 Peijun Zhang
Peijun Zhang Cuomaoji Zhang
Cuomaoji Zhang Bixin Zheng1
Bixin Zheng1 Yuntao Liu
Yuntao Liu Dingkun Zhang
Dingkun Zhang Hong Xiao
Hong Xiao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 10 March 2025
Sec. Headache and Neurogenic Pain
Volume 16 - 2025 | https://doi.org/10.3389/fneur.2025.1535136
Postherpetic neuralgia (PHN), a representative type of neuropathic pain, has attracted much research on its diagnosis and therapy at the molecular level. Interestingly, this study based on the brain-gut axis provided a novel point of view to interpret the mechanism of PHN. Past neuroanatomical and neuroimaging studies of pain suggest that the prefrontal cortex, anterior cingulate cortex, amygdala, and other regions of the brain may play crucial roles in the descending inhibition of PHN. Dominant bacterial species in patients with PHN, such as Lactobacillus, generate short-chain fatty acids, including butyrate. Evidence indicates that disturbance of some metabolites (such as butyrate) is closely related to the development of hyperalgesia. In addition, tryptophan and 5-HT in the intestinal tract act as neurotransmitters that regulate the descending transmission of neuropathic pain signals. Concurrently, the enteric nervous system establishes close connections with the central nervous system through the vagus nerve and other pathways. This review aims to investigate and elucidate the molecular mechanisms associated with PHN, focusing on the interplay among PHN, the gut microbiota, and relevant metabolites while scrutinizing its pathogenesis.
Postherpetic neuralgia (PHN) is defined as pain persisting for more than three months following the onset or healing of herpes zoster according to the International Association for the Study of Pain (IASP) among the latest classifications of chronic pain (1). The occurrence of herpes zoster is due to the reactivation of the Varicella Zoster Virus (VZV) that has been latent in the ganglion when immune function declines, leading to ganglioneuritis and hemorrhagic necrosis with associated neuritis, and even leptomeningitis and unilateral segmental poliomyelitis. Inflammatory damage to neurons is the cause of neuropathic pain. PHN is the state that results from the chronicization of this neuropathic pain (2, 3). As a typical refractory and debilitating form of neuropathic pain, PHN decreases the quality of life of patients (4, 5). The prevalence of PHN in adults has varied from 2.3% to 5% in China over the past four years (6, 7). A global epidemiology survey reported that the incidence of PHN in people over 50 years old is 18%. Among people over the age of 80, 33% are diagnosed (8). PHN manifests as persistent and refractory neuropathic pain, which has adverse effects on patients' physical and psychiatric health. Patients suffer from multiple types of pain, including constant deep, aching, or burning pain; paroxysmal, lancinating pain; hyperalgesia; and allodynia (9). In addition to intractable pain, patients with PHN often have comorbidities, such as insomnia, depression, or anxiety (10). Patients with long-term PHN appear to have a worse prognosis (11), which makes the complete eradication and elimination of PHN a considerable challenge. However, due to the complexity and individual differences of PHN, the mechanism underlying its chronicity remains unclear and has become a significant problem to be solved in PHN treatment. Therefore, it is necessary and urgent to explore the potential mechanism of PHN to better understand and guide future theranostic innovations and drug development.
The brain-gut axis, also called the gut-brain axis, microbiota-gut-brain axis, or brain-gut interaction, refers to bidirectional neurohumoral interactions among the brain, gut, and gut microbiome of the host (12, 13). Such bidirectional neurohumoural interactions always occur through neural and humoral pathways. Specifically, the neural pathways of the brain-gut axis include the enteric nervous system (ENS), vagus, sympathetic, and spinal nerves, while humoral pathways include cytokines, hormones, and neuropeptides (13, 14). In 1980s, researchers proposed the concept of the gut-brain axis based on the discovery that several polypeptide hormones originate from the gastroenteric tract and are present both in nerves and the brain (15). Previous studies have focused on the role of peptides such as cholecystokinin octapeptide, somatostatin, and bombesin in central and peripheral nerves (16) that coexist in the gut and brain. Comparatively, more researchers have focused on the types of molecules involved in brain-gut interactions (17, 18). The biological role of the brain-gut axis has been demonstrated in multiple disease models, including stress ulcers (19), functional dyspepsia (20), irritable bowel syndrome (21), inflammatory bowel diseases (22) [including Crohn's disease (23), ulcerative colitis (24)] and obesity (25). The results showed that stress is crucial for exacerbating gastrointestinal inflammation, reducing immunity, and increasing digestive tract vulnerability (22). In addition, researchers have explored potential therapeutic mechanisms based on the brain-gut axis (26, 27), such as monitoring the neurological and etiological effects of ingesting specific strains of bacteria (28, 29). Psychotropic drugs have been proposed for use in treating digestive diseases such as irritable bowel syndrome (27). In the last decade, scientists have investigated the relationships between the brain-gut axis and neuropsychiatric illnesses such as autism (30), anxiety, fear (31), bipolar disorder (32), depression, anxiety (33, 34), obsessive-compulsive disorder (35) and neurodegenerative diseases (36) (including Parkinson's disease (37), Alzheimer's disease (38) and central nervous system demyelination (39)). The diversity of models involved in the above brain-gut axis studies needs to be improved. Research on the nervous system in the brain-gut axis has focused mainly on psychiatric rather than neurological diseases.
Interestingly, recent studies revealed that the brain-gut axis is involved in the development of visceral hypersensitivity and pain (40–42), especially in individuals with irritable bowel syndrome (43). Neural sensitization may be driven by peripheral mechanisms within the intestinal wall, encompassing an interplay between immunocytes, enterochromaffin cells, resident macrophages, neurons, and smooth muscles. In the brain, neuronal synaptic changes and enhanced release of neurotransmitters in the spinal cord and brain lead to central sensitization (44). Alterations in the brain-gut axis originating from small intestinal bacterial overgrowth or subclinical intestinal infections such as giardiasis may be significant causes of cormobidity of fibromyalgia and irritable bowel syndrome (45). Several therapeutic approaches have been suggested for the brain-gut axis, such as dietary modifications, oral probiotics, antibiotics, and anti-inflammatory agents (46, 47). These therapeutic approaches have shown some efficacy in gastrointestinal diseases, but their effects under other conditions still need to be clarified. Alternatively, developing the brain-gut axis theory provides a new method for managing refractory pain. Peripheral pain from both visceral and somatosensory nerves is transmitted to the brain via spinal dorsal horn neurons (48, 49). Therefore, it is reasonable to speculate that the brain-gut axis is involved in the occurrence and development of neuropathic pain, such as PHN, to some extent. Potential therapeutic methodologies based on the brain-gut axis are expected to be used to treat PHN and improve patients' quality of life.
