 Yingquan Liu
Yingquan Liu Peijia Hu2*
Peijia Hu2* Hongliang Cheng
Hongliang Cheng Fangyuan Xu
Fangyuan Xu Yu Ye
Yu Ye
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 24 January 2025
Sec. Stroke
Volume 16 - 2025 | https://doi.org/10.3389/fneur.2025.1514394
This article is part of the Research TopicExploring Immune Cell Roles in Cardiac Repair and RemodelingView all 15 articles
Ischemic stroke (IS), a leading cause of disability and mortality worldwide, remains a significant challenge due to its complex pathogenesis. Glycolysis, a central metabolic pathway, plays a critical role in bridging the gap between metabolic dysfunction and neurological impairment. During ischemic conditions, glycolysis replaces oxidative phosphorylation as the primary energy source for brain tissue. However, in the ischemia–reperfusion state, neuronal cells show a particular reliance on aerobic glycolysis. Immune cells, such as monocytes, also contribute to atheromatous plaque formation and thrombi through increased aerobic glycolysis. Given glycolysis’s involvement in various pathological stages of IS, it offers the potential for improved diagnosis, treatment, and prevention. This review comprehensively explores the role of glycolysis in different phases of IS, addresses existing controversies, and discusses its diagnostic and therapeutic applications. By elucidating the intricate relationship between glycolysis and IS, this review aims to provide novel insights for future research and clinical advancements.
Stroke, a prevalent cerebrovascular disease characterized by sudden onset neurological deficits, is a significant global health concern. Primarily caused by damage to cerebral blood vessels, strokes result in focal or diffuse brain tissue injury. With over 10 million new cases and 6.55 million deaths annually, stroke ranks as the second leading cause of death worldwide, surpassed only by ischemic heart disease (1–3). The enduring impact of stroke extends beyond mortality, as up to 50% of survivors experience long-term disability, making it the third leading cause of disability globally (4, 5). Strokes are categorized into ischemic strokes (IS), resulting from disrupted blood supply to the brain, and hemorrhagic strokes, caused by bleeding from a ruptured blood vessel. Ischemic strokes account for approximately 85% of all stroke cases (6).
The underlying pathological mechanism of IS is the formation of thrombus in blood vessels. This leads to disruption of the blood supply to an area of the brain, leading to localized necrosis of brain tissue and neuronal damage. Three main factors are known to cause IS: (1) Cerebrovascular atherosclerotic plaques and their rupture (in nearly half of the cases); (2) Cardiogenic cerebral infarction (in about 20% of the cases); (3) Cavernous cerebral infarction caused by small-vessel lesions (in about 20% of the cases) (7). In addition, other factors, such as vasculitis, extracranial arterial entrapment, and pathogenic infections, can lead to IS (8, 9). Current management of acute ischemic stroke (AIS) primarily involves recombinant tissue plasminogen activator (rt-PA) intravenous thrombolysis and/or mechanical thrombectomy (10). While these approaches can be practical, their success is limited to a small proportion of patients due to narrow time windows and other factors (11). The urgent need for innovative therapeutic strategies to address ischemic stroke is underscored by these limitations.
The primary metabolic pathways for glucose sugars include glycolysis, the pentose phosphate pathway (PPP), and oxidative phosphorylation (OXPHOS). Under normal conditions, the brain primarily relies on OXPHOS for energy, accounting for approximately 95% of the ATP required to maintain its function (12). However, in AIS, OXPHOS is inhibited, leading to disrupted ATP production and glucose metabolism. During this period, neurons predominantly depend on anaerobic metabolic processes like glycolysis for energy (13, 14). Glycolysis degrades glucose and glycogen into pyruvate, generating ATP. In the presence of adequate oxygenation, pyruvate can produce significant amounts of ATP through the tricarboxylic acid (TCA) cycle. Nevertheless, during hypoxia or periods of high energy expenditure, pyruvate is reduced to lactate (15). While excessive lactic acid accumulation in ischemic brain tissue can contribute to increased neurological damage (15, 16). Recent studies have demonstrated that lactate can have a protective effect on mouse neurons during ischemia and hypoxia in AIS (17).
Glycolysis is a metabolic pathway primarily occurring under anaerobic or hypoxic conditions (18). Notably, during ischemia–reperfusion (IR), when blood oxygenation to ischemic tissue is restored, neurons exhibit a preference for glycolysis as their primary energy source, even in the presence of adequate oxygen supply (19, 20). This metabolic shift in glucose utilization was initially observed in cancer cells and is referred to as the Warburg effect (also known as aerobic glycolysis, which involves the conversion of glucose to lactate in the presence of oxygen) (21, 22). While previous studies suggested that aerobic glycolysis following IR might have detrimental effects on neurons, including exacerbated neuroinflammation and enhanced nerve damage (23, 24). Recent research has revealed that enhanced aerobic glycolysis can be protective for nerve cells and promote nerve damage repair in a mouse model of focal ischemic stroke (25).
The role of glycolysis in the pathophysiology of IS remains a subject of ongoing debate. This review aims to comprehensively examine recent research on glycolysis and IS, exploring its involvement in the progression of ischemic stroke, its diagnostic value, and its potential therapeutic applications. We will also address current controversies and offer our perspective. By synthesizing this information, we hope to provide valuable insights for future scientific research and clinical approaches to the diagnosis and treatment of IS.
The pathogenesis of IS remains incompletely understood. However, current research suggests it primarily involves inflammatory responses, oxidative stress, excitotoxicity, apoptosis, and other related factors (26). Following IS, focal ischemia triggers a robust inflammatory response, primarily mediated by the activation of microglia and astrocytes (27). These cells subsequently release pro-inflammatory mediators such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), and matrix metalloproteinases (MMPs) from injured tissues, which in turn promote the overexpression of genes involved in inflammation, neurotoxicity, and glycolysis (28, 29). Notably, this is different from the inflammatory response caused by IS due to pathogen infection. Bacterial virulence factors and pathogens directly breach the blood–brain barrier, subsequently triggering leukocytes to respond to infection (9). Cytokines released in this way cause a series of immune injuries called pathogen-associated molecular patterns (PAMPs). Metabolism-related sterile inflammation induced by damaged tissue-derived danger signals after stroke onset is termed damage-associated molecular patterns (DAMPs) (30). The high-mobility box group 1 (HMGB1) is recognized as the representative DAMPs (31). It is released upon cell and tissue necrosis and is actively produced by immune cells (32). In the core region of cerebral infarction, severe ischemia and hypoxia-induced metabolic disturbances lead to an increased release and impaired reuptake of glutamate, the primary excitatory neurotransmitter of the nervous system. This excessive glutamate accumulation in ischemic brain regions subsequently activates the glutamate-gated ion channel N-methyl-D-aspartate receptor (NMDAR), resulting in the influx of calcium and the activation of downstream cell death signaling pathways. This, in turn, leads to the aberrant activation of numerous calcium-dependent enzymes in the brain, initiating neuronal necrosis, apoptosis, and autophagy processes (33). During cerebral IR, neuronal mitochondrial dysfunction and excessive reactive oxygen species (ROS) accumulation generate oxidative stress and release large amounts of free radicals (e.g., nitric oxide (NO), peroxynitrite), further exacerbating excitotoxicity and leading to neuronal apoptosis and death (26).
In the brain, nerve cells are divided into two main categories: neurons and glial cells. Glial cells are further divided into astrocytes, microglia, and oligodendrocytes (34). They all play a crucial role in the course of IS. However, glial cells may have either protective or damaging effects on neurons in IS (given the relative paucity of studies of oligodendrocytes in IS and glycolysis, they will not be addressed in this review). Figure 1 summarises the main relationships between neurons and glial cells.
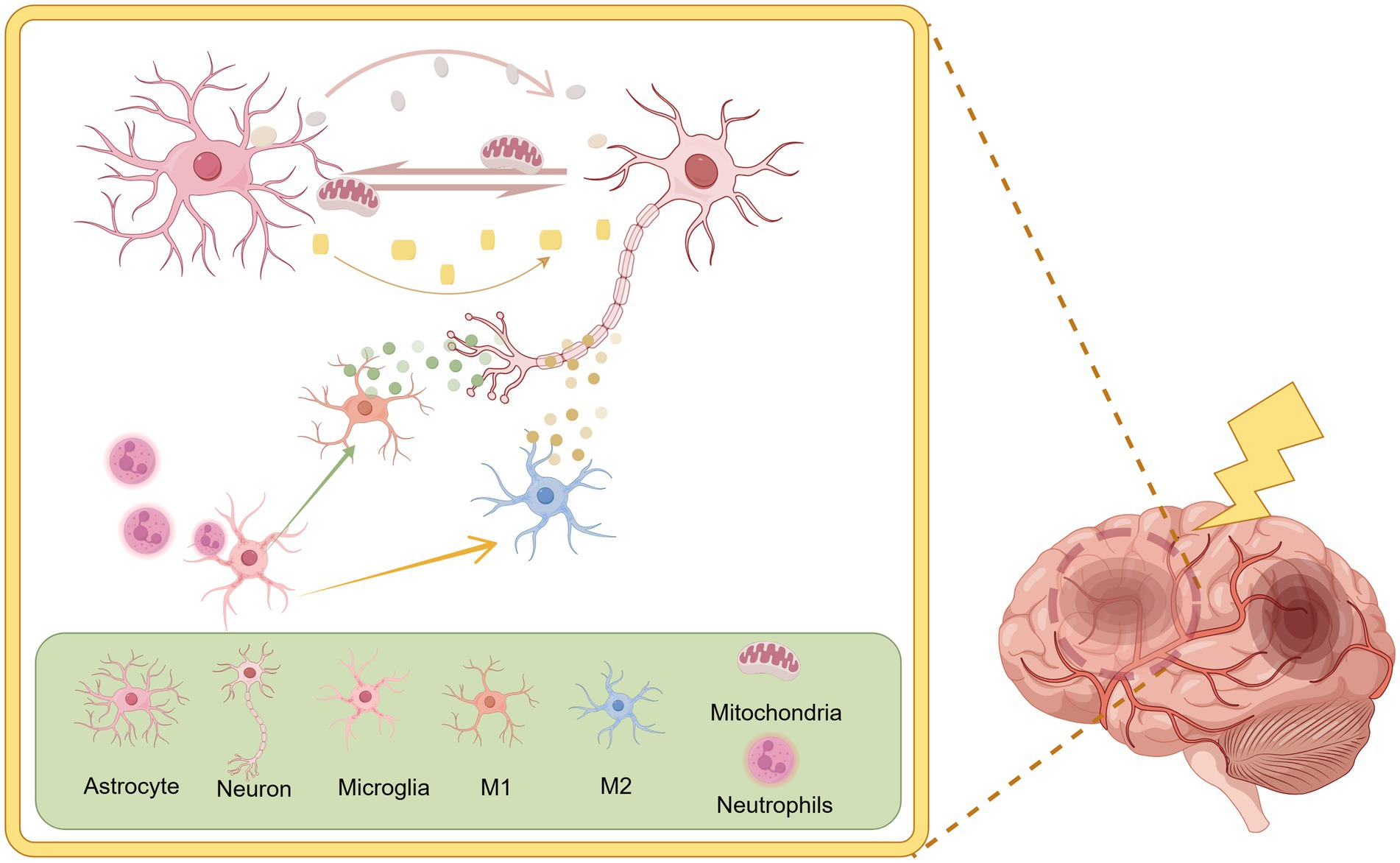
Figure 1. There is a complex regulatory relationship between neurons and glial cells. Activated microglia can differentiate into pro-inflammatory phenotypes (M1) and anti-inflammatory phenotypes (M2). Microglia also possess immunophagocytic and surveillance functions, resulting in different regulatory effects on neurons. Astrocytes have been shown to facilitate the transfer of energy and antioxidants to neurons, thereby providing crucial support for their function. By Figdraw.
Microglia are the main nerve cells in the brain responsible for immunity and are important for normal brain functioning (35). During the initiation and progression of acute IS, the neuroinflammatory response induced by microglial activation is of primary concern (36). Activated microglia exhibit two distinct phenotypes. One is the pro-inflammatory type (M1), which triggers neuroinflammatory responses and tissue damage by secreting pro-inflammatory factors. The other is the anti-inflammatory type (M2), which releases anti-inflammatory factors to provide protection to nerves and promote the repair of damaged tissues (37). Microglia in IS usually present an M1 phenotype, a state in which they have a destructive effect on neurons and are detrimental to the recovery of brain tissue functions (38). Besides, microglia have immunosurveillance and phagocytosis functions and are responsible for removing pro-inflammatory cells that are over-activated in IS (39).
Astrocytes are also the major glycogen storage units in the brain. Physiologically they consume large amounts of glucose and produce lactate. Strong aerobic glycolysis is present regardless of oxygen availability (29, 40). Thus, compared to neurons, astrocytes exhibit greater tolerance to nutrient depletion and oxidative stress (29). Reactive astrocytes (activated astrocytes) after IS onset rely on glycolysis to survive in large numbers and to play a regulatory role (41). The survival of neuronal cells after IS onset is closely related to the regulatory role of astrocytes. This regulatory role is mainly reflected in the following aspects: (1) Glycolytic processes are increased in morpho glial cells during hypoxia, producing small concentrations of lactate that are delivered to neurons for their utilization (42). This process is called the astrocyte-neuron lactate shuttle (ANLS) (43). (2) Neurons can release damaged mitochondria and transfer them to astrocytes for processing, followed by the release of healthy mitochondria used to maintain the health of the holding neuron (44). However, when ischemic damage is severe, astrocytes also release damaged mitochondria to neurons, which in turn affects neuronal health (45). (3) Glutathione (GSH) concentration in astrocytes is twice that of neurons and maintains neuronal resistance to oxidative stress during ischemia and hypoxia (29). (4) Reactive astrocytes release neuroprotective agents during the onset of cerebral ischemia. Whereas overproliferating astrocytes form a scar in the later stages of cerebral ischemia and physically impede recovery from neurological injury (46).
How to regulate the phenotypic transition of microglia and the functional role of astrocytes on neurons. Thus, utilization of these cells with dual roles has become the focus of current research.
It is well-established that impaired glucose and oxygen delivery during cerebral ischemia disrupts oxidative phosphorylation. Consequently, the brain rapidly transitions its primary metabolic strategy from oxidative phosphorylation to glycolysis to fulfill the energy demands of the ischemic region. However, the increased glycolysis during cerebral ischemia has been associated with detrimental effects on salvageable tissue survival, as it can lead to lactate accumulation and ROS production (47). This phenomenon is prevalent in both clinical and experimental settings, particularly in stroke patients with hyperglycemia (16).
There is an inter-regulatory relationship between glucose metabolism and the immune system (48, 49). Aerobic glycolysis was found to be an important factor in activating microglia and shifting them to the M1-polarized state, promoting their pro-inflammatory effects (24, 50). In addition, there is evidence that microglia phagocytosis is critical for counteracting neutrophil tissue damage during the reperfusion period of IS. Microglia will preferentially utilize aerobic glycolysis for energy supply over OXPHOS after reperfusion. Reduced microglial phagocytosis due to a relative lack of ATP supply (51). Overproliferation of reactive astrocytes after ischemia leads to the production of glial scar. Glycolysis may accelerate this process (46, 52).
It is now understood that glutamate- or NMDA-mediated excitotoxicity triggers neuronal death by stimulating the expression of relevant apoptotic proteins (53). This process is accompanied by the activation of glycolysis (54). In ischemic tissues, excessive accumulation of calcium ions in mitochondria leads to depolarization and dysfunction of their membranes (53). Mitochondrial autophagy is crucial for maintaining neuronal bioenergetic homeostasis by effectively removing damaged mitochondria. However, highly active glycolysis following ischemia can disrupt this repair mechanism (55).
The aerobic glycolysis pathway is no longer exclusively associated with cancer cells. Emerging evidence suggests that stem cells, immune cells, and other cell types also utilize aerobic glycolysis for their physiological or pathological functions (56). Neural progenitor cells (NPCs) have been shown to have limited capacity for aerobic glycolysis in hypoxic environments, leading to their preferential differentiation into glial cells (57). However, by inhibiting aerobic glycolysis, NPCs can be effectively promoted to utilize pyruvate for energy production and drive the oxidative phosphorylation process, thereby favoring their differentiation toward neurons (58). This suggests a potential role for aerobic glycolysis in nerve damage repair after stroke. Additionally, aerobic glycolysis is significantly involved in the formation of atherosclerotic plaques and arterial and venous thrombi through its regulation of immune cell proliferation and activation, primarily involving monocytes/macrophages, neutrophils, and platelets (39, 59–62). Consequently, aerobic glycolysis profoundly influences the probability of ischemic stroke occurrence and recurrence.
The glycolytic pathway is regulated by three key rate-limiting enzymes: Hexokinase (HK), Phosphofructokinase (PFK), and Pyruvate kinase (PK), which catalyze irreversible reactions (18). While pyruvate production marks the end of the glycolytic pathway, the process does not necessarily cease. Lactate dehydrogenase (LDH) subsequently regulates the reversible conversion of pyruvate to lactate (63). Although pyruvate dehydrogenase (PDH) and pyruvate dehydrogenase kinase (PDK) are located in the mitochondria, they influence glycolysis by regulating the flux of pyruvate into the TCA cycle, which in turn affects glycolytic activity (64). Growing evidence suggests that these enzymes and their products play significant roles in the pathophysiological processes of IS through complex regulatory mechanisms. Figure 2 summarises the contributions of glycolysis-related enzymes and products to IS.
