 Marianna Gabriella Rispoli
Marianna Gabriella Rispoli Maria Vittoria De Angelis
Maria Vittoria De Angelis Donato Melchionda
Donato Melchionda Gabriele Manente
Gabriele Manente
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Neurol. , 07 April 2025
Sec. Headache and Neurogenic Pain
Volume 16 - 2025 | https://doi.org/10.3389/fneur.2025.1569361
Migraine is a common primary and often disabling neurological disorder, whose pathophysiology is still debated. It does not appear to be an isolated event of head pain but the consequence of recurrent disruption of healthy homeostasis in some brain functions. We propose a new theoretical model, focused on the existence of a “high-risk area” for migraine attacks, which can represent a potential target of non-pharmacologic treatment and prevention. We suggest that migraine arises from the combined effects of three primary factors, namely depressive or unstable mood, unrestful sleep and sympathetic-parasympathetic imbalance with parasympathetic prevalence, alongside with their temporal variability, potentially through dysfunction of homeostatic hypothalamic networks in susceptible individuals. Moreover, these three primary factors contribute to a state of low brain energy, that contains the high-risk area and represents the condition in which migraine attacks rise up. Wearable devices, self-administered questionnaires and clinical tools (i.e., polysomnography, pupillary light reflex, plasma catecholamines dosage) may be used to monitor autonomic nervous system function, mood and sleep and demonstrate the existence of the high-risk area. This will be helpful for patients to understand when they are about to enter in the high-risk area, in order to implement strategies to prevent migraine attacks. This approach would provide a significant advantage in terms of prevention and early treatment.
Migraine is a common primary and often disabling neurological disorder, experienced by approximately 10% of the population (1, 2). According to the International Headache Society (IHS), migraine attacks are recurrent and last 4–72 h. Typical characteristics of migraine attacks include unilateral location, pulsating quality, moderate or severe pain intensity, aggravation by habitual physical activity and an association with digestive symptoms (e.g., nausea, vomiting) and/or photo- and phonophobia (3). Migraine attacks can vary in intensity of pain and patterns of associated symptoms, demonstrating a broad variability in the clinical presentation (4). The classic migraine attack consists of three phases: the premonitory phase, the headache phase, and the postdrome (5). The premonitory phase may occur hours to days before an attack and is characterized by fatigue, irritability, concentration impairment, neck stiffness, sensitivity to light and noise, nausea, food cravings, difficulties in speaking and reading, mood disturbances and yawning (6). According to premonitory symptoms, some patients can correctly predict migraine attacks (7). Patients may experience postdromes, which include fatigue, concentration impairment, neck stiffness, and sensibility to light and noise, for days after the headache. Up to one-third of migraine patients suffer migraine aura, i.e., fully reversible focal visual, sensory, language and/or brainstem symptoms that gradually spread over 5 to 60 min and then disappear (8). The aura phase commonly precedes or accompanies headache (9). Even if the description of migraine attacks as a succession of stages over time is useful from a didactic point of view, the phases of an attack can be overlapping and variable (9).
A wide range of acute medications are now available for migraine treatment. However, they are mainly aimed at resolving pain, which is just one component, even if the most bothersome, of migraine attacks. Moreover, many patients have a lot of side effects, and the severity of their headaches does not improve significantly (10).
The pathophysiology of migraine is still debated (Table 1) and has yet to be fully elucidated (11). It was first considered a vascular disorder originating from dilated meningeal vessels, due to characteristic throbbing pain (12, 13). However, there is conflicting evidence about how intracranial and extracranial vessels are ictally and interictally affected in migraine patients (14, 15). Likewise, migraine provocation studies have provided contrasting results on the role of vasodilators in the generation of migraine attacks (16–19). For example, vasoactive intestinal polypeptide (VIP) demonstrated migraine-inducing properties only after prolonged infusion (19). Subsequently, a series of laboratory experiments have suggested that migraine pain may be due to a sterile neurogenically driven inflammation of the dura mater (20). Therefore, the first branch of the trigeminal nerve, connecting peripheral nociceptors in the meninges to their terminations in the brainstem, was hypothesized as responsible for migraine attacks in susceptible individuals (2), according to the trigemino-vascular theory (21). Indeed, in experimental models, a mechanical, electrical or chemical activation of afferent C-fibers meningeal nociceptors in vivo leads to the release of vasoactive proinflammatory peptides, such as calcitonin gene-related peptide (CGRP) and substance P, which produce a vasodilation of meningeal blood vessels, plasma extravasation, and local activation of dural mast cells, with consequent neurogenic inflammation (21–23). CGRP emerged as a crucial mediator of migraine attacks, and an important therapeutic target. The infusion of CGRP triggers delayed migraine attacks in susceptible individuals (24). However, contradictory results have been reported regarding interictal CGRP levels: some studies have identified elevated CGRP levels in the peripheral blood of both episodic (25, 26) and chronic migraine patients (27) compared to healthy controls, while other studies have found no differences in serum CGRP levels between migraine patients and healthy individuals (28, 29). These discrepancies may partly be attributed to methodological factors (30). Since some migraine patients do not respond to treatments that either target CGRP or its receptor, other substrates have been explored for their potential implication in migraine pathogenesis: among them, other neuropeptides (i.e., pituitary adenylate cyclase-activating polypeptide, VIP, amylin, and adrenomedullin), nitric oxide, and phosphodiesterase-3 and -5 appeared able to induce migraine-like attacks through vasodilation of intracranial arteries in human provocation studies (31).
