 Shakevia Johnson1
Shakevia Johnson1 Shawna Tazik1 Deyin Lu1 Chandra Johnson1 Moussa B. H. Youdim2,3 Junming Wang4 Grazyna Rajkowska1
Shawna Tazik1 Deyin Lu1 Chandra Johnson1 Moussa B. H. Youdim2,3 Junming Wang4 Grazyna Rajkowska1 Xiao-Ming Ou1*
Xiao-Ming Ou1*
- 1 Department of Psychiatry and Human Behavior, University of Mississippi Medical Center, Jackson, MS, USA
- 2 Eve Topf Center, Technion-Rappaport Family Faculty of Medicine, Haifa, Israel
- 3 Department of Biology, Yonsei University, Seoul, South Korea
- 4 Department of Pathology, University of Mississippi Medical Center, Jackson, MS, USA
Stress detrimentally affects the brain and body and can lead to or be accompanied by depression. Although stress and depression may contribute to each other, the exact molecular mechanism underlying the effects is unclear. However, there is a correlation between stress and an increase in glucocorticoid secretion which causes a subsequent increase in monoamine oxidase (MAO) activity during stress. Consequently, MAO inhibitors have been used as traditional antidepressant drugs. Cellular treatment with the synthetic glucocorticoid, dexamethasone (a cellular stressor), has been reported to markedly increase both MAO A and MAO B catalytic activities, as well as apoptosis. This study compares the neuroprotective abilities of M30 (a new generation inhibitor of both MAO A and MAO B) with rasagiline (Azilect®, another new MAO B inhibitor) and selegiline (Deprenyl®, a traditional MAO B inhibitor) in the prevention of dexamethasone-induced brain cell death and MAO activity in human neuroblastoma cells, SH-SY5Y. M30 demonstrated the highest inhibitory effect on MAO A; however, M30 showed the lowest inhibitory effect on MAO B enzymatic activity in comparison to rasagiline and selegiline. Although, M30 exhibited the greatest neuroprotective effect by decreasing cell death rates and apoptotic DNA damage compared to rasagiline and selegiline, these neuroprotective effects of M30 were, overall, similar to rasagiline. Summarily, M30 has a generally greater impact on neuroprotection than the MAO B inhibitors, selegiline and rasagiline. Our results suggest that M30 may have great potential in alleviating disorders involving increases in both MAO A and MAO B, such as stress-induced disorders.
Introduction
Stress encompasses the specific responses that affect the normal physiological state of the body. The chief organ that responds to stress is the brain. Stress interferes with the emotional, social, physiological, mental and physical aspects of health, and well being and often leads to depression. According to the World Health Organization, major depression is among the most burdensome diseases in the world. Depression is a major public health concern that costs the United States 83 billion dollars, annually. Furthermore, the point prevalence of depression is approximately 3–5% for males and 8–10% for females, and has a lifetime prevalence about twice that of the point prevalence (Lyness et al., 2009). A major response to stress is the production of glucocorticoids which are steroid hormones secreted from the adrenal gland. Glucocorticoids have a significant role in neuronal cell death and depression related to stressors as an abnormal increase of glucocorticoid levels has been associated with atrophy of the hippocampus (Lee et al., 2002) and major depression (Duval et al., 2006). Due to an improved understanding of the cellular changes that occur during stressful events, antidepressants such as monoamine oxidase (MAO) inhibitors are a traditional drug class used for treatment.
Glucocorticoids are involved in many cellular functions that also involve MAO as MAO plays a role in stress, behavioral adaptation, and mood (de Kloet et al., 1990). MAO, appearing as two isoforms: MAO A and MAO B, is located on the outer membranes of mitochondria in neuronal, glial, and other cells and catalyzes the oxidative deamination of monoamine neurotransmitters (Shih et al., 1999). MAO (Meyer et al., 2006, 2009; Sacher et al., 2010) and glucocorticoid hypersecretion (Duval et al., 2006) are associated with depression. The synthetic glucocorticoid, dexamethasone, has been documented to increase MAO A activity in human neuroblastoma and glioblastoma cells through its role as a cellular stressor (Ou et al., 2006). More specifically, dexamethasone has been shown to increase MAO A mRNA, protein and enzymatic activity in human skeletal muscle cells (Manoli et al., 2005) and increases MAO A in the dorsal raphe nucleus in rats (Jahng et al., 2008). Additionally, dexamethasone has been reported to induce MAO B expression and activity in both neuronal cells (Tazik et al., 2009) and astrocytes (Carlo et al., 1996) and reduces the number of viable brain cells (Yu et al., 2010). Elevated levels of MAO degrade serotonin and produce reactive oxygen species (ROS, such as hydrogen peroxide) (Carlsson et al., 1980) that may cause cell death and, as a result, an MAO inhibitor prevents cell death related to this manner of toxicity (Haynes et al., 2004; Lu et al., 2008).
MAO B inhibitors, such as rasagiline (Azilect®) and selegiline (Deprenyl®), are effectively used for the treatment of several neuropsychiatric and neurodegenerative diseases such as Parkinson’s disease (Youdim et al., 2005). Additionally, these drugs have also exhibited neuroprotective properties by increasing cellular proliferation rates at low concentrations (Malorni et al., 1998; Youdim et al., 2001). The neuroprotective mechanism of MAO B inhibitors has been suggested through the blockage of the translocation of the GAPDH complex with the transcriptional activator, transforming growth factor-beta-inducible early gene 2 (TIEG2) into the nucleus, and, secondarily, inhibiting MAO B gene expression (Ou et al., 2009b).