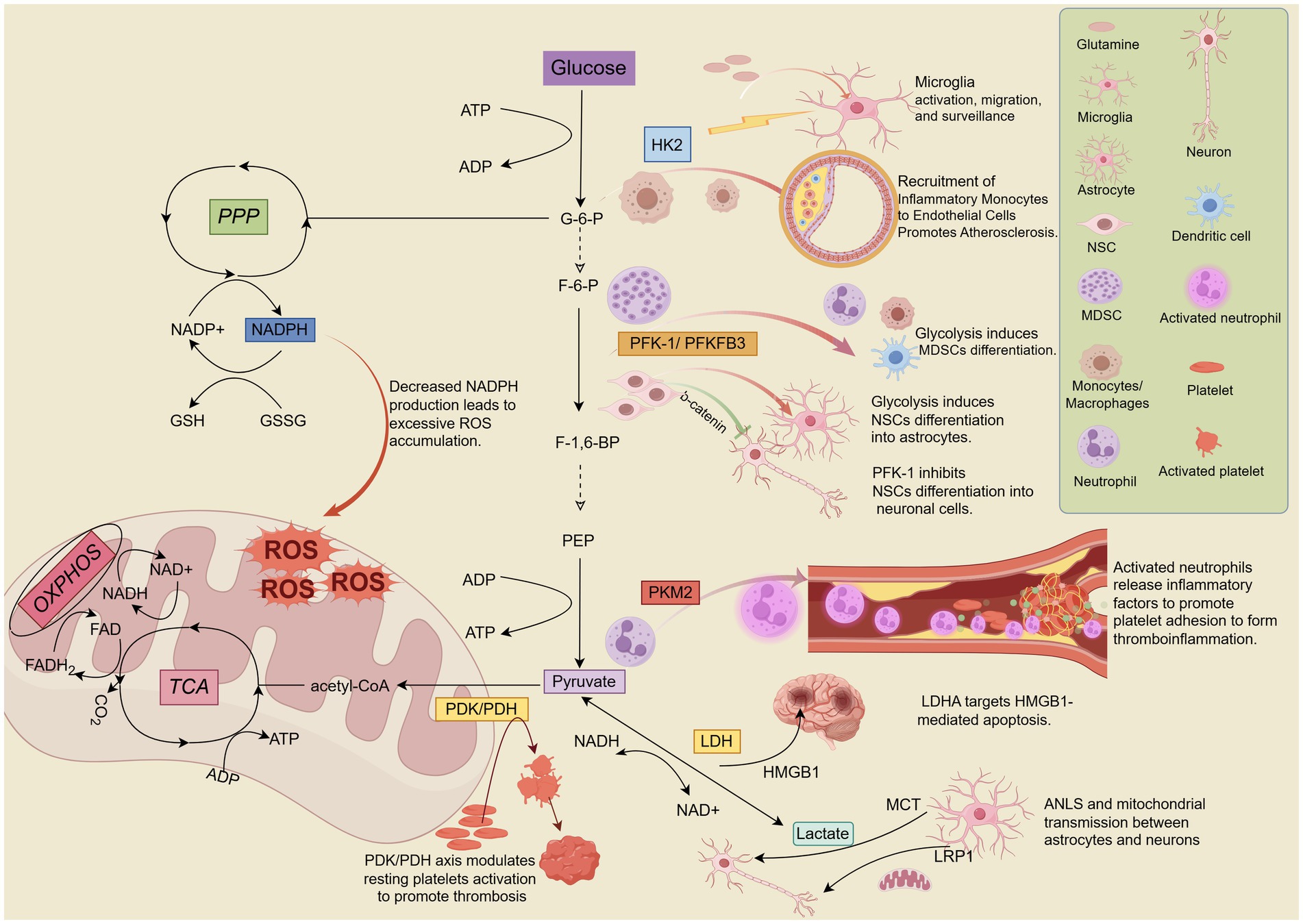
Figure 2. Hexokinase 2 (HK2), phosphofructokinase-1/6 (PFK-1/PFKFB3), pyruvate kinase M2 (PKM2), pyruvate dehydrogenase kinase/pyruvate dehydrogenase (PDK/PDH), and lactate dehydrogenase A (LDHA) are key enzymes and kinases involved in the regulation of glycolysis. These enzymes play crucial roles in the pathophysiological processes of IS, including neuroinflammation, mitochondrial oxidative stress, neuronal differentiation, apoptosis, and others. Lactate, the end product of glycolysis, directly or indirectly influences lactate shuttling and mitochondrial transport between neurons and astrocytes, contributing significantly to the progression of pathological mechanisms in IS. By Figdraw.
Hexokinase is the initial enzyme in the glucose metabolic pathway and the first key rate-limiting enzyme in glycolysis. It catalyzes the phosphorylation of glucose, consuming ATP to produce glucose-6-phosphate (G-6-P) (65). The formation of G-6-P initiates the major pathways of glucose utilization: glycolysis, PPP, and OXPHOS. Therefore, HK is considered a key regulator of glucose metabolism (66). Mammalian tissues express five hexokinase isozymes: HK1, HK2, HK3, HK4, and HK structural domain component 1 (HKDC1), each with distinct tissue distribution, biochemical profiles, and catalytic activities (67, 68). HK1 is widely expressed in mammalian tissues, particularly the brain, and is closely linked to the activation of inflammatory pathways in neurological diseases. It is also known as “brain hexokinase” but is not significantly regulated by most physiological, hormonal, and metabolic factors (68–70). While HK2 has limited expression in most normal tissues, it is frequently overexpressed in various cancer tissues and has recently been found to be overexpressed in ischemic stroke brain tissues (68, 71, 72). HK3 is highly expressed in bone marrow, lung, liver, and kidney (73). HK4 is predominantly expressed in hepatic and pancreatic β-cells, regulating insulin secretion and hepatic glycogen synthesis and catabolism (74, 75). HKDC1, primarily expressed in the kidney and intestine, is a novel HK isoform most similar to HK1 (76). Notably, HK4 has only one kinase structure active site domain with enzymatic activity, whereas HK1, HK2, and HK3 possess two kinase structurally active site domains (n- and c-domains). While both domains of HK1 and HK2 are enzymatically active only at the c-terminus, HK2’s n- and c-terminal domains are both enzymatically active (74). This confers a higher glucose affinity and catalytic efficiency to HK2 than other HK isoforms. Interestingly, HK2’s unique n-terminal catalytic structural domain, which binds to the voltage-dependent anion channel (VDAC) of the mitochondrial outer membrane, enables it to further regulate cell activity and apoptosis (77). Additionally, HK2 is the most potent isoform in promoting aerobic glycolysis among all HK isoforms (78). These properties suggest that HK2 plays a unique role in glucose metabolism under various physiological and pathological conditions and may be a significant regulated isoform in cells of a wide range of tissues.
Neuroinflammatory responses are a prominent pathological feature of ischemic stroke during acute ischemia and/or reperfusion (79). Cell culture studies have demonstrated that microglia-mediated neuroinflammation under hypoxic conditions requires elevated glycolysis and HK2 induction. Upregulation of HK2 leads to the accumulation of acetyl-coenzyme A, contributing to histone acetylation and IL-1β transcriptional activation. However, inhibiting HK2 in vivo has been shown to prevent microglia activation, IL-1β production, and ischemic brain injury (80). It is generally believed that inhibiting HK2 can reduce glycolytic flux, potentially alleviating immune cell activation and pathological processes in inflammatory diseases (49). Interestingly, a recent study found that HK2 knockdown exacerbated neuroinflammation and worsened brain injury in a photothrombotic stroke model. This was attributed to HK2 knockout inhibiting microglia’s glycolytic flux and energy production, limiting their proliferation, and reducing surveillance and injury-triggered migration (72). However, other studies have shown that microglia are metabolically flexible and can rapidly adapt to utilize glutamine as an alternative energy source to support their immunosurveillance functions in the brain parenchyma, even when glucose is limited (81). Besides, inhibition of aerobic glycolysis in microglia in a mouse model of middle cerebral artery occlusion promoted glutamine solubilization in microglia, thereby providing fuel for OXPHOS (51). It has also been reported that glutamine utilization also promotes macrophage phagocytosis and M2-type activation, exerting anti-inflammatory effects (82, 83). The discrepancies in these findings may be related to the different models used and the adoption of various interventions, warranting further investigation. Atherosclerosis is a chronic inflammatory disease of the vascular wall (84). It is not only a common cause of stroke but also a significant risk factor for recurrent stroke (85). Initially, monocytes invade and accumulate in the dilated arterial wall, differentiating into macrophages. The pro-inflammatory processes mediated by these macrophages are the primary drivers of plaque formation. Macrophage proliferation is another critical factor in further plaque growth (84, 86). The study reveals that levels of HK2 are significantly increased in monocytes from patients with IS as well as IS model mice. HK2 exacerbated vascular inflammation and accelerated the process of atherosclerosis by promoting the recruitment of inflammatory monocytes to endothelial cells and upregulating Il-1β expression (59).
Phosphofructokinase is the second key rate-limiting enzyme in glycolysis, catalyzing the irreversible conversion of fructose-6-phosphate (F-6-P) to fructose-1,6-bisphosphate (F-1,6-BP), a crucial control point in regulating glycolytic flux (87). PFK-1 is a tetrameric protein encoded by three genes: PFK-M (muscle), PFK-L (liver), and PFK-P (platelet or brain) (88, 89). The activity of PFK-1 is modulated by various cytoplasmic substances, including adenosine triphosphate (ATP), adenosine diphosphate (ADP), F-6-P, and fructose-2,6-bisphosphate (F-2,6-BP). Among these, F-2,6-BP is the most potent allosteric activator of PFK-1, regulating increased glycolytic flux (90). The synthesis and degradation of F-2,6-BP are catalyzed by the bifunctional enzyme 6-phosphofructose kinase/fructose-2,6-bisphosphatase (PFKFB). PFKFB possesses both kinase and phosphatase activities, with the kinase domain located at the N-terminus and the bisphosphatase domain at the C-terminus. Despite an 85% overlap in the core sequences of these domains, they exhibit distinct functional properties due to differences in surrounding residues and the active site (87, 90, 91). The four genes encoding PFKFB isoforms, PFKFB1, PFKFB2, PFKFB3, and PFKFB4, exhibit varying kinase/phosphatase activities and tissue expression profiles (87). PFKFB1 is predominantly expressed in liver, muscle, and fetal tissues. PFKFB2 is mainly found in the heart, kidney, and specific cancer cells. PFKFB3 is predominantly expressed in the brain, placenta, and adipose tissue and is also the most abundant isoform in proliferating cells and cancer cells. PFKFB4 is primarily distributed in the testis but also in various tumor cells (87, 92). Notably, among the PFKFB members, PFKFB3 exhibits a significantly higher kinase/phosphatase ratio (730/1), representing more significant enzymatic activity. This increased sensitivity for F-2,6-BP production allows PFKFB3 to tightly control the rate of glycolysis under both normal and pathophysiological conditions (91, 93).
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells characterized by immunosuppressive properties and elevated glycolytic activity in inflammatory diseases (49). However, MDSCs can differentiate into mature inflammatory cells (neutrophils, macrophages, dendritic cells) with pro-inflammatory effects (94). There is evidence that MDSC significantly accumulated in acute IS patients with ischemic stroke mouse models. Ischemic brain injury can be ameliorated by knockdown or inhibition of PFKFB3 is able to block MDSC differentiation and increase endogenous MDSCs, thereby suppressing the activation state of T effector cell subpopulations (95). Glycolysis is not only closely linked to the maturation and differentiation of immune cells but also plays a crucial role in the differentiation of neural stem cells (NSCs). NSCs are typically found in the dentate gyrus and subventricular zone of the mammalian hippocampus and maintain lifelong neurogenesis by proliferating and producing neurons and glial cells, contributing to the repair of brains damaged by neurodegenerative diseases and/or ischemic injury (96–98). Studies have shown that NSCs differentiation and maturation are marked by a transition from aerobic glycolysis to oxidative phosphorylation (58, 98). The availability of oxygen and the shift in energy metabolism influence cell proliferation and phenotypic differentiation (99). A recent study found that inhibiting PFK-1 expression enhanced neuronal differentiation and dendritic development, improving spatial memory recovery after stroke. Interestingly, these effects of PFK-1 are closely associated with its promotion of β-catenin nuclear translocation and activation of downstream signaling, independent of the Wnt signaling pathway (100). Normally, differentiated mature neurons maintain neuronal redox status through the PPP with low PFKFB3 abundance (101, 102). However, during cerebral IR, neuronal redox homeostasis is disrupted due to a shift from the PPP pathway to aerobic glycolysis, accompanied by a significant increase in PFKFB3 expression and a decrease in NADPH production, an essential cofactor for reducing glutathione disulfide (55). This leads to neuronal mitochondrial dysfunction, excessive ROS accumulation, increased oxidative stress, excitotoxicity, and ultimately, neuronal apoptosis and death (20, 102). Inhibiting PFKFB3 activity in an in vitro IR model blocks glycolytic activation, forcing neurons to switch to the PPP metabolic pathway, thereby realizing a neuroprotective effect (54).
Pyruvate kinase is the key rate-limiting enzyme in the final step of the glycolytic pathway, catalyzing the conversion of phosphoenolpyruvate (PEP) to phosphoenolpyruvate (ADP) to produce pyruvate and ATP (103, 104). In mammals, PK is encoded by two distinct genes, PKLR and PKM, which can generate two different isoforms. This results in four distinct isoforms: PKR, PKL, PKM1, and PKM2 (105). The expression of these PK isoforms is tissue-specific, influenced by the tissue’s energy requirements and nutrient availability (106, 107). PKM1 is predominantly expressed in adult tissues with high ATP demands, such as the heart, skeletal muscle, and brain (105). PKM2 is expressed in tissues at all stages of embryonic development (108). However, in most cases, PKM2 is gradually replaced by other PK isoforms during tissue growth (109). While PKM2 is primarily expressed in embryonic tissues, it has also been found in other tissues, including malignant tumors, lungs, kidneys, liver (110, 111) and various cell types such as stem cells, neuronal cells, endothelial cells, platelets, neutrophils, macrophages, T cells, etc. (39, 62, 112–116). Notably, PKM1 is the most abundantly expressed isoform in neuronal cells under normal physiological conditions, whereas PKM2 is relatively less expressed. PKM2 is primarily expressed in embryonic cells and NPCs in the hippocampus, cerebellum, and subventricular region (117). Furthermore, PKM2 is overexpressed in various cancers, conferring a growth and survival advantage to cancer cells and promoting tumor cell proliferation and metastasis (118). Beyond its classical functions, emerging evidence suggests that PKM2 plays a significant role in neurological disorders (105).
PKM2 is a complexly regulated protein involved in multiple signaling pathways. In addition to regulating glycolysis and promoting tumor cell proliferation, PKM2 also has biological functions in regulating inflammatory responses, oxidative stress, apoptosis, and mitochondrial dysfunction (105, 119). Leukocytes, represented by neutrophils in ischemic tissues, are recruited by cell adhesion molecules on endothelial cells, leading to tissue inflammatory infiltration damage. Simultaneously, platelets adhere and are activated at the site of ischemic stroke vascular lesions, increasing the risk of secondary thrombotic events. The intricate relationship between thrombosis and inflammatory pathways during stroke pathology has shaped the concept of thromboinflammation (79, 120–122). PKM2 is a modulator of systemic inflammation (123). Nuclear PKM2 is upregulated in peripheral neutrophils after ischemic stroke episodes in humans and mice, resulting in their hyperactivation (39). It was found that the expression of inflammatory factors (TNF-α, IL-1β, and IL-6) in neutrophils after cerebral ischemia or reperfusion was significantly reduced in mice with deletion of the PKM2 gene in myeloid cells (39). Additionally, medulla-specific PKM2-deficient mice exhibited improved local cerebral blood flow, reduced infarct size, and enhanced long-term sensorimotor recovery. This is attributed to PKM2’s involvement in post-ischemic cerebral thrombotic inflammation through its regulation of fibrinogen, platelet [CD41] deposition, neutrophil infiltration, and inflammatory factors in the brain (39). A series of oxidative stress and inflammatory responses are the main pathological processes during cerebral ischemia/reperfusion (I/R) (124). PKM2 was upregulated after cerebral I/R injury in both in vivo middle cerebral artery occlusion (MCAO) or in vitro oxygen–glucose deprivation and reoxygenation (OGD/R) models of cerebral I/R injury. Knockdown of PKM2 improves neuronal cell viability and suppresses neuroinflammation, ultimately protecting neurological function from cerebral I/R damage (125). In addition, PKM2 knockdown significantly reduced the expression levels of oxidative stress factors (malondialdehyde (MDA), lactate dehydrogenase (LDH), and nitric oxide (NO)) (126). Knockdown of PKM2 expression levels has been reported to lead to growth inhibition and apoptosis of tumor cells (127). In non-tumor tissues, such as brain tissues subjected to hypoxia and ischemia, down-regulation of PKM2 expression also reduces the activity of the pro-apoptotic protein caspase-3, which inhibits the resulting apoptosis of neurons in the rat brain cortex (128). Mitochondrial dysfunction is a critical factor in the overdependence of neurons on glycolytic function during acute ischemia and/or reperfusion in ischemic stroke (129). Thus, protection of mitochondrial oxidative and autophagic functions, maintenance of mitochondrial homeostasis, and inhibition of glycolysis may play a key role in the brain tissue energy crisis during acute ischemia in AIS, versus the final extent of brain tissue injury after reperfusion (130).
The PDH complex catalyzes the conversion of pyruvate, the end product of glycolysis, to acetyl-coenzyme A (acetyl-CoA), which is further oxidized in the TCA cycle and ultimately utilizes mitochondrial OXPHOS to produce ATP (131). Therefore, the PDH complex serves as a gateway between glycolysis and the TCA cycle (132). The PDH complex consists of three major subunits: the PDH (E1), dihydrolipoic acid transacetylase (E2), and dihydrolipoic acid dehydrogenase (E3) (133). The activity of the PDH complex is tightly regulated by PDK, which phosphorylates serine residues on the α-subunit (E1α) of PDH, inhibiting its activity (134). This shift in pyruvate flux from OXPHOS to aerobic glycolysis is crucial in ischemic neurological disorders closely related to energy metabolism. Humans and rodents possess four PDK isozymes (PDK1-4), with PDK2 and PDK4 (PDK2/4) exhibiting a more generalized tissue distribution and being significantly associated with metabolic disorders compared to PDK1 and PDK3 (135, 136). Consequently, the PDK/PDH axis plays a pivotal role in ischemic neurological disorders.
