 Xuan Zhang
Xuan Zhang Bo Zhang
Bo Zhang Zhiming Tao
Zhiming Tao Jianmin Liang
Jianmin Liang- 1Department of Pediatric Neurology, Children's Medical Center, First Hospital of Jilin University, Changchun, China
- 2Jilin Provincial Key Laboratory of Pediatric Neurology, Changchun, China
- 3Neuromedical Center, First Hospital of Jilin University, Changchun, China
Mitochondria is the cell’s powerhouse. Mitochondrial disease refers to a group of clinically heterogeneous disorders caused by dysfunction in the mitochondrial respiratory chain, often due to mutations in mitochondrial DNA (mtDNA) or nuclear DNA (nDNA) that encodes mitochondrial proteins. This dysfunction can lead to a variety of clinical phenotypes, particularly affecting organs with high energy demands, such as the brain and muscles. Epilepsy is a prevalent neurological disorder in children and is also a frequent manifestation of mitochondrial disease. The exact mechanisms underlying epilepsy in mitochondrial disease remain unclear and are thought to involve multiple contributing factors. This review explores common mitochondrial diseases associated with epilepsy, focusing on their prevalence, seizure types, EEG features, therapeutic strategies, and outcomes. It also summarizes the relationship between the molecular genetics of mitochondrial respiratory chain components and the development of epilepsy.
Highlights
• The incidence of epilepsy in children with mitochondrial disease is higher than in adults.
• The pathogenesis of mitochondrial-associated epilepsy in children is complex, involving various genetic mutations.
• Understanding the molecular genetics of mitochondrial respiratory chain components can aid in diagnosing and treating epilepsy for diagnosis and treatment.
• While no cure currently exists for mitochondrial diseases, gene therapy is a promising potential treatment option.
1 Mitochondrial physiology and mitochondrial diseases
Mitochondria, essential organelles in eukaryotic cells, produce energy by generating adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS). This energy production occurs within the mitochondrial respiratory chain (MRC) situated in the inner mitochondrial membrane. The MRC consists of five enzyme complexes (complexes I to V) encoded by both mitochondrial DNA (mtDNA) and nuclear DNA (nDNA), along with two electron transport proteins, coenzyme Q10, and cytochrome C, which are both encoded by DNA. Therefore, mitochondrial diseases can result from both maternally inherited mtDNA mutations and classical genetic mutations in nDNA (1).
Dysfunction of the MRC in mitochondrial diseases is often caused by mutations in mtDNA or nDNA. mtDNA mutations include point mutations, deletions, duplications, and depletion of copy number. nDNA mutations impact several processes, such as the formation of MRC subunits, assembly factors, intergenomic signaling, mitochondrial protein synthesis, lipid metabolism, mitochondrial dynamics (including fusion and fission), and coenzyme Q10 homeostasis (2). ATP is essential for all cells, but MRC dysfunction disproportionately affects those with high energy demands, such as neurons, skeletal muscles, and retinal ganglion cells. Beyond energy production, mitochondria play key roles in intracellular calcium regulation, reactive oxygen species (ROS) production, apoptosis, and neurotransmitter synthesis.
2 Mechanism of epilepsy caused by mitochondrial dysfunction
Epilepsy, a prevalent neurological disorder, partly arises due to the central nervous system’s substantial energy demands. When mitochondrial function is compromised, energy deficits can precipitate seizures. Zsurka et al. suggest that the pathogenesis of mitochondrial-associated epilepsy involves processes such as neuronal energy depletion, oxidative stress, impaired calcium signaling, neurotransmitter imbalances, β-oxidation deficiency, and glial cell dysfunction (3). Additional factors, such as changes in cerebral blood flow, structural abnormalities in the brain, and immune-mediated damage, also contribute to the development of mitochondria-related epilepsy.
Mutations in specific mtDNA or nDNA genes encoding the mitochondrial respiratory chain are closely related to epilepsy. These gene mutations prevent the oxidative phosphorylation process in the mitochondria from proceeding normally, resulting in a decrease in intracellular ATP levels. This disruption increases neuronal excitability by damaging the activity of sodium and potassium ATP enzymes and decreasing membrane potential (4). After the occurrence of mitochondrial-associated epilepsy, the energy demand of neurons that have been metabolically damaged increases. This energy exhaustion will further lead to the production of reactive oxygen species (ROS), apoptosis, and abnormal calcium homeostasis, which will promote epilepsy, thus forming a vicious circle (5). ROS is mainly produced in mitochondria. High concentrations of ROS can oxidize mitochondrial proteins, lipids, and nucleic acids, cause cell dysfunction, damage the ability of cells to maintain energy levels, cause energy exhaustion, damage and death of neurons, and change the excitability of neurons, thus reducing the threshold of seizures (6, 7). The imbalance of calcium homeostasis caused by mitochondrial dysfunction can increase the excitability of neurons and induce seizures. Levetiracetam can play a partial antiepileptic effect by regulating the level of intracellular calcium (8). The relative imbalance of excitatory and inhibitory neurotransmitters may cause seizures. Disorders of receptor and ion channel gene mutation/protein function lead to abnormal stimulus transmission and epileptic discharges. Disorders of neurotransmitters such as glutamate, γ-aminobutyric acid (GABA), acetylcholine (Ach), dopamine, 5-hydroxytryptamine, norepinephrine, histamine, melatonin, and nitric oxide are involved in the pathogenesis of epilepsy (9). Epilepsy is often accompanied by neuronal overexcitation. Repeated seizures can cause oxidative stress, inflammation, and excitotoxic damage and eventually lead to neuronal death, including apoptosis, autophagy, necrotizing apoptosis, scorch death, and iron death (10, 11). The changes in ion channels, transporters, and metabolism of astrocytes are also closely related to the occurrence of epilepsy (9). Astrocytes are rich in mitochondrial glutamate carrier SLC25A22, which controls glutamate uptake. Its functional loss will lead to extracellular glutamate imbalance and activation of extrasynaptic glutamate receptors, increase neuronal excitability, and cause epilepsy (12, 13). Congenital metabolic defects, such as fatty acid β oxidation disorder, make it impossible for the liver to use fatty acids as a source of energy, resulting in a decrease in blood glucose levels (14). Hypoglycemia results in seizures through excitatory neurotoxicity mediated by the N-methyl-D-aspartate receptor (NMDAR), increasing the production of mitochondrial free radicals, initiating apoptosis, and changing brain energy production (15).
Energy deficiency can stimulate the proliferation of mitochondria of smooth muscle and small vascular endothelial cells and cause angiopathy (such as MELAS syndrome), resulting in damage to microvascular hemoperfusion and insufficient energy supply to the brain (16). ATP deficiency can cause astrocyte dysfunction and excitotoxicity, leading to nerve cell death. In addition, hemodynamics and metabolic stress enhance the mobilization of nitric oxide (NO) and decrease the level of circulating NO. Both mechanisms are involved in epilepsy (17). Mitochondrial disease-related epilepsy may also be related to immune dysfunction (18). Recently, it has been reported that MELAS syndrome is complicated by autoimmune injury, but the mechanism between it and autoimmune disease is not clear and needs further study (19). The RARS2 gene is the pathogenic gene of cerebellopontine dysplasia type 6 (PCH6). MRI is characterized by cerebellopontine dysplasia or progressive cerebellopontine and cerebellar cortex atrophy (20). MT-TL1 gene mutations in mtDNA can involve the cortex, mostly located in the occipital lobe and parietal lobe, and may be accompanied by cerebellar and cerebellar atrophy (21). Brain structural abnormalities caused by RARS2 and MT-TL1 gene mutations are common seizures. It can be seen that the cause of brain structural venereal disease is also one of the pathogenesis of mitochondrial-related epilepsy. The pathways of cell death associated with epilepsy are shown in Figure 1. The mechanisms of epilepsy caused by various mitochondrial diseases are different, and understanding its underlying pathological mechanism is very important to develop reasonable strategies for the treatment of mitochondrial-related epilepsy.
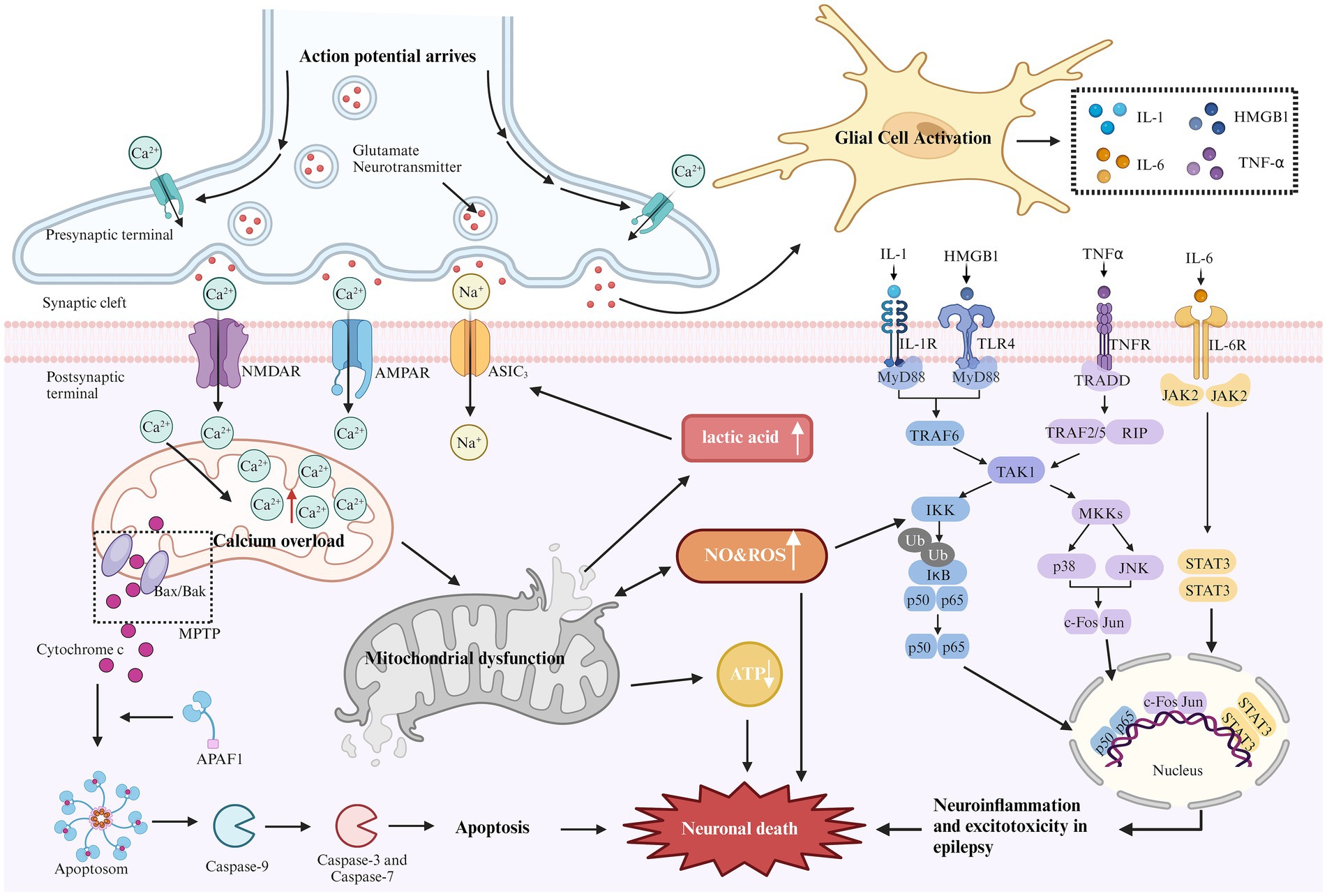
Figure 1. Mechanism of epilepsy-related cell death. During epilepsy, excitability increases due to the loss of inhibitory neurons. The elevated glutamate levels or mutations in metabolic receptors (e.g., AMPA, NMDA) can prolong the activation time of glutamatergic receptors, increase excitatory postsynaptic potential, lead to excitotoxicity, and trigger downstream apoptotic cascades. Excessive activation of NMDA and AMPA receptors can cause a sharp influx of sodium and calcium ions. Excessive calcium influx leads to calcium overload in the mitochondrial matrix, resulting in mitochondrial dysfunction. This dysfunction opens the mitochondrial permeability transition pore (MPTP), leading to the release of cytochrome C into the cytoplasm. Cytochrome C binds with apoptosis-related factor 1 (APAF1), promoting caspase-9 binding to form apoptosomes, thereby activating other caspases and initiating the cascade of apoptosis, ultimately inducing neuronal cell death. Continuous brain discharges increase energy demand and decrease oxygen levels, leading to increased anaerobic glycolysis. Lactic acid, as a by-product of anaerobic glycolysis, appears to activate acid-sensitive ion channels (ASIC3), causing further calcium and sodium ion influx, which aggravates excitotoxicity. In addition, mitochondrial dysfunction reduces ATP production and increases reactive oxygen species (ROS) generation. Long-term seizures often activate the innate immune response, leading to glial cell activation and promoting the release of cytokines (e.g., IL-1, HMGB1, TNF-α, and IL-6). These cytokines activate several inflammatory pathways, such as NF-kB, p38, JNK, and JAK–STAT, regulating the expression of pro-inflammatory mediators and transcription factors, resulting in neuronal apoptosis.
Although the mechanisms of epilepsy vary across different mitochondrial diseases, Figure 1 outlines common pathways of cell death associated with epilepsy. Understanding these underlying pathological mechanisms is crucial for developing effective strategies to treat mitochondrial-related epilepsy.
