

- 1 Physiopathologie des Maladies Psychiatriques, Centre de Psychiatrie et Neurosciences,INSERM U894, Centre Hospitalier Sainte-Anne, Paris, France
- 2 Université Paris Descartes, Paris, France
From our research and literature search we propose an understanding of the mechanism of action of antidepressants treatments (ADTs) that should lead to increase efficacy and tolerance. We understand that ADTs promote synaptic plasticity and neurogenesis. This promotion is linked with stimulation of dopaminergic receptors. Previous evidence shows that all ADTs (chemical, electroconvulsive therapy, repetitive transcranial magnetic stimulation, sleep deprivation) increase at least one monoamine neurotransmitter serotonin (5-HT), noradrenaline (NA) or dopamine (DA); this article focuses on DA release or turn-over in the frontal cortex. DA increased dopaminergic activation promotes synaptic plasticity with an inverted U shape dose–response curve. Specific interaction between DA and glutamate is mediated by D1 receptor subtypes and Glutamate (NMDA) receptors with neurotrophic factors likely to play a modulatory role. With the understanding that all ADTs have a common, final, DA-ergic stimulation that promotes synaptic plasticity we can predict that (1) AD efficiency is related to the compound strength for inducing DA-ergic stimulation. (2) ADT efficiency presents a therapeutic window that coincides with the inverted U shape DA response curve. (3) ADT delay of action is related to a “synaptogenesis and neurogenesis delay of action.” (4) The minimum efficient dose can be found by starting at a low dosage and increasing up to the patient response. (5) An increased tolerance requires a concomitant prescription of a few ADTs, with different or opposite adverse effects, at a very low dose. (6) ADTs could improve all diseases with cognitive impairments and synaptic depression by increasing synaptic plasticity and neurogenesis.
Introduction
Original antidepressants treatments (ADTs) were discovered by serendipity and their mechanism of action is still unclear. There is a delay of action between chronic ADT administration and patient’s response. Side effects differ between ADTs but not with efficiency. All chemical ADTs increase at least one monoamine neuromodulator (serotonin, noradrenaline (NA), DA). Assessment of depression intensity shows that all items of depression scales are strongly inter-correlated. The response does not differ with different ADTs or with different clinical profiles.
Depression involves emotion, cognition, and physical symptoms. Clinical depression is characterized in the DSM-IV-R (APA, 2004) by low mood (sadness, loss of motivation, worthlessness/guilt, and suicidal ideas), reduced cognition (low psychomotor activity, fatigue, concentration/attention deficit), and body symptoms like retardation, appetite, and sleep changes. Depression is characterized by a reduction of confidence in one-self, the world, and the future. Depression is often preceded by a period of acute or chronic stress.
Animal models of depression induce low mood (e.g., anhedonia can be expressed by reduction in sucrose preference) and psychomotor retardation (e.g., reduced reactivity can be expressed by immobility in the Porsolt test or escape deficit in the learned helplessness model). To induce depression-like behavior, animals are submitted to intense stress (unavoidable electric shocks), or chronic mild stress (2 weeks of unpredictable changes in the environment). Moreover, stress induces DA release in the prefrontal cortex (Del Arco and Mora, 2001).
In our working hypothesis, depression is a state of synaptic depression brought by a low rate of dopaminergic transmission with D1 receptor up-regulation that can be reversed by an increase in DA transmission. We propose that D1 receptors regulate mood. We see depression in a temporal dimension: in stressful situations, the organism releases DA in the brain as a way to adapt to stressful situations and cortisol release contributes to this adaptation. If this fighting phase is successful the person will heal from his stress. In depression this “fighting phase” persists and ends in a “loosing phase” in which DA cannot be released anymore and D1 receptors are up-regulated. We choose to study our hypothesis at the level of the prefrontal cortex since this brain area is at the intersection of mood and cognition and is an essential component for personality integration. The prefrontal cortex, receives the projections from the limbic system, may serve to integrate the cognitive and emotional information. This area is modulated by NA, serotonin, and DA neurotransmission.
Depression as a Hypodopaminergic State
At the pharmacological level, depression is a state involving low levels of DA transmission resulting in D1 receptor up-regulation. In the rat model of depression, a 4 weeks stress session induces a depressive state and reduces DA concentration in the prefrontal cortex for 3 months (Mizoguchi et al., 2008b). Moreover, adrenalectomy induces a depressive state with impaired working memory (Mizoguchi et al., 2004) and a hypodopaminergic state with a D1 receptor up-regulation (Mizoguchi et al., 2004). This hypodopaminergic function can be reversed by (1) D1 agonists, (2) DA release, and (3) ADT administration.
(1) Infusion of a D1 agonist, SKF81297, in the rat prefrontal cortex, reverses the working memory impairment due to adrenalectomy (Mizoguchi et al., 2004), and the rotarod impairment due to both chronic stress (Mizoguchi et al., 2002) and adrenalectomy (Mizoguchi et al., 2008a). Furthermore, working memory is enhanced in naïve, non-depressed rats, 12 h after learning (Floresco and Phillips, 2001). Other specific D1 receptor agonists A68930 (D’Aquila et al., 1994), and the partial agonist SKF38393 (D’Aquila et al., 1994; Gambarana et al., 1995; Takamori et al., 2001), reverse, after the first administration, the escape deficit induced by the learned helplessness model of depression. SKF38393 also reduces the spontaneous escape deficit of rats not exposed to the inescapable shocks (Gambarana et al., 1995). When given repeatedly this D1 agonist produces tolerance to its own protective effect. Indeed, long term administration of this selective D1 agonist is known to down-regulate D1 receptors number in the prefrontal cortex (Gambarana et al., 1995). The specific D1 receptor antagonist SCH23390 reverses the effects of SKF 38393, (Takamori et al., 2001) and completely antagonized the effect of a chronic-2-weeks-imipramine treatment in the learned helplessness model of depression (D’Aquila et al., 1994). It is of interest to see that ADT is suppressed by D1 antagonist.
(2) DA challenge, with 30 mg of d-amphetamine induces a larger mood elevating effect, in unmedicated depressed patients when compared to controls. Controls are healthy volunteers without history of axis I disorders in DSM-IV-R (Tremblay et al., 2005). This mood enhancing DA challenge could indicate that D1 receptors are up-regulated during depression and more sensitive to DA increase.
(3) Chronic ADT treatment with imipramine reverses the escape deficit induced by the learned helplessness model of depression (D’Aquila et al., 1994; Gambarana et al., 1995) and down regulates D1 receptor number in the prefrontal cortex (Gambarana et al., 1995).
At the cellular level depression may be a state of depressed synaptic plasticity that can be reversed by (1) D1 agonist administration, (2) DA release, and (3) AD treatments.
(1) The selective D1 receptor agonist, SKF81297 at an optimal dose facilitates long term potentiation (LTP) in the hippocampal–prefrontal cortex pathway whereas the D1 antagonist SCH23390 caused a dose-related impairment of its induction (Gurden et al., 2000).
(2) DA released from DArgic axon terminals in the prefrontal cortex facilitates synaptic plasticity whereas a depletion of cortical DA levels generates a dramatic decrease in this LTP (Gurden et al., 1999). Magnitude of this prefrontal LTP is also enhanced by clozapine and this effect is reversed by the D1 receptor antagonist SCH23390 (Matsumoto et al., 2008).
(3) Administration of ADTs, tianeptine and to a lesser extent fluoxetine, reverses the long lasting inhibition of in vivo LTP induced by stress in rats (Rocher et al., 2004). In contrast, the selective D1 antagonist SKF83566 combined with high-frequency stimulation by electrode implanted in the corpus callosum prevented prefrontal cortex LTP and resulted in long term depression (Coppa-Hopman et al., 2009). Together these data indicates that D1 receptor activation is necessary for the induction of medial prefrontal cortex glutamate-based LTP (Gurden et al., 2000; Coppa-Hopman et al., 2009).
All Antidepressants Increase Dopamine Levels
We performed a literature search, retrieving the articles that measure DA release in the prefrontal cortex using chemical and non-chemical antidepressant treatments. The studies show that, in rat, mouse, monkey, and man electroconvulsive therapy (Glue et al., 1990; Yoshida et al., 1998; Inoue et al., 2003), repetitive transcranial magnetic stimulation (Lisanby and Belmaker, 2000; Ohnishi et al., 2004), sleep deprivation (Lara-Lemus et al., 1998; Gillin et al., 2001; Wu et al., 2001), and all classes of chemical antidepressants, increase DA release in the prefrontal cortex (see Table 1). Administration of ADTs was either through ip, po, or intra-cortical route. Assessments of DA levels were mostly done through in vivo microdialysis.
The following ADTs induce DA release in PFC:
– From the “tricyclic” class: imipramine (Jordan et al., 1994; Tanda et al., 1994; Valentini et al., 2005), clomipramine (Tanda et al., 1994; Owen and Whitton, 2006), desipramine (Carboni et al., 1990; Tanda et al., 1994; Gresch et al., 1995; Carlson et al., 1996; Shoblock et al., 2004; Valentini et al., 2004; Bongiovanni et al., 2005), amoxapine (Kobayashi et al., 1992), amitriptiline (Kihara and Ikeda, 1995), nortryptiline (Carlson et al., 1996), maproptyline (Kihara and Ikeda, 1995).