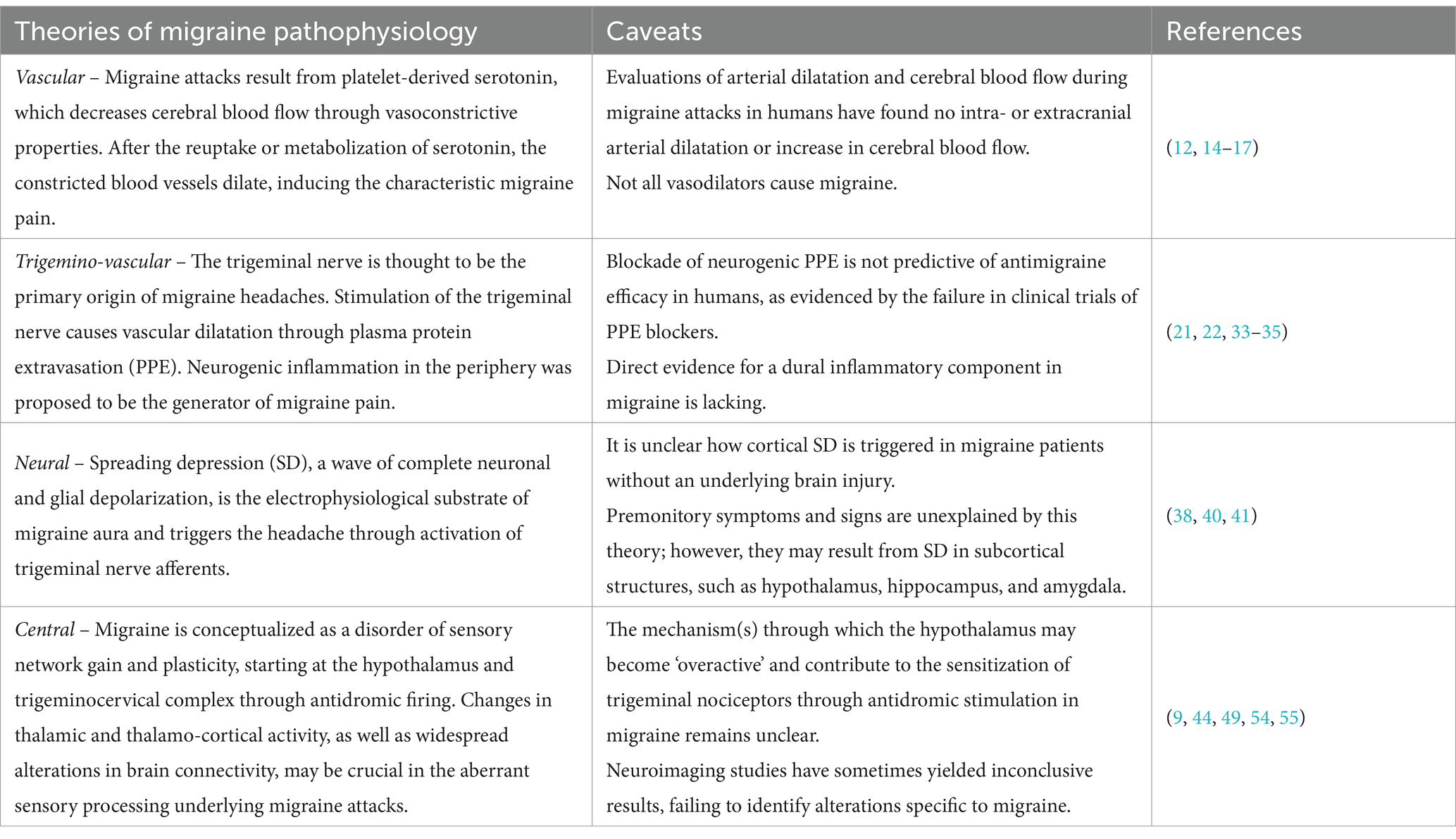
Table 1. Theories about migraine pathophysiology over time.
The classical view suggests that nociceptive signaling involves peripheral generation and orthodromic propagation of the spikes to the brain centers (32). Although trigeminal activation with subsequent neurogenic inflammation continues to be discussed (33), there is no direct evidence supporting a peripheral inflammatory component in migraine. Therefore, the possibility of the functionally opposite, antidromic signal conduction in the meningeal system was previously considered (34, 35). There is bidirectional nociceptive traffic in meningeal afferents implicated in the generation of migraine pain: beyond orthodromic propagation, a spontaneous antidromic activity of the trigeminal nerve in meninges, essentially originating from the trigeminal ganglion, has been demonstrated (36). Similarly to orthodromic activation, antidromic signal conduction leads to the release of CGRP from C-fibers, dilation of meningeal vessels and degranulation of local mast cells (34), activating Aδ-fibers and, in turn, trigeminal ganglions, the trigeminocervical complex, and axonal projections ascending to the midbrain, thalamic and hypothalamic nuclei. Thus, the focus was set on the cortical wave of spreading depolarization, which causes the aura phase and releases molecules affecting neurons, glial cells, and blood vessels (37). These mediators may diffuse to the overlying leptomeninges, activate the trigemino-vascular system, and lead to the typical head pain of migraine attacks (38). Infections like COVID-19 have been suggested as possible modifiers of brain bioelectrical activity through still unknown mechanisms: preliminary observations suggest that they may be responsible for cortical spreading depression (CSD) and subsequent appearance of visual aura by increasing neuronal activity, especially in the occipital lobe (39). Other possible internal and external factors of CSD include hormonal changes in women, changes of day-night rhythm, strong sensory stimuli, hunger, stress or intense physical activity (40). Although CSD has been demonstrated to cause migraine aura and headache in the animal model, the evidence of a causal link between these entities is still lacking in humans (41). Moreover, most migraine attacks do not include the aura phase and migraine aura can occur without headache, indicating that aura is neither necessary nor sufficient for headache onset. Therefore, CSD may be absent or “silent” during most migraine attacks. “Silent” CSD may result from the depolarization of a non-eloquent cortex, or produce symptoms not typically classified as aura (42). Spreading depression in brain regions different from the cortex or disruption of normal thalamocortical oscillations by CSD have been suggested as an alternative model to canonical CSD (43).
