 Marcos Delgado
Marcos Delgado Reto A. Schuepbach
Reto A. Schuepbach Jan Bartussek
Jan Bartussek
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Neurol. , 09 April 2025
Sec. Dementia and Neurodegenerative Diseases
Volume 16 - 2025 | https://doi.org/10.3389/fneur.2025.1556456
Oxidative stress arises from the excessive production of toxic free radicals, known as reactive oxygen species (ROS), regularly generated as byproducts of mitochondrial ATP production. When the balance between ROS generation and antioxidant defenses, such as glutathione, is disrupted, ROS will accumulate, leading to cellular damage (1). Oxidative stress leads to lipid peroxidation of neuronal membranes, protein damage, and DNA/RNA oxidation, triggering cellular apoptosis and promoting the release of inflammatory cytokines, which play a key role in the pathogenesis of neurocognitive disorders (2). These free radicals cause neuronal protein and genetic dysfunctions, leading to protein aggregation (such as Lewy bodies, amyloid plaques, or neurofibrillary tangles) and iron accumulation in the brain—common pathological features of neurocognitive disorders like Parkinson's disease (PD), Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), and prion diseases (2).
Smoking is a major public health concern due to its association with high morbidity and mortality. Numerous studies have demonstrated its strong correlation with cardiovascular, respiratory, and cancer-related diseases, ultimately increasing mortality rates and healthcare costs (3). However, this contrasts with extensive epidemiological research suggesting an inverse correlation between smoking and the incidence of neurodegenerative diseases like PD and AD, while not negating the higher mortality rate among smokers (4, 5). This observation suggests that certain compounds in tobacco smoke may offer potential therapeutic benefits.
Nicotine is the most abundant and extensively studied components of tobacco. Numerous studies have shown that neurodegenerative diseases are associated with a reduction in nicotinic acetylcholine receptors (nAChRs) (6).
Nicotine's interaction with α7 and α4β2-nAChRs, leading to their upregulation and an increase in binding sites, may underlie its neuroprotective effects in patients with PD and AD. This mechanism could contribute to both symptomatic relief and improved cognitive function (7). Radiological and postmortem studies have shown a reduction in nAChR density in individuals with these neurodegenerative diseases (6, 8). These findings support the hypothesis that enhancing nAChR expression, may be one mechanism by which nicotine provides neuroprotection, potentially reducing or delaying disease onset.
A substantial body of research has investigated the antioxidant properties of nicotine. Most of these studies are based on preclinical trials at the cellular, molecular, or animal level. However, also clinical, epidemiological, and postmortem studies in humans highlight the significant therapeutic potential of nicotine. A mechanistic biochemical representation of the major nicotine-related pathways that are thought to be involved in reducing oxidative stress is depicted in Figure 1.