In this review, we collected papers published in PHN-related fields over the last 20 years to explore and summarize PHN-related molecular mechanisms among PHN, the gut microbiota, and relevant metabolites. And we examined the pathogenesis and development of PHN from the perspective of the brain-gut axis and identified potential targets for improvement of gastrointestinal motility treatment of PHN in the future.
We conducted searches using the keywords “pain”, “neuropathic pain”, “postherpetic neuralgia”, “pathway”, “mechanism”, and “fMRI” to find articles concerning the involvement of the brain in PHN. Similarly, we utilized the keywords “intestinal”, “gut”, “bacterium”, “microorganism”, and “postherpetic neuralgia” to explore research on the intestinal microbiota of PHN patients. Based on the predominant bacterial species suggested by relevant studies, we searched for the relationship between their metabolites and pain modulation. Using “brain-gut axis” and “brain-gut interaction” as keywords, we searched for the main mechanisms and pathways confirmed in current research, further exploring the bidirectional effects of PHN on the intestine and brain.
As the body's most complex nervous system organ, the brain perceives pain, directing the body's response. Regardless of whether the pain originates from herpes zoster, patients with various types of pain show high similarity in pain transduction pathways. The spinothalamic tract projects the stimulation it receives to the ventroposterolateral nucleus of the contralateral thalamus. Superior neurons transmit pain signals to relevant somatosensory cortical regions, including the primary somatosensory cortex, secondary somatosensory cortex, posterior insula, medial parietal operculum, orbitofrontal cortex, dorsal-lateral prefrontal cortex, extended amygdala, and cingulate cortex. Parts of pain signals are transferred between the anterior and posterior insular cortex. The final pain experience combines sensory input and behavioral and cognitive interpretations of pain (50–52). The pathway is regulated by the descending modulation system, which descends from the brain to the spinal cord and modifies incoming somatosensory information to alter the perception and reactions to somatosensory stimuli, resulting in increased or decreased pain (5). The descending inhibitory pathway consists of the amygdala, the prefrontal cortex, the periaqueductal gray (PAG), and the rostral ventromedial medulla (RVM) from top to bottom (53). Finally, the RVM sends diffuse bilateral projections to the dorsal horn, terminating at multiple levels (54). Here, we list a few brain structural regions related to pain (Supplementary Tables S1, S2).
PHN has unique characteristics of the central nervous system. Some functional imaging studies conducted on PHN patients have shown significant changes in gray matter volume, functional connections, regional homogeneity (ReHo), fractional aptitude of low-frequency fluctuation (fALFF), mean diffusivity (MD), and other parameters in multiple brain regions (such as the temporal lobe, insula, frontal lobe, cingulate gyrus, amygdala, and thalamus) compared with those in healthy controls (Supplementary Table S3). However, there are significant differences between the results of different studies, possibly because of the following reasons: 1. The affected side was not classified; 2. There is a lack of extensive sample size studies; 3. Differences in instruments and equipment. Since the completion of resting-state fMRI requires the subject to stop thinking activities, stay awake, and keep the head still, good cooperation is required during the examination. The above factors will confound the research results if the combination is not good. In addition, a study has shown that neurovascular aging can affect fMRI imaging in some aspects (55). Furthermore, functional changes in brain regions on fMRI can only indicate that they are relay stations in the pain transmission pathway and cannot clarify the signaling pathways and regulatory mechanisms involved. However, further primary studies are needed to verify its role in pain transduction pathways.
In summary, within the central nervous system of PHN patients, the thalamus is responsible for the ascending transmission of pain information; the somatosensory cortex is involved in pain perception; the insular cortex is like a relay station of pain signal and related to pain hypersensitivity (56); the ACC, PFC, and amygdala contribute to the formation of unpleasant emotions; and the PAG and RVM participate in the descending inhibition of pain signals. The specific roles of other cortical areas, the precuneus, and the brainstem in PHN require further investigation.
Research indicates that inflammatory bowel disease can increase the risk of developing herpes zoster (57). Dysregulation of the immune response of the gut microbiota may be involved in the development of postherpetic neuralgia (58). However, there is only one study on the gut microbiota in patients with PHN (59). Since there is a need for more relevant studies in other regions, its generalizability is controversial. Therefore, based on the limited research results, we speculate on categorizing predominant gut bacteria in patients with PHN and healthy controls.
As a major resident bacterium in the human gut, Lactobacillus breaks down intestinal polysaccharides and starch into glucose and maltose. This process provides substrates for bacteria lacking this function, such as Eubacterium ramulus, indirectly promoting the production of butyrate (60) As a short-chain fatty acid (SCFA), butyrate may relieve neuropathic pain in obese mice by regulating gene expression and immune function in the peripheral nervous system (61). Its immunomodulatory effects may be achieved by suppressing proinflammatory cytokines (62). In an animal model of chronic compression injury, repeated administration of butyrate alleviated mechanical and thermal hyperalgesia, thereby mitigating neuropathic pain (63). One study suggested that butyrate alleviates neuropathic pain by increasing withdrawal thresholds and reducing hyperalgesia (64). This may be achieved through the induction of regulatory T cells, the reduction of proinflammatory cytokines, and the downregulation of Toll-like receptor (TLR) 4 receptors (65).
The Lachnospiraceae family is a significant branch of the Firmicutes phylum, which, along with the Bacteroidetes phylum, comprises the two most crucial microbial groups in the human gut. Lachnospiraceae species breakdown starch and various sugars, generating SCFAs. The Roseburia species primarily synthesizes butyrate as the primary SCFA, in addition to acetate (66). Supplementation with Roseburia hominis significantly increased cecal butyrate levels, alleviated visceral hypersensitivity and prevented the decreased expression of occludin in the colon of rats (67). In patients with PHN, there is a decrease in the abundance of bacterial species that produce butyrate in the gut (59). However, the underlying mechanism remains unknown, suggesting butyrate may take part in endogenous analgesia.