Rasagiline is reportedly more effective than selegiline due to the differences in metabolite formation. Aminoindan, the metabolite of rasagiline, reinforces the neuroprotectivity of rasagiline as it, in fact, possesses its own neuroprotective properties (Lu et al., 2008; Bar-Am et al., 2009; Ou et al., 2009a). In contrast, the metabolite of selegiline, methamphetamine, counteracts the neuroprotectivity afforded by selegiline (Chen and Ly, 2006). A novel therapeutic agent, M30, has been introduced as a potential drug to be used for the treatment of neurodegenerative disorders (Youdim, 2006). M30, an iron-chelator, possesses the same neuroprotective propargylamine moiety as rasagiline; however, M30 is a brain selective MAO A and B inhibitor that does not cause a cheese effect in response to tyramine (Gal et al., 2005, 2009; Zheng et al., 2005). Studies have demonstrated that M30 exhibits a wide range of pharmacological activities including neuroprotection against ROS-induced neurotoxicity (specifically caused by increased hydrogen peroxide production) in mouse motor neurons (Kupershmidt et al., 2009) and neurorestoration in lactacystin- and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced models of Parkinson’s disease (Zhu et al., 2007; Gal et al., 2009).
In the present study, we have extended our comparative analyses to describe the neuroprotective effects of rasagiline, selegiline, and M30 in the human neuroblastoma SH-SY5Y cells treated with dexamethasone in charcoal-stripped serum.
Materials and Methods
Cell Lines and Reagents
The human neuroblastoma cell line, SH-SY5Y, was purchased from ATCC. The cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 2 mm l-glutamine, 100 units/ml penicillin, 10 μg/ml streptomycin, and 10% fetal bovine serum (Invitrogen). Rasagiline was synthesized by a doctoral student, Hailin Zheng, in the laboratory of Dr. Youdim (Teva Pharmaceutical Co., Haifa, Israel). M30 was also developed in Dr. Youdim’s laboratory. Selegiline was purchased from Sigma-Aldrich, USA.
Cell Culture and Treatments
SH-SY5Y cells were seeded into 24-well plates or 10-cm dishes and cultured overnight in medium. Cells were supplemented with charcoal-stripped, steroid-free fetal calf serum for ∼6 h for acclimation. The medium was then replaced with medium containing 2 μM of dexamethasone and with or without 0.25 nM of rasagiline, M30 or selegiline in the presence of charcoal-stripped fetal calf serum. The treatments were performed daily for 3 days.
MAO Catalytic Activity Assay
SH-SY5Y cells were seeded in 10-cm dishes. After 24 h, cells were treated with dexamethasone (2 μM) with or without 0.25 nM M30, rasagiline, or selegiline for 72 h. Cells were then harvested and washed with phosphate-buffered saline (PBS).
MAO A catalytic activity assay
For determining the catalytic activity of MAO A, 100 μg of total proteins was incubated with 100 μl of 14C-labeled 5-hydroxytryptamine (New England Nuclear Corporation) in the assay buffer (50 mM sodium phosphate buffer, pH of 7.4) at 37°C for 20 min and terminated by the addition of 100 μl of 6 N HCl (Ou et al., 2006). The reaction products were extracted with ethyl acetate/benzene (1:1) and centrifuged at room temperature for 7 min. The organic phase containing the reaction product was extracted from each sample and its radioactivity was quantified by liquid scintillation spectroscopy (Ou et al., 2006).
MAO B catalytic activity assay
For measuring the catalytic activity of MAO B, 100 μg of total proteins was incubated with 10 μl of 14C-labeled phenylethylamine (Amersham Biosciences) in the assay buffer at 37oC for 20 min and terminated by the addition of 100 μl of 6 N HCl. The reaction products were extracted with ethyl acetate/toluene (1:1) and centrifuged at room temperature for 7 min. The organic phase containing the reaction product was extracted from each sample and its radioactivity was quantified by liquid scintillation spectroscopy (Ou et al., 2004).
MTT Assay
The survival and proliferation rates of the cells were measured using MTT assays. MTT, 3-[4.5-dimethyl-thiazol-2-yl]-2,5-diphenyl tetrazolium bromide, is a yellow salt that is metabolized within the mitochondria of the cells forming a purple formazan crystal, which can be dissolved with the use of a detergent. The intensity of the dissolved purple color allows for the measurement of the solution’s light absorbance. For the 24-well plates, 100 μl of MTT dye (0.5 mg/ml) was added to each well and the cells were then incubated at 37°C for 4–5 h. During the incubation period, the yellow dye was converted into a purple formazan crystal by the mitochondria of the viable cells. The purple crystals were dissolved by the addition of 250 μl dimethyl sulfoxide (DMSO). The NanoDrop Spectrophotometer was used to determine the optical density of each well at 572 nm (Alexander-Kaufman et al., 2006; Lu et al., 2008).
Tunel Assay
The terminal deoxynucleotidyl transferase (TdT)-mediated dUTP Nick End Labeling (TUNEL) assay was used to assess the extent of apoptosis in treated cells. Briefly, cells were plated on a four-well chamber slide on the day preceding the experiment, and treated with or without 2 μM dexamethasone and/or 0.25 nM of M30, rasagiline or selegiline for 3 days. Cells were then washed with PBS and fixed using 4% paraformaldehyde in PBS. The slides were again washed with PBS and fragmented DNA was detected in apoptotic cells by adding Fluorescein 12-dUTP to nicked ends of DNA (In Situ Cell Death Detection Kit, Roche). Slides were incubated for 1 h at 37oC in the dark, followed by a wash with PBS three times and stained with DAPI (for the nucleus), and then visualized with a fluorescent light microscope. Green fluorescence was correlated with DNA fragmentation. Experiments were done in three independent sets of duplicates and the percentages of TUNEL-positive cells were determined directly under a fluorescence microscope using a camera lucida with an excitation wavelength in the range of 450–500 nm and detection in the range of 515–565 nm (green) (Tazik et al., 2009).