– From the “Noradrenalin Serotonin Reuptake Inhibitor” class: duloxetine (Carlson et al., 1996; Gobert et al., 1997a,c), milnacipran (Muneoka et al., 2009), venlafaxine (Weikop et al., 2004).
– From the “specific serotonin reuptake inhibitor” (SSRI) class: fluoxetine (Jordan et al., 1994; Tanda et al., 1994; Gobert et al., 1997a,b,c, 1999; Pozzi et al., 1999; Millan et al., 2000; Sakaue et al., 2000; Bymaster et al., 2002; Koch et al., 2002), citalopram (Pozzi et al., 1999; Valentini et al., 2005), paroxetine (Carlson et al., 1996; Owen and Whitton, 2006), fluvoxamine (Jordan et al., 1994).
– From the “MonoAmine Oxidase Inhibitor” class: deprenyl (Lakshmana et al., 1998), moclobemide (Kan et al., 1987), IMAO-A (Inoue et al., 2003).
– From “Other ADs” class: mianserine (Tanda et al., 1996a; Valentini et al., 2004), mirtazapine (Devoto et al., 2004; Nakayama et al., 2004), tianeptine (Louilot et al., 1990; Sacchetti et al., 1993), amineptine (Invernizzi et al., 1992; Garattini, 1997), agomelatine (Millan et al., 2003), bupropion (Li et al., 2002; Inoue et al., 2003), reboxetine (Invernizzi et al., 2001; Linner et al., 2001; Page and Lucki, 2002; Kitaichi et al., 2004; Valentini et al., 2004; Carboni et al., 2006; Owen and Whitton, 2006).
Table 1 indicates that all ADTs, irrespective of their mechanism of action, (electroconvulsive therapy, repetitive transcranial magnetic stimulation, sleep deprivation, and all chemical class of antidepressants), induce DA release in the prefrontal cortex. They also increase DA in other brain areas like the limbic system, the nucleus accumbens, the striatum, and other cortical regions, but the increase was not as systematic as in the frontal cortex. DA increases with the first ADT administration and is kept at this increased level during chronic treatment. Acute ADT treatment releases DA in the same range as a 2–3 weeks chronic treatment (Tanda et al., 1996b; Page and Lucki, 2002). DA release is increased by 50–600%, depending on the type of ADT and on the dosage of ADT that is administered. From these data, we could predict the “strength” of an ADT (speed/efficacy) on its capacity to increase DA levels in the prefrontal cortex. In some studies using SSRI, it was found that a low ADT dosage does not produce the expected increase in DA, while higher dosages of ADTs do induce the increase in DA (Invernizzi et al., 1992; Pozzi et al., 1999; Valentini et al., 2005). As mentioned above, the increase in DA could be a direct pharmacological effect (e.g., imipramine is an inhibitor of DA reuptake) or could result from an indirect mechanism. For example, sleep deprivation increases the release of thyroid and estrogen hormones (Baumgartner et al., 1993), which in turn can enhance DA release, and SSRI can release DA through 5HT3 receptors activation in prefrontal cortex (Tanda et al., 1995).

Table 1. Prefrontal dopamine release from antidepressant treatment.
Some receptors are specifically implicated in this DA increase. Pharmacological experiments show that DA release can be attributed to specific receptor activation or deactivation. Table 2 shows, in animals, that alpha 2, 5HT3, 5HT1A, 5HT2C, D2 receptors can mediate the release of DA. DA increases when blocking Alpha 2, 5HT2C, and D2 receptors at a low dose. DA is also increased when activating the 5HT1A, and maybe the 5HT3 receptors. In contrast agonists of Alpha 2, 5HT2C, and D2 receptors and antagonists of 5HT1A and 5HT3 receptors reduce DA release.
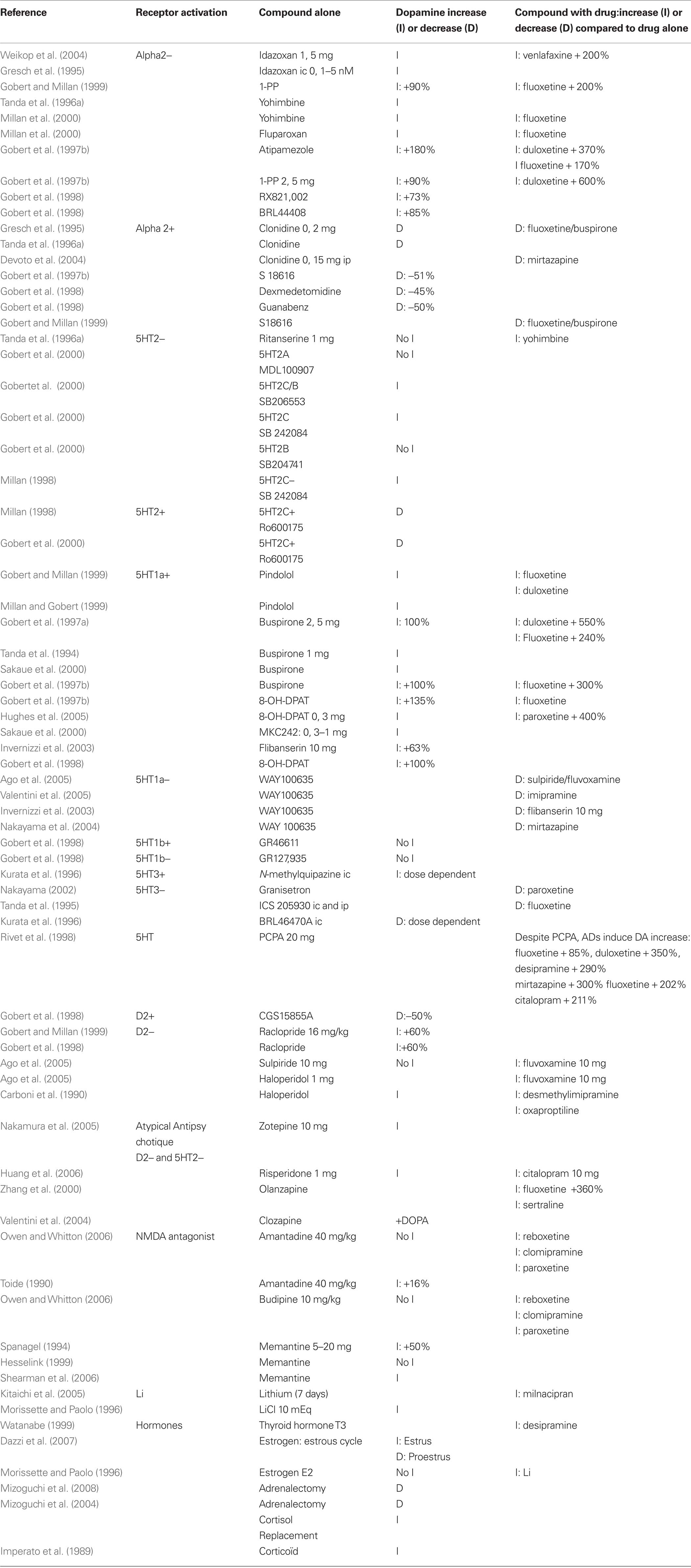
Table 2. Prefrontal dopamine release from compounds.
Table 2 indicates:
– Antagonists of the Alpha 2 receptor, idazoxan (Gresch et al., 1995; Weikop et al., 2004), yohimbine (Tanda et al., 1996a; Millan et al., 2000), fluparoxan (Millan et al., 2000), atipemazole (Gobert et al., 1997c), 1-(2-pyrimidinyl)piperazine (Gobert et al., 1997c, 1999), RX821002 (Gobert et al., 1998), BRL44408 (Gobert et al., 1998) increase prefrontal DA release. Conversely, receptor agonists: clonidine (Gresch et al., 1995; Tanda et al., 1996a; Devoto et al., 2004), demedetomidine (Gobert et al., 1998), guanabenz (Gobert et al., 1998), S18616 (Gobert et al., 1997c) reduce prefrontal DA release.
– Antagonist of the 5HT2C receptor, SB206553 (Gobert et al., 2000), SB242084 (Millan et al., 1998; Gobert et al., 2000) increase prefrontal DA, while 5HT2C receptor agonists decrease prefrontal DA (Millan et al., 1998; Gobert et al., 2000).
– Selective or mixed receptors antagonists of the D2 receptor increase prefrontal DA: raclopride (Gobert et al., 1998, 1999) haloperidol (Carboni et al., 1990). The atypical antipsychotic drug risperidone, a multireceptor antagonist, which lacks 5-HT6 receptor antagonist properties, at doses of 0.1, 0.3, and 1.0 mg/kg, produces a bell-shaped dose response effect on DA efflux in the prefrontal (Li et al., 2007). Conversely, the selective D2/3 agonist CGS 15855A decreases DA by 50% (Gobert et al., 1998). Interestingly, neuroleptic augmentation of ADT treatment has been used in severely depressed patients with good results. The explanation might be that neuroleptic (D2 antagonists) increase DA release and therefore are likely to augment ADT response. Indeed the studies show that sulpiride (Ago et al., 2005) and haloperidol (Carboni et al., 1990; Ago et al., 2005) enhance fluvoxamine-induced DA release. In addition, haloperidol (Carboni et al., 1990) enhances desmethylimipramine and oxaproptiline-induced DA release. All antipsychotics that share a common D2 and 5HT2C receptor antagonism should increase DA release. Indeed, zotepine (Nakamura et al., 2005), risperidone (Huang et al., 2006), and clozapine (Zhang et al., 2000) enhance prefrontal DA concentration. Risperidone (Huang et al., 2006) and olanzapine (Zhang et al., 2000) enhance AD-induced DA release. The olanzapine + fluoxetine combination (Zhang et al., 2000) increases prefrontal DA by 360% compared to baseline. This combination was significantly greater than either drug alone.
