 YiYing Wang1†
YiYing Wang1† Hui Wang
Hui Wang JingSi Jiang
JingSi Jiang Le Mao
Le Mao YanXi Heng
YanXi Heng Min Deng
Min Deng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 18 March 2025
Sec. Neurogenetics
Volume 16 - 2025 | https://doi.org/10.3389/fneur.2025.1438207
This article is part of the Research Topic Genetics in Rare Neurological Diseases: From Discovery to Targeted Treatment View all 12 articles
Introduction: Amyotrophic lateral sclerosis (ALS) is a severe neurodegenerative disease characterized by a progressive loss of motor neurons and muscle atrophy. Genetic factors are known to play important roles in ALS and concomitant presence of rare variants in ALS patients have been increasingly reported.
Methods: In order to explore the genetic variants in ALS patients within the context of oligogenic inheritance and to elucidate the clinical heterogeneity observed in these patients, we conducted whole-genome sequencing on 34 familial ALS (FALS) probands.
Results: In one proband, we identified a CHCHD10 p.Gly66Val variant, along with three additional variants: UNC13A p.Leu1034Val, SUSD1 p.Trp704Ser, and SQSTM1 p.His359del. This patient exhibited a slow disease progression and a prolonged survival duration, consistent with the clinical features of ALS patients with CHCHD10 variants. This suggests that the CHCHD10 p.Gly66Val variant may play a predominant role in shaping the patient's phenotype, while the other variants may primarily contribute to ALS occurrence.
Discussion: Variants in CHCHD10 have been found in ALS and other neurodegenerative diseases, exhibiting significant clinical variability. However, the combinatorial effect of CHCHD10 and other ALS-related gene variants has not been fully studied. Our findings suggest that the combined impact of these four variants contributes to this patient's ALS phenotype, distinguishing it from other, less severe neuromuscular disorders associated with CHCHD10 mutations. Overall, this study further supports the oligogenic pathogenic basis of ALS and offers new insights into understanding the intricate clinical presentations associated with CHCHD10 variants.
Amyotrophic lateral sclerosis (ALS) is a severe neurodegenerative disease that affects both upper and lower motor neurons, leading to gradual degeneration and loss of these cells. Patients with ALS exhibit highly heterogeneous clinical manifestations marked by progressive muscular atrophy, and often die from respiratory failure within 2–4 years after the symptom initiation (1). Typically, ALS can be classified as the familial (FALS) or sporadic (SALS) form. Research has revealed that heredity plays a significant role in ALS, especially in familial cases. However, patients from different ethnicities exhibit a distinct genetic basis. In the Chinese ALS population, the most prevalent mutation causing ALS is located in the SOD1 gene, representing around 21.9% of familial cases and 1.90% of sporadic cases, followed by FUS and TARDBP, accounting for 6.3 and 3.1% of FALS patients, respectively (2). Currently, despite the growing identification of pathogenic genes linked to ALS, its etiology remains largely unknown.
The CHCHD10 gene encodes a protein containing coiled-coil-helix-coiled-coil-helix domains, which is implicated in maintaining mitochondrial cristae morphology and mitochondrial morphological remodeling (3). Since 2014, many mutations in CHCHD10 have been discovered in ALS patients, solidifying its status as a definitive ALS gene (4). Subsequent studies have also unveiled a wide range of CHCHD10-related disorders, including late-onset spinal motor neuronopathy (LOSMoN/SMAJ) (5), axonal Charcot-Marie-Tooth disease type 2 (CMT2) (6), Alzheimer's disease (7), and myopathy (8). Consequently, patients with the same CHCHD10 variant can exhibit a wide variety of clinical phenotypes, suggesting the complexity of the underlying pathogenesis mechanism, possibly involving other contributing factors.
Recent studies have also unveiled the oligogenic pathogenic pattern of ALS, where multiple mutations in ALS-related genes can be identified within the same patient, contributing collectively to disease onset (9–11). This model arose due to the observation of incomplete penetrance in FALS cases with a clear autosomal dominant inheritance pattern, suggesting the potential involvement of additional genetic factors (12). Several studies have reported on the prevalence of oligogenic carriers across different ALS cohorts. For example, Naruse et al. identified multiple variants in ALS-related genes through whole-exome sequencing (WES) in 7/56 FALS probands and 8/87 SALS patients (9). Additionally, Scarlino et al. discovered that 17% of ALS patients in an Italian cohort carried multi-gene variants through next-generation sequencing (NGS) (13). The combined effect of these variants contributes to the onset of the disease and often influences phenotypic characteristics, such as a more severe clinical phenotype, which may include earlier age of onset (9, 14) and poorer prognosis (10, 13). Therefore, the oligogenic model provides new insights into the clinical heterogeneity observed in ALS patients. While the number of newly identified genes related to ALS is increasing, mutation screenings remain predominantly centered around common ALS-associated genes, with limited researches employing comprehensive mutation screening across all potential ALS-related genes, especially in Chinese population. In light of this, there is an increasing need for thorough genetic screening of ALS patients.
Therefore, we conducted whole-genome sequencing (WGS) on 34 Chinese FALS probands to gain a thorough understanding of the genetic landscape of these patients. Given the intricate clinical phenotypes observed in patients carrying CHCHD10 variants, we specifically focused on this gene in this study. We explored whether CHCHD10 variants co-occurred with additional variants in the other 112 ALS-associated genes, aiming to provide further insights into the potential role of oligogenic pathogenesis in ALS.
In total, 127 probands diagnosed with familial amyotrophic lateral sclerosis (FALS), meeting the criteria for definite or probable ALS as per the EI criteria (15), were recruited from the Department of Neurology at Peking University Third Hospital during 2003 and 2018. Additionally, some of the family members were also enrolled for subsequent segregation analysis. Written informed consent for involvement in both clinical and genetic research was obtained from all participants during their hospital visits. This project received approval from the Peking University Third Hospital Institutional Ethics Committee (No. IRB00006761-L2010055), and was conducted in accordance with the principles of the Declaration of Helsinki.
Detailed clinical features information of these patients was collected, including age of onset, initial symptoms, family history, and medication usage. Additionally, regular telephonic follow-ups were conducted. Disease severity was evaluated using the ALS Function Rating Scale (ALSFRS).
We first screened these 127 FALS probands for common mutations in the four most prevalent ALS genes (SOD1, FUS, TARDBP, and C9orf72). Mutations in SOD1, FUS, and TARDBP were identified using polymerase chain reaction (PCR) and Sanger sequencing. The repeat length of the pathogenic C9orf72 G4C2 repeat expansion was examined by repeat-primed PCR combined with fragment length analysis using fluorescently labeled primers as previously reported (16). Patients with negative results in these four genes were considered more likely to harbor rare or novel variants in other ALS-related genes. To further elucidate the mutation status of these patients, we conducted whole-genome sequencing on probands from 34 pedigrees. The selection of these probands for WGS was based on a comprehensive evaluation of factors including the completeness of clinical phenotypes and follow-up information, the quality of preserved DNA samples, and the availability of DNA from additional family members.