Clostridium in the gut is an anaerobic bacterium, with only a few species being pathogenic. It can form endospores and has extensive applications in medical and bioengineering fields. For instance, it can be utilized for treating tumors and antibiotic-associated diarrhea. Some species within Clostridium can generate acetate, propionate, butyrate, butanol, acetone, formate, and other compounds through various metabolic pathways. Hence, they are utilized to produce organic solvents (68, 69). Clostridia sourced from humans can boost the population and efficacy of colonic Treg cells (a specialized subpopulation of T cells that could suppress immune responses to keep immunological homeostasis (70)) in colonized rodents, consequently reducing symptoms of experimental allergic diarrhea and colitis (71). Research indicates that the dysregulation of Treg cells and effector T cells (which could secrete interleukin 1 or 17) can lead to an overly active immune response and elevated levels of neurological inflammation and can promote the progression of conditions such as neuropathic pain disorders. PHN represents a classic form of peripheral neuropathic pain, from which we can infer that an increase in gut Treg cells contributes to the advancement of this condition (72).
In conclusion, predominant bacterial species in the gut of PHN patients may regulate the levels of neuroinflammation, enhance pain thresholds, and alleviate tactile hypersensitivity to some extent by participating in the synthesis of short-chain fatty acids, thereby modulating the quantity of T-cell subsets.
Due to the exclusive tropism of the varicella-zoster virus for humans, many rodent-based PHN models currently fail to accurately reflect authentic pathological and physiological conditions. The lack of researches focused on the gut-brain mechanism of PHN in humans leads to many mechanisms being speculative. Current mainstream studies have shown that the gut-brain interaction is primarily manifested in the following aspects: the connection between the enteric nervous system and the central nervous system and the impact of gut metabolites with neurotransmitter or neuromodulator properties on the central nervous system (73).
The ENS spans from the upper esophagus to the internal anal sphincter, establishing connections with the biliary tract, liver, gallbladder, and pancreas. The input and output of ENS signals are primarily mediated through the vagus nerve and splanchnic nerves. Research has shown that the vagal neural crest of mice enters the foregut at approximately embryonic days 7-9.5. The majority of the ENS originates from the vagal neural crest, with a smaller portion originating from the sacral neural crest (74). The vagus nerve collects mechanical and chemical sensations from the intestine, influencing the movement of smooth muscles. Splanchnic nerves can transmit pain signals and regulate glucose metabolism. The ENS can be divided into two neural plexuses: the myenteric plexus and the submucosal plexus. The myenteric plexus is situated between the smooth muscles of the intestine and governs the movement of the intestinal smooth muscles. The submucosal plexus is located between the mucosa and smooth muscle of the intestines, dominating intestinal absorption and secretion and comprising various types of neurons, including intrinsic primary afferent neurons, motor neurons, enteric glia, and interneurons (75, 76).
Vagus nerve stimulation (VNS) is a technique that relies on surgical implantation, where electrodes are placed on the surface of the vagus nerve, and a pulse generator connected to it is implanted under the skin. The pulse generator releases pulsed electrical currents to control epileptic seizures (77). VNS was once considered an adjunctive treatment for patients aged four years and older with medically refractory partial-onset seizures (78). Moreover, the medication pregabalin, used to treat neuropathic pain, including PHN, is also an antiepileptic drug. Therefore, we have reason to suspect a potential connection between the vagus nerve and neuropathic pain, including PHN. Intermittent vagus nerve stimulation enhances the expression of neuronal fos in the medullary vagal complex, locus coeruleus, and various thalamic and hypothalamic nuclei Fos serves as an indicator of heightened metabolic activity. Additionally, VNS leads to the upregulation of brain-derived neurotrophic factor and fibroblast growth factor in the hippocampus and cerebral cortex, a reduction in nerve growth factor mRNA abundance in the hippocampus, and an increase in the norepinephrine concentration in the prefrontal cortex (79). Pregabalin binds to the α2 − δ subunit of voltage-gated calcium channels on the neocortex, amygdala, hippocampus, striatum, dorsal horn of the spinal cord, cerebellum, and habenula, reducing the influx of calcium ions into cells. Pregabalin can decrease the release of glutamate and norepinephrine in the trigeminal nucleus and neocortex while simultaneously reducing synaptic potentials and intrasynaptic currents (80). This appears to contradict the effects of VNS. Currently, there is a lack of direct research on the functionality of the vagus nerve, neuropathic pain, and antiepileptic drugs, perhaps suggesting a potential avenue for future investigations.
Tryptophan is an essential amino acid in the human body that is absorbed through food intake and is primarily metabolized via various pathways: 1. Conversion to kynurenine in the liver, 2. Transformation to 5-HT in enterochromaffin cells in the intestine; 3. Iron is converted to indole under the influence of intestinal microorganisms. Kynurenine mainly affects the immune system. Kynurenine regulates colonic epithelial cell immunity by activating colonic aryl hydrocarbon receptors. It actively enters the central nervous system and is metabolized into kynurenic acid, quinolinic acid, and anthranilic acid. Kynurenic acid is an antagonist of N-methyl-D-aspartate (NMDA) receptors in the CNS. Additionally, it opposes nicotinic α−7 receptors expressed on macrophages and lymphocytes, thereby increasing the release of TNFα from macrophages in the reticuloendothelial system (81). L-4-Chlorokynurenine attenuates overactive glutamatergic transmission via NMDA receptors by blocking the allosteric glycine B coagonist site on the receptor (82). L-4-Chlorokynurenine inhibits central sensitization, making it a potential therapeutic option for neuropathic pain, including PHN.