3 Mitochondrial diseases and epilepsy in children
Mitochondrial diseases in children can present at any age and often affect multiple systems. In these conditions, seizures tend to originate in the occipital lobe or posterior brain regions, with seizure types ranging from epileptic spasms (as seen in infantile epileptic spasm syndrome), focal seizures (with or without secondary generalization), generalized seizures, myoclonic seizures, and refractory status epilepticus (22). In pediatric patients, myoclonic seizures are the most frequently observed seizure type associated with mitochondrial diseases (23). The incidence of epilepsy in children with mitochondrial disease ranges from 25 to 100%, in contrast to approximately 14% in adults. Children with mitochondrial-related epilepsy exhibit a higher prevalence of focal seizures and nDNA mutations compared to adults. Their clinical presentation aligns more closely with electro-clinical syndromes and mitochondrial disease-related syndromes (24).
Mitochondrial diseases can be genetically categorized based on mutations in mtDNA or nDNA, which encode mitochondrial proteins. mtDNA mutations are implicated in various disorders, including mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS), maternally inherited Leigh syndrome (MILS), myoclonic epilepsy with ragged red fibers (MERRF), Leber’s hereditary optic neuropathy (LHON), Kearns-Sayre syndrome, Pearson syndrome, and chronic progressive external ophthalmoplegia (CPEO). In contrast, nuclear gene mutations encode mitochondrial proteins, resulting in a more complex pathogenic mechanism. Although the incidence of nDNA mutations in mitochondrial disease is lower, they are associated with conditions such as Leigh syndrome, Alpers-Huttenlocher syndrome (AHS), GRACILE syndrome, and Bjornstad syndrome. A wide variety of these mitochondrial disorders are associated with epilepsy, as outlined below.
4 MELAS and epilepsy
The pathogenesis of MELAS is associated with gene mutations, vascular disease, nitric oxide (NO) dysregulation, and energy deficiency. The most commonly implicated genetic mechanism is the mutation of the MT-TL1 gene, with the most frequent mutation being m.3243A > G (25). In MELAS, seizures occur with a frequency ranging from 71 to 96% (26), and typically present as generalized tonic–clonic seizures, focal seizures, or generalized status epilepticus (27).
Electroencephalograms (EEGs) in patients with MELAS typically show diffuse background slowing, often accompanied by slow waves mixed with sharp waves, especially in the bilateral anterior regions (28). The most common EEG findings are non-specific diffuse or focal slow-wave activity in the bilateral occipital and anterior head regions. Epileptic discharges are typically focal but may become widespread and are frequently associated with seizures (27). Regardless of the genotype or presence of status epilepticus, about 30% of patients exhibit transient, non-specific, periodic unilateral epileptic discharges during the acute phase, though these are rarely observed in the chronic phase (29). Additionally, some MELAS patients experience loss of fixation sensitivity (FOS), an EEG phenomenon in which epileptic discharges are triggered by loss of central vision or gaze, characterized by idiopathic or generalized epileptic discharges following eye closure for more than 3 s (30).
Magnetic resonance imaging (MRI) in MELAS patients typically reveals cortical lesions, primarily in the occipital and parietal lobes, presenting as low signal intensity on T1-weighted imaging (WI) and high signal intensity on T2WI. These lesions do not follow the typical vascular distribution of cerebral arteries and may be accompanied by cerebellar or cerebral atrophy (21). Newer imaging modalities, such as oxygen uptake fraction measurements, transcranial Doppler ultrasound, and magnetoencephalography, can provide additional insight into the affected brain tissue and aid in prognosis prediction (31). MRI findings from a pediatric MELAS patient are shown in Figure 2.
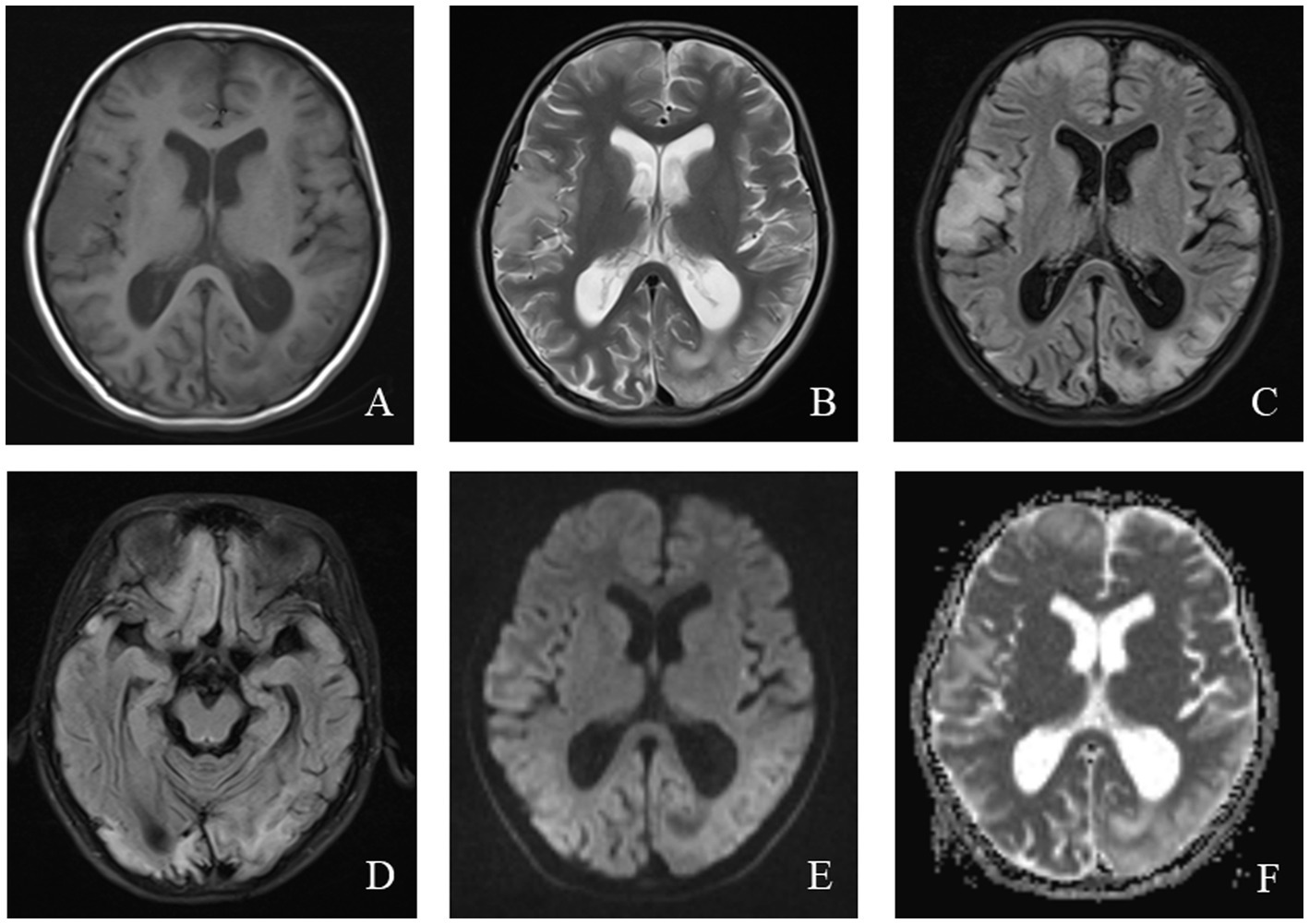
Figure 2. An 11-year-old female was diagnosed with MELAS syndrome due to intermittent convulsions for 3.5 years, which worsened over the last 7 days. Head MRI revealed: (A) Low signal intensity in the bilateral frontal, parietal lobes on axial T1WI. (B–D) On axial T2WI and T2/FLAIR, the brain tissue of the bilateral frontal parietal-occipital lobes was swollen, showing strip-like and gyrus-like high signal intensity. (E) DWI showed high signal intensity in the gyrus of the lesion area. (F) High signal intensity of the corresponding lesions was observed on the ADC.
5 LS and epilepsy
Leigh syndrome (LS), also known as subacute necrotizing encephalomyelopathy, is associated with approximately 100 different mtDNA or nDNA gene defects (32). LS can result from mitochondrial tRNA mutations in Cox I, II, III, IV, V, and PDH. Based on the age of onset, LS is classified into early-onset (before 2 years of age) and late-onset (after 2 years). A meta-analysis of 385 LS cases revealed that nDNA mutations were more common than mtDNA mutations (38% vs. 32%), and earlier onset of symptoms was more frequently associated with nDNA mutations. Additionally, 80% of patients had defects in respiratory chain enzyme complexes, with 35% displaying isolated complex I defects (33).
The typical clinical presentation of LS includes motor retardation, seizures, reduced exercise tolerance, and lethargy (34). Seizures are the second most common symptom, with 13.3% of cases initially manifesting as epilepsy (35). A report of 110 LS cases from Korea found that focal seizures with impaired consciousness were the most prevalent seizure type, followed by generalized myoclonic, tonic, atonic, and tonic–clonic seizures. While seizure types did not significantly differ between early- and late-onset LS, focal and generalized tonic–clonic seizures were most common in late-onset cases (34, 36).
EEG findings in LS are variable. Most patients exhibit a decreased and disorganized background rhythm, along with focal or multifocal sharp waves during the interictal period (37). In one case of late-onset LS, diffuse spike–wave complexes at 3 ~ 4 Hz were observed (38). Another case of LS caused by an MT-ND1 mutation demonstrated decreased background activity, multifocal epileptic discharges, and significant irregularity (39).
MRI findings in LS often help elucidate the underlying etiology, with classic abnormalities involving the basal ganglia, particularly the bilateral putamen, periaqueductal gray, and brainstem (40). In patients with SURF-1 mutations, symmetrical involvement of the bilateral brainstem and subthalamic nuclei is common, and some patients exhibit basal ganglia abnormalities. In contrast, patients without SURF mutations show symmetrical bilateral basal ganglia involvement without significant white matter or brainstem abnormalities (41). The MRI findings of a pediatric patient with LS are shown in Figure 3.
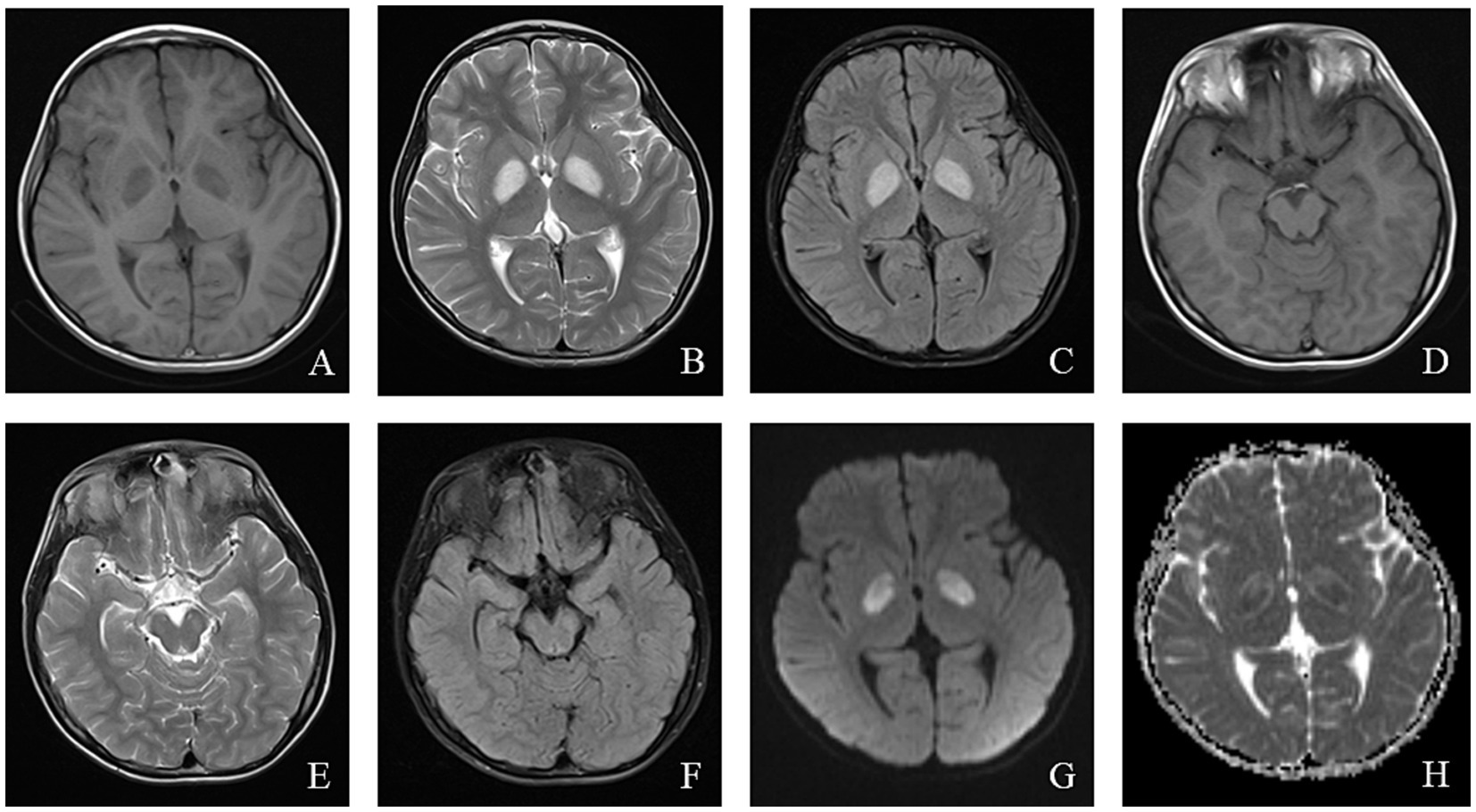
Figure 3. An 8-year-old male who experienced intermittent fever and limb weakness for over 6 years and drowsiness for 1 day was diagnosed with Leigh syndrome. Head MRI showed the following: (A–F) On axial T1WI, patchy low signal intensity was seen in the bilateral globus pallidus and midbrain, while patchy high signal intensity was observed in the bilateral globus pallidus and midbrain on axial T2WI and T2/FLAIR. (G) High signal intensity in the focal area was observed on DWI. (H) ADC showed high and low signal intensity in the corresponding lesions.