DNA samples were extracted from 5 ml peripheral blood collected from the participants using a QIAamp® DNA Blood Mini Kit (Qiagen, Valencia, CA, USA) and quantified using the Qubit 3.0 fluorometer (Life Technologies, Paisley, UK). Genomic DNA was broken up into DNA fragments between 50 and 800 bp on Covaris E220 (Covaris, Brighton, UK), which were further narrowed down using AMPure XP beads (AGENCOURT) to between 100 and about 300 bp for construction of the sequencing library by NanoBalls (DNB) technology.
Whole-genome sequencing was performed using the BGISEQ-500 protocol provided by BGI (Beijing Genomics Institution), incorporating DNA nanoball and probe-anchor synthesis technologies for library construction, quality control, pooling, and sequencing. The filtering and screening process for whole-genome sequencing analysis is illustrated in Figure 1. SOAPnuke (version 2.0.1) was utilized for filtering raw reads, removing those containing a fraction of gaps (N) or low-quality bases (Q < 20) exceeding a threshold of >0.1. The resulting clean reads were aligned to the human reference genome (hg38) using the Burrows-Wheeler Aligner (version 0.7.15), and the alignment summaries were generated by samtools (version 1.15.1).
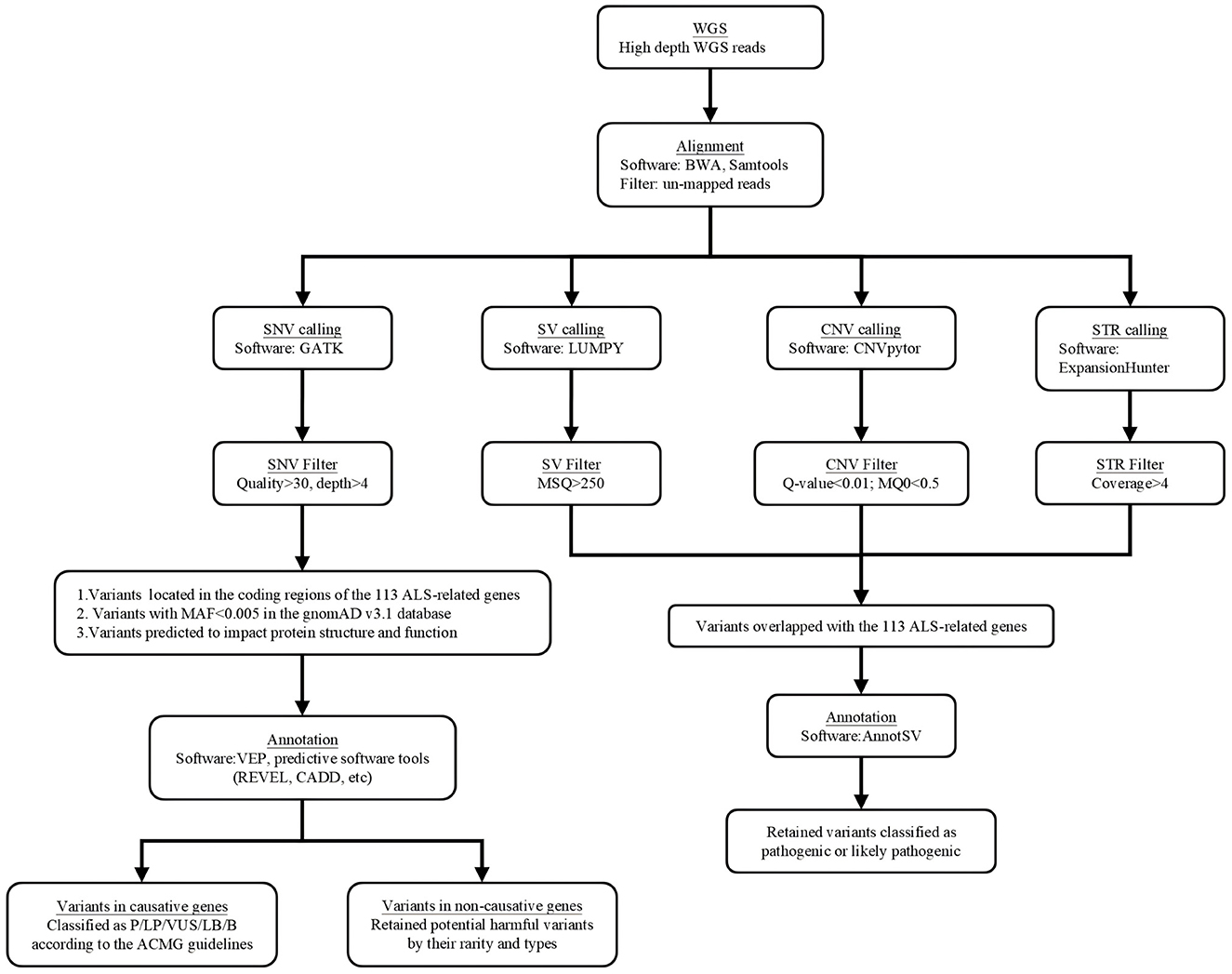
Figure 1. The filtering and screening process of whole genome sequencing analysis.
Different filtering criteria were applied based on variant types. SNVs and INDELs were identified using the Genome Analysis Tool Kit (v3.6) and filtered with the following QC criteria: read depth < 4, mapping quality < 55, variant quality < 30, and genotype quality < 30. SVs were detected with LUMPY (v0.2.13) and SVTyper (v0.1.4), and excluded if: split-read and paired-end counts < 10%, mean sample quality < 150, deletion size < sequencing library insert size, or copy number estimates (CNVnator) >0.5 for deletions or < 1.5 for duplications. CNVs were detected using CNVnator (v0.4, 100 bp bins), merged with bedtools (v2.30.0, 1 bp overlap), and excluded if Q-value > 0.05, zero map quality reads > 50%, gaps > 0, or size < 1,000 bp, or > 100,000 bp. Repeat expansions were called with ExpansionHunter (v5.0.0), excluding variants with quality < 30 or coverage depth < 10×.
To comprehensively analyze the potential disease-related variants in these patients, we systematically collected ALS-related genes from OMIM (https://www.omim.org/), PubMed (https://pubmed.ncbi.nlm.nih.gov/), Google Scholar (https://scholar.google.com/), the MGI database (https://www.informatics.jax.org/), and ALSoD (https://alsod.ac.uk/). Aiming to obtain a balance between the breadth of gene inclusion and the feasibility of data analysis, we included all 113 genes that have been previously associated with ALS in cellular or familial studies (Supplementary Table S1). Variants with MAF > 0.005 in the gnomAD v4.1.0 database were excluded. Variants located in the coding region of these 113 ALS-associated genes and were predicted to impact protein structure and function were retained for further evaluation. For causative genes, we scored and categorized the variants based on the American College of Medical Genetics (ACMG) guidelines. For variants located on non-causative genes, we filtered rare variants through frequency (MAF < 0.005 in gnomAD database), selected potentially harmful variants by variant type, and ranked the variants in conjunction with the results from software tools that predicts harmfulness. We used VEP (version 107) software, which integrates tools like SIFT (17) and PolyPhen-2 (18) for predicting the impact of variants on protein function. Additionally, we employed other in silico prediction tools—CADD (19), FATHMM (20), MutationTaster (21), M-CAP Score (22), and REVEL (23)—to further evaluate their pathogenic potential. PhastCons (24) and phyloP (25) scores were also used to assess the evolutionary conservation of these variants. Variants predicted to be harmful by multiple tools were given priority under equivalent conditions.