The regulation of pain by 5-HT is comprehensive. 5-HT is secreted by enterochromaffin cells on the intestinal surface and released into the submucosa to act on intrinsic serotonergic neurons in the submucosal plexus of the ENS (81). Specifically, upon receiving information about intestinal distension, enterochromaffin cells secrete 5-HT, activating 5-HT3 and 5-HT4 receptors on submucosal splanchnic nerves and the vagus nerve. Subsequently, these signals ascend through the dorsal horn of the spinal cord, conveying nociceptive signals to the thalamus and projecting to the cortex. In this process, 5-HT participates in hyperalgesia through the mediation of 5-HT1 and 5-HT2 receptors in the spinal cord, a process that is regulated by the serotonin transporter (83). 5-HT1A receptors is one of six subtypes of 5-HT1 receptors, a large number of distributions in raphe nucleus, amygdala, cingulate cortex, insula, prefrontal cortex and spinal dorsal horn. The 5-HT1A receptor functions as an inhibitory autoreceptor, presynaptically suppressing nociceptive transmission into laminae I and II of the dorsal horn. This effect is mediated by the upregulation and downregulation of 5-HT1A receptors in the rostral ventromedial medulla. 5-HT2 receptor is coupled to the G q/11 protein. And 5-HT2 receptors which are located on peripheral neurons induce primary thermal hyperalgesia and secondary mechanical allodynia (84). 5-HT3 receptor is a ligand-gated cation channel.Antagonists of 5-HT3 receptors can block nonspecific ion channels, restricting the influx of Na+ and Ca2+ and efflux of Cl−, thereby interrupting the transmission of pain signals and mitigating visceral hypersensitivity to some extent (85). A portion of the 5-HT produced in the gut enters the bloodstream and is absorbed and stored by platelets. In the central nervous system, 5-HTergic neurons originating from the nucleus raphe magnus regulate the descending transmission of neuropathic pain signals, including PHN. Among these receptors, the 5-HT3 receptor couples with cAMP (cyclic adenosine monophosphate), enhancing nociceptive sensations, tactile allodynia and thermal hyperalgesia. While the 5-HT7 receptor can increase the release of endorphins or GABA (gamma-amino-butyric acid), exerting a pain-relieving effect. It also plays a critical role in opiate-induced antinociception, probably through activation of descending inhibition (86, 87).The administration of systemic antidepressants such as selective serotonin reuptake inhibitors (SSRIs) can increase the concentration of 5-HT in the spinal cord, producing anti-hypersensitive effects in rodent models of neuropathic pain (88). Studies have indicated that 5-HT receptor subtypes play distinct roles in neuropathic pain. 5-HT2A and 5-HT4 may promote neuropathic pain, while 5-HT1A, 5-HT1B/1D, 5-HT2C, and 5-HT7 may exert inhibitory effects. The role of 5-HT3 in neuropathic pain could be dual (89, 90). In fact, the actions of these receptors are likely to be synergistic. The summary of the roles of 5-HT receptors in neuropathic pain does not account for all research findings. Several factors, such as the specific cause of neuropathic pain and the concentration of 5-HT in the body, may influence which receptor plays a dominant role.
Gut and brain interactions manifest in multiple aspects and at multiple levels (Figure 1). In PHN patients, pain is modulated through the ENS and intestinal metabolites, while the disease also affects inflammation levels in the nervous system and intestinal metabolite levels. However, it must be noted that this interaction is complex and subject to individual differences. The specific underlying mechanisms require further research.
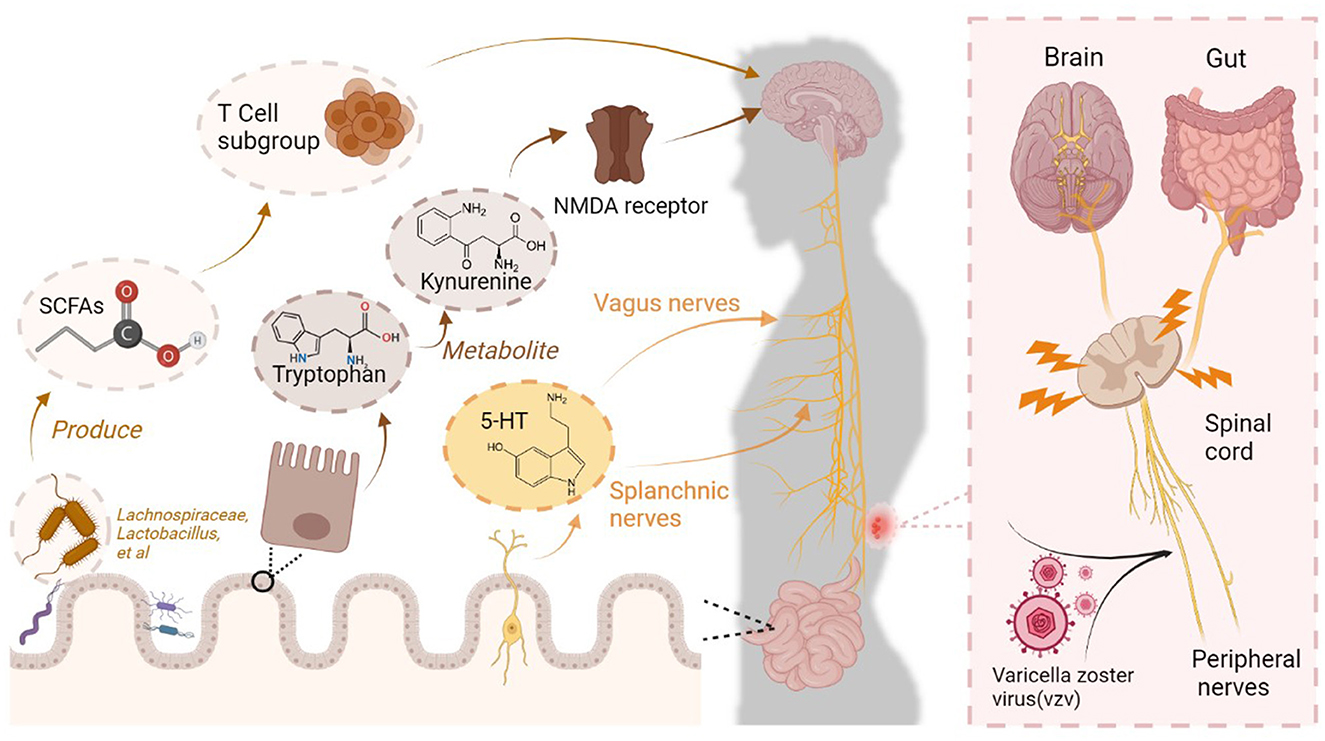
Figure 1. Complex connections between the brain and intestine in PHN patients. Varicella zoster virus (VZV) invades and damages peripheral nerves, ascending to the brain through the spinal cord. Central sensitization in the brain contributes to PHN becoming a refractory condition. This process is accompanied by changes in gut microbiota colonization, which involves mechanisms for responding to pain. Specific bacteria in the gut produce short-chain fatty acids (SCFAs), which regulate the level of neuroinflammation by modulating T-cell subsets, thereby adjusting pain sensitivity. The enteric nervous system (ENS) receives neurotransmitters such as 5-HT, which transmit nociceptive signals to the central nervous system via the vagus and splanchnic nerves. The gut metabolite kynurenine enters the central nervous system via the circulation and acts as an N-methyl-D-aspartate (NMDA) receptor antagonist, exerting analgesic effects.