6 AHS and epilepsy
AHS is an autosomal recessive genetic disorder caused by biallelic mutations in the POLG gene, which encodes a DNA polymerase responsible for the replication and repair of mtDNA. These mutations result in mitochondrial dysfunction, including mtDNA mutations, deletions, or depletion. The most common mutation associated with AHS is A467T (42, 43).
Approximately 50% of children with AHS present with focal, multifocal, or myoclonic seizures at the onset of epilepsy. As the disease progresses, most patients develop recurrent seizures, including status epilepticus and persistent focal epilepsy (epilepsia partialis continua, EPC) (44, 45). EPC is a hallmark feature of AHS, and in some cases, multi-site EPC is observed early, aiding in early diagnosis.
In AHS, EEGs typically show reduced background activity along with high-amplitude slow waves in the occipital region. This is often accompanied by occasional spike–wave or intermittent to continuous spike–wave or multi-spike–wave activity, known as rhythmic high-amplitude delta with superimposed (poly)spikes (RHADS) (44). The presence of RHADS in patients with status epilepticus may indicate AHS (46). If RHADS is not detected on the initial EEG, extended monitoring may increase the likelihood of identification. RHADS generally appears in the early stages of AHS and is not affected by anti-seizure medications (ASMs). It is frequently associated with high-energy gamma (γ) oscillations, which may reflect active epileptic lesions (47).
Computed tomography (CT) in AHS typically reveals low-density areas in the gray and white matter of the posterior temporal and occipital lobes, which may progress to diffuse cerebral atrophy in later stages (48). MRI shows high signal intensity on T2WI or FLAIR sequences, particularly in the deep nuclei of the occipital lobes, thalamus, basal ganglia, and cerebellum (49). The EEG and MRI findings from a pediatric patient with AHS are illustrated in Figures 4, 5.
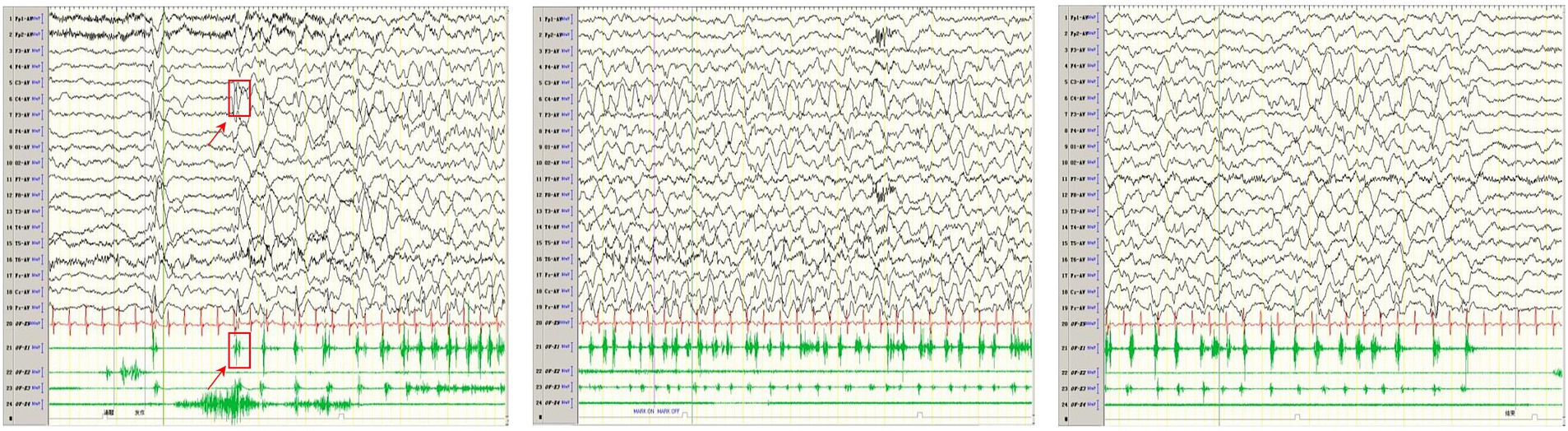
Figure 4. In one patient with Alpers syndrome, EEG showed a 2-3 Hz slow-wave complex (multiple) spike sign in the right central region, that is, the characteristic rhythmic high-amplitude δ (RHADS) of superposition (multiple) spike waves, and simultaneous left-side limb jitter (EPC). One time of jitter is shown in the arrow and box in the figure. The top arrow indicates the EEG performance, and the bottom arrow indicates the same myoelectric performance. Each subsequent jitter is the same as this one.
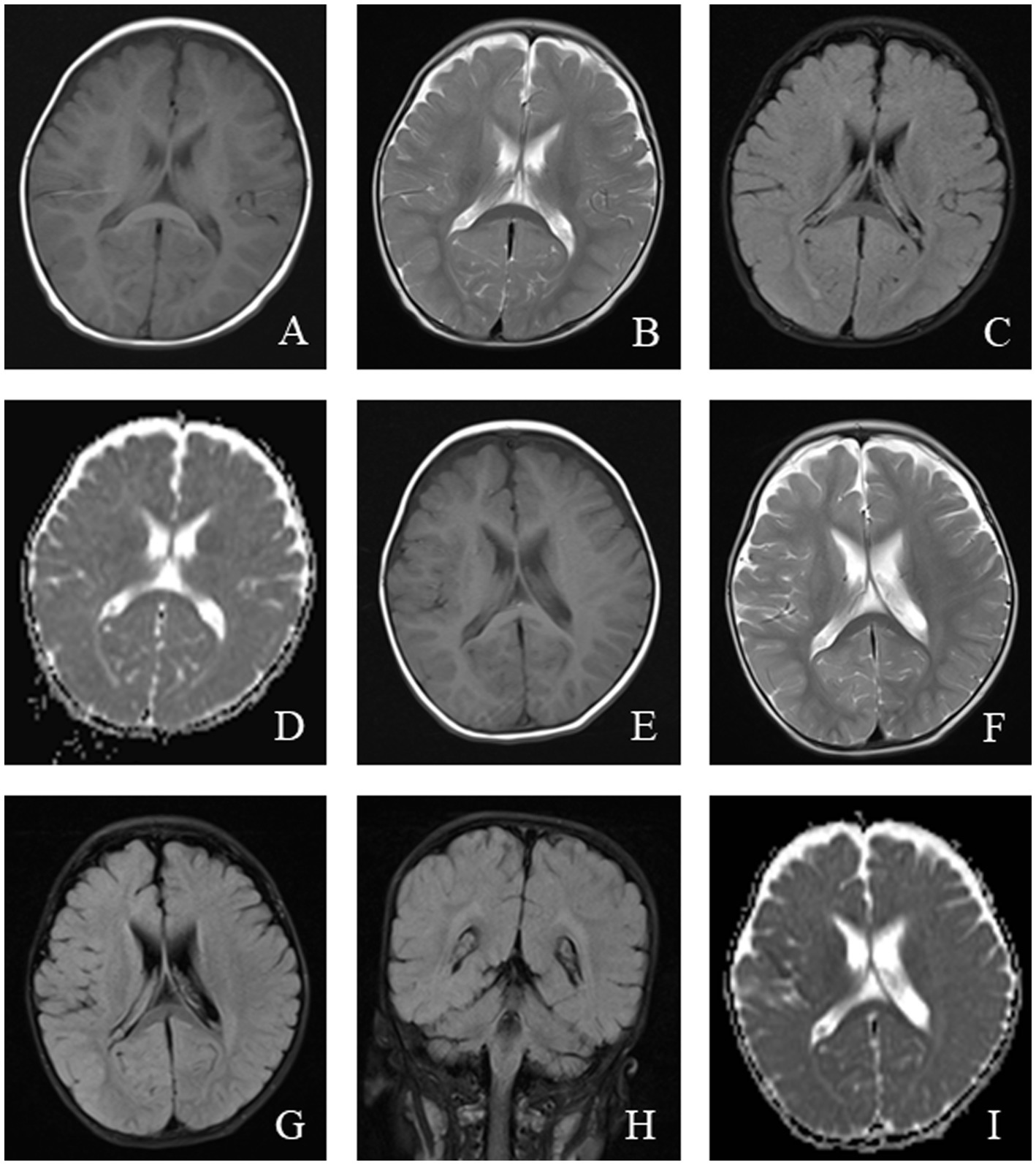
Figure 5. An 11-month-old male was diagnosed with Alpers syndrome due to persistent left limb convulsions for 4 h. Initial head MRI showed: (A–C) No abnormal signals in T1WI, T2WI, and dark-fluid images, small ventricles, and no displacement of midline structures in the brain tissue. (D) Bilateral symmetrical low signal intensity was observed on the ADC map of the caudate nucleus. Reexamination after 3 months: (E–H) Patchy abnormal signals were observed around the bilateral ventricles. T1WI showed iso-signal, while T2WI and fluid-attenuated inversion recovery (FLAIR) images showed slightly high signal intensity. (I) Bilateral symmetrical low signal intensity was still visible on the ADC images of the caudate nucleus.
7 MERRF and epilepsy
MERRF is a rare mitochondrial disorder caused by pathogenic mutations in mtDNA. Approximately 80% of cases are attributed to the m.8344A > G point mutation in the MT-TK gene, leading to reduced complex I and IV activity, decreased respiratory rate and compromised mitochondrial membrane potential (50). This mutation can also result in an overlap syndrome involving MELAS, MERRF, and LS.
MERRF affects multiple systems and typically presents with myoclonus as the initial symptom, followed by generalized seizures, ataxia, fatigue, motor intolerance, and dementia. Seizure frequency in MERRF patients varies widely, ranging from 33 to100% (51). Generalized myoclonic seizures are the most common, followed by focal atonic seizures, focal clonic seizures, generalized tonic–clonic seizures, myoclonic absence seizures, typical absence seizures, and status epilepticus (52, 53). There is ongoing debate regarding whether the motor symptoms in MERRF represent myoclonic seizures or cerebellar or spinal cord symptoms, with some researchers proposing the term “myoclonic ataxia” rather than “myoclonic epilepsy” (51).
EEGs in MERRF patients lack specificity but are generally characterized by background slowing, generalized epileptic discharges, and focal epileptic discharges. The EEG patterns may vary depending on the specific mutation. For example, patients with the m.8344A > G mutation in MT-TK may exhibit widespread sharp waves, sharp-slow wave complexes, or slow waves, while those with the m.3291 T > C mutation show widespread or focal multi-spike slow waves. Similarly, the m.4279A > G mutation can produce extensive spike–wave or multi-spike slow-wave complexes (51).
MRI findings in MERRF often reveal cerebellar and cerebral atrophy, focal white matter abnormalities, and bilateral symmetrical lesions in the brainstem, subthalamic nuclei, and basal ganglia (54). MRS frequently shows a decreased cerebellar N-acetyl aspartate/creatine ratio, with no significant increase in lactate levels (55). The EEG and MRI findings from a pediatric MERRF patient are shown in Figures 6, 7.
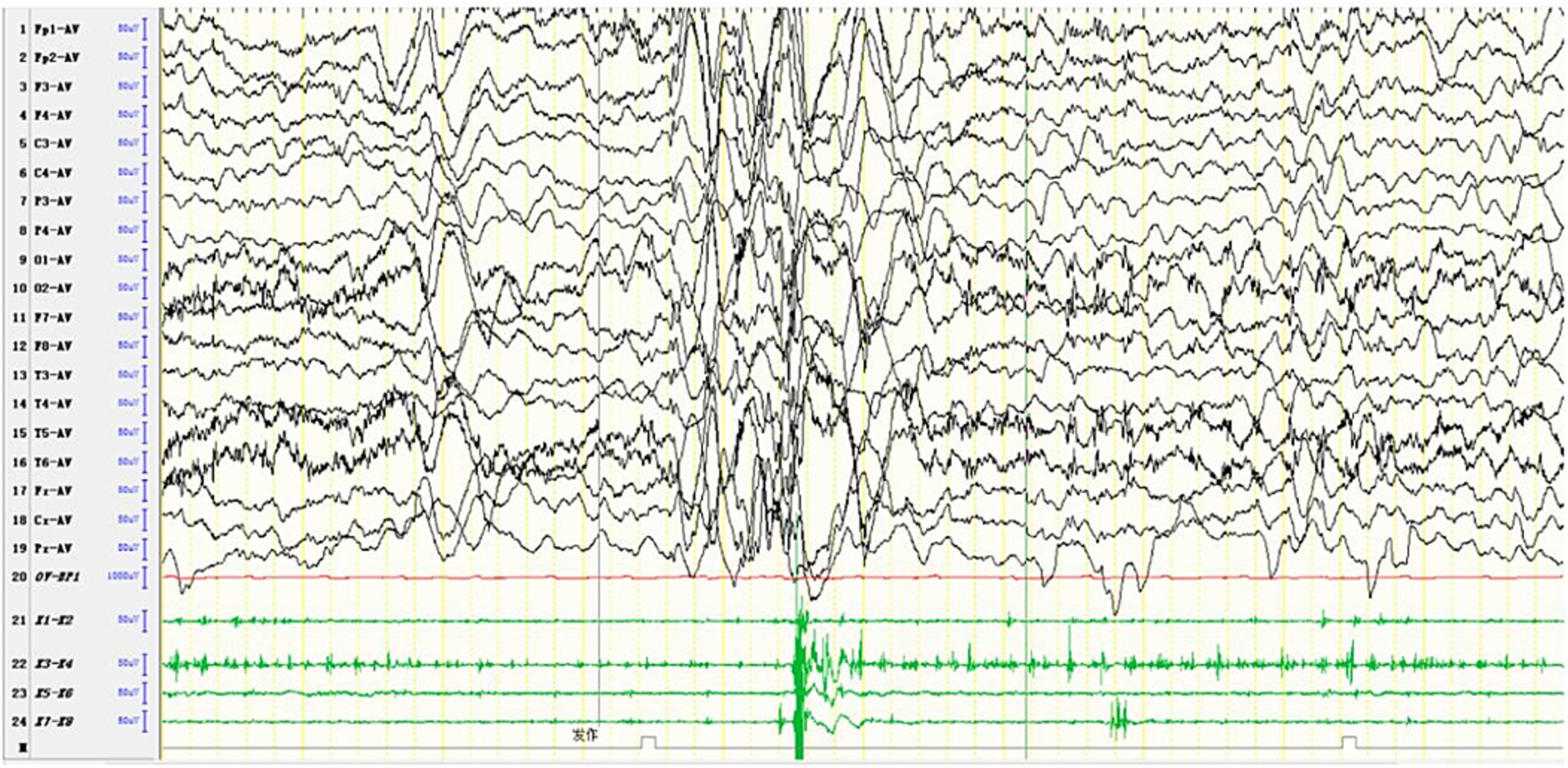
Figure 6. In one patient diagnosed with MERRF, an episode was detected during the awake period, showing limb tremors. The EEG showed widespread 3–4 Hz moderate to high-amplitude spike and slow waves for 1–2 s, with a transient EMG burst lasting 200 ms occurring simultaneously.