Annotation and Ranking of Human Structural Variations (AnnotSV) was employed to provide comprehensive annotations and scores for SVs, CNVs, and STRs, with scores ranging from 1 to 5 corresponding to P, LP, VUS, LB, and B. Variants meeting two conditions were preserved: those classified as pathogenic or likely pathogenic by AnnotSV, and those where the variant's genomic region overlapped with any of the 113 ALS-associated genes.
All variants identified were validated through PCR and Sanger sequencing. Based on the reference genomic sequences of the human genome from GenBank at NCBI, primer pairs were designed for the candidate loci (Supplementary Table S2).
Demographic and clinical characteristics of all 127 patients included in this study are presented in Table 1. Among the 34 probands who underwent whole-genome sequencing, only one variant located in the CHCHD10 gene, CHCHD10 Gly66Val, was identified in the proband of Family 43 (Figure 2A). Additionally, this patient harbored three additional rare variants in other ALS-associated genes that met our variant filtering criteria: UNC13A p.Leu1034Val, SUSD1 p.Trp704Ser, and SQSTM1 p.His359del, which may have collectively contributed to the development of ALS in this pedigree (Table 2). All the variants have been verified through Sanger sequencing (Figure 2B). Multiple tools were employed to predict the pathogenicity of these variants (Table 3).
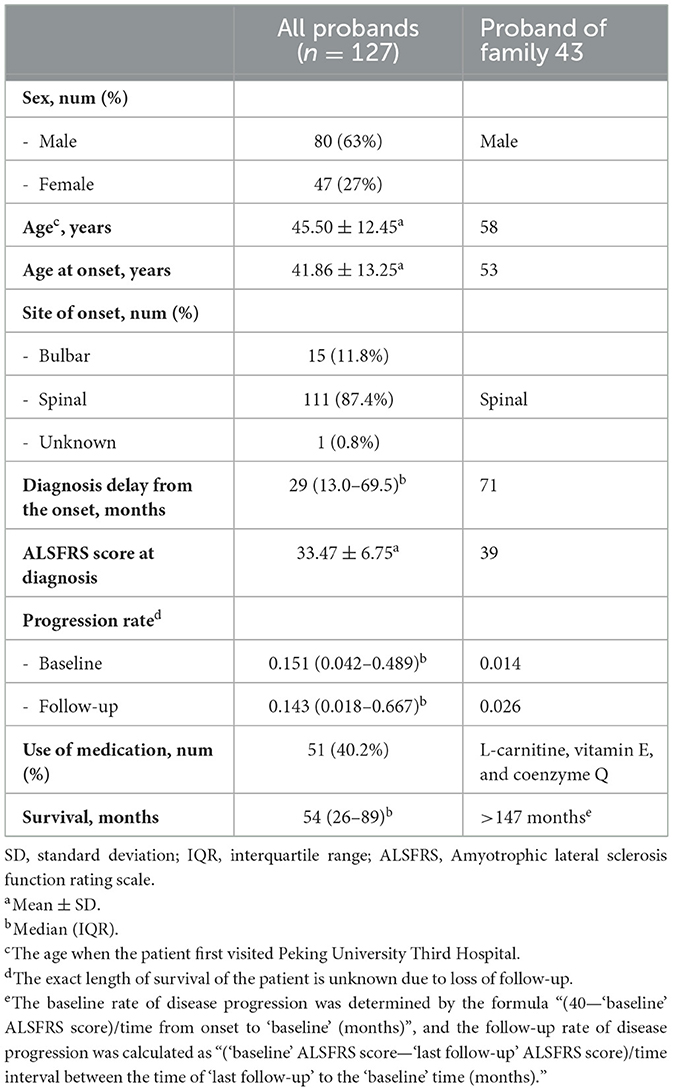
Table 1. Demographic and clinical characteristics of FALS patients included in this study.
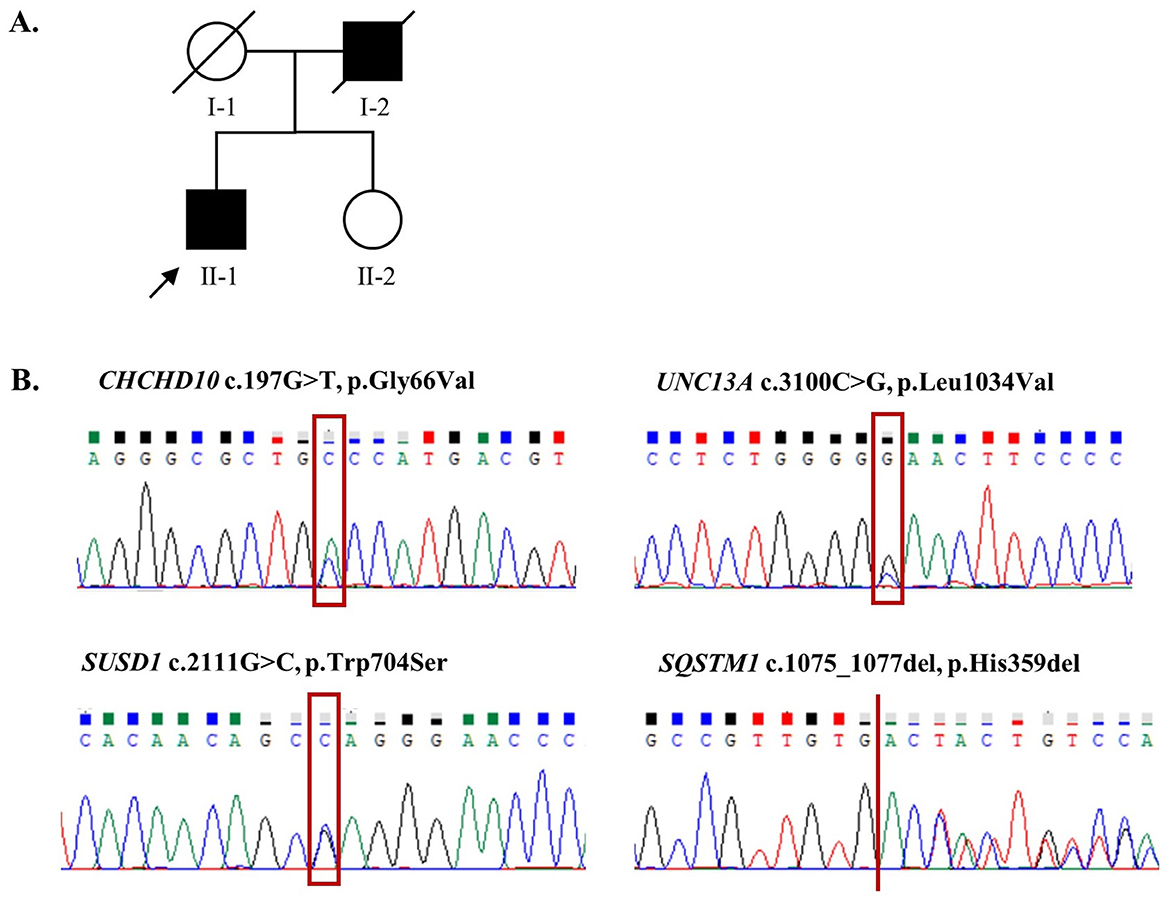
Figure 2. Pedigree of family 43 and the four ALS-related gene mutations identified in the proband. (A) Pedigree chart of Family 43. The proband of this family is marked with an arrow; filled and empty symbols indicate individuals with ALS and without ALS, respectively; circles, females; squares, males; diagonal line, deceased individual; the survival status of II-1 and II-2 is unknown due to loss of follow-up. (B) Sanger sequencing for the variants in CHCHD10, UNC13A, SUSUD1, and SQSTM1.