Nowadays, migraine is conceptualized as a disorder of sensory network gain and plasticity, starting at the hypothalamus and trigeminocervical complex (2, 44). Since migraine may display a diurnal periodicity, be triggered by altered homeostasis and include yawning, food cravings and fatigue among prodromal symptoms, the hypothalamus has been investigated as a potential generator of migraine attacks (38, 45). According to PET and functional MRI studies of triggered and spontaneous migraine attacks, migraine patients show stronger functional connections between the hypothalamus and brain areas regulating sympathetic and parasympathetic functions in the hours preceding a headache attack (46, 47), explaining some of the hypothalamic-mediated autonomic symptoms accompanying migraine, such as facial flushing, lacrimation and nasal congestion (48). The mechanism(s) through which the hypothalamus may become ‘overactive’ and contribute to the sensitization of trigeminal nociceptors in migraine remains unclear (49). More complex midbrain/brainstem networks might contribute to the generation of migraine attacks. Physiological changes like stress, sleep deprivation and hypoglycaemia may lead to synaptic and network hyperactivity in brainstem-cortical and thalamo-cortical networks of susceptible individuals, altering cortical excitability via glutamate increase (23, 50–53). Indeed, electrophysiological studies have shown changes in the function of thalamo-cortical circuits during the premonitory phase (50). In addition, structural and functional imaging studies have revealed differences in thalamic and thalamo-cortical activity in migraine patients versus controls, both during migraine attacks and the interictal phase (52, 53). Therefore, changes in thalamic and thalamo-cortical activity may be crucial in the aberrant sensory processing underlying migraine attacks (9). Furthermore, resting-state functional MRI studies during interictal phase revealed widespread alterations in brain connectivity of migraine patients, consistent with changes in the function of multiple overlapping sensory and pain-processing circuits involving the cortex, thalamus, hypothalamus, brainstem, amygdala and cerebellum (9). However, neuroimaging studies have sometimes yielded inconclusive results (54), failing to identify alterations specific to migraine (55), highlighting the need for further research on the topic.
Two key aspects of migraine management should be carefully considered: (1) early intervention is more effective; (2) there is a significant need for alternative, non-pharmacologic approaches for both acute and preventive treatments (56). A recent observational study suggested that incident-morning migraine attacks may result from poorer sleep quality and decreased energy during the prior day. On the contrary, increased energy and greater average stress may lead to headache onset later in the day. Despite different patterns of predictors related to morning and later-day incident headaches, circadian rhythms seem to play a major role in the manifestations of headache (57). Since the most common triggers of migraine are stress, fasting, atmospheric changes, sleep-related factors, hormonal and mood fluctuations (58, 59), non-pharmacologic treatment strategies (i.e., setting routines, relaxation therapies, aerobic exercise, hydration, avoidance of fasting and stressful situations) have proven effective in reducing migraine burden or days (60). Recently, situational prevention—treating patients during the interictal phase before symptoms develop, in situations of increased risk for migraine attacks—is becoming more widespread, as the small molecule CGRP antagonists (gepants) are indicated for both the acute and preventive treatment of migraine (61). However, this approach exposes asymptomatic patients to medication and potentially to unnecessary risks. Moreover, there is no clear definition of periods of increased risk to date (62). Lastly, treatment strategies will only be effective if they focus on correcting the complex pathophysiological alterations underlying the disease before its onset.
On this matter, we propose a new theoretical model focused on the existence of a “high-risk area” for migraine attacks onset, which can represent a potential target of non-pharmacologic treatment and prevention. We suggest that migraine arises from the combined effects of three primary factors, namely depressive or unstable mood, unrestful sleep and sympathetic-parasympathetic imbalance with parasympathetic prevalence, alongside with their temporal variability, potentially through dysfunction of homeostatic hypothalamic networks in susceptible individuals. Moreover, these three primary factors contribute to a state of low brain energy, that contains the high-risk area and represents the condition in which migraine attacks rise up (Figure 1).
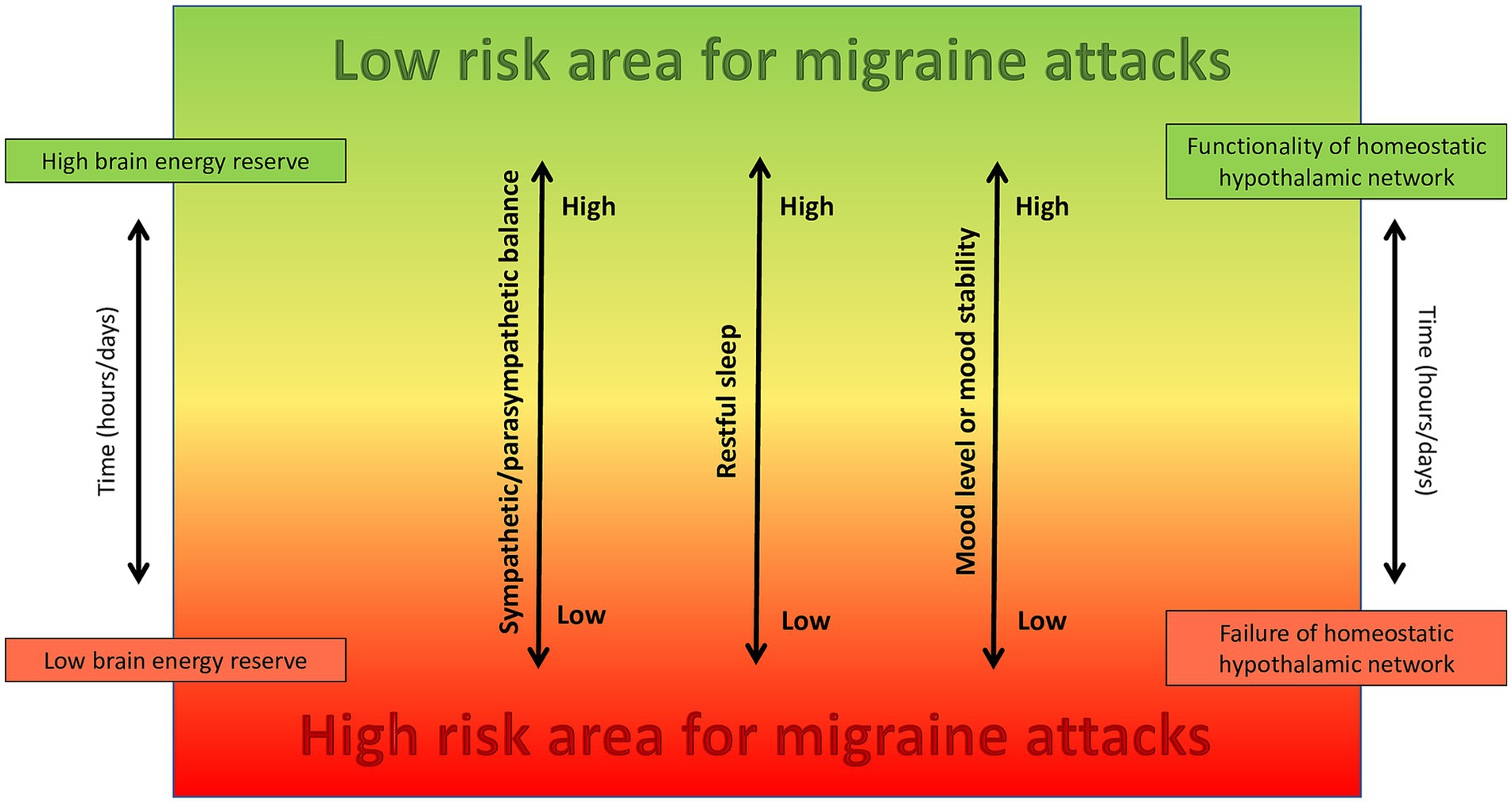
Figure 1. The combined effect of sympathetic/parasympathetic balance, restful sleep, and mood tone and stability, which are all regulated by the hypothalamus and associated neural networks, play a crucial role in determining brain energy levels and the likelihood of reaching the high-risk area for developing migraine attacks in susceptible individuals.