– Selective or mixed agonists of the 5HT1A receptor increase prefrontal DA release: pindolol (Gobert and Millan, 1999), buspirone (Tanda et al., 1994; Gobert et al., 1997a; Sakaue et al., 2000), 8-OH-DPAT (Gobert et al., 1998, 1999; Hughes et al., 2005), flibanserin (Invernizzi et al., 2003). In contrast, specific 5HT1A antagonists WAY100635, decreases prefrontal DA that was previously increased either by imipramine (Valentini et al., 2005), mirtazapine (Nakayama et al., 2004), or by the association of sulpiride and fluvoxamine (Ago et al., 2005).
– Direct perfusion of the 5-HT3 agonist, N-methylquipazine, in the anterior medial prefrontal cortex produces a concentration-dependent increase in extracellular DA level (Kurata et al., 1996). A recent review (Rajkumar and Mahesh, 2010) proposed that 5-HT3 receptor could mediate the SSRI effect. 5HT3 antagonist, ICS205930, prevents the DA increase due to fluoxetine or desipramine when infused in the prefrontal cortex or when administered systemically (Tanda et al., 1995) and Granisetron reduces the DA increase due to paroxetine (Nakayama, 2002). The 5HT3 antagonist, BRL4647OA, produces a dose dependent decrease of DA when infused in the anterior medial prefrontal cortex (Kurata et al., 1996).
Drugs known to increase the speed or efficiency of AD response also increase DA in the prefrontal cortex: lithium (Morissette and Paolo, 1996; Kitaichi et al., 2005), pindolol (Gobert and Millan, 1999; Millan and Gobert, 1999), idazoxan (Gresch et al., 1995; Weikop et al., 2004), buspirone (Tanda et al., 1994; Gobert et al., 1997a; Sakaue et al., 2000), yohimbine (Tanda et al., 1996a; Millan et al., 2000). Those drugs, also potentiate the DA increases due to ADs: chronic lithium increases milnacipran-induced DA release (Kitaichi et al., 2005), pindolol potentiates fluoxetine- and duloxetine-induced DA release (Gobert and Millan, 1999), yohimbine increases fluoxetine-induced DA release (Millan et al., 2000). Buspirone augments duloxetine-induced DA up to 550% (Gobert et al., 1997a) and fluoxetine-induced DA up to 300% (Gobert et al., 1997a; Gobert et al., 1999). Idazoxan with venlafaxine increases DA up to 200% (Weikop et al., 2004). These compounds also act to block their respective 5HT and NE autoreceptors.
Other drugs or hormones increase DA in the prefrontal cortex and present AD efficiency. This is the case for cortisol (Imperato et al., 1989; Mizoguchi et al., 2004, 2008a), estrogen, (Dazzi et al., 2007) thyroid hormones (Watanabe, 1999), substance P (Cador et al., 1989), and nicotine. Nicotine (Shearman et al., 2005; Tsukada et al., 2005) and anticholinesterase drugs such as galantamine (Noda et al., 2010; Schilstrom et al., 2007) and donepezil (Shearman et al., 2006), enhance extracellular levels of DA in the medial prefrontal cortex. In addition, anticholinesterase treatment presents AD properties (Tanaka et al., 2004; Rozzini et al., 2007; Cummings et al., 2008) and anticholinesterase augmentation of AD treatment improves the response in depressed patients (Pelton et al., 2008) and the release of DA in the prefrontal cortex (Wang et al., 2007a). The pro-cognitive effect of anticholinesterase drugs could rely on the synaptic potentiation of the D1/NMDA activation, since the D1 antagonist, SCH23390, and not the muscarinic acetylcholine receptor antagonist, scopolamine, reverses the cognitive improvement due to galantamine in an animal model of Alzheimer’s disease (Wang et al., 2007b). The D1 antagonist, SCH23390 also abolishes the pro-cognitive effect of the galantamine–risperidone association in a chronic phencyclidine model of cognitive deficit (Wang et al., 2007a).
Dopamine Modulation of Neuroplasticity in Depression
Pharmacological evidence for a role of D1 receptors in the bidirectional modulation of synaptic plasticity in the prefrontal cortex has been demonstrated in the past few years by different groups including ours. DA through D1 receptors increases NMDA currents and this synergism which occurs at the postsynaptic level appears to be mediated through both a PKA and Ca2+-dependent mechanisms (Jay et al., 1998; Gurden et al., 1999, 2000; Jay, 2003; Kruse et al., 2009). In addition, DA through D1 receptors appears to control the rate of phosphorylation/dephosphorylation of NMDA and AMPA receptor subunits which is required for a functional NMDA receptor (Sun et al., 2005; Gao and Wolf, 2008). D1 receptors may facilitate LTP by increasing the AMPA receptor pool available for synaptic insertion. Conversely, stimulation of D2 receptors decreased surface and synaptic GluR1 expression. Abnormal engagement of such mechanism could account for the maladaptive plasticity of prefrontal cortex, an area that we identified to be a target for intervention in stress-relates disorders like depression (Rocher et al., 2004; Cerqueira et al., 2007; Caudal et al., 2010). Emerging findings reveal that ADT treatment enhance membrane expression of AMPA receptors and phosphorylation of GluR1 subunit in the prefrontal cortex and hippocampus (Martinez-Turrillas et al., 2002, 2007; Qi et al., 2009). The mechanisms involved in the synaptic targeting of AMPA receptors by desipramine and paroxetine, ADs which differentially affect monoamine reuptake (NA and 5-HT), are similar but not identical. The mechanisms activated by these ADs could lead to an enhanced neuronal plasticity probably underlying, at least in part, the clinical efficacy of ADs treatment. A coordinated activation of DA D1 and NMDA systems is also an important feature of adaptive behavior. The processing of working memory in the mPFC involves DA D1 receptors that depend, at least in part, of NMDA receptors activity in this cortical area.
In our understanding of the AD-induced DA changes, the DA enhancement provides an AD effect through its interaction with NMDA receptors. The direct action on the NMDA receptors could bypass the DA increase and induce a strong AD response (Pacher et al., 2001; Skolnick et al., 2009; aan het Rot et al., 2010). On the other hand, the DArgic-antidepressant mechanism could provide a regulation that may be protective for the neuron: the DA inverted U dose–response curve of NMDA effects could bring some regulation and prevent toxic response.
Working Hypothesis: Depression as a Synaptic Long Term Depression State
As we have seen in the prefrontal cortex, DA-induced activation of NMDA receptors could be the final common pathway for AD treatments (Lavergne and Jay, 2009). LTP is facilitated, in naïve rats, when DA is released (Jay et al., 2004) or when D1 agonist is infused in the prefrontal cortex (Gurden et al., 2000; Matsumoto et al., 2008). Conversely D1 antagonist reverses the D1 agonist effect on LTP (Gurden et al., 2000; Matsumoto et al., 2008; Coppa-Hopman et al., 2009). D1/NMDA activation is required for synaptic plasticity. Our working hypothesis proposes that depression is a state of synaptic depression and that AD induces synaptic potentiation. Both emotional and cognitive aspects of depression rely on synaptic plasticity. The hypothesis predicts several characteristics for AD activity: the AD response should (1) be non-specific (same efficacy for all types of depressions), (2) be global (all items of the depression scales should improve together), (3) be regulated inside a physiological range, (changes in synaptic plasticity are kept under strict homeostatic regulation), (4) needs time to archive the changes in synaptic plasticity (delay of action of AD), and (5) depends on a correct dosage (too little or too much AD should be ineffective, they should be a therapeutic window). All those five points are demonstrated in the literature.
(1) Non-specific
(a) All characterized types of depression respond the same way to an AD treatment (Lavergne et al., 2005). ADs improve identically the different forms of clinically characterized depression (melancholy, atypical depression, recurrent depression, post partum depression, seasonal depression, previous suicide attempts, bipolar spectrum), only severe depression with psychotic symptoms responded less to 30 mg/day of mirtazapine and possibly did not reach the synaptic potentiation state. (b) The healing process in depression is identical under AD treatment compared to placebo. Patients, with and without AD treatment, present the same response profile when healing. With placebo only the less depressed patients respond (Rabkin et al., 1987). ADs increase the speed of response and allows response in severely depressed patients.
(2) Global
Clinicians know that mood and psychomotor retardation improve together when the patient respond. The global AD effect is demonstrated by the fact that all ADs, despite different lateral effects, induce a parallel decrease of all depression scale items in clinical trials. Before and during treatment all depression items are strongly inter-related.