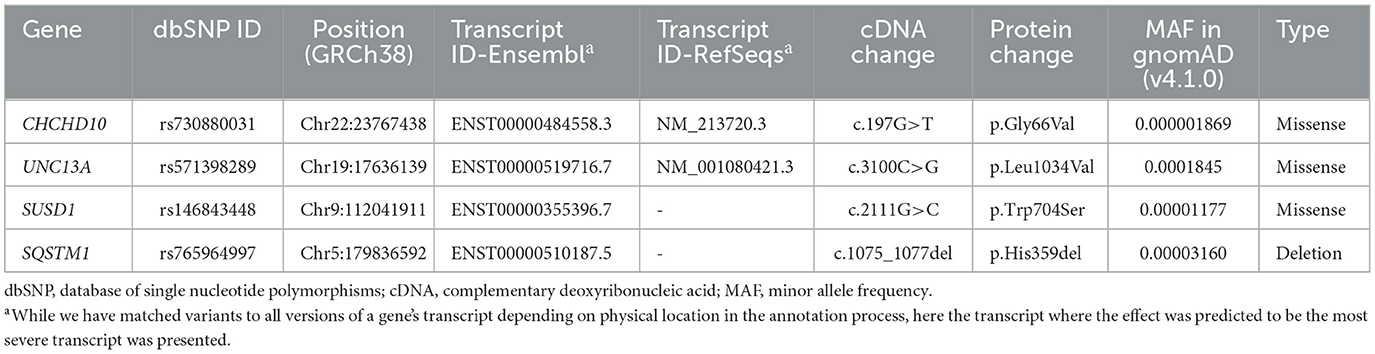
Table 2. Variants detected in the proband of family 43.
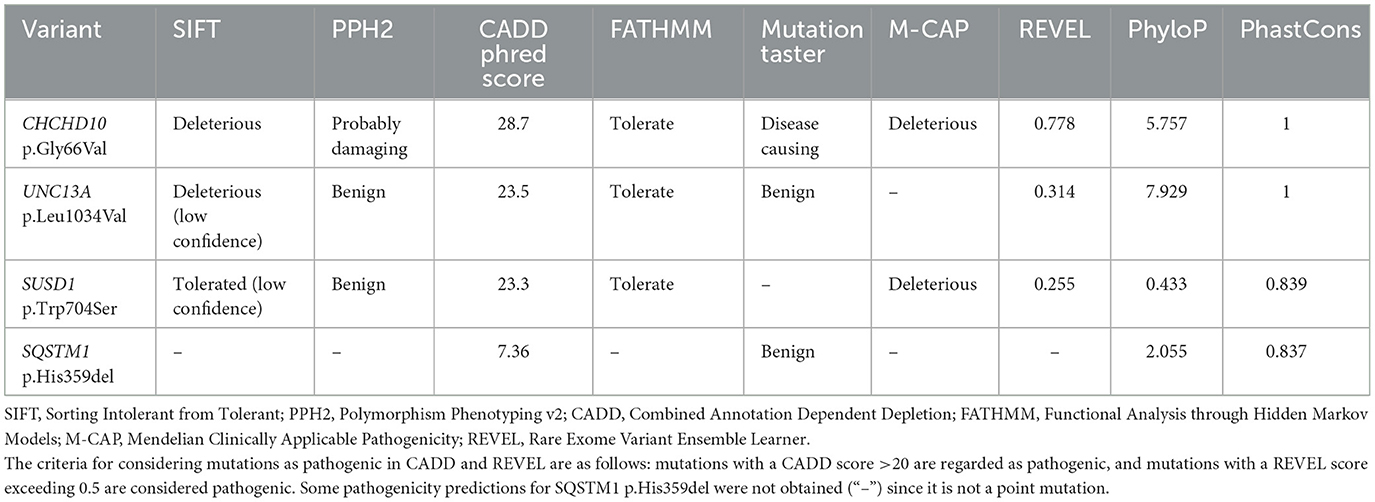
Table 3. Pathogenicity prediction of variants detected in the proband of family 43.
In gnomAD v4.1.0 and the ExAC database, the CHCHD10 p.Gly66Val variant is absent in the East Asian population, with a minor allele frequency (MAF) of 0.000001869 in total population. This variant led to the replace of glycine with valine at position 66 in the second exon of the CHCHD10 gene. According to the Uniprot database, this amino acid site is highly conserved across various species. Predictions from SIFT, CADD, PPH2 Mutation, Taster, and M-cap all indicate that this variant is damaging or probably damaging. Additionally, scores from PhyloP and PhastCons suggest a high level of conservation. When considered collectively, these findings suggest that CHCHD10 Gly66Val has potential pathogenicity in ALS.
The other variants in UNC13A, SUSD1, and SQSTM1 also deserve attention. SUSD1 p.Trp704Ser and SQSTM1 p.His359del were rare in the East Asian population and exhibit extremely low MAFs in the gnomAD database (v4.1.0). UNC13A p.Leu1034Val was also rare (MAF = 0.0001845 in gnomAD v4.1.0), but the allele frequency is higher in the East Asian population (Allele Frequency = 0.005925). Considering the transcript that was predicted to have the most severe effect, these variants may impact the structure of the encoded proteins, potentially affecting their physiological functions. Combined with the predictions from in silico tools (Table 3), these factors together further support that all three variants passed our filtering criteria. Among them, SQSTM1 p.His359del is recorded in the ClinVar database, classified as a variant of uncertain significance for frontotemporal dementia and/or amyotrophic lateral sclerosis 1 (FTD-ALS1) and Paget disease of bone 2, early-onset (PDB2). However, it has not been reported in the literature in individuals with SQSTM1-related conditions. The remaining two variants are novel and have not been documented in ClinVar. According to annotations from the InterPro database, both variants affected amino acids located within or near important structural domains. Specifically, the amino acid affected by UNC13A p.Leu1034Val resides in the MUN domain, which is implicated in regulating membrane trafficking, suggesting its potential role in membrane-related processes. The amino acid affected by SUSD1 p.Trp704Ser is located near the fibronectin type 3 domain of the receptor-type tyrosine-protein phosphatase U (Fn3_RPTPU) domain of the SUSD1 protein, which is close to the transmembrane region, suggesting its potential involvement in interactions with the cell membrane or extracellular components. Based on the ACMG guidelines, we currently classify these variants as variants of uncertain significance due to insufficient evidence. However, given the complex genetic landscape and pathogenic mechanisms in ALS patients, we cannot rule out their potential involvement in ALS.