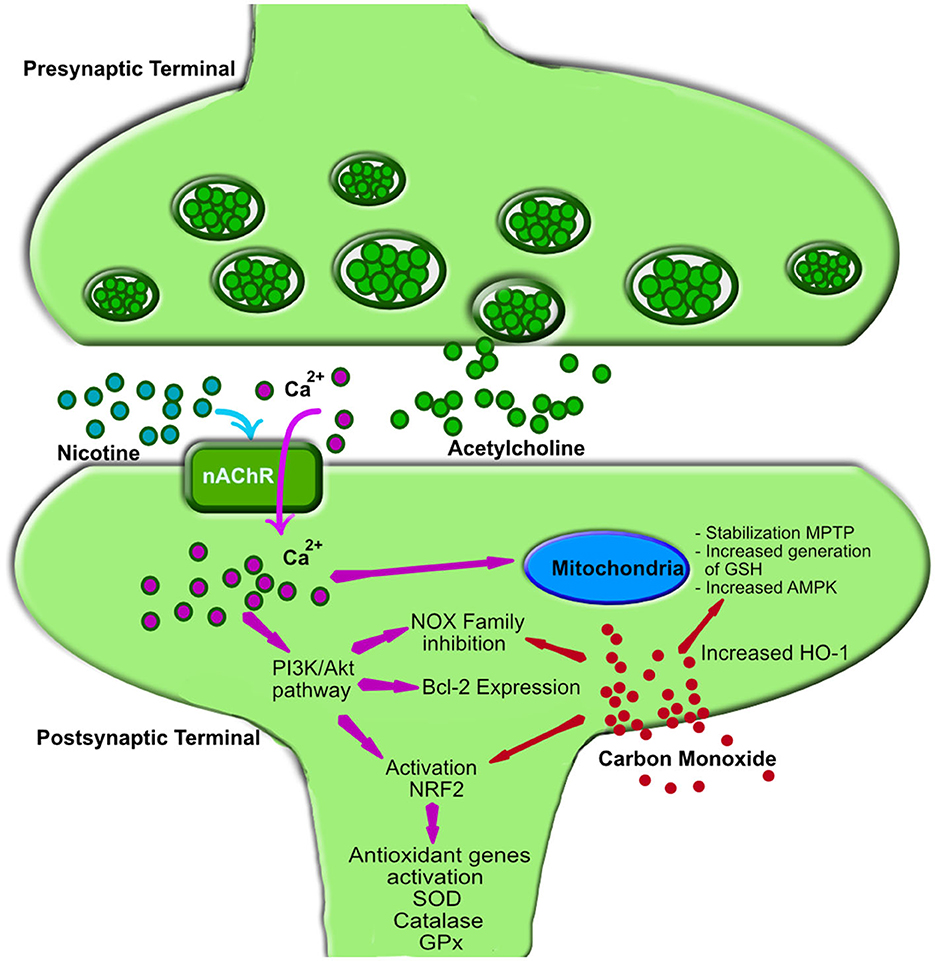
Figure 1. Mechanism of action of nicotine and carbon monoxide in oxidative stress reduction. Nicotine interacts with nicotinic acetylcholine receptors (nAChRs), triggering calcium (Ca2+) influx into the postsynaptic membrane of neurons (32). The increased intracellular Ca2+ activates the phosphoinositide 3-kinase (PI3K) and protein kinase B (PKB or Akt) signaling pathways (32), leading to the following effects: (1) Activation of Nuclear Factor Erythroid 2-Related Factor 2 (NRF2): This upregulates antioxidant genes such as superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPx), reducing free radical accumulation (33). (2) Upregulation of B-cell lymphoma 2 (Bcl-2): Bcl-2, a key anti-apoptotic protein, protects neurons from oxidative damage by preventing mitochondrial dysfunction and inhibiting cytochrome C release (34). (3) Inhibition of NADPH oxidase (NOX) enzymes: This decreases reactive oxygen species (ROS) production, reducing oxidative stress (35). Additionally, the Ca2+ influx is the starting point for modulation on mitochondrial energy metabolism through: (4) Activation of AMP-activated protein kinase (AMPK): This enhances ATP production efficiency and minimizes excessive ROS generation by optimizing electron transport chain function (32). (5) Regulation of mitochondrial membrane potential, through Bcl-2 and AMPK activity: Nicotine prevents the opening of the mitochondrial permeability transition pore (MPTP), thereby mitigating uncontrolled ROS release (36). (6) NRF2-dependent mitochondrial glutathione (GSH) synthesis, through NRF2 Activation: This enhances mitochondrial antioxidant capacity and protects against oxidative damage (37). Carbon monoxide (CO), a lipophilic molecule, readily enters cells and triggers several intracellular responses, primarily leading to the upregulation of heme oxygenase 1 (HO-1) (38). (1) Similar to nicotine, CO stabilizes NRF2 (39) and activates AMPK (40), which in turn promotes HO-1 expression. (2) Additionally, CO inhibits the NOX family of enzymes, thereby reducing ROS formation. (3) The increased HO-1 production exerts antioxidant effects by degrading free heme, lowering free iron levels, and enhancing neurotrophins activity (41). nAChR, nicotinic acetylcholine receptor; PI3K, phosphoinositide 3-kinase; Akt, protein kinase B (PKB); NRF2, Nuclear Factor Erythroid 2-Related Factor 2; SOD, superoxide dismutase; GPx, glutathione peroxidase; Bcl-2, B-cell lymphoma 2; NOX, NADPH oxidase enzymes family; AMPK, AMP-activated protein kinase; MPTP, mitochondrial permeability transition pore; ROS, reactive oxygen species; GSH, mitochondrial glutathione; CO, Carbon monoxide; HO-1, heme oxygenase 1.