Resting platelets primarily rely on OXPHOS and aerobic glycolysis to meet their energy demands (137). However, activated platelets require a high energy drive and exhibit a preferential glycolytic phenotype due to their limited mitochondrial density. This transition from a resting to an activated state is associated with an increased propensity for thrombosis (138). Arterial thrombosis, particularly in the carotid arteries, is a major contributor to the development and recurrence of ischemic stroke (139). The PDK/PDH axis has been shown to play a vital role in regulating aerobic glycolysis during the transition of platelets from a resting to an activated state. Combined genetic ablation of PDK2 and PDK4 inhibited platelet activation, reduced susceptibility to arterial thrombosis, and did not adversely affect hemostasis (60). Furthermore, deep vein thrombosis (DVT) is associated with an increased risk of ischemic stroke (140). The development of DVT requires continuous cooperation between neutrophils and platelets. The process of thrombosis triggers the formation of neutrophil extracellular traps (NETs), promoting platelet activation and intravascular coagulation (141). NET formation is an energy-intensive process that depends on aerobic glycolysis. Targeting PDK2/4 inhibits NET formation by reducing aerobic glycolysis, thereby further preventing DVT (61).
LDH is an intracellular oxidoreductase-like enzyme that catalyzes the reversible conversion of pyruvate to lactate in the glycolytic pathway and the reversible conversion of NADH to NAD+ (63). LDH is encoded by four genes: LDHA, LDHB, LDHC, and LDHD. LDHA, LDHB, and LDHC are L isomers, while LDHD is a D isomer. The predominant enantiomer in vertebrates is the L isomer, which functions to use or produce L-lactic acid. Vertebrate L-LDHs are primarily composed of two identified structural isoforms: heart (H-type, encoded by LDHB) and muscle (M-type, encoded by LDHA) (142, 143). LDHB, which is more abundant in aerobic tissues, converts lactate to pyruvate, playing a significant role in lactate oxidation. When body tissues and cells consume large amounts of oxygen or experience inadequate oxygen supply (e.g., strenuous exercise, inflammation, ischemia, and tumors), the TCA cycle becomes inhibited, resulting in insufficient ATP production by OXPHOS. This activates the glycolytic pathway to compensate for the lack of ATP. During this process, lactate dehydrogenase (LDHA) reduces pyruvate and NADH to lactate and NAD+, respectively, to maintain the glycolytic pathway (144, 145).
HMGB1 was reported to be upregulated in both OGD/R-treated N2a cells and MCAO model rats. Meanwhile, Hgmb1 was found to be involved in the up-regulation of the histone H3 lysine 18 lactylation (H3K18lac) induced neuronal apoptosis by LDHA (146). The phenotype of microglia is regulated by LDHA. It was found that inhibition of LDHA promotes the release of anti-inflammatory factors (Arg1, Ym1, CD206) from microglia toward M2 polarization to exert anti-inflammatory effects, which in turn protects against rat cerebral I/R injury (147). Furthermore, a reduction in cerebral infarct volume 24 h after cerebral IR has been associated with the downregulation of LDHA expression in mice (148).
Lactic acid, once mistakenly considered a “metabolic waste”, is now recognized as a crucial byproduct of glycolysis (149). Recent research has highlighted at least three significant roles for lactate. Firstly, lactate produced in active muscle tissue can be reused for glucose formation in the liver, completing the Cori cycle and enabling gluconeogenesis (149). Secondly, circulating lactate is a supplementary glucose source when blood glucose levels are insufficient to directly support neuronal activity. Moreover, while glucose primarily contributes indirectly to the TCA cycle, circulating lactate directly contributes to the production of TCA cycle intermediates, considered the primary energy source (150, 151). Thirdly, lactate can function as both an energy substrate and a signaling molecule, engaging in “lactate shuttling” (intra- or intercellular lactate exchange). The cytoplasmic (glycolysis)-mitochondrial (oxidative metabolism) exchange is a classic example of intracellular lactate shuttling. In the brain, the release of lactate from astrocytes to intercellular compartments via the monocarboxylic acid transporter protein (MCT)4 and its subsequent uptake by peripheral neurons through MCT2 for metabolism has been termed the astrocyte-neuron lactate shuttle (43, 152). Furthermore, a growing body of evidence supports lactate’s role as a signaling molecule that binds to and activates the specific receptor G protein-coupled receptor 81 (GPR81) for signaling (153–155).
Localized acute inflammatory responses or inadequate tissue perfusion are often accompanied by lactate accumulation (117). However, the contribution of lactate accumulation to ischemic brain injury remains controversial. Theoretically, lactate can function as an energy substrate during the ischemic phase, shuttling from astrocytes to neurons to provide energy and protect neuronal cells from ischemic damage. This hypothesis is supported by animal model studies (42, 156–159). However, the effect of lactate on nerve cells appears to be concentration-dependent. During cell culture, lower concentrations (5 mmol/L) of L-lactate had minimal effects on neuronal cell viability in vitro. At concentrations of 10 mmol/L, L-lactate produced results similar to the neuroprotection induced by MCT2-mediated lactate release through optogenetic stimulation of astrocytes (42). However, lactate concentrations below 10 mmol/L were insufficient to compensate for the energy crisis during cerebral ischemia. Even lactate concentrations up to 20 mmol/L only partially rescued the decreased ATP levels in N2A cells or primary neurons (160). Notably, higher concentrations (20 mmol/L) of L-lactate were neurotoxic, leading to an increase in apoptotic and dead cells (42, 157). The absence of partial pressure of oxygen (PO2) during the ischemic phase results in insufficient oxidative capacity to metabolize the accumulating lactate, which cannot be utilized as an energy substrate by neurons (161). Excessive lactate accumulation causes a significant decrease in cytoplasmic pH, leading to lactic acidosis (145, 162). This may be a more plausible explanation for the adverse effects of lactate accumulation. Additionally, recent evidence suggests that excessive lactate accumulation, particularly during ischemia, drives protein lactation (Kla) formation in mouse brain neurons, exacerbating brain injury (148). Therefore, while inhibiting lactate accumulation during ischemia might seem beneficial for neural recovery, excessive inhibition of glycolysis can lead to counterintuitive results. It has been found that during ischemia, when lactate accumulates at relatively low concentrations (approximately 1-3 mmol/L), lactate may exacerbate ischemic injury by activating GPR81 (160).
Studies have demonstrated that astrocyte activation induces the release of lactate into the extracellular space (42, 159). However, it cannot be ruled out that astrocytes may be activated through other mechanisms and subsequently participate in protecting neurons. Recently, some researchers have utilized in vitro cell cultures and animal models to observe that astrocyte lipoprotein receptor-related protein 1 (LRP1), a multifunctional transmembrane receptor, facilitates the transfer of healthy mitochondria from astrocytes to neurons by inhibiting glucose uptake, glycolysis, and lactic acid production, thereby preventing brain I /R damage. Furthermore, it was found that incubating astrocytes with graded doses of lactate (ranging from 0 to 50 mmol/L) for 2 h resulted in a dose-dependent decrease in extracellular mitochondria and ATP levels (163).
In conclusion, the spatiotemporal expression profile of lactate within the brain is dynamic and evolves in tandem with the progression of ischemic stroke. The impact of lactate on neuronal cells is multifaceted, encompassing both deleterious and protective effects, suggesting a complex regulatory mechanism. This regulation appears to be concentration-dependent, potentially accounting for the discrepancies observed in previous studies investigating the functional role of lactate.
During the cerebral ischemia and/or reperfusion phase, the glycolytic pathway is overly relied upon as the primary mode of energy supply. This results in the activation or inhibition of a number of molecular/signaling pathways associated with the regulation of glycolysis, which are involved in neuroinflammation, oxidative stress, excitotoxicity, as well as cellular damage and apoptosis. Figure 3 summarises the contribution of glycolysis and related molecules/signaling pathways to IS.
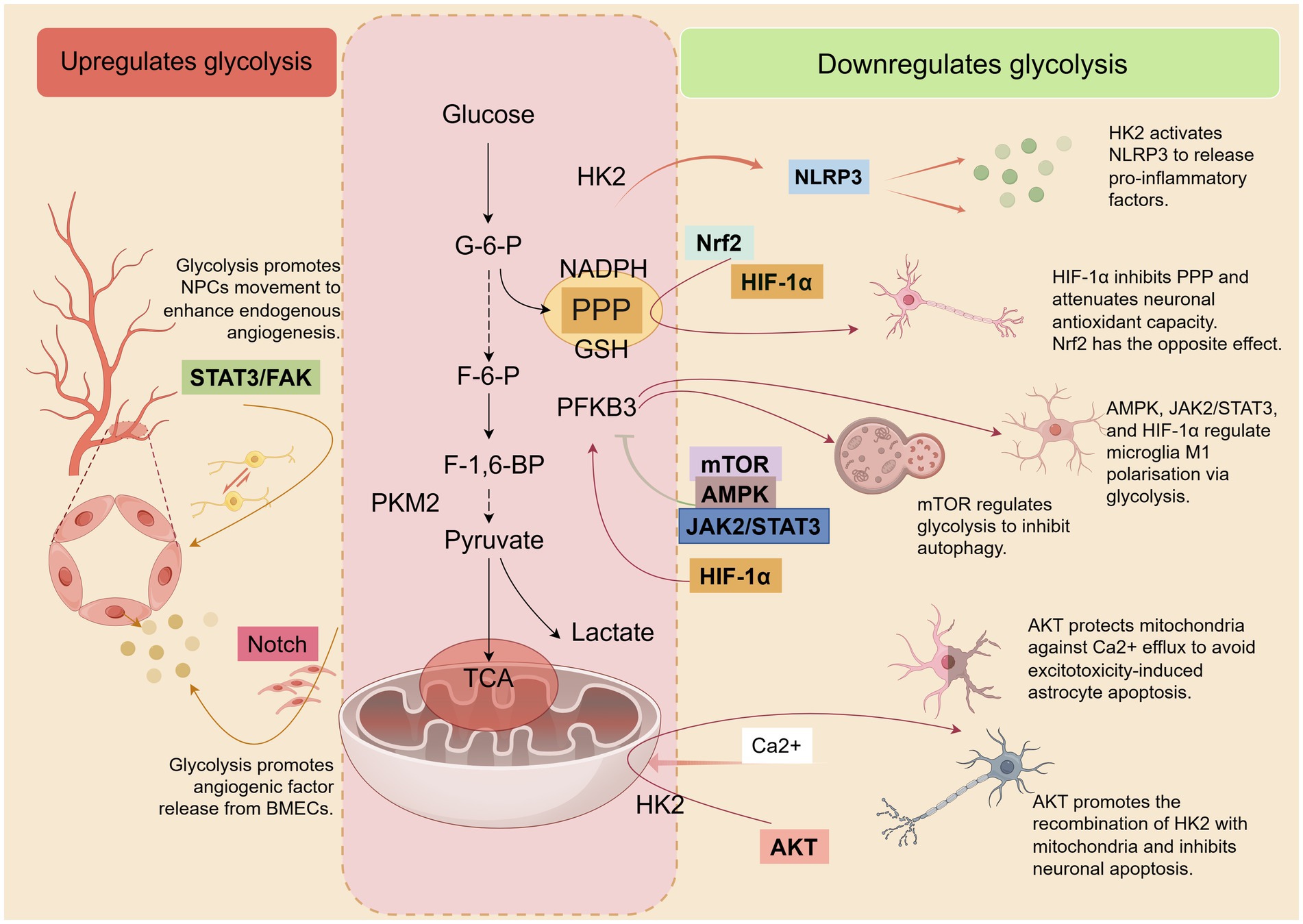
Figure 3. After the onset of IS, several molecules/signaling pathways involved in inflammation, oxidative stress, excitotoxicity, cell damage, and apoptosis are activated or inhibited. These pathways directly or indirectly regulate glycolysis and are also modulated by glycolysis, which in turn affects the pathological process of IS. Some of these pathways positively affect IS by decreasing glycolytic activity. Concurrently, there are also some pathways that have a positive effect on IS by elevating glycolytic activity. By Figdraw.
The NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammatory vesicles are a complex of multiple proteins that play key roles in regulating the innate immune system and inflammatory signaling processes (164). Some studies have shown that when the NLRP3 inflammasome is activated, it releases downstream pro-inflammatory mediators such as IL-1β and IL-18, which become one of the key factors triggering neuroinflammation after cerebral ischemia–reperfusion (164, 165). In addition, glycolysis (mainly involving HK2 activity) is involved in the regulation of inflammatory vesicle activation and to play a crucial role in the activation of NLRP3 (166). An increase in glycolytic flux has been reported under hypoxic conditions, accompanied by a significant upregulation of HK1 and HK2. This leads to inhibition of the pentose phosphate pathway, reducing the conversion of pyruvate to acetyl coenzyme A, which in turn allows more pyruvate to be converted to lactate. In this environment, NLRP3 inflammatory vesicles are activated and release pro-inflammatory mediators, which trigger a neuroinflammatory response (167).
Adenosine monophosphate-activated protein kinase (AMPK) is a central regulator of cellular responses to energy metabolic stress. Its function is to sense changes in cellular energy status and maintain the balance between energy production and expenditure by regulating various metabolic and immune-related activities (168). AMPK has been reported to be an important protective factor against cerebral ischemic injury during cerebral ischemia or hypoxia (169). AMPK exerts an endogenous defense mechanism, possibly by promoting the reprogramming of energy metabolism in microglia (from glycolysis to OXPHOS) to enhance their immunophagocytosis and thus attenuate neuroinflammation and microglia M1 polarization in IS (51). A study found that the knockdown of protein kinase N1 (PKN1), a key brain developmental regulator, in an in vivo or in vitro model of stroke resulted in the improved coupling of the pyruvate and TCA cycles despite an increase in glycolytic fluxes, contributing to enhanced mitochondrial uptake of pyruvate. This helps to cope with energy metabolic stress and protects nerve cells from ischemic damage. However, when AMPK activation was inhibited by BAY-3827, the neuroprotective effect of PKN1 knockdown was counteracted. This suggests that in the absence of PKN1, activated AMPK exerts its protective effects more effectively in stroke models (170). Besides, studies have revealed the role of glycolysis in regulating cerebral angiogenesis after IS. This is mainly achieved through the activation of the key rate-limiting enzyme PFKFB3 as well as the signaling pathway represented by AMPK (171).
Mechanistic target of rapamycin (mTOR) is a key regulator of cellular metabolism, capable of integrating multiple signals from nutrients, growth factors, energy status, and oxygen levels. mTOR forms two protein complexes, mTORC1 and mTORC2, but mTORC1 signaling has broader effects on the growth and homeostasis of key groups of immune cells and organs (172, 173). It was found that downregulation of PFKFB3-driven glycolysis partially reduced lactate-mediated activation of mTORC1, thereby inhibiting MDSC differentiation to mature inflammatory cells to attenuate acute ischemic brain injury (95). Autophagy is activated after I/R, but there is still debate on whether autophagy activation is a pro-survival or pro-death mechanism in IS (174). It has been shown that TP53-induced glycolysis and apoptosis regulator (TIGAR) reduces I/R-induced autophagic activity in mouse brain and primary cortical neurons in both in vivo and in vitro experiments. In addition to the possible mechanism of ROS reduction, TIGAR inhibits autophagy by regulating the mTOR-S6KP70 signaling pathway, thereby preventing neuronal damage (174).
Signal transducer and activator of transcription 3 (STAT3) is important for vertebrate development and the functional performance of mature tissues. It is also involved in the regulation of inflammatory and immune responses (175). Overexpression and/or aberrant activation of the STAT3 signaling pathway has been previously observed in a variety of human diseases, such as cancer, autoimmune diseases, and inflammatory diseases (176). Recent studies have found that glycolysis can increase inflammatory responses to cerebral ischemia by up-regulating the phosphorylation of STAT3, which in turn enhances the infiltration of peripheral blood neutrophils and the expression of inflammatory cytokines (39). Janus kinase 2 (JAK2) is expressed in a variety of tissues and cell types, and it has important effects on cell differentiation, apoptosis, and immune regulation. The JAK2/STAT3 signaling pathway is a classical signaling module that plays a key role in several aspects of the mammalian immune system (177). It was found that both JAK2/STAT3 expression was dramatically increased in microglia treated with OGD/R in vitro. By blocking the JAK2/STAT3 signaling pathway, the OGD/R-induced aerobic glycolysis process in microglia could be reduced and its polarization toward M2-type could be promoted (178). When the stroke occurs, astrocytes normally produce protective mechanisms for neurons (ANLS, mitochondrial transmission, and antioxidant support as mentioned above in this review). However, the mechanism of astrocyte-mediated neurotoxicity after stroke has also been reported recently. In an OGD/R-treated astrocyte-neuron indirect co-culture system, astrocytes in an ischemic state activate the STAT3 signaling pathway, which leads to an upregulation of the lactate-directed glycolytic process and is accompanied by impairment of mitochondrial function. These damaged mitochondria are delivered to healthy neurons by astrocytes, which may be a potential mechanism of toxicity leading to synaptic degeneration in nonischemic neurons. However, if STAT3 inhibition in ischemic astrocytes prevents neuronal synaptic degeneration, this may be due to the restoration of mitochondrial function in astrocytes (45). Focal adhesion kinase (FAK) is a regulator involved in cell survival, proliferation, and migration (179). Activation of the STAT3/FAK signaling pathway promotes NPCs migration and enhances endogenous angiogenesis and neurogenesis in ischemic brain injury (180). This suggests that STAT3 activation after cerebral ischemia is not the only negative effect of neural tissue damage.