The patient initially presented with bilateral lower limb weakness at the age of 53, with the right lower limb being more severely affected. He subsequently encountered difficulties when ascending and descending stairs. During the next 5 years, there was minimal progression in the patient's condition. However, at the age of 58, the weakness began to spread, and numbness developed in his left foot. Physical examination revealed both upper motor neuron and lower motor neuron signs at the lumbar level. Neurophysiological testing indicated evidence of chronic denervation in the cervical, thoracic, and lumbar segments. There were no observed bulbar symptoms or cognitive impairment. The patient received L-carnitine, vitamin E, and coenzyme Q as part of his management plan. After 71 months from disease onset, the ALSFRS-R score was 39. During his last follow-up in April 2015, the patient was still alive, with an ALSFRS score of 37. He was lost to follow-up afterwards, with a total disease duration exceeding 144 months.
The proband's father is also diagnosed with ALS. According to the proband's memory, the father exhibited symptoms around the age of 50, with an onset in the lower limbs, and the progression of the disease was relatively slow. However, specific details about the illness are not available. The proband's sister does not show any ALS-related symptoms but has a history of tumors. Unfortunately, we were unable to obtain blood samples from these and other family members, preventing further pedigree segregation analysis.
In this study, we conducted whole genome sequencing in 34 FALS probands to identify their potential pathogenic variants and explore the genotype-phenotype associations within an oligogenic context. In one proband, we identified a p.Gly66Val variant in CHCHD10, along with three VUS in UNC13A, SUSD1 and SQSTM1. We believe that the cumulative effect of these four variants together contributes to the clinical manifestation of ALS in this patient. This finding provides evidence for potential oligogenic pathogenicity and advances our understanding of the complex clinical manifestations in CHCHD10 variant carriers.
Variants in CHCHD10 gene have been linked to a wide spectrum of disorders including ALS, FTD, SMAJ (5), CMT2 (6) as well as myopathy (8), with a higher prevalence observed in ALS and FTD patients. In China, screenings for the CHCHD10 gene in ALS patients have revealed a lower variant frequency compared with European populations, with variants detected only in sporadic cases (3, 26, 27).
The CHCHD10 Gly66Val variant is commonly found in relatively mild motor neuron diseases, including late-onset spinal motor neuronopathy (spinal muscular atrophy Jokela type, SMAJ) (5, 28) and type 2 Charcot-Marie-Tooth disease (CMT2) (6), both prevalent in the Finnish population. However, this variant has not yet been reported in Chinese populations. Compared to ALS, these two diseases generally exhibit milder progression and more favorable prognosis. Although infrequently reported, a single case of this variant in ALS has been identified. In 2014, Müller et al. (29) described a familial ALS patient carrying the CHCHD10 Gly66Val variant. Compared to classic ALS, this patient exhibited a slow-progressing, spinal-onset form of ALS with extended survival, consistent with the phenotype observed in our proband. Slow progression and longer survival are frequently reported among ALS patients with CHCHD10 variants (29). Actually, research has indicated that the clinical phenotype associated with CHCHD10 p.Gly66Val exhibits significant heterogeneity, with individuals carrying the same variant within the same family receiving different diagnoses (30). CHCHD10-related diseases show clinical overlap, and therefore comprehensive diagnostic assessments may be required for a definitive diagnosis. The complex clinical phenotypes of these patients suggest that they may not be exclusively attributable to a single genetic variant; instead, the possibility of a convergence of multiple genetic factors is implicated.
Several studies have explored the role of the p.Gly66Val variant in motor neuron disease. Using bioinformatics, homology modeling, and multiple molecular dynamics simulations, Alici et al. reported that the α-helix and β-sheet formations of wild-type CHCHD10 are disrupted by the Gly66Val variant, affecting its overall structure (31). The Gly66Val variant leads to weaker intramolecular interactions and greater protein flexibility compared to the wild-type protein, potentially facilitating degradation by cellular machinery (31). Brockmann et al. (32) also demonstrated that CHCHD10 p.Gly66Val exhibits an increased degradation rate and contributes to motor neuron disease via haploinsufficiency in patient-derived cell lines and an in vivo zebrafish model. In addition, CHCHD10-related mutations have also demonstrated an impact on TDP-43 pathology and mitochondrial dysfunction (33, 34). Genin et al. (34) observed that fibroblasts harboring the CHCHD10 Ser59Leu or Gly66Val variants show comparable mitochondrial accumulation of phosphorylated TDP-43. However, mitochondrial fragmentation and cell death are significantly less pronounced in Gly66Val cells compared to Ser59Leu cells (34). This difference may explain the clinical differences between these variants: p.Ser59Leu is associated with a severe form of motor neuron disease, characterized by ataxia, dementia, and an ALS-like presentation (4), whereas p.Gly66Val is more commonly linked to a benign form of lower motor neuron disease, as mentioned above.
Therefore, based on the currently available evidence and ACMG guidelines, we tend to classify CHCHD10 p.Gly66Val as a pathogenic variant in ALS for the following reasons: (1) Well-established in vitro functional studies have demonstrated a damaging effect of this variant on the gene product, as discussed above (PS3). (2) The variant has been observed in unrelated patients with similar phenotypes, including the patient described by Müller et al. and the proband in our study, and it is extremely rare in public databases (MAF = 0.000001869 in gnomAD v4.1.0) (PS4). (3) Computational evidence supports its deleterious effect on the gene or gene product, as detailed in Table 3 (PP3).
Studies have shown the potential impact of VUS on ALS patients, indicating that focusing solely on pathogenic mutations may not fully capture ALS's complex genetic architecture (35). Approximately 21% of ALS patients are reported to carry VUS (36). The effects of the three other rare variants detected in this proband remain uncertain compared to CHCHD10 p.Gly66Val. However, it's worth noting that variants that are found to lack penetrance or sufficiency for the development of the disease may still act as genetic risk factors, so their potential role in ALS cannot be dismissed.
UNC13A is mainly expressed in neural tissues and plays a role in synaptic transmission by initiating and anchoring synaptic vesicles (37). It is a prominent candidate gene for ALS, with UNC13A SNPs being significant genetic markers associated with both frontotemporal dementia (FTD) and ALS in genome-wide association studies (38). These SNPs are primarily located in intronic regions. The mechanisms linking these variants to ALS may involve the loss of TDP-43 function, which leads to the inclusion of a cryptic exon in UNC13A, resulting in nonsense-mediated decay and loss of UNC13A protein (39, 40). In contrast, the p.Leu1034Val variant we detected is a missense variant located in exon 26 of UNC13A. While missense variants in UNC13A are not frequently reported in ALS, the p.Pro814Leu variant identified in a patient with dyskinetic movement disorder and autism has been associated with elevated initial synaptic vesicle release likelihood and abnormal short-term synaptic plasticity (41). As synaptic abnormalities are also observed in ALS (42), the pathogenic mechanism of p.Leu1034Val may also be related to this process. This variant affects an amino acid located in the MUN domain of the UNC13A protein, which contains several evolutionarily conserved domains involved in synaptic transmission, as well as synaptic vesicle docking and priming (43).