In 1999, Linert et al. (9) investigated the potential antioxidant actions of nicotine in both in vitro and in vivo models. They analyzed the effect of iron in the substantia nigra pars compacta when combined with hydrogen peroxide (H2O2) to induce the Fenton reaction, a process that amplifies oxidative stress and is considered a key mechanism underlying Parkinson's disease. Their results suggest that nicotine can bind iron and potentially reduce its redox activity; however, direct evidence for its inhibition of the Fenton reaction remains limited. Nonetheless, this interaction could represent one pathway through which nicotine exerts antioxidative and, consequently, neuroprotective effects.
A study conducted by Guan et al. in (10) investigated the effects of varying concentrations of nicotine, along with oxidative and antioxidative substances, on the viability of a rat pheochromocytoma cell line. Cell survival was assessed using the MTT assay (3-(4,5-methylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay), a method that evaluates cellular metabolic activity based on mitochondrial function. A decrease in MTT assay readings was interpreted as indicative of cell destruction through oxidative stress mechanisms. The results demonstrated that cell viability remained stable when exposed to low concentrations of nicotine. In contrast, higher nicotine doses resulted in a reduction in cell survival. But, the co-administration of antioxidants with high nicotine doses mitigated this effect, preserving cell viability at levels comparable to control samples. Similarly, the addition of oxidative agents to the culture, combined with low-dose nicotine, maintained cell viability comparable to that of the control group. The study also explored the impact of amyloid beta peptide (Aβ25-35), a neurotoxic molecule implicated in Alzheimer's disease due to its oxidative properties. Exposure to Aβ25-35 significantly reduced cell viability; however, the administration of low-dose nicotine restored cell survival to normal levels. In contrast, increasing the nicotine dose led to a marked reduction in cell viability. These findings underscore the dual role of nicotine, with low concentrations exerting a protective effect under conditions of oxidative stress, while higher doses are cytotoxic.
Dong and collaborators (11) conducted an interesting study evaluating the effect of low-dose nicotine on hippocampal neurons under the hypothesis of a protective effect against Alzheimer's disease. In their research, they demonstrated that nicotine suppresses neuronal damage induced by H2O2 in hippocampal cells, reducing the generation of ROS through the activation of α7-nAchR and upregulation of Erk 1 and 2 (extracellular signal-Regulated Kinases 1 and 2), essential in promoting cell survival and proliferation and prevention of apoptosis.
Another recent trial conducted by Boiangiu et al. (12) using rat models of AD showed that cotinine and 6-hydroxy-L-nicotine, both nicotinic derivatives, improve memory and reduce oxidative stress by modulating nicotinic acetylcholine receptors (nAChRs). These substances enhance spatial and recognition memory, decrease acetylcholinesterase activity, and restore antioxidant defenses in the hippocampus, suggesting their potential as treatments for cognitive deterioration.
For ethical reasons, clinical trials assessing nicotine at different doses have not been possible. However, the reviews by Fratiglioni (4), Allam (5) or Picciotto (13) provide a comprehensive summary of several epidemiological, imaging, and postmortem studies investigating the mechanisms of nicotine's neuroprotection. Some of these trials are summarized in Table 1. Gorell et al. (14) reported a clear negative association between smoking and PD, with a dose-dependent neuroprotective effect that diminishes over time after smoking cessation. While the neuroprotective effect of nicotine in PD is well established, its role in AD remains controversial. Although preclinical studies suggest a protective effect in AD, this is challenged by the cerebrovascular ischemia associated with smoking (4) and other studies suggesting neuroinflammatory, oxidative stress mechanism and mitochondrial dysfunction in the pathophysiology of AD (8). In contrast, postmortem studies comparing elderly smokers and non-smokers have shown a significant reduction in amyloid-beta plaques in smokers (15), suggesting a potential protective effect against AD. This finding may also be linked to the upregulation of nAChRs in the brains of smokers with AD (16).