Hypoxia inducible factor-1α (HIF-1α) is a is a sensitive regulator of oxygen homeostasis. It regulates endogenous adaptive programs such as glycolytic pathways, inflammatory responses, and oxidative stress by modulating multiple signaling pathways and targeting downstream genes under hypoxic conditions (181, 182). HIF-1α expression is rapidly induced when tissues or cells are hypoxic/ischemic (182). It was found that when microglia converge to the M1 phenotype during AIS, the shift of OXPHOS to aerobic glycolysis becomes a prominent energy metabolism feature, a process that is dependent on the activation of the AMPK/mTOR/HIF-1α signaling pathway (183). In addition, numerous studies revealed that HIF-1α significantly increased the glycolytic flux of microglia by mediating the activity of glycolysis-related enzymes such as HK2, PKM2, and LDHA, which promoted their conversion to the M1 phenotype and triggered neuroinflammation (59, 147, 184–186). It is worth mentioning that activation of HIF-1α during AIS may help to attenuate nerve tissue damage and dysfunction induced by ischemia, but this depends on the different types of nerve cells. A recent study found that timely activation of HIF-1α in astrocytes in the early stages of ischemia and enhancement of ischemic tolerance in astrocytes can improve the prognosis of acute ischemic stroke. However, if HIF-1α is overactivated in neurons, it triggers a HIF-1α-dependent increase in PFKFB3 expression, leading to a shift of F-6-P from the PPP pathway to the glycolytic pathway. This process is accompanied by a decrease in NADPH production in the PPP pathway, which in turn weakens the already low antioxidant defenses of neurons and makes them more vulnerable to damage from ischemic stress. Interestingly, in an environment where neurons are co-cultured with astrocytes, the sensitivity of neurons to ischemic stress triggered by hyperactivation of HIF-1α is reduced. This may be due to the strong antioxidant support that neurons receive from their surrounding astrocytes (55).
Nuclear factor-E2-related factor 2 (Nrf2) is an important transcription factor for the maintenance of redox homeostasis, which regulates cellular defenses against toxicity and oxidative damage through the expression of genes involved in oxidative stress response and drug detoxification (187). The study revealed that aerobic glycolysis was significantly enhanced during I/R, while the activity of antioxidant enzymes was inhibited. This process leads to impaired mitochondrial function and the release of large amounts of reactive oxygen species (ROS), which in turn disrupts the blood–brain barrier and exacerbates brain damage. However, by activating downstream antioxidant proteins associated with Nrf2, oxidative metabolic homeostasis can be restored, blood–brain barrier integrity can be protected, and neuronal death can be reduced (14). NADPH provided by the PPP pathway plays a crucial role in the defense against ischemia-induced oxidative stress in neurons. TP53-induced glycolysis and apoptosis regulator (TIGAR) was found to activate Nrf2 in ischemic neurons by promoting autophagic processes, thereby shifting glucose metabolism to the PPP pathway to reduce oxidative stress and prevent ischemic neuronal injury (188).
The kinase AKT (protein kinase B), is a key signaling molecule that regulates cell survival, proliferation, tumor development, and angiogenesis (189). The diminished activity of AKT is strongly associated with cellular metabolic disorders and apoptosis (190). It was found that a mechanism dependent on AKT (p-AKT) inactivation induced neuronal apoptosis after PKM2 upregulation during brain tissue ischemia and hypoxia (128). Cerebral ischemia triggers neuronal cell apoptosis through multiple mechanisms, of which mitochondrial dysfunction is particularly critical. Glutamate excitotoxicity leads to the dissociation of HK2 from mitochondria, which in turn disrupts the structural and functional integrity of mitochondria. This process is manifested by the opening of the mitochondrial permeability transition pore, the collapse of the mitochondrial membrane potential, and a decrease in the neuronal mitochondrial oxygen consumption ratio, which ultimately leads to apoptosis of the neuronal cell. However, promoting the recombination of HK2 with mitochondria through the activation of AKT and protecting HK activity by improving the glycolytic process can effectively protect neurons from apoptosis (191). Although astrocytes exhibit a greater ability to tolerate ischemic and hypoxic environments than neurons, exposure of astrocytes to acidic pH (6.8, 6.0) environments during ischemia when the glycolytic product lactate accumulates to a certain level leads to the release and inward flow of Ca2+, which in turn triggers mitochondrial hyperpolarization, ultimately leading to astrocyte death. This process is accompanied by activation of p38 MAPK and inhibition of AKT. However, SC79, an AKT activator, prevents mitochondrial hyperpolarization in astrocytes (192). Activation of AKT is usually achieved through close association with activated kinases of PI3K and is involved in the regulation of glycolysis-related enzymes (193). However, there seems to be a lack of studies on the effects of neurocellular glycolysis and cell survival after stroke via the PI3K/AKT pathway, suggesting that further exploration may be needed in the future.
In addition to the above, several pathways related to the regulation of glycolysis in IS have been reported. For example, the Wnt/β-catenin pathway, which has recently been reported to be involved in IS progression by affecting the several enzymes of glycolysis, NADPH/NADP, and the levels of ATP, induces ferroptosis of tract astrocytes (194). Notch signaling pathway (closely related to angiogenesis) can be activated by PFKFB3 to promote the formation of brain microvascular endothelial cells (BMECs) after stroke (195). AK4 enhances neuronal adaptive responses to hypoxia/ischemia by promoting Parkin-mediated PKM2 degradation (130). Knockdown of PKM2 increases neuronal cell viability and inhibits inflammatory cytokines (IL-1β, IL-6, and TNF-α) released by HMGB1-mediated TLR4/MyD88/TRAF6 expression, ultimately protecting neurological function from cerebral I/R damage (125).
Glycolytic metabolic processes, particularly enzymes like HK2, PFKFB3, PKM2, PDH, LHD, and metabolites such as lactate, have emerged as potential predictors of stroke risk, development, and prognosis (39, 59, 196–199). This suggests their potential diagnostic value. HK2, specifically expressed in microglia, is upregulated in activated microglia and tightly controls glycolytic flux. Assessing HK2 levels can dynamically evaluate microglial glycolysis and bioenergetic responses, making it a promising marker for early diagnosis of IS (72). Neutrophils, among the first responders to ischemic injury (121, 122). Exhibit elevated levels of nuclear PKM2 in ischemic stroke patients treated with mechanical thrombolysis compared to healthy controls (39). This suggests that continuous monitoring of PKM2 marker levels could reflect clinical outcomes after thrombolysis, though further studies are needed. Additionally, research has implicated the fatty acid-related PPAR signaling pathway and PKM2 as key markers of neuronal cells responding to OGD/R, potentially aiding in early diagnosis (200). The pathogenesis of IS involves significant immune infiltration and the participation of multiple genes. PFKFB3, identified as a core functional gene through clinical sample validation, has emerged as a potential early diagnostic marker for ischemic stroke (201).
Conventional clinical tools such as computed tomography (CT) and magnetic resonance imaging (MRI) are currently employed to diagnose IS. While these methods enable rapid assessment of infarct location and volume, they are less effective in determining the underlying cause of infarcts (202). A dearth of research on imaging biomarkers that reflect dynamic changes in ischemic brain tissue has hindered the ability to sensitively assess tissue viability within the ischemic penumbra (199). Recent investigations have demonstrated the potential of hyperpolarized [1-13C]pyruvate as an imaging biomarker to characterize changes in tissue viability during reperfusion and subsequent brain tissue recovery (203). By monitoring the conversion of hyperpolarized [1-13C]pyruvate to [1-13C]lactate and [13C]bicarbonate, researchers have quantified the rates of LDH and PDH enzymes. The results revealed a significant increase in the LDH/PDH enzyme activity ratio during IR, followed by a decrease to pre-ischemic levels during recovery. These findings suggest that this ratio could serve as a potential indicator for detecting changes in IS disease (199).
Glycolysis-related enzymes and metabolites, including LDH, lactate, and pyruvate, have been observed at elevated levels in the plasma and cerebrospinal fluid (CSF) of IS patients, indicating active glycolysis (204–206). AIS is often accompanied by impaired consciousness, the underlying causes can also be attributed to non-structural brain tissue changes, such as intoxication, infection, or metabolic disorders (204). CSF-LDH values have been found to differentiate between structural and non-structural etiologies in cases of acute alterations in consciousness, especially in the absence of trauma or access to conventional imaging tools (204). This holds promise for providing a reliable diagnostic measure in regions with limited medical resources. In a study involving 5,129 patients with AIS, high LDH levels were associated with an increased risk of dysfunction and recurrence, partially explained by stroke recurrence (207). Another multicenter retrospective study of 527 AIS patients revealed that elevated serum LDH levels during the first 3 days after intravenous thrombolytic therapy were strongly predictive of adverse clinical outcomes, including hemorrhagic conversion, neurological deterioration, disability, and others, 3 months post-thrombolysis (196). Additionally, LDH levels have been shown to predict the risk of DVT during rehabilitation in stroke patients (208). LDH demonstrates significant potential in predicting the risk of IS recurrence and clinical outcomes. Among thrombolytic adverse outcomes, hemorrhagic conversion poses the most significant limitation to the clinical use of rt-PA. The endogenous metabolite lactate has been identified as a potential biomarker for early prediction of hemorrhagic conversion (198). Overall, these biomarkers offer the potential to improve diagnostic accuracy, predict the risk of adverse outcomes, and inform clinical decision-making (Table 1).
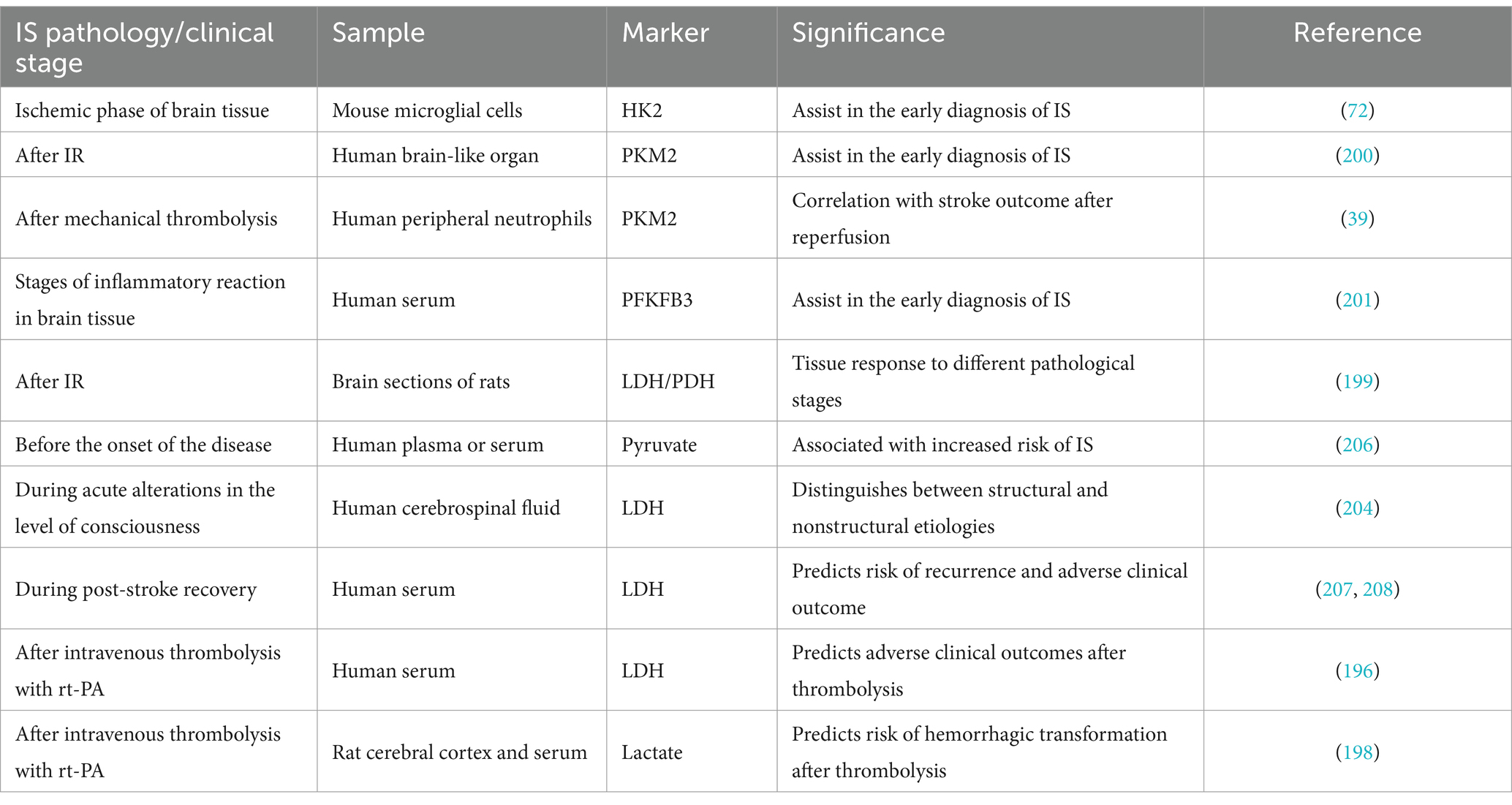
Table 1. Diagnosis of IS by glycolysis.
Beyond accurate diagnosis, precise treatment is paramount in improving the prognosis of IS and preventing potentially severe complications. Glycolytic metabolic pathways are gaining recognition as promising therapeutic targets for neurological disorders, not only in tumor cells but also across multiple cell types.
AZ67, a potent and selective inhibitor of PFKFB3, has been shown to protect neurons from excitotoxicity following IR by inhibiting PFKFB3 activity (54). Pretreatment of mice with oxalate, an LDHA inhibitor, has been reported to significantly reduce neuronal death and infarct volume 24 h after reperfusion in ischemic stroke rats (148). Natural bioactive products, known for their high efficacy and safety compared to enzyme inhibitors, have been increasingly studied for inflammation-related diseases. Tetrahydroxy stilbene glucoside (TSG), a small molecule natural bioactive ingredient, has been shown to reverse the energy metabolism profile of M1 macrophages to the M2 state, exerting potent anti-inflammatory and neuroprotective properties. This effect of TSG is closely linked to the conformational change of PKM2 (209). Benzoxepane derivatives 10a exhibit anti-inflammatory effects both in vitro and in vivo by inhibiting PKM2-mediated glycolysis and NLRP3 (an inflammation-activating protein) activation, leading to beneficial effects on ischemic stroke (210).
Chlorpromazine and promethazine (C + P) can induce depressive or hibernation-like effects on brain activity, leading to pharmacological hypothermia, which reduces HIF-1α and glycolytic enzyme (PFK-1, LDH) levels and promotes neuroprotection after ischemic stroke (211). Combining dihydrocapsaicin (DHC) with C + P not only inhibits glycolytic and gluconeogenic enzyme activities but also reduces neurological damage caused by oxidative stressors, such as ROS and NADPH oxidase (NOX) (212). However, the most problematic side effect of pharmacological hypothermia is the risk of increased blood viscosity at low temperatures, leading to platelet aggregation and potential microcirculatory obstruction in the brain and heart (213). Intravenous administration of ethanol (1.0 g/kg) has been reported to achieve similar hyperglycolytic inhibition with pharmacological hypothermia (33°C), suggesting a potentially more straightforward and safer treatment option after stroke (213). Arginine, an essential amino acid with anti-inflammatory properties, has been shown to reduce HIF-1α/LDHA-mediated neuronal death and improve functional recovery in stroke animals after brain IR injury when injected into the ventricles (147). Leptin, administered via tail vein injection in mice after transient ischemia, has been shown to significantly reduce LDH levels, the lactate/pyruvate ratio, neurological deficits, infarct volume, and cerebral edema (214). Dichloroacetate (DCA), a small molecule that activates PDH and crosses the blood–brain barrier (BBB), reduces neurotoxic lactic acidosis during cerebral ischemia and protects BBB integrity (14, 215, 216). Recently, researchers have synthesized fasudil dichloroacetate (FDCA), a novel drug that combines the mechanisms of action of fasudil and dichloroacetate, resulting in superior efficacy in reducing cerebral ischemic injury compared to their individual or combined use (217). Celastrol is a naturally occurring bioactive compound that inhibits glycolysis via the HIF-1α/PDK1 axis and provides neuroprotection in cerebral I/R injury (218).
