczki and Fekete, 2009).
Other PDE1 Inhibitors
One of the biggest problems to assess the efficacy of PDE1 inhibitors is the lack of specificity of currently used drugs. In fact, many of these drugs have been used because of other pharmacological properties (Table 1). Interestingly, despite these pharmacological differences, many of these drugs showed beneficial effects on learning and memory, a process that requires neuronal plasticity. It is possible that these beneficial results are also influenced by PDE1 inhibition and the increase of cAMP/cGMP levels.

Table 1. Non-specific properties of some PDE1 inhibitors.
In Parkinson’s disease there is a alteration in cAMP/cGMP levels and it has been shown that the activity of PDE1B is increased in the 6-hydroxydopamine hemi-Parkinson model (Sancesario et al., 2004). In fact, the application of Zaprinast (a PDE5 and PDE1 inhibitor), reduced the severity levodopa-induced dyskinesias in this model (Giorgi et al., 2008). Interestingly, some classic anti- Parkinson drugs, such as deprenyl (selegiline) and amantadine (Chen and Swope, 2007) preferentially inhibit PDE1A2 (present in cortex and hippocampus) with weak or none action on PDE1B1 (present in striatum and nucleus accumbens; Kakkar et al., 1996, 1997).
In Alzheimer’s accumulation of the amyloid-β protein may lead to a reduction on CREB phosphorylation (Vitolo et al., 2002), which could be related to the cognitive deficits seen in this condition. Recently, it was demonstrated that increasing cAMP levels by PDE4 inhibition (Vitolo et al., 2002) or cGMP levels by PDE5 inhibition (Puzzo et al., 2009) restored neuronal plasticity in models of Alzheimer’s. Surprisingly, in the latter study the PDE1 inhibitor IC354 did not show an effect. Unfortunately, little is known about the capacity of IC354 to raise levels of both cGMP and cAMP (as other PDE1 inhibitors do) and what the effect of this compound on the PDE1 subunits present in the brain.
Caffeine, at concentrations of 0.04 and 0.1 mM, leads to inhibition of 50 and 90% of adenosine receptors, respectively. However, at higher concentrations, it can also inhibit PDE1 (20 and 80% inhibition at 0.1 and 1 mM respectively; Fredholm et al., 1999). Caffeine have been showing promising results in improving learning and memory in animal models of different conditions such as stress, diabetes-triggered dementia, Alzheimer’s, Parkinson’s, and sleep deprivation (see Cunha and Agostinho, 2010 for review). However, because of the doses used in these studies it is likely that the caffeine effects were due only to its effects on adenosine receptors, however an influence of PDE inhibition was not discarded (Cunha and Agostinho, 2010).
A recent clinical study showed that perinatal caffeine, which was used to improve respiratory function, was able to improve survival rate and ameliorate cognitive deficits resulted from prematurity (Schmidt et al., 2007; Stevenson, 2007). Remarkably, the effects of caffeine on the respiratory function were able to explain only half of the effect of caffeine treatment (Schmidt et al., 2007; Stevenson, 2007). Interestingly, in this study a dose of 20 mg/kg (followed by a daily maintenance dose of 5 mg/kg) was used. This high initial dose, combined to the long half-life of caffeine in premature babies (over 100 h; Parsons and Neims, 1981) might be sufficient to reach the levels needed for PDE1 inhibition.
Nimodipine is another example of a non-specific PDE1 inhibitor (Schachtelle et al., 1987). This drug is largely used as an L-type voltage-gated calcium channel blocker and has shown to facilitate LTP and improve learning (Martin et al., 2004; Person and Raman, 2010). However, numerous studies established that L-type channels are implicated in neuronal plasticity (Perrier et al., 2002; Striessnig et al., 2006; Ziburkus et al., 2009). Therefore, whether PDE1 inhibition contributes for the effects of nimodipine on neuronal plasticity is not clear.
In conclusion, the therapeutic efficiency of PDE1 inhibitors in neurological conditions it is still poorly known. While the mechanism of action of PDE1 inhibitors strongly suggest a therapeutic potential, the development of more specific drugs would be crucial to elucidate whether this class of drugs could be relevant to clinical use. The development of these drugs may play important therapeutic roles in cases of mental retardation (such as in FASD and Cerebral-palsy) and in degenerative disorders such as Alzheimer’s and Parkinson’s.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Atkins, C. M., Selcher, J. C., Petraitis, J. J., Trzaskos, J. M., and Sweatt, J. D. (1998). The MAPK cascade is required for mammalian associative learning. Nat. Neurosci. 1, 602–609.