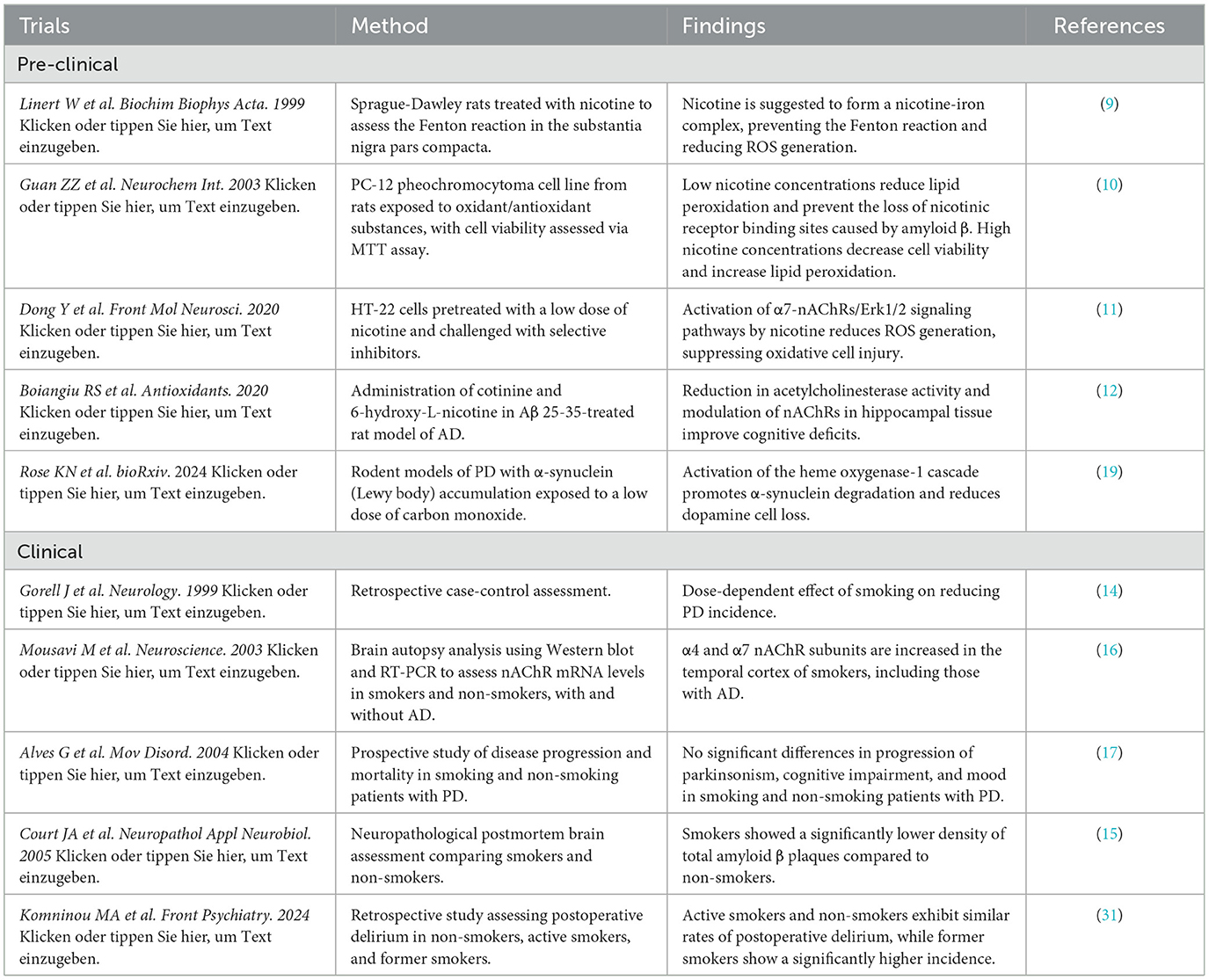
Table 1. Overview of selected studies.
Despite these findings on nicotine in neurocognitive diseases, its role remains unclear regarding dose safety, administration frequency, intervals, and its preventive or therapeutic effects. This uncertainty is highlighted by Alves et al. (17), who found no benefit of smoking on PD progression once the disease was already established, and by Nielsen et al. (18), who demonstrated that malondialdehyde—a well-established biomarker of oxidative stress and a product of lipid peroxidation—is present at significantly higher levels in the plasma of active smokers compared to non-smokers.