Statistical Analysis
The statistical significance was analyzed using a one-way ANOVA to test for differences between groups. A value of P < 0.05 was considered significant.
Results
The expression of MAO, subsequent to treatment with MAO inhibitors after dexamethasone exposure, was comparatively investigated in human neuroblastoma SH-SY5Y cells. Cells were seeded in 10-cm dishes. After 24 h, cells were treated with dexamethasone (2 μM) and/or 0.25 nM M30, rasagiline or selegiline for 72 h. The catalytic activities were determined for MAO A (Figure 1A) and MAO B (Figure 1B) after treatment with the MAO inhibitors. With regards to MAO A, the results indicated that M30 significantly (*P < 0.05) decreased MAO A catalytic activity by 37% (Figure 1A, lanes 2 vs. 1); there was a 26% decrease due to rasagiline (Figure 1A, lanes 4 vs. 3) and a 24% decrease in MAO A enzymatic activity after treatment with selegiline (Figure 1A, lanes 6 vs. 5).
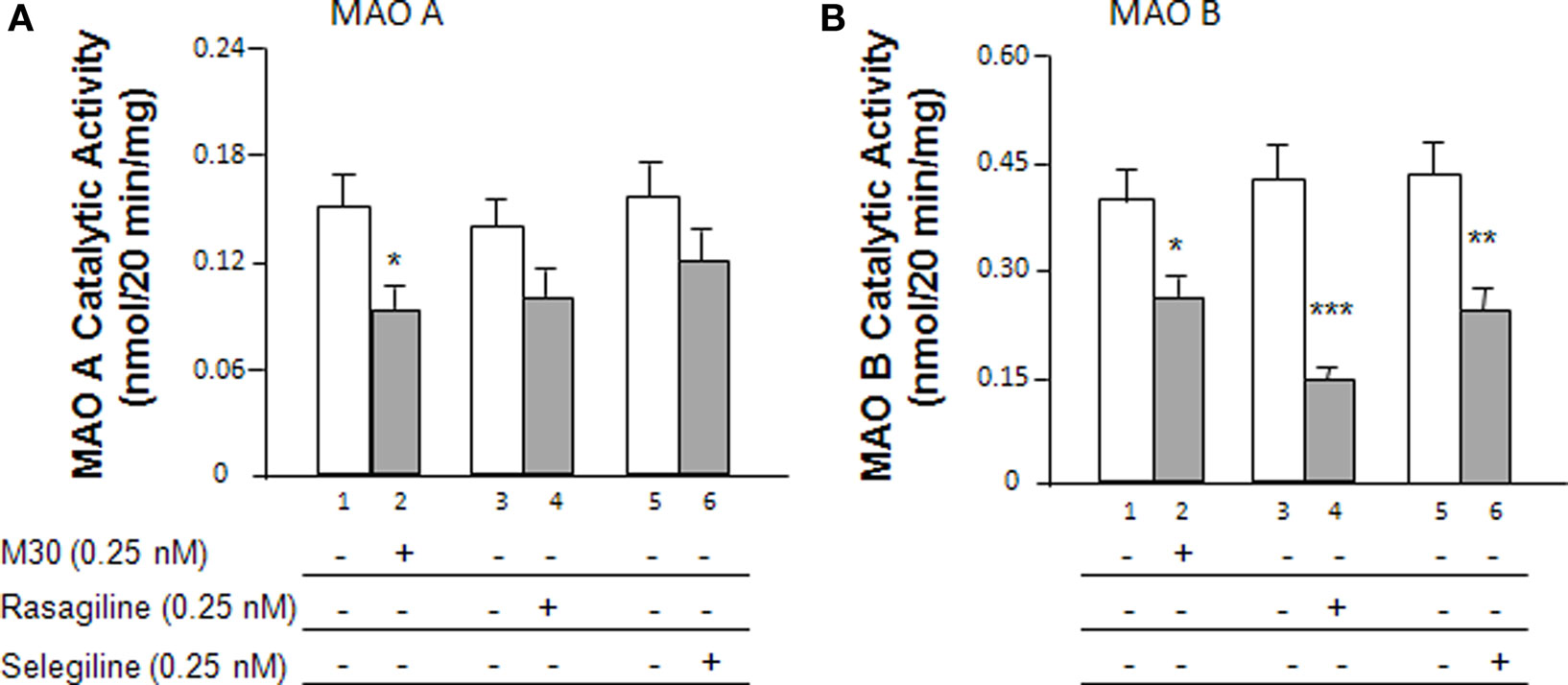
Figure 1. Effects of the MAO inhibitors, M30, rasagiline and selegiline, on MAO catalytic activity in SH-SY5Y cells. (A) Cells were treated with the MAO inhibitors (0. 25 nM) for 72 h and the MAO A catalytic activities were determined. (B) Cells were treated with the MAO inhibitors (0.25 nM) for 72 h and the MAO B catalytic activities were determined. *P < 0.05, **P < 0.01, and ***P < 0.002 compared to untreated controls, respectively.
Figure 1B demonstrates the effects of the MAO inhibitors on MAO B catalytic activity. Rasagiline and selegiline showed the greatest impact in MAO B catalytic activity decreasing the activities by 66% (P < 0.002) and 48% (P < 0.01), respectively (Figure 1B, lanes 4 vs. 3 and 6 vs. 5). M30 decreased MAO B enzymatic activity by 34% (Figure 1B, lanes 2 vs. 1, P < 0.05). Although M30 showed a statistically significant decrease in MAO B catalytic activity, its inhibitory effect was lower than those of rasagiline and selegiline.