(3) Regulated inside a physiological range
The range goes from synaptic depression to synaptic potentiation. The outside of the D1/NMDA inversed U curve corresponds to a state of synaptic depression; the inside of the curve corresponds to a state of synaptic potentiation. An argument to believe that AD efficacy is limited inside a physiological range is the limitation of the AD response. At the end of a 6 weeks-AD treatment (mirtazapine), the remission rate (stable response with > 50% MADRS score decrease) is only 30.9% (Lavergne et al., 2005). The benefit of treatment is to facilitate a state of synaptic potentiation. ADs cannot modulate mood higher. The state of synaptic potentiation is, with time, reduced by D1 receptors down-regulation. The slowdown in improvement observed after the first 2 weeks of treatment presumably results from homeostatic counter-regulation. In fact most of the AD response occurs before the counter-regulation. All the ADs studied in the mirtazapine French regulatory “dossier de transparence” were pooled in a meta-analysis. At 2-week the score decrease was 60% of the end-point score decrease (Lavergne and De Mouzon, 2000), indicating that most of the score decrease occurs before the second week of treatment. The AD activity is limited by the D1/NMDA inversed U curve and by the D1 receptors down-regulation. Subsequent improvement relies on the new positive life experiences that are now possible in a state of synaptic potentiation.
Another argument to believe that AD activity is kept within homeostatic regulation is the lack of perception of an AD effect. Patients and healthy volunteers perceive the AD effect like a global positive mood change “more good days than usual” and do not perceives a “high” like with alcohol, cocaine…
(4) Time needed to archive synaptic potentiation
Changes, from synaptic depression to synaptic potentiation, takes time, and likely more time when the patient is in a profound state of synaptic depression. This would explain the AD delay of action. Animals and healthy volunteers respond to ADs within 3 h, while it takes longer for depressed patients to respond to ADs. In animal models of depression, the behavioral effects can be measured 20 min after AD administration and the increased synaptogenesis and neurogenesis can be measured after 2–3 h. Behavioral changes, in healthy volunteers, are noticed 3 h after administration of the ADs (Harmer et al., 2003a,b; Tse and Bond, 2003). In all clinical trials with ADs, the depression scales’ score decrease is larger in the first half of treatment compared to the second half. The speed of improvement, measured as the score change per day, is maximal in the first week of treatment (2.65%/day, on the Montgomery Asberg Depression Rating Scale). The improvement period (rate of improvement > 2%/day) lasted only 2 weeks. Subsequent improvement was much slower (<0.5%/day). The AD activity is not delayed but is time-limited (Berlin and Lavergne, 1998). In clinical trials the AD improvement is observed after 4 days. In the Zurich meta-analysis, Angst and coworkers (Stassen et al., 1993, 1996) found that the score decrease in the AD group differs from the placebo group starting on the fourth day of treatment. After 1 week clinicians can usually observe the patient’s improvement. In a mirtazapine study, 50% of the 4771 depressed patients present, a “little improvement” after 1-week treatment (one point increase or more with the Clinical Global Impression Scale) and a 18.5% score decrease with the Montgomery Asberg Depression Rating Scale. Another argument that points toward early AD effect is that an improvement predicts the final response. A “one point increase or more” on the Clinical Global Impression-Improvement scale was found to allow correct classification of responders in 71.6% of the patients when the response was defined as a “50% score reduction in the Montgomery Asberg Depression Rating Scale” (Lavergne et al., 2005).
(5) Therapeutic window
The D1/NMDA inversed U response curve implies a minimum and a maximum therapeutic dose. It predicts a therapeutic window for the AD response. It is intuitive that too low dosage would be inefficacious and less intuitive and that high dosage could be detrimental on efficacy. Too low dosage could be archived by interrupting the monoamine pathway. Catecholamine and tyrosine depletion induce a depressive relapse, within a few hours, in recently remitted depressed patients (Heninger et al., 1996; Delgado et al., 2002). We speculate that an interruption of transmission in the monoamine pathways or administration of a D1 antagonism could treat a manic episode. Tyrosine depletion decreases the release of DA produced by amphetamine (McTavish et al., 2001). The same amino acid mixture lacking tyrosine and its precursor phenylalanine lowers both subjective and objective measures of the psychostimulant effect of methamphetamine (McTavish et al., 2001) and impairs spatial recognition memory and spatial working memory in volunteers (Harmer et al., 2001). It also reduces mania ratings in bipolar patients (McTavish et al., 2001). High AD dosage or high blood concentration of AD in patients with low drug metabolism, may compromise the response. High blood concentration of amitriptyline compromises the therapeutic effect in depressed patients. The data support the existence of a therapeutic window (70–220 ng/ml) for amitriptyline serum level (Ulrich et al., 2001). Moreover high blood concentration of paroxetine and desipramine compromises the benefit of treatment in patients with panic disorder (Watanabe et al., 2007) and the analgesia in chronic back pain (Atkinson et al., 2007). Future research is needed to more precisely test the hypothesis that high dosage of D1 agonist compromises AD activity in an animal model of depression.
New Prescription Strategy for Antidepressant Treatment
The D1/NMDA inversed U dose–response curve predicts a therapeutic window. To reach the therapeutic window we recommend to start the prescription at a low dosage (from 1/10 to 1/3 of the usually recommended dosage) and to increase the dosage, every 4–7 days, until the patient presents a qualitative positive change in his mood and functioning. This progressively incremental dosage will find the “minimal individual effective dosage.” The progressive increase will likely reduce the adverse effects intensity and might reduce the speed of the D1 receptors down-regulation, providing a longer D1/NMDA activation. In our practice we expect a small improvement after 3–7 days of treatment in mildly depressed young patients and expect the improvement to take longer in severely older depressed patients. However, the AD dosage should start at a higher level in severely depressed patients compared to less depressed patients.
One can take advantages of the fact that different monoamines pathways can increase DA release. A new strategy for AD treatment is to mix low doses of AD to augment efficiency and tolerance. This strategy can increase efficacy if the mixed ADs have synergetic effects on DA release and increases tolerance if the ADs have opposite side effects. Efficacy: AD of different mechanisms of action, and drugs known to enhance speed and efficacy of AD response, have synergistic effects on DA release. See Table 2. For example, monoamine reuptake inhibitors could be co-prescribed with one or two AD with other mechanism of action (monoamine-oxidase inhibitor, 5-HT2C antagonist, alpha 2 antagonist, 5-HT1A agonist), to promote DA increase. The benefit of AD association has been tested. Mianserine augmentation of fluoxetine treatment increases response in depressed patients, previously not responding to fluoxetine alone (Ferreri et al., 2001). Tolerance: Increasing tolerance requires concomitant prescription of several ADs, with different or opposite side effects. The prescribed ADs should have different – and ideally opposite – types of side effect. For example, three ADs each given at a third of the recommended dosage should reduce the adverse effects intensity and possibly increase the type of adverse effects. The strategy becomes interesting when ADs have opposite side effects. For example, sedative antidepressants can be co-prescribed with stimulating ADs, nausea inducing ADs can be prescribed with appetite increasing ADs, libido decreasing ADs can be prescribed with libido increasing ADs. The new strategy predicts that mixing different ADs increases efficacy and tolerance.
Conclusion
Our research and literature search indicate that (1) D1 receptors activation (D1 stimulation, DA availability in the prefrontal cortex), is the common denominator of all AD treatments (2) depression appears as a state of D1 receptor deactivation (D1 up-regulation with little DA availability). In our working hypothesis depression is a state of synaptic depression that can be reversed by AD treatment. Indeed ADs promote synaptic potentiation. All ADs enhance DA release in the prefrontal cortex. Activation of D1 receptors induces NMDA activation and an enhancement of synaptic potentiation through an inverted U dose–response curve that could represent a form of mood regulation. The mood improvement, with ADs, has to be in a physiological range, being maximal at the start of treatment and reduced thereafter because of D1 receptors down-regulation.
Based on this understanding, we propose a new strategy for prescribing Ads which consists on a progressive escalating dosage of an association of ADs. Mixing ADs with opposite side effects should increase tolerance. Mixing ADs with synergistic mechanism on DA release should increase efficacy. The strength of an AD can be assessed by the DA release in the prefrontal cortex. The benefit of AD treatment can be valuable in all states of cognitive deficits, even in non-depressed patients.
Conflict of Interest Statement:
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
aan het Rot, M, Collins, K. A., Murrough, J. W., Perez, A. M., Reich, D. L., Charney, D. S., and Mathew, S. J. (2010). Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol. Psychiatry 67, 139–145.
Ago, Y., Nakamura, S., Baba, A., and Matsuda, T. (2005). Sulpiride in combination with fluvoxamine increases in vivo dopamine release selectively in rat prefrontal cortex. Neuropsychopharmacology 30, 43–51.
Atkinson, J. H., Slater, M. A., Capparelli, E. V., Wallace, M. S., Zisook, S., Abramson, I., Matthews, S. C., and Garfin, S. R. (2007). Efficacy of noradrenergic and serotonergic antidepressants in chronic back pain: a preliminary concentration-controlled trial. J. Clin. Psychopharmacol. 27, 135–142.
Baumgartner, A., Dietzel, M., Saletu, B., Wolf, R., Campos-Barros, A., Graf, K. J., Kurten, I., and Mannsmann, U. (1993). Influence of partial sleep deprivation on the secretion of thyrotropin, thyroid hormones, growth hormone, prolactin, luteinizing hormone, follicle stimulating hormone, and estradiol in healthy young women. Psychiatry Res. 48, 153–178.