As mentioned earlier, due to the specific infection of VZV in humans, it is impossible to replicate patients' pathological and physiological states in rodent models. Clinical studies based on patient data also have certain limitations. Further in-depth research requires support from primary experimental results. Consequently, research on PHN is subject to certain limitations. PHN is the only type of neuropathic pain involving one to two peripheral nerve segments caused by viral infection. However, in many studies on the central mechanisms of neuropathic pain, PHN has yet to be studied separately. The generalizability of new drugs developed based on related research on neuropathic pain in PHN patients is unknown. Therefore, it is necessary to conduct large-scale studies on neuroimaging, neurophysiology, neuroimmunology, and other related aspects of PHN to fill the gaps in this field. Currently, more research on the gut microbiota of PHN patients is needed, leading to a suboptimal representation of the microbiological characteristics of patients' intestines. Additionally, there is a lack of mechanistic studies on the interaction between the gut and central nervous system in PHN patients. Many of the mechanisms mentioned in the current literature are speculative and derived from neuropathic pain-based hypotheses, and their validity needs further experimental design and research.
This article elucidated the brain-gut regulatory mechanisms in PHN patients by reviewing the pathogenic mechanisms, neurophysiology, neuroimaging, and dominant bacterial strains associated with PHN. At the brain level, the thalamus is responsible for the ascending transmission of pain information in PHN patients; the somatosensory cortex is involved in pain perception; the ACC, PFC, and amygdala contribute to the formation of unpleasant emotions; and the PAG and RVM exert descending inhibition on pain signals. At the gut level, predominant bacterial species in the gut of PHN patients may regulate the levels of neuroinflammation, enhance pain thresholds, and alleviate tactile hypersensitivity to some extent by participating in the synthesis of short-chain fatty acids, thereby modulating the quantity of T-cell subsets. A study on PHN at the brain-gut level revealed that interactions between the gut and the brain manifest at multiple aspects and levels. In PHN patients, pain is modulated through the ENS and intestinal metabolites, while the disease also affects inflammation levels in the nervous system and intestinal metabolite levels. Although this review has certain limitations in that the role of other brain regions and bacteria in PHN lacks satisfactory interpretation due to insufficient supplementation of existing works, this review could provide a theoretical basis for proposing new treatment methods and direct future research endeavors toward PHN. Future studies investigating the brain-gut mechanisms of PHN could provide potential therapeutic strategies for disease management and improvement of patient quality of life.
PZ: Conceptualization, Data curation, Formal analysis, Writing – original draft. CZ: Conceptualization, Data curation, Funding acquisition, Writing – original draft. BZ: Investigation, Funding acquisition, Writing – review & editing. YL: Methodology, Project administration, Writing – review & editing. DZ: Conceptualization, Supervision, Validation, Writing – review & editing. HX: Funding acquisition, Resources, Supervision, Validation, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Sichuan Science and Technology Program (2023ZYD01680), National Natural Science Foundation of China (82201363), Science and Technology Department of Sichuan Province (No. 2023NSFSC1065), and Sichuan Provincial Administration of Traditional Chinese Medicine for Scientific and Technological Research (2023MS618).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2025.1535136/full#supplementary-material
1. Scholz J, Finnerup NB, Attal N, Aziz Q, Baron R, Bennett MI, et al. The IASP classification of chronic pain for ICD-11: chronic neuropathic pain. Pain. (2019) 160:53–9. doi: 10.1097/j.pain.0000000000001365
2. Birlea M, Nagel MA, Khmeleva N, Choe A, Kleinschmidt-Demasters B, Hevner R, et al. Varicella-zoster virus trigeminal ganglioneuritis without rash. Neurology. (2014) 82:90–2. doi: 10.1212/01.wnl.0000438228.48470.86
3. Nagel MA, Gilden D. Complications of varicella zoster virus reactivation. Curr Treat Options Neurol. (2013) 15:439–53. doi: 10.1007/s11940-013-0246-5
4. Gruver C, Guthmiller KB. Postherpetic Neuralgia. Treasure Island (FL): StatPearls Publishing LLC. (2025).
5. Cohen SP, Mao J. Neuropathic pain: mechanisms and their clinical implications. BMJ. (2014) 348:f7656. doi: 10.1136/bmj.f7656
6. Yang F, Yu S, Fan B, Liu Y, Chen YX, Kudel I, et al. The epidemiology of herpes zoster and postherpetic neuralgia in china: results from a cross-sectional study. Pain Ther. (2019) 8:249–59. doi: 10.1007/s40122-019-0127-z
7. Chan PKS, Wong MCS, Chan M, Ching K, Giannelos N, Ng C. Public health impact of herpes zoster vaccination on older adults in Hong Kong. Hum Vaccin Immunother. (2023) 19:2176065. doi: 10.1080/21645515.2023.2176065
8. Yawn BP, Gilden D. The global epidemiology of herpes zoster. Neurology. (2013) 81:928–30. doi: 10.1212/WNL.0b013e3182a3516e
9. Hadley GR, Gayle JA, Ripoll J, Jones MR, Argoff CE, Kaye RJ, et al. Post-herpetic neuralgia: a review. Curr Pain Headache Rep. (2016) 20:17. doi: 10.1007/s11916-016-0548-x
10. Jeon H, Lee S, Kim SA, Lee U, Lee S. Oral herbal medicine for treatment of postherpetic neuralgia: a protocol for systematic review and meta-analysis. Medicine. (2022)101:e32484. doi: 10.1097/md.0000000000032484
11. Watson PNC, Watt VR, Chipman M, Birkett N, Evans RJ. The prognosis with postherpetic neuralgia. Pain. (1991) 46:195–9. doi: 10.1016/0304-3959(91)90076-A
12. Mayer EA, Nance K, Chen S. The gut-brain axis. Annu Rev Med. (2022) 73:439–53. doi: 10.1146/annurev-med-042320-014032
13. Collins SM, Surette M, Bercik P. The interplay between the intestinal microbiota and the brain. Nat Rev Microbiol. (2012) 10:735–42. doi: 10.1038/nrmicro2876
14. Bercik P, Collins SM, Verdu EF. Microbes and the gut-brain axis. Neurogastroenterol Motil. (2012) 24:405–13. doi: 10.1111/j.1365-2982.2012.01906.x
16. Pappas TN, Taché Y, Debas HT. Opposing central and peripheral actions of brain-gut peptides: a basis for regulation of gastric function. Surgery. (1985) 98:183–90.