It was suggested that migraine happens when the brain workload exceeds its energy reserve (63, 64). Indeed, the connectivity between hypothalamus and limbic areas, processing internal and external information, gradually increases toward migraine attacks (65). At some point, the hypothalamus loses its control over limbic structures, and this leads to a sensory overload of the deep brain regions (65). We hypothesize that this sensory overload could produce a spreading depression-like phenomenon, which involves non-cortical brain structures and activates migraine pain through antidromic pathways. This phenomenon may also be influenced by the activation state of some brainstem nuclei that modulate trigeminal nociceptive inputs (22, 45). For example, according to a diffusion tensor imaging study, the brainstem displays microstructural alterations throughout the migraine cycle. In particular, immediately before a migraine attack, mean diffusivity (MD) decreased in the spinal trigeminal nucleus, dorsomedial/dorsolateral pons, and midbrain periaqueductal gray matter/nucleus cuneiform. These nuclei influence the activity of the trigeminocervical complex and play a role in pain transmission. MD then increased again immediately following the migraine attack (66), which may reflect an increase in endogenous analgesic ability.
According to biochemical and functional imaging studies, a hypothalamic and brainstem dysfunction may also underlie sleep disorders (67) and depression (68, 69). Beyond controlling alertness and awareness, the concentration of monoamines in synaptic gaps is reduced in the depressive state (70). Serotonin, which mediates several types of human behavior including sleep, mood (depression and anxiety), appetite, sexual function, and pain, may be playing a cross-cutting function (71). Similarly, the hypothalamic–pituitary–adrenal axis, which is required for appropriate sleep regulation, is also involved in depression (69).
With regard to the autonomic nervous system (ANS), multiple afferents (i.e., lateral hypothalamus, stria terminalis, periaqueductal gray, paraventricular hypothalamic nucleus and piriform cortex) carry parasympathetic signals to the superior salivatory nucleus, which can in turn activate postganglionic parasympathetic neurons in the sphenopalatine ganglion, meningeal nociceptors and the trigemino-vascular pathway (38). An increased parasympathetic tone due to altered physiological or emotional homeostasis may activate these nociceptive pathways (38, 72). Therefore, migraine is more than an isolated event of head pain and should be seen as a bio-behavioral response to restore a disturbed brain homeostasis (73) in genetically predisposed individuals (74). We believe the ANS plays a major role in migraine pathophysiology, by modulating the internal homeostasis and using an existing defensive neuronal pathway, the trigeminovascular system, to alert the individual through pain when there is an imbalance. Regarding this aspect, some patients experience more migraine attacks on weekends (75, 76), possibly due to a sudden shift in the balance between sympathetic and parasympathetic tone and their ability to adapt to a more relaxed state.
The glymphatic system, a network of perivascular channels that helps clear waste products and toxic solutes from the brain, primarily during sleep (77), may play a key role in the pathogenesis of migraine. Even though the exact mechanisms underlying this relationship remains to be fully elucidated, it should be noted that migraine patients present a poor sleep endophenotype (i.e., poor sleep quality, shorter self-reported sleep duration, poorer sleep health score, a modestly lower percentage of rapid eye movement sleep) (78, 79). Poor sleep can produce an impairment in the glymphatic flow which, in turn, leads to the accumulation of neuroexcitatory and pro-inflammatory chemicals involved in the development of migraine (80, 81). Beyond sleep deprivation, preclinical studies have reported that body posture, stress, and adrenergic tone may also modulate the activity of the glymphatic system (82, 83).
In our theoretical model, migraine results from the combined effect of unrestful sleep, sympathetic/parasympathetic imbalance with parasympathetic prevalence and an unstable or depressive mood, all of which contribute to a state of low mental energy, potentially through the exhaustion of hypothalamic homeostatic networks. If these biological factors are strictly monitored, their prompt recognition and correction may avoid migraine progression into the painful phase. Probably, the faster the correction, the lower the probability of developing migraine attacks.