Berlin, I., and Lavergne, F. (1998). Early predictors of two month response with mianserin and selective serotonin reuptake inhibitors and influence of definition of outcome on prediction. Eur. Psychiatry 13, 138–142.
Bongiovanni, R., Kirkbride, B., Walmire, P., and Jaskiw, G. E. (2005). Tyrosine administration does not affect desipramine-induced dopamine levels as measured in vivo in prefrontal cortex. Brain Res. 1054, 203–206.
Bymaster, F. P., Zhang, W., Carter, P. A., Shaw, J., Chernet, E., Phebus, L., Wong, D. T., and Perry, K. W. (2002). Fluoxetine, but not other selective serotonin uptake inhibitors, increases norepinephrine and dopamine extracellular levels in prefrontal cortex. Psychopharmacology (Berl.) 160, 353–361.
Cador, M., Rivet, J. M., Kelley, A. E., Le Moal, M., and Stinus, L. (1989). Substance P, neurotensin and enkephalin injections into the ventral tegmental area: comparative study on dopamine turnover in several forebrain structures. Brain Res. 486, 357–363.
Carboni, E., Silvagni, A., Vacca, C., and Di Chiara, G. (2006). Cumulative effect of norepinephrine and dopamine carrier blockade on extracellular dopamine increase in the nucleus accumbens shell, bed nucleus of stria terminalis and prefrontal cortex. J. Neurochem. 96, 473–481.
Carboni, E., Tanda, G. L., Frau, R., and Di Chiara, G. (1990). Blockade of the noradrenaline carrier increases extracellular dopamine concentrations in the prefrontal cortex: evidence that dopamine is taken up in vivo by noradrenergic terminals. J. Neurochem. 55, 1067–1070.
Carlson, J. N., Visker, K. E., Nielsen, D. M., Keller, R. W. Jr., and Glick, S. D. (1996). Chronic antidepressant drug treatment reduces turning behavior and increases dopamine levels in the medial prefrontal cortex. Brain Res. 707, 122–126.
Caudal, D., Mailliet, F., Bergerot, D., Watilliaux, A, and Jay, T. M. (2010). “PO113.2: acute stess induces opposite changes in AMPA receptor subunits phosphorylation in prefontal cortex and amygdala,” in 7th Forum of European Neuroscience (Amsterdam), 167.
Cerqueira, J. J., Mailliet, F., Almeida, O. F., Jay, T. M., and Sousa, N. (2007). The prefrontal cortex as a key target of the maladaptive response to stress. J. Neurosci. 27, 2781–2787.
Coppa-Hopman, R., Galle, J., and Pimkine, D. (2009). D1 receptor antagonist-induced long-term depression in the medial prefrontal cortex of rat, in vivo: an animal model of psychiatric hypofrontality. J. Psychopharmacol. 23, 672–685.
Cummings, J. L., Mackell, J., and Kaufer, D. (2008). Behavioral effects of current Alzheimer’s disease treatments: a descriptive review. Alzheimers Dement. 4, 49–60.
D’Aquila, P. S., Collu, M., Pani, L., Gessa, G. L., and Serra, G. (1994). Antidepressant-like effect of selective dopamine D1 receptor agonists in the behavioural despair animal model of depression. Eur. J. Pharmacol. 262, 107–111.
Dazzi, L., Seu, E., Cherchi, G., Barbieri, P. P., Matzeu, A., and Biggio, G. (2007). Estrous cycle-dependent changes in basal and ethanol-induced activity of cortical dopaminergic neurons in the rat. Neuropsychopharmacology 32, 892–901.
Del Arco, A., and Mora, F. (2001). Dopamine release in the prefrontal cortex during stress is reduced by the local activation of glutamate receptors. Brain Res. Bull. 56, 125–130.
Delgado, P. L., Moreno, F. A., Onate, L., and Gelenberg, A. J. (2002). Sequential catecholamine and serotonin depletion in mirtazapine-treated depressed patients. Int. J. Neuropsychopharmacol. 5, 63–66.
Devoto, P., Flore, G., Pira, L., Longu, G., and Gessa, G. L. (2004). Mirtazapine-induced corelease of dopamine and noradrenaline from noradrenergic neurons in the medial prefrontal and occipital cortex. Eur. J. Pharmacol. 487, 105–111.
Ferreri, M., Lavergne, F., Berlin, I., Payan, C., and Puech, A. J. (2001). Benefits from mianserin augmentation of fluoxetine in patients with major depression non-responders to fluoxetine alone. Acta Psychiatr. Scand. 103, 66–72.
Floresco, S. B., and Phillips, A. G. (2001). Delay-dependent modulation of memory retrieval by infusion of a dopamine D1 agonist into the rat medial prefrontal cortex. Behav. Brain Res. 115, 934–939.
Gambarana, C., Ghiglieri, O., and Graziella de Montis, M. (1995). Desensitization of the D1 dopamine receptors in rats reproduces a model of escape deficit reverted by imipramine, fluoxetine and clomipramine. Prog. Neuropsychopharmacol. Biol. Psychiatry 19, 741–755.
Gao, C., and Wolf, M. E. (2008). Dopamine receptors regulate NMDA receptor surface expression in prefrontal cortex neurons. J. Neurochem. 106, 2489–2501.
Garattini, S. (1997). Pharmacology of amineptine, an antidepressant agent acting on the dopaminergic system: a review. Int. Clin. Psychopharmacol. 12(Suppl. 3), S15–S19.
Gillin, J. C., Buchsbaum, M., Wu, J., Clark, C., and Bunney, W. Jr. (2001). Sleep deprivation as a model experimental antidepressant treatment: findings from functional brain imaging. Depress. Anxiety 14, 37–49.
Glue, P., Costello, M. J., Pert, A., Mele, A., and Nutt, D. J. (1990). Regional neurotransmitter responses after acute and chronic electroconvulsive shock. Psychopharmacology (Berl.) 100, 60–65.
Gobert, A., and Millan, M. J. (1999). Modulation of dialysate levels of dopamine, noradrenaline, and serotonin (5-HT) in the frontal cortex of freely-moving rats by (−)-pindolol alone and in association with 5-HT reuptake inhibitors: comparative roles of beta-adrenergic, 5-HT1A, and 5-HT1B receptors. Neuropsychopharmacology 21, 268–284.
Gobert, A., Rivet, J. M., Audinot, V., Newman-Tancredi, A., Cistarelli, L., and Millan, M. J. (1998). Simultaneous quantification of serotonin, dopamine and noradrenaline levels in single frontal cortex dialysates of freely-moving rats reveals a complex pattern of reciprocal auto- and heteroreceptor-mediated control of release. Neuroscience 84, 413–429.
Gobert, A., Rivet, J. M., Cistarelli, J. M., and Millan, M. J. (1997a). Buspirone enhances duloxetine- and fluoxetine-induced increases in dialysate levels of dopamine and noradrenaline, but not serotonin, in the frontal cortex of freely moving rats. J. Neurochem. 68, 1326–1329.
Gobert, A., Rivet, J. M., Cistarelli, L., and Millan, M. J. (1997b). Potentiation of the fluoxetine-induced increase in dialysate levels of serotonin (5-HT) in the frontal cortex of freely moving rats by combined blockade of 5-HT1A and 5-HT1B receptors with WAY 100,635 and GR 127,935. J. Neurochem. 68, 1159–1163.
Gobert, A., Rivet, J. M., Cistarelli, L., Melon, C., and Millan, M. J. (1997c). Alpha2-adrenergic receptor blockade markedly potentiates duloxetine- and fluoxetine-induced increases in noradrenaline, dopamine, and serotonin levels in the frontal cortex of freely moving rats. J. Neurochem. 69, 2616–2619.
Gobert, A., Rivet, J. M., Cistarelli, L., Melon, C., and Millan, M. J. (1999). Buspirone modulates basal and fluoxetine-stimulated dialysate levels of dopamine, noradrenaline and serotonin in the frontal cortex of freely moving rats: activation of serotonin1A receptors and blockade of alpha2-adrenergic receptors underlie its actions. Neuroscience 93, 1251–1262.
Gobert, A., Rivet, J. M., Lejeune, F., Newman-Tancredi, A., Adhumeau-Auclair, A., Nicolas, J. P., Cistarelli, L., Melon, C., and Millan, M. J. (2000). Serotonin(2C) receptors tonically suppress the activity of mesocortical dopaminergic and adrenergic, but not serotonergic, pathways: a combined dialysis and electrophysiological analysis in the rat. Synapse 36, 205–221.
Gresch, P. J., Sved, A. F., Zigmond, M. J., and Finlay, J. M. (1995). Local influence of endogenous norepinephrine on extracellular dopamine in rat medial prefrontal cortex. J. Neurochem. 65, 111–116.
Gurden, H., Takita, M., and Jay, T. M. (2000). Essential role of D1 but not D2 receptors in the NMDA receptor-dependent long-term potentiation at hippocampal-prefrontal cortex synapses in vivo. J. Neurosci. 20, RC106.
Gurden, H., Tassin, J. P., and Jay, T. M. (1999). Integrity of the mesocortical dopaminergic system is necessary for complete expression of in vivo hippocampal-prefrontal cortex long-term potentiation. Neuroscience 94, 1019–1027.