Additionally, cellular survival rates of SH-SY5Y cells were evaluated among the different treatment groups by MTT assay (Figure 2). Cells were seeded in 24-well plates and, after overnight incubation, treated with 2 μM dexamethasone with or without 0.25 nM M30, rasagiline or selegiline for 72 h. The vehicle-treated, control cells were taken as 100%. Treatment with dexamethasone alone had a negative effect on cell viability with a survival rate of 60%. The effects of the drugs were compared to the survival rates of cells that underwent stress induction by dexamethasone. M30 significantly increased cell viability to ∼90% after exposure to dexamethasone (Figure 2, lanes 3 vs. 2, P < 0.02). Rasagiline and selegiline increased cell viability to 85 and 70% (Figure 2, lanes 4 vs. 2 and 5 vs. 2, P < 0.02 and P < 0.05, respectively). Although selegiline (Deprenyl) demonstrated a statistically significant increase in cell viability, its neuroprotective effect was significantly lower than those of M30 and rasagiline (Figure 2, lanes 5 vs. 3 and 4, P < 0.05).
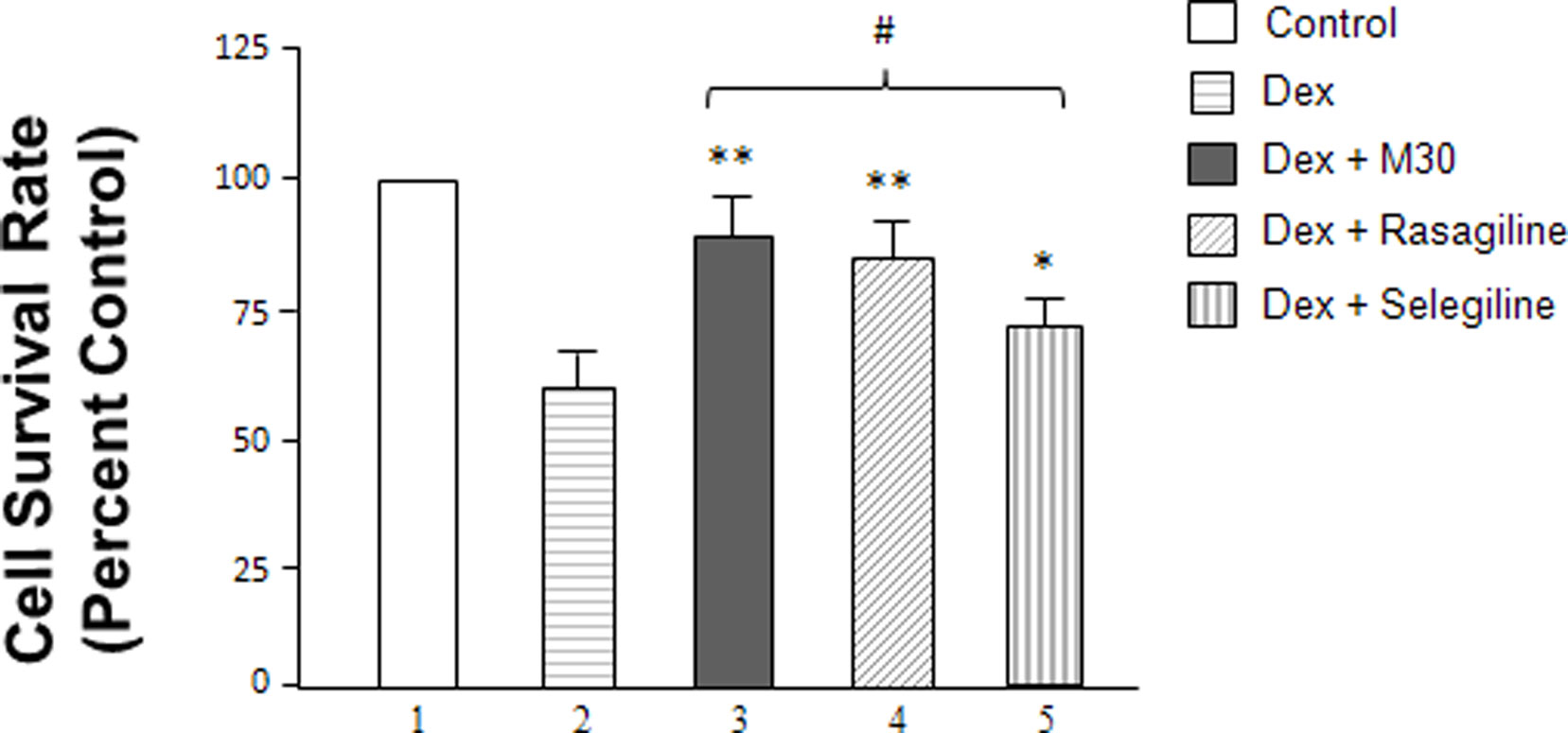
Figure 2 Effects of dexamethasone and MAO inhibitors on cell survival rates (cell proliferation rates) of SH-SY5Y cells. Cells were treated with dexamethasone (2 μM) with and without the MAO inhibitors (0. 25 nM) for 72 h. The cell survival rates were determined by MTT assay. *P < 0.05 and **P < 0.02 compared to cells treated with dexamethasone alone. #P < 0.05 compared lanes 5 vs. 3 and 4.
