



- Yale Child Study Center, Yale University School of Medicine, New Haven, CT, USA
There is increasing appreciation for the neurodevelopmental underpinnings of many psychiatric disorders. Disorders that begin in childhood such as autism, language disorders or mental retardation as well as adult-onset mental disorders may have origins early in neurodevelopment. Neural stem cells (NSCs) can be defined as self-renewing, multipotent cells that are present in both the embryonic and adult brain. Several recent research findings demonstrate that psychiatric illness may begin with abnormal specification, growth, expansion and differentiation of embryonic NSCs. For example, candidate susceptibility genes for schizophrenia, autism and major depression include the signaling molecule Disrupted In Schizophrenia-1 (DISC-1), the homeodomain gene engrailed-2 (EN-2), and several receptor tyrosine kinases, including brain-derived growth factor and fibroblast growth factors, all of which have been shown to play important roles in NSCs or neuronal precursors. We will discuss here stem cell biology, signaling factors that affect these cells, and the potential contribution of these processes to the etiology of neuropsychiatric disorders. Hypotheses about how some of these factors relate to psychiatric disorders will be reviewed.
Introduction
There is a growing appreciation for the neurodevelopmental underpinnings of many psychiatric disorders. While the importance of the growth and structuring of the brain has long been recognized for disorders that begin in childhood such as autism, language disorders or mental retardation, there is consensus building that adult-onset mental disorders also have origins early in neurodevelopment. In order to expand our understanding of these origins, the National Institute of Mental Health (NIMH) is investing in studies of developmental neurobiology, choosing one of its strategies to be the support of “research to improve our basic understanding of the development, structure, and function of neural circuits, with a focus on those most relevant to mental disorders” (NAMHC, 2008). Forwarding this agenda requires a better understanding of the neurobiology of neural stem cells (NSCs) and the factors that regulate them in the developing brain.
Neural stem cells can be defined as self-renewing, multipotent cells that are present in both the embryonic and adult brain. Several recent research findings demonstrate that psychiatric illness may begin with abnormal specification, growth, expansion and differentiation of embryonic NSCs. For example, candidate susceptibility genes for schizophrenia, autism and major depression include those specifying the signaling molecule Disrupted In Schizophrenia-1 (DISC-1), the homeodomain gene engrailed-2 (EN-2), and several receptor tyrosine kinases (RTKs), including brain-derived neurotrophic factor (BDNF) and fibroblast growth factors (FGF), all of which have been shown to play important roles in NSCs or neuronal precursors. Hypotheses about how some of these factors relate to psychiatric disorders are now the focus of much neurodevelopmental work (see several reviews: Arnold et al., 2005; Buckley et al., 2007; Eisch et al., 2008; Thomas and Peterson, 2008; Jaaro-Peled et al., 2009; Vaccarino et al., 2009).
This review will lay out some aspects of the new frontier represented by stem cells for understanding the origins of mental illness. In section I, we will review embryonic central nervous system (CNS) development, focusing on forebrain NSCs and the role of the RTKs, such as FGF receptors (FGFRs), in the earliest developmental processes. RTKs are transmembrane proteins that, upon binding a ligand presented or secreted by nearby cells, phosphorylate intracellular signaling molecules and transduce cell-to-cell signaling in the brain. FGF receptors (FGFRs) are among the earliest RTKs expressed in the developing brain, but also continue to be expressed in the mature brain.
Following this review of NSCs and FGFs in embryonic development, in section II we will describe neurobiological findings in clinical populations that support disrupted embryonic stem cell activity. In section III, adult NSC functioning and connections with mental illness will be reviewed. In section IV, we will discuss how new research with inducible pluripotent stem cells derived from patients with neuropsychiatric disorders may further our understanding of developmental aspect of psychopathology and reveal potential targets for psychiatric treatment.
Embryonic CNS Development
An Introduction to Neurogenic Stem Cells (NSCs)
Perturbations in early development of the CNS may increase an individual’s susceptibility to neuropsychiatric disorders. These perturbations may occur at the earliest stages of development, when the primordium of the brain is a sheet of neuroepithelial stem cells. The functioning of NSCs is tightly regulated by both intrinsic and extrinsic factors (Johnson et al., 2009a). Intrinsic factors that shape the development of NSCs include transcription factors, non-coding RNAs and covalent modifications of chromatin in the nucleus (epigenetic modifications). All of these factors, a description of which is outside the scope of this review, can influence stem cells and their proliferation, or the migration and terminal differentiation of their progeny into defined neural cell types with specific connectivity (Figure 1).
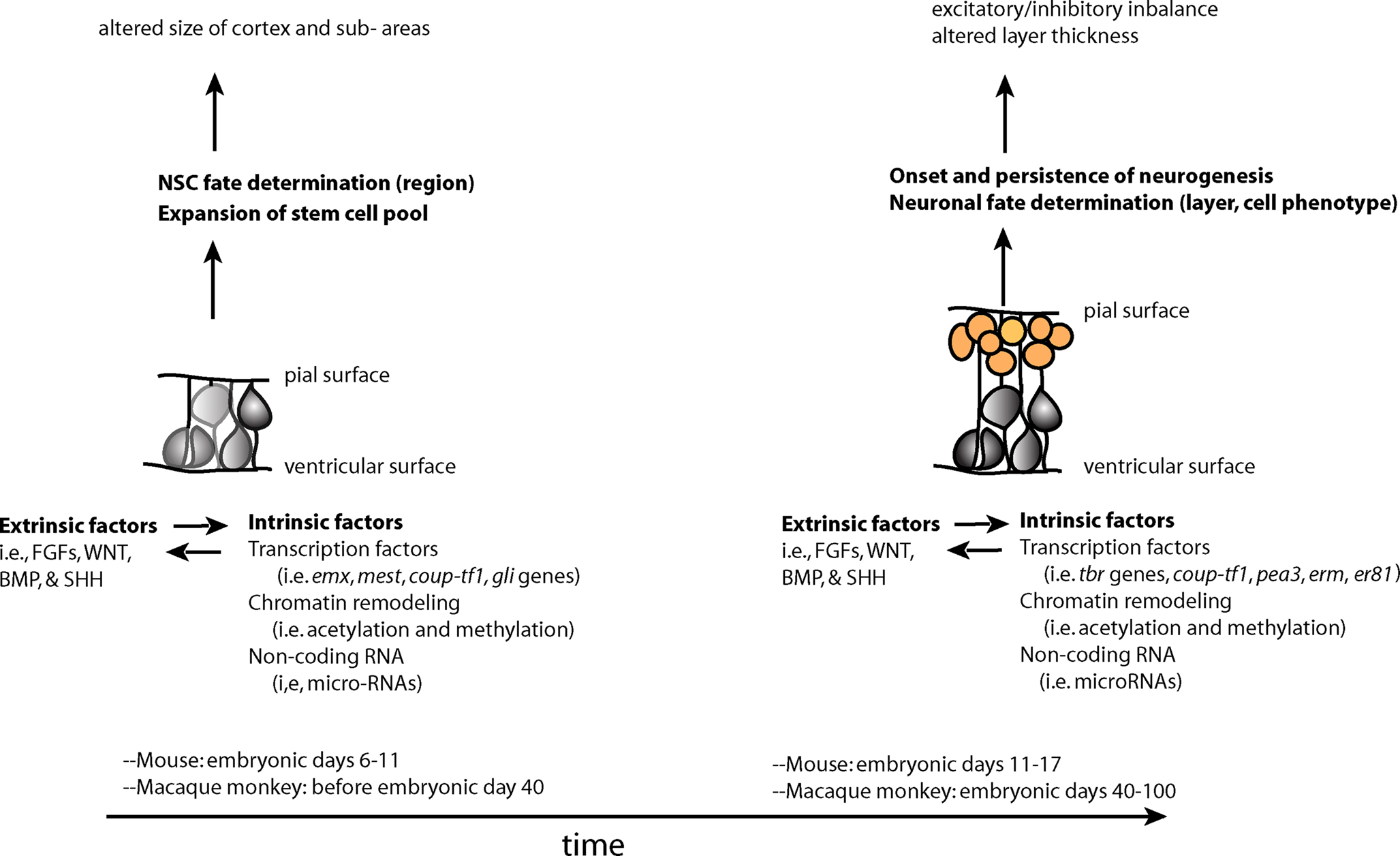
Figure 1. Two overlapping phases of NSCs – expansion of the stem cell pool and the subsequent switch to the neurogenic period–during embryonic development. Changes in stem and progenitor cells (gray cells) and neural precursors (orange cells) during these phases may contribute to later cortical phenotypes found in neuropsychiatric disorders. These changes are driven by both extrinsic and intrinsic factors to stem and progenitor cells, which influence each other in complex manner.
The specific “mix” of intrinsic factors that characterizes distinct neural stem cell function and identity acts in conjunction with a local “mix” of extracellular signaling molecules bathing NSCs. There are four major classes of secreted extracellular signaling molecules that are expressed in the developing brain during embryogenesis and that participate in the patterning of the nervous system – FGFs, WNTs, Sonic Hedgehog (SHH) and Bone Morphogenetic Proteins (BMP). FGFs diffuse from the anterior neural ridge, a region corresponding later in development to the commissural plate, which is the foremost rostral boundary of the telencephalon; WNTs and BMPs emanate from the cortical hem, comprising the medial margin of each hemisphere; BMPs are secreted from the roof plate, the dorsal region in between the cerebral hemispheres; and SHH diffuses from the ventral portion of the neural tube or floor plate (Figure 2). In addition to these long-range signals, the balance between Notch ligands and Notch receptors, which are membrane bound, strongly influences neural stem cell fate (Johnson et al., 2009a). Extracellular signals shape CNS morphogenesis and regulate cell fate by influencing the specific “mix” of intrinsic factors present at specific locations and times in the developing CNS (Figure 1).
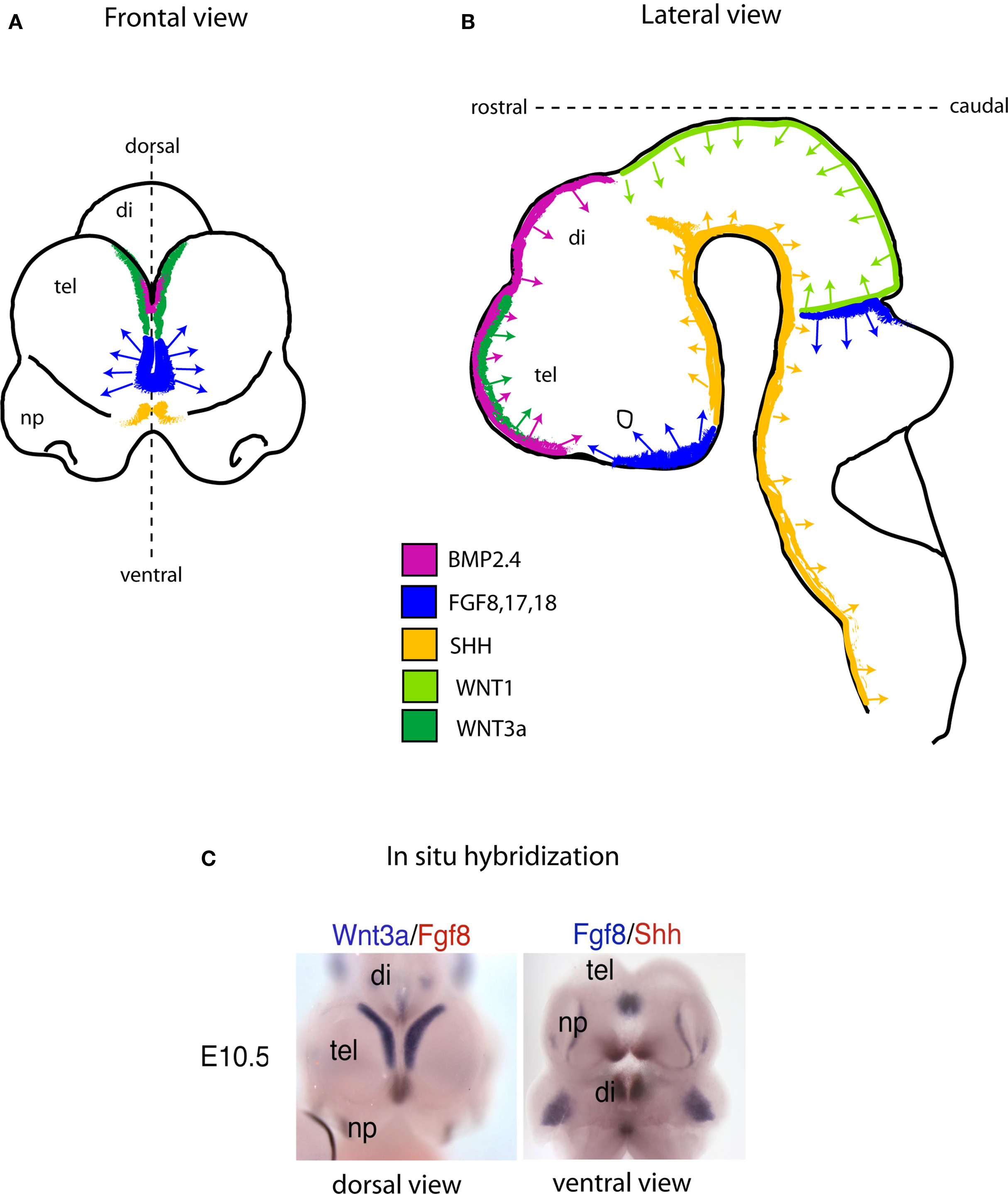
Figure 2. Schematic showing the location of embryonic signaling centers at embryonic day (E) 10.5 in the mouse brain. The four families of secreted signaling molecules are positioned mainly at and near the midline region. In the telencephalon (Frontal view; A) the most prominent source of Fgf activity is positioned between the cortical hem (green; a source of WNT and BMP), and the ventral SHH source (yellow). As in the forebrain, the same or similar signaling molecules direct neural tube patterning and NSC development in the remainder of the CNS (B). (C), Dual probe in situ hybridization data exemplifying the schematic drawn in (A) in a mouse embryo at the age of E10.5. Abbreviations: tel, telencephalon; di, diencephalon; np, nasal processes.