Although nicotine remains the primary focus of research on neuroprotection in neurocognitive disorders, a recent study by Rose et al. (19) provides valuable new insights of carbon monoxide (CO), another component of tobacco smoke, that open new therapeutic avenues in Parkinson's disease. Oxidative stress in PD underlies the formation and spread of α-synuclein (αSyn)-rich aggregates, known as Lewy bodies, which are thought to induce degeneration of dopaminergic neurons in the substantia nigra pars compacta, a hallmark feature of the Parkinson's disease. Rose et al. used rodent models of Parkinson's disease driven by αSyn accumulation and oxidative stress to demonstrate that CO reduces neurodegeneration and αSyn pathology by activating heme oxygenase-1 (HO-1)-mediated pathways (Figure 1). These pathways limit oxidative stress and promote the degradation of αSyn aggregates, mitigating key drivers of Parkinson's disease pathology. A particularly striking aspect of the study is the emphasis on using low doses of carbon monoxide, carefully maintained well below neurotoxic levels, to achieve these protective effects.
Consequently, low-dose carbon monoxide emerges as a potential protective factor against the destruction of dopaminergic neurons in the substantia nigra pars compacta, providing a novel therapeutic avenue for neuroprotection in Parkinson's disease.
Unlike the gradual, progressive decline seen in neurodegenerative diseases, delirium is characterized by an acute onset and fluctuating course, yet both conditions may involve disruptions in neurotransmitter systems and inflammatory pathways, providing a possible common ground for investigating protective effects. Delirium is widely recognized as a multifactorial syndromic manifestation that remains challenging to categorize comprehensively. It is a frequent and life-threatening complication, particularly in Intensive Care Units (ICU), where it commonly arises in post-operative patients (20). Despite its acute and often reversible nature, delirium carries significant morbidity and mortality risks, underscoring the urgency of understanding its pathophysiological mechanisms and identifying effective preventive and therapeutic strategies (21). Growing evidence suggests that oxidative stress may underlie the molecular mechanisms responsible for neuronal dysfunction in delirium (22). In the postoperative and ICU setting, oxidative stress can be triggered by a variety of factors, including hypoxemia (23), ischemia-reperfusion injury during surgery (24), hyperoxemia induced by mechanical ventilation or extracorporeal membrane oxygenation (25), systemic inflammation (26), and the metabolic effects of critical illness (27, 28), all of which may contribute to neuronal damage and cognitive dysfunction.
Both nicotine and CO are toxic substances; however, at low doses, they induce effects opposite to those typically associated with their toxicity. This phenomenon, known as hormesis (29), contributes to cellular survival. Therefore, it can be postulated that nicotine and CO may act synergistically to exert a neuroprotective effect in neurodegenerative diseases. This neuroprotective effect may also be apparent in delirium, a condition that, while distinct from chronic neurodegenerative diseases like PD and AD, shares overlapping features in its impact on cognition and potential mechanisms of neuronal dysfunction (7, 30).
Recently, we published a study in which we evaluated the possible relationship between nicotine consumption and the development of delirium in postoperative ICU patients (31). In contrast to previous studies, we differentiated between former smokers and non-smokers—defined as individuals who reported never having smoked in their lifetime—and compared their incidence of postoperative delirium with that of active smokers. Surprisingly, our results demonstrated that active smokers did not exhibit a higher incidence of postoperative delirium than non-smokers, despite being in a withdrawal phase. Instead, former smokers showed a significantly higher incidence of delirium. Although the underlying mechanisms were not directly investigated in our study, we hypothesize that the desensitization and upregulation of nAChRs commonly observed in nicotine users (13) may have contributed to increased neuronal vulnerability in both active and former smokers due to receptor overexpression. However, in active smokers, the presence of nicotine during the perioperative period may have exerted a neuroprotective effect, potentially mitigating the risk of delirium observed in the former smoker group.