Results from the MAO A and B enzymatic activity assays and the MTT assay were further validated by the TUNEL assay (Figure 3) which uses TUNEL staining to measure fragmented DNA due to cellular apoptosis caused by an MAO catalytic activity-linked increase in H2O2 production (Phillips, 2003). Cells were plated on a four-well chamber slide and, after overnight incubation, treated with 2 μM dexamethasone with or without 0.25 nM of M30, rasagiline or selegiline for 3 days. Figure 3A depicts a significant increase in green fluorescent-labeled DNA fragmentation in SH-SY5Y cells that were treatment with dexamethasone compared to the untreated cells [(b) vs. (a)]. Figure 3 also indicates that treatment with M30, rasagiline or selegiline significantly decreases the occurrence of fragmented DNA compared to the dexamethasone-treated group [(c) vs. (b)]. An immunohistological representation of the effect of M30 on DNA fragmentation is shown in Figure 3A(c); rasagiline and selegiline exhibited similar effects. M30 caused a 34% decrease in the percentage of TUNEL-positive cells in dexamethasone-treated SH-SY5Y cells (P < 0.02; Figure 3B, lanes 6 vs. 5). Rasagiline and selegiline decreased the amount of fragmented DNA in cells treated with dexamethasone by 31 and 20% (P < 0.02 and P < 0.05; Figure 3B, lanes 7 vs. 5 and 8 vs. 5, respectively).
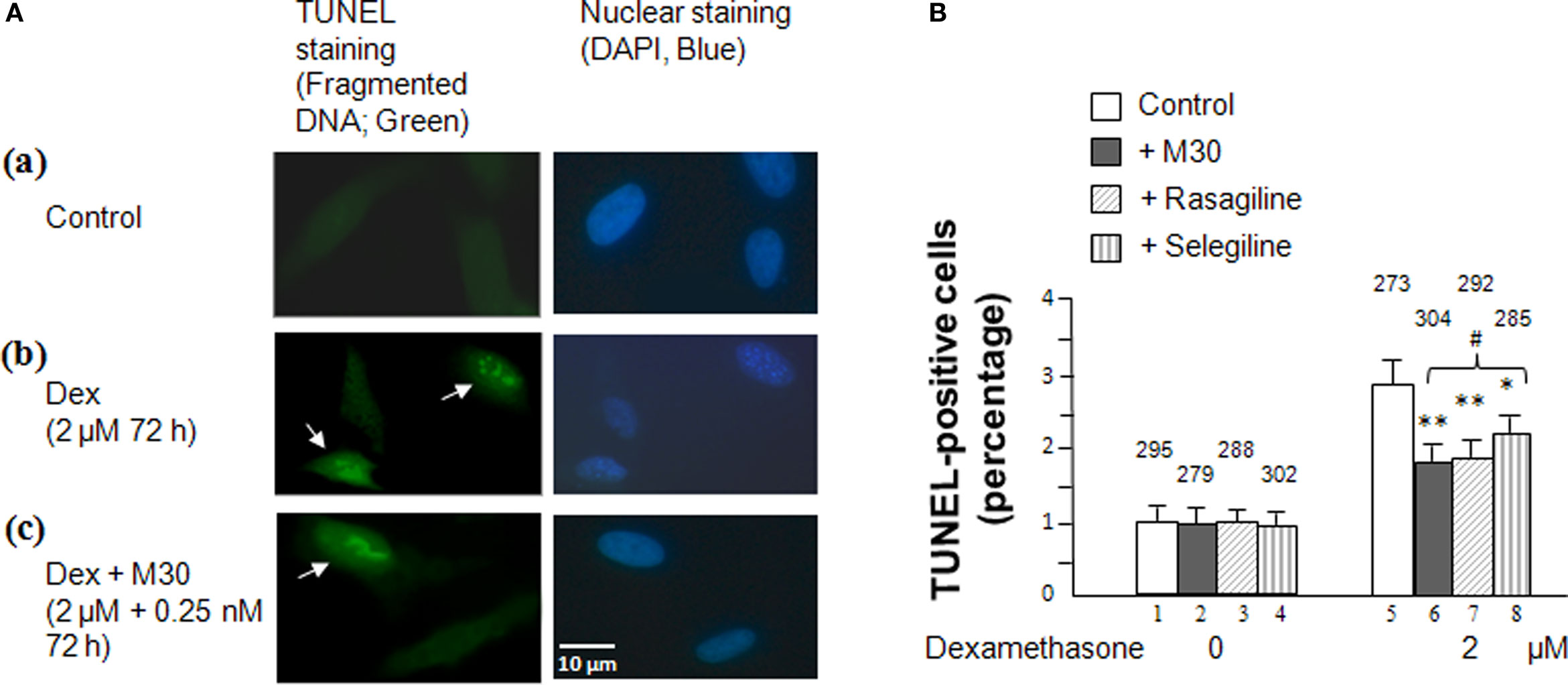
Figure 3 Effects of dexamethasone and MAO inhibitors on cell apoptosis in SHSY5Y cells. (A). Immunofluorescence showing TUNEL(+) and TUNEL(−) cells in (a) control cells, (b) cells treated with 2 μM dexamethasone for 72 h, and (c) cells treated with 2 μM dexamethasone and 0.25 nM M30 for 72 h. Photomicrographs show representative cells from each experimental group and the arrows indicate apoptotic cells. (B) Percentage of cells that contain damaged DNA as revealed by the TUNEL assay in each group. TUNEL-labeled DNA fragmentation correlates with green fluorescence. Experiments were done in duplicates, three times. The bar graph represents the average percentage of TUNEL-positive cells counted from each experimental group manually under a fluorescence microscope using a camera lucida. The counted cell numbers are shown at the top of each group. *P < 0.05 and **P < 0.02 compared to cells treated with dexamethasone alone. #P < 0.05 compared lanes 8 vs. 6 and 7.