One mechanism by which changes in embryonic NSCs could lead to behavioral symptoms include an imbalance between production of specific types of excitatory and inhibitory neurons, resulting in abnormal levels of activation in cortical circuits (Rubenstein and Merzenich, 2003). A second mechanism may involve impairments in the relative size of cortical areas receiving specific thalamic inputs or sending projections to subcortical stations that play an important role in emotional/behavioral regulation (Figure 1).
WNT, SHH, and BMP decrease in expression with age, indicating that their primary function is in establishing early identities of NSCs. Continued expression of WNT and SHH in the adult NSC niches regulates stem cell proliferation (Lai et al., 2003; Lie et al., 2005; Palma et al., 2005). In contrast, FGFs continue to be widely expressed and play a role not only in adult NSC niches (Zheng et al., 2004) but also in the maturation of the postnatal cerebral cortex.
FGF Ligands, Stem Cell Amplification and Cortical Neurogenesis
Fibroblast growth factor ligands are peptides that act both intracellularly and through secretion into the extracellular space. There are 22 known FGFs which act upon the four membrane bound FGFRs. Amongst the FGF ligands, 13 are known to be expressed in the CNS during embryonic development (Fgf1,2, 3,7,8, 9,10,13,15,16,17,18,22) in specific regions of the neuroepithelium (Figure 2). Three of the receptors, FGFR1, FGFR2 and FGFR3 are present in the embryonic brain. Indeed, FGFRs are among the earliest RTKs expressed in brain development.
Two FGF ligand molecules must bind a receptor dimer in order to cause receptor activation. FGF receptors, akin to other members of the RTK family of proteins, cross-phosphorylate their partner upon ligand binding, triggering the activation of three main intracellular pathways, the Ras/MAP Kinase, PI3 kinase, and PLCγ/Protein Kinase C (Schlessinger, 2000). The cascades eventually impinge upon the transcriptional machinery in the cell nucleus. Although RAS/MAPK and PI3K pathways are known to be important mediators of FGF signaling in the developing CNS, the relative role of each of these signaling pathways and of the other putative nuclear functions of FGF signaling for transcriptional regulation in stem/progenitor cells and biological functions are still unclear.
Concurrently with patterning in the developing dorsal telencephalon, NSCs expand in number. Through a developmental switch not yet fully understood, after the majority of this expansion has occurred, stem cells then begin to generate neuronal precursors in a neurogenic phase that lasts for approximately 6 days in rodents and 10–12 weeks in primates (Caviness et al., 1995; Rakic, 1995) (Figure 1). Cortical excitatory neurons are derived from NSC that line the dorsal telencephalic ventricle. The primary stem cells in this ventricular zone (VZ) are called radial glia because of their expression of glial markers such as GFAP and GLAST, and their cellular morphology. Radial glial cells have an apical end foot attachment at the ventricle, a cell body that is near the ventricle, and a long radial process that is attached at the pial surface (Levitt et al., 1981). Radial glia can undergo self-renewing cell divisions, or asymmetric cell divisions that directly give rise to neurons (Noctor et al., 2001). Another product of radial glial division are committed neurogenic progenitors that migrate to the subventricular zone (SVZ), above the VZ, where they in turn proliferate to give rise to neurons. The committed neuronal progenitors of the SVZ, referred to as intermediate progenitor cells (IPCs) express the transcription factor TBR2 and lack the self-renewal properties of true stem cells (Pontious et al., 2008). However, their proliferation is important for the expansion of cortical layers, as demonstrated by the decrease in cortical surface area and thickness in mice lacking tbr2 (Arnold et al., 2008; Sessa et al., 2008).
Fibroblast growth factor signaling is important for the regulation of neurogenesis in the developing cortex. Studies in vitro originally suggested that the ability of a cortical NSC to stop self-renewing and begin the differentiation process was somehow delayed by increased FGF signaling, resulting in an expanded stem cell pool (Kilpatrick and Bartlett, 1993). The first in vivo demonstration was provided by injection of an FGF ligand, FGF2, in rat embryonic brain ventricles, which resulted in an expanded cortex with increased excitatory neuron production (Vaccarino et al., 1999). Conversely, the deletion of the fgf2 gene resulted in a cortex with reduced numbers of glutamatergic excitatory neurons, particularly in the anterior neocortex (Raballo et al., 2000; Korada et al., 2002). This was not due to a change in the cell cycle or by alterations in cell survival, suggesting that FGF signaling might affect the early amplification of stem cells or their immediate descendants (Raballo et al., 2000). This was confirmed by later work performed on FGF receptor knockout mice (see FGFs and the Developing Dorsal Telencephalon).
Similarly, mice with reduced fgf8 gene expression have decreased proliferation and increased numbers of apoptotic cells in the developing telencephalon (Fukuchi-Shimogori and Grove, 2001; Garel et al., 2003; Storm et al., 2006). However, reducing the gene dosages of fgf15 has opposite effects (Borello et al., 2008) with fgf15 expression in the telencephalon promoting cell differentiation, inhibiting proliferation, and promoting the expression of the coup-tf1 transcription factor, which plays a role in the development of layer four neurons and posterior cortex (Gimeno et al., 2003; Borello et al., 2008). Therefore, the combination of different FGFs and other cell extrinsic signaling proteins expressed in the neurogenic period may regulate the behavior of stem cells and the production of neuroblasts in a precise sequence, resulting in the establishment of a cortex with the correct number of neurons.
FGFs and the Developing Dorsal Telencephalon
An essential feature of secreted morphogens during development is their ability to regulate the neurogenic process in a spatially restricted manner. Two distinct areas of the dorsal telencephalon are of relevance to the pathophysiology of neuropsychiatric disorders, the cerebral cortex and the hippocampus, the first developing primarily prenatally, and the second both prenatally and postnatally. In this section we will focus more on the prenatal role of FGF receptors on cortical development. Later sections will discuss the role of FGF on hippocampal development and relate these phenomena to neuropsychiatric disorders.
FGF8, FGF17 and FGF18 are expressed in the anterior telencephalon (Figure 2), and evidence suggests that this high rostral and low caudal gradient of FGF signaling contributes to prefrontal cortex (PFC) development (Fukuchi-Shimogori and Grove, 2001; Cholfin and Rubenstein, 2007). A given amount of FGFs in the developing cortical field can determine whether stem cells will form specific sub-regions such as somatosensory cortex or PFC (Fukuchi-Shimogori and Grove, 2001). Alterations in the level of FGFs have been shown to lead to abnormal behaviors in animals (Scearce-Levie et al., 2008). FGF8 is negatively regulated by BMP (Crossley et al., 2001; Ohkubo et al., 2002) and in turn, FGF8 restricts WNT3a expression, confining it into the hem region (Shimogori et al., 2004). WNT3a expression in posterior hem regions is important for the formation of the hippocampus (Lee et al., 2000).
If we consider the secreted signaling molecules expressed by NSCs in the cortical primordium, only disruptions in the FGF pathways have thus far resulted in major defects in cortical development (Hebert et al., 2003; Cheng et al., 2006). Although overexpression of a stabilized β-catenin, which is downstream of the WNT pathway, increases cortical size (Chenn and Walsh, 2002), the disruption of the WNT pathway affects the hippocampus rather than the cortex (Galceran et al., 2000). Thus, normal embryonic cortical development primarily depends on FGF signaling, and in order to better understand the mechanisms mediating these roles, several FGF receptor mutant models have been generated.
Mice that transiently overexpress a dominant negative fgfr1 gene that interferes with normal functioning of all receptor types during early embryogenesis have a smaller cortex, particularly in the frontal and temporal regions, and reduced numbers of excitatory neurons (Shin et al., 2004). Furthermore, these mice exhibited hyperactivity. Specific receptor knockouts have subsequently shown the relative contribution of each FGF receptor to cortical development. In order to avoid embryonic lethality of fgfr1 and fgfr2 systemic gene knockouts, fgfr alleles containing loxP site have been recombined in vivo with a variety of Cre lines, including the foxg1 knock-in Cre, nestin-Cre and hgfap-Cre lines. Mice lacking fgfr1 in radial glial progenitors driven by hgfap-Cre (which targets the dorsal telencephalon and hippocampal anlage), exhibited severe reduction of hippocampal volume, almost complete absence of midline telencephalic commissures due to abnormal development of midline glia, and decreased inhibitory interneuron number in the cortex and hippocampus (Ohkubo et al., 2004; Smith et al., 2006; Muller Smith et al., 2008). In contrast, the number of cortical excitatory neurons was not decreased in mice lacking fgfr1 alone (Muller Smith et al., 2008). However, mice lacking fgfr2 alone or in combination with fgfr1, via hgfap-Cre mediated recombination, showed a decrease in cortical excitatory neurons and volume, both of which were more pronounced in the medial prefrontal area of the cortex (Stevens et al., 2010). The mechanism of these abnormalities leading to a loss of cortical excitatory neurons resides in the ability of FGFR2 signaling to induce radial glial stem cells to re-enter the cell cycle, particularly in anterior regions. Therefore, FGFR2 support prefrontal cortical development by promoting the self-renewal or maintenance of cortical stem cells. These data converge with previous work demonstrating that PFC size is reduced by knockout of FGF17, a ligand for FGFR2 (Cholfin and Rubenstein, 2008).
Consistent with the decreased number of excitatory projection neurons in PFC in conditional knockouts for fgfr2 driven by the hgfap-Cre transgene, glutamatergic synapses in the bed nuclei of the stria terminalis (BST), an area that receives projections from the PFC and in turn projects to the hypothalamus, were also reduced in these mice. A decreased number of GABAergic neurons in the BST was also noted in these animals, which is likely to be secondary to decreased glutamate input, as FGFR2 was not targeted in this region (Stevens et al., 2010). These changes may have important consequences for the correct functioning of limbic circuitry.
Mice constitutively lacking FGFR3 through the germline have skeletal defects but have not been reported to have abnormal cortical morphogenesis. (Colvin et al., 1996; Oh et al., 2003). On the other hand, mice with activating mutations of fgfr3 have an increase in the rate of the cell cycle of cortical stem cells in early neurogenesis and an increase in the generation of TBR2+ IPCs at later stages, leading to increased cortical size and cortical cell number (Inglis-Broadgate et al., 2005; Thomson et al., 2007, 2009). The caudal and lateral areas of the cortex were most affected, reflecting the natural gradient of fgfr3 expression. Thus, FGFR2 and FGFR3 both appear to influence the appropriate proliferation of stem cells in different regions of cortex (anterior and posterior, respectively), suggesting that the regulation of signaling by each of these receptors individually and in combination may be critical for the appropriate expansion of different cortical areas. Compound mutation for fgfr1, fgfr2 and fgfr3 in the early anterior neural tube (driven by foxg1-Cre) results in an almost complete agenesis of both dorsal and ventral telencephalic regions (Gutin et al., 2006; Hebert and Fishell, 2008).