Harmer, C. J., Bhagwagar, Z., Perrett, D. I., Vollm, B. A., Cowen, P. J., and Goodwin, G. M. (2003b). Acute SSRI administration affects the processing of social cues in healthy volunteers. Neuropsychopharmacology 28, 148–152.
Harmer, C. J., Hill, S. A., Taylor, M. J., Cowen, P. J., and Goodwin, G. M. (2003a). Toward a neuropsychological theory of antidepressant drug action: increase in positive emotional bias after potentiation of norepinephrine activity. Am. J. Psychiatry 160, 990–992.
Harmer, C. J., McTavish, S. F., Clark, L., Goodwin, G. M., and Cowen, P. J. (2001). Tyrosine depletion attenuates dopamine function in healthy volunteers. Psychopharmacology (Berl.) 154, 105–111.
Heninger, G. R., Delgado, P. L., and Charney, D. S. (1996). The revised monoamine theory of depression: a modulatory role for monoamines, based on new findings from monoamine depletion experiments in humans. Pharmacopsychiatry 29, 2–11.
Hesselink, M. B., De Boer, B. G., Breimer, D. D., and Danysz, W. (1999). Brain penetration and in vivo recovery of NMDA receptor antagonists amantadine and memantine: a quantitative microdialysis study. Pharm. Res. 16, 637–642.
Huang, M., Ichiwaka, J., Li, Z., Dai, J., and Meltzer, H. Y. (2006). Augmentation by citalopram of risperidone-induced monoamine release in rat prefrontal cortex. Psychopharmacology (Berl.) 185, 274–281.
Hughes, Z. A., Starr, K. R., Langmead, C. J., Hill, M., Bartoszyk, G. D., Hagan, J. J., Middlemiss, D. N., and Dawson, L. A. (2005). Neurochemical evaluation of the novel 5-HT1A receptor partial agonist/serotonin reuptake inhibitor, vilazodone. Eur. J. Pharmacol. 510, 49–57.
Imperato, A., Puglisi-Allegra, S., Casolini, P., Zocchi, A., and Angelucci, L. (1989). Stress-induced enhancement of dopamine and acetylcholine release in limbic structures: role of corticosterone. Eur. J. Pharmacol. 165, 337–338.
Inoue, T., Kitaichi, Y., and Koyama, T. (2003). Treatment strategy of refractory depression and its presynaptic mechanism of action. Nihon Shinkei Seishin Yakurigaku Zasshi 23, 11–20.
Invernizzi, R., Pozzi, L., Vallebuona, F., Bonini, I., Sacchetti, G., and Samanin, R. (1992). Effect of amineptine on regional extracellular concentrations of dopamine and noradrenaline in the rat brain. J. Pharmacol. Exp. Ther. 262, 769–774.
Invernizzi, R. W., Parini, S., Sacchetti, G., Fracasso, C., Caccia, S., Annoni, K., and Samanin, R. (2001). Chronic treatment with reboxetine by osmotic pumps facilitates its effect on extracellular noradrenaline and may desensitize alpha(2)-adrenoceptors in the prefrontal cortex. Br. J. Pharmacol. 132, 183–188.
Invernizzi, R. W., Sacchetti, G., Parini, S., Acconcia, S., and Samanin, R. (2003). Flibanserin, a potential antidepressant drug, lowers 5-HT and raises dopamine and noradrenaline in the rat prefrontal cortex dialysate: role of 5-HT(1A) receptors. Br. J. Pharmacol. 139, 1281–1288.
Jay, T. M. (2003). Dopamine: a potential substrate for synaptic plasticity and memory mechanisms. Prog. Neurobiol. 69, 375–390.
Jay, T. M., Gurden, H., and Yamaguchi, T. (1998). Rapid increase in PKA activity during long-term potentiation in the hippocampal afferent fibre system to the prefrontal cortex in vivo. Eur. J. Neurosci. 10, 3302–3306.
Jay, T. M., Rocher, C., Hotte, M., Naudon, L., Gurden, H., and Spedding, M. (2004). Plasticity at hippocampal to prefrontal cortex synapses is impaired by loss of dopamine and stress: importance for psychiatric diseases. Neurotox. Res. 6, 233–244.
Jordan, S., Kramer, G. L., Zukas, P. K., Moeller, M, and Petty, F. (1994) In vivo biogenic amine efflux in medial prefrontal cortex with imipramine, fluoxetine, and fluvoxamine. Synapse 18, 294–297.
Kan, J. P., Steinberg, R., Mouget-Goniot, C., Worms, P., and Biziere, K. (1987). SR 95191, a selective inhibitor of type A monoamine oxidase with dopaminergic properties. II. Biochemical characterization of monoamine oxidase inhibition. J. Pharmacol. Exp. Ther. 240, 251–258.
Kihara, T., and Ikeda, M. (1995). Effects of duloxetine, a new serotonin and norepinephrine uptake inhibitor, on extracellular monoamine levels in rat frontal cortex. J. Pharmacol. Exp. Ther. 272, 177–183.
Kitaichi, Y., Inoue, T., Nakagawa, S., Izumi, T., and Koyama, T. (2004). Effect of co-administration of lithium and reboxetine on extracellular monoamine concentrations in rats. Eur. J. Pharmacol. 489, 187–191.
Kitaichi, Y., Inoue, T., Nakagawa, S., Izumi, T., and Koyama, T. (2005). Effect of milnacipran on extracellular monoamine concentrations in the medial prefrontal cortex of rats pre-treated with lithium. Eur. J. Pharmacol. 516, 219–226.
Kobayashi, A., Fujita, K., and Nakazawa, K. (1992). In rat brain amoxapine enhances dopamine metabolism: pharmacokinetic variations of the effect. Eur. J. Pharmacol. 215, 43–49.
Koch, S., Perry, K. W., Nelson, D. L., Conway, R. G., Threlkeld, P. G., and Bymaster, F. P. (2002). R-fluoxetine increases extracellular DA, NE, as well as 5-HT in rat prefrontal cortex and hypothalamus: an in vivo microdialysis and receptor binding study. Neuropsychopharmacology 27, 949–959.
Kruse, M. S., Premont, J., Krebs, M. O., and Jay, T. M. (2009). Interaction of dopamine D1 with NMDA NR1 receptors in rat prefrontal cortex. Eur. Neuropsychopharmacol. 19, 296–304.
Kurata, K., Ashby, C. R. Jr., Oberlender, R., Tanii, Y., Kurachi, M., Rini, N. J., and Strecker, R. E. (1996). The characterization of the effect of locally applied N-methylquipazine, a 5-HT3 receptor agonist, on extracellular dopamine levels in the anterior medial prefrontal cortex in the rat: an in vivo microdialysis study. Synapse 24, 313–321.
Lakshmana, M. K., Rao, B. S., Dhingra, N. K., Ravikumar, R., Govindaiah, Sudha, S., Meti, B. L., and Raju, T. R. (1998). Role of monoamine oxidase type A and B on the dopamine metabolism in discrete regions of the primate brain. Neurochem. Res. 23, 1031–1037.
Lara-Lemus, A., Drucker-Colin, R., Mendez-Franco, J., Palomero-Rivero, M., and Perez de la Mora, M. (1998) .Biochemical effects induced by REM sleep deprivation in naive and in D-amphetamine treated rats. Neurobiology (Bp) 6, 13–22.
Lavergne, F., Berlin, I., Gamma, A., Stassen, H., and Angst, J. (2005). Onset of improvement and response to mirtazapine in depression: a multicenter naturalistic study of 4771 patients. Neuropsychiatr. Dis. Treat. 1, 59–68.
Lavergne, F., and De Mouzon, J. (2000). “Antidepressant efficacy at two weeks,” in 1st ECNP Workshop (Neuropsychopharmacology E, ed), p S26. Nice, France: Elsevier.
Lavergne, F., and Jay, T. M. (2009). “P-29-012: a unifying theory of antidepressant action,” in 9th World Congress of Biological Psychiatry (Paris: Informaworld), 385.
Li, S. X., Perry, K. W., and Wong, D. T. (2002). Influence of fluoxetine on the ability of bupropion to modulate extracellular dopamine and norepinephrine concentrations in three mesocorticolimbic areas of rats. Neuropharmacology 42, 181–190.
Li, Z., Huang, M., Prus, A. J., Dai, J., and Meltzer, H. Y. (2007). 5-HT6 receptor antagonist SB-399885 potentiates haloperidol and risperidone-induced dopamine efflux in the medial prefrontal cortex or hippocampus. Brain Res. 1134, 70–78.
Linner, L., Endersz, H., Ohman, D., Bengtsson, F., Schalling, M., and Svensson, T. H. (2001). Reboxetine modulates the firing pattern of dopamine cells in the ventral tegmental area and selectively increases dopamine availability in the prefrontal cortex. J. Pharmacol. Exp. Ther. 297, 540–546.
Lisanby, S. H., and Belmaker, R. H. (2000). Animal models of the mechanisms of action of repetitive transcranial magnetic stimulation (RTMS): comparisons with electroconvulsive shock (ECS). Depress. Anxiety 12, 178–187.