Both nicotine and carbon monoxide in active smokers may contribute to neurofunctional stability through their antioxidant properties, potentially offering protection against both delirium and neurodegenerative diseases. Given the relatively short half-life of carbon monoxide (4 to 6 h), its levels in active smokers were likely reduced in the postoperative period during our trial. However, it is important to note that a major period of oxidative stress likely occurs during surgery, when carbon monoxide levels might still be elevated, potentially exerting protective effects at a critical time. Moreover, nicotine and its metabolites, such as cotinine, have a much longer half-life, enabling them to remain in the bloodstream for several hours to days, potentially extending their neuroprotective effects throughout the postoperative period, including during prolonged stays in the ICU.
Nicotine and carbon monoxide, despite their distinct mechanisms, share antioxidative properties and exhibit neuroprotective effects at low doses. Given the links between oxidative stress, delirium, and neurodegenerative diseases, these findings suggest potential therapeutic applications for preventing and managing neurodegenerative diseases and delirium. Randomized controlled trials are warranted to evaluate their controlled use in mitigating oxidative stress and exploring their broader clinical implications.
MD: Conceptualization, Writing – original draft, Writing – review & editing. RS: Supervision, Validation, Visualization, Writing – review & editing. JB: Conceptualization, Supervision, Validation, Visualization, Writing – review & editing.
The author(s) declare that no financial support was received for the research and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that Gen AI was used in the creation of this manuscript. GenAI was solely used to improve writing style.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Cacciatore I, Baldassarre L, Fornasari E, Mollica A, Pinnen F. Recent advances in the treatment of neurodegenerative diseases based on GSH delivery systems. Oxid Med Cell Longev. (2012) 2012:240146. doi: 10.1155/2012/240146
2. Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. (2006) 97:1634–58. doi: 10.1111/j.1471-4159.2006.03907.x
3. Jha P, Ramasundarahettige C, Landsman V, Rostron B, Thun M, Anderson RN, et al. 21st-century hazards of smoking and benefits of cessation in the United States. N Engl J Med. (2013) 368:341–50. doi: 10.1056/NEJMsa1211128
4. Fratiglioni L, Wang HX. Smoking and Parkinson's and Alzheimer's disease: review of the epidemiological studies. Behav Brain Res. (2000) 113:117–20. doi: 10.1016/S0166-4328(00)00206-0
5. Allam MF, Campbell MJ, Hofman A, Del Castillo AS, Fernández-Crehuet Navajas R. Smoking and Parkinson's disease: systematic review of prospective studies. Mov Disord. (2004) 19:614–21. doi: 10.1002/mds.20029
6. Perry EK, Martin-Ruiz CM, Court JA. Nicotinic receptor subtypes in human brain related to aging and dementia. Alcohol. (2001) 24:63–8. doi: 10.1016/S0741-8329(01)00130-6
7. Xu J, Mendrek A, Cohen MS, Monterosso J, Rodriguez P, Simon SL, et al. Brain activity in cigarette smokers performing a working memory task: effect of smoking abstinence. Biol Psychiatry. (2005) 58:143–50. doi: 10.1016/j.biopsych.2005.03.028
8. Song T, Song X, Zhu C, Patrick R, Skurla M, Santangelo I, et al. Mitochondrial dysfunction, oxidative stress, neuroinflammation, and metabolic alterations in the progression of Alzheimer's disease: a meta-analysis of in vivo magnetic resonance spectroscopy studies. Ageing Res Rev. (2021) 72:101503. doi: 10.1016/j.arr.2021.101503