The downstream targets that mediate FGF regulated cellular events are still under investigation. Relevant to the early patterning processes in the cortex, FGF8-mediated activation of FGFRs is known to repress genes that specify dorsal and posterior cell fate including coup-tf1, emx2, and wnt8b (Crossley et al., 2001; Garel et al., 2003; Storm et al., 2006). FGF and other signaling factors also induce RAS/MAPK pathways, supporting NSC proliferation and self-renewal, and PI3K/AKT pathways, supporting cell survival. Several transcription factor genes are also activated by FGF signaling in NSCs as well as neuronal progeny, including pea3, erm (etv5), and er81 (etv1) some of which are anteriorly expressed (Hasegawa et al., 2004; Cholfin and Rubenstein, 2008) and may be involved in cell differentiation or fate. The multiplicity of targets demonstrates how master regulatory factors such as FGF may exert multifaceted roles in stem cells and their progenies.
Cortical Interneuron Development
The ganglionic eminences of the ventral telencephalon give rise to the striatum which emerges from the lateral ganglionic eminence (LGE), and to cortical and hippocampal GABA interneurons which are born in the medial and caudal ganglionic eminences (MGE and CGE). Parvalbumin (PV)+ and somatostatin (Sst)+ interneurons arise from the MGE, while calretinin+ interneurons arise largely from the CGE (Wonders et al., 2008). The interneurons from the MGE and CGE travel to the cortex via tangential migration during embryogenesis and progressively integrate into the cortical circuitry. Postnatally they undergo a protracted maturation and develop into their mature interneuron subtypes through processes that are largely unknown.
FGFR1 and FGFR2 appear to regulate early patterning of the ganglionic eminences before neurogenesis (Gutin et al., 2006). We shown that loss of fgfr1 or fgfr2 in the cerebral cortex via hgfap-Cre mediated recombination, which does not target the ganglionic eminences, results in a decrease in PV+ and Sst+ interneurons within the cortex (Smith et al., 2008). Work in our lab suggests that this deficit develops postnatally, and does not arise from defects in the patterning or proliferation of stem cells and progenitors in the ventral telencephalon. FGFR1 may act in the developing postnatal neocortex to support the survival and maturation of GABA interneurons.
Clinical Studies
Biological mechanisms of neuropsychiatric disorders can be studied in clinical populations using multiple lines of inquiry. Structural brain imaging can reveal changes in brain region volume while functional imaging can demonstrate altered functioning of brain regions, both of which may originate in early developmental processes, particularly when found in patients presenting early in the course of illness or young subjects at high risk for psychiatric disorders. More specific mechanisms relevant to stem cell biology may be examined in the post mortem brain as well as genetic analysis of patients. Converging evidence from populations of patients with autism, schizophrenia and affective disorders suggests that stem cell biology is implicated in neuropsychiatric etiology and pathophysiology.
Autism
The brain structure of patients with autism spectrum disorders (ASD) including autism, Asperger’s Disorder (AD) and pervasive development disorder not otherwise specified (PDD-NOS) has been found to be abnormal using several different approaches. It is now widely believed that at least some of the deficits are present very early in life and that abnormal embryonic brain development may be a contributor to later structural deficits.
The head circumference of some patients with autism has long been known to be larger (Davidovitch et al., 1996; Woodhouse et al., 1996; Fombonne et al., 1999; Miles et al., 2000) because of abnormal acceleration in growth in early infancy (Courchesne et al., 2003). While no studies have been published on the neuroanatomy of high-risk individuals before a diagnosis of autism is made, retrospective data have shown that children with macrocephaly and autism do not have increased head circumference at birth (Lainhart et al., 1997; Courchesne et al., 2003; Dementieva et al., 2005; Redcay and Courchesne, 2005) but begin to show larger head measures at about 4 months of age (Gillberg and de Souza, 2002; Courchesne et al., 2005; Redcay and Courchesne, 2005). These findings suggest that autism may be underlain by either problems in early postnatal life and/or processes of embryonic development on which these postnatal events depend. Structural imaging of individuals with autism has shown that differences in brain volume, including both white and gray matter, diminish after the age of 5 years (Hazlett et al., 2005), although some studies have reported increases in gray matter volume in adolescents and adults with ASD (Lotspeich et al., 2004; Palmen et al., 2005; Hazlett et al., 2006), particularly in PFC (Mitchell et al., 2009).
Although there is no clear mechanism accounting for the dysregulation in the trajectory of brain growth in ASD, one hypothesis stipulates that it is the consequence of altered regulation of neural stem cell proliferation or differentiation arising before birth (Vaccarino et al., 2009). Further evidence that early embryonic developmental events are implicated in the pathophysiology of autism comes from post mortem studies demonstrating a fundamental change in cortical structure. Patients with autism were shown to have an increased packing density of mini-columns, which are vertical (radial) assemblies of neurons thought to be anatomically and functionally interconnected (Casanova et al., 2003, 2006).
Several underlying mechanisms could explain macrocephaly and minicolumn pathology, all based on altered embryonic cortical development (Figure 3). The first is an increase in the number of radial units in the embryonic cerebral cortex, which in turn is thought to depend upon an increase in the number of “founder” NSCs in the cortical primordium (Rakic, 1995) (Figure 3). This mechanism is supported by the occurrence of mutations in pten, a gene that regulates embryonic stem cell proliferation (Eng, 2003) in a small number of autistic patients with macrocephaly (Butler et al., 2005). Abnormal expression of this gene in NSCs would likely result in an intrinsic alteration of stem cells. Interestingly, an animal model of pten mutations shows increased brain size and social deficits (Kwon et al., 2006), although this mutation was in differentiated neurons, not intrinsically affecting NSCs. In Fragile X syndrome, which frequently presents with symptoms of autism, fetal NSCs have been shown to differentiate into neurons at greater rates (Castren et al., 2005) and to misexpress multiple genes involved in proliferation and differentiation (Bhattacharyya et al., 2008). Mutant embryonic NSCs isolated from mice lacking the fragile X mental retardation protein (FMRP) due to a deletion in the fmr1 gene differentiate in greater numbers into immature neurons (Castren et al., 2005). These findings are similar to those obtained in Drosophila germline stem cells lacking an ortholog of the fmr1 gene (Yang et al., 2009). Thus, the pten and fmr1 mouse models of ASD support the hypothesis that an intrinsic abnormality in NSC is responsible for features of these disorders. Two members of the TF-II family of transcription factors involved in Williams syndrome, another disorder with abnormal social behavior, have been shown in mice to regulate specific gene targets that may be involved in embryonic stem cell differentiation (Makeyev and Bayarsaihan, 2009).
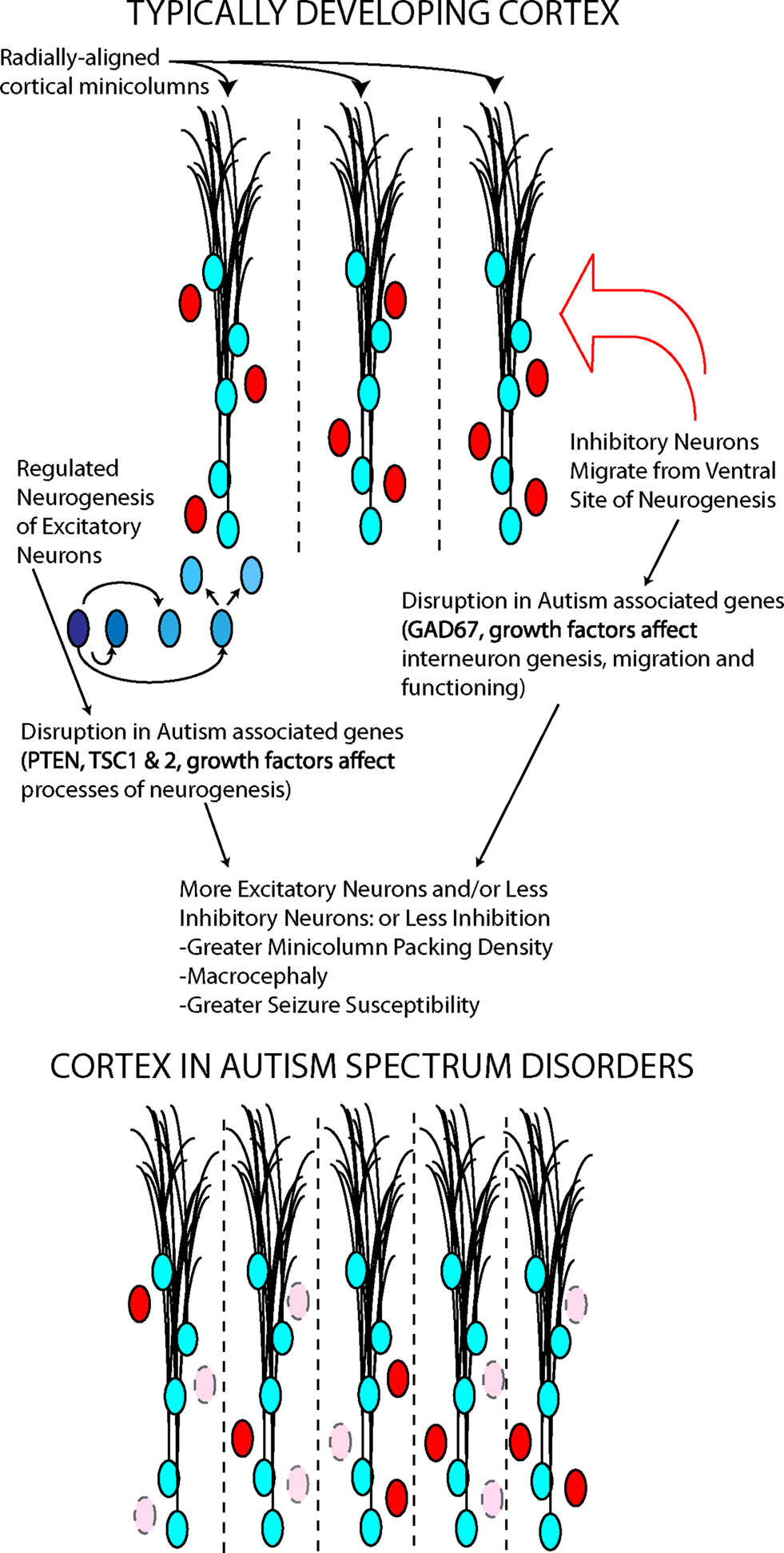
Figure 3. Two neuronal cell types, excitatory (blue) and inhibitory (red), contribute to minicolumn structure and functional balance in cortex. Autism associated genes play roles in the processes underlying the generation and migration of these cortical neurons. The disruption of these genes may determine some cortical abnormalities in autism.
GABA interneurons surround each minicolumn, and some genetic evidence and post mortem data suggest a GABAergic abnormality in ASD (Fatemi et al., 2002). Multiple genes involved in the development and function of the GABAergic system, including dlx5, have been involved in Rett syndrome, a developmental abnormality with autistic features (Horike et al., 2005); furthermore, alterations in the development of GABAergic neuron circuitry have been found in mice lacking the Methyl-CpG binding protein 2 (MeCP2) gene, whose mutations are responsible for Rett syndrome (Medrihan et al., 2008; Zhang et al., 2010). Lastly, a large number of synaptic-related genes have been implicated in small subsets of patients with ASDs (in total, accounting for probably less than 3–4% of the cases) (Buxbaum, 2009; Radyushkin et al., 2009). Thus, it appears that abnormalities in both the early-determined size and scaffolding of the cerebral cortex and later developing synaptic connections may play a role in individual cases of autism.
Schizophrenia
In schizophrenia, retrospective studies have suggested that head circumference is decreased at birth and developmental delays are present in early childhood, both of which implicate prenatal and early postnatal alterations in forebrain development (Cannon et al., 2002). The occurrence of prodromal symptoms in the majority of cases (Hafner et al., 1994), as well as the presence both prior to and after onset of illness of neuropsychological deficits (Crespo-Facorro et al., 2007) also implicates disruption of forebrain development. Neuropsychological dysfunction implicates the PFC in patients with schizophrenia, the cortical region that has expanded most extensively in mammalian evolution and that has been suggested to rely on key evolutionary developments in stem cell function (Martinez-Cerdeno et al., 2006).