With similar results pertaining to cell survival rates (MTT assay), although selegiline (Deprenyl) showed a statistically significant increase in cell viability, its neuroprotective effect was significantly lower than those exhibited by M30 and rasagiline (Figure 3B, lanes 8 vs. 6 and 7, P < 0.05).
Discussion
The results of this study indicate that the new generation of MAO inhibitors (M30 and rasagiline) provides increased neuroprotection to cells which are subject to the toxic and damaging effects of increased glucocorticoid secretion and MAO activity, compared to the traditional drug, selegiline. Rasagiline and selegiline are currently being used as pharmaceutical therapies for neurodegenerative diseases and mental disorders such as Parkinson’s Disease (Fernandez and Chen, 2007; Hughes, 2008), depression (Youdim and Weinstock, 2002) and Alzheimer’s Disease (senile dementia) (Sano et al., 1997). The neuroprotective effects of these drugs from glucocorticoid-induced toxicity has only recently been examined demonstrating that rasagiline and selegiline both increase cell proliferation rates after dexamethasone-induced toxicity, with the former providing significantly greater protection (Tazik et al., 2009). However, the neuroprotectivity of M30 after glucocorticoid-induced toxicity had not yet been studied.
This study provides the initial documentation that M30, rasagiline, and selegiline significantly reduce MAO catalytic activity and prevent dexamethasone-induced cellular death in human neuroblastoma cells. Among the three MAO inhibitors, M30 had the highest neuroprotectivity by providing the greatest decrease in cell death rates and MAO A activity, in addition to significantly decreasing MAO B catalytic activity. Rasagiline and selegiline, which are irreversible inhibitors of MAO B, produced the most significant decreases in MAO B catalytic activity. Furthermore, the inhibitory effects of these drugs on apoptotic DNA fragmentation were also examined by TUNEL staining. M30 exhibited the highest prevention of apoptosis (demonstrated by the least amount of TUNEL-positive cells with DNA fragment damage) compared to rasagiline and selegiline.
The neuroprotective effects of these three drugs in the present study show that M30 has a generally greater impact on neuroprotection than the traditional MAO B inhibitor, selegiline. However, M30 displays a similar neuroprotective effect against dexamethasone-induced brain cell apoptosis as compared to the neuroprotection afforded by rasagiline. M30 also exhibits neurorestorative activity in post MPTP and lactacystin models of Parkinson’s disease (Zhu et al., 2007; Gal et al., 2010). These data suggest that, although M30 has a lower inhibitory effect on MAO B than does rasagiline, the similarities in the neuroprotection exhibited by both M30 and rasagiline may be due the additional inhibitory effect of M30 on MAO A. Therefore, the best neuroprotective effect for M30 would be optimal in a situation in which both MAO A and MAO B are elevated, such as in disorders involving stress and depression, in which the glucocorticoid levels are abnormally increased (Duval et al., 2006; Kieran et al., 2010). The synthetic glucocorticoid, dexamethasone is been documented to increase oxidative stress and the expression of MAO A (Ou et al., 2006) and MAO B (Carlo et al., 1996; Tazik et al., 2009) in dopaminergic neurons; this dexamethasone-induced neurodegeneration was prevented by an MAO inhibitor (Tazik et al., 2009) which suggest that the toxic effects of dexamethasone and other glucocorticoids is mediated by MAO (Arguelles et al., 2010). Furthermore, aged rats given dexamethasone showed robust induction of both MAO A and MAO B in the frontal and parietal cortices (Slotkin et al., 1998). More relevantly, MAO A and MAO B activity levels increased considerably in brains of mice exposed to chronic unpredictable stress for 24 days (Mao et al., 2009).
Summarily, M30 has demonstrated its effectiveness in providing neuroprotection by significantly decreasing the levels of enzymatic activity of both MAO A and MAO B in human neuroblastoma cells, in addition to decreasing the amount of fragmented DNA due to ROS production and increasing cell viability in stressful environments. The results of this distinctive study have identified a potential pharmacological agent, M30. This new generation of MAO inhibitor may be considered as a potential drug candidate for treating disorders involving chronic stressful situations which cause increases in both MAO A and MAO B.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by a small grant from Public Health Service Grants P20 RR17701, a NARSAD Young Investigator Award and an Intramural Research Support grant from the University of Mississippi Medical Center. We thank Varinel Inc. for samples of M30. Dr. Youdim is the Scientific Founder of Varinel Inc and has financial interest in M30.
References
Alexander-Kaufman, K., James, G., Sheedy, D., Harper, C., and Matsumoto, I. (2006). Differential protein expression in the prefrontal white matter of human alcoholics: a proteomics study. Mol. Psychiatry 11, 56–65.
Arguelles, S., Herrera, A. J., Carreno-Muller, E., de Pablos, R. M., Villaran, R. F., Espinosa-Oliva, A. M., Machado, A., and Cano, J. (2010). Degeneration of dopaminergic neurons induced by thrombin injection in the substantia nigra of the rat is enhanced by dexamethasone: role of monoamine oxidase enzyme. Neurotoxicology 31, 55–66.
Bar-Am, O., Weinreb, O., Amit, T., and Youdim, M. B. (2009). The neuroprotective mechanism of 1-(R)- aminoindan, the major metabolite of the anti-parkinsonian drug rasagiline. J. Neurochem. 112, 1131–1137.
Carlo, P., Violani, E., Del Rio, M., Olasmaa, M., Santagati, S., Maggi, A., and Picotti, G. B. (1996). Monoamine oxidase B expression is selectively regulated by dexamethasone in cultured rat astrocytes. Brain Res. 711, 175–183.