Bagetta, V., Ghiglieri, V., Sgobio, C., Calabresi, P., and Picconi, B. (2010). Synaptic dysfunction in Parkinson’s disease. Biochem. Soc. Trans. 38, 493–497.
Barco, A., Alaarcon, J. M., and Kandel, E. (2002). Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell 35, 345–353.
Beavo, J. A. (1995). Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms. Physiol. Rev. 75, 725–748.
Bereczki, D., and Fekete, I. (2009). Vinpocetine for acute ischaemic stroke. Cochrane Database Syst. Rev. 23, CD000480.
Blaha, L., Erzigkeit, H., Adamczyk, A., Freytag, S., and Schaltenbrand, R. (1989). Clinical evidence of the effectiveness of vinpocetine in the treatment of organic psychosyndrome. Hum. Psychopharmacol. 4, 103–111.
Bourtchouladze, R., Lidge, R., Catapano, R., Stanley, J., Gossweiler, S., Romashko, D., Scott, R., and Tully, T. (2003). A mouse model of Rubinstein–Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc. Natl. Acad. Sci. U.S.A. 100, 10518–10522.
Brostom, C. O., Huang, Y. C., Breckenridge, B., and Wolff, D. J. (1975). Identification of a calcium-binding protein as a calcium-dependent regulatory of brain adenylate cyclase. Proc. Natl. Acad. Sci. U.S.A. 73, 64–68.
Chen, J. J., and Swope, D. M. (2007). Pharmacotherapy for Parkinson’s disease. Pharmacotherapy 27, 161s–173s.
Chen, R. W., Williams, A. J., Liao, Z., Yao, C., Tortella, F. C., and Dave, J. R. (2007). Broad spectrum neuroprotection profile of phosphodiesterase inhibitors as related to modulation of cell-cycle elements and caspase-3 activation. Neurosci. Lett. 418, 165–169.
Cunha, R. A., and Agostinho, P. M. (2010). Chronic caffeine consumption prevents memory disturbance in different animal models of memory decline. J. Alzheimers Dis. 20, S95–S116.
DeNoble, V. J. (1987). Vinpocetine enhances retrieval of a step-through passive avoidance response in rats. Pharmacol. Biochem. Behav. 26, 183–186.
Deshmukh, R., Sharma, V., Mehan, S., Sharma, N., and Bedi, K. L. (2009). Amelioration of intracerebroventricular streptozotocin induced cognitive dysfunction and oxidative stress by vinpocetine – a PDE1 inhibitor. Eur. J. Pharmacol. 620, 49–56.
Dyke, H. J., and Montana, J. G. (1999). The therapeutic potential of PDE4 inhibitors. Expert Opin. Investig. Drugs 8, 1301–1325.
Etkin, A., Alarcon, J. M., Weisberg, S. P., Touzani, K., Huang, Y. Y., Nordheim, A., and Kandel, E. R. (2006). A role in learning for SRF: deletion in the adult forebrain disrupts LTD and the formation of an immediate memory of a novel context. Neuron 50, 127–143.
Fenzl, E., Apecechea, M., Schaltenbrand, R., and Friedel, R. (1986). “Long-term study concerning tolerance and efficacy of vinpocetine in elderly patients suffering from a mild to moderate organic psychosyndrome,” in Senile Dementias: Early Detection, eds A. Bes, J. Cahn, R. Cahn, S. Hoyer, J. P. Marc-Vergnes, and H. M. Wisniewski (London: John Libby Eurotext), 580–585.
Ferguson, G. D., and Storm, D. R. (2004). Why calcium-stimulated adenylyl cyclases? Physiology 19, 271–276.
Filgueiras, C. C., Krahe, T. E., and Medina, A. E. (2010). Phosphodiesterase type 1 inhibition improves learning in rats exposed to alcohol during the third trimester equivalent of human gestation. Neurosci. Lett. 473, 202–207.
Frank, D. A., and Greenberg, M. E. (1994). CREB: a mediator of long-term memory from mollusks to mammals. Cell 79, 5–8.
Fredholm, B. B., Battig, K., Holmen, J., Nehlig, A., and Zvartau, E. E. (1999). Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol. Rev. 51, 83–133.
Giorgi, M., D’Angelo, V., Esposito, Z., Nuccetelli, V., Sorge, R., Martorana, A., Stefani, A., Berardi, G., and Sancesario, G. (2008). Lowered cAMP and cGMP signalling in the brain during levedopa-induced dyskinesias in hemiParkinsonian rats: new aspects in the pathogenic mechanisms. Eur. J. Neurosci. 28, 941–950.