Altered hippocampal development has also been implicated in the pathogenesis of schizophrenia (Kobayashi, 2009). Total brain gray matter and hippocampal volumes are reduced at the time of first episode in schizophrenia (Ohnuma et al., 1997; Gur et al., 1999) and at times later in the disease course, volume reductions have been observed in PFC, hippocampus and temporal lobe (Shenton et al., 2001). Longitudinal studies with structural imaging show that both the PFC and the medial temporal lobe become progressively smaller during the period when psychosis develops and that dorsal prefrontal regions experience even more loss as illness progresses, consistent with a neurodegenerative process (Pantelis et al., 2007). Post mortem studies of the cerebral cortex of patients with schizophrenia have shown reductions in neuropil (Selemon et al., 1998), as well as in GABAergic cells expressing PV and reelin (Guidotti et al., 2000; Fatemi et al., 2001; Hashimoto et al., 2008). Post mortem analysis of the hippocampus also shows evidence for an abnormal GABAergic system and suggests that important signaling pathways are altered, including WNT and TGFbeta (Benes et al., 1998, 2007; Todtenkopf and Benes, 1998; Arnold et al., 2005). While these post mortem findings may reflect changes in the adult brain due to disease progression and medication effects, alterations in cortical neurodevelopmental processes could also create vulnerabilities that develop later in life.
Genes that are implicated in stem cell regulation, including neuregulin, disc-1, wnt related genes, bdnf and fgfr1, have been associated with schizophrenia in genetic association and post mortem studies, suggesting NSCs dysregulation in at least some cases. Mouse models lacking fgfr1 embryonically have smaller hippocampi and cortical interneuron deficits similar to those in patients with psychosis (Ohkubo et al., 2004). Similarly, deficient bdnf, neuregulin and disc-1 genes in mice mimic various aspects of the disorder (Ayhan et al., 2009; Brandon et al., 2009; Meyer and Morris, 2009). Conversely, mutations found in some patients with schizophrenia have significant effects on cortical stem cell development. Mice lacking the genes within the 22q11 mutation (velocardiofacial syndrome, a known chromosomal abnormality predisposing to schizophrenia) have abnormal neurogenesis, specifically affecting upper cortical layers (Meechan et al., 2009). As in autism, patients with schizophrenia are likely heterogenous, with pathology in some determined by a component of neuronal functioning that arises postnatally and in others, determined by earlier disruptions of patterning and neurogenesis.
Affective Disorders
Patients with major depression have been found to have smaller hippocampi (Videbech and Ravnkilde, 2004) and smaller anterior cingulate cortical regions (Caetano et al., 2006). Connectivity and functioning of prefrontal and cingulate cortices have also been shown to be abnormal in individuals with major depression (Drevets, 2000; Grimm et al., 2008; Vasic et al., 2009). A recent longitudinal study of people at high familial risk for developing depression show thinning of the cerebral cortex, particularly in right-sided frontal and parietal areas (Peterson et al., 2009). This thinning was present even in children and adolescents prior to the onset of any mood episode, suggesting that structural changes that result from abnormal early cortical pruning during infancy or adolescence may be a predisposing factor in depression. In patients with bipolar disorder with psychotic symptoms, neuroanatomical and neuropsychological findings are similar to those found in patients with schizophrenia (Murray et al., 2004). In non-psychotic bipolar disorder, patients presenting at the first episode of illness have shown decreased volume in prefrontal and temporal cortex (Hirayasu et al., 1999; Strakowski et al., 1999; Kasai et al., 2003). Bipolar disease progression is associated with ventricular enlargement and stronger reductions of the PFC which correlate with impaired functioning during manic episodes (Bearden et al., 2001; Blumberg et al., 2003).
Post mortem studies have demonstrated that prefrontal cortical regions have abnormal densities of pyramidal, GABAergic and glial cells in patients with depression. Medium to large pyramidal neurons appear to be lower in density and smaller in soma size (Rajkowska et al., 1999, 2005; Law and Harrison, 2003) while smaller neurons and those in layers III, V may be increased in density (Chana et al., 2003; Rajkowska, 2003). Glial cell density in patients with depression appears significantly reduced (Ongur et al., 1998; Rajkowska et al., 1999; Cotter et al., 2001, 2002; Uranova et al., 2004). These findings suggest that abnormal prenatal development of stem/progenitor cells that generate the neurons populating different cortical layers and abnormal development of glial elements may account for structural brain abnormalities in depression.
Post mortem work has also examined the levels of signaling factors in patients with depression. In frontal cortical regions, FGF ligand and receptor levels as well as BDNF receptor levels were shown to be altered in patients with depression. Findings generally show reduced expression (Evans et al., 2004; Sibille et al., 2004) but some point to upregulated receptor levels (Tochigi et al., 2008). Altered gene expression in the mature cortex may suggest a role for FGF and other important mediators of neuronal growth in mood disorders in the adult brain. However, they may also reflect a genetic defect that may have contributed to an earlier altered developmental trajectory. Parallel findings in model animal systems (Chen et al., 2008) and patients with mood disorders (Rajkowska et al., 2001; Valentine and Sanacora, 2009) suggest a role for FGF in both embryonic and mature brain functions (Figure 4).
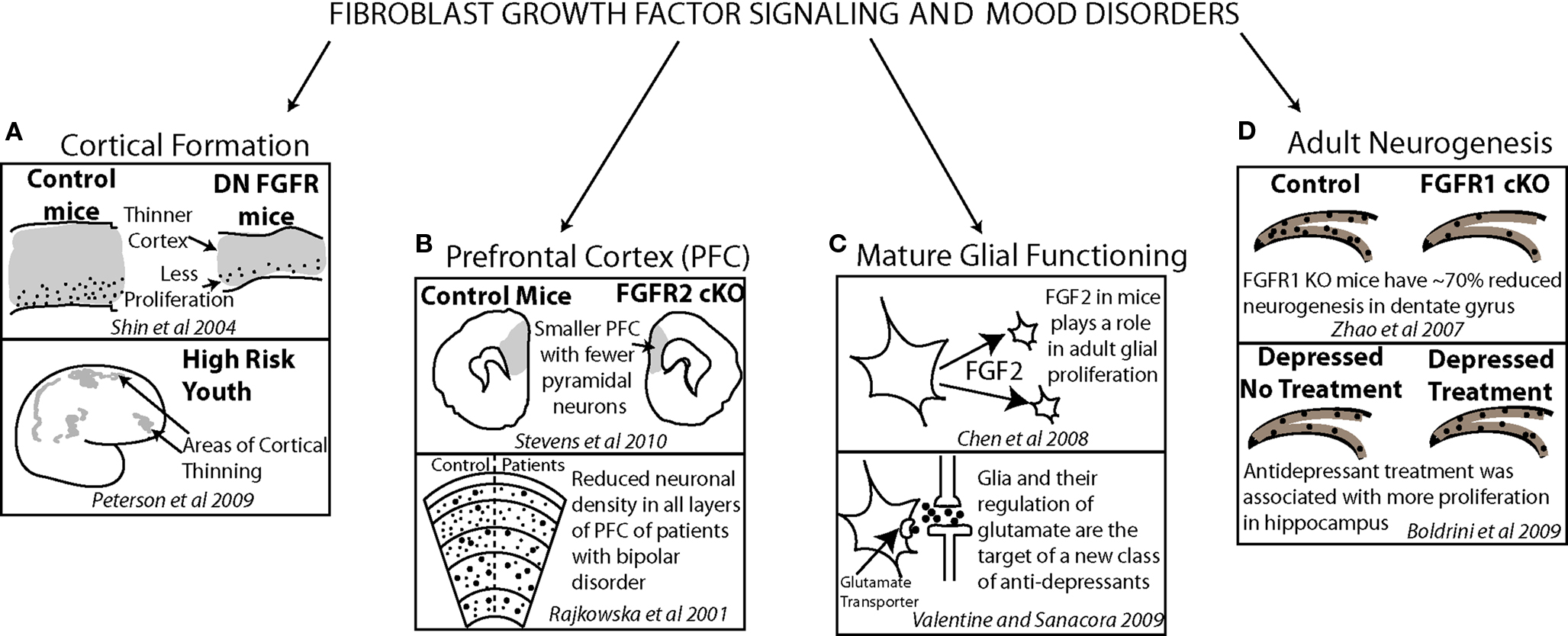
Figure 4. Evidence from model animal systems on fibroblast growth factor, cortical anatomy and functioning (top of each frame) converges with results from clinical studies, cortical anatomy and functioning (bottom of each frame) demonstrating how FGF may have a critical role in affective disorder psychopathology. FGF signaling contributes to normal cortical thickness and youth at risk for depression show early deficits in cortical thickness (A). The density of neurons in prefrontal cortex is reduced in both mice lacking FGF receptors and patients with bipolar disorder (B). A new target for antidepressant treatment is glial functioning in which FGF signaling plays a significant role (C). FGF receptor 1 plays a role in dentate gyrus adult neurogenesis in mice and, in patients with depression, antidepressant treatment has been correlated with increased dentate gyrus proliferation (D).
Adult Neural Stem Cells
Neural Stem Cells in the adult brain are present in regions around the ventricles, specifically in the SVZ, which gives rise to neurons that migrate to the olfactory bulb via the rostral migratory stream. A region in the hippocampal dentate gyrus (DG), the subgranular zone (SGZ), also contains NSCs which generate DG granule cells. Interestingly, over 80% of the interneurons in the olfactory bulb and 10% of granule cells in the DG are replaced by this mechanism in the period of about 1 year, suggesting that a portion of these neurons are under constant turnover (Lemasson et al., 2005; Ninkovic et al., 2007; Imayoshi et al., 2008).
The contribution of adult stem cells to regeneration and neuroplasticity in adult patients with mental illness has received relatively more attention than the role of embryonic NSCs in these disorders. Interestingly, as more is understood about the intrinsic and extrinsic regulation of both embryonic and adult NSCs, common mechanisms are being identified which may underlie disruption in both types of NSCs in disease states (Bordey, 2006).
While the identity of NSCs in the adult brain is not clearly documented, it appears likely that they have many features characteristic of astroglia (Imura et al., 2003; Alvarez-Buylla and Lim, 2004; Liu et al., 2006). Like embryonic radial glia, adult SVZ NSCs have GABAA receptors which limit their proliferation in response to GABA released by neuroblasts, suggesting negative feed-back regulation via non-synaptic GABAergic signaling between neuroblasts and GFAP-expressing NSCs (Liu et al., 2005).
Neuronal stem cells in the adult brain pass through several stages from quiescent multipotent cell (astroglial in nature, slowly dividing) to amplifying neural progenitors which rapidly proliferate and generate immature neurons, which finally mature into granule cells or olfactory interneurons (Ma et al., 2009a). A large proportion of postnatally generated neurons die and only some are incorporated into the existing circuitry. It has been proposed that this pace may be adaptive in order to enhance the sensitivity of the system while the new neurons fully integrate. Environmental and activity-dependant factors may be critical for regulating how these cells pass appropriately through these stages and hence how newly born neurons will participate in normal brain functioning and potentially compensate for pathological states.
Like embryonic NSCs and radial glial cells, adult astroglial NSCs and progenitor cells in the SVZ and SGZ express several RTKs including fgfr1, fgfr2 and fgfr3, bmpr-1a/alk3 as well as egfr, and have active SHH and Notch signaling systems. Adult NSC maintenance and self-renewal depends on secreted FGF, BMP, SHH, and EGF, acting in conjunction with the cell-attached Notch signaling system (Machold et al., 2003; Zheng et al., 2004; Lie et al., 2005; Mira et al., 2010). For example, the number of proliferating cells in the adult SVZ is reduced by 40% in fgf2 KO mice (Zheng et al., 2004). FGF2 increases cell cycle re-entry and BMP promotes quiescence in adult NSCs, and thus, as in the embryo, the balance of these 2 extrinsic factors is likely to be important for proper regulation of adult NSC proliferation and differentiation (Ming and Song, 2005; Mira et al., 2010).