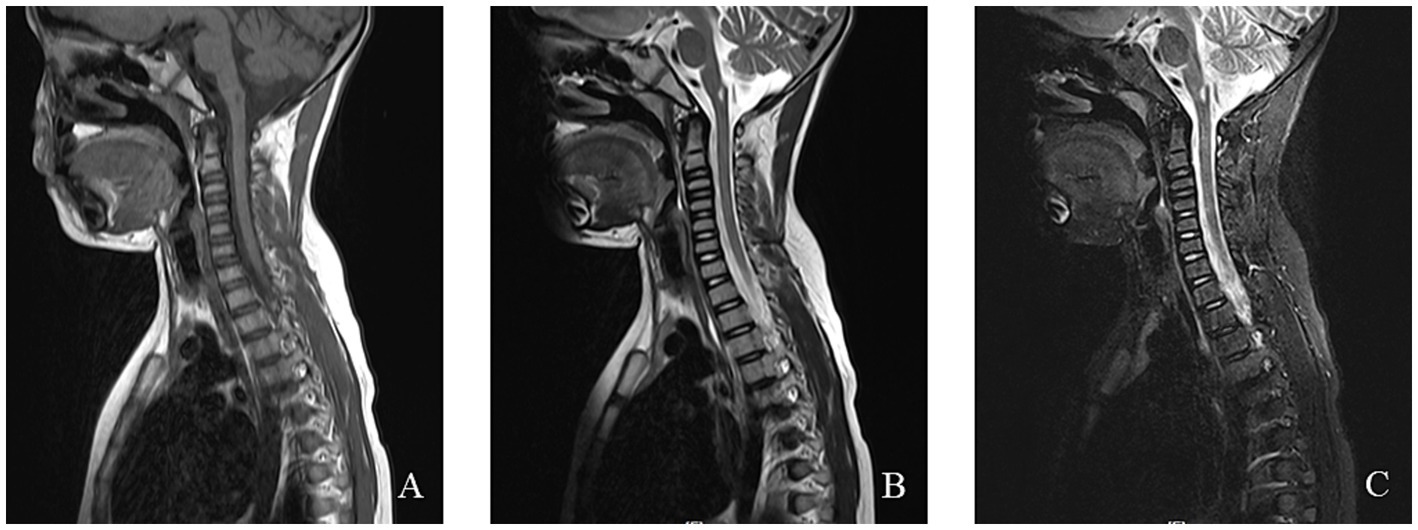
Figure 7. An 8-year-old female was diagnosed with MERRF syndrome due to limb tremors for more than 10 months. (A–C) Cervical MRI showed a patchy abnormal signal in the medulla oblongata. T1WI showed a low signal, T2WI showed a high signal, and the fat-suppression image showed a slightly high signal.
8 Epilepsy characteristics caused by different mitochondrial complex dysfunctions
The classification of mitochondrial diseases is based on several factors, including the affected tissues and organs, the dysfunction of respiratory chain enzyme complexes, and the types of gene mutations involved. The MRC consists of five enzyme complexes, along with two key electron transport proteins: coenzyme Q10 and cytochrome C. These components are essential for the proper functioning of the electron transport chain (ETC). Figure 8 illustrates the five MRC complexes and the composition of the electron transport proteins while listing common diseases caused by mutations in these complexes. Additionally, Table 1 presents the mitochondrial and nuclear genes that encode the proteins of the mitochondrial electron transport chain.
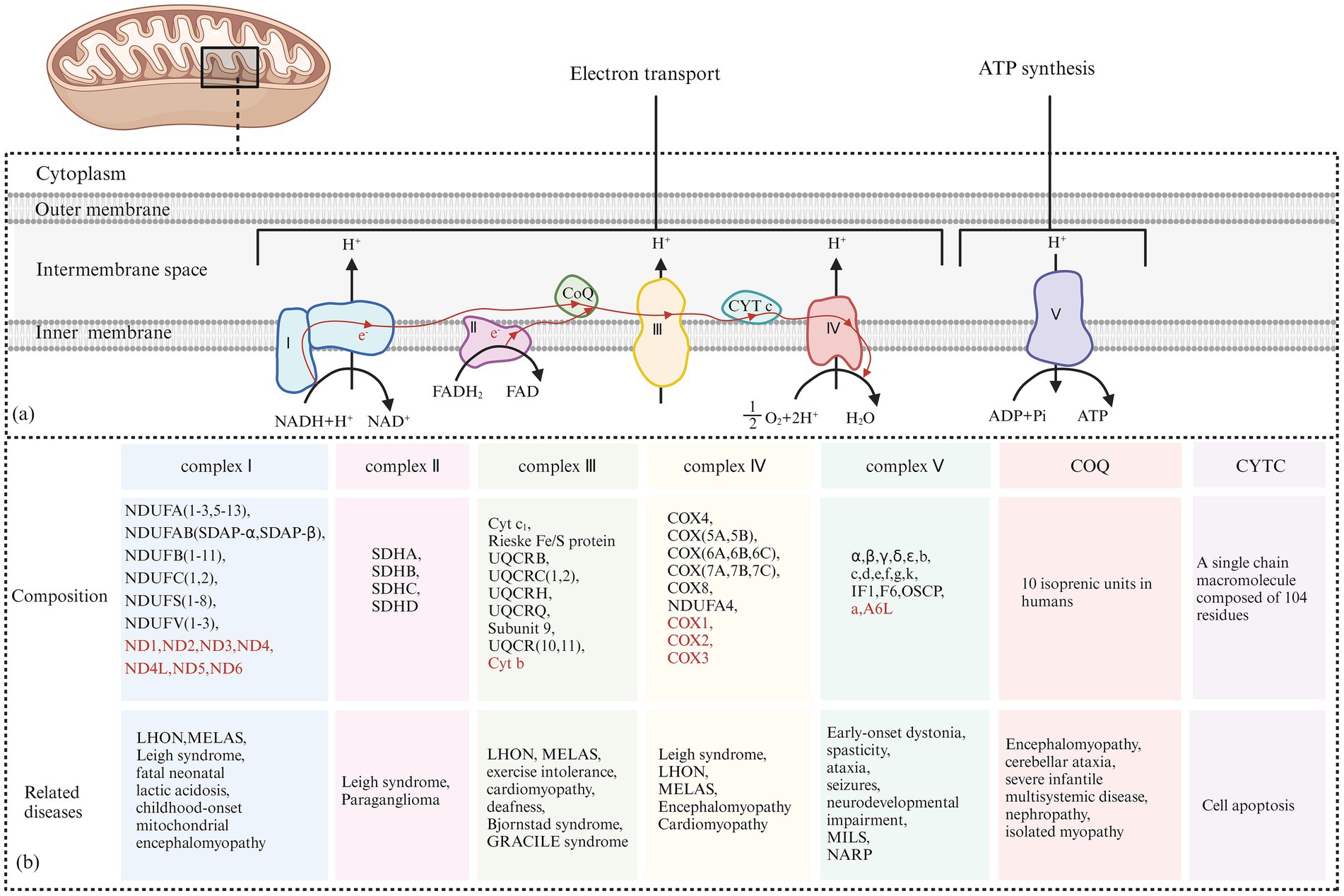
Figure 8. (A) Electron Transfer Process of the Mitochondrial Respiratory Chain: Complex I receives electrons from NADH + H+ and transfers them to coenzyme Q. The released energy pumps H+ from the matrix side of the mitochondria to the intermembrane side. Complex II receives electrons from FADH₂ and transfers them to coenzyme Q, but the energy released during this electron transfer is insufficient to pump H+ out. Coenzyme Q collects electrons from Complex I and Complex II, and the resulting QH₂ shuttles to Complex III. Complex III transfers electrons from QH₂ to CytC through a “Q cycle.” The energy released during the electron transfer in Complex III can pump H+ from the matrix side of the mitochondria to the intermembrane side. Complex IV receives electrons from CytC and transfers them to O₂ to produce H₂O, and the energy released during this electron transfer can also pump H+ to the intermembrane side. The H+ pumped into the intermembrane space forms an electrochemical gradient of H+, which is transformed into a proton-motive force, driving H+ back into the matrix along the concentration gradient and releasing stored potential energy. This stored energy is fully utilized by Complex V to catalyze the formation of ATP from ADP and Pi. (B) The Composition of the Five Complexes of the Mitochondrial Respiratory Chain and Two Types of Electron Transport Proteins (Coenzyme Q10 and Cytochrome C): In addition, typical cases of diseases caused by their respective genetic mutations are presented. The sources of each subunit are classified according to whether they are encoded by mitochondrial or nuclear genes. Subunits encoded by nuclear DNA (nDNA) are marked in red, while subunits encoded by mitochondrial DNA (mtDNA) are marked accordingly.
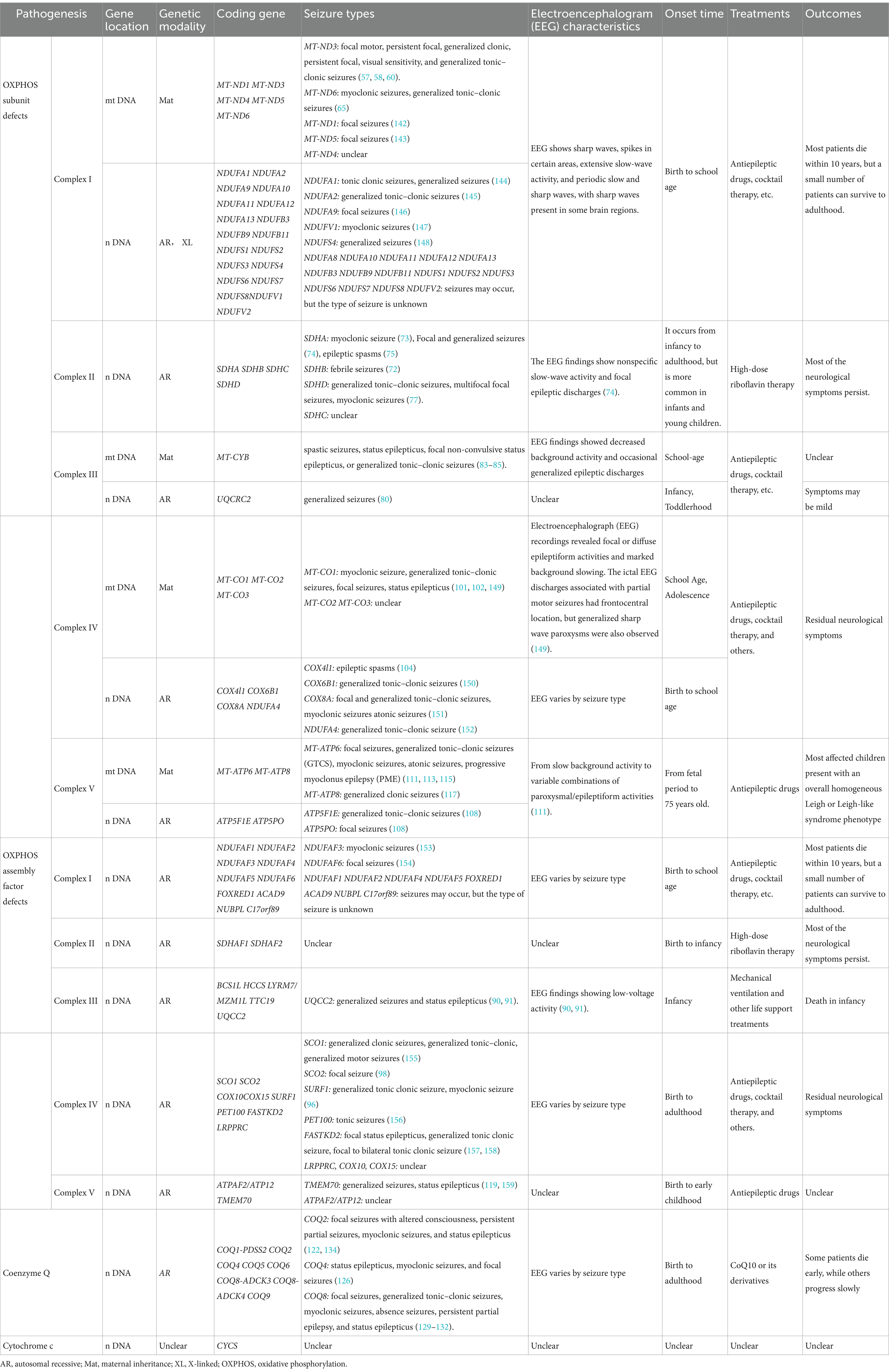
Table 1. Summary of the genes, seizure types, and EEG characteristics of mitochondrial disease-related epilepsy.