The SUSD1 gene encodes the sushi domain-containing protein 1 precursor, known for its involvement in protein-protein interactions. This protein is believed to raise the risk of venous thromboembolism by interacting with factors in the coagulation pathway via its sushi domain (44). In 2007, a whole-genome gene association study on sporadic ALS patients initially established an association between SUSD1 and ALS (45). However, the specific impact of single-site variants on disease risk remains uncertain. Further studies revealed an upregulated circular RNA within this gene in ALS patients' white blood cells, potentially serving as a biomarker (46). While the variant we detected provides direct evidence of SUSD1's involvement in ALS, its functional effect requires further investigation.
The SQSTM1 gene encodes p62, a protein crucial for protein degradation via both proteasome and autophagy pathways. SQSTM1-positive inclusions often co-localize with ubiquitin and TARDBP, and are commonly found in ALS and FTD patients (47). Since 2011, several SQSTM1 variants have been identified in ALS patients (48, 49), with lower frequencies reported in Chinese populations (50). Some SQSTM1 variants are also linked to Paget's disease of bone (PDB), indicating potential interactions with other genetic or environmental factors influencing ALS risk or disease phenotype (48). The SQSTM1 variant we identified results in the deletion of histidine at position 359 in the protein, with a MAF of 0.00003160 in the gnomAD database. In addition to the existing records in ClinVar, our report provides new evidence supporting the association of this variant with ALS. Further functional studies are needed to evaluate its impact on protein structure or function.
The phenotypic complexity seen in carriers of CHCHD10 variants were similar to that of C9orf72. Initially identified in ALS-FTD, C9orf72 has since been associated with a wide range of neurodegenerative and psychiatric conditions, including Alzheimer's, Parkinson's, Huntington-like phenotypes, and schizophrenia (11). One hypothesis explaining the variability in carriers of C9orf72 variants is that the existence additional genetic variants may influence the phenotype, supported by evidence for an oligogenic model (11). A similar explanation could apply to CHCHD10 variants, where the combined contribution of multiple genetic variants could lead to the diverse clinical presentations.
ALS is considered to arise through a multistep process involving both genetic and environmental factors (51). Given the oligogenic pathogenesis of ALS and the characteristics of the CHCHD10 p.Gly66Val discussed previously, we suggest that this variant alone may not be adequate to trigger ALS. Instead, it could be the interaction between different genetic variants that determined the final ALS phenotype, rather than showing the relatively mild phenotypes associated with CHCHD10 p.Gly66Val. The cumulative genetic burden from ALS-related genes might increase the patient's risk of developing a more severe form of the disease, as other studies on oligogenic ALS have shown (14, 52). This could help explain the variation in clinical phenotypes among patients with the same mutation. However, further research is needed to clarify how these variants contribute to ALS.
For the proband reported in this study, it is possible that the onset of ALS is due to the combined effect of CHCHD10 p.Gly66Val and the VUS in three other genes. In contrast, the Finnish ALS patient carrying CHCHD10 p.Gly66Val, as reported by Müller et al. (29), did not have any additional ALS-related gene variants detected through whole exome sequencing at the time. However, with new ALS-related genes being discovered in recent studies, it is challenging to completely rule out the possibility of other contributing variants. Larger-scale genetic screenings in broader populations may help validate this hypothesis.
The four variants carried by this proband have not been frequently reported to co-occur with other ALS-related gene variants. This may be partly explained by the lack of comprehensive genetic screening or stricter variant filtering criteria. One study reported the co-occurrence of CHCHD10 and C9orf72 variants in an ALS-FTD patient (53), and another reported a person carrying both TBK1 and SQSTM1 variants, who was diagnosed with non-fluent variant primary progressive aphasia (nfv-PPA) (54). However, the specific phenotypic characteristics of these patients remain unclear. Consequently, the simultaneous presence of these four rare gene variants in a single patient makes our case particularly remarkable.
While previous research often associates an increased burden of rare variants with reduced survival (10, 36), our proband's clinical characteristics deviated from this pattern, showing slower disease progression and extended survival compared to the cohort's average. The inconsistency may be attributed to the fact that the clinical phenotype of patients with multiple gene variants represent statistical patterns, which differs by individual cases depending on the combined effect of the exact variants. Interestingly, this patient's clinical phenotype aligned with typical features of ALS patients carrying CHCHD10 variants. We propose that the cumulative genetic burden of the four variants contributed to the development and specific phenotype of ALS, with CHCHD10 p.Gly66Val playing the main role, and the other three VUS potentially acting as modifying factors. Additionally, the presence of multiple variants may have an impact on the age of disease onset, as some CHCHD10 variant carriers in China showed a later average onset (27). However, we cannot yet draw definitive conclusions regarding the exact effects of these variants in this patient currently.
Our study has several limitations. First, due to the high mortality rate of ALS, we were unable to obtain blood samples from additional family members, which prevented us from performing segregation analysis. Second, the small sample size and rarity of these gene variants meant that variants in these four genes were identified in only one proband. As a result, we cannot provide compelling genetic or statistical evidence for the proposed oligogenic pathogenic mechanism related to CHCHD10 Gly66Val. Currently, no definitive conclusions can be drawn regarding the pathogenicity of the three variants in UNC13A, SQSTM1, and SUSD1. We plan to conduct functional studies using cell or animal models to further clarify the roles of these variants and their interactions. At this stage, this study primarily reports the screening results for the CHCHD10 gene in the Chinese FALS population. While we have not extensively explored the pathogenic mechanisms of each variant, our hypothesis that CHCHD10 Gly66Val is involved in ALS through interactions with other variants is reasonable, given previous reports on oligogenic mechanisms in ALS. This also provides insights into the complex phenotypes associated with CHCHD10 variants. Future screenings in larger cohorts will help provide more robust evidence for this hypothesis.
In summary, our study identified the CHCHD10 p.Gly66Val variant in Chinese FALS patients for the first time, and revealed its coexistence with three VUS in other ALS-related genes (UNC13A p.Leu1034Val, SUSD1 p.Trp704Ser, and SQSTM1 p.His359del) through whole-genome sequencing. We suppose that the interplay between these variants led to typical ALS symptoms in this patient, rather than other milder neuromuscular disorders caused by CHCHD10 p.Gly66Val. Our research further supports the oligogenic pathogenesis of ALS. Future investigations focused on unraveling the interactions between these genetic variants can deepen our understanding of the intricate mechanisms underlying ALS.
The original contributions presented in the study are included in the article and the Supplementary material; further inquiries can be directed to the corresponding author.