Much evidence has been found linking major neuropsychiatric disorders to altered regulation of adult NSCs and to the molecular systems involved in their maintenance and differentiation. The SGZ of the DG has been the region of the most research focus. DG functioning underlies mood, memory, and reward, which are impaired in schizophrenia, major depression and bipolar disorder. In animal models of depression, schizophrenia, and bipolar disorder, the activity of SGZ stem cells is reduced. For example, chronic stress leads to a depression-like phenotype in rodents, suppresses neurogenesis in the SGZ and reduces FGF2 expression in hippocampus (Warner-Schmidt and Duman, 2006). Findings from post mortem studies have demonstrated that numbers of proliferating cells in the DG are decreased in psychotic disorders (Reif et al., 2006); furthermore, abnormal FGF expression has been documented in the post mortem hippocampus of depressed and schizophrenic patients (Gaughran et al., 2006). In this study, neither antidepressant treatment nor the presence of affective disorders were found to be correlated with altered numbers of hippocampal dividing cells. However, other post mortem work has demonstrated that patients who received antidepressant treatment did show a greater number of neural progenitor and dividing cells in the DG than untreated patients with depression (Boldrini et al., 2009). Furthermore, antidepressant treatment increases hippocampal neurogenesis in both rodents (Malberg et al., 2000) and non-human primates (Perera et al., 2007). Chronic antidepressant administration to rodents has been shown to increase FGF2 expression in astrocytes and neurons within and outside of the hippocampus, suggesting that FGF signaling may be involved in the neurogenic action of antidepressants. In support of this idea, the infusion of FGF2 into the ventricles of adult rats increases cell proliferation and reduces depression-like behavior in rodents (Turner et al., 2008). Also, conditional knockout of fgfr1 in neuronal stem cells by nestin-Cre results in reduced proliferation and neurogenesis in the adult DG (Zhao et al., 2007). New data suggest FGF2 may be specifically involved in the modulation of anxiety/fear behavior in rodents (Perez et al., 2009). However, it is currently controversial whether ongoing hippocampal neurogenesis is required for the therapeutic action of antidepressants. Some have proposed that hippocampal neurogenesis may be limited to mediating the beneficial action of antidepressants on cognition. One problem is the limitations of animal models of depression in which behavior may rely on mechanisms that are peripheral to human disease (Santarelli et al., 2003; Sahay and Hen, 2007; Holick et al., 2008; David et al., 2009). Hence, the specific role of FGF2 on the development and function of systems that subserve affective and cognitive functions in humans is still somewhat unclear.
Gene products implicated with neuropsychiatric illnesses via association studies have been shown to affect hippocampal neurogenesis, most likely by working intrinsically within stem and progenitors cells. The gene with the strongest association to schizophrenia, disc-1, has been shown to regulate the maturation of newborn neurons in the mature brain. When expression of disc-1 is suppressed, neurons mature too quickly (Kim et al., 2009). This may involve a disruption of the balance between those signaling pathways that maintain neuronal stem cells, such as FGF, and those that advance their differentiation into neurons, such as BMP2/4. A role in progenitor cell maintenance and neuronal differentiation within hippocampal NSCs has been also identified for systems that regulate gene expression by methylating or acetylating specific histone proteins in stem and progenitor cells (Wu and Sun, 2009; Yu et al., 2009). A gene that has been associated with some forms of autism, gadd45b, may also play a role in these pathways as it increases progenitors in the DG after electroconvulsive treatment, perhaps by controlling fgf gene expression by DNA methylation (Ma et al., 2009b). In summary, neuropsychiatric disorders may have links to stem cell biology in the mature as well as the developing brain.
The Future of Stem Cells and Interventions: Integrating Human and Animal Knowledge
Work in animal models reveals fundamental processes of brain development that are important to generate and test hypotheses concerning the pathogenesis of human mental disorders. This opens the possibility of manipulating stem cells in clinical populations. Avenues of research are utilizing general knowledge about stem cells to (1) use manipulated stem cells in a rehabilitative capacity in patients and (2) use stem cells from patients to understand pathophysiology and test candidate drugs for reversing abnormal cellular functions. We will review briefly here how knowledge about stem cell biology from both humans and animal models is being applied to develop these new research directions.
Research on the use of stem cells for rehabilitation was originally motivated by the potential for direct development of new neurons from stem cells that might replace lost neurons that are at the center of pathophysiology. For example, in degenerative illnesses such as Parkinson’s disease and Amyotrophic Lateral Sclerosis, specific populations of neurons, dopaminergic cells and spinal motor neurons respectively, are progressively lost. Stem cell research has sought to use either embryonic or other types of stem cells to generate replacement neurons that could be transplanted and incorporated into the CNS of patients (for a Review, see Hynes and Rosenthal, 2000). This work has required knowledge acquired from animal models of the transcription and signaling factors that determine dopaminergic and spinal motor neuron fate and maturation. As researchers have pursued this goal, it has been shown that stem cells in animal model transplant recipients may have an indirect trophic role, i.e., reducing cell death, increasing metabolic functions and protecting remaining neurons from insults (Jung et al., 2004; Rafuse et al., 2005; Yasuhara et al., 2006). These findings may hold promise for future application of stem cell-derived therapies to neuropsychiatric disorders. Disorders such as schizophrenia and depression may have a neurodegenerative component, similar to neurological disorders for which the supportive role of stem cells has been demonstrated. The regions of the brain involved may be different for major affective and psychotic disorders than degenerative motor diseases – e.g., cortex and hippocampus – but these regions may be even better candidates for the application of stem cell support therapy given their nearby neurogenic zones. Extrinsic factors that influence the stem cell niches, like FGF and BMPs, will likely have an important role to play in these potential therapies.
Induced pluripotent stem cells (iPSCs) also hold great promise in their application to neuropsychiatric disorders. These pluripotent stem cells can be derived from skin fibroblasts or blood lymphocytes by introducing a combination of genes that confer stem cell like properties, essentially reprogramming the differentiated cells to behave like embryonically derived stem cells (Takahashi et al., 2007; Yu et al., 2007; Muller et al., 2008). Takahashi and colleagues reported the first generation of human iPSCs in 2007. They achieved this by introducing the oct3/4, sox2, klf4, and c-myc genes into fibroblasts with retroviral vectors. As iPSCs reproduce the essential features of human embryonic stem cells (hESC) they can be coaxed to give rise to neurons. It has already been shown that hESC lines can give rise to mature, physiologically active neurons using different protocols, and many of these protocols have already been successfully applied to iPSC (Johnson et al., 2007; Chambers et al., 2009; Kriks and Studer, 2009; Soldner et al., 2009). Furthermore, by using different combinations of secreted patterning gene products such as FGFs, SHH and WNTs, different subtypes of neurons can be generated such as forebrain specific neurons, midbrain dopaminergic or GABAergic neurons (Yan et al., 2005; Eiraku et al., 2008; Chambers et al., 2009). Here again, decades of animal research in neurodevelopment is paying off, in that the accumulated knowledge in patterning and cell fate specification events in normal development will aid in the development of in vitro differentiation paradigms for generating specific neuron subtypes. More needs to be done and human and animal research must proceed in parallel.
Our inability to examine the direct consequences of gene sequence variations in the living CNS at the transcript and biological levels is a formidable challenge to the investigation of the genetic pathophysiology of psychiatric disorders. The derivation of iPSCs may allow us to investigate basic neurobiological aspects of neuropsychiatric disorders using patient-derived cell lines. For the first time, scientists will be able to use patient-derived cells to investigate developmental properties of the nervous system, such as neural stem cell proliferation, neural differentiation, and synapse formation either in vitro by using cell culture techniques, or in vivo by transplantation into a host animal, such as the developing rodent brain. One of the most promising aspects of iPSCs is the potential to examine the effects of specific human gene variants upon neural differentiation and function, by creating iPSCs from patients with particular genetic mutations. Furthermore, the gene expression profiles of neurons derived from such patients-derived neural stem cell lines can be examined. One potential complication of such studies is the large number of genetic variants present in the human genome. While on the one hand it is useful to begin to catalog such variants, associating them with particular phenotypes will require sophisticated statistical analyses and a large number of samples. Prior to the development of iPSCs, the standard approach for investigating the role of individual genes in brain development was to create a transgenic mouse model with either gene inactivation, gene overexpression, or mutations that mimic the mutations found in human disease. This remains a powerful approach, and indeed, animal work has provided useful experimental frameworks in which the functional impact of specific gene mutations can be tested. However, there are important differences in the development of the rodent brain and the human brain, which are reflected in the diversity of gene splice variants expressed during animal and human brain development (Johnson et al., 2009b). While work in animals continues to represent an essential reference point against which differences in gene expression and function found in humans can be compared, the use of iPSCs will allow us to test hypotheses about the effects of specific gene variants and mutations in developing human cells. Using iPSC models, we should be able to ask questions such as, how mutations in the disc-1 gene, together with a specific constellation of gene mutations/variants present an individual patient, lead to abnormal cell proliferation or neuronal differentiation in human NSCs? We may also be able to identify abnormalities in stem cell development in patients for which no specific genetic mutation has yet been identified. Such research may also provide means by which to test candidate pharmacological agents for their ability to normalize the function of neural cells derived from patients that may have abnormal functions.
The study of NSCs in multiple model systems will continue to inform our understanding of normal brain structure and function. The application of this information to patient populations is slowly shedding light on the pathophysiology of major neuropsychiatric diseases. The advent of new techniques for manipulating human cells may accelerate this process of discovery and may hold real promise for improving the lives of patients with mental illness.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alvarez-Buylla, A., and Lim, D. A. (2004). For the long run: maintaining germinal niches in the adult brain. Neuron 41, 683–686.
Arnold, S. E., Talbot, K., and Hahn, C. G. (2005). Neurodevelopment, neuroplasticity, and new genes for schizophrenia. Prog. Brain Res. 147, 319–345.
Arnold, S. J., Huang, G. J., Cheung, A. F., Era, T., Nishikawa, S., Bikoff, E. K., Molnar, Z., Robertson, E. J., and Groszer, M. (2008). The T-box transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes Dev. 22, 2479–2484.
Ayhan, Y., Sawa, A., Ross, C. A., and Pletnikov, M. V. (2009). Animal models of gene-environment interactions in schizophrenia. Behav. Brain Res. 204, 274–281.
Bearden, C. E., Hoffman, K. M., and Cannon, T. D. (2001). The neuropsychology and neuroanatomy of bipolar affective disorder: a critical review. Bipolar Disord. 3, 106–150; discussion 151–103.
Benes, F. M., Kwok, E. W., Vincent, S. L., and Todtenkopf, M. S. (1998). A reduction of nonpyramidal cells in sector CA2 of schizophrenics and manic depressives. Biol. Psychiatry 44, 88–97.
Benes, F. M., Lim, B., Matzilevich, D., Walsh, J. P., Subburaju, S., and Minns, M. (2007). Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc. Natl. Acad. Sci. U.S.A. 104, 10164–10169.
Bhattacharyya, A., McMillan, E., Wallace, K., Tubon, T. C. Jr., Capowski, E. E., and Svendsen, C. N. (2008). Normal neurogenesis but abnormal gene expression in human fragile X cortical progenitor cells. Stem Cells Dev. 17, 107–117.
Blumberg, H. P., Leung, H. C., Skudlarski, P., Lacadie, C. M., Fredericks, C. A., Harris, B. C., Charney, D. S., Gore, J. C., Krystal, J. H., and Peterson, B. S. (2003). A functional magnetic resonance imaging study of bipolar disorder: state- and trait-related dysfunction in ventral prefrontal cortices. Arch. Gen. Psychiatry 60, 601–609.
Boldrini, M., Underwood, M. D., Hen, R., Rosoklija, G. B., Dwork, A. J., John Mann, J., and Arango, V. (2009). Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 34, 2376–2389.
Borello, U., Cobos, I., Long, J. E., McWhirter, J. R., Murre, C., and Rubenstein, J. L. (2008). FGF15 promotes neurogenesis and opposes FGF8 function during neocortical development. Neural Dev. 3, 17.
Brandon, N. J., Millar, J. K., Korth, C., Sive, H., Singh, K. K., and Sawa, A. (2009). Understanding the role of DISC1 in psychiatric disease and during normal development. J. Neurosci. 29, 12768–12775.
Buckley, P. F., Mahadik, S., Pillai, A., and Terry, A. Jr. (2007). Neurotrophins and schizophrenia. Schizophr. Res. 94, 1–11.
Butler, M. G., Dasouki, M. J., Zhou, X. P., Talebizadeh, Z., Brown, M., Takahashi, T. N., Miles, J. H., Wang, C. H., Stratton, R., Pilarski, R., and Eng, C. (2005). Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 42, 318–321.
Buxbaum, J. D. (2009). Multiple rare variants in the etiology of autism spectrum disorders. Dialogues Clin. Neurosci. 11, 35–43.
Caetano, S. C., Kaur, S., Brambilla, P., Nicoletti, M., Hatch, J. P., Sassi, R. B., Mallinger, A. G., Keshavan, M. S., Kupfer, D. J., Frank, E., and Soares, J. C. (2006). Smaller cingulate volumes in unipolar depressed patients. Biol. Psychiatry 59, 702–706.
Cannon, M., Jones, P. B., and Murray, R. M. (2002). Obstetric complications and schizophrenia: historical and meta-analytic review. Am. J. Psychiatry 159, 1080–1092.