8.1 Mitochondrial complex I and epilepsy
Mitochondrial complex I, also known as NADH dehydrogenase or NADH-Q reductase, consists of 45 structural subunits. Deficiency in mitochondrial complex I is associated with mutations in various structural subunits and assembly factors, resulting in a wide range of clinical manifestations, including LHON, infantile LS, MELAS, and epilepsy (56).
Among the core subunits of complex I, mutations in MT-ND3 and MT-ND5 are more common. Point mutations in MT-ND3, such as m.10191 T > C, m.10158 T > C, and m.10197G > A, have been widely studied. More than 30 patients with the MT-ND3 m.10191 T > C mutation have been reported, with approximately 90% developing epilepsy, typically between the ages of 8 and 10. Seizure types include focal motor, persistent focal, and generalized tonic–clonic seizures (57, 58).
EEG findings in patients with the MT-ND3 m.10191 T > C mutation typically display interictal focal epileptic discharges, sharp waves, spikes in certain areas, and extensive slow-wave activity, with sharp waves present in some brain regions (57, 59). Imaging often reveals LS-like changes, such as involvement of the basal ganglia, thalamus, and brainstem, in about 50% of patients, with some exhibiting white matter-dominant polyencephalic lesions (57).
The MT-ND3 m.10158 T > C mutation is occasionally associated with isolated and recurrent stroke-like seizures. These seizures typically occur in middle age or later, manifesting as generalized clonic seizures, persistent focal seizures, and visual sensitivity seizures (60). EEG has periodic slow and sharp waves (61). MRI findings usually indicate lesions in the posterior cortex of the supratentorial area (62).
The common MT-ND5 m.13513G > A mutation is linked to multiple syndromes, including LS, MELAS, MERRF, LHON, and MELAS-LS overlap syndrome (63, 64). Some patients with this mutation also experience seizures. Dermaut et al. reported that a patient with the MT-ND6 m.14487 T > C mutation presented with myoclonic seizures, generalized tonic–clonic seizures, decreased EEG background activity, spike waves, and multi-spike waves (65). Patients with NDUFV1 mutations may exhibit myoclonic seizures and absence seizures (66, 67). Seizures are also linked to mutations in nDNA genes, including NDUFS2, NDUFS4, NDUFS8, NDUFA1, and NDUFA8 (68–70).
Currently, there is no effective treatment for mitochondrial complex I deficiency. Clinical management primarily focuses on enhancing mitochondrial energy metabolism, increasing antioxidant capacity, providing nutritional support, and using ASMs to control seizures. Approximately 60% of patients with the MT-ND3 m.10191 T > C mutation survive into puberty or later, which is a better prognosis than other complex I-related gene defects. A report from Japan indicated that homologous expression of codon-optimized MT-ND3 in patients with MT-ND3 mutations can increase ATP production and improve mitochondrial function, showing promise as a potential new treatment strategy (71).
8.2 Mitochondrial complex II and epilepsy
Mitochondrial complex II, also known as succinate dehydrogenase (SDH) or succinate-ubiquinone oxidoreductase (SQR), Comprises four structural subunits: SDHA, SDHB, SDHC, and SDHD, all encoded by nDNA. Additionally, four assembly factors, SDHAF1, SDHAF2, SDHAF3, and SDHAF4, are involved in its assembly and function (72). Clinical manifestations of complex II deficiency vary and include LS, leukoencephalopathy, optic nerve atrophy, and epilepsy. Among 61 patients with complex II deficiency reported by Fullerton et al., five had seizures (72).
Mutations in SDHA are the most common cause of complex II deficiency and are mainly associated with LS. Neurological symptoms include epileptic encephalomyopathy, external ophthalmoplegia, and myoclonic seizures (73). In one case of an SDHA mutation, the patient experienced focal and generalized seizures, with EEG findings showing nonspecific slow-wave activity and focal epileptic discharges (74). Another case presented with Lennox–Gastaut syndrome and highly irregular EEG patterns (75). Both cases demonstrated imaging features resembling LS.
Biallelic variations in SDHB frequently result in complex II deficiency. Approximately 10 patients with LS have been identified with pathogenic SDHB variations, most exhibiting neurodegenerative symptoms, seizures, and dystonia, with one patient showing heat sensitivity (72). The clinical manifestations of complex II deficiency related to SDHB mutations are similar to LS but typically do not involve the basal ganglia, which can help differentiate the two conditions (76).
A review by SiyingLin et al. identified six patients with SDHD mutations, four of whom experienced generalized tonic–clonic seizures, multifocal focal seizures, and myoclonic seizures (77). Of the four known SDH assembly factors, only mutations in SDHAF1 have been linked to complex II deficiency. Andreas et al. reported five patients with homozygous SDHAF1 mutations, all of whom had motor degeneration and spastic paraplegia, with only one patient experiencing seizures (78).
Since complex II deficiency is rarer than other mitochondrial diseases, limited reports exist on its treatment and prognosis. Riboflavin has been shown to improve clinical manifestations in patients with complex II deficiency, potentially preventing disease progression and even reversing symptoms. In particular, patients with riboflavin transporter deficiency may benefit from high-dose riboflavin therapy (up to 70 mg/kg/day) (79).
8.3 Mitochondrial complex III and epilepsy
Mitochondrial complex III, also known as ubiquinone-cytochrome c reductase or cytochrome bc1 complex, consists of 11 structural subunits. Cytochrome b is encoded by mitochondrial DNA (mtDNA), while the remaining 10 subunits are encoded by nuclear DNA (nDNA). Additionally, BCS1L, TTC19, and UQCC2, encoded by nDNA, are involved in the assembly of complex III (80). The Leucine zipper EF-hand domain transmembrane protein 1 (LETM1) is located in the inner mitochondrial membrane and plays a critical role in regulating the synthesis of cytochrome b. Downregulation of LETM1 reduces MT-CYB expression and increases susceptibility to seizures (81).
Mutations in cytochrome b can manifest in skeletal muscle involvement, exercise intolerance, MELAS, LS, and seizures (82). Seizures in these cases may present as spastic seizures, status epilepticus, focal non-convulsive status epilepticus, or generalized tonic–clonic seizures (83, 84). Scott et al. described a patient with a cytochrome b mutation who exhibited both Parkinson’s syndrome and MELAS, along with status epilepticus and tonic–clonic seizures. EEG findings showed decreased background activity and occasional generalized epileptic discharges (85).
BCS1L mutations are the most common cause of complex III deficiency and are linked to GRACILE syndrome, Björstand syndrome, liver disease, encephalopathy, dyskinesia, and epilepsy. Neurological symptoms, such as dyskinesia and seizures, generally manifest after the age of 1 month, with an epilepsy incidence of 33%. Patients with the BCS1L c.232A > G mutation have typically developed GRACILE syndrome, although epilepsy is often absent (86). Erika et al. described a case of rapid progression in a patient with a BCS1L mutation, characterized by growth retardation, epileptic spasms, and frequent seizures in later stages. MRI findings showed involvement of the thalamus and supratentorial white matter (87). Helen et al. described a patient with a novel BCS1L mutation who had slower disease progression, presenting with myopathy, focal motor seizures, and optic nerve atrophy (88). Hikmat et al. reported that 6 out of 33 patients with BCS1L mutations had seizures (86).
Mutations in TTC19, UQCC2, and UQCRC2 are also associated with seizures. Patients with missense TTC19 mutations (c.971 T > C and c.554 T > C) may present with tonic seizures and refractory epilepsy (89). Patients with UQCC2 mutations may experience generalized seizures and status epilepticus, with EEG findings showing low-voltage activity (90, 91). There have also been reports suggesting that mutations in the UQCRC2 gene can lead to generalized seizures (80). MRI findings in patients with complex III defects vary widely. Some resemble those seen in LS, with focal and bilateral symmetrical lesions in the basal ganglia, thalamus, and brain stem, while others show brain atrophy or underdeveloped myelin.
There is no specific therapy for respiratory chain complex III deficiency. Treatment focuses on managing symptoms and slowing disease progression. In one case, clinical symptoms improved following treatment with vitamin K3 and vitamin C (92). Patients with TTC19 mutations often present with early psychomotor retardation, followed by progressive neurological decline and poor prognosis (89). The survival rate for patients with homozygous or compound heterozygous BCS1L c.232A > G mutations is significantly lower compared to patients with other BCS1L mutations, underscoring the importance of genotype–phenotype correlation, prognosis evaluation, and genetic counseling (86). Early disease onset is generally associated with faster progression and higher mortality.
8.4 Mitochondrial complex IV and epilepsy
Mitochondrial complex IV, also known as cytochrome c oxidase (COX), is the terminal enzyme in the electron transport chain. It comprises 14 subunits, with 3 encoded by mitochondrial DNA (mtDNA) and the remaining 11 by nuclear DNA (nDNA). Several nuclear-encoded assembly factors, including SURF1, SCO1, SCO2, COX10, COX15, and LRPPRC, are involved in the assembly of complex IV (93). Cytochrome c oxidase deficiency affects the brain, heart, and skeletal muscle, leading to disorders such as LS and cardiomyopathy (94).
SURF1 deficiency is a monogenic mitochondrial disorder and the most common cause of cytochrome c oxidase-deficient LS. Approximately 80% of SURF1 mutations are truncating, caused by abnormal splicing, frameshift deletions, or nonsense mutations. Clinical features include weight loss, dystonia, stunted growth, and epilepsy, with an epilepsy incidence of 33.3%, most commonly presenting as generalized tonic–clonic and myoclonic seizures (95). MRI findings often show LS-like features, with symmetrical necrotic lesions in the basal ganglia and brainstem (96). In comparison, patients with SCO2 mutations tend to have earlier-onset symptoms that progress more rapidly, including hypertrophic cardiomyopathy, dystonia, and seizures. EEG findings commonly show focal epileptic discharges in the central and temporal regions (97, 98). The COX15 p.R217W mutation is associated with lactic acidosis, ataxia, and hypotonia, with epilepsy occurring in 71.4% of cases. Status epilepticus has also been reported, though specific seizure types have not been well-documented. EEG findings typically include multifocal slow-wave discharge (99, 100).
In addition to mutations in assembly factor genes, mutations in the three COX subunits encoded by mtDNA are also linked to epilepsy. MT-CO1 mutations can lead to a wide range of seizure types, including myoclonic seizures associated with the m.6480G > A and m.6930G > A missense mutations (101, 102). The m.7402delC mutation can cause non-convulsive status epilepticus (102). The missense mutation m.7023G > A is associated with seizures such as mid-wind, generalized tonic–clonic, and focal seizures. Patients with the m.6489G > A mutation display persistent partial and myoclonic seizures, generalized tonic–clonic seizures, recurrent status epilepticus, and diffuse epileptic activity on EEG, often accompanied by significantly reduced background activity. MT-CO3 subunit mutations have been reported in patients with recurrent MELAS-like seizures (103).
In mammals, COX4 has two isoforms: COX4I1 and COX4I2. The COX4I1 c.454C > A missense mutation has been linked to infantile spasms, high EEG irregularity, and multifocal spikes (104).
Treatment options for mitochondrial complex IV deficiency are limited and vary depending on the specific mutation. Arginine may help alleviate stroke-like episodes in patients with COX-deficient MELAS, while coenzyme Q10 has shown promise in slowing disease progression in COX4I1 deficiency (16, 104). EPI-743, a coenzyme Q10 derivative, has been found to prevent disease progression and improve quality of life and motor function in some SURF1-related LS cases (105). Additional treatments, such as the Pan-PPAR agonist bezafibrate and the AMPK agonist AICAR, have shown the potential to partially restore COX function in human and animal models (106). Intrathecal delivery of an adeno-associated viral vector serotype 9 (AAV9)-human SURF1 (hSURF1) has effectively corrected biochemical abnormalities in mouse models of SURF1 deficiency (107). Emerging therapies such as mitochondrial biogenesis and gene replacement also hold promise for future treatment strategies.
8.5 Mitochondrial complex V and epilepsy
Mitochondrial complex V, also known as ATP synthase, is essential for mitochondrial energy production and comprises two functional domains: F1 and F0. The F1 domain contains five subunits (α, β, γ, δ, and ε), while the F0 domain consists of subunits a, b, c, d, e, f, g, I, k, A6L, F6, and the oligomycin-sensitive protein (OSCP) (108). Subunit, an and A6L of the F0 domain, are encoded by the MT-ATP6 and MT-ATP8 genes in mtDNA, while the remaining subunits, along with assembly factors (ATP assembly factors 1 and 2), coupling factors (inhibitor IF1 and coupling factor B), and ATP synthase-related proteins (MLQ and DAPIT), are encoded by nDNA (109). Mutations in MT-ATP6, MT-ATP8, ATPAF2, TMEM70, and ATP5E can lead to complex V deficiencies.