Glazewski, S., Barth, A. L., Wallace, H., McKenna, M., Silva, A., and Fox, K. (1999). Impaired experience- dependent plasticity in barrel cortex of mice lacking the alpha and delta isoforms of CREB. Cereb. Cortex 9, 249–256.
Goraya, T. E., and Cooper, D. M. (2005). Ca2+-calmodulin-dependent phosphodiesterase (PDE1): current perspectives. Cell. Signal. 17, 789–797.
Hindmarch, I., Fuchs, H. H., and Erzigkeit, H. (1991). Efficacy and tolerance of vinpocetine in ambulant patients suffering from mild to moderate organic psychosyndromes. Int. Clin. Psychopharmacol. 6, 31–43.
Jeon, K.-I., Xu, X., Aizawa, T., Lim, J. H., Jono, H., Kwon, D.-S., Abe, J.-I., Berk, B., Li, J.-D., and Yan, C. (2010). Vinpocetine inhibits NF-kB-dependent inflammation via an IKK-dependent but PDE-independent mechanism. Proc. Natl. Acad. Sci. U.S.A. 107, 9795–9800.
Josselyn, S. A., and Nguyen, P. V. (2005). CREB, synapses and memory disorders: past progress and future challenges. Curr. Drug. Targets CNS Neurol. Disord. 4, 481–497.
Kakkar, R., Raju, R. V. S., Rajput, A., and Sharma, R. K. (1996). Inhibition of bovine brain calmodulin-dependent cyclic nucleotide phosphodiesterase isozymes by deprenyl. Life Sci. 59, 337–341.
Kakkar, R., Raju, R. V. S., Rajput, A., and Sharma, R. K. (1997). Amantadine: an antiParkinsonian agent inhibits bovine 60 kDa calmodulin-dependent cyclic nucleotide phosphodiesterase isozyme. Brain Res. 749, 294.
Kakkar, R., Raju, R. V. S., and Sharma, R. K. (1999). Calmodulin-dependent cyclic nucleotide phosphodiesterase (PDE1). Cell. Mol. Life Sci. 55, 1164–1186.
Kitagawa, K. (2007). CREB and cAMP response element-mediated gene expression in the ischemic brain. FEBS Lett. 274, 3210–3217.
Krahe, T. E., Wang, W., and Medina, A. E. (2008). Vinpocetine increase phosphorylation of CREB and restores orientation selectivity in a ferret model of fetal alcohol spectrum disorder. Int. J. Dev. Neurosci. 26, 890.
Krahe, T. E., Wang, W., and Medina, A. E. (2009). Phosphodiesterase inhibition increases CREB phosphorylation and restores orientation selectivity in a model of fetal alcohol spectrum disorders. PLoS ONE 4, e6643. doi: 10.1371/journal.pone.0006643
Lakics, V., Karran, E. H., and Boess, F. G. (2010). Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 59, 367–374.
Lavaur, J., Bernard, F., Trifilieff, P., Pascoli, V., Kappes, V., Pages, C., Vanhoutte, P., and Caboche, J. (2008). A TAT-DEF-Elk-1 peptide regulates the cytonuclear trafficking of Elk-1 and controls cytoskeleton dynamics. J. Neurosci. 27, 14448–14458.
Lendvai, B., Zelles, T., Rozsa, B., and Vizi, E. S. (2003). A vinca alkaloid enhances morphological dynamics of dendritic spines of neocortical layer 2/3 pyramidal cells. Brain Res. Bull. 59, 257–260.
Lonze, B. E., and Ginty, D. D. (2002). Function and regulation of CREB family transcription factors in the nervous system. Neuron 35, 605–623.
Malinow, R., and Malenka, R. C. (2002). AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126.
Marcello, E., Epis, R., and Di Luca, M. (2008). Amyloid flirting with synaptic failure: towards a comprehensive view of Alzheimer’s disease pathogenesis. Eur. J. Pharmacol. 585, 109–118.
Martin, L. J., Fournier, N. M., Galic, M. A., and Emond, M. H. (2004). Chronic administration of the L-type calcium channel blocker nimodipine can facilitate the acquisition of sequence learning in a radial-arm maze. Behav. Pharmacol. 15, 133–139.
Mayer, M. L., Westbrook, G. L., and Guthrie, P. B. (1984). Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 309, 261–263.
Medina, A. E. (2010). Vinpocetine as a potent antiinflammatory agent. Proc. Natl. Acad. Sci. U.S.A. 107, 9921–9922.
Medina, A. E., and Krahe, T. E. (2008). Neocortical plasticity deficits in fetal alcohol spectrum disorders: lessons from barrel and visual cortex. J. Neurosci. Res. 86, 256–263.