9. Linert W, Bridge MH, Huber M, Bjugstad KB, Grossman S, Arendash GW. In vitro and in vivo studies investigating possible antioxidant actions of nicotine: relevance to Parkinson's and Alzheimer's diseases. Biochim Biophys Acta. (1999) 1454:143–52. doi: 10.1016/S0925-4439(99)00029-0
10. Guan ZZ, Yu WF, Nordberg A. Dual effects of nicotine on oxidative stress and neuroprotection in PC12 cells. Neurochem Int. (2003) 43:243–9. doi: 10.1016/S0197-0186(03)00009-3
11. Dong Y, Bi W, Zheng K, Zhu E, Wang S, Xiong Y, et al. Nicotine prevents oxidative stress-induced hippocampal neuronal injury through α7-nAChR/Erk1/2 signaling pathway. Front Mol Neurosci. (2020) 13:557647. doi: 10.3389/fnmol.2020.557647
12. Boiangiu RS, Mihasan M, Gorgan DL, Stache BA, Petre BA, Hritcu L. Cotinine and 6-hydroxy-L-nicotine reverses memory deficits and reduces oxidative stress in Aβ25-35-induced rat model of Alzheimer's disease. Antioxidants. (2020) 9:768. doi: 10.3390/antiox9080768
13. Picciotto MR, Zoli M. Neuroprotection via nAChRs: the role of nAChRs in neurodegenerative disorders such as Alzheimer's and Parkinson's disease. Front Biosci. (2008) 13:492–504. doi: 10.2741/2695
14. Gorell J, Rybicki B, Jonson C, Peterson EL. Smoking and Parkinson's disease: a dose-response relationship. Neurology. (1999) 52:115–9. doi: 10.1212/WNL.52.1.115
15. Court JA, Johnson M, Religa D, Keverne J, Kalaria R, Jaros E, et al. Attenuation of abeta deposition in the entorhinal cortex of normal elderly individuals associated with tobacco smoking. Neuropathol Appl Neurobiol. (2005) 31:522–35. doi: 10.1111/j.1365-2990.2005.00674.x
16. Mousavi M, Hellström-Lindahl E, Guan ZZ, Shan KR, Ravid R, Nordberg A. Protein and mRNA levels of nicotinic receptors in brain of tobacco using controls and patients with Alzheimer's disease. Neuroscience. (2003) 122:515–20. doi: 10.1016/S0306-4522(03)00460-3
17. Alves G, Kurz M, Lie SA, Larsen JP. Cigarette smoking in Parkinson's disease: influence on disease progression. Mov Disord. (2004) 19:1087–92. doi: 10.1002/mds.20117
18. Nielsen F, Mikkelsen BB, Nielsen JB, Andersen HR, Grandjean P. Plasma malondialdehyde as biomarker for oxidative stress: reference interval and effects of life-style factors. Clin Chem. (1997) 43:1209–14. doi: 10.1093/clinchem/43.7.1209
19. Rose KN, Zorlu M, Xue X, Fassini A, Cai W, Lin S, et al. Neuroprotection of low dose carbon monoxide in Parkinson's disease models commensurate with the reduced risk of Parkinson's among smokers. bioRxiv. (2024). doi: 10.1101/2023.05.27.542565
20. Kotfis K, Marra A, Ely EW. ICU delirium—a diagnostic and therapeutic challenge in the intensive care unit. Anaesthesiol Intensive Ther. (2018) 50:160–7. doi: 10.5603/AIT.a2018.0011
21. Morandi A, Brummel NE, Ely EW. Sedation, delirium and mechanical ventilation: the “ABCDE” approach. Curr Opin Crit Care. (2011) 17:43–9. doi: 10.1097/MCC.0b013e3283427243
22. Pang Y, Li Y, Zhang Y, Wang H, Lang J, Han L, et al. Effects of inflammation and oxidative stress on postoperative delirium in cardiac surgery. Front Cardiovasc Med. (2022) 9:1049600. doi: 10.3389/fcvm.2022.1049600
23. Smith KA, Waypa GB, Schumacker PT. Redox signaling during hypoxia in mammalian cells. Redox Biol. (2017) 13:228–34. doi: 10.1016/j.redox.2017.05.020
24. Caputo M, Mokhtari A, Rogers CA, Panayiotou N, Chen Q, Ghorbel MT, et al. The effects of normoxic vs. hyperoxic cardiopulmonary bypass on oxidative stress and inflammatory response in cyanotic pediatric patients undergoing open cardiac surgery: a randomized controlled trial. J Thorac Cardiovasc Surg. (2009) 138:206–14. doi: 10.1016/j.jtcvs.2008.12.028
25. Tigano S, Caruso A, Liotta C, LaVia L, Vargas M, Romagnoli S, et al. Exposure to severe hyperoxemia worsens survival and neurological outcome in patients supported by veno-arterial extracorporeal membrane oxygenation: a meta-analysis. Resuscitation. (2024) 194:110071. doi: 10.1016/j.resuscitation.2023.110071
26. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. (2014) 20:1126–67. doi: 10.1089/ars.2012.5149
27. Rahal A, Kumar A, Singh V, Yadav B, Tiwari R, Chakraborty S, et al. Oxidative stress, prooxidants, and antioxidants: the interplay. Biomed Res Int. (2014) 2014:761264. doi: 10.1155/2014/761264
28. Terraneo L, Samaja M. Comparative response of brain to chronic hypoxia and hyperoxia. Int J Mol Sci. (2017) 18:1914. doi: 10.3390/ijms18091914
30. Valentine G, Sofuoglu M. Cognitive effects of nicotine: recent progress. Curr Neuropharmacol. (2018) 16:403–14. doi: 10.2174/1570159X15666171103152136
31. Komninou MA, Egli S, Rossi A, Ernst J, Krauthammer M, Schuepbach RA, et al. Former smoking, but not active smoking, is associated with delirium in postoperative ICU patients: a matched case-control study. Front Psychiatry. (2024) 15:1347071. doi: 10.3389/fpsyt.2024.1347071
32. Xu ZQ, Zhang WJ, Su DF, Zhang GQ, Miao CY. Cellular responses and functions of α7 nicotinic acetylcholine receptor activation in the brain: a narrative review. Ann Transl Med. (2021) 9:509. doi: 10.21037/atm-21-273
33. Cao Y, Sun J, Wang X, Zhang X, Tian H, Huang L, et al. The double-edged nature of nicotine: toxicities and therapeutic potentials. Front Pharmacol. (2024) 15:1427314. doi: 10.3389/fphar.2024.1427314
34. Hua Y, Yang B, Chen Q, Zhang J, Hu J, Fan Y. Activation of α7 nicotinic acetylcholine receptor protects against 1-methyl-4-phenylpyridinium-induced astroglial apoptosis. Front Cell Neurosci. (2019) 13:507. doi: 10.3389/fncel.2019.00507
35. Noda M, Kobayashi AI. Nicotine inhibits activation of microglial proton currents via interactions with α7 acetylcholine receptors. J Physiol Sci. (2017) 67:235–45. doi: 10.1007/s12576-016-0460-5
36. Malińska D, Wieckowski MR, Michalska B, Drabik K, Prill M, Patalas-Krawczyk P, et al. Mitochondria as a possible target for nicotine action. J Bioenerg Biomembr. (2019) 51:259–76. doi: 10.1007/s10863-019-09800-z
37. Lee C. Therapeutic modulation of virus-induced oxidative stress via the NRF2-dependent antioxidative pathway. Oxid Med Cell Longev. (2018) 2018:6208067. doi: 10.1155/2018/6208067
38. Otterbein LE, Foresti R, Motterlini R. Heme oxygenase-1 and carbon monoxide in the heart: the balancing act between danger signaling and pro-survival. Circ Res. (2016) 118:1940–59. doi: 10.1161/CIRCRESAHA.116.306588
39. Wang B, Cao W, Biswal S, Doré S. Carbon monoxide-activated NRF2 pathway leads to protection against permanent focal cerebral ischemia. Stroke. (2011) 42:2605–10. doi: 10.1161/STROKEAHA.110.607101
40. Lin CC, Yang CC, Hsiao LD, Chen SY, Yang CM. Heme oxygenase-1 induction by carbon monoxide releasing molecule-3 suppresses interleukin-1β-mediated neuroinflammation. Front Mol Neurosci. (2017) 10:387. doi: 10.3389/fnmol.2017.00387
Keywords: delirium, oxidative stress, nicotine, carbon monoxide, nicotinic receptors, neurodegenerative diseases
Citation: Delgado M, Schuepbach RA and Bartussek J (2025) Opinion: exploring alternative pathways to neuroprotection—nicotine and carbon monoxide as antioxidative factors in neurodegeneration and delirium. Front. Neurol. 16:1556456. doi: 10.3389/fneur.2025.1556456
Received: 06 January 2025; Accepted: 24 March 2025;
Published: 09 April 2025.
Edited by:
Robert Weissert, University of Regensburg, GermanyReviewed by:
Lorenzo More, University of Central Lancashire, United KingdomCopyright © 2025 Delgado, Schuepbach and Bartussek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcos Delgado, bWFyY29zLmRlbGdhZG9AdXN6LmNo
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.