The studies involving humans were approved by Peking University Third Hospital Institutional Ethics Committee (No. IRB00006761-L2010055). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
YW: Data curation, Formal analysis, Investigation, Validation, Writing – original draft, Writing – review & editing. YM: Data curation, Formal analysis, Investigation, Validation, Writing – review & editing. HW: Data curation, Formal analysis, Investigation, Validation, Writing – review & editing. JJ: Data curation, Writing – review & editing. LM: Data curation, Writing – review & editing. YH: Data curation, Writing – review & editing. XL: Data curation, Writing – review & editing. MD: Data curation, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing, Conceptualization, Formal analysis, Investigation, Methodology, Visualization.
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by the National Natural Science Foundation of China (No. 82273915) and the Beijing Nova Programme Interdisciplinary Cooperation Project (20240484624).
We appreciate all cohort individuals and their families for their participation and cooperation in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2025.1438207/full#supplementary-material
1. Goutman SA, Hardiman O, Al-Chalabi A, Chió A, Savelieff MG, Kiernan MC, et al. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. (2022) 21:465–79. doi: 10.1016/S1474-4422(21)00414-2
2. Chen YP, Yu SH, Wei QQ, Cao B, Gu XJ, Chen XP, et al. Role of genetics in amyotrophic lateral sclerosis: a large cohort study in Chinese mainland population. J Med Genet. (2022) 59:840–9. doi: 10.1136/jmedgenet-2021-107965
3. Li XL, Shu S, Li XG, Liu Q, Liu F, Cui B, et al. CHCHD10 is not a frequent causative gene in Chinese ALS patients. Amyotroph Lateral Scler Frontotemporal Degener. (2016) 17:458–60. doi: 10.3109/21678421.2016.1170151
4. Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, Fragaki K, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain. (2014) 137:2329–45. doi: 10.1093/brain/awu138
5. Penttilä S, Jokela M, Bouquin H, Saukkonen AM, Toivanen J, Udd B. Late onset spinal motor neuronopathy is caused by mutation in CHCHD10. Ann Neurol. (2015) 77:163–72. doi: 10.1002/ana.24319
6. Auranen M, Ylikallio E, Shcherbii M, Paetau A, Kiuru-Enari S, Toppila JP, et al. CHCHD10 variant p(Gly66Val) causes axonal Charcot-Marie-Tooth disease. Neurol Genet. (2015) 1:e1. doi: 10.1212/NXG.0000000000000003
7. Xiao T, Jiao B, Zhang W, Pan C, Wei J, Liu X, et al. Identification of CHCHD10 mutation in Chinese patients with Alzheimer disease. Mol Neurobiol. (2017) 54:5243–7. doi: 10.1007/s12035-016-0056-3
8. Ajroud-Driss S, Fecto F, Ajroud K, Lalani I, Calvo SE, Mootha VK, et al. Mutation in the novel nuclear-encoded mitochondrial protein CHCHD10 in a family with autosomal dominant mitochondrial myopathy. Neurogenetics. (2015) 16:1–9. doi: 10.1007/s10048-014-0421-1
9. Naruse H, Ishiura H, Mitsui J, Takahashi Y, Matsukawa T, Tanaka M, et al. Burden of rare variants in causative genes for amyotrophic lateral sclerosis (ALS) accelerates age at onset of ALS. J Neurol Neurosurg Psychiatry. (2019) 90:537–42. doi: 10.1136/jnnp-2018-318568
10. Pang SY, Hsu JS, Teo KC, Li Y, Kung MHW, Cheah KSE, et al. Burden of rare variants in ALS genes influences survival in familial and sporadic ALS. Neurobiol Aging. (2017). 58:238.e9–e15. doi: 10.1016/j.neurobiolaging.2017.06.007
11. Dekker AM, Seelen M, van Doormaal PT, van Rheenen W, Bothof RJ, van Riessen T, et al. Large-scale screening in sporadic amyotrophic lateral sclerosis identifies genetic modifiers in C9orf72 repeat carriers. Neurobiol Aging. (2016). 39:220.e9–15. doi: 10.1016/j.neurobiolaging.2015.12.012
12. van Blitterswijk M, van Es MA, Hennekam EA, Dooijes D, van Rheenen W, Medic J, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet. (2012) 21:3776–84. doi: 10.1093/hmg/dds199
13. Scarlino S, Domi T, Pozzi L, Romano A, Pipitone GB, Falzone YM, et al. Burden of rare variants in ALS and axonal hereditary neuropathy genes influence survival in ALS: insights from a next generation sequencing study of an Italian ALS cohort. Int J Mol Sci. (2020) 21:3346. doi: 10.3390/ijms21093346
14. Cady J, Allred P, Bali T, Pestronk A, Goate A, Miller TM, et al. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann Neurol. (2015) 77:100–13. doi: 10.1002/ana.24306
15. Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) 1:293–9. doi: 10.1080/146608200300079536
16. Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. (2011) 72:257–68. doi: 10.1016/j.neuron.2011.09.010
17. Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. (2012) 40:W452–7. doi: 10.1093/nar/gks539
18. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
19. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. (2019) 47:D886–d94. doi: 10.1093/nar/gky1016
20. Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. (2013) 34:57–65. doi: 10.1002/humu.22225
21. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. (2014) 11:361–2. doi: 10.1038/nmeth.2890
22. Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. (2016) 48:1581–6. doi: 10.1038/ng.3703
23. Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. (2016) 99:877–85. doi: 10.1016/j.ajhg.2016.08.016
24. Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. (2005) 15:1034–50. doi: 10.1101/gr.3715005
25. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. (2010) 20:110–21. doi: 10.1101/gr.097857.109
26. Shen S, He J, Tang L, Zhang N, Fan D. CHCHD10 mutations in patients with amyotrophic lateral sclerosis in Mainland China. Neurobiol Aging. (2017) 54:214.e7–.e10. doi: 10.1016/j.neurobiolaging.2017.02.011
27. Zhou Q, Chen Y, Wei Q, Cao B, Wu Y, Zhao B, et al. Mutation screening of the CHCHD10 gene in Chinese patients with amyotrophic lateral sclerosis. Mol Neurobiol. (2017) 54:3189–94. doi: 10.1007/s12035-016-9888-0
28. Shammas MK, Huang TH, Narendra DP. CHCHD2 and CHCHD10-related neurodegeneration: molecular pathogenesis and the path to precision therapy. Biochem Soc Trans. (2023) 51:797–809. doi: 10.1042/BST20221365
29. Müller K, Andersen PM, Hübers A, Marroquin N, Volk AE, Danzer KM, et al. Two novel mutations in conserved codons indicate that CHCHD10 is a gene associated with motor neuron disease. Brain J Neurol. (2014) 137:e309. doi: 10.1093/brain/awu227