Targeted regulation of key glycolytic enzymes by active ingredients from traditional Chinese medicine represents a promising approach for the effective prevention and treatment of IS in the future. Regulatory T cells (Tregs), crucial regulators of inflammation, play a vital role in maintaining immune tolerance and homeostasis (219). Ginkgo biloba extract (EGb) has been shown to protect neurons from inflammatory damage by inhibiting the HIF-1α/HK2 signaling pathway and promoting the proliferation and differentiation of Tregs (184). Salvianolic acid C (SAC), an active ingredient in Salvia miltiorrhiza, inhibits the M1 polarization of microglia through the glycolytic pathway (down-regulation of PKM2), reducing the expression of inflammatory factors TNF-α, IL-6, and IL-1β, and protecting against neurological injury in a MCAO model (220). Panax notoginseng saponins (PNS) administration has been found to inhibit microglia activation-induced HIF-1α/PKM2/STAT3 signaling activation, counteracting ischemic brain injury (221). Icariin has been reported to inhibit neuronal cell inflammation and apoptosis by down-regulating PKM2 overexpression after OGD in neuronal cells (222). Baicalin improves PDK2-PDH function during IR by inhibiting succinate dehydrogenase (SDH)-mediated oxidative stress and increasing pyruvate flow to the mitochondria, supplying neurons with sufficient energy rather than relying on aerobic glycolysis (132). In addition, ginsenoside Rd. (Rd) likewise increases pyruvate flow to mitochondria, reducing the release of cytochrome c (CytoC) and apoptosis-inducing factor (AIF) from mitochondria, thus maximizing the inhibition of neuronal apoptosis (223). Astragaloside IV (AIV), an active ingredient in Astragalus, helps to counteract ischemia-induced neuronal apoptosis and DNA damage by activating the Akt signaling pathway, protecting mitochondrial HK2, and reducing the release of pro-apoptotic proteins and AIF (191). AIV was able to regulate the expression of key glycolytic enzymes (HK2, PFKFB3, PKM2) and energy transporter proteins (GLUT1, MCT1, MCT4) through activation of the AMPK pathway and inhibition of the mTOR/HIF-1α pathway, which promotes microglial cell polarization toward the M2-type and inhibits M1-type polarization (224). Upregulation of glycolysis after IS onset does not necessarily imply a negative effect on brain tissue recovery. BMECs have been reported to rely primarily on glycolytic function, whereas other vascular cells rely on aerobic respiration (225). Hydroxysafflor Yellow A (HSYA), a major bioactive constituent of saffron, was found to enhance angiogenesis in BMECs after OGD/R by promoting glycolysis (195). Moreover, classical Chinese herbal remedies, which have the advantage of multi-target and multi-pathway regulation, have also been applied in the treatment of IS. Taohong Siwu Decoction (THSWD) can accelerate mature angiogenesis by promoting the expression of mature angiogenic factors (VEGFA, Ang1, and PDGFB) through the activation of glycolysis (225). Buyang Huanwu decoction (BHD) is a classic prescription for promoting angiogenesis after IS by maintaining moderate levels of glycolysis in ischemic brain tissue, as well as promoting angiogenesis (171). Mailuo Shutong Pill (MLST) suppresses brain tissue inflammation by modulating glucose metabolism disorders, interfering with immunometabolic reprogramming, inhibiting microglia infiltration, and down-regulating the NLRP3 inflammatory vesicle signaling pathway, ultimately ameliorating cerebral ischemia–reperfusion injury (167) (Table 2).
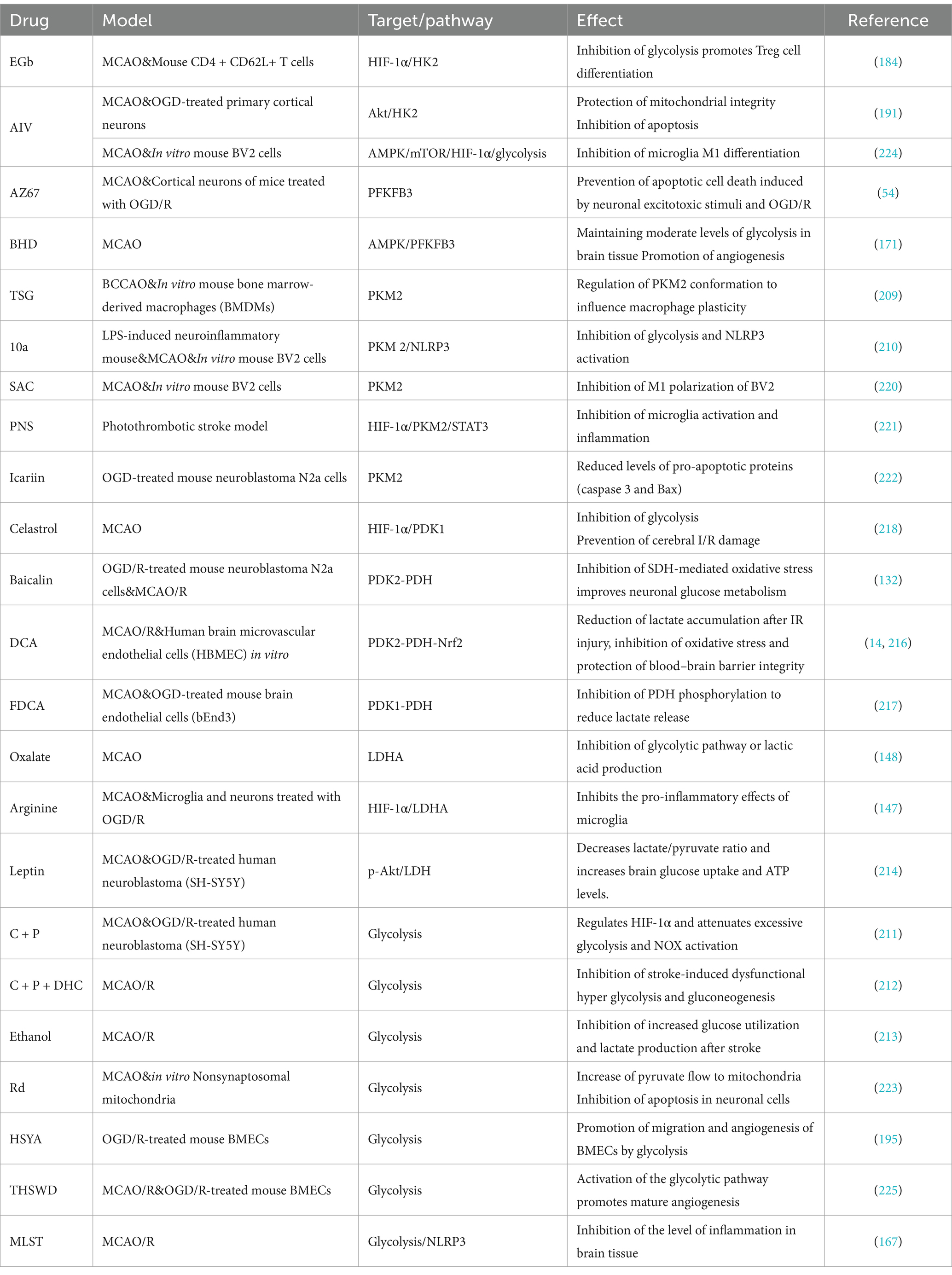
Table 2. Treatment of IS by glycolysis.
Overall, modulation of glycolytic processes in immune cells, neuronal cells, and endothelial cells through multiple pathways to influence the onset and progression of IS has become a relatively well-established research strategy. However, we seem to have some previous uncertainty as to whether glycolysis should be promoted or inhibited in IS. However, after careful combing of the literature, we found that among the key cells involved in the pathomechanisms of IS, inhibition of glycolytic activity resulted in many cells exhibiting positive effects on brain tissue recovery. These cells include monocytes/macrophages, neutrophils, platelets that promote inflammation in arterial plaques and thrombi, and microglia that induce neuroinflammation. However, in vascular endothelial cells, increased glycolytic flux contributes to the promotion of neovascularization, which also has a positive effect on brain tissue recovery in IS. Interestingly, in tumor tissues, elevated glycolytic activity of vascular endothelial cells tends to lead to overgrowth of tumor tissues and, therefore, its activity needs to be inhibited (46). It follows that the variability of the functions played by glycolysis in different disease environments as well as in cells of different tissues determines whether its up-regulation is beneficial or not. However, it is important to note that the current study has not clarified the extent to which glycolysis is up- or down-regulated in different cells, which may require further exploration in the future. In related studies, the development and prognosis of the disease can be predicted by monitoring the levels of glycolysis-related enzymes as well as metabolites.
Monitoring glycolysis-related enzymes and metabolites can aid in predicting disease development and prognosis. Targeting these enzymes and metabolites has shown potential in reducing neuronal damage and promoting neuronal function recovery in both experimental and clinical settings. However, the study of glycolysis and IS faces several limitations: (1) The mechanisms underlying glycolysis and IS are complex, involving intricate regulatory interactions among neurons, microglia, and astrocytes. A comprehensive understanding requires a broader perspective beyond the effects of glycolysis on microglial immunosurveillance or astrocyte lactate delivery. (2) Assessing reactive glycolytic activity can be challenging due to the limitations of existing indices. Lactate accumulation may not accurately reflect glycolytic activity, as other pathways (e.g., glutamine and pentose phosphate pathways) also contribute to lactate production. (3) The lack of uniformity in animal models, cellular models, and interventions can lead to inconsistent or even contradictory results for the same enzyme, as the expression of the same enzyme in different cell types is often specific. (4) The use of glycolysis-related enzymes and metabolites as diagnostic indicators remains relatively broad, lacking objective, precise, and specific quantitative criteria. (5) The primary use of intraperitoneal injection in animal models may not fully represent the clinical administration route, requiring further investigation to determine the relevance of the findings to the actual clinical setting. Future research can address these limitations by (1) Considering the complex intercellular functions and regulatory mechanisms to avoid one-sided conclusions. (2) Standardizing and unifying experimental modeling, intervention methods, and observation indices to ensure cell specificity and minimize interference from other factors. (3) Providing detailed descriptions of the trends in index changes and corresponding disease stages for diagnostic purposes. (4) Exploring intranasal administration as a promising drug delivery method for treating IS, offering advantages such as convenience, non-invasiveness, bypassing the blood–brain barrier, rapid delivery to the brain and spinal cord, minimal systemic exposure, and reduced side effects (226, 227). In conclusion, both diagnosing/predicting IS through glycolysis and treating IS by targeting glycolytic pathways represent promising research approaches that warrant further in-depth investigation in the context of ischemic stroke.
YL: Conceptualization, Methodology, Writing – original draft, Data curation. PH: Methodology, Funding acquisition, Project administration, Writing – review & editing. HC: Funding acquisition, Project administration, Supervision, Writing – review & editing. FX: Writing – original draft, Data curation. YY: Writing – original draft, Data curation.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by Anhui Clinical Medical Research Transformation Project (Grant number: 202304295107020123).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. Krishnamurthi, RV, Ikeda, T, and Feigin, VL. Global, regional and country-specific burden of Ischaemic stroke, intracerebral Haemorrhage and subarachnoid Haemorrhage: a systematic analysis of the global burden of disease study 2017. Neuroepidemiology. (2020) 54:171–9. doi: 10.1159/000506396
3. Feigin, VL, Stark, BA, Johnson, CO, Roth, GA, Bisignano, C, Abady, GG, et al. Global, regional, and national burden of stroke and its risk factors, 1990–2019: a systematic analysis for the global burden of disease study 2019. Lancet Neurol. (2021) 20:795–820. doi: 10.1016/S1474-4422(21)00252-0
4. Donkor, ES. Stroke in the 21st century: a snapshot of the burden, epidemiology, and quality of life. Stroke Res Treat. (2018) 2018:1–10. doi: 10.1155/2018/3238165
5. Feigin, VL, Brainin, M, Norrving, B, Martins, S, Sacco, RL, Hacke, W, et al. World stroke organization (WSO): global stroke fact sheet 2022. Int J Stroke. (2022) 17:18–29. doi: 10.1177/17474930211065917
6. Jayaraj, RL, Azimullah, S, Beiram, R, Jalal, FY, and Rosenberg, GA. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation. (2019) 16:142. doi: 10.1186/s12974-019-1516-2
7. Bailey, EL, Smith, C, Sudlow, CLM, and Wardlaw, JM. Pathology of lacunar ischemic stroke in humans—a systematic review. Brain Pathol. (2012) 22:583–91. doi: 10.1111/j.1750-3639.2012.00575.x
8. Warlow, C, Sudlow, C, Dennis, M, Wardlaw, J, and Sandercock, P. Stroke. Lancet. (2003) 362:1211–24. doi: 10.1016/S0140-6736(03)14544-8
9. Siegel, JL. Acute bacterial meningitis and stroke. Neurol Neurochir Pol. (2019) 53:242–50. doi: 10.5603/PJNNS.a2019.0032
10. Catanese, L, Tarsia, J, and Fisher, M. Acute ischemic stroke therapy overview. Circ Res. (2017) 120:541–58. doi: 10.1161/CIRCRESAHA.116.309278
11. Prabhakaran, S, Ruff, I, and Bernstein, RA. Acute stroke intervention. JAMA. (2015) 313:1451. doi: 10.1001/jama.2015.3058
12. Mergenthaler, P, Lindauer, U, Dienel, GA, and Meisel, A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. (2013) 36:587–97. doi: 10.1016/j.tins.2013.07.001
13. Geng, J, Aa, J, Feng, S, Wang, S, Wang, P, Zhang, Y, et al. Exploring the neuroprotective effects of ginkgolides injection in a rodent model of cerebral ischemia–reperfusion injury by GC–MS based metabolomic profiling. J Pharm Biomed Anal. (2017) 142:190–200. doi: 10.1016/j.jpba.2017.04.031
14. Zhao, X, Li, S, Mo, Y, Li, R, Huang, S, Zhang, A, et al. DCA protects against oxidation injury attributed to cerebral ischemia-reperfusion by regulating glycolysis through PDK2-PDH-Nrf2 Axis. Oxidative Med Cell Longev. (2021) 2021:5173035. doi: 10.1155/2021/5173035
15. Beppu, K, Sasaki, T, Tanaka, KF, Yamanaka, A, Fukazawa, Y, Shigemoto, R, et al. Optogenetic countering of glial acidosis suppresses glial glutamate release and ischemic brain damage. Neuron. (2014) 81:314–20. doi: 10.1016/j.neuron.2013.11.011
16. Wass, CT, and Lanier, WL. Glucose modulation of ischemic brain injury: review and clinical recommendations. Mayo Clin Proc. (1996) 71:801–12. doi: 10.1016/S0025-6196(11)64847-7
17. Geiseler, SJ, Hadzic, A, Lambertus, M, Forbord, KM, Sajedi, G, Liesz, A, et al. L-lactate treatment at 24 h and 48 h after acute experimental stroke is neuroprotective via activation of the L-lactate receptor HCA1. Int J Mol Sci. (2024) 25:1232. doi: 10.3390/ijms25021232
18. Ganapathy-Kanniappan, S, and Geschwind, J-FH. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. (2013) 12:152. doi: 10.1186/1476-4598-12-152
19. Zhang, X-C, Gu, A-P, Zheng, C-Y, Li, Y-B, Liang, H-F, Wang, H-J, et al. YY1/LncRNA GAS5 complex aggravates cerebral ischemia/reperfusion injury through enhancing neuronal glycolysis. Neuropharmacology. (2019) 158:107682. doi: 10.1016/j.neuropharm.2019.107682
20. Li, Z, Zhang, B, Yao, W, Zhang, C, Wan, L, and Zhang, Y. APC-Cdh1 regulates neuronal apoptosis through modulating glycolysis and pentose-phosphate pathway after oxygen-glucose deprivation and reperfusion. Cell Mol Neurobiol. (2019) 39:123–35. doi: 10.1007/s10571-018-0638-x
21. Wang, H, Liu, S, Sun, Y, Chen, C, Hu, Z, Li, Q, et al. Target modulation of glycolytic pathways as a new strategy for the treatment of neuroinflammatory diseases. Ageing Res Rev. (2024) 101:102472. doi: 10.1016/j.arr.2024.102472
22. Fendt, S-M. 100 years of the Warburg effect: a cancer metabolism endeavor. Cell. (2024) 187:3824–8. doi: 10.1016/j.cell.2024.06.026
23. Qian, Y, Yang, L, Chen, J, Zhou, C, Zong, N, Geng, Y, et al. SRGN amplifies microglia-mediated neuroinflammation and exacerbates ischemic brain injury. J Neuroinflammation. (2024) 21:35. doi: 10.1186/s12974-024-03026-6
24. Cao, W, Feng, Z, Zhu, D, Li, S, Du, M, Ye, S, et al. The role of PGK1 in promoting ischemia/reperfusion injury-induced microglial M1 polarization and inflammation by regulating glycolysis. NeuroMolecular Med. (2023) 25:301–11. doi: 10.1007/s12017-023-08736-3
25. Zeng, X, Liu, N, Zhang, J, Wang, L, Zhang, Z, Zhu, J, et al. Inhibition of miR-143 during ischemia cerebral injury protects neurones through recovery of the hexokinase 2-mediated glucose uptake. Biosci Rep. (2017) 37:BSR20170216. doi: 10.1042/BSR20170216
26. Moskowitz, MA, Lo, EH, and Iadecola, C. The science of stroke: mechanisms in search of treatments. Neuron. (2010) 67:181–98. doi: 10.1016/j.neuron.2010.07.002
27. Gong, L, Liang, J, Xie, L, Zhang, Z, Mei, Z, and Zhang, W. Metabolic reprogramming in Gliocyte post-cerebral ischemia/reperfusion: from pathophysiology to therapeutic potential. Curr Neuropharmacol. (2024) 22:1672–96. doi: 10.2174/1570159X22666240131121032
28. Maida, CD, Norrito, RL, Daidone, M, Tuttolomondo, A, and Pinto, A. Neuroinflammatory mechanisms in ischemic stroke: focus on Cardioembolic stroke, background, and therapeutic approaches. Int J Mol Sci. (2020) 21:6454. doi: 10.3390/ijms21186454
29. Patabendige, A, Singh, A, Jenkins, S, Sen, J, and Chen, R. Astrocyte activation in neurovascular damage and repair following Ischaemic stroke. Int J Mol Sci. (2021) 22:4280. doi: 10.3390/ijms22084280