Medina, A. E., Krahe, T. E., and Ramoa, A. S. (2006). Restoration of neuronal plasticity by a phosphodiesterase type 1 inhibitor in a model of fetal alcohol exposure. J. Neurosci. 26, 1057–1060.
Menniti, F. S., Faraci, W. S., and Schmidt, C. J. (2006). Phosphodiesterases in the CNS: targets for drug development. Nat. Rev. Drug Discov. 5, 660–670.
Molnar, P., and Gaal, L. (1992). Effect of different subtypes of cognition enhancers on long-term potentiation in the rat dentate gyrus in vivo. Eur. J. Pharmacol. 215, 17–22.
Molnar, P., Gaal, L., and Horvath, C. (1994). The impairment of long-term potentiation in rats with medial septal lesion and its restoration by cognition enhancers. Neurobiology 2, 255–266.
Morgante, F., Espay, A. J., Gunraj, C., Lang, A. E., and Chen, R. (2006). Motor cortex plasticity in Parkinson’s disease and levodopa-induced dyskinesias. Brain 129, 1059–1069.
Mower, A. F., Liao, D. S., Nestler, E. J., Neve, R. L., and Ramoa, A. S. (2002). cAMP/Ca2+ response element-binding protein function is essential for ocular dominance plasticity. J. Neurosci. 22, 2237–2245.
Nalbantoglu, J., Tirado-Santiago, G., Lahsaini, A., Poirier, J., Goncalves, O., Verge, G., Momoli, F., Welner, S. A., Masicotte, G., Julien, J. P., and Shapiro, M. L. (1997). Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature 387, 457–458.
Nicholson, C. D. (1990). Pharmacology of nootropics and metabolically active compounds in relation to their use in dementia. Psychopharmacology (Berl.) 101, 147–159.
Nithianantharajah, J., and Hannan, A. J. (2006). Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat. Rev. Neurosci. 7, 697–709.
Nowak, L., Bregestovski, P., Ascher, P., Herbet, A., and Prochiantz, A. (1984). Magnesium gates glutamate-activated channels in mouse central neurons. Nature 307, 462–465.
Parsons, W. D., and Neims, A. H. (1981). Prolonged half-life of caffeine in healthy term newborn infants. J. Pediatr. 98, 640–641.
Perrier, J. F., Alaburda, A., and Hounsgaard, J. (2002). Spinal plasticity mediated by postsynaptic L-type Ca2+ channels. Brain Res. Brain Res. Rev. 40, 223–229.
Person, A., and Raman, I. M. (2010). Deactivation of L-type Ca current by inhibition controls LTP at excitatory synapses in the cerebellar nuclei. Neuron 66, 550–559.
Pintchovski, S. A., Peebles, C. L., Kim, H. J., Verdin, E., and Finkbeiner, S. (2009). The serum response factor and a putative novel transcription factor regulate expression of the immediate-early gene Arc/Arg3.1 in neurons. J. Neurosci. 29, 1525–1537.
Puzzo, D., Staniszewski, A., Xiandeng, S., Privitera, L., Leznik, E., Liu, S., Zhang, H., Feng, Y., Palmeri, A., Landry, D. W., and Arancio, O. (2009). Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-load in an Alzheimer’s disease mouse model. J. Neurosci. 29, 8075–8086.
Raff, M. C., Barres, B. E., Burne, J. F., Coles, H. S., Ishizaki, Y., and Jacobson, M. D. (1993). Programmed cell death and the control of cell survival: lessons from the nervous system. Science 262, 695–700.
Ramanan, N., Shen, Y., Sarsfield, S., Lemberger, T., Schutz, G., Linden, D. J., and Ginty, D. D. (2005). SRF mediates activity-induced gene expression and synaptic plasticity but not neuronal viability. Nat. Neurosci. 8, 759–767.
Rose, G. M., Hopper, A., De Vivo, M., and Tehim, A. (2005). Phosphodiesterase inhibitors for cognitive enhancement. Curr. Pharm. Des. 11, 3329–3334.
Sancesario, G., Giorgi, M., D’Angelo, V., Modica, A., Martorana, A., Morello, M., Bengtson, C. P., and Bernardi, G. (2004). Down-regulation of nitrergic transmission in the rat striatum after chronic nigrostriatal deafferentation. Eur. J. Neurosci. 20, 989–1000.
Schachtelle, C., Wagner, B., and Marme, D. (1987). Stereoselective inhibition of calmodulin-dependent cAMP phosphodiesterase from bovine heart by (+)- and (−)-nimodipine. Naunyn Schmiedebergs Arch. Pharmacol. 335, 340–343.