Casanova, M. F., Buxhoeveden, D., and Gomez, J. (2003). Disruption in the inhibitory architecture of the cell minicolumn: implications for autisim. Neuroscientist 9, 496–507.
Casanova, M. F., van Kooten, I. A., Switala, A. E., van Engeland, H., Heinsen, H., Steinbusch, H. W., Hof, P. R., Trippe, J., Stone, J., and Schmitz, C. (2006). Minicolumnar abnormalities in autism. Acta Neuropathol. (Berl.) 112, 287–303.
Castren, M., Tervonen, T., Karkkainen, V., Heinonen, S., Castren, E., Larsson, K., Bakker, C. E., Oostra, B. A., and Akerman, K. (2005). Altered differentiation of neural stem cells in fragile X syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 17834–17839.
Caviness, V. S., Takahashi, T., and Nowakowski, R. S. (1995). Numbers, time and neocortical neurogenesis: a general developmental and evolutionary model. Trends Neurosci. 18, 379–383.
Chambers, S. M., Fasano, C. A., Papapetrou, E. P., Tomishima, M., Sadelain, M., and Studer, L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280.
Chana, G., Landau, S., Beasley, C., Everall, I. P., and Cotter, D. (2003). Two-dimensional assessment of cytoarchitecture in the anterior cingulate cortex in major depressive disorder, bipolar disorder, and schizophrenia: evidence for decreased neuronal somal size and increased neuronal density. Biol. Psychiatry 53, 1086–1098.
Chen, K., Ohkubo, Y., Shin, D., Doetschman, T., Sanford, L. P., Li, H., and Vaccarino, F. M. (2008). Decrease in excitatory neurons, astrocytes and proliferating progenitors in the cerebral cortex of mice lacking exon 3 from the Fgf2 gene. BMC Neurosci. 9, 94.
Cheng, X., Hsu, C. M., Currle, D. S., Hu, J. S., Barkovich, A. J., and Monuki, E. S. (2006). Central roles of the roof plate in telencephalic development and holoprosencephaly. J. Neurosci. 26, 7640–7649.
Chenn, A., and Walsh, C. A. (2002). Regulation of cerebral cortical size by control of cell cycle exit in neural precursors [comment]. Science 297, 365–369.
Cholfin, J. A., and Rubenstein, J. L. (2007). Genetic regulation of prefrontal cortex development and function. Novartis Found. Symp. 288, 165–173; discussion 173–167, 276–181.
Cholfin, J. A., and Rubenstein, J. L. (2008). Frontal cortex subdivision patterning is coordinately regulated by Fgf8, Fgf17, and Emx2. J. Comp. Neurol. 509, 144–155.
Colvin, J. S., Bohne, B. A., Harding, G. W., McEwen, D. G., and Ornitz, D. M. (1996). Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat. Genet. 12, 390–397.
Cotter, D., Mackay, D., Chana, G., Beasley, C., Landau, S., and Everall, I. P. (2002). Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb. Cortex 12, 386–394.
Cotter, D., Mackay, D., Landau, S., Kerwin, R., and Everall, I. (2001). Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch. Gen. Psychiatry 58, 545–553.
Courchesne, E., Carper, R., and Akshoomoff, N. (2003). Evidence of brain overgrowth in the first year of life in autism. JAMA 290, 337–344.
Courchesne, E., Redcay, E., Morgan, J. T., and Kennedy, D. P. (2005). Autism at the beginning: microstructural and growth abnormalities underlying the cognitive and behavioral phenotype of autism. Dev. Psychopathol. 17, 577–597.
Crespo-Facorro, B., Barbadillo, L., Pelayo-Teran, J. M., and Rodriguez-Sanchez, J. M. (2007). Neuropsychological functioning and brain structure in schizophrenia. Int. Rev. Psychiatry 19, 325–336.
Crossley, P. H., Martinez, S., Ohkubo, Y., and Rubenstein, J. L. (2001). Coordinate expression of Fgf8, Otx2, Bmp4, and Shh in the rostral prosencephalon during development of the telencephalic and optic vesicles. Neuroscience 108, 183–206.
David, D. J., Samuels, B. A., Rainer, Q., Wang, J. W., Marsteller, D., Mendez, I., Drew, M., Craig, D. A., Guiard, B. P., Guilloux, J. P., Artymyshyn, R. P., Gardier, A. M., Gerald, C., Antonijevic, I. A., Leonardo, E. D., and Hen, R. (2009). Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 62, 479–493.
Davidovitch, M., Patterson, B., and Gartside, P. (1996). Head circumference measurements in children with autism. J. Child Neurol. 11, 389–393.
Dementieva, Y. A., Vance, D. D., Donnelly, S. L., Elston, L. A., Wolpert, C. M., Ravan, S. A., DeLong, G. R., Abramson, R. K., Wright, H. H., and Cuccaro, M. L. (2005). Accelerated head growth in early development of individuals with autism. Pediatr. Neurol. 32, 102–108.
Drevets, W. C. (2000). Functional anatomical abnormalities in limbic and prefrontal cortical structures in major depression. Prog. Brain Res. 126, 413–431.
Eiraku, M., Watanabe, K., Matsuo-Takasaki, M., Kawada, M., Yonemura, S., Matsumura, M., Wataya, T., Nishiyama, A., Muguruma, K., and Sasai, Y. (2008). Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 3, 519–532.
Eisch, A. J., Cameron, H. A., Encinas, J. M., Meltzer, L. A., Ming, G. L., and Overstreet-Wadiche, L. S. (2008). Adult neurogenesis, mental health, and mental illness: hope or hype? J. Neurosci. 28, 11785–11791.
Evans, S. J., Choudary, P. V., Neal, C. R., Li, J. Z., Vawter, M. P., Tomita, H., Lopez, J. F., Thompson, R. C., Meng, F., Stead, J. D., Walsh, D. M., Myers, R. M., Bunney, W. E., Watson, S. J., Jones, E. G., and Akil, H. (2004). Dysregulation of the fibroblast growth factor system in major depression. Proc. Natl. Acad. Sci. U.S.A. 101, 15506–15511.
Fatemi, S. H., Halt, A. R., Stary, J. M., Kanodia, R., Schulz, S. C., and Realmuto, G. R. (2002). Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol. Psychiatry 52, 805–810.
Fatemi, S. H., Kroll, J. L., and Stary, J. M. (2001). Altered levels of Reelin and its isoforms in schizophrenia and mood disorders. Neuroreport 12, 3209–3215.
Fombonne, E., Roge, B., Claverie, J., Courty, S., and Fremolle, J. (1999). Microcephaly and macrocephaly in autism. J. Autism Dev. Disord. 29, 113–119.
Fukuchi-Shimogori, T., and Grove, E. A. (2001). Neocortex patterning by the secreted signaling molecule FGF8. Science 294, 1071–1074.
Galceran, J., Miyashita-Lin, E. M., Devaney, E., Rubenstein, J. L., and Grosschedl, R. (2000). Hippocampus development and generation of dentate gyrus granule cells is regulated by LEF1. Development 127, 469–482.
Garel, S., Huffman, K. J., and Rubenstein, J. L. (2003). Molecular regionalization of the neocortex is disrupted in Fgf8 hypomorphic mutants. Development 130, 1903–1914.
Gaughran, F., Payne, J., Sedgwick, P. M., Cotter, D., and Berry, M. (2006). Hippocampal FGF-2 and FGFR1 mRNA expression in major depression, schizophrenia and bipolar disorder. Brain Res. Bull. 70, 221–227.
Gillberg, C., and de Souza, L. (2002). Head circumference in autism, Asperger syndrome, and ADHD: a comparative study. Dev. Med. Child Neurol. 44, 296–300.
Gimeno, L., Brulet, P., and Martinez, S. (2003). Study of Fgf15 gene expression in developing mouse brain. Gene Expr. Patterns 3, 473–481.
Grimm, S., Beck, J., Schuepbach, D., Hell, D., Boesiger, P., Bermpohl, F., Niehaus, L., Boeker, H., and Northoff, G. (2008). Imbalance between left and right dorsolateral prefrontal cortex in major depression is linked to negative emotional judgment: an fMRI study in severe major depressive disorder. Biol. Psychiatry 63, 369–376.
Guidotti, A., Auta, J., Davis, J. M., Gerevini, V. D., Dwivedi, Y., Grayson, D. R., Impagnatiello, F., Pandey, G., Pesold, C., Sharma, R., Uzunov, D., and Costa, E. (2000). Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study [In Process Citation]. Arch. Gen. Psychiatry 57, 1061–1069.
Gur, R. E., Turetsky, B. I., Bilker, W. B., and Gur, R. C. (1999). Reduced gray matter volume in schizophrenia. Arch. Gen. Psychiatry 56, 905–911.
Gutin, G., Fernandes, M., Palazzolo, L., Paek, H., Yu, K., Ornitz, D. M., McConnell, S. K., and Hebert, J. M. (2006). FGF signalling generates ventral telencephalic cells independently of SHH. Development 133, 2937–2946.
Hafner, H., Maurer, K., Loffler, W., Fatkenheuer, B., an der Heiden, W., Riecher-Rossler, A., Behrens, S., and Gattaz, W. F. (1994). The epidemiology of early schizophrenia. Influence of age and gender on onset and early course. Br. J. Psychiatry Suppl. 23, 29–38.
Hasegawa, H., Ashigaki, S., Takamatsu, M., Suzuki-Migishima, R., Ohbayashi, N., Itoh, N., Takada, S., and Tanabe, Y. (2004). Laminar patterning in the developing neocortex by temporally coordinated fibroblast growth factor signaling. J. Neurosci. 24, 8711–8719.
Hashimoto, T., Bazmi, H. H., Mirnics, K., Wu, Q., Sampson, A. R., and Lewis, D. A. (2008). Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am. J. Psychiatry 165, 479–489.
Hazlett, H. C., Poe, M., Gerig, G., Smith, R. G., Provenzale, J., Ross, A., Gilmore, J., and Piven, J. (2005). Magnetic resonance imaging and head circumference study of brain size in autism: birth through age 2 years. Arch. Gen. Psychiatry 62, 1366–1376.
Hazlett, H. C., Poe, M. D., Gerig, G., Smith, R. G., and Piven, J. (2006). Cortical gray and white brain tissue volume in adolescents and adults with autism. Biol. Psychiatry 59, 1–6.
Hebert, J. M., Fishell, G. (2008). The genetics of early telencephalon patterning: some assembly required. Nat. Rev. Neurosci. 9, 678–685.
Hebert, J. M., Lin, M., Partanen, J., Rossant, J., and McConnell, S. K. (2003). FGF signaling through FGFR1 is required for olfactory bulb morphogenesis. Development 15, 1101–1111.
Hirayasu, Y., Shenton, M. E., Salisbury, D. F., Kwon, J. S., Wible, C. G., Fischer, I. A., Yurgelun-Todd, D., Zarate, C., Kikinis, R., Jolesz, F. A., and McCarley, R. W. (1999). Subgenual cingulate cortex volume in first-episode psychosis. Am. J. Psychiatry 156, 1091–1093.
Holick, K. A., Lee, D. C., Hen, R., and Dulawa, S. C. (2008). Behavioral effects of chronic fluoxetine in BALB/cJ mice do not require adult hippocampal neurogenesis or the serotonin 1A receptor. Neuropsychopharmacology 33, 406–417.
Horike, S., Cai, S., Miyano, M., Cheng, J. F., and Kohwi-Shigematsu, T. (2005). Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 37, 31–40.
Imayoshi, I., Sakamoto, M., Ohtsuka, T., Takao, K., Miyakawa, T., Yamaguchi, M., Mori, K., Ikeda, T., Itohara, S., and Kageyama, R. (2008). Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat. Neurosci. 11, 1153–1161.
Imura, T., Kornblum, H. I., and Sofroniew, M. V. (2003). The predominant neural stem cell isolated from postnatal and adult forebrain but not early embryonic forebrain expresses GFAP. J. Neurosci. 23, 2824–2832.
Inglis-Broadgate, S. L., Thomson, R. E., Pellicano, F., Tartaglia, M. A., Pontikis, C. C., Cooper, J. D., and Iwata, T. (2005). FGFR3 regulates brain size by controlling progenitor cell proliferation and apoptosis during embryonic development. Dev. Biol. 279, 73–85.
Jaaro-Peled, H., Hayashi-Takagi, A., Seshadri, S., Kamiya, A., Brandon, N. J., and Sawa, A. (2009). Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci. 32, 485–495.
Johnson, M. A., Ables, J. L., and Eisch, A. J. (2009a). Cell-intrinsic signals that regulate adult neurogenesis in vivo: insights from inducible approaches. BMB Rep. 42, 245–259.