In order to demonstrate this theoretical model, wearable devices may be used to monitor heart rate variability, which provides information on ANS and the balance between sympathetic and parasympathetic activity (84). Similarly, they may be useful in monitoring sleep quality and quantity (85), coupled with self-administered questionnaires, like the Pittsburgh Sleep Quality Index (86), which has already been used to assess poor sleep quality in migraine patients (59, 79). Patients should also undergo repeated polysomnographies for comprehensive assessment of sleep and evaluation of sleep disorders (87, 88). This diagnostic tool has already been used to evaluate the possible association between migraine, bruxism, sleep apnoea and other sleep disorders (88). Monitoring of pupillary light reflex and plasma catecholamines at rest and during calibrated challenges may be used as proxies of ANS balance (89). Self-administered questionnaires, for example the Beck’s Depression Inventory II (90), or the Patient Health Questionnaire-9 (91), could be employed to evaluate mood disorders.
Regular monitoring of ANS function, mood, and sleep would allow migraine patients to identify when they are nearing or already inside the high-risk area for migraine attacks.
Our theoretical model, based on repeated clinical and instrumental observations, remains at the moment a pure hypothesis, which requires validation through ongoing evidence that will be the subject of upcoming research. In the near future, the first step is to confirm the existence of the high-risk area, the second step is to identify non-pharmacological strategies in order to avoid this condition, or eventually, recuperate a well state of being, through reversing the underlying pathophysiological dysfunction. Thus, preventing migraine attacks, and enhancing the understanding of their complex pathophysiology.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
MGR: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. MVDA: Data curation, Supervision, Writing – original draft, Writing – review & editing. DM: Methodology, Supervision, Writing – original draft, Writing – review & editing. GM: Conceptualization, Data curation, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Stovner, L, Hagen, K, Jensen, R, Katsarava, Z, Lipton, R, Scher, A, et al. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia. (2007) 27:193–210. doi: 10.1111/j.1468-2982.2007.01288.x
2. Rapoport, AM, and Edvinsson, L. Some aspects on the pathophysiology of migraine and a review of device therapies for migraine and cluster headache. Neurol Sci. (2019) 40:75–80. doi: 10.1007/s10072-019-03828-0
3. (IHS) HCCotIHS. The international classification of headache disorders. Cephalalgia. (2013) 33:629–808. doi: 10.1177/0333102413485658
4. Lipton, RB, and Silberstein, SD. Episodic and chronic migraine headache: breaking down barriers to optimal treatment and prevention. Headache. (2015) 55:103–22. doi: 10.1111/head.12505_2
5. Fraser, CL, Hepschke, JL, Jenkins, B, and Prasad, S. Migraine Aura: pathophysiology, mimics, and treatment options. Semin Neurol. (2019) 39:739–48. doi: 10.1055/s-0039-1700525
6. Goadsby, PJ, Holland, PR, Martins-Oliveira, M, Hoffmann, J, Schankin, C, and Akerman, S. Pathophysiology of Migraine: a disorder of sensory processing. Physiol Rev. (2017) 97:553–622. doi: 10.1152/physrev.00034.2015
7. Giffin, NJ, Ruggiero, L, Lipton, RB, Silberstein, SD, Tvedskov, JF, Olesen, J, et al. Premonitory symptoms in migraine: an electronic diary study. Neurology. (2003) 60:935–40. doi: 10.1212/01.WNL.0000052998.58526.A9
8. Lucas, C. Migraine with aura. Rev Neurol (Paris). (2021) 177:779–84. doi: 10.1016/j.neurol.2021.07.010
9. Charles, A. The pathophysiology of migraine: implications for clinical management. Lancet Neurol. (2018) 17:174–82. doi: 10.1016/S1474-4422(17)30435-0
10. Ducros, A, de Gaalon, S, Roos, C, Donnet, A, Giraud, P, Guégan-Massardier, E, et al. Revised guidelines of the French headache society for the diagnosis and management of migraine in adults. Part 2: pharmacological treatment. Rev Neurol (Paris). (2021) 177:734–52. doi: 10.1016/j.neurol.2021.07.006
11. Raggi, A, Leonardi, M, Arruda, M, Caponnetto, V, Castaldo, M, Coppola, G, et al. Hallmarks of primary headache: part 1- migraine. J Headache Pain. (2024) 25:189. doi: 10.1186/s10194-024-01889-x
12. Graham, JR, and Wolff, HG. Mechanism of migraine headache and action of ergotamine tartrate. Arch Neurol Psychiatr. (1938) 39:737–63. doi: 10.1001/archneurpsyc.1938.02270040093005
13. Ziegeler, C, Mehnert, J, Asmussen, K, and May, A. Central effects of erenumab in migraine patients: an event-related functional imaging study. Neurology. (2020) 95:e2794–802. doi: 10.1212/WNL.0000000000010740
14. Asghar, MS, Hansen, AE, Amin, FM, van der Geest, RJ, Koning, P, Larsson, HB, et al. Evidence for a vascular factor in migraine. Ann Neurol. (2011) 69:635–45. doi: 10.1002/ana.22292
15. Amin, FM, Asghar, MS, Hougaard, A, Hansen, AE, Larsen, VA, de Koning, PJ, et al. Magnetic resonance angiography of intracranial and extracranial arteries in patients with spontaneous migraine without aura: a cross-sectional study. Lancet Neurol. (2013) 12:454–61. doi: 10.1016/S1474-4422(13)70067-X