17. Glavin GB, Szabo S. Dopamine in gastrointestinal disease. Dig Dis Sci. (1990) 35:1153–61. doi: 10.1007/BF01537589
18. Jaworek J, Brzozowski T, Konturek SJ. Melatonin as an organoprotector in the stomach and the pancreas. J Pineal Res. (2005) 38:73–83. doi: 10.1111/j.1600-079X.2004.00179.x
19. Glavin GB, Murison R, Overmier JB, Pare WP, Bakke HK, Henke PG, et al. The neurobiology of stress ulcers. Brain Res Brain Res Rev. (1991) 16:301–43. doi: 10.1016/0165-0173(91)90012-W
20. Mearin F, Cucala M, Azpiroz F, Malagelada JR. The origin of symptoms on the brain-gut axis in functional dyspepsia. Gastroenterology. (1991) 101:999–1006. doi: 10.1016/0016-5085(91)90726-2
21. Kilkens TO, Honig A, van Nieuwenhoven MA, Riedel WJ, Brummer RJ. Acute tryptophan depletion affects brain-gut responses in irritable bowel syndrome patients and controls. Gut. (2004) 53:1794–800. doi: 10.1136/gut.2004.041657
22. Hollander D. Inflammatory bowel diseases and brain-gut axis. J Physiol Pharmacol. (2003) 54 Suppl 4:183–90.
23. Stasi C, Orlandelli E. Role of the brain-gut axis in the pathophysiology of Crohn's disease. Dig Dis. (2008) 26:156–66. doi: 10.1159/000116774
24. Agostini A, Filippini N, Cevolani D, Agati R, Leoni C, Tambasco R, et al. Brain functional changes in patients with ulcerative colitis: a functional magnetic resonance imaging study on emotional processing. Inflamm Bowel Dis. (2011) 17:1769–77. doi: 10.1002/ibd.21549
25. Suzuki K, Jayasena CN, Bloom SR. Obesity and appetite control. Exp Diabetes Res. (2012) 2012:824305. doi: 10.1155/2012/824305
26. Camilleri M, Di Lorenzo C. Brain-gut axis: from basic understanding to treatment of IBS and related disorders. J Pediatr Gastroenterol Nutr. (2012) 54:446–53. doi: 10.1097/MPG.0b013e31823d34c3
27. Grover M, Drossman DA. Centrally acting therapies for irritable bowel syndrome. Gastroenterol Clin North Am. (2011) 40:183–206. doi: 10.1016/j.gtc.2010.12.003
28. Bravo JA, Forsythe P, Chew MV, Escaravage E, Savignac HM, Dinan TG, et al. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci U S A. (2011) 108:16050–5. doi: 10.1073/pnas.1102999108
29. Ohland CL, Kish L, Bell H, Thiesen A, Hotte N, Pankiv E, et al. Effects of Lactobacillus helveticus on murine behavior are dependent on diet and genotype and correlate with alterations in the gut microbiome. Psychoneuroendocrinology. (2013) 38:1738–47. doi: 10.1016/j.psyneuen.2013.02.008
30. Mayer EA, Padua D, Tillisch K. Altered brain-gut axis in autism: comorbidity or causative mechanisms? Bioessays. (2014) 36:933–9. doi: 10.1002/bies.201400075
31. Klarer M, Arnold M, Günther L, Winter C, Langhans W, Meyer U. Gut vagal afferents differentially modulate innate anxiety and learned fear. J Neurosci. (2014) 34:7067–76. doi: 10.1523/JNEUROSCI.0252-14.2014
32. Hamdani N, Boukouaci W, Hallouche MR, Charron D, Krishnamoorthy R, Leboyer M, et al. Resolution of a manic episode treated with activated charcoal: Evidence for a brain-gut axis in bipolar disorder. Aust N Z J Psychiatry. (2015) 49:1221–3. doi: 10.1177/0004867415595873
33. Slyepchenko A, Carvalho AF, Cha DS, Kasper S, McIntyre RS. Gut emotions - mechanisms of action of probiotics as novel therapeutic targets for depression and anxiety disorders. CNS Neurol Disord Drug Targets. (2014) 13:1770–86. doi: 10.2174/1871527313666141130205242
34. Luna RA, Foster JA. Gut brain axis: diet microbiota interactions and implications for modulation of anxiety and depression. Curr Opin Biotechnol. (2015) 32:35–41. doi: 10.1016/j.copbio.2014.10.007
35. Turna J, Grosman Kaplan K, Anglin R, Van Ameringen M. “What's bugging the gut in OCD? A review of the gut microbiome in obsessive-compulsive disorder. Depress Anxiety. (2016) 33:171–8. doi: 10.1002/da.22454
36. Rueda-Ruzafa L, Cruz F, Cardona D, Hone AJ, Molina-Torres G, Sánchez-Labraca N, et al. Opioid system influences gut-brain axis: Dysbiosis and related alterations. Pharmacol Res. (2020) 159:104928. doi: 10.1016/j.phrs.2020.104928
37. Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E, et al. Gut microbiota are related to Parkinson's disease and clinical phenotype. Mov Disord. (2015) 30:350–8. doi: 10.1002/mds.26069
38. Pei Y, Lu Y, Li H, Jiang C, Wang L. Gut microbiota and intestinal barrier function in subjects with cognitive impairments: a cross-sectional study. Front Aging Neurosci. (2023) 15:1174599. doi: 10.3389/fnagi.2023.1174599
39. Joscelyn J, Kasper LH. Digesting the emerging role for the gut microbiome in central nervous system demyelination. Mult Scler. (2014) 20:1553–9. doi: 10.1177/1352458514541579
40. O'Mahony SM, Bulmer DC, Coelho AM, Fitzgerald P, Bongiovanni C, Lee K, et al. 5-HT(2B) receptors modulate visceral hypersensitivity in a stress-sensitive animal model of brain-gut axis dysfunction. Neurogastroenterol Motil. (2010) 22:573–8, e124. doi: 10.1111/j.1365-2982.2009.01432.x
41. Sarnelli G, Vandenberghe J, Tack J. Visceral hypersensitivity in functional disorders of the upper gastrointestinal tract. Dig Liver Dis. (2004) 36:371–6. doi: 10.1016/j.dld.2004.01.018