MT-ATP6 mutations are the most common and widely studied causes of complex V deficiency, characterized by a reduction in ATP synthesis and basal oxygen consumption. This can lead to clinical manifestations such as ataxia, cognitive impairment, neuropathy, and epilepsy, with an incidence of 37%. MILS and NARP syndrome are associated with seizure rates of 86 and 44%, respectively, with generalized seizures being the most frequent type (110). Common mutations in MT-ATP6-related epilepsy include m.8993 T > G, m.8993 T > C, m.9032 T > C, m.9058A > G, and m.8921G > A (111–114). The m.8993 T > G mutation is particularly associated with a high incidence of generalized tonic–clonic, myoclonic, and progressive myoclonic seizures. EEGs in patients with this mutation typically show slow background activity and widespread sharp-slow complexes, making EEG a key tool for assessing disease severity In contrast, the m. 8,993 T > C mutation generally leads to milder symptoms and later onset, though it is still associated with generalized tonic–clonic seizures. Patients with the m.9032 T > C mutation may experience focal seizures, and EEG findings often show sharp waves in the central parietal region (115).
Mutations in MT-ATP8, such as m.8502A > T and m.8411A > G, have been identified in patients with epilepsy (116, 117). The former is closely associated with medial temporal lobe epilepsy, while the latter is linked to generalized tonic–clonic seizures. Mutations in ATP5F1E, ATPAF2, ATP5PO, and TMEM70, all encoded by nDNA, are also implicated in seizure disorders (108, 118, 119). Homozygous mutations in ATP5F1E (c.35A > G), which affect the ε subunit of the F1 domain, commonly result in generalized tonic–clonic seizures. A patient with an ATP5PO mutation encoding OSCP recently died from refractory status epilepticus, and a separate case involving a TMEM70 mutation also presented with status epilepticus (119).
Treatment for complex V deficiency is largely symptomatic, focusing on the use of coenzyme Q10, vitamin B1, and vitamin C. α-ketoglutarate and aspartic acid have shown potential in increasing mitochondrial substrate phosphorylation, thereby normalizing ATP levels in cells with the m.8993 T > G mutation and reducing cell death (120). N-acetylcysteine and dihydrolipoic acid also hold therapeutic potential for patients with the m.8993 T > G mutation (121).
8.6 Coenzyme Q10 and epilepsy
Coenzyme Q (CoQ or ubiquinone) plays a crucial role in mitochondrial electron transfer, shuttling electrons from complex I or II to complex III. Coenzyme Q deficiency can be classified into primary and secondary forms based on etiology. Secondary CoQ deficiency is not directly related to the biosynthesis of CoQ, while primary CoQ deficiency results from mutations in genes responsible for CoQ biosynthesis. The Clinical manifestations are highly heterogeneous (122).
The CoQ biosynthesis pathway is complex and involves at least 15 genes, including PDSS1, COQ1-PDSS2, COQ2, COQ3, COQ4, COQ5, COQ6, COQ7, COQ8-ADCK3, COQ8-ADCK4, COQ9, COQ10a, COQ10b, FDX1L, and FDXR. Additionally, three other genes, ADCK1, ADCK2, and ADCK5, are also believed to play roles in CoQ biosynthesis (123). The incidence of primary CoQ10 deficiency is estimated at approximately 1 in 50,000, and while it can occur at any age, it is more common in children. When the central nervous system (CNS) is involved, the clinical phenotype is diverse, ranging from growth retardation and encephalopathy to intellectual disability and epilepsy. Several CoQ biosynthesis genes have been implicated in epilepsy including COQ1-PDSS2, COQ2, COQ4, COQ5, COQ6, COQ8-ADCK3, COQ8-ADCK4, and COQ9, with COQ2, COQ4, and COQ8-ADCK3 mutations being more frequently associated with seizures (124).
COQ4 mutations have the highest incidence of epilepsy, reaching up to 69%, with a higher prevalence among females. The age of onset ranges from birth to 9 years, and clinical symptoms may appear as early as the first week of life. These symptoms include growth retardation, epilepsy, skeletal muscle involvement, and cardiomyopathy (125). Seizure types include status epilepticus, myoclonic seizures, and focal seizures, with or without altered consciousness (126). EEG findings typically show reduced background activity and focal seizures, with occasional sharp waves, particularly in the left parietal region during REM sleep (127, 128).
Mutations in the COQ8A gene (also known as CABC1 or ADCK3) represent the most common form of primary coenzyme Q10 deficiency. Clinical manifestations include early-onset cerebellar ataxia, various motor disorders, cognitive impairment, motor intolerance, and epilepsy, which occurs in approximately 32% of cases. Epilepsy phenotypes associated with COQ8A mutations include focal seizures, generalized tonic–clonic seizures, myoclonic seizures, absence seizures, persistent partial epilepsy, and status epilepticus (129–132). EEG findings typically show frequent focal epileptic discharges and δ waves. During the interictal period, multi-spike wave or spike–wave rhythms are observed in the bioccipital region but may also be seen in the parietal, temporal, and frontal lobes. Some patients exhibit paroxysmal discharges in response to medium- and high-frequency (10-30 Hz) light stimulation (133). Cranial MRI frequently reveals cerebellar atrophy, though other regions may also be affected, including the brainstem and supratentorial areas such as the parietal and frontal insular lobes. Additionally, T2-weighted imaging often shows high signal intensity in the dentate nucleus and dorsal pons (129).
Primary CoQ deficiency was initially reported in association with COQ2 mutations, characterized by encephalopathy, cerebellar dysplasia, and epilepsy, with a seizure incidence of approximately 40%. Seizure types in COQ2 deficiency include focal seizures with altered consciousness, persistent partial seizures, myoclonic seizures, and status epilepticus (122, 134, 135).
Therapeutic response to coenzyme Q10 or its derivatives varies by genetic mutation. Patients with COQ4 mutations may benefit from coenzyme Q10 supplementation, while those with PDSS2, COQ8A, and COQ9 mutations tend to respond less effectively. For patients with COQ2 or COQ6 mutations, supplementation with 4-hydroxybenzoic acid (4-HBA) may enhance endogenous CoQ10 synthesis (136, 137). In preclinical studies, the antioxidant probucol has shown greater efficacy in treating renal and metabolic complications in PDSS2 mutant mice but compared to coenzyme Q10 supplementation (138), this treatment remains under investigation.
8.7 Cytochrome C and epilepsy
Cytochrome c (CytC) is a water-soluble protein encoded by Ndna and composed of 104 amino acid residues. Its primary function is to facilitate electron transport between complex III and complex IV in the mitochondrial electron transport chain. Additionally, CytC plays a crucial role in apoptosis induction (139). During seizures, the production of harmful substances in neurons can increase the permeability of the mitochondrial inner membrane. CytC is subsequently released into the cytoplasm, where it binds to apoptosis-related factor 1(Apaf-1), promoting the formation of apoptotic bodies by binding with caspase-9. This process activates other caspases, leading to neuronal apoptosis. Mitochondrial damage exacerbates the susceptibility to seizures, creating a vicious circle that promotes further neuronal damage (140). Luan et al. found that a ketogenic diet can reduce the release of cytochrome c, thus playing a neuroprotective role (141).
9 Summary and prospects
When we encounter children with mitochondrial disease, it is necessary to conduct a series of relevant medical examinations. These auxiliary examinations that help diagnose mitochondrial diseases mainly include serum lactate levels, neuroelectrophysiological analysis, imaging diagnosis, and pathological examinations of muscles and brain, as well as diagnosis of gene mutations. Given that there is currently no definite treatment for mitochondrial disease, symptomatic treatment is particularly critical for patients. Some drugs may cause abnormalities in mitochondria or energy metabolism, so special care should be taken when using them. For example, sodium valproate may have significant side effects on the liver, so patients with mitochondrial disease should use it with caution. Although most of the drugs are still in development and trials, we outline in Table 2 the various modification therapies for mitochondrial diseases, such as small molecule therapy, gene therapy, and reproductive therapy.
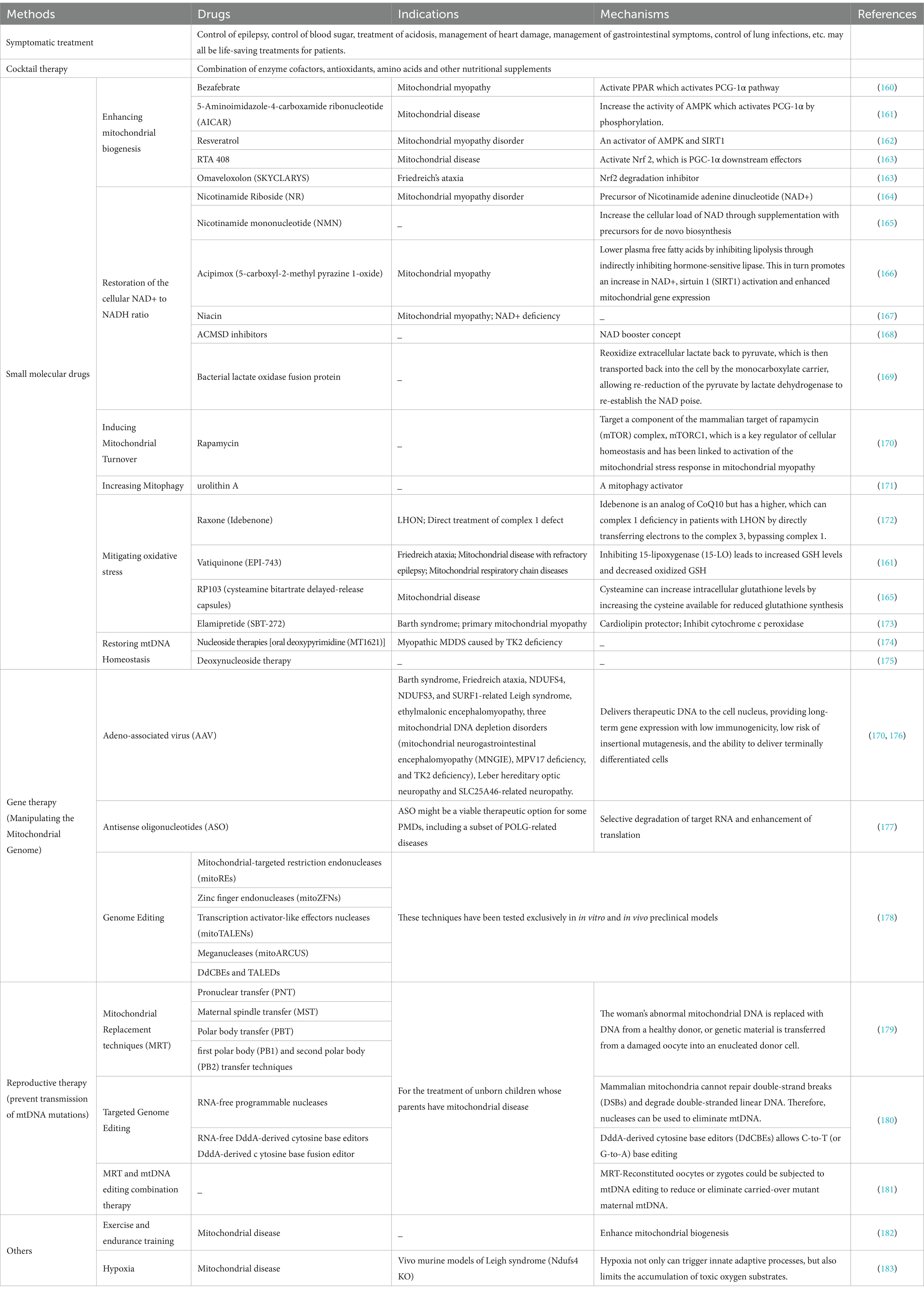
Table 2. Disease-modifying treatments for mitochondrial diseases.
The pathogenesis of childhood mitochondrial-associated epilepsy is highly complex, involving a range of genetic mutations that lead to various seizure types, often resulting in drug-refractory epilepsy. As research in mitochondrial-associated epilepsy advances, new gene mutations continue to be discovered. While significant progress has been made in understanding some aspects of the condition, many questions remain unanswered. The advent of next-generation sequencing (NGS) and the growing focus on precision medicine has facilitated the identification of mitochondrial genes linked to epilepsy. These discoveries have become integral to diagnosing hereditary epilepsy and have advanced the fields of personalized medicine and genetic counseling.
Currently, there are no effective treatments to completely cure mitochondrial diseases, and gene therapy remains in the experimental stages. However, both gene therapy and cell therapy are promising areas of research, offering potential for new and effective treatments. There is hope that future advancements in these therapies may ultimately lead to a cure for mitochondrial diseases.
Author contributions
XZ: Data curation, Software, Visualization, Writing – original draft. BZ: Formal analysis, Methodology, Writing – review & editing. ZT: Investigation, Software, Writing – review & editing. JL: Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Nature and Science Foundation of China (nos. 82271509 and 81771396), the Foundation of Jilin Provincial Key Laboratory of Pediatric Neurology (no. YDZJ202102CXJD021), the Project of Jilin Provincial Science and Technology Development Plan (No. YDZJ202201ZYTS676), and the Project of Jilin Medical and Health Talents (nos. JLSWSRCZX2023-20 and JLSWSRCZX2023-84).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wallace, DC, Fan, W, and Procaccio, V. Mitochondrial energetics and therapeutics. Annu Rev Pathol. (2010) 5:297–348. doi: 10.1146/annurev.pathol.4.110807.092314
2. Molnar, MJ, and Kovacs, GG. Mitochondrial diseases. Handb Clin Neurol. (2018) 145:147–55. doi: 10.1016/B978-0-12-802395-2.00010-9
3. Zsurka, G, and Kunz, WS. Mitochondrial dysfunction and seizures: the neuronal energy crisis. Lancet Neurol. (2015) 14:956–66. doi: 10.1016/S1474-4422(15)00148-9
4. Bindoff, LA, and Engelsen, BA. Mitochondrial diseases and epilepsy. Epilepsia. (2012) 53:92–7. doi: 10.1111/j.1528-1167.2012.03618.x
5. Hikmat, O, Eichele, T, Tzoulis, C, and Bindoff, LA. Understanding the epilepsy in POLG related disease. Int J Mol Sci. (2017) 18:1845. doi: 10.3390/ijms18091845
6. Waldbaum, S, and Patel, M. Mitochondrial dysfunction and oxidative stress: a contributing link to acquired epilepsy? J Bioenerg Biomembr. (2010) 42:449–55. doi: 10.1007/s10863-010-9320-9
7. Hari, AS, Walker, MC, and Patel, M. Role of reactive oxygen species in epilepsy In: JL Noebels, M Avoli, MA Rogawski, A Vezzani, and AV Delgado-Escueta, editors. Jasper's basic mechanisms of the epilepsies. edn ed. New York: Oxford University Press (2024). 633–64.