16. Rahmann, A, Wienecke, T, Hansen, JM, Fahrenkrug, J, Olesen, J, and Ashina, M. Vasoactive intestinal peptide causes marked cephalic vasodilation, but does not induce migraine. Cephalalgia. (2008) 28:226–36. doi: 10.1111/j.1468-2982.2007.01497.x
17. Petersen, KA, Birk, S, Kitamura, K, and Olesen, J. Effect of adrenomedullin on the cerebral circulation: relevance to primary headache disorders. Cephalalgia. (2009) 29:23–30. doi: 10.1111/j.1468-2982.2008.01695.x
18. Ghanizada, H, Al-Karagholi, MA, Arngrim, N, Mørch-Rasmussen, M, Walker, CS, Hay, DL, et al. Effect of Adrenomedullin on Migraine-like attacks in patients with Migraine: a randomized crossover study. Neurology. (2021) 96:e2488–99. doi: 10.1212/WNL.0000000000011930
19. Pellesi, L, Al-Karagholi, MA, Chaudhry, BA, Lopez, CL, Snellman, J, Hannibal, J, et al. Two-hour infusion of vasoactive intestinal polypeptide induces delayed headache and extracranial vasodilation in healthy volunteers. Cephalalgia. (2020) 40:1212–23. doi: 10.1177/0333102420937655
20. Moskowitz, MA, and Cutrer, FM. SUMATRIPTAN: a receptor-targeted treatment for migraine. Annu Rev Med. (1993) 44:145–54. doi: 10.1146/annurev.me.44.020193.001045
22. Pietrobon, D, and Moskowitz, MA. Pathophysiology of migraine. Annu Rev Physiol. (2013) 75:365–91. doi: 10.1146/annurev-physiol-030212-183717
23. Harriott, AM, Takizawa, T, Chung, DY, and Chen, SP. Spreading depression as a preclinical model of migraine. J Headache Pain. (2019) 20:45. doi: 10.1186/s10194-019-1001-4
24. Ashina, M, Hansen, JM, and Olesen, J. Pearls and pitfalls in human pharmacological models of migraine: 30 years' experience. Cephalalgia. (2013) 33:540–53. doi: 10.1177/0333102412475234
25. Ashina, M, Bendtsen, L, Jensen, R, Schifter, S, and Olesen, J. Evidence for increased plasma levels of calcitonin gene-related peptide in migraine outside of attacks. Pain. (2000) 86:133–8. doi: 10.1016/S0304-3959(00)00232-3
26. Fusayasu, E, Kowa, H, Takeshima, T, Nakaso, K, and Nakashima, K. Increased plasma substance P and CGRP levels, and high ACE activity in migraineurs during headache-free periods. Pain. (2007) 128:209–14. doi: 10.1016/j.pain.2006.09.017
27. Jang, MU, Park, JW, Kho, HS, Chung, SC, and Chung, JW. Plasma and saliva levels of nerve growth factor and neuropeptides in chronic migraine patients. Oral Dis. (2011) 17:187–93. doi: 10.1111/j.1601-0825.2010.01717.x
28. Tvedskov, JF, Lipka, K, Ashina, M, Iversen, HK, Schifter, S, and Olesen, J. No increase of calcitonin gene-related peptide in jugular blood during migraine. Ann Neurol. (2005) 58:561–8. doi: 10.1002/ana.20605
29. Lee, MJ, Lee, SY, Cho, S, Kang, ES, and Chung, CS. Feasibility of serum CGRP measurement as a biomarker of chronic migraine: a critical reappraisal. J Headache Pain. (2018) 19:53. doi: 10.1186/s10194-018-0883-x
30. Gárate, G, Pascual, J, Pascual-Mato, M, Madera, J, Martín, MM, and González-Quintanilla, V. Untangling the mess of CGRP levels as a migraine biomarker: an in-depth literature review and analysis of our experimental experience. J Headache Pain. (2024) 25:69. doi: 10.1186/s10194-024-01769-4
31. Al-Hassany, L, Boucherie, DM, Creeney, H, van Drie, RWA, Farham, F, Favaretto, S, et al. Future targets for migraine treatment beyond CGRP. J Headache Pain. (2023) 24:76. doi: 10.1186/s10194-023-01567-4
32. Basbaum, AI, and Woolf, CJ. Pain. Curr Biol. (1999) 9:R429–31. doi: 10.1016/S0960-9822(99)80273-5
33. Ramachandran, R. Neurogenic inflammation and its role in migraine. Semin Immunopathol. (2018) 40:301–14. doi: 10.1007/s00281-018-0676-y
34. Geppetti, P, Rossi, E, Chiarugi, A, and Benemei, S. Antidromic vasodilatation and the migraine mechanism. J Headache Pain. (2012) 13:103–11. doi: 10.1007/s10194-011-0408-3
35. Dimitriadou, V, Buzzi, MG, Moskowitz, MA, and Theoharides, TC. Trigeminal sensory fiber stimulation induces morphological changes reflecting secretion in rat dura mater mast cells. Neuroscience. (1991) 44:97–112. doi: 10.1016/0306-4522(91)90253-K
36. Gafurov, O, Koroleva, K, and Giniatullin, R. Antidromic spike propagation and dissimilar expression of P2X, 5-HT, and TRPV1 channels in peripheral vs. Central Sensory Axons in Meninges Front Cell Neurosci. (2020) 14:623134. doi: 10.3389/fncel.2020.623134
38. Dodick, DW. A phase-by-phase review of Migraine pathophysiology. Headache. (2018) 58:4–16. doi: 10.1111/head.13300
39. Waliszewska-Prosół, M, and Budrewicz, S. The unusual course of a migraine attack during COVID-19 infection - case studies of three patients. J Infect Public Health. (2021) 14:903–5. doi: 10.1016/j.jiph.2021.04.013
40. Diener, HC. What triggers migraine aura? Cephalalgia. (2016) 36:501–2. doi: 10.1177/0333102415607177
41. Charles, A. The Migraine Aura. Continuum (Minneap Minn). Headache. (2018) 24:1009–22. doi: 10.1212/CON.0000000000000627
42. Charles, A, and Hansen, JM. Migraine aura: new ideas about cause, classification, and clinical significance. Curr Opin Neurol. (2015) 28:255–60. doi: 10.1097/WCO.0000000000000193
43. Bolay, H, Vuralli, D, and Goadsby, PJ. Aura and Head pain: relationship and gaps in the translational models. J Headache Pain. (2019) 20:94. doi: 10.1186/s10194-019-1042-8