42. Kirkup AJ, Brunsden AM, Grundy D. Receptors and transmission in the brain-gut axis: potential for novel therapies. I Receptors on visceral afferents. Am J Physiol Gastrointest Liver Physiol. (2001) 280:G787–94. doi: 10.1152/ajpgi.2001.280.5.G787
43. Theodorou V, Ait Belgnaoui A, Agostini S, Eutamene H. Effect of commensals and probiotics on visceral sensitivity and pain in irritable bowel syndrome. Gut Microbes. (2014) 5:430–6. doi: 10.4161/gmic.29796
44. Vermeulen W, De Man JG, Pelckmans PA, De Winter BY. Neuroanatomy of lower gastrointestinal pain disorders. World J Gastroenterol. (2014) 20:1005–20. doi: 10.3748/wjg.v20.i4.1005
45. Slim M, Calandre EP, Rico-Villademoros F. An insight into the gastrointestinal component of fibromyalgia: clinical manifestations and potential underlying mechanisms. Rheumatol Int. (2015) 35:433–44. doi: 10.1007/s00296-014-3109-9
46. Lee KN, Lee OY. Intestinal microbiota in pathophysiology and management of irritable bowel syndrome. World J Gastroenterol. (2014) 20:8886–97. doi: 10.3748/wjg.v20.i10.2456
47. Quigley EM, Shanahan F. The future of probiotics for disorders of the brain-gut axis. Adv Exp Med Biol. (2014) 817:417–32. doi: 10.1007/978-1-4939-0897-4_19
48. Finnerup NB, Kuner R, Jensen TS. Neuropathic pain: from mechanisms to treatment. Physiol Rev. (2021) 101:259–301. doi: 10.1152/physrev.00045.2019
49. Grundy L, Erickson A, Brierley SM. Visceral pain. Annu Rev Physiol. (2019) 81:261–84. doi: 10.1146/annurev-physiol-020518-114525
50. Bourne S, Machado AG, Nagel SJ. Basic anatomy and physiology of pain pathways. Neurosurg Clin N Am. (2014) 25:629–38. doi: 10.1016/j.nec.2014.06.001
51. Garcia-Larrea L, Peyron R. Pain matrices and neuropathic pain matrices: a review. Pain. (2013) 154 Suppl 1:S29–s43. doi: 10.1016/j.pain.2013.09.001
52. Labrakakis C. The role of the insular cortex in pain. Int J Mol Sci. (2023) 24:5736. doi: 10.3390/ijms24065736
53. Huang J, Gadotti VM, Chen L, Souza IA, Huang S, Wang D, et al. A neuronal circuit for activating descending modulation of neuropathic pain. Nat Neurosci. (2019) 22:1659–68. doi: 10.1038/s41593-019-0481-5
54. Chen Q, Heinricher MM. Descending control mechanisms and chronic pain. Curr Rheumatol Rep. (2019) 21:13. doi: 10.1007/s11926-019-0813-1
55. Dai H, Jiang C, Wu G, Huang R, Jin X, Zhang Z, et al. A combined DTI and resting state functional MRI study in patients with postherpetic neuralgia. Jpn J Radiol. (2020) 38:440–50. doi: 10.1007/s11604-020-00926-4
56. Harris RE, Sundgren PC, Craig AD, Kirshenbaum E, Sen A, Napadow V, et al. Elevated insular glutamate in fibromyalgia is associated with experimental pain. Arthritis Rheum. (2009) 60:3146–52. doi: 10.1002/art.24849
57. Ning L, Liu R, Li S, Shan G, Du H, Zhang J, et al. Increased risk of herpes zoster infection in patients with inflammatory bowel disease: a meta-analysis of cohort studies. Eur J Clini Microbiol Infect Dis. (2020) 39:219–27. doi: 10.1007/s10096-019-03706-9
58. Côté-Daigneault J, Peerani F, MacMahon E, Delaporte E, Rahier JF, Colombel JF. Management and prevention of herpes zoster in the immunocompromised inflammatory bowel disease patient: a clinical quandary. Inflamm Bowel Dis. (2016) 22:2538–47. doi: 10.1097/MIB.0000000000000902
59. Jiao B, Cao X, Zhang C, Zhang W, Yu S, Zhang M, et al. Alterations of the gut microbiota in patients with postherpetic neuralgia. AMB Express. (2023) 13:108. doi: 10.1186/s13568-023-01614-y
60. Zafar H, Saier JMH. Gut Bacteroides species in health and disease. Gut Microbes. (2021) 13:1–20. doi: 10.1080/19490976.2020.1848158
61. Bonomo RR, Cook TM, Gavini CK, White CR, Jones JR, Bovo E, et al. Fecal transplantation and butyrate improve neuropathic pain, modify immune cell profile, and gene expression in the PNS of obese mice. Proc Natl Acad Sci U S A. (2020) 117:26482–93. doi: 10.1073/pnas.2006065117
62. Nozu T, Miyagishi S, Nozu R, Takakusaki K, Okumura T. Butyrate inhibits visceral allodynia and colonic hyperpermeability in rat models of irritable bowel syndrome. Sci Rep. (2019) 9:19603. doi: 10.1038/s41598-019-56132-4
63. Russo R, De Caro C, Avagliano C, Cristiano C, La Rana G, Mattace Raso G, et al. Sodium butyrate and its synthetic amide derivative modulate nociceptive behaviors in mice. Pharmacol Res. (2016) 103:279–91. doi: 10.1016/j.phrs.2015.11.026
64. Lanza M, Filippone A, Casili G, Giuffrè L, Scuderi SA, Paterniti I, et al. Supplementation with SCFAs re-establishes microbiota composition and attenuates hyperalgesia and pain in a mouse model of NTG-induced migraine. Int J Mol Sci. (2022) 23:4847. doi: 10.3390/ijms23094847
65. Portune KJ, Benítez-Péez A, Del Pulgar EM, Cerrudo V, Sanz Y. Gut microbiota, diet, and obesity-related disorders-The good, the bad, and the future challenges. Mol Nutr Food Res. (2017) 61:1. doi: 10.1002/mnfr.201600252
66. Vacca M, Celano G, Calabrese FM, Portincasa P, Gobbetti M, De Angelis M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms. (2020) 8:573. doi: 10.3390/microorganisms8040573
67. Zhang J, Song L, Wang Y, Liu C, Zhang L, Zhu S, et al. Beneficial effect of butyrate-producing Lachnospiraceae on stress-induced visceral hypersensitivity in rats. J Gastroenterol Hepatol. (2019) 34:1368–76. doi: 10.1111/jgh.14536