30. Fann, DY-W, Lee, S-Y, Manzanero, S, Chunduri, P, Sobey, CG, and Arumugam, TV. Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res Rev. (2013) 12:941–66. doi: 10.1016/j.arr.2013.09.004
31. Nishibori, M, Wang, D, Ousaka, D, and Wake, H. High mobility group Box-1 and blood-brain barrier disruption. Cells. (2020) 9:2650. doi: 10.3390/cells9122650
32. Castiglioni, A, Canti, V, Rovere-Querini, P, and Manfredi, AA. High-mobility group box 1 (HMGB1) as a master regulator of innate immunity. Cell Tissue Res. (2011) 343:189–99. doi: 10.1007/s00441-010-1033-1
33. Shen, Z, Xiang, M, Chen, C, Ding, F, Wang, Y, Shang, C, et al. Glutamate excitotoxicity: potential therapeutic target for ischemic stroke. Biomed Pharmacother. (2022) 151:113125. doi: 10.1016/j.biopha.2022.113125
34. García-Cabezas, MÁ, John, YJ, Barbas, H, and Zikopoulos, B. Distinction of neurons, glia and endothelial cells in the cerebral cortex: An algorithm based on cytological features. Front Neuroanat. (2016) 10:107. doi: 10.3389/fnana.2016.00107
35. Borst, K, Dumas, AA, and Prinz, M. Microglia: immune and non-immune functions. Immunity. (2021) 54:2194–208. doi: 10.1016/j.immuni.2021.09.014
36. Hu, Y, Mai, W, Chen, L, Cao, K, Zhang, B, Zhang, Z, et al. mTOR-mediated metabolic reprogramming shapes distinct microglia functions in response to lipopolysaccharide and ATP. Glia. (2020) 68:1031–45. doi: 10.1002/glia.23760
37. Butovsky, O, and Weiner, HL. Microglial signatures and their role in health and disease. Nat Rev Neurosci. (2018) 19:622–35. doi: 10.1038/s41583-018-0057-5
38. Kanazawa, M, Ninomiya, I, Hatakeyama, M, Takahashi, T, and Shimohata, T. Microglia and monocytes/macrophages polarization reveal novel therapeutic mechanism against stroke. Int J Mol Sci. (2017) 18:2135. doi: 10.3390/ijms18102135
39. Dhanesha, N, Patel, RB, Doddapattar, P, Ghatge, M, Flora, GD, Jain, M, et al. PKM2 promotes neutrophil activation and cerebral thromboinflammation: therapeutic implications for ischemic stroke. Blood. (2022) 139:1234–45. doi: 10.1182/blood.2021012322
40. Takahashi, S. Neuroprotective function of high glycolytic activity in astrocytes: common roles in stroke and neurodegenerative diseases. Int J Mol Sci. (2021) 22:6568. doi: 10.3390/ijms22126568
41. Liu, Z, and Chopp, M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog Neurobiol. (2016) 144:103–20. doi: 10.1016/j.pneurobio.2015.09.008
42. Cerina, M, Levers, M, Keller, JM, and Frega, M. Neuroprotective role of lactate in a human in vitro model of the ischemic penumbra. Sci Rep. (2024) 14:7973. doi: 10.1038/s41598-024-58669-5
43. Pellerin, L, and Magistretti, PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci. (1994) 91:10625–9. doi: 10.1073/pnas.91.22.10625
44. Hayakawa, K, Esposito, E, Wang, X, Terasaki, Y, Liu, Y, Xing, C, et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. (2016) 535:551–5. doi: 10.1038/nature18928
45. Borbor, M, Yin, D, Brockmeier, U, Wang, C, Doeckel, M, Pillath-Eilers, M, et al. Neurotoxicity of ischemic astrocytes involves <scp>STAT3</scp> -mediated metabolic switching and depends on glycogen usage. Glia. (2023) 71:1553–69. doi: 10.1002/glia.24357
46. Lv, Y, Zhang, B, Zhai, C, Qiu, J, Zhang, Y, Yao, W, et al. PFKFB3-mediated glycolysis is involved in reactive astrocyte proliferation after oxygen-glucose deprivation/reperfusion and is regulated by Cdh1. Neurochem Int. (2015) 91:26–33. doi: 10.1016/j.neuint.2015.10.006
47. Kochanski, R, Peng, C, Higashida, T, Geng, X, Hüttemann, M, Guthikonda, M, et al. Neuroprotection conferred by post-ischemia ethanol therapy in experimental stroke: an inhibitory effect on hyperglycolysis and NADPH oxidase activation. J Neurochem. (2013) 126:113–21. doi: 10.1111/jnc.12169
48. O’Neill, LAJ, and Pearce, EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. (2016) 213:15–23. doi: 10.1084/jem.20151570
49. Soto-Heredero, G, de Las, G, Heras, MM, Gabandé-Rodríguez, E, Oller, J, and Mittelbrunn, M. Glycolysis – a key player in the inflammatory response. FEBS J. (2020) 287:3350–69. doi: 10.1111/febs.15327
50. Orihuela, R, McPherson, CA, and Harry, GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. (2016) 173:649–65. doi: 10.1111/bph.13139
51. Li, L, Cheng, S-Q, Sun, Y-Q, Yu, J-B, Huang, X-X, Dong, Y-F, et al. Resolvin D1 reprograms energy metabolism to promote microglia to phagocytize neutrophils after ischemic stroke. Cell Rep. (2023) 42:112617. doi: 10.1016/j.celrep.2023.112617
52. Li, F, Sami, A, Noristani, HN, Slattery, K, Qiu, J, Groves, T, et al. Glial metabolic rewiring promotes axon regeneration and functional recovery in the central nervous system. Cell Metab. (2020) 32:767–785.e7. doi: 10.1016/j.cmet.2020.08.015
53. Sekerdag, E, Solaroglu, I, and Gursoy-Ozdemir, Y. Cell death mechanisms in stroke and novel molecular and cellular treatment options. Curr Neuropharmacol. (2018) 16:1396–415. doi: 10.2174/1570159X16666180302115544
54. Burmistrova, O, Olias-Arjona, A, Lapresa, R, Jimenez-Blasco, D, Eremeeva, T, Shishov, D, et al. Targeting PFKFB3 alleviates cerebral ischemia-reperfusion injury in mice. Sci Rep. (2019) 9:11670. doi: 10.1038/s41598-019-48196-z
55. Madai, S, Kilic, P, Schmidt, RM, Bas-Orth, C, Korff, T, Büttner, M, et al. Activation of the hypoxia-inducible factor pathway protects against acute ischemic stroke by reprogramming central carbon metabolism. Theranostics. (2024) 14:2856–80. doi: 10.7150/thno.88223
56. Kawano, I, Bazila, B, Ježek, P, and Dlasková, A. Mitochondrial dynamics and cristae shape changes during metabolic reprogramming. Antioxid Redox Signal. (2023) 39:684–707. doi: 10.1089/ars.2023.0268
57. Xie, Y, Zhang, J, Lin, Y, Gaeta, X, Meng, X, Wisidagama, DRR, et al. Defining the role of oxygen tension in human neural progenitor fate. Stem Cell Reports. (2014) 3:743–57. doi: 10.1016/j.stemcr.2014.09.021
58. Zheng, X, Boyer, L, Jin, M, Mertens, J, Kim, Y, Ma, L, et al. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. eLife. (2016) 5:e13374. doi: 10.7554/eLife.13374
59. Sun, Y, Zhang, L, Cao, Y, Li, X, Liu, F, Cheng, X, et al. Stroke-induced hexokinase 2 in circulating monocytes exacerbates vascular inflammation and atheroprogression. J Thromb Haemost. (2023) 21:1650–65. doi: 10.1016/j.jtha.2023.02.021
60. Flora, GD, Nayak, MK, Ghatge, M, Kumskova, M, Patel, RB, and Chauhan, AK. Mitochondrial pyruvate dehydrogenase kinases contribute to platelet function and thrombosis in mice by regulating aerobic glycolysis. Blood Adv. (2023) 7:2347–59. doi: 10.1182/bloodadvances.2023010100
61. Flora, GD, Ghatge, M, Nayak, MK, Barbhuyan, T, Kumskova, M, and Chauhan, AK. Deletion of pyruvate dehydrogenase kinases reduces susceptibility to deep vein thrombosis in mice. Blood Adv. (2024) 8:3906–13. doi: 10.1182/bloodadvances.2024013199
62. Doddapattar, P, Dev, R, Ghatge, M, Patel, RB, Jain, M, Dhanesha, N, et al. Myeloid cell PKM2 deletion enhances Efferocytosis and reduces atherosclerosis. Circ Res. (2022) 130:1289–305. doi: 10.1161/CIRCRESAHA.121.320704
63. Khan, AA, Allemailem, KS, Alhumaydhi, FA, Gowder, SJT, and Rahmani, AH. The biochemical and clinical perspectives of lactate dehydrogenase: An enzyme of active metabolism. Endocr Metab Immune Disord Drug Targets. (2020) 20:855–68. doi: 10.2174/1871530320666191230141110
64. Fernie, AR, Carrari, F, and Sweetlove, LJ. Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr Opin Plant Biol. (2004) 7:254–61. doi: 10.1016/j.pbi.2004.03.007
65. Wilson, JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. (2003) 206:2049–57. doi: 10.1242/jeb.00241
66. Lee, H-J, Li, C-F, Ruan, D, He, J, Montal, ED, Lorenz, S, et al. Non-proteolytic ubiquitination of hexokinase 2 by HectH9 controls tumor metabolism and cancer stem cell expansion. Nat Commun. (2019) 10:2625. doi: 10.1038/s41467-019-10374-y
67. Chen, J, Li, G, Sun, D, Li, H, and Chen, L. Research progress of hexokinase 2 in inflammatory-related diseases and its inhibitors. Eur J Med Chem. (2024) 264:115986. doi: 10.1016/j.ejmech.2023.115986
68. Okur, V, Cho, MT, van Wijk, R, van Oirschot, B, Picker, J, Coury, SA, et al. De novo variants in HK1 associated with neurodevelopmental abnormalities and visual impairment. Eur J Hum Genet. (2019) 27:1081–9. doi: 10.1038/s41431-019-0366-9
69. Zhang, X, Wang, R, Hu, D, Sun, X, Fujioka, H, Lundberg, K, et al. Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer’s disease. Sci Adv. (2020) 6:eabb8680. doi: 10.1126/sciadv.abb8680
70. Purich, DL, and Fromm, HJ. The kinetics and regulation of rat brain hexokinase. J Biol Chem. (1971) 246:3456–63. doi: 10.1016/S0021-9258(18)62152-0
71. Roberts, DJ, and Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. (2015) 22:248–57. doi: 10.1038/cdd.2014.173
72. Hu, Y, Cao, K, Wang, F, Wu, W, Mai, W, Qiu, L, et al. Dual roles of hexokinase 2 in shaping microglial function by gating glycolytic flux and mitochondrial activity. Nat Metab. (2022) 4:1756–74. doi: 10.1038/s42255-022-00707-5
73. Lin, Y-H, Wu, Y, Wang, Y, Yao, Z-F, Tang, J, Wang, R, et al. Spatio-temporal expression of Hexokinase-3 in the injured female rat spinal cords. Neurochem Int. (2018) 113:23–33. doi: 10.1016/j.neuint.2017.11.015
74. Xu, S, and Herschman, HR. A tumor agnostic therapeutic strategy for hexokinase 1-null/hexokinase 2-positive cancers. Cancer Res. (2019) 79:5907–14. doi: 10.1158/0008-5472.CAN-19-1789
75. John, S, Weiss, JN, and Ribalet, B. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS One. (2011) 6:e17674. doi: 10.1371/journal.pone.0017674
76. Ludvik, AE, Pusec, CM, Priyadarshini, M, Angueira, AR, Guo, C, Lo, A, et al. HKDC1 is a novel hexokinase involved in whole-body glucose use. Endocrinology. (2016) 157:3452–61. doi: 10.1210/en.2016-1288
77. Ciscato, F, Ferrone, L, Masgras, I, Laquatra, C, and Rasola, A. Hexokinase 2 in Cancer: a Prima Donna playing multiple characters. Int J Mol Sci. (2021) 22:4716. doi: 10.3390/ijms22094716
78. Gong, L, Cui, Z, Chen, P, Han, H, Peng, J, and Leng, X. Reduced survival of patients with hepatocellular carcinoma expressing hexokinase II. Med Oncol. (2012) 29:909–14. doi: 10.1007/s12032-011-9841-z
79. Laridan, E, Denorme, F, Desender, L, François, O, Andersson, T, Deckmyn, H, et al. Neutrophil extracellular traps in ischemic stroke thrombi. Ann Neurol. (2017) 82:223–32. doi: 10.1002/ana.24993
80. Li, Y, Lu, B, Sheng, L, Zhu, Z, Sun, H, Zhou, Y, et al. Hexokinase 2-dependent hyperglycolysis driving microglial activation contributes to ischemic brain injury. J Neurochem. (2018) 144:186–200. doi: 10.1111/jnc.14267
81. Bernier, L-P, York, EM, Kamyabi, A, Choi, HB, Weilinger, NL, and MacVicar, BA. Microglial metabolic flexibility supports immune surveillance of the brain parenchyma. Nat Commun. (2020) 11:1559. doi: 10.1038/s41467-020-15267-z
82. Lu, T, Li, Q, Lin, W, Zhao, X, Li, F, Ji, J, et al. Gut microbiota-derived glutamine attenuates liver ischemia/reperfusion injury via macrophage metabolic reprogramming. Cell Mol Gastroenterol Hepatol. (2023) 15:1255–75. doi: 10.1016/j.jcmgh.2023.01.004
83. Merlin, J, Ivanov, S, Dumont, A, Sergushichev, A, Gall, J, Stunault, M, et al. Non-canonical glutamine transamination sustains efferocytosis by coupling redox buffering to oxidative phosphorylation. Nat Metab. (2021) 3:1313–26. doi: 10.1038/s42255-021-00471-y
84. Hansson, GK, and Libby, P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. (2006) 6:508–19. doi: 10.1038/nri1882
85. Banerjee, C, and Chimowitz, MI. Stroke caused by atherosclerosis of the major intracranial arteries. Circ Res. (2017) 120:502–13. doi: 10.1161/CIRCRESAHA.116.308441
86. Robbins, CS, Hilgendorf, I, Weber, GF, Theurl, I, Iwamoto, Y, Figueiredo, J-L, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. (2013) 19:1166–72. doi: 10.1038/nm.3258
87. Bartrons, R, Simon-Molas, H, Rodríguez-García, A, Castaño, E, Navarro-Sabaté, À, Manzano, A, et al. Fructose 2,6-bisphosphate in Cancer cell metabolism. Front Oncol. (2018) 8:331. doi: 10.3389/fonc.2018.00331
88. Dunaway, GA, Kasten, TP, Sebo, T, and Trapp, R. Analysis of the phosphofructokinase subunits and isoenzymes in human tissues. Biochem J. (1988) 251:677–83. doi: 10.1042/bj2510677
89. Vora, S, Oskam, R, and Staal, GE. Isoenzymes of phosphofructokinase in the rat. Demonstration of the three non-identical subunits by biochemical, immunochemical and kinetic studies. Biochem J. (1985) 229:333–41. doi: 10.1042/bj2290333
90. Lu, L, Chen, Y, and Zhu, Y. The molecular basis of targeting PFKFB3 as a therapeutic strategy against cancer. Oncotarget. (2017) 8:62793–802. doi: 10.18632/oncotarget.19513
91. Kotowski, K, Rosik, J, Machaj, F, Supplitt, S, Wiczew, D, Jabłońska, K, et al. Role of PFKFB3 and PFKFB4 in Cancer: genetic basis, impact on disease development/progression, and potential as therapeutic targets. Cancers (Basel). (2021) 13:909. doi: 10.3390/cancers13040909
92. Rider, MH, Bertrand, L, Vertommen, D, Michels, PA, Rousseau, GG, and Hue, L. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J. (2004) 381:561–79. doi: 10.1042/BJ20040752
93. Sakakibara, R, Kato, M, Okamura, N, Nakagawa, T, Komada, Y, Tominaga, N, et al. Characterization of a human placental Fructose-6-phosphate, 2-kinase/fructose- 2,6-Bisphosphatase. J Biochem. (1997) 122:122–8. doi: 10.1093/oxfordjournals.jbchem.a021719
94. Gabrilovich, DI, and Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
95. Yan, J, Li, A, Chen, X, Cao, K, Song, M, Guo, S, et al. Glycolysis inhibition ameliorates brain injury after ischemic stroke by promoting the function of myeloid-derived suppressor cells. Pharmacol Res. (2022) 179:106208. doi: 10.1016/j.phrs.2022.106208
96. Gonçalves, JT, Schafer, ST, and Gage, FH. Adult neurogenesis in the Hippocampus: from stem cells to behavior. Cell. (2016) 167:897–914. doi: 10.1016/j.cell.2016.10.021
97. Matsubara, S, Matsuda, T, and Nakashima, K. Regulation of adult mammalian neural stem cells and neurogenesis by cell extrinsic and intrinsic factors. Cells. (2021) 10:1145. doi: 10.3390/cells10051145
98. Boese, AC, Le, Q-SE, Pham, D, Hamblin, MH, and Lee, J-P. Neural stem cell therapy for subacute and chronic ischemic stroke. Stem Cell Res Ther. (2018) 9:154. doi: 10.1186/s13287-018-0913-2
99. Gothié, JD, Sébillot, A, Luongo, C, Legendre, M, Nguyen Van, C, Le Blay, K, et al. Adult neural stem cell fate is determined by thyroid hormone activation of mitochondrial metabolism. Mol Metab. (2017) 6:1551–61. doi: 10.1016/j.molmet.2017.08.003
100. Zhang, F, Li, Q, Liang, H, and Zhang, Y. Phosphofructokinase-1 inhibition promotes neuronal differentiation of neural stem cells and functional recovery after stroke. Neuroscience. (2021) 459:27–38. doi: 10.1016/j.neuroscience.2021.01.037