44. Brennan, KC, and Pietrobon, D. A systems neuroscience approach to Migraine. Neuron. (2018) 97:1004–21. doi: 10.1016/j.neuron.2018.01.029
45. Burstein, R, Noseda, R, and Borsook, D. Migraine: multiple processes, complex pathophysiology. J Neurosci. (2015) 35:6619–29. doi: 10.1523/JNEUROSCI.0373-15.2015
46. Maniyar, FH, Sprenger, T, Monteith, T, Schankin, C, and Goadsby, PJ. Brain activations in the premonitory phase of nitroglycerin-triggered migraine attacks. Brain. (2014) 137:232–41. doi: 10.1093/brain/awt320
47. Schulte, LH, and May, A. Functional neuroimaging in Migraine: chances and challenges. Headache. (2016) 56:1474–81. doi: 10.1111/head.12944
48. Moulton, EA, Becerra, L, Johnson, A, Burstein, R, and Borsook, D. Altered hypothalamic functional connectivity with autonomic circuits and the locus coeruleus in migraine. PLoS One. (2014) 9:e95508. doi: 10.1371/journal.pone.0095508
49. Puledda, F, Silva, EM, Suwanlaong, K, and Goadsby, PJ. Migraine: from pathophysiology to treatment. J Neurol. (2023) 270:3654–66. doi: 10.1007/s00415-023-11706-1
50. de Tommaso, M, Ambrosini, A, Brighina, F, Coppola, G, Perrotta, A, Pierelli, F, et al. Altered processing of sensory stimuli in patients with migraine. Nat Rev Neurol. (2014) 10:144–55. doi: 10.1038/nrneurol.2014.14
51. Coppola, G, Di Renzo, A, Tinelli, E, Di Lorenzo, C, Di Lorenzo, G, Parisi, V, et al. Thalamo-cortical network activity during spontaneous migraine attacks. Neurology. (2016) 87:2154–60. doi: 10.1212/WNL.0000000000003327
52. Coppola, G, Di Renzo, A, Tinelli, E, Lepre, C, Di Lorenzo, C, Di Lorenzo, G, et al. Thalamo-cortical network activity between migraine attacks: insights from MRI-based microstructural and functional resting-state network correlation analysis. J Headache Pain. (2016) 17:100. doi: 10.1186/s10194-016-0693-y
53. Russo, A, Silvestro, M, Tessitore, A, and Tedeschi, G. Recent insights in Migraine with Aura: a narrative review of advanced neuroimaging. Headache. (2019) 59:637–49. doi: 10.1111/head.13512
54. Sheng, L, Zhao, P, Ma, H, Yuan, C, Zhong, J, Dai, Z, et al. A lack of consistent brain grey matter alterations in migraine. Brain. (2020) 143:e45. doi: 10.1093/brain/awaa123
55. Chen, ZH, Cui, YL, Sun, JT, Li, YT, Zhang, C, Zhang, YM, et al. The brain structure and function abnormalities of migraineurs: a systematic review and neuroimaging meta-analysis. Front Neurol. (2022) 13:1022793. doi: 10.3389/fneur.2022.1022793
56. Puledda, F, and Shields, K. Non-pharmacological approaches for Migraine. Neurotherapeutics. (2018) 15:336–45. doi: 10.1007/s13311-018-0623-6
57. Lateef, TM, Dey, D, Leroux, A, Cui, L, Xiao, M, Zipunnikov, V, et al. Association between electronic diary-rated sleep, mood, energy, and stress with incident headache in a community-based sample. Neurology. (2024) 102:e208102. doi: 10.1212/WNL.0000000000208102
58. Kelman, L. The triggers or precipitants of the acute migraine attack. Cephalalgia. (2007) 27:394–402. doi: 10.1111/j.1468-2982.2007.01303.x
59. Duan, S, Ren, Z, Xia, H, Wang, Z, Zheng, T, Li, G, et al. Associations between anxiety, depression with migraine, and migraine-related burdens. Front Neurol. (2023) 14:1090878. doi: 10.3389/fneur.2023.1090878
60. Aguilar-Shea, AL, Membrilla Md, JA, and Diaz-de-Teran, J. Migraine review for general practice. Aten Primaria. (2022) 54:102208. doi: 10.1016/j.aprim.2021.102208
61. Lipton, RB, Ailani, J, Mullin, K, Pavlovic, JM, Tepper, SJ, Dodick, DW, et al. Situational prevention: pharmacotherapy during periods of increased risk for migraine attacks. Headache. (2024) 64:859–64. doi: 10.1111/head.14775
62. Pellesi, L, and Martelletti, P. Situational prevention in migraine: are we doing the right thing? J Headache Pain. (2024) 25:137. doi: 10.1186/s10194-024-01841-z
63. Pensato, U, Cevoli, S, Pierangeli, G, and Cortelli, P. The evolutionary meaning of migraine. Cephalalgia. (2023) 43:3331024231209303. doi: 10.1177/03331024231209303
64. Gross, EC, Lisicki, M, Fischer, D, Sándor, PS, and Schoenen, J. The metabolic face of migraine - from pathophysiology to treatment. Nat Rev Neurol. (2019) 15:627–43. doi: 10.1038/s41582-019-0255-4
65. Stankewitz, A, Keidel, L, Rehm, M, Irving, S, Kaczmarz, S, Preibisch, C, et al. Migraine attacks as a result of hypothalamic loss of control. Neuroimage Clin. (2021) 32:102784. doi: 10.1016/j.nicl.2021.102784
66. Marciszewski, KK, Meylakh, N, Di Pietro, F, Macefield, VG, Macey, PM, and Henderson, LA. Fluctuating regional brainstem diffusion imaging measures of microstructure across the Migraine cycle. eNeuro. (2019) 6:ENEURO.0005–19.2019. doi: 10.1523/ENEURO.0005-19.2019
67. Tiseo, C, Vacca, A, Felbush, A, Filimonova, T, Gai, A, Glazyrina, T, et al. Migraine and sleep disorders: a systematic review. J Headache Pain. (2020) 21:126. doi: 10.1186/s10194-020-01192-5
68. Cui, L, Li, S, Wang, S, Wu, X, Liu, Y, Yu, W, et al. Major depressive disorder: hypothesis, mechanism, prevention and treatment. Signal Transduct Target Ther. (2024) 9:30. doi: 10.1038/s41392-024-01738-y
69. Murphy, MJ, and Peterson, MJ. Sleep disturbances in depression. Sleep Med Clin. (2015) 10:17–23. doi: 10.1016/j.jsmc.2014.11.009