Carlsson, A., Adolfsson, R., Aquilonius, S. M., Gottfries, C. G., Oreland, L., Svennerholm, L., and Winblad, B. (1980). Biogenic amines in human brain in normal aging, senile dementia, and chronic alcoholism. Adv. Biochem. Psychopharmacol. 23, 295–304.
Chen, J. J., and Ly, A. V. (2006). Rasagiline: a second-generation monoamine oxidase type-B inhibitor for the treatment of Parkinson’s disease. Am. J. Health Syst. Pharm. 63, 915–928.
de Kloet, E. R., Reul, J. M., and Sutanto, W. (1990). Corticosteroids and the brain. J. Steroid Biochem. Mol. Biol. 37, 387–394.
Duval, F., Mokrani, M. C., Monreal-Ortiz, J. A., Fattah, S., Champeval, C., Schulz, P., and Macher, J. P. (2006). Cortisol hypersecretion in unipolar major depression with melancholic and psychotic features: dopaminergic, noradrenergic and thyroid correlates. Psychoneuroendocrinology 31, 876–888.
Fernandez, H. H., and Chen, J. J. (2007). Monamine oxidase inhibitors: current and emerging agents for Parkinson disease. Clin. Neuropharmacol. 30, 150–168.
Gal, S., Abassi, Z. A., and Youdim, M. B. (2009). Limited potentiation of blood pressure in response to oral tyramine by the anti-Parkinson brain selective multifunctional monoamine oxidase-AB inhibitor, M30. Neurotox. Res. 18, 143–150.
Gal, S., Zheng, H., Fridkin, M., and Youdim, M. B. (2005). Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases. In vivo selective brain monoamine oxidase inhibition and prevention of MPTP-induced striatal dopamine depletion. J. Neurochem. 95, 79–88.
Gal, S., Zheng, H., Fridkin, M., and Youdim, M. B. (2010). Restoration of nigrostriatal dopamine neurons in post-MPTP treatment by the novel multifunctional brain-permeable iron chelator-monoamine oxidase inhibitor drug, M30. Neurotox. Res. 17, 15–27.
Haynes, L. E., Barber, D., and Mitchell, I. J. (2004). Chronic antidepressant medication attenuates dexamethasone-induced neuronal death and sublethal neuronal damage in the hippocampus and striatum. Brain Res. 1026, 157–167.
Hughes, B. (2008). New hope for Parkinson’s disease progression delay. Nat. Rev. Drug Discov. 7, 791.
Jahng, J. W., Kim, N. Y., Ryu, V., Yoo, S. B., Kim, B. T., Kang, D. W., and Lee, J. H. (2008). Dexamethasone reduces food intake, weight gain and the hypothalamic 5-HT concentration and increases plasma leptin in rats. Eur. J. Pharmacol. 581, 64–70.
Kieran, N., Ou, X. M., and Iyo, A. H. (2010). Chronic social defeat downregulates the 5-HT1A receptor but not Freud-1 or NUDR in the rat prefrontal cortex. Neurosci. Lett. 469, 380–384.
Kupershmidt, L., Weinreb, O., Amit, T., Mandel, S., Carri, M. T., and Youdim, M. B. (2009). Neuroprotective and neuritogenic activities of novel multimodal iron-chelating drugs in motor-neuron-like NSC-34 cells and transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. 23, 3766–3779.
Lee, A. L., Ogle, W. O., and Sapolsky, R. M. (2002). Stress and depression: possible links to neuron death in the hippocampus. Bipolar Disord. 4, 117–128.
Lu, D., Johnson, C., Johnson, S., Tazik, S., and Ou, X. M. (2008). The neuroprotective effect of antidepressant drug via inhibition of TIEG2-MAO B mediated cell death. Drug Discov. Ther. 2, 289–295.
Lyness, J. M., Yu, Q., Tang, W., Tu, X., and Conwell, Y. (2009). Risks for depression onset in primary care elderly patients: potential targets for preventive interventions. Am. J. Psychiatry 166, 1375–1383.
Malorni, W., Giammarioli, A. M., Matarrese, P., Pietrangeli, P., Agostinelli, E., Ciaccio, A., Grassilli, E., and Mondovi, B. (1998). Protection against apoptosis by monoamine oxidase A inhibitors. FEBS Lett. 426, 155–159.
Manoli, I., Le, H., Alesci, S., McFann, K. K., Su, Y. A., Kino, T., Chrousos, G. P., and Blackman, M. R. (2005). Monoamine oxidase-A is a major target gene for glucocorticoids in human skeletal muscle cells. FASEB J. 19, 1359–1361.
Mao, Q. Q., Ip, S. P., Ko, K. M., Tsai, S. H., Xian, Y. F., and Che, C. T. (2009). Effects of peony glycosides on mice exposed to chronic unpredictable stress: further evidence for antidepressant-like activity. J. Ethnopharmacol. 124, 316–320.
Meyer, J. H., Ginovart, N., Boovariwala, A., Sagrati, S., Hussey, D., Garcia, A., Young, T., Praschak-Rieder, N., Wilson, A. A., and Houle, S. (2006). Elevated monoamine oxidase a levels in the brain: an explanation for the monoamine imbalance of major depression. Arch. Gen. Psychiatry 63, 1209–1216.
Meyer, J. H., Wilson, A. A., Sagrati, S., Miler, L., Rusjan, P., Bloomfield, P. M., Clark, M., Sacher, J., Voineskos, A. N., and Houle, S. (2009). Brain monoamine oxidase A binding in major depressive disorder: relationship to selective serotonin reuptake inhibitor treatment, recovery, and recurrence. Arch. Gen. Psychiatry 66, 1304–1312.