Schmidt, B., Roberts, R. S., Davis, P., Doyle, L. W., Barrington, K. J., Ohlsson, A., Solimano, A., and Tin, W. (2007). Long-term effects of caffeine therapy for apnea of prematurity. N. Engl. J. Med. 357, 1893–1902.
Serulle, Y., Zhang, S., Ninan, I., Puzzo, D., McCarthy, M., Khatri, L., Arancio, O., and Ziff, E. B. (2007). A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron 56, 670–688.
Siarey, R. J., Stoll, J., Rapoport, S. I., and Galdzicki, Z. (1997). Altered long-term potentiation in the young and old Ts65Dn mouse, a model for Down syndrome. Neuropharmacology 36, 1549–1554.
Siarey, R. J., Villar, A. J., Epstein, C. J., and Galdzicki, Z. (2005). Abnormal synaptic plasticity in the Ts1Cje segmental trisomy 16 mouse model of Down syndrome. Neuropharmacology 49, 122–128.
Silva, A. J., Kogan, J. H., Frankland, P. W., and Kida, S. (1998). CREB and memory. Annu. Rev. Neurosci. 21, 127–148.
St-Pierre, J., Drori, S., Uldry, M., Silvaggi, J. M., Rhee, J., Jager, S., Handschin, C., Zheng, K., Lin, J., Yang, W., Simon, D. K., Bachoo, R., and Spiegelman, B. M. (2006). Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127, 397–408.
Striessnig, J., Koschak, A., Sinnegger-Brauns, M. J., Hetzenauer, A., Nguyen, N. K., Busquet, P., Pelster, G., and Singewald, N. (2006). Role of voltage-gated L-type Ca2+ channel isoforms for brain function. Biochem. Soc. Trans. 34, 903–909.
Suzuki, S., Zhou, H., Neumaier, J. F., and Pham, T. A. (2007). Opposing functions of CREB and MKK1 synergistically regulate the geometry of dendritic spines in visual cortex. J. Comp. Neurol. 503, 605–617.
Szatmari, S. Z., and Whitehouse, P. J. (2003). Vinpocetine for cognitive impairment and dementia. Cochrane Database Syst. Rev. 1, CD003119.
Tyan, S. W., Tsai, M. C., Lin, C. L., Ma, Y. L., and Lee, E. H. (2008). Serum- and glucocorticoid-inducible kinase 1 enhances zif268 expression through the mediation of SRF and CREB1 associated with spatial memory formation. J. Neurochem. 105, 820–832.
Vereczkey, L. (1985). Pharmacokinetics and metabolism of vincamine and related compounds. Eur. J. Drug Metab. Pharmacokinet. 10, 89–103.
Vitolo, O. V., Sant’Angelo, A., Costanzo, V., Battaglia, F., Arancio, O., and Shelanski, M. (2002). Amyloid beta-peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibilty by drugs that enhance cAMP signaling. Proc. Natl. Acad. Sci. U.S.A. 99, 13217–13221.
Volakakis, N., Kadkhodaei, B., Joodmardi, E., Wallis, K., Panman, L., Silvaggi, J., Spiegelman, B. M., and Perlmann, T. (2010). NR4A orphan nuclear receptors as mediators of CREB-dependent neuroprotection. Proc. Natl. Acad. Sci. U.S.A. 107, 12317–12322.
Yashiro, K., Riday, T. T., Condon, K. H., Roberts, A. C., Bernardo, D. R., Prakash, R., Weinberg, R. J., Ehlers, M. D., and Philpot, B. D. (2009). Ube3a is required for experience-dependent maturation of the neocortex. Nat. Neurosci. 12, 777–783.
Keywords: phosphodiesterase, vinpocetine, fetal alcohol spectrum disorders, Alzheimer, Parkinson, drug development, cAMP, cGMP
Citation: Medina AE (2011) Therapeutic utility of phosphodiesterase type I inhibitors in neurological conditions. Front. Neurosci. 5:21. doi: 10.3389/fnins.2011.00021
Received: 18 November 2010;
Paper pending published: 19 January 2011;
Accepted: 09 February 2011;
Published online: 18 February 2011.
Edited by:
Ian M. Stanford, Aston University, UKReviewed by:
Muzamil Ahmad, Indian Institute of Integrative Medicine, IndiaAmiel Rosenkranz, Rosalind Franklin University, USA
Copyright: © 2011 Medina. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Alexandre E. Medina, Department of Anatomy and Neurobiology, Virginia Commonwealth University Medical Center, 1101 E Marshall Street 23298-0709, Richmond, VA 1101, USA. e-mail:YW1lZGluYUB2Y3UuZWR1