70. Hirschfeld, RM. History and evolution of the monoamine hypothesis of depression. J Clin Psychiatry. (2000) 61:4–6.
71. Vgontzas, A, and Pavlović, JM. Sleep disorders and Migraine: review of literature and potential pathophysiology mechanisms. Headache. (2018) 58:1030–9. doi: 10.1111/head.13358
72. Maleki, N, Becerra, L, and Borsook, D. Migraine: maladaptive brain responses to stress. Headache. (2012) 52:102–6. doi: 10.1111/j.1526-4610.2012.02241.x
73. Cortelli, P, Pierangeli, G, and Montagna, P. Is migraine a disease? Neurol Sci. (2010) 31:29–31. doi: 10.1007/s10072-010-0270-0
74. Montagna, P, Pierangeli, G, and Cortelli, P. The primary headaches as a reflection of genetic darwinian adaptive behavioral responses. Headache. (2010) 50:273–89. doi: 10.1111/j.1526-4610.2009.01584.x
75. Nattero, G, De Lorenzo, C, Biale, L, Allais, G, Torre, E, and Ancona, M. Psychological aspects of weekend headache sufferers in comparison with migraine patients. Headache. (1989) 29:93–9. doi: 10.1111/j.1526-4610.1989.hed2902093.x
76. Torelli, P, Cologno, D, and Manzoni, GC. Weekend headache: a retrospective study in migraine without aura and episodic tension-type headache. Headache. (1999) 39:11–20. doi: 10.1046/j.1526-4610.1999.3901011.x
77. Lee, S, Yoo, RE, Choi, SH, Oh, SH, Ji, S, Lee, J, et al. Contrast-enhanced MRI T1 mapping for quantitative evaluation of putative dynamic Glymphatic activity in the human brain in sleep-wake states. Radiology. (2021) 300:661–8. doi: 10.1148/radiol.2021203784
78. Vgontzas, A, Li, W, Mostofsky, E, Mittleman, MA, and Bertisch, SM. Baseline sleep quality, stress, and depressive symptoms, and subsequent headache occurrence in a six-week prospective cohort study of patients with episodic migraine. Headache. (2021) 61:727–33. doi: 10.1111/head.14105
79. Stanyer, EC, Creeney, H, Nesbitt, AD, Holland, PR, and Hoffmann, J. Subjective sleep quality and sleep architecture in patients with Migraine: a Meta-analysis. Neurology. (2021) 97:e1620–31. doi: 10.1212/WNL.0000000000012701
80. Piantino, JA, Iliff, JJ, and Lim, MM. The bidirectional link between sleep disturbances and traumatic brain injury symptoms: a role for Glymphatic dysfunction? Biol Psychiatry. (2022) 91:478–87. doi: 10.1016/j.biopsych.2021.06.025
81. Vittorini, MG, Sahin, A, Trojan, A, Yusifli, S, Alashvili, T, Bonifácio, GV, et al. The glymphatic system in migraine and other headaches. J Headache Pain. (2024) 25:34. doi: 10.1186/s10194-024-01741-2
82. Lee, H, Xie, L, Yu, M, Kang, H, Feng, T, Deane, R, et al. The effect of body posture on brain Glymphatic transport. J Neurosci. (2015) 35:11034–44. doi: 10.1523/JNEUROSCI.1625-15.2015
83. Wei, F, Song, J, Zhang, C, Lin, J, Xue, R, Shan, LD, et al. Chronic stress impairs the aquaporin-4-mediated glymphatic transport through glucocorticoid signaling. Psychopharmacology. (2019) 236:1367–84. doi: 10.1007/s00213-018-5147-6
84. Hinde, K, White, G, and Armstrong, N. Wearable devices suitable for monitoring twenty four hour heart rate variability in military populations. Sensors (Basel). (2021) 21:1061. doi: 10.3390/s21041061
85. Miller, DJ, Sargent, C, and Roach, GD. A validation of six wearable devices for estimating sleep, heart rate and heart rate variability in healthy adults. Sensors (Basel). (2022) 22:6317. doi: 10.3390/s22166317
86. Buysse, DJ, Reynolds, CF, Monk, TH, Berman, SR, and Kupfer, DJ. The Pittsburgh sleep quality index: a new instrument for psychiatric practice and research. Psychiatry Res. (1989) 28:193–213. doi: 10.1016/0165-1781(89)90047-4
87. Rundo, JV, and Downey, R. Polysomnography. Handb Clin Neurol. (2019) 160:381–92. doi: 10.1016/B978-0-444-64032-1.00025-4
88. Błaszczyk, B, Waliszewska-Prosół, M, Smardz, J, Więckiewicz, M, Wojakowska, A, and Martynowicz, H. Exploring the associations of sleep bruxism and obstructive sleep apnea with migraine among patients with temporomandibular disorder: a polysomnographic study. Headache. (2025) 65:242–57. doi: 10.1111/head.14892
89. Roche, F, Pichot, V, Mouhli-Gasmi, L, Monier, M, Barthélémy, JC, Berger, M, et al. Anatomy and physiology of the autonomic nervous system: implication on the choice of diagnostic/monitoring tools in 2023. Rev Neurol (Paris). (2024) 180:42–52. doi: 10.1016/j.neurol.2023.12.003
90. Beck, AT, Steer, RA, and Brown, GK. Manual for the beck depression inventory-II. San Antonio, TX: Psychological Corporation. (1996); 1: 1037
Keywords: migraine pathophysiology, homeostasis, hypothalamic dysfunction, antidromic stimulation, trigeminovascular system, high-risk area
Citation: Rispoli MG, De Angelis MV, Melchionda D and Manente G (2025) High-risk area for migraine attacks — a new concept in migraine pathophysiology. Front. Neurol. 16:1569361. doi: 10.3389/fneur.2025.1569361
Edited by:
Parisa Gazerani, Oslo Metropolitan University, NorwayReviewed by:
Lanfranco Pellesi, University of Southern Denmark, DenmarkCopyright © 2025 Rispoli, De Angelis, Melchionda and Manente. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marianna Gabriella Rispoli, bWFyaWFubmFyaXNwb2xpOTJAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.