Ou, X. M., Chen, K., and Shih, J. C. (2004). Dual functions of transcription factors, transforming growth factor-beta-inducible early gene (TIEG)2 and Sp3, are mediated by CACCC element and Sp1 sites of human monoamine oxidase (MAO) B gene. J. Biol. Chem. 279, 21021–21028.
Ou, X. M., Chen, K., and Shih, J. C. (2006). Glucocorticoid and androgen activation of monoamine oxidase A is regulated differently by R1 and Sp1. J. Biol. Chem. 281, 21512–21525.
Ou, X. M., Lu, D., Johnson, C., Chen, K., Youdim, M. B., Rajkowska, G., and Shih, J. C. (2009a). Glyceraldehyde-3-phosphate dehydrogenase-monoamine oxidase B-mediated cell death-induced by ethanol is prevented by rasagiline and 1-R-aminoindan. Neurotox. Res. 16, 148–159.
Ou, X. M., Stockmeier, C. A., Meltzer, H. Y., Overholser, J. C., Jurjus, G. J., Dieter, L., Chen, K., Lu, D., Johnson, C., Youdim, M. B., Austin, M. C., Luo, J., Sawa, A., May, W., and Shih, J. C. (2009b). A novel role for glyceraldehyde-3-phosphate dehydrogenase and monoamine oxidase B cascade in ethanol-induced cellular damage. Biol. Psychiatry. 67, 855–863.
Phillips, A. J., Sudbery, I., Ramsdale, M. (2003). Apoptosis induced by environmental stresses and amphotericin B in Candida albicans. Proc. Natl. Acad. Sci. U. S. A. 100, 14327–14332.
Sacher, J., Wilson, A. A., Houle, S., Rusjan, P., Hassan, S., Bloomfield, P. M., Stewart, D. E., and Meyer, J. H. (2010). Elevated brain monoamine oxidase A binding in the early postpartum period. Arch. Gen. Psychiatry 67, 468–474.
Sano, M., Ernesto, C., Thomas, R. G., Klauber, M. R., Schafer, K., Grundman, M., Woodbury, P., Growdon, J., Cotman, C. W., Pfeiffer, E., Schneider, L. S., and Thal, L. J. (1997). A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N. Engl. J. Med. 336, 1216–1222.
Shih, J. C., Chen, K., and Ridd, M. J. (1999). Monoamine oxidase: from genes to behavior. Annu. Rev. Neurosci. 22, 197–217.
Slotkin, T. A., Seidler, F. J., and Ritchie, J. C. (1998). Effects of aging and glucocorticoid treatment on monoamine oxidase subtypes in rat cerebral cortex: therapeutic implications. Brain Res. Bull. 47, 345–348.
Tazik, S., Johnson, S., Lu, D., Johnson, C., Youdim, M. B., Stockmeier, C. A., and Ou, X. M. (2009). Comparative neuroprotective effects of rasagiline and aminoindan with selegiline on dexamethasone-induced brain cell apoptosis. Neurotox. Res. 15, 284–290.
Youdim, M. B. (2006). The path from anti Parkinson drug selegiline and rasagiline to multifunctional neuroprotective anti Alzheimer drugs ladostigil and m30. Curr. Alzheimer Res. 3, 541–550.
Youdim, M. B., and Weinstock, M. (2002). Novel neuroprotective anti-Alzheimer drugs with anti-depressant activity derived from the anti-Parkinson drug, rasagiline. Mech. Ageing Dev. 123, 1081–1086.
Youdim, M. B., Gross, A., and Finberg, J. P. (2001). Rasagiline [N-propargyl-1R( + )-aminoindan], a selective and potent inhibitor of mitochondrial monoamine oxidase B. Br. J. Pharmacol. 132, 500–506.
Youdim, M. B., Bar Am, O., Yogev-Falach, M., Weinreb, O., Maruyama, W., Naoi, M., and Amit, T. (2005). Rasagiline: neurodegeneration, neuroprotection, and mitochondrial permeability transition. J. Neurosci. Res. 79, 172–179.
Yu, S., Patchev, A. V., Wu, Y., Lu, J., Holsboer, F., Zhang, J. Z., Sousa, N., and Almeida, O. F. (2010). Depletion of the neural precursor cell pool by glucocorticoids. Ann. Neurol. 67, 21–30.
Zheng, H., Gal, S., Weiner, L. M., Bar-Am, O., Warshawsky, A., Fridkin, M., and Youdim, M. B. (2005). Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases: in vitro studies on antioxidant activity, prevention of lipid peroxide formation and monoamine oxidase inhibition. J. Neurochem. 95, 68–78.
Keywords: M30, monoamine oxidase inhibitor, rasagiline, selegiline, glucocorticoids, apoptosis, neuroprotection, neuroblastoma
Citation: Johnson S, Tazik S, Lu D, Johnson C, Youdim MBH, Wang J, Rajkowska G and Ou X-M (2010). The new inhibitor of monoamine oxidase, M30, has a neuroprotective effect against dexamethasone-induced brain cell apoptosis. Front. Neurosci. 4:180. doi: 10.3389/fnins.2010.00180
Received: 01 June 2010;
Paper pending published: 07 September 2010;
Accepted: 05 October 2010;
Published online: 02 November 2010.
Edited by:
Nick Andrews, Pfizer, UKReviewed by:
Jason B. Wu, University of Southern California, USACopyright: © 2010 Johnson, Tazik, Lu, Johnson, Youdim, Wang, Rajkowska and Ou. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Xiao-Ming Ou, Division of Neurobiology and Behavioral Research, Department of Psychiatry and Human Behavior (G-109), University of Mississippi Medical Center, 2500 N. State Street, Jackson, MS 39216, USA. e-mail:eG91QHVtYy5lZHU=