8. Niespodziany, I, Klitgaard, H, and Margineanu, DG. Levetiracetam inhibits the high-voltage-activated ca(2+) current in pyramidal neurones of rat hippocampal slices. Neurosci Lett. (2001) 306:5–8. doi: 10.1016/S0304-3940(01)01884-5
9. Akyuz, E, Polat, AK, Eroglu, E, Kullu, I, Angelopoulou, E, and Paudel, YN. Revisiting the role of neurotransmitters in epilepsy: an updated review. Life Sci. (2021) 265:118826. doi: 10.1016/j.lfs.2020.118826
10. Ferriero, DM. Protecting neurons. Epilepsia. (2005) 46:45–51. doi: 10.1111/j.1528-1167.2005.00302.x
11. Mao, XY, Zhou, HH, and Jin, WL. Redox-related neuronal death and crosstalk as drug targets: focus on epilepsy. Front Neurosci. (2019) 13:512. doi: 10.3389/fnins.2019.00512
12. Lemattre, C, Imbert-Bouteille, M, Gatinois, V, Benit, P, Sanchez, E, Guignard, T, et al. Report on three additional patients and genotype-phenotype correlation in SLC25A22-related disorders group. Eur J Hum Genet. (2019) 27:1692–700. doi: 10.1038/s41431-019-0433-2
13. Goubert, E, Mircheva, Y, Lasorsa, FM, Melon, C, Profilo, E, Sutera, J, et al. Inhibition of the mitochondrial glutamate carrier SLC25A22 in astrocytes leads to intracellular glutamate accumulation. Front Cell Neurosci. (2017) 11:149. doi: 10.3389/fncel.2017.00149
14. Tein, I. Disorders of fatty acid oxidation. Handb Clin Neurol. (2013) 113:1675–88. doi: 10.1016/B978-0-444-59565-2.00035-6
15. Arhan, E, Öztürk, Z, Serdaroğlu, A, Aydın, K, Hirfanoğlu, T, and Akbaş, Y. Neonatal hypoglycemia: A wide range of electroclinical manifestations and seizure outcomes. Eur J Paediatr Neurol. (2017) 21:738–44. doi: 10.1016/j.ejpn.2017.05.009
16. El-Hattab, AW, Adesina, AM, Jones, J, and Scaglia, F. MELAS syndrome: clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab. (2015) 116:4–12. doi: 10.1016/j.ymgme.2015.06.004
17. Tetsuka, S, Ogawa, T, Hashimoto, R, and Kato, H. Clinical features, pathogenesis, and management of stroke-like episodes due to MELAS. Metab Brain Dis. (2021) 36:2181–93. doi: 10.1007/s11011-021-00772-x
18. Harris, MO, Walsh, LE, Hattab, EM, and Golomb, MR. Is it ADEM, POLG, or both? Arch Neurol. (2010) 67:493–6. doi: 10.1001/archneurol.2010.36
19. Zhao, M, Zuo, C, Hao, H, Xing, X, Zhao, L, and Li, N. A patient with MELAS syndrome combined with autoimmune abnormalities: a case report. Front Neurol. (2023) 14:1239664. doi: 10.3389/fneur.2023.1239664
20. Zhang, J, Zhang, Z, Zhang, Y, and Wu, Y. Distinct magnetic resonance imaging features in a patient with novel RARS2 mutations: A case report and review of the literature. Exp Ther Med. (2018) 15:1099–104. doi: 10.3892/etm.2017.5491
21. Cheng, W, Zhang, Y, and He, L. MRI features of stroke-like episodes in mitochondrial Encephalomyopathy with lactic acidosis and stroke-like episodes. Front Neurol. (2022) 13:843386. doi: 10.3389/fneur.2022.843386
22. Khurana, D, Salganicoff, L, Melvin, J, Hobdell, E, Valencia, I, Hardison, H, et al. Epilepsy and respiratory chain defects in children with mitochondrial encephalopathies. Neuropediatrics. (2008) 39:8–13. doi: 10.1055/s-2008-1076737
23. Caietta, E, Cano, A, Halbert, C, Hugonenq, C, Mancini, J, Milh, M, et al. Épilepsie et cytopathies mitochondriales: étude rétrospective de 53 enfants épileptiques. Arch Pediatr. (2012) 19:794–802. doi: 10.1016/j.arcped.2012.05.004
24. Finsterer, J, and Zarrouk Mahjoub, S. Mitochondrial epilepsy in pediatric and adult patients. Acta Neurol Scand. (2013) 128:141–52. doi: 10.1111/ane.12122
25. Meseguer, S, Navarro-González, C, Panadero, J, Villarroya, M, Boutoual, R, Sánchez-Alcázar, JA, et al. The MELAS mutation m. 3243A> G alters the expression of mitochondrial tRNA fragments. Biochimica et Biophysica Acta (BBA)-molecular Cell Res. (2019) 1866:1433–49. doi: 10.1016/j.bbamcr.2019.06.004
26. Finsterer, J. Manifestations of the mitochondrial A3243G mutation. Int J Cardiol. (2009) 137:60–2. doi: 10.1016/j.ijcard.2008.04.089
27. Li, J, Zhang, W, Cui, Z, Li, Z, Jiang, T, and Meng, H. Epilepsy associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. Front Neurol. (2021) 12:675816. doi: 10.3389/fneur.2021.675816
28. Chevallier, JA, Von Allmen, GK, and Koenig, MK. Seizure semiology and EEG findings in mitochondrial diseases. Epilepsia. (2014) 55:707–12. doi: 10.1111/epi.12570
29. Ng, YS, Lax, NZ, Blain, AP, Erskine, D, Baker, MR, Polvikoski, T, et al. Forecasting stroke-like episodes and outcomes in mitochondrial disease. Brain. (2022) 145:542–54. doi: 10.1093/brain/awab353
30. Lee, S, Oh, D-A, and Bae, E-K. Fixation-off sensitivity in mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome. Seizure-European J Epilepsy. (2019) 64:6–7. doi: 10.1016/j.seizure.2018.11.010
31. Malhotra, K, and Liebeskind, DS. Imaging of MELAS. Curr Pain Headache Rep. (2016) 20:1–8. doi: 10.1007/s11916-016-0583-7
32. Rahman, S. Mitochondrial disease in children. J Intern Med. (2020) 287:609–33. doi: 10.1111/joim.13054
33. Chang, X, Wu, Y, Zhou, J, Meng, H, Zhang, W, and Guo, J. A meta-analysis and systematic review of Leigh syndrome: clinical manifestations, respiratory chain enzyme complex deficiency, and gene mutations. Medicine. (2020) 99:e18634. doi: 10.1097/MD.0000000000018634
34. Hong, C-M, Na, J-H, Park, S, and Lee, Y-M. Clinical characteristics of early-onset and late-onset Leigh syndrome. Front Neurol. (2020) 11:267. doi: 10.3389/fneur.2020.00267
35. Sofou, K, De Coo, IF, Isohanni, P, Ostergaard, E, Naess, K, De Meirleir, L, et al. A multicenter study on Leigh syndrome: disease course and predictors of survival. Orphanet J Rare Dis. (2014) 9:52–16. doi: 10.1186/1750-1172-9-52
36. Wei, Y, Cui, L, and Peng, B. Mitochondrial DNA mutations in late-onset Leigh syndrome. J Neurol. (2018) 265:2388–95. doi: 10.1007/s00415-018-9014-5
37. Lee, S, Na, J-H, and Lee, Y-M. Epilepsy in Leigh syndrome with mitochondrial DNA mutations. Front Neurol. (2019) 10:496. doi: 10.3389/fneur.2019.00496
38. Monden, Y, Mori, M, Kuwajima, M, Goto, T, Yamagata, T, and Momoi, MY. Late-onset Leigh syndrome with myoclonic epilepsy with ragged-red fibers. Brain and Development. (2013) 35:582–5. doi: 10.1016/j.braindev.2012.08.006
39. Wray, CD, Friederich, MW, du Sart, D, Pantaleo, S, Smet, J, Kucera, C, et al. A new mutation in MT-ND1 m. 3928G> C p. V208L causes Leigh disease with infantile spasms. Mitochondrion. (2013) 13:656–61. doi: 10.1016/j.mito.2013.09.004
40. Alves, CAPF, Gonçalves, FG, Grieb, D, Lucato, LT, Goldstein, AC, and Zuccoli, G. Neuroimaging of mitochondrial cytopathies. Top Magn Reson Imaging. (2018) 27:219–40. doi: 10.1097/RMR.0000000000000173
41. Senthilvelan, S, Sekar, SS, Kesavadas, C, and Thomas, B. Neuromitochondrial disorders: genomic basis and an algorithmic approach to imaging diagnostics. Clin Neuroradiol. (2021) 31:559–74. doi: 10.1007/s00062-021-01030-4
42. Qian, Y, Ziehr, JL, and Johnson, KA. Alpers disease mutations in human DNA polymerase gamma cause catalytic defects in mitochondrial DNA replication by distinct mechanisms. Front Genet. (2015) 06:134388. doi: 10.3389/fgene.2015.00135
43. Desguerre, I, Hully, M, Rio, M, and Nabbout, R. Mitochondrial disorders and epilepsy. Rev Neurol. (2014) 170:375–80. doi: 10.1016/j.neurol.2014.03.010
44. Li, H, Wang, W, Han, X, Zhang, Y, Dai, L, Xu, M, et al. Clinical attributes and electroencephalogram analysis of patients with varying Alpers’ syndrome genotypes. Front Pharmacol. (2021) 12:669516. doi: 10.3389/fphar.2021.669516
45. Saneto, RP, Cohen, BH, Copeland, WC, and Naviaux, RK. Alpers-huttenlocher syndrome. Pediatr Neurol. (2013) 48:167–78. doi: 10.1016/j.pediatrneurol.2012.09.014
46. van Westrhenen, A, Cats, EA, van den Munckhof, B, van der Salm, SM, Teunissen, NW, Ferrier, CH, et al. Specific EEG markers in POLG1 Alpers’ syndrome. Clin Neurophysiol. (2018) 129:2127–31. doi: 10.1016/j.clinph.2018.07.016
47. Melani, F, Zelmann, R, Dubeau, F, and Gotman, J. Occurrence of scalp-fast oscillations among patients with different spiking rate and their role as epileptogenicity marker. Epilepsy Res. (2013) 106:345–56. doi: 10.1016/j.eplepsyres.2013.06.003
48. Gordon, N. Alpers syndrome: progressive neuronal degeneration of children with liver disease. Dev Med Child Neurol. (2006) 48:1001–3. doi: 10.1111/j.1469-8749.2006.tb01275.x
49. Saneto, RP, Friedman, SD, and Shaw, DW. Neuroimaging of mitochondrial disease. Mitochondrion. (2008) 8:396–413. doi: 10.1016/j.mito.2008.05.003
50. James, AM, Sheard, PW, Wei, YH, and Murphy, MP. Decreased ATP synthesis is phenotypically expressed during increased energy demand in fibroblasts containing mitochondrial tRNA mutations: implications for neurodegenerative and mitochondrial DNA diseases. Eur J Biochem. (1999) 259:462–9. doi: 10.1046/j.1432-1327.1999.00066.x
51. Finsterer, J. Pharmacotherapeutic management of epilepsy in MERRF syndrome. Expert Opin Pharmacother. (2019) 20:1289–97. doi: 10.1080/14656566.2019.1609941
52. Finsterer, J, and Zarrouk-Mahjoub, S. Management of epilepsy in MERRF syndrome. Seizure. (2017) 50:166–70. doi: 10.1016/j.seizure.2017.06.010
53. Sitburana, O, Witoonpanich, R, Phudhichareonrat, S, Lertrit, P, and Supavilai, R. Seizures in myoclonic epilepsy with ragged-red fibers detected by DNA analysis: a case report. J Med Assoc Thailand= Chotmaihet Thangphaet. (2001) 84:1051–5.