Louilot, A., Mocaer, E., Simon, H., and Le Moal, M. (1990). Difference in the effects of the antidepressant tianeptine on dopaminergic metabolism in the prefrontal cortex and the nucleus accumbens of the rat. A voltammetric study. Life Sci. 47, 1083–1089.
Martinez-Turrillas, R., Del Rio, J., and Frechilla, D. (2007). Neuronal proteins involved in synaptic targeting of AMPA receptors in rat hippocampus by antidepressant drugs. Biochem. Biophys. Res. Commun. 353, 750–755.
Martinez-Turrillas, R., Frechilla, D., and Del Rio, J. (2002). Chronic antidepressant treatment increases the membrane expression of AMPA receptors in rat hippocampus. Neuropharmacology 43, 1230–1237.
Matsumoto, M., Shikanai, H., Togashi, H., Izumi, T., Kitta, T., Hirata, R., Yamaguchi, T., and Yoshioka, M. (2008). Characterization of clozapine-induced changes in synaptic plasticity in the hippocampal-mPFC pathway of anesthetized rats. Brain Res. 1195, 50–55.
McTavish, S. F., McPherson, M. H., Harmer, C. J., Clark, L., Sharp, T., Goodwin, G. M., and Cowen, P. J. (2001). Antidopaminergic effects of dietary tyrosine depletion in healthy subjects and patients with manic illness. Br. J. Psychiatry 179, 356–360.
Millan, M. J., Dekeyne, A., and Gobert, A. (1998). Serotonin (5-HT)2C receptors tonically inhibit dopamine (DA) and noradrenaline (NA), but not 5-HT, release in the frontal cortex in vivo. Neuropharmacology 37, 953–955.
Millan, M. J., and Gobert, A. (1999). (−)-Pindolol increases dialysate concentrations of dopamine and noradrenaline, but not serotonin, in the frontal cortex of freely-moving rats. Neuropharmacology 38, 909–912.
Millan, M. J., Gobert, A., Lejeune, F., Dekeyne, A., Newman-Tancredi, A., Pasteau, V., Rivet, J. M., and Cussac, D. (2003). The novel melatonin agonist agomelatine (S20098) is an antagonist at 5-hydroxytryptamine2C receptors, blockade of which enhances the activity of frontocortical dopaminergic and adrenergic pathways. J. Pharmacol. Exp. Ther. 306, 954–964.
Millan, M. J., Newman-Tancredi, A., Audinot, V., Cussac, D., Lejeune, F., Nicolas, J. P., Coge, F., Galizzi, J. P., Boutin, J. A., Rivet, J. M., Dekeyne, A., and Gobert, A. (2000). Agonist and antagonist actions of yohimbine as compared to fluparoxan at alpha(2)-adrenergic receptors (AR)s, serotonin (5-HT)(1A), 5-HT(1B), 5-HT(1D) and dopamine D(2) and D(3) receptors. Significance for the modulation of frontocortical monoaminergic transmission and depressive states. Synapse 35, 79–95.
Mizoguchi, K., Ikeda, R., Shoji, H., Tanaka, Y., and Tabira, T. (2008a). Suppression of glucocorticoid secretion induces a behaviorally depressive state in rotarod performance in rat. Pharmacol. Biochem. Behav. 90, 730–734.
Mizoguchi, K., Shoji, H., Ikeda, R., Tanaka, Y., and Tabira, T. (2008b). Persistent depressive state after chronic stress in rats is accompanied by HPA axis dysregulation and reduced prefrontal dopaminergic neurotransmission. Pharmacol. Biochem. Behav. 91, 170–175.
Mizoguchi, K., Ishige, A., Takeda, S., Aburada, M., and Tabira, T. (2004). Endogenous glucocorticoids are essential for maintaining prefrontal cortical cognitive function. J. Neurosci. 24, 5492–5499.
Mizoguchi, K., Yuzurihara, M., Nagata, M., Ishige, A., Sasaki, H., and Tabira, T. (2002). Dopamine-receptor stimulation in the prefrontal cortex ameliorates stress-induced rotarod impairment. Pharmacol. Biochem. Behav. 72, 723–728.
Morissette, M., and Paolo, T. D. (1996). Acute effect of 17 beta-estradiol and lithium on ovariectomized rat brain biogenic amines metabolism. J. Psychiatr. Res. 30, 95–107.
Muneoka, K., Shirayama, Y., Takigawa, M., and Shioda, S. (2009). Brain region-specific effects of short-term treatment with duloxetine, venlafaxine, milnacipran and sertraline on monoamine metabolism in rats. Neurochem. Res. 34, 542–555.
Nakamura, S., Ago, Y., Itoh, S., Koyama, Y., Baba, A., and Matsuda, T. (2005). Effect of zotepine on dopamine, serotonin and noradrenaline release in rat prefrontal cortex. Eur. J. Pharmacol. 528, 95–98.
Nakayama, K. (2002). Effect of paroxetine on extracellular serotonin and dopamine levels in the prefrontal cortex. Naunyn Schmiedebergs Arch. Pharmacol. 365, 102–105.
Nakayama, K., Sakurai, T., and Katsu, H. (2004). Mirtazapine increases dopamine release in prefrontal cortex by 5-HT1A receptor activation. Brain Res. Bull. 63, 237–241.
Noda, Y., Mouri, A., Ando, Y., Waki, Y., Yamada, S. N., Yoshimi, A., Yamada, K., Ozaki, N., Wang, D., Nabeshima, T. (2010). Galantamine ameliorates the impairment of recognition memory in mice repeatedly treated with methamphetamine: involvement of allosteric potentiation of nicotinic acetylcholine receptors and dopaminergic-ERK1/2 systems. Int. J. Neuropsychopharmacol. 13, 1343–1354.
Ohnishi, T., Hayashi, T., Okabe, S., Nonaka, I., Matsuda, H., Iida, H., Imabayashi, E., Watabe, H., Miyake, Y., Ogawa, M., Teramoto, N., Ohta, Y., Ejima, N., Sawada, T., and Ugawa, Y. (2004). Endogenous dopamine release induced by repetitive transcranial magnetic stimulation over the primary motor cortex: an [11C]raclopride positron emission tomography study in anesthetized macaque monkeys. Biol. Psychiatry 55, 484–489.
Owen, J. C., and Whitton, P. S. (2006). Effects of amantadine and budipine on antidepressant drug-evoked changes in extracellular dopamine in the frontal cortex of freely moving rats. Brain Res. 1117, 206–212.
Pacher, P., Kohegyi, E., Kecskemeti, V., and Furst, S. (2001). Current trends in the development of new antidepressants. Curr. Med. Chem. 8, 89–100.
Page, M. E., and Lucki, I. (2002). Effects of acute and chronic reboxetine treatment on stress-induced monoamine efflux in the rat frontal cortex. Neuropsychopharmacology 27, 237–247.
Pelton, G. H., Harper, O. L., Tabert, M. H., Sackeim, H. A., Scarmeas, N., Roose, S. P., and Devanand, D. P. (2008). Randomized double-blind placebo-controlled donepezil augmentation in antidepressant-treated elderly patients with depression and cognitive impairment: a pilot study. Int. J. Geriatr. Psychiatry 23, 670–676.
Pozzi, L., Invernizzi, R., Garavaglia, C., and Samanin, R. (1999). Fluoxetine increases extracellular dopamine in the prefrontal cortex by a mechanism not dependent on serotonin: a comparison with citalopram. J. Neurochem. 73, 1051–1057.
Qi, H., Mailliet, F., Spedding, M., Rocher, C., Zhang, X., Delagrange, P., McEwen, B., Jay, T. M., and Svenningsson, P. (2009). Antidepressants reverse the attenuation of the neurotrophic MEK/MAPK cascade in frontal cortex by elevated platform stress; reversal of effects on LTP is associated with GluA1 phosphorylation. Neuropharmacology 56, 37–46.
Rabkin, J. G., Stewart, J. W., McGrath, P. J., Markowitz, J. S., Harrison, W., and Quitkin, F. M. (1987). Baseline characteristics of 10-day placebo washout responders in antidepressant trials. Psychiatry Res. 21, 9–22.
Rajkumar, R., and Mahesh, R. (2010). The auspicious role of the 5-HT3 receptor in depression: a probable neuronal target? J. Psychopharmacol. 24, 455–469.
Rivet, J. M., Gobert, L., Cistarelli, C., Melon, C., and Millan, M. J. (1998). Antidepressants (ADs) differentially modify dialysate serotonin (5HT), dopamine (DA) and noradrenaline (NAD) levels in rats. Soc. Neurosci. Abstr. 24, 440.9.
Rocher, C., Spedding, M., Munoz, C., and Jay, T. M. (2004). Acute stress-induced changes in hippocampal/prefrontal circuits in rats: effects of antidepressants. Cereb. Cortex 14, 224–229.
Rozzini, L., Vicini Chilovi, B., Bertoletti, E., Trabucchi, M., and Padovani, A. (2007). Acetylcholinesterase inhibitors and depressive symptoms in patients with mild to moderate Alzheimer’s disease. Aging Clin. Exp. Res. 19, 220–223.
Sacchetti, G., Bonini, I., Waeterloos, G. C., and Samanin, R. (1993). Tianeptine raises dopamine and blocks stress-induced noradrenaline release in the rat frontal cortex. Eur. J. Pharmacol. 236, 171–175.