Johnson, M. B., Kawasawa, Y. I., Mason, C. E., Krsnik, Z., Coppola, G., Bogdanovic, D., Geschwind, D. H., Mane, S. M., State, M. W., and Sestan, N. (2009b). Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62, 494–509.
Johnson, M. A., Weick, J. P., Pearce, R. A., and Zhang, S. C. (2007). Functional neural development from human embryonic stem cells: accelerated synaptic activity via astrocyte coculture. J. Neurosci. 27, 3069–3077.
Jung, C. G., Hida, H., Nakahira, K., Ikenaka, K., Kim, H. J., and Nishino, H. (2004). Pleiotrophin mRNA is highly expressed in neural stem (progenitor) cells of mouse ventral mesencephalon and the product promotes production of dopaminergic neurons from embryonic stem cell-derived nestin-positive cells. FASEB J. 18, 1237–1239.
Kasai, K., Shenton, M. E., Salisbury, D. F., Onitsuka, T., Toner, S. K., Yurgelun-Todd, D., Kikinis, R., Jolesz, F. A., and McCarley, R. W. (2003). Differences and similarities in insular and temporal pole MRI gray matter volume abnormalities in first-episode schizophrenia and affective psychosis. Arch. Gen. Psychiatry 60, 1069–1077.
Kilpatrick, T. J., and Bartlett, P. F. (1993). Cloning and growth of multipotential neural precursors: requirements for proliferation and differentiation. Neuron 10, 255–265.
Kim, J. Y., Duan, X., Liu, C. Y., Jang, M. H., Guo, J. U., Pow-anpongkul, N., Kang, E., Song, H., and Ming, G. L. (2009). DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron 63, 761–773.
Kobayashi, K. (2009). Targeting the hippocampal mossy fiber synapse for the treatment of psychiatric disorders. Mol. Neurobiol. 39, 24–36.
Korada, S., Zheng, W., Basilico, C., Schwartz, M. L., and Vaccarino, F. M. (2002). Fgf2 is necessary for the growth of glutamate projection neurons in the anterior neocortex. J. Neurosci. 22, 863–875.
Kriks, S., and Studer, L. (2009). Protocols for generating ES cell-derived dopamine neurons. Adv. Exp. Med. Biol. 651, 101–111.
Kwon, C. H., Luikart, B. W., Powell, C. M., Zhou, J., Matheny, S. A., Zhang, W., Li, Y., Baker, S. J., and Parada, L. F. (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388.
Lai, K., Kaspar, B. K., Gage, F. H., and Schaffer, D. V. (2003). Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat. Neurosci. 6, 21–27.
Lainhart, J., Piven, J., Wzorek, M., Landa, R., Santangelo, S. L., Coon, H., and Folstein, S. (1997). Macrocephaly in children and adults with autism. J. Am. Acad. Child Adolesc. Psychiatry 36, 282–290.
Law, A. J., and Harrison, P. J. (2003). The distribution and morphology of prefrontal cortex pyramidal neurons identified using anti-neurofilament antibodies SMI32, N200 and FNP7. Normative data and a comparison in subjects with schizophrenia, bipolar disorder or major depression. J. Psychiatr. Res. 37, 487–499.
Lee, S. M., Tole, S., Grove, E., and McMahon, A. P. (2000). A local Wnt-3a signal is required for development of the mammalian hippocampus. Dev. Suppl. 127, 457–467.
Lemasson, M., Saghatelyan, A., Olivo-Marin, J. C., and Lledo, P. M. (2005). Neonatal and adult neurogenesis provide two distinct populations of newborn neurons to the mouse olfactory bulb. J. Neurosci. 25, 6816–6825.
Levitt, P., Cooper, M. L., and Rakic, P. (1981). Coexistence of neuronal and glial precursor cells in the cerebral ventricular zone of the fetal monkey:an ultrastructural immunoperoxidase analysis. J. Neurosci. 1, 27–39.
Lie, D. C., Colamarino, S. A., Song, H. J., Desire, L., Mira, H., Consiglio, A., Lein, E. S., Jessberger, S., Lansford, H., Dearie, A. R., and Gage, F. H. (2005). Wnt signalling regulates adult hippocampal neurogenesis. Nature 437, 1370–1375.
Liu, X., Bolteus, A. J., Balkin, D. M., Henschel, O., and Bordey, A. (2006). GFAP-expressing cells in the postnatal subventricular zone display a unique glial phenotype intermediate between radial glia and astrocytes. Glia 54, 394–410.
Liu, X., Wang, Q., Haydar, T. F., and Bordey, A. (2005). Nonsynaptic GABA signaling in postnatal subventricular zone controls proliferation of GFAP-expressing progenitors. Nat. Neurosci. 8, 1179–1187.
Lotspeich, L. J., Kwon, H., Schumann, C. M., Fryer, S. L., Goodlin-Jones, B. L., Buonocore, M. H., Lammers, C. R., Amaral, D. G., and Reiss, A. L. (2004). Investigation of neuroanatomical differences between autism and Asperger syndrome. Arch. Gen. Psychiatry 61, 291–298.
Ma, D. K., Bonaguidi, M. A., Ming, G. L., and Song, H. (2009a). Adult neural stem cells in the mammalian central nervous system. Cell Res. 19, 672–682.
Ma, D. K., Jang, M. H., Guo, J. U., Kitabatake, Y., Chang, M. L., Pow-Anpongkul, N., Flavell, R. A., Lu, B., Ming, G. L., and Song, H. (2009b). Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 323, 1074–1077.
Machold, R., Hayashi, S., Rutlin, M., Muzumdar, M. D., Nery, S., Corbin, J. G., Gritli-Linde, A., Dellovade, T., Porter, J. A., Rubin, L. L., Dudek, H., McMahon, A. P., and Fishell, G. (2003). Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron 39, 937–950.
Makeyev, A. V., and Bayarsaihan, D. (2009). New TFII-I family target genes involved in embryonic development. Biochem. Biophys. Res. Commun. 386, 554–558.
Malberg, J. E., Eisch, A. J., Nestler, E. J., and Duman, R. S. (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 20, 9104–9110.
Martinez-Cerdeno, V., Noctor, S. C., and Kriegstein, A. R. (2006). The role of intermediate progenitor cells in the evolutionary expansion of the cerebral cortex. Cereb. Cortex 16(Suppl. 1), i152–i161.
Medrihan, L., Tantalaki, E., Aramuni, G., Sargsyan, V., Dudanova, I., Missler, M., and Zhang, W. (2008). Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J. Neurophysiol. 99, 112–121.
Meechan, D. W., Tucker, E. S., Maynard, T. M., and LaMantia, A. S. (2009). Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome. Proc. Natl. Acad. Sci. U.S.A. 106, 16434–16445.
Meyer, K. D., and Morris, J. A. (2009). Disc1 regulates granule cell migration in the developing hippocampus. Hum. Mol. Genet. 18, 3286–3297.
Miles, J. H., Hadden, L. L., Takahashi, T. N., and Hillman, R. E. (2000). Head circumference is an independent clinical finding associated with autism. Am. J. Med. Genet. 95, 339–350.
Ming, G. L., and Song, H. (2005). Adult neurogenesis in the mammalian central nervous system. Annu. Rev. Neurosci. 28, 223–250.
Mira, H., Andreu, Z., Suh, H., Lie, D. C., Jessberger, S., Consiglio, A., San Emeterio, J., Hotiguela, R., Marques-Torrejon, M. A., Nakashima, K., Colak, D., Gotz, M. Farinas, I., and Gage, F. H. (2010). Signaling through BMPR-IA regulates quiescence and long-term activity of neural stem cells in the adult hippocampus. Cell Sten Cell 7, 78–89.
Mitchell, S. R., Reiss, A. L., Tatusko, D. H., Ikuta, I., Kazmerski, D. B., Botti, J. A., Burnette, C. P., and Kates, W. R. (2009). Neuroanatomic alterations and social and communication deficits in monozygotic twins discordant for autism disorder. Am. J. Psychiatry 166, 917–925.
Muller, F. J., Laurent, L. C., Kostka, D., Ulitsky, I., Williams, R., Lu, C., Park, I. H., Rao, M. S., Shamir, R., Schwartz, P. H., Schmidt, N. O., and Loring, J. F. (2008). Regulatory networks define phenotypic classes of human stem cell lines. Nature 455, 401–405.
Muller Smith, K., Fagel, D. M., Stevens, H. E., Rabenstein, R. L., Maragnoli, M. E., Ohkubo, Y., Picciotto, M. R., Schwartz, M. L., and Vaccarino, F. M. (2008). Deficiency in inhibitory cortical interneurons associates with hyperactivity in fibroblast growth factor receptor 1 mutant mice. Biol. Psychiatry 63, 953–962.
Murray, R. M., Sham, P., Van Os, J., Zanelli, J., Cannon, M., and McDonald, C. (2004). A developmental model for similarities and dissimilarities between schizophrenia and bipolar disorder. Schizophr. Res. 71, 405–416.
National Advisory Mental Health Council (NAMHC). (2008). Transformative Neurodevelopmental Research in Mental Illness. Report of the National Advisory Mental Health Council’s Workgroup on Neurodevelopment. Bethesda, MD: National Institute of Mental Health.
Ninkovic, J., Mori, T., and Gotz, M. (2007). Distinct modes of neuron addition in adult mouse neurogenesis. J. Neurosci. 27, 10906–10911.
Noctor, S. C., Flint, A. C., Weissman, T. A., Dammerman, R. S., and Kriegstein, A. R. (2001). Neurons derived from radial glial cells establish radial units in neocortex. Nature 409, 714–720.
Oh, L. Y., Denninger, A., Colvin, J. S., Vyas, A., Tole, S., Ornitz, D. M., and Bansal, R. (2003). Fibroblast growth factor receptor 3 signaling regulates the onset of oligodendrocyte terminal differentiation. J. Neurosci. 23, 883–894.
Ohkubo, Y., Chiang, C., and Rubenstein, J. L. (2002). Coordinate regulation and synergistic actions of BMP4, SHH and FGF8 in the rostral prosencephalon regulate morphogenesis of the telencephalic and optic vesicles. Neuroscience 111, 1–17.
Ohkubo, Y., Uchida, A. O., Shin, D., Partanen, J., and Vaccarino, F. M. (2004). Fibroblast growth factor receptor 1 is required for the proliferation of hippocampal progenitor cells and for hippocampal growth in mouse. J. Neurosci. 24, 6057–6069.
Ohnuma, T., Kimura, M., Takahashi, T., Iwamoto, N., and Arai, H. (1997). A magnetic resonance imaging study in first-episode disorganized-type patients with schizophrenia. Psychiatry Clin. Neurosci. 51, 9–15.
Ongur, D., Drevets, W. C., and Price, J. L. (1998). Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl. Acad. Sci. U.S.A. 95, 13290–13295.
Palma, V., Lim, D. A., Dahmane, N., Sanchez, P., Brionne, T. C., Herzberg, C. D., Gitton, Y., Carleton, A., Alvarez-Buylla, A., and Ruiz i Altaba, A. (2005). Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development 132, 335–344.
Palmen, S. J., Hulshoff Pol, H. E., Kemner, C., Schnack, H. G., Durston, S., Lahuis, B. E., Kahn, R. S., and Van Engeland, H. (2005). Increased gray-matter volume in medication-naive high-functioning children with autism spectrum disorder. Psychol. Med. 35, 561–570.
Pantelis, C., Velakoulis, D., Wood, S. J., Yucel, M., Yung, A. R., Phillips, L. J., Sun, D. Q., and McGorry, P. D. (2007). Neuroimaging and emerging psychotic disorders: the Melbourne ultra-high risk studies. Int. Rev. Psychiatry 19, 371–381.
Perera, T. D., Coplan, J. D., Lisanby, S. H., Lipira, C. M., Arif, M., Carpio, C., Spitzer, G., Santarelli, L., Scharf, B., Hen, R., Rosoklija, G., Sackeim, H. A., and Dwork, A. J. (2007). Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. J. Neurosci. 27, 4894–4901.
Perez, J. A., Clinton, S. M., Turner, C. A., Watson, S. J., and Akil, H. (2009). A new role for FGF2 as an endogenous inhibitor of anxiety. J. Neurosci. 29, 6379–6387.
Peterson, B. S., Warner, V., Bansal, R., Zhu, H., Hao, X., Liu, J., Durkin, K., Adams, P. B., Wickramaratne, P., and Weissman, M. M. (2009). Cortical thinning in persons at increased familial risk for major depression. Proc. Natl. Acad. Sci. U.S.A. 106, 6273–6278.