68. Dürre P. Physiology and sporulation in clostridium. Microbiol Spectr. (2014) 2:Tbs-0010-2012. doi: 10.1128/microbiolspec.TBS-0010-2012
69. Gheshlaghi R, Scharer JM, Moo-Young M, Chou CP. Metabolic pathways of clostridia for producing butanol. Biotechnol Adv. (2009) 27:764–81. doi: 10.1016/j.biotechadv.2009.06.002
70. Bittner S, Hehlgans T, Feuerer M. Engineered Treg cells as putative therapeutics against inflammatory diseases and beyond. Trends Immunol. (2023) 44:468–83. doi: 10.1016/j.it.2023.04.005
71. Narushima S, Sugiura Y, Oshima K, Atarashi K, Hattori M, Suematsu M, et al. Characterization of the 17 strains of regulatory T cell-inducing human-derived Clostridia. Gut Microbes. (2014) 5:333–9. doi: 10.4161/gmic.28572
72. Olson KE, Mosley RL, Gendelman HE. The potential for treg-enhancing therapies in nervous system pathologies. Clin Exp Immunol. (2023) 211:108–21. doi: 10.1093/cei/uxac084
73. Corriero A, Giglio M, Inchingolo F, Moschetta A, Varrassi G, Puntillo F. Gut microbiota modulation and its implications on neuropathic pain: a comprehensive literature review. Pain Ther. (2023). doi: 10.1007/s40122-023-00565-3
74. Lake JI, Heuckeroth RO. Enteric nervous system development: migration, differentiation, and disease. Am J Physiol Gastrointest Liver Physiol. (2013) 305:G1–24. doi: 10.1152/ajpgi.00452.2012
75. Sharkey KA, Mawe GM. The enteric nervous system. Physiol Rev. (2023) 103:1487–564. doi: 10.1152/physrev.00018.2022
76. Nguyen TT, Baumann P, Tüscher O, Schick S, Endres K. The aging enteric nervous system. Int J Mol Sci. (2023) 24:9471. doi: 10.3390/ijms24119471
77. Toffa DH, Touma L, El Meskine T, Bouthillier A, Nguyen DK. Learnings from 30 years of reported efficacy and safety of vagus nerve stimulation (VNS) for epilepsy treatment: a critical review. Seizure. (2020) 83:104–23. doi: 10.1016/j.seizure.2020.09.027
78. González HFJ, Yengo-Kahn A, Englot DJ. Vagus nerve stimulation for the treatment of epilepsy. Neurosurg Clin N Am. (2019) 30:219–30. doi: 10.1016/j.nec.2018.12.005
79. Lulic D, Ahmadian A, Baaj AA, Benbadis SR, Vale FL. Vagus nerve stimulation. Neurosurg Focus. (2009) 27:E5. doi: 10.3171/2009.6.FOCUS09126
80. Taylor CP, Angelotti T, Fauman E. Pharmacology and mechanism of action of pregabalin: the calcium channel alpha2-delta (alpha2-delta) subunit as a target for antiepileptic drug discovery. Epilepsy Res. (2007) 73:137–50. doi: 10.1016/j.eplepsyres.2006.09.008
81. Roth W, Zadeh K, Vekariya R, Ge Y, Mohamadzadeh M. Tryptophan metabolism and gut-brain homeostasis. Int J Mol Sci. (2021) 22:2973. doi: 10.3390/ijms22062973
82. Tajti J, Szok D, Csáti A, Szabó, Tanaka M, Vécsei L. Exploring novel therapeutic targets in the common pathogenic factors in migraine and neuropathic Pain. Int J Mol Sci. (2023) 24:4114. doi: 10.3390/ijms24044114
83. Brito RG, Rasmussen LA, Sluka KA. Regular physical activity prevents development of chronic muscle pain through modulation of supraspinal opioid and serotonergic mechanisms. Pain Rep. (2017) 2:e618. doi: 10.1097/PR9.0000000000000618
84. Cortes-Altamirano JL, Olmos-Hernandez A, Jaime HB, Carrillo-Mora P, Bandala C, Reyes-Long S, et al. Review: 5-HT1, 5-HT2, 5-HT3 and 5-HT7 receptors and their role in the modulation of pain response in the central nervous system. Curr Neuropharmacol. (2018) 16:210–21. doi: 10.2174/1570159X15666170911121027
85. Crowell MD. Role of serotonin in the pathophysiology of the irritable bowel syndrome. Br J Pharmacol. (2004) 141:1285–93. doi: 10.1038/sj.bjp.0705762
86. Petroianu GA, Aloum L, Adem A. Neuropathic pain: Mechanisms and therapeutic strategies. Front Cell Dev Biol. (2023) 11:1072629. doi: 10.3389/fcell.2023.1072629
87. Dogrul A, Ossipov MH, Porreca F. Differential mediation of descending pain facilitation and inhibition by spinal 5HT-3 and 5HT-7 receptors. Brain Res. (2009) 1280:52–9. doi: 10.1016/j.brainres.2009.05.001
88. Obata H. Analgesic mechanisms of antidepressants for neuropathic pain. Int J Mol Sci. (2017) 18:2483. doi: 10.3390/ijms18112483
89. Viguier F, Michot B, Hamon M, Bourgoin S. Multiple roles of serotonin in pain control mechanisms-implications of 5-HT and other 5-HT receptor types. Eur J Pharmacol. (2013) 716:8–16. doi: 10.1016/j.ejphar.2013.01.074
Keywords: postherpetic neuralgia, brain-gut axis, mechanism, bacteria, microbiota, enteric nervous system
Citation: Zhang P, Zhang C, Zheng B, Liu Y, Zhang D and Xiao H (2025) The “brain-gut” mechanism of postherpetic neuralgia: a mini-review. Front. Neurol. 16:1535136. doi: 10.3389/fneur.2025.1535136
Received: 26 November 2024; Accepted: 17 February 2025;
Published: 10 March 2025.
Edited by:
Rainer Viktor Haberberger, University of Adelaide, AustraliaReviewed by:
Ulises Coffeen, National Institute of Psychiatry Ramon de la Fuente Muñiz (INPRFM), MexicoCopyright © 2025 Zhang, Zhang, Zheng, Liu, Zhang and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Xiao, eGlhb2hvbmdodWF4aUAxMjYuY29t; Dingkun Zhang, emhhbmdkazE0QHRzaW5naHVhLm9yZy5jbg==
†These authors have contributed equally to this work and share first authorship
‡ORCID: Yuntao Liu orcid.org/0009-0006-8778-2480
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.