Sakaue, M., Somboonthum, P., Nishihara, B., Koyama, Y., Hashimoto, H., Baba, A., and Matsuda, T. (2000). Postsynaptic 5-hydroxytryptamine(1A) receptor activation increases in vivo dopamine release in rat prefrontal cortex. Br. J. Pharmacol. 129, 1028–1034.
Schilstrom, B., Ivanov, V. B., Wiker, C., and Svensson, T. H. (2007). Galantamine enhances dopaminergic neurotransmission in vivo via allosteric potentiation of nicotinic acetylcholine receptors. Neuropsychopharmacology 32, 43–53.
Shearman, E., Rossi, S., Sershen, H., Hashim, A., and Lajtha, A. (2005). Locally administered low nicotine-induced neurotransmitter changes in areas of cognitive function. Neurochem. Res. 30, 1055–1066.
Shearman, E., Rossi, S., Szasz, B., Juranyi, Z., Fallon, S., Pomara, N., Sershen, H., and Lajtha, A. (2006). Changes in cerebral neurotransmitters and metabolites induced by acute donepezil and memantine administrations: a microdialysis study. Brain Res. Bull. 69, 204–213.
Shoblock, J. R., Maisonneuve, I. M., and Glick, S. D. (2004). Differential interactions of desipramine with amphetamine and methamphetamine: evidence that amphetamine releases dopamine from noradrenergic neurons in the medial prefrontal cortex. Neurochem. Res. 29, 1437–1442.
Skolnick, P., Popik, P., and Trullas, R. (2009). Glutamate-based antidepressants: 20 years on. Trends Pharmacol. Sci. 30, 563–569.
Spanagel, R., Eilbacher, B., and Wilke, R. (1994). Memantine-induced dopamine release in the prefrontal cortex and striatum of the rat–a pharmacokinetic microdialysis study. Eur. J. Pharmacol. 262, 21–26.
Stassen, H. H., Angst, J., and Delini-Stula, A. (1996). Delayed onset of action of antidepressant drugs? Survey of results of Zurich meta-analyses. Pharmacopsychiatry 29, 87–96.
Stassen, H. H., Delini-Stula, A., and Angst, J. (1993). Time course of improvement under antidepressant treatment: a survival-analytical approach. Eur. Neuropsychopharmacol. 3, 127–135.
Sun, X., Zhao, Y., and Wolf, M. E. (2005). Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J. Neurosci. 25, 7342–7351.
Takamori, K., Yoshida, S., and Okuyama, S. (2001). Repeated treatment with imipramine, fluvoxamine and tranylcypromine decreases the number of escape failures by activating dopaminergic systems in a rat learned helplessness test. Life Sci. 69, 1919–1926.
Tanaka, M., Namiki, C., Thuy, D. H., Yoshida, H., Kawasaki, K., Hashikawa, K., Fukuyama, H., and Kita, T. (2004). Prediction of psychiatric response to donepezil in patients with mild to moderate Alzheimer’s disease. J. Neurol. Sci. 225, 135–141.
Tanda, G., Bassareo, V., and Di Chiara, G. (1996a). Mianserin markedly and selectively increases extracellular dopamine in the prefrontal cortex as compared to the nucleus accumbens of the rat. Psychopharmacology (Berl.) 123, 127–130.
Tanda, G., Frau, R., and Di Chiara, G. (1996b). Chronic desipramine and fluoxetine differentially affect extracellular dopamine in the rat prefrontal cortex. Psychopharmacology (Berl.) 127, 83–87.
Tanda, G., Carboni, E., Frau, R., and Di Chiara, G. (1994). Increase of extracellular dopamine in the prefrontal cortex: a trait of drugs with antidepressant potential? Psychopharmacology (Berl.) 115, 285–288.
Tanda, G., Frau, R., and Di Chiara, G. (1995). Local 5HT3 receptors mediate fluoxetine but not desipramine-induced increase of extracellular dopamine in the prefrontal cortex. Psychopharmacology (Berl.) 119, 15–19.
Toide, K. (1990). Effects of amantadine on dopaminergic neurons in discrete regions of the rat brain. Pharm. Res. 7, 670–672.
Tremblay, L. K., Naranjo, C. A., Graham, S. J., Herrmann, N., Mayberg, H. S., Hevenor, S., and Busto, U. E. (2005). Functional neuroanatomical substrates of altered reward processing in major depressive disorder revealed by a dopaminergic probe. Arch. Gen. Psychiatry 62, 1228–1236.
Tse, W. S., and Bond, A. J. (2003). Reboxetine promotes social bonding in healthy volunteers. J. Psychopharmacol. 17, 189–195.
Tsukada, H., Miyasato, K., Nishiyama, S., Fukumoto, D., Kakiuchi, T., and Domino, E. F. (2005). Nicotine normalizes increased prefrontal cortical dopamine D1 receptor binding and decreased working memory performance produced by repeated pretreatment with MK-801: a PET study in conscious monkeys. Neuropsychopharmacology 30, 2144–2153.
Ulrich, S., Northoff, G., Wurthmann, C., Partscht, G., Pester, U., Herscu, H., and Meyer, F. P. (2001). Serum levels of amitriptyline and therapeutic effect in non-delusional moderately to severely depressed in-patients: a therapeutic window relationship. Pharmacopsychiatry 34, 33–40.
Valentini, V., Cacciapaglia, F., Frau, R., and Di Chiara, G. (2005). Selective serotonin reuptake blockade increases extracellular dopamine in noradrenaline-rich isocortical but not prefrontal areas: dependence on serotonin-1A receptors and independence from noradrenergic innervation. J. Neurochem. 93, 371–382.
Valentini, V., Frau, R., and Di Chiara, G. (2004). Noradrenaline transporter blockers raise extracellular dopamine in medial prefrontal but not parietal and occipital cortex: differences with mianserin and clozapine. J. Neurochem. 88, 917–927.
Wang, D., Noda, Y., Zhou, Y., Nitta, A., Furukawa, H., and Nabeshima, T. (2007a). Synergistic effect of combined treatment with risperidone and galantamine on phencyclidine-induced impairment of latent visuospatial learning and memory: Role of nAChR activation-dependent increase of dopamine D1 receptor-mediated neurotransmission. Neuropharmacology 53, 379–389.
Wang, D., Noda, Y., Zhou, Y., Mouri, A., Mizoguchi, H., Nitta, A., Chen, W., and Nabeshima, T. (2007b). The allosteric potentiation of nicotinic acetylcholine receptors by galantamine ameliorates the cognitive dysfunction in beta amyloid25-35 i.c.v.-injected mice: involvement of dopaminergic systems. Neuropsychopharmacology 32, 1261–1271.
Watanabe, A. (1999). The influence of L-triiodothyronine on the action of desipramine on beta and serotonin 2A receptor, monoamines in the rat brain. Nihon Shinkei Seishin Yakurigaku Zasshi 19, 139–146.
Watanabe, T., Ueda, M., Saeki, Y., Hirokane, G., Morita, S., Okawa, M., Akiyama, K., and Shimoda, K. (2007). High plasma concentrations of paroxetine impede clinical response in patients with panic disorder. Ther. Drug. Monit. 29, 40–44.
Weikop, P., Kehr, J., and Scheel-Kruger, J. (2004). The role of alpha1- and alpha2-adrenoreceptors on venlafaxine-induced elevation of extracellular serotonin, noradrenaline and dopamine levels in the rat prefrontal cortex and hippocampus. J. Psychopharmacol. 18, 395–403.
Wu, J. C., Buchsbaum, M., and Bunney, W. E. Jr. (2001). Clinical neurochemical implications of sleep deprivation’s effects on the anterior cingulate of depressed responders. Neuropsychopharmacology 25, S74–S78.
Yoshida, K., Higuchi, H., Kamata, M., Yoshimoto, M., Shimizu, T., and Hishikawa, Y. (1998). Single and repeated electroconvulsive shocks activate dopaminergic and 5-hydroxytryptaminergic neurotransmission in the frontal cortex of rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 22, 435–444.
Zhang, W., Perry, K. W., Wong, D. T., Potts, B. D., Bao, J., Tollefson, G. D., and Bymaster, F. P. (2000). Synergistic effects of olanzapine and other antipsychotic agents in combination with fluoxetine on norepinephrine and dopamine release in rat prefrontal cortex. Neuropsychopharmacology 23, 250–262.
Keywords: antidepressant, dopamine, prefrontal cortex, major depression, synaptic plasticity
Citation: Lavergne F and Jay TM (2010) A new strategy for antidepressant prescription. Front. Neurosci. 4:192. doi: 10.3389/fnins.2010.00192
Received: 19 July 2010;
Paper pending published: 07 September 2010;
Accepted: 01 November 2010;
Published online: 19 November 2010
Edited by:
Xiao-Ming Ou, University of Mississippi Medical Center, USAReviewed by:
Xiao-Ming Ou, University of Mississippi Medical Center, USAMichael F. O'Neil, Eolas Biosciences Ltd, UK
Copyright: © 2010 Lavergne and Jay. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Thérèse M Jay, Centre de Psychiatrie et de Neurosciences, INSERM U894, 2ter rue d’Alésia, 75014 Paris, France. e-mail:dGhlcmVzZS5qYXlAaW5zZXJtLmZy