101. Herrero-Mendez, A, Almeida, A, Fernández, E, Maestre, C, Moncada, S, and Bolaños, JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol. (2009) 11:747–52. doi: 10.1038/ncb1881
102. Rodriguez-Rodriguez, P, Fernandez, E, Almeida, A, and Bolaños, JP. Excitotoxic stimulus stabilizes PFKFB3 causing pentose-phosphate pathway to glycolysis switch and neurodegeneration. Cell Death Differ. (2012) 19:1582–9. doi: 10.1038/cdd.2012.33
103. Israelsen, WJ, Dayton, TL, Davidson, SM, Fiske, BP, Hosios, AM, Bellinger, G, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. (2013) 155:397–409. doi: 10.1016/j.cell.2013.09.025
104. Zhu, S, Guo, Y, Zhang, X, Liu, H, Yin, M, Chen, X, et al. Pyruvate kinase M2 (PKM2) in cancer and cancer therapeutics. Cancer Lett. (2021) 503:240–8. doi: 10.1016/j.canlet.2020.11.018
105. Zhang, X, Lei, Y, Zhou, H, Liu, H, and Xu, P. The role of PKM2 in multiple signaling pathways related to neurological diseases. Mol Neurobiol. (2024) 61:5002–26. doi: 10.1007/s12035-023-03901-y
106. Yamada, K, and Noguchi, T. Nutrient and hormonal regulation of pyruvate kinase gene expression. Biochem J. (1999) 337:1–11. doi: 10.1042/bj3370001
107. Puckett, DL, Alquraishi, M, Chowanadisai, W, and Bettaieb, A. The role of PKM2 in metabolic reprogramming: insights into the regulatory roles of non-coding RNAs. Int J Mol Sci. (2021) 22:1171. doi: 10.3390/ijms22031171
108. Zhang, Z, Deng, X, Liu, Y, Liu, Y, Sun, L, and Chen, F. PKM2, function and expression and regulation. Cell Biosci. (2019) 9:52. doi: 10.1186/s13578-019-0317-8
109. Gupta, V, and Bamezai, RNK. Human pyruvate kinase M2: A multifunctional protein. Protein Sci. (2010) 19:2031–44. doi: 10.1002/pro.505
110. Zhou, Q, Yin, Y, Yu, M, Gao, D, Sun, J, Yang, Z, et al. GTPBP4 promotes hepatocellular carcinoma progression and metastasis via the PKM2 dependent glucose metabolism. Redox Biol. (2022) 56:102458. doi: 10.1016/j.redox.2022.102458
111. Li, H, Guo, H, Huang, Q, Wang, S, Li, X, and Qiu, M. Circular RNA P4HB promotes glycolysis and tumor progression by binding with PKM2 in lung adenocarcinoma. Respir Res. (2023) 24:252. doi: 10.1186/s12931-023-02563-7
112. Alquraishi, M, Puckett, DL, Alani, DS, Humidat, AS, Frankel, VD, Donohoe, DR, et al. Pyruvate kinase M2: a simple molecule with complex functions. Free Radic Biol Med. (2019) 143:176–92. doi: 10.1016/j.freeradbiomed.2019.08.007
113. Palsson-McDermott, EM, Curtis, AM, Goel, G, Lauterbach, MAR, Sheedy, FJ, Gleeson, LE, et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the Warburg effect in LPS-activated macrophages. Cell Metab. (2015) 21:65–80. doi: 10.1016/j.cmet.2014.12.005
114. Zhang, Y, Chen, K, Sloan, SA, Bennett, ML, Scholze, AR, O’Keeffe, S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. (2014) 34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014
115. Dang, G, Li, T, Yang, D, Yang, G, Du, X, Yang, J, et al. T lymphocyte-derived extracellular vesicles aggravate abdominal aortic aneurysm by promoting macrophage lipid peroxidation and migration via pyruvate kinase muscle isozyme 2. Redox Biol. (2022) 50:102257. doi: 10.1016/j.redox.2022.102257
116. Christensen, DR, Calder, PC, and Houghton, FD. GLUT3 and PKM2 regulate OCT4 expression and support the hypoxic culture of human embryonic stem cells. Sci Rep. (2015) 5:17500. doi: 10.1038/srep17500
117. Tech, K, Tikunov, AP, Farooq, H, Morrissy, AS, Meidinger, J, Fish, T, et al. Pyruvate kinase inhibits proliferation during postnatal cerebellar neurogenesis and suppresses Medulloblastoma formation. Cancer Res. (2017) 77:3217–30. doi: 10.1158/0008-5472.CAN-16-3304
118. Luo, W, and Semenza, GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab. (2012) 23:560–6. doi: 10.1016/j.tem.2012.06.010
119. Gui, DY, Lewis, CA, and Vander Heiden, MG. Allosteric regulation of PKM2 allows cellular adaptation to different physiological states. Sci Signal. (2013) 6:pe7. doi: 10.1126/scisignal.2003925
120. Stoll, G, Kleinschnitz, C, and Nieswandt, B. Molecular mechanisms of thrombus formation in ischemic stroke: novel insights and targets for treatment. Blood. (2008) 112:3555–62. doi: 10.1182/blood-2008-04-144758
121. Nieswandt, B, Kleinschnitz, C, and Stoll, G. Ischaemic stroke: a thrombo-inflammatory disease? J Physiol. (2011) 589:4115–23. doi: 10.1113/jphysiol.2011.212886
122. De Meyer, SF, Denorme, F, Langhauser, F, Geuss, E, Fluri, F, and Kleinschnitz, C. Thromboinflammation in stroke brain damage. Stroke. (2016) 47:1165–72. doi: 10.1161/STROKEAHA.115.011238
123. Alves-Filho, JC, and Pålsson-McDermott, EM. Pyruvate kinase M2: a potential target for regulating inflammation. Front Immunol. (2016) 7:145. doi: 10.3389/fimmu.2016.00145
124. Zhao, J-J, Xiao, H, Zhao, W-B, Zhang, X-P, Xiang, Y, Ye, Z-J, et al. Remote ischemic Postconditioning for ischemic stroke: a systematic review and Meta-analysis of randomized controlled trials. Chin Med J. (2018) 131:956–65. doi: 10.4103/0366-6999.229892
125. Zhang, B, Shen, J, Zhong, Z, and Zhang, L. PKM2 aggravates cerebral ischemia reperfusion-induced Neuroinflammation via TLR4/MyD88/TRAF6 signaling pathway. Neuroimmunomodulation. (2021) 28:29–37. doi: 10.1159/000509710
126. Feng, Y, Li, X, Wang, J, Huang, X, Meng, L, and Huang, J. Pyruvate kinase M2 (PKM2) improve symptoms of post-ischemic stroke depression by activating VEGF to mediate the MAPK/ERK pathway. Brain Behav. (2022) 12:e2450. doi: 10.1002/brb3.2450
127. Qin, X, Du, Y, Chen, X, Li, W, Zhang, J, and Yang, J. Activation of Akt protects cancer cells from growth inhibition induced by PKM2 knockdown. Cell Biosci. (2014) 4:20. doi: 10.1186/2045-3701-4-20
128. Wu, Q, Ge, W, Chen, Y, Kong, X, and Xian, H. PKM2 involved in neuronal apoptosis on hypoxic-ischemic encephalopathy in neonatal rats. Neurochem Res. (2019) 44:1602–12. doi: 10.1007/s11064-019-02784-7
129. Wang, P, Cui, Y, Liu, Y, Li, Z, Bai, H, Zhao, Y, et al. Mitochondrial ferritin alleviates apoptosis by enhancing mitochondrial bioenergetics and stimulating glucose metabolism in cerebral ischemia reperfusion. Redox Biol. (2022) 57:102475. doi: 10.1016/j.redox.2022.102475
130. Zhong, Y, Jia, B, Xie, C, Hu, L, Liao, Z, Liu, W, et al. Adenylate kinase 4 promotes neuronal energy metabolism and mitophagy in early cerebral ischemia via Parkin/PKM2 pathway. Exp Neurol. (2024) 377:114798. doi: 10.1016/j.expneurol.2024.114798
131. Sugden, MC, and Holness, MJ. Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch Physiol Biochem. (2006) 112:139–49. doi: 10.1080/13813450600935263
132. Liu, K, Zhou, Y, Song, X, Zeng, J, Wang, Z, Wang, Z, et al. Baicalin attenuates neuronal damage associated with SDH activation and PDK2-PDH axis dysfunction in early reperfusion. Phytomedicine. (2024) 129:155570. doi: 10.1016/j.phymed.2024.155570
133. Korotchkina, LG, and Patel, MS. Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J Biol Chem. (2001) 276:37223–9. doi: 10.1074/jbc.M103069200
134. Tuganova, A, Boulatnikov, I, and Popov, KM. Interaction between the individual isoenzymes of pyruvate dehydrogenase kinase and the inner lipoyl-bearing domain of transacetylase component of pyruvate dehydrogenase complex. Biochem J. (2002) 366:129–36. doi: 10.1042/BJ20020301
135. Gudi, R, Bowker-Kinley, MM, Kedishvili, NY, Zhao, Y, and Popov, KM. Diversity of the pyruvate dehydrogenase kinase gene family in humans. J Biol Chem. (1995) 270:28989–94. doi: 10.1074/jbc.270.48.28989
136. Sugden, MC, and Holness, MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol Metab. (2003) 284:E855–62. doi: 10.1152/ajpendo.00526.2002
137. Ravi, S, Chacko, B, Sawada, H, Kramer, PA, Johnson, MS, Benavides, GA, et al. Metabolic plasticity in resting and thrombin activated platelets. PLoS One. (2015) 10:e0123597. doi: 10.1371/journal.pone.0123597
138. Aibibula, M, Naseem, KM, and Sturmey, RG. Glucose metabolism and metabolic flexibility in blood platelets. J Thromb Haemost. (2018) 16:2300–14. doi: 10.1111/jth.14274
139. Spagnoli, LG, Mauriello, A, Sangiorgi, G, Fratoni, S, Bonanno, E, Schwartz, RS, et al. Extracranial thrombotically active carotid plaque as a risk factor for ischemic stroke. JAMA. (2004) 292:1845–52. doi: 10.1001/jama.292.15.1845
140. Koohi, F, Harshfield, EL, Shatunov, A, and Markus, HS. Does thrombosis play a causal role in lacunar stroke and cerebral small vessel disease? Stroke. (2024) 55:934–42. doi: 10.1161/STROKEAHA.123.044937
141. Kaiser, R, Escaig, R, Erber, J, and Nicolai, L. Neutrophil-platelet interactions as novel treatment targets in cardiovascular disease. Front Cardiovasc Med. (2021) 8:824112. doi: 10.3389/fcvm.2021.824112
142. Ciotti, MM, Kaplan, NO, and Stolzenbach, FE. Reaction of pyridine nucleotide analogues with dehydrogenases. J Biol Chem. (1956) 221:833–44. doi: 10.1016/S0021-9258(18)65197-X
143. Kaplan, NO, Ciotti, MM, Bieber, RE, and Hamolsky, M. Molecular heterogeneity and evolution of enzymes. Science (1979). (1960) 131:392–7. doi: 10.1126/science.131.3398.392
144. Fang, Y, Li, Z, Yang, L, Li, W, Wang, Y, Kong, Z, et al. Emerging roles of lactate in acute and chronic inflammation. Cell Commun Signal. (2024) 22:276. doi: 10.1186/s12964-024-01624-8
145. Pasti, AP, Rossi, V, Di Stefano, G, Brigotti, M, and Hochkoeppler, A. Human lactate dehydrogenase a undergoes allosteric transitions under pH conditions inducing the dissociation of the tetrameric enzyme. Biosci Rep. (2022) 42:BSR20212654. doi: 10.1042/BSR20212654
146. Yao, X, and Li, C. Lactate dehydrogenase a mediated histone lactylation induced the pyroptosis through targeting HMGB1. Metab Brain Dis. (2023) 38:1543–53. doi: 10.1007/s11011-023-01195-6
147. Chen, S-F, Pan, M-X, Tang, J-C, Cheng, J, Zhao, D, Zhang, Y, et al. Arginine is neuroprotective through suppressing HIF-1α/LDHA-mediated inflammatory response after cerebral ischemia/reperfusion injury. Mol Brain. (2020) 13:63. doi: 10.1186/s13041-020-00601-9
148. Xiong, X-Y, Pan, X-R, Luo, X-X, Wang, Y-F, Zhang, X-X, Yang, S-H, et al. Astrocyte-derived lactate aggravates brain injury of ischemic stroke in mice by promoting the formation of protein lactylation. Theranostics. (2024) 14:4297–317. doi: 10.7150/thno.96375
149. Chen, A-N, Luo, Y, Yang, Y-H, Fu, J-T, Geng, X-M, Shi, J-P, et al. Lactylation, a novel metabolic reprogramming code: current status and prospects. Front Immunol. (2021) 12:688910. doi: 10.3389/fimmu.2021.688910
150. Li, X, Yang, Y, Zhang, B, Lin, X, Fu, X, An, Y, et al. Lactate metabolism in human health and disease. Signal Transduct Target Ther. (2022) 7:305. doi: 10.1038/s41392-022-01151-3
151. Wyss, MT, Jolivet, R, Buck, A, Magistretti, PJ, and Weber, B. In vivo evidence for lactate as a neuronal energy source. J Neurosci. (2011) 31:7477–85. doi: 10.1523/JNEUROSCI.0415-11.2011
152. Li, R, Yang, Y, Wang, H, Zhang, T, Duan, F, Wu, K, et al. Lactate and Lactylation in the brain: current Progress and perspectives. Cell Mol Neurobiol. (2023) 43:2541–55. doi: 10.1007/s10571-023-01335-7
153. Sun, S, Li, H, Chen, J, and Qian, Q. Lactic acid: no longer an inert and end-product of glycolysis. Physiology. (2017) 32:453–63. doi: 10.1152/physiol.00016.2017
154. Certo, M, Llibre, A, Lee, W, and Mauro, C. Understanding lactate sensing and signalling. Trends Endocrinol Metab. (2022) 33:722–35. doi: 10.1016/j.tem.2022.07.004
155. Tassinari, ID, Rodrigues, F d S, Bertram, C, Mendes-da-Cruz, DA, Guedes, RP, Paz, AH, et al. Lactate protects microglia and neurons from oxygen-glucose deprivation/Reoxygenation. Neurochem Res. (2024) 49:1762–81. doi: 10.1007/s11064-024-04135-7
156. Castillo, X, Rosafio, K, Wyss, MT, Drandarov, K, Buck, A, Pellerin, L, et al. A probable dual mode of action for both L- and D-lactate neuroprotection in cerebral ischemia. J Cereb Blood Flow Metab. (2015) 35:1561–9. doi: 10.1038/jcbfm.2015.115
157. Berthet, C, Lei, H, Thevenet, J, Gruetter, R, Magistretti, PJ, and Hirt, L. Neuroprotective role of lactate after cerebral ischemia. J Cereb Blood Flow Metab. (2009) 29:1780–9. doi: 10.1038/jcbfm.2009.97
158. Berthet, C, Castillo, X, Magistretti, PJ, and Hirt, L. New evidence of neuroprotection by lactate after transient focal cerebral Ischaemia: extended benefit after Intracerebroventricular injection and efficacy of intravenous administration. Cerebrovasc Dis. (2012) 34:329–35. doi: 10.1159/000343657
159. Hirayama, Y, Le, HPN, Hashimoto, H, Ishii, I, Koizumi, S, and Anzai, N. Preconditioning-induced facilitation of lactate release from astrocytes is essential for brain ischemic tolerance. eNeuro. (2024) 11:ENEURO.0494–23.2024. doi: 10.1523/ENEURO.0494-23.2024
160. Shen, Z, Jiang, L, Yuan, Y, Deng, T, Zheng, Y-R, Zhao, Y-Y, et al. Inhibition of G protein-coupled receptor 81 (GPR81) protects against ischemic brain injury. CNS Neurosci Ther. (2015) 21:271–9. doi: 10.1111/cns.12362
161. Magistretti, PJ, and Allaman, I. Lactate in the brain: from metabolic end-product to signalling molecule. Nat Rev Neurosci. (2018) 19:235–49. doi: 10.1038/nrn.2018.19
162. Regli, L, Anderson, RE, and Meyer, FB. Effects of intermittent reperfusion on brain pHi, rCBF, and NADH during rabbit focal cerebral ischemia. Stroke. (1995) 26:1444–52. doi: 10.1161/01.STR.26.8.1444
163. Zhou, J, Zhang, L, Peng, J, Zhang, X, Zhang, F, Wu, Y, et al. Astrocytic LRP1 enables mitochondria transfer to neurons and mitigates brain ischemic stroke by suppressing ARF1 lactylation. Cell Metab. (2024) 36:2054–2068.e14. doi: 10.1016/j.cmet.2024.05.016
164. Blevins, HM, Xu, Y, Biby, S, and Zhang, S. The NLRP3 Inflammasome pathway: a review of mechanisms and inhibitors for the treatment of inflammatory diseases. Front Aging Neurosci. (2022) 14:879021. doi: 10.3389/fnagi.2022.879021
165. Barrington, J, Lemarchand, E, and Allan, SM. A brain in flame; do inflammasomes and pyroptosis influence stroke pathology? Brain Pathol. (2017) 27:205–12. doi: 10.1111/bpa.12476
166. Chen, C, Zhou, Y, Ning, X, Li, S, Xue, D, Wei, C, et al. Directly targeting ASC by lonidamine alleviates inflammasome-driven diseases. J Neuroinflammation. (2022) 19:315. doi: 10.1186/s12974-022-02682-w
167. Guan, Y, Pan, L, Niu, D, Li, X, Li, S, Cheng, G, et al. Mailuo Shutong pills inhibit neuroinflammation by regulating glucose metabolism disorders to protect mice from cerebral ischemia-reperfusion injury. J Ethnopharmacol. (2024) 335:118621. doi: 10.1016/j.jep.2024.118621
168. Yan, Y, Mukherjee, S, Harikumar, KG, Strutzenberg, TS, Zhou, XE, Suino-Powell, K, et al. Structure of an AMPK complex in an inactive, ATP-bound state. Science (1979). (2021) 373:413–9. doi: 10.1126/science.abe7565