Pontious, A., Kowalczyk, T., Englund, C., and Hevner, R. F. (2008). Role of intermediate progenitor cells in cerebral cortex development. Dev. Neurosci. 30, 24–32.
Raballo, R., Rhee, J., Lyn-Cook, R., Leckman, J. F., Schwartz, M. L., and Vaccarino, F. M. (2000). Basic Fibroblast Growth Factor (Fgf2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J. Neurosci. 20, 5012–5023.
Radyushkin, K., Hammerschmidt, K., Boretius, S., Varoqueaux, F., El-Kordi, A., Ronnenberg, A., Winter, D., Frahm, J., Fischer, J., Brose, N., and Ehrenreich, H. (2009). Neuroligin-3-deficient mice: model of a monogenic heritable form of autism with an olfactory deficit. Genes Brain Behav. 8, 416–425.
Rafuse, V. F., Soundararajan, P., Leopold, C., and Robertson, H. A. (2005). Neuroprotective properties of cultured neural progenitor cells are associated with the production of sonic hedgehog. Neuroscience 131, 899–916.
Rajkowska, G. (2003). Depression: what we can learn from postmortem studies. Neuroscientist. 9, 273–284.
Rajkowska, G., Halaris, A., and Selemon, L. D. (2001). Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder. Biol. Psychiatry 49, 741–752.
Rajkowska, G., Miguel-Hidalgo, J. J., Dubey, P., Stockmeier, C. A., and Krishnan, K. R. (2005). Prominent reduction in pyramidal neurons density in the orbitofrontal cortex of elderly depressed patients. Biol. Psychiatry 58, 297–306.
Rajkowska, G., Miguel-Hildago, J. J., Wei, J., Dilley, G., Pittman, S. D., Melzer, H. Y., Overholser, J. C., Roth, B. L., and Stockeimer, C. A. (1999). Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol. Psychiatry 45, 1085–1098.
Rakic, P. (1995). A small step for the cell, a giant leap for mankind: A hypothesis of neocortical expansion during evolution. Trends Neurosci. 18, 383–388.
Redcay, E., and Courchesne, E. (2005). When is the brain enlarged in autism? A meta-analysis of all brain size reports. Biol. Psychiatry 58, 1–9.
Reif, A., Fritzen, S., Finger, M., Strobel, A., Lauer, M., Schmitt, A., and Lesch, K. P. (2006). Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol. Psychiatry 11, 514–522.
Rubenstein, J. L., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267.
Sahay, A., and Hen, R. (2007). Adult hippocampal neurogenesis in depression. Nat. Neurosci. 10, 1110–1115.
Santarelli, L., Saxe, M., Gross, C., Surget, A., Battaglia, F., Dulawa, S., Weisstaub, N., Lee, J., Duman, R., Arancio, O., Belzung, C., and Hen, R. (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301, 805–809.
Scearce-Levie, K., Roberson, E. D., Gerstein, H., Cholfin, J. A., Mandiyan, V. S., Shah, N. M., Rubenstein, J. L., and Mucke, L. (2008). Abnormal social behaviors in mice lacking Fgf17. Genes Brain Behav. 7, 344–354.
Selemon, L. D., Rajkowska, G., and Goldman-Rakic, P. S. (1998). Elevated neuronal density in prefrontal area 46 in brains from schizophrenic patients: application of a three-dimensional, stereologic counting method. J. Comp. Neurol. 392, 402–412.
Sessa, A., Mao, C. A., Hadjantonakis, A. K., Klein, W. H., and Broccoli, V. (2008). Tbr2 directs conversion of radial glia into basal precursors and guides neuronal amplification by indirect neurogenesis in the developing neocortex. Neuron 60, 56–69.
Shenton, M. E., Dickey, C. C., Frumin, M., and McCarley, R. W. (2001). A review of MRI findings in schizophrenia. Schizophr. Res. 49, 1–52.
Shimogori, T., Banuchi, V., Ng, H. Y., Strauss, J. B., and Grove, E. A. (2004). Embryonic signaling centers expressing BMP, WNT and FGF proteins interact to pattern the cerebral cortex. Development 131, 5639–5647.
Shin, D. M., Korada, S., Raballo, R., Shashikant, C. S., Simeone, A., Taylor, J. R., and Vaccarino, F. (2004). Loss of glutamatergic pyramidal neurons in frontal and temporal cortex resulting from attenuation of FGFR1 signaling is associated with spontaneous hyperactivity in mice. J. Neurosci. 24, 2247–2258.
Sibille, E., Arango, V., Galfalvy, H. C., Pavlidis, P., Erraji-Benchekroun, L., Ellis, S. P., and John Mann, J. (2004). Gene expression profiling of depression and suicide in human prefrontal cortex. Neuropsychopharmacology 29, 351–361.
Smith, K. M., Ohkubo, Y., Maragnoli, M. E., Rasin, M. R., Schwartz, M. L., Sestan, N., and Vaccarino, F. M. (2006). Midline radial glia translocation and corpus callosum formation require FGF signaling. Nat. Neurosci. 9, 787–797.
Soldner, F., Hockemeyer, D., Beard, C., Gao, Q., Bell, G. W., Cook, E. G., Hargus, G., Blak, A., Cooper, O., Mitalipova, M., Isacson, O., and Jaenisch, R. (2009). Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136, 964–977.
Stevens, H. E., Smith, K. M., Maragnoli, M. E., Fagel, D., Borok, E., Shanabrough, M., Horvath, T. L., and Vaccarino, F. M. (2010). Fgfr2 is required for the development of the medial prefrontal cortex and its connections with limbic circuits. J. Neurosci. 30, 5590–5602.
Storm, E. E., Garel, S., Borello, U., Hebert, J. M., Martinez, S., McConnell, S. K., Martin, G. R., and Rubenstein, J. L. (2006). Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development 133, 1831–1844.
Strakowski, S. M., DelBello, M. P., Sax, K. W., Zimmerman, M. E., Shear, P. K., Hawkins, J. M., and Larson, E. R. (1999). Brain magnetic resonance imaging of structural abnormalities in bipolar disorder. Arch. Gen. Psychiatry 56, 254–260.
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., and Yamanaka, S. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872.
Thomas, R. M., and Peterson, D. A. (2008). Even neural stem cells get the blues: evidence for a molecular link between modulation of adult neurogenesis and depression. Gene Expr. 14, 183–193.
Thomson, R. E., Kind, P. C., Graham, N. A., Etherson, M. L., Kennedy, J., Fernandes, A. C., Marques, C. S., Hevner, R. F., and Iwata, T. (2009). Fgf receptor 3 activation promotes selective growth and expansion of occipitotemporal cortex. Neural Dev. 4, 4.
Thomson, R. E., Pellicano, F., and Iwata, T. (2007). Fibroblast growth factor receptor 3 kinase domain mutation increases cortical progenitor proliferation via mitogen-activated protein kinase activation. J. Neurochem. 100, 1565–1578.
Tochigi, M., Iwamoto, K., Bundo, M., Sasaki, T., Kato, N., and Kato, T. (2008). Gene expression profiling of major depression and suicide in the prefrontal cortex of postmortem brains. Neurosci. Res. 60, 184–191.
Todtenkopf, M. S., and Benes, F. M. (1998). Distribution of glutamate decarboxylase65 immunoreactive puncta on pyramidal and nonpyramidal neurons in hippocampus of schizophrenic brain. Synapse 29, 323–332.
Turner, C. A., Gula, E. L., Taylor, L. P., Watson, S. J., and Akil, H. (2008). Antidepressant-like effects of intracerebroventricular FGF2 in rats. Brain Res. 1224, 63–68.
Uranova, N. A., Vostrikov, V. M., Orlovskaya, D. D., and Rachmanova, V. I. (2004). Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: a study from the Stanley Neuropathology Consortium. Schizophr. Res. 67, 269–275.
Vaccarino, F. M., Grigorenko, E. L., Smith, K. M., and Stevens, H. E. (2009). Regulation of cerebral cortical size and neuron number by fibroblast growth factors: implications for autism. J. Autism Dev. Disord. 39, 511–520.
Vaccarino, F. M., Schwartz, M. L., Raballo, R., Nilsen, J., Rhee, J., Zhou, M., Doetschman, T., Coffin, J. D., Wyland, J. J., and Hung, Y. T. (1999). Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat. Neurosci. 2, 246–253.
Valentine, G. W., and Sanacora, G. (2009). Targeting glial physiology and glutamate cycling in the treatment of depression. Biochem. Pharmacol. 78, 431–439.
Vasic, N., Walter, H., Sambataro, F., and Wolf, R. C. (2009). Aberrant functional connectivity of dorsolateral prefrontal and cingulate networks in patients with major depression during working memory processing. Psychol. Med. 39, 977–987.
Videbech, P., and Ravnkilde, B. (2004). Hippocampal volume and depression: a meta-analysis of MRI studies. Am. J. Psychiatry 161, 1957–1966.
Warner-Schmidt, J. L., and Duman, R. S. (2006). Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment. Hippocampus 16, 239–249.
Wonders, C. P., Taylor, L., Welagen, J., Mbata, I. C., Xiang, J. Z., and Anderson, S. A. (2008). A spatial bias for the origins of interneuron subgroups within the medial ganglionic eminence. Dev. Biol. 314, 127–136.
Woodhouse, W., Bailey, A., Rutter, M., Bolton, P., Baird, G., and Le Couteur, A. (1996). Head circumference in autism and other pervasive developmental disorders. J. Child. Psychol. Psychiatry 37, 665–671.
Wu, H., and Sun, Y. E. (2009). Reversing DNA methylation: new insights from neuronal activity-induced Gadd45b in adult neurogenesis. Sci. Signal. 2, pe17.
Yan, Y., Yang, D., Zarnowska, E. D., Du, Z., Werbel, B., Valliere, C., Pearce, R. A., Thomson, J. A., and Zhang, S. C. (2005). Directed differentiation of dopaminergic neuronal subtypes from human embryonic stem cells. Stem Cells 23, 781–790.
Yang, Y., Xu, S., Xia, L., Wang, J., Wen, S., Jin, P., and Chen, D. (2009). The bantam microRNA is associated with drosophila fragile X mental retardation protein and regulates the fate of germline stem cells. PLoS Genet. 5, e1000444. doi:10.1371/journal.pgen.1000444.
Yasuhara, T., Matsukawa, N., Hara, K., Yu, G., Xu, L., Maki, M., Kim, S. U., and Borlongan, C. V. (2006). Transplantation of human neural stem cells exerts neuroprotection in a rat model of Parkinson’s disease. J. Neurosci. 26, 12497–12511.
Yu, J., Vodyanik, M. A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J. L., Tian, S., Nie, J., Jonsdottir, G. A., Ruotti, V., Stewart, R., Slukvin, II, and Thomson, J. A. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920.
Yu, I. T., Park, J. Y., Kim, S. H., Lee, J. S., Kim, Y. S., and Son, H. (2009). Valproic acid promotes neuronal differentiation by induction of proneural factors in association with H4 acetylation. Neuropharmacology 56, 473–480.
Zhang, Z. W., Zak, J. D., and Liu, H. (2010). MeCP2 is required for normal development of GABAergic circuits in the thalamus. J. Neurophysiol. 103, 2470–2481.
Zhao, M., Li, D., Shimazu, K., Zhou, Y. X., Lu, B., and Deng, C. X. (2007). Fibroblast growth factor receptor-1 is required for long-term potentiation, memory consolidation, and neurogenesis. Biol. Psychiatry 62, 381–390.
Keywords: Neural stem cell, cerebral cortex, fibroblast growth factor, autism, schizophrenia, depression, bipolar disorder
Citation: Stevens HE, Smith KM, Rash BG and Vaccarino FM (2010) Neural stem cell regulation, fibroblast growth factors, and the developmental origins of neuropsychiatric disorders. Front. Neuroscience 4:59. doi: 10.3389/fnins.2010.00059
Received: 21 March 2010;
Paper pending published: 19 May 2010;
Accepted: 20 July 2010;
Published online: 01 September 2010
Edited by:
Ronald S. Duman, Yale University School of Medicine, USAReviewed by:
Amelia J. Eisch, University of Texas Southwestern Medical Center, USACopyright: © 2010 Stevens, Smith, Rash and Vaccarino. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Flora M. Vaccarino, Child Study Center and Program in Neurodevelopment and Regeneration, Yale University School of Medicine, 230 South Frontage Road, New Haven, CT 06520, USA. e-mail:ZmxvcmEudmFjY2FyaW5vQHlhbGUuZWR1