30. Pasanen P, Myllykangas L, Pöyhönen M, Kiuru-Enari S, Tienari PJ, Laaksovirta H, et al. Intrafamilial clinical variability in individuals carrying the CHCHD10 mutation Gly66Val. Acta Neurol Scand. (2016) 133:361–6. doi: 10.1111/ane.12470
31. Alici H, Uversky VN, Kang DE, Woo JA, Coskuner-Weber O. Effects of the Jokela type of spinal muscular atrophy-related G66V mutation on the structural ensemble characteristics of CHCHD10. Proteins. (2023) 91:739–49. doi: 10.1002/prot.26463
32. Brockmann SJ, Freischmidt A, Oeckl P, Müller K, Ponna SK, Helferich AM, et al. CHCHD10 mutations pR15L and pG66V cause motoneuron disease by haploinsufficiency. Hum Mol Genet. (2018) 27:706–15. doi: 10.1093/hmg/ddx436
33. Woo JA, Liu T, Trotter C, Fang CC, De Narvaez E, LePochat P, et al. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat Commun. (2017) 8:15558. doi: 10.1038/ncomms15558
34. Genin EC, Bannwarth S, Lespinasse F, Ortega-Vila B, Fragaki K, Itoh K, et al. Loss of MICOS complex integrity and mitochondrial damage, but not TDP-43 mitochondrial localisation, are likely associated with severity of CHCHD10-related diseases. Neurobiol Dis. (2018) 119:159–71. doi: 10.1016/j.nbd.2018.07.027
35. Dong S, Yin X, Wang K, Yang W, Li J, Wang Y, et al. Presence of rare variants is associated with poorer survival in Chinese patients with amyotrophic lateral sclerosis. Phenomics. (2023) 3:167–81. doi: 10.1007/s43657-022-00093-8
36. Shepheard SR, Parker MD, Cooper-Knock J, Verber NS, Tuddenham L, Heath P, et al. Value of systematic genetic screening of patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. (2021) 92:510–8. doi: 10.1136/jnnp-2020-325014
37. Willemse SW, Harley P, van Eijk RPA, Demaegd KC, Zelina P, Pasterkamp RJ, et al. UNC13A in amyotrophic lateral sclerosis: from genetic association to therapeutic target. J Neurol Neurosurg Psychiatry. (2023) 94:649–56. doi: 10.1136/jnnp-2022-330504
38. van Es MA, Veldink JH, Saris CG, Blauw HM, van Vught PW, Birve A, et al. Genome-wide association study identifies 19p133 (UNC13A) and 9p212 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. (2009) 41:1083–7. doi: 10.1038/ng.442
39. Brown AL, Wilkins OG, Keuss MJ, Hill SE, Zanovello M, Lee WC, et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature. (2022) 603:131–7. doi: 10.1038/s41586-022-04436-3
40. Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, et al. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature. (2022) 603:124–30. doi: 10.1038/s41586-022-04424-7
41. Lipstein N, Verhoeven-Duif NM, Michelassi FE, Calloway N, van Hasselt PM, Pienkowska K, et al. Synaptic UNC13A protein variant causes increased neurotransmission and dyskinetic movement disorder. J Clin Invest. (2017) 127:1005–18. doi: 10.1172/JCI90259
42. Meszlényi V, Patai R, Polgár TF, Nógrádi B, Körmöczy L, Kristóf R, et al. Passive transfer of sera from ALS patients with identified mutations evokes an increased synaptic vesicle number and elevation of calcium levels in motor axon terminals, similar to sera from sporadic patients. Int J Mol Sci. (2020) 21:5566. doi: 10.3390/ijms21155566
43. Jusyte M, Blaum N, Böhme MA, Berns MMM, Bonard AE, Vámosi Á B, et al. Unc13A dynamically stabilizes vesicle priming at synaptic release sites for short-term facilitation and homeostatic potentiation. Cell Rep. (2023) 42:112541. doi: 10.1016/j.celrep.2023.112541
44. Tang W, Teichert M, Chasman DI, Heit JA, Morange PE, Li G, et al. A genome-wide association study for venous thromboembolism: the extended cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Genet Epidemiol. (2013) 37:512–21. doi: 10.1002/gepi.21731
45. Schymick JC, Scholz SW, Fung HC, Britton A, Arepalli S, Gibbs JR, et al. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. (2007) 6:322–8. doi: 10.1016/S1474-4422(07)70037-6
46. Dolinar A, Koritnik B, Glavač D, Ravnik-Glavač M. Circular RNAs as potential blood biomarkers in amyotrophic lateral sclerosis. Mol Neurobiol. (2019) 56:8052–62. doi: 10.1007/s12035-019-1627-x
47. Chua JP, De Calbiac H, Kabashi E, Barmada SJ. Autophagy and ALS: mechanistic insights and therapeutic implications. Autophagy. (2022) 18:254–82. doi: 10.1080/15548627.2021.1926656
48. Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. (2011) 68:1440–6. doi: 10.1001/archneurol.2011.250
49. Rubino E, Rainero I, Chiò A, Rogaeva E, Galimberti D, Fenoglio P, et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. (2012) 79:1556–62. doi: 10.1212/WNL.0b013e31826e25df
50. Yang Y, Tang L, Zhang N, Pan L, Hadano S, Fan D. Six SQSTM1 mutations in a Chinese amyotrophic lateral sclerosis cohort. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 16:378–84. doi: 10.3109/21678421.2015.1009466
51. Al-Chalabi A, Calvo A, Chio A, Colville S, Ellis CM, Hardiman O, et al. Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol. (2014) 13:1108–13. doi: 10.1016/S1474-4422(14)70219-4
52. Kuuluvainen L, Kaivola K, Mönkäre S, Laaksovirta H, Jokela M, Udd B, et al. Oligogenic basis of sporadic ALS: the example of SOD1 pAla90Val mutation. Neurol Genet. (2019) 5:e335. doi: 10.1212/NXG.0000000000000335
53. Dobson-Stone C, Shaw AD, Hallupp M, Bartley L, McCann H, Brooks WS, et al. Is CHCHD10 Pro34Ser pathogenic for frontotemporal dementia and amyotrophic lateral sclerosis? Brain J Neurol. (2015) 138:e385. doi: 10.1093/brain/awv115
54. Dols-Icardo O, García-Redondo A, Rojas-García R, Borrego-Hernández D, Illán-Gala I, Muñoz-Blanco JL, et al. Analysis of known amyotrophic lateral sclerosis and frontotemporal dementia genes reveals a substantial genetic burden in patients manifesting both diseases not carrying the C9orf72 expansion mutation. J Neurol Neurosurg Psychiatry. (2018) 89:162–8. doi: 10.1136/jnnp-2017-316820
Keywords: amyotrophic lateral sclerosis, CHCHD10, oligogenic pathogenesis, whole-genome sequencing, genotype-phenotype analysis
Citation: Wang Y, Mi Y, Wang H, Jiang J, Mao L, Heng Y, Li X and Deng M (2025) Combined impact of CHCHD10 p.Gly66Val and three other variants suggests oligogenic contributions to ALS. Front. Neurol. 16:1438207. doi: 10.3389/fneur.2025.1438207
Received: 25 May 2024; Accepted: 28 February 2025;
Published: 18 March 2025.
Edited by:
Huifang Shang, Sichuan University, ChinaReviewed by:
Chiara Reale, IRCCS Carlo Besta Neurological Institute Foundation, ItalyCopyright © 2025 Wang, Mi, Wang, Jiang, Mao, Heng, Li and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Deng, ZGVuZ21pbjA0QDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.