169. Qin, C, Yang, S, Chu, Y-H, Zhang, H, Pang, X-W, Chen, L, et al. Signaling pathways involved in ischemic stroke: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther. (2022) 7:215. doi: 10.1038/s41392-022-01064-1
170. Zur Nedden, S, Safari, MS, Weber, D, Kuenkel, L, Garmsiri, C, Lang, L, et al. Protein kinase N1 deficiency results in upregulation of cerebral energy metabolism and is highly protective in in vivo and in vitro stroke models. Metabolism. (2024) 161:156039. doi: 10.1016/j.metabol.2024.156039
171. Tian, F, Yi, J, Liu, Y, Chen, B, Wang, X, Ouyang, Y, et al. Integrating network pharmacology and bioinformatics to explore and experimentally verify the regulatory effect of Buyang Huanwu decoction on glycolysis and angiogenesis after cerebral infarction. J Ethnopharmacol. (2024) 319:117218. doi: 10.1016/j.jep.2023.117218
172. Szwed, A, Kim, E, and Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol Rev. (2021) 101:1371–426. doi: 10.1152/physrev.00026.2020
173. Goul, C, Peruzzo, R, and Zoncu, R. The molecular basis of nutrient sensing and signalling by mTORC1 in metabolism regulation and disease. Nat Rev Mol Cell Biol. (2023) 24:857–75. doi: 10.1038/s41580-023-00641-8
174. Zhang, D-M, Zhang, T, Wang, M-M, Wang, X-X, Qin, Y-Y, Wu, J, et al. TIGAR alleviates ischemia/reperfusion-induced autophagy and ischemic brain injury. Free Radic Biol Med. (2019) 137:13–23. doi: 10.1016/j.freeradbiomed.2019.04.002
175. Qin, J, Shen, X, Zhang, J, and Jia, D. Allosteric inhibitors of the STAT3 signaling pathway. Eur J Med Chem. (2020) 190:112122. doi: 10.1016/j.ejmech.2020.112122
176. Hillmer, EJ, Zhang, H, Li, HS, and Watowich, SS. STAT3 signaling in immunity. Cytokine Growth Factor Rev. (2016) 31:1–15. doi: 10.1016/j.cytogfr.2016.05.001
177. Hu, X, Li, J, Fu, M, Zhao, X, and Wang, W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. (2021) 6:402. doi: 10.1038/s41392-021-00791-1
178. Liu, C, and Zhou, X. TREM2 impairs glycolysis to interrupt microglial M1 polarization and inflammation via JAK2/STAT3 Axis. Cell Biochem Biophys. [Online ahead of print] (2024). doi: 10.1007/s12013-024-01520-5
179. Lv, P-C, Jiang, A-Q, Zhang, W-M, and Zhu, H-L. FAK inhibitors in Cancer, a patent review. Expert Opin Ther Pat. (2018) 28:139–45. doi: 10.1080/13543776.2018.1414183
180. Chen, D, Wei, L, Liu, Z-R, Yang, JJ, Gu, X, Wei, ZZ, et al. Pyruvate kinase M2 increases angiogenesis, neurogenesis, and functional recovery mediated by upregulation of STAT3 and focal adhesion kinase activities after ischemic stroke in adult mice. Neurotherapeutics. (2018) 15:770–84. doi: 10.1007/s13311-018-0635-2
181. Eltzschig, HK, Bratton, DL, and Colgan, SP. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat Rev Drug Discov. (2014) 13:852–69. doi: 10.1038/nrd4422
182. He, Q, Ma, Y, Liu, J, Zhang, D, Ren, J, Zhao, R, et al. Biological functions and regulatory mechanisms of hypoxia-inducible factor-1α in ischemic stroke. Front Immunol. (2021) 12:801985. doi: 10.3389/fimmu.2021.801985
183. Ma, W, Wu, Q, Wang, S, Wang, H, Ye, J, Sun, H, et al. A breakdown of metabolic reprogramming in microglia induced by CKLF1 exacerbates immune tolerance in ischemic stroke. J Neuroinflammation. (2023) 20:97. doi: 10.1186/s12974-023-02779-w
184. Hui, W, Huang, W, Zheng, Z, Li, Y, Li, P, and Yang, H. Ginkgo biloba extract promotes Treg differentiation to ameliorate ischemic stroke via inhibition of HIF-1α/HK2 pathway. Phytother Res. (2023) 37:5821–36. doi: 10.1002/ptr.7988
185. Gao, J, Liu, R, Tang, J, Pan, M, Zhuang, Y, Zhang, Y, et al. Suppressing nuclear translocation of microglial PKM2 confers neuroprotection via downregulation of neuroinflammation after mouse cerebral ischemia–reperfusion injury. Int Immunopharmacol. (2024) 141:112880. doi: 10.1016/j.intimp.2024.112880
186. Kaushik, DK, Bhattacharya, A, Mirzaei, R, Rawji, KS, Ahn, Y, Rho, JM, et al. Enhanced glycolytic metabolism supports transmigration of brain-infiltrating macrophages in multiple sclerosis. J Clin Invest. (2019) 129:3277–92. doi: 10.1172/JCI124012
187. He, F, Ru, X, and Wen, T. NRF2, a transcription factor for stress response and beyond. Int J Mol Sci. (2020) 21:4777. doi: 10.3390/ijms21134777
188. Liu, M, Zhou, X, Li, Y, Ma, S, Pan, L, Zhang, X, et al. TIGAR alleviates oxidative stress in brain with extended ischemia via a pentose phosphate pathway-independent manner. Redox Biol. (2022) 53:102323. doi: 10.1016/j.redox.2022.102323
189. Zhang, Y, Wang, X, Yang, H, Liu, H, Lu, Y, Han, L, et al. Kinase <scp>AKT</scp> controls innate immune cell development and function. Immunology. (2013) 140:143–52. doi: 10.1111/imm.12123
190. Franke, TF. Intracellular signaling by Akt: bound to be specific. Sci Signal. (2008) 1:pe29. doi: 10.1126/scisignal.124pe29
191. Li, Y, Yang, Y, Zhao, Y, Zhang, J, Liu, B, Jiao, S, et al. Astragaloside IV reduces neuronal apoptosis and parthanatos in ischemic injury by preserving mitochondrial hexokinase-II. Free Radic Biol Med. (2019) 131:251–63. doi: 10.1016/j.freeradbiomed.2018.11.033
192. Wu, K, Cheng, K, Wang, Y, Chen, Y, Wong, K, Su, T, et al. Perturbation of Akt signaling, mitochondrial potential, and ADP/ATP ratio in acidosis-challenged rat cortical astrocytes. J Cell Biochem. (2017) 118:1108–17. doi: 10.1002/jcb.25725
193. Xie, Y, Shi, X, Sheng, K, Han, G, Li, W, Zhao, Q, et al. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (review). Mol Med Rep. (2018) 19:783–91. doi: 10.3892/mmr.2018.9713
194. Wu, L, Cheng, Y, Wang, R, Sun, S, Ma, B, and Zhang, Z. NDRG2 regulates glucose metabolism and ferroptosis of OGD/R-treated astrocytes by the Wnt/β-catenin signaling. J Biochem Mol Toxicol. (2024) 38:e23827. doi: 10.1002/jbt.23827
195. Ruan, J, Wang, L, Wang, N, Huang, P, Chang, D, Zhou, X, et al. Hydroxysafflor yellow a promotes angiogenesis of brain microvascular endothelial cells from ischemia/reperfusion injury via glycolysis pathway in vitro. J Stroke Cerebrovasc Dis. (2025) 34:108107. doi: 10.1016/j.jstrokecerebrovasdis.2024.108107
196. Jin, H, Bi, R, Hu, J, Xu, D, Su, Y, Huang, M, et al. Elevated serum lactate dehydrogenase predicts unfavorable outcomes after rt-PA thrombolysis in ischemic stroke patients. Front Neurol. (2022) 13:816216. doi: 10.3389/fneur.2022.816216
197. Anan, M, Nagai, Y, Fudaba, H, and Fujiki, M. Lactate and lactate dehydrogenase in cistern as biomarkers of early brain injury and delayed cerebral ischemia of subarachnoid hemorrhage. J Stroke Cerebrovasc Dis. (2020) 29:104765. doi: 10.1016/j.jstrokecerebrovasdis.2020.104765
198. Kong, L-L, Gao, L, Wang, K-X, Liu, N-N, Liu, C, Ma, G-D, et al. Pinocembrin attenuates hemorrhagic transformation after delayed t-PA treatment in thromboembolic stroke rats by regulating endogenous metabolites. Acta Pharmacol Sin. (2021) 42:1223–34. doi: 10.1038/s41401-021-00664-x
199. Sapir, G, Shaul, D, Lev-Cohain, N, Sosna, J, Gomori, MJ, and Katz-Brull, R. LDH and PDH activities in the ischemic brain and the effect of reperfusion-An ex vivo MR study in rat brain slices using hyperpolarized [1-13C]pyruvate. Meta. (2021) 11:210. doi: 10.3390/metabo11040210
200. Iwasa, N, Matsui, TK, Iguchi, N, Kinugawa, K, Morikawa, N, Sakaguchi, YM, et al. Gene expression profiles of human cerebral organoids identify PPAR pathway and PKM2 as key markers for oxygen-glucose deprivation and Reoxygenation. Front Cell Neurosci. (2021) 15:605030. doi: 10.3389/fncel.2021.605030
201. Zhang, D, Jia, N, Hu, Z, Keqing, Z, Chenxi, S, Chunying, S, et al. Bioinformatics identification of potential biomarkers and therapeutic targets for ischemic stroke and vascular dementia. Exp Gerontol. (2024) 187:112374. doi: 10.1016/j.exger.2024.112374
202. Makris, K, Haliassos, A, Chondrogianni, M, and Tsivgoulis, G. Blood biomarkers in ischemic stroke: potential role and challenges in clinical practice and research. Crit Rev Clin Lab Sci. (2018) 55:294–328. doi: 10.1080/10408363.2018.1461190
203. Shaul, D, Grieb, B, Sapir, G, Uppala, S, Sosna, J, Gomori, JM, et al. The metabolic representation of ischemia in rat brain slices: a hyperpolarized 13 C magnetic resonance study. NMR Biomed. (2021) 34:e4509. doi: 10.1002/nbm.4509
204. Vázquez, JA, Adducci, MDC, Godoy Monzón, D, and Iserson, KV. Lactic dehydrogenase in cerebrospinal fluid may differentiate between structural and non-structural central nervous system lesions in patients with diminished levels of consciousness. J Emerg Med. (2009) 37:93–7. doi: 10.1016/j.jemermed.2008.04.032
205. Baheerathan, A, Pitceathly, RD, Curtis, C, and Davies, NW. CSF lactate. Pract Neurol. (2020) 20:320–3. doi: 10.1136/practneurol-2019-002191
206. Vojinovic, D, Kalaoja, M, Trompet, S, Fischer, K, Shipley, MJ, Li, S, et al. Association of Circulating Metabolites in plasma or serum and risk of stroke: Meta-analysis from 7 prospective cohorts. Neurology. (2021) 96:e1110–23. doi: 10.1212/WNL.0000000000011236
207. He, Q, Wang, M, Zhu, H, Xiao, Y, Wen, R, Liu, X, et al. Mediation effect of stroke recurrence in the association between post-stroke lactate dehydrogenase and functional disability. Front Aging Neurosci. (2024) 16:1450863. doi: 10.3389/fnagi.2024.1450863
208. Pambianco, G, Orchard, T, and Landau, P. Deep vein thrombosis: prevention in stroke patients during rehabilitation. Arch Phys Med Rehabil. (1995) 76:324–30. doi: 10.1016/S0003-9993(95)80657-1
209. Li, M, Lu, W, Meng, Y, Zhang, W, Wang, F, Sun, L, et al. Tetrahydroxy stilbene glucoside alleviates ischemic stroke by regulating conformation-dependent intracellular distribution of PKM2 for M2 macrophage polarization. J Agric Food Chem. (2022) 70:15449–63. doi: 10.1021/acs.jafc.2c03923
210. Gao, C-L, Hou, G-G, Liu, J, Ru, T, Xu, Y-Z, Zhao, S-Y, et al. Synthesis and target identification of Benzoxepane derivatives as potential anti-Neuroinflammatory agents for ischemic stroke. Angew Chem Int Ed Engl. (2020) 59:2429–39. doi: 10.1002/anie.201912489
211. Guo, S, Cosky, E, Li, F, Guan, L, Ji, Y, Wei, W, et al. An inhibitory and beneficial effect of chlorpromazine and promethazine (C + P) on hyperglycolysis through HIF-1α regulation in ischemic stroke. Brain Res. (2021) 1763:147463. doi: 10.1016/j.brainres.2021.147463
212. Guan, L, Lee, H, Geng, X, Li, F, Shen, J, Ji, Y, et al. Neuroprotective effects of pharmacological hypothermia on Hyperglycolysis and gluconeogenesis in rats after ischemic stroke. Biomol Ther. (2022) 12:851. doi: 10.3390/biom12060851
213. Cai, L, Stevenson, J, Peng, C, Xin, R, Rastogi, R, Liu, K, et al. Adjuvant therapies using normobaric oxygen with hypothermia or ethanol for reducing hyperglycolysis in thromboembolic cerebral ischemia. Neuroscience. (2016) 318:45–57. doi: 10.1016/j.neuroscience.2016.01.010
214. Zhang, J, Deng, Z, Liao, J, Song, C, Liang, C, Xue, H, et al. Leptin attenuates cerebral ischemia injury through the promotion of energy metabolism via the PI3K/Akt pathway. J Cereb Blood Flow Metab. (2013) 33:567–74. doi: 10.1038/jcbfm.2012.202
215. Marangos, PJ, Turkel, CC, Dziewanowska, ZE, and Fox, AW. Dichloroacetate and cerebral ischaemia therapeutics. Expert Opin Investig Drugs. (1999) 8:373–82. doi: 10.1517/13543784.8.4.373
216. Chang, LH, Shimizu, H, Abiko, H, Swanson, RA, Faden, AI, James, TL, et al. Effect of dichloroacetate on recovery of brain lactate, phosphorus energy metabolites, and glutamate during reperfusion after complete cerebral ischemia in rats. J Cereb Blood Flow Metab. (1992) 12:1030–8. doi: 10.1038/jcbfm.1992.140
217. Guan, X, Wei, D, Liang, Z, Xie, L, Wang, Y, Huang, Z, et al. FDCA attenuates Neuroinflammation and brain injury after cerebral ischemic stroke. ACS Chem Neurosci. (2023) 14:3839–54. doi: 10.1021/acschemneuro.3c00456
218. Chen, M, Liu, M, Luo, Y, Cao, J, Zeng, F, Yang, L, et al. Celastrol protects against cerebral ischemia/reperfusion injury in mice by inhibiting glycolysis through targeting HIF-1α/PDK1 Axis. Oxidative Med Cell Longev. (2022) 2022:1–14. doi: 10.1155/2022/7420507
219. Göschl, L, Scheinecker, C, and Bonelli, M. Treg cells in autoimmunity: from identification to Treg-based therapies. Semin Immunopathol. (2019) 41:301–14. doi: 10.1007/s00281-019-00741-8
220. Shen, H, Pei, H, Zhai, L, Guan, Q, and Wang, G. Salvianolic acid C improves cerebral ischemia reperfusion injury through suppressing microglial cell M1 polarization and promoting cerebral angiogenesis. Int Immunopharmacol. (2022) 110:109021. doi: 10.1016/j.intimp.2022.109021
221. Gao, J, Yao, M, Zhang, W, Yang, B, Yuan, G, Liu, J-X, et al. Panax notoginseng saponins alleviates inflammation induced by microglial activation and protects against ischemic brain injury via inhibiting HIF-1α/PKM2/STAT3 signaling. Biomed Pharmacother. (2022) 155:113479. doi: 10.1016/j.biopha.2022.113479
222. Chen, S, Zou, R, Si, J, Shi, Q, Zhang, L, Kang, L, et al. Icariin inhibits apoptosis in OGD-induced neurons by regulating M2 pyruvate kinase. IBRO Neurosci Rep. (2024) 16:535–41. doi: 10.1016/j.ibneur.2024.04.005
223. Ye, R, Zhang, X, Kong, X, Han, J, Yang, Q, Zhang, Y, et al. Ginsenoside Rd attenuates mitochondrial dysfunction and sequential apoptosis after transient focal ischemia. Neuroscience. (2011) 178:169–80. doi: 10.1016/j.neuroscience.2011.01.007
224. Li, M, Jia, J, Wang, Y, Zhuang, Y, Wang, H, Lin, Z, et al. Astragaloside IV promotes cerebral tissue restoration through activating AMPK- mediated microglia polarization in ischemic stroke rats. J Ethnopharmacol. (2024) 334:118532. doi: 10.1016/j.jep.2024.118532
225. Tang, L, Liu, Z, Ji, Z, Zhang, X, Zhao, M, Peng, D, et al. Promotion of mature angiogenesis in ischemic stroke by Taohong Siwu decoction through glycolysis activation. Front Pharmacol. (2024) 15:1395167. doi: 10.3389/fphar.2024.1395167
226. Dhuria, SV, Hanson, LR, and Frey, WH. Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. (2010) 99:1654–73. doi: 10.1002/jps.21924
Keywords: ischemic stroke, glycolysis, glucose metabolism, pathomechanism, diagnosis, treatment
Citation: Liu Y, Hu P, Cheng H, Xu F and Ye Y (2025) The impact of glycolysis on ischemic stroke: from molecular mechanisms to clinical applications. Front. Neurol. 16:1514394. doi: 10.3389/fneur.2025.1514394
Edited by:
Vinay Kumar, The Pennsylvania State University, United StatesReviewed by:
Akanksha Agrawal, Indiana University-Purdue University Indianapolis, United StatesCopyright © 2025 Liu, Hu, Cheng, Xu and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peijia Hu, emp5eWhwakAxNjMuY29t; Hongliang Cheng, Y2hsLjc1ODExQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.