54. Catteruccia, M, Sauchelli, D, Della Marca, G, Primiano, G, Cuccagna, C, Bernardo, D, et al. “Myo-cardiomyopathy” is commonly associated with the A8344G “MERRF” mutation. J Neurol. (2015) 262:701–10. doi: 10.1007/s00415-014-7632-0
55. Ito, S, Shirai, W, Asahina, M, and Hattori, T. Clinical and brain MR imaging features focusing on the brain stem and cerebellum in patients with myoclonic epilepsy with ragged-red fibers due to mitochondrial A8344G mutation. Am J Neuroradiol. (2008) 29:392–5. doi: 10.3174/ajnr.A0865
56. Fiedorczuk, K, and Sazanov, LA. Mammalian mitochondrial complex I structure and disease-causing mutations. Trends Cell Biol. (2018) 28:835–67. doi: 10.1016/j.tcb.2018.06.006
57. Li, T-R, Wang, Q, and Liu, M-M. Lv R-J: A Chinese family with adult-onset leigh-like syndrome caused by the heteroplasmic m. 10191T> C mutation in the mitochondrial MTND3 gene. Front Neurol. (2019) 10:347. doi: 10.3389/fneur.2019.00347
58. Na, J-H, Lee, MJ, Lee, CH, and Lee, Y-M. Association between epilepsy and leigh syndrome with MT-ND3 mutation, particularly the m. 10191T> C point mutation. Front Neurol. (2021) 12:752467. doi: 10.3389/fneur.2021.752467
59. Nesbitt, V, Morrison, PJ, Crushell, E, Donnelly, DE, Alston, CL, He, L, et al. The clinical spectrum of the m. 10191T> C mutation in complex I-deficient Leigh syndrome. Dev Med Child Neurol. (2012) 54:500–6. doi: 10.1111/j.1469-8749.2012.04224.x
60. Werner, KG, Morel, CF, Kirton, A, Benseler, SM, Shoffner, JM, Addis, JB, et al. Rolandic mitochondrial encephalomyelopathy and MT-ND3 mutations. Pediatr Neurol. (2009) 41:27–33. doi: 10.1016/j.pediatrneurol.2009.02.010
61. Mezuki, S, Fukuda, K, Matsushita, T, Fukushima, Y, and Matsuo, R. Goto Y-i, Yasukawa T, Uchiumi T, Kang D, Kitazono T: isolated and repeated stroke-like episodes in a middle-aged man with a mitochondrial ND3 T10158C mutation: a case report. BMC Neurol. (2017) 17:1–5. doi: 10.1186/s12883-017-1001-4
62. Fu, X-L, Zhou, X-X, Shi, Z, and Zheng, W-C. Adult-onset mitochondrial encephalopathy in association with the MT-ND3 T10158C mutation exhibits unique characteristics: A case report. World J Clin Cases. (2019) 7:1066–72. doi: 10.12998/wjcc.v7.i9.1066
63. Blok, M, Spruijt, L, De Coo, I, Schoonderwoerd, K, Hendrickx, A, and Smeets, H. Mutations in the ND5 subunit of complex I of the mitochondrial DNA are a frequent cause of oxidative phosphorylation disease. J Med Genet. (2007) 44:e74–4. doi: 10.1136/jmg.2006.045716
64. Finsterer, J, and Hayman, J. Mitochondrial encephalopathy, lactic acidosis and stroke-like episodes/Leigh overlap syndrome due to variant m. 13513G> A in MT-ND5. Cureus. (2022) 14:e24746. doi: 10.7759/cureus.24746
65. Dermaut, B, Seneca, S, Dom, L, Smets, K, Ceulemans, L, Smet, J, et al. Progressive myoclonic epilepsy as an adult-onset manifestation of Leigh syndrome due to m. 14487T> C. J Neurol Neurosurg Psychiatry. (2010) 81:90–3. doi: 10.1136/jnnp.2008.157354
66. Vilain, C, Rens, C, Aeby, A, Balériaux, D, Van Bogaert, P, Remiche, G, et al. A novel NDUFV1 gene mutation in complex I deficiency in consanguineous siblings with brainstem lesions and Leigh syndrome. Clin Genet. (2012) 82:264–70. doi: 10.1111/j.1399-0004.2011.01743.x
67. Kiss, S, Christodoulou, J, Thorburn, DR, Freeman, JL, Kornberg, AJ, Mandelstam, S, et al. A cryptic pathogenic NDUFV1 variant identified by RNA-seq in a patient with normal complex I activity in muscle and transient magnetic resonance imaging changes. Am J Med Genet A. (2023) 191:1599–606. doi: 10.1002/ajmg.a.63170
68. Bindu, PS, Sonam, K, Govindaraj, P, Govindaraju, C, Chiplunkar, S, Nagappa, M, et al. Outcome of epilepsy in patients with mitochondrial disorders: phenotype genotype and magnetic resonance imaging correlations. Clin Neurol Neurosurg. (2018) 164:182–9. doi: 10.1016/j.clineuro.2017.12.010
69. Tuppen, HA, Hogan, VE, He, L, Blakely, EL, Worgan, L, Al-Dosary, M, et al. The p. M292T NDUFS2 mutation causes complex I-deficient Leigh syndrome in multiple families. Brain. (2010) 133:2952–63. doi: 10.1093/brain/awq232
70. Yatsuka, Y, Kishita, Y, Formosa, LE, Shimura, M, Nozaki, F, Fujii, T, et al. A homozygous variant in NDUFA8 is associated with developmental delay, microcephaly, and epilepsy due to mitochondrial complex I deficiency. Clin Genet. (2020) 98:155–65. doi: 10.1111/cge.13773
71. Borna, NN, Kishita, Y, Shimura, M, Murayama, K, Ohtake, A, and Okazaki, Y. Identification of a novel MT-ND3 variant and restoring mitochondrial function by allotopic expression of MT-ND3 gene. Mitochondrion. (2024) 76:101858. doi: 10.1016/j.mito.2024.101858
72. Fullerton, M, McFarland, R, Taylor, RW, and Alston, CL. The genetic basis of isolated mitochondrial complex II deficiency. Mol Genet Metab. (2020) 131:53–65. doi: 10.1016/j.ymgme.2020.09.009
73. Ürey, BC, Ceylan, AC, Çavdarlı, B, Çıtak Kurt, AN, and Köylü, OK. YÜrek B, Kasapkara CidS: two patients diagnosed as succinate dehydrogenase deficiency: case report. Molecular Syndromol. (2023) 14:171–4. doi: 10.1159/000527538
74. Horváth, R, Abicht, A, Holinski-Feder, E, Laner, A, Gempel, K, Prokisch, H, et al. Leigh syndrome caused by mutations in the flavoprotein (Fp) subunit of succinate dehydrogenase (SDHA). J Neurol Neurosurg Psychiatry. (2006) 77:74–6. doi: 10.1136/jnnp.2005.067041
75. Renkema, GH, Wortmann, SB, Smeets, RJ, Venselaar, H, Antoine, M, Visser, G, et al. SDHA mutations causing a multisystem mitochondrial disease: novel mutations and genetic overlap with hereditary tumors. Eur J Hum Genet. (2015) 23:202–9. doi: 10.1038/ejhg.2014.80
76. Kaur, P, Sharma, S, Kadavigere, R, and Girisha, KM. Novel variant p. (Ala102Thr) in SDHB causes mitochondrial complex II deficiency: case report and review of the literature. Ann Hum Genet. (2020) 84:345–51. doi: 10.1111/ahg.12377
77. Lin, S, Fasham, J, Al-Hijawi, F, Qutob, N, Gunning, A, Leslie, JS, et al. Consolidating biallelic SDHD variants as a cause of mitochondrial complex II deficiency. Eur J Hum Genet. (2021) 29:1570–6. doi: 10.1038/s41431-021-00887-w
78. Ohlenbusch, A, Edvardson, S, Skorpen, J, Bjornstad, A, Saada, A, Elpeleg, O, et al. Leukoencephalopathy with accumulated succinate is indicative of SDHAF1 related complex II deficiency. Orphanet J Rare Dis. (2012) 7:69–5. doi: 10.1186/1750-1172-7-69
79. Nimmo, GA, Ejaz, R, Cordeiro, D, Kannu, P, and Mercimek-Andrews, S. Riboflavin transporter deficiency mimicking mitochondrial myopathy caused by complex II deficiency. Am J Med Genet A. (2018) 176:399–403. doi: 10.1002/ajmg.a.38530
80. Smith, PM, Fox, JL, and Winge, DR. Reprint of: biogenesis of the cytochrome bc1 complex and role of assembly factors. Biochimica et Biophysica Acta (BBA)-Bioenergetics. (2012) 1817:872–82. doi: 10.1016/j.bbabio.2012.03.003
81. Zhang, X, Chen, G, Lu, Y, Liu, J, Fang, M, Luo, J, et al. Association of mitochondrial letm1 with epileptic seizures. Cereb Cortex. (2014) 24:2533–40. doi: 10.1093/cercor/bht118
82. Meunier, B, Fisher, N, Ransac, S, Mazat, J-P, and Brasseur, G. Respiratory complex III dysfunction in humans and the use of yeast as a model organism to study mitochondrial myopathy and associated diseases. Biochimica Et Biophysica Acta (BBA)-Bioenergetics. (2013) 1827:1346–61. doi: 10.1016/j.bbabio.2012.11.015
83. Emmanuele, V, Sotiriou, E, Rios, PG, Ganesh, J, Ichord, R, Foley, AR, et al. A novel mutation in the mitochondrial DNA cytochrome b gene (MTCYB) in a patient with mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes syndrome. J Child Neurol. (2013) 28:236–42. doi: 10.1177/0883073812445787
84. Rahman, MM, and Fatema, K. Genetic diagnosis in children with epilepsy and developmental disorders by targeted gene panel analysis in a developing country. J Epilepsy Res. (2021) 11:22–31. doi: 10.14581/jer.21004
85. De Coo, I, Renier, W, Ruitenbeek, W, Ter Laak, H, Bakker, M, Schägger, H, et al. A 4–base pair deletion in the mitochondrial cytochrome b gene associated with parkinsonism/MELAS overlap syndrome. Ann Neurol: Official J American Neurolog Assoc Child Neurol Society. (1999) 45:130–3. doi: 10.1002/1531-8249(199901)45:1<130::AID-ART21>3.0.CO;2-Z
86. Hikmat, O, Isohanni, P, Keshavan, N, Ferla, MP, Fassone, E, Abbott, MA, et al. Expanding the phenotypic spectrum of BCS1L-related mitochondrial disease. Ann Clin Transl Neurol. (2021) 8:2155–65. doi: 10.1002/acn3.51470
87. Fernandez-Vizarra, E, Bugiani, M, Goffrini, P, Carrara, F, Farina, L, Procopio, E, et al. Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy. Hum Mol Genet. (2007) 16:1241–52. doi: 10.1093/hmg/ddm072
88. Tuppen, HA, Fehmi, J, Czermin, B, Goffrini, P, Meloni, F, Ferrero, I, et al. Long-term survival of neonatal mitochondrial complex III deficiency associated with a novel BCS1L gene mutation. Mol Genet Metab. (2010) 100:345–8. doi: 10.1016/j.ymgme.2010.04.010
89. Koch, J, Freisinger, P, Feichtinger, RG, Zimmermann, FA, Rauscher, C, Wagentristl, HP, et al. Mutations in TTC19: expanding the molecular, clinical and biochemical phenotype. Orphanet J Rare Dis. (2015) 10:1–12. doi: 10.1186/s13023-015-0254-5
90. Feichtinger, RG, Brunner-Krainz, M, Alhaddad, B, Wortmann, SB, Kovacs-Nagy, R, Stojakovic, T, et al. Combined respiratory chain deficiency and UQCC2 mutations in neonatal Encephalomyopathy: defective Supercomplex assembly in complex III deficiencies. Oxidative Med Cell Longev. (2017) 2017:7202589. doi: 10.1155/2017/7202589
91. Miyake, N, Yano, S, Sakai, C, Hatakeyama, H, Matsushima, Y, Shiina, M, et al. Mitochondrial complex III deficiency caused by a homozygous UQCRC2 mutation presenting with neonatal-onset recurrent metabolic decompensation. Hum Mutat. (2013) 34:446–52. doi: 10.1002/humu.22257
92. Argov, Z, Bank WJMaris, J, Eleff, S, Kennaway, NG, Olson, RE, et al. Treatment of mitochondrial myopathy due to complex III deficiency with vitamins K3 and C: a 31P-NMR follow-up study. Ann Neurol: Official J American Neurolog Assoc Child Neurol Society. (1986) 19:598–602. doi: 10.1002/ana.410190615
93. Timón-Gómez, A, Nývltová, E, Abriata, LA, Vila, AJ, Hosler, J, and Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: recent developments. Semin Cell Dev Biol. (2018) 76:163–78. doi: 10.1016/j.semcdb.2017.08.055
94. Brischigliaro, M, and Zeviani, M. Cytochrome c oxidase deficiency. Biochim Biophys Acta Bioenerg. (2021) 1862:148335. doi: 10.1016/j.bbabio.2020.148335
95. Khan, TR, Leprince, I, Messahel, S, Minassian, BA, and Kayani, S. Natural history of SURF1 deficiency: A retrospective chart review. Pediatr Neurol. (2023) 140:40–6. doi: 10.1016/j.pediatrneurol.2022.12.002
96. Wedatilake, Y, Brown, RM, McFarland, R, Yaplito-Lee, J, Morris, AA, Champion, M, et al. SURF1 deficiency: a multi-Centre natural history study. Orphanet J Rare Dis. (2013) 8:1–13. doi: 10.1186/1750-1172-8-96
97. Sue, C, Karadimas, C, Checcarelli, N, Tanji, K, Papadopoulou, L, Pallotti, F, et al. Differential features of patients with mutations in two COX assembly genes, SURF-1 and SCO2. Ann Neurol. (2000) 47:589–95. doi: 10.1002/1531-8249(200005)47:5<589::AID-ANA6>3.0.CO;2-D
98. Papadopoulou, LC, Sue, CM, Davidson, MM, Tanji, K, Nishino, I, Sadlock, JE, et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat Genet. (1999) 23:333–7. doi: 10.1038/15513
99. Valnot, I, von Kleist-Retzow, J-C, Barrientos, A, Gorbatyuk, M, Taanman, J-W, Mehaye, B, et al. A mutation in the human heme A: farnesyltransferase gene (COX10) causes cytochrome c oxidase deficiency. Hum Mol Genet. (2000) 9:1245–9. doi: 10.1093/hmg/9.8.1245
100. de Oliveira, MG, Tengan, C, Micheletti, C, de Macedo, PR, Cernach, MCSP, Cavole, TR, et al. A novel variant in the COX15 gene causing a fatal infantile cardioencephalomyopathy: A case report with clinical and molecular review. Eur J Med Genet. (2021) 64:104195. doi: 10.1016/j.ejmg.2021.104195
101. Jaksch, M, Hofmann, S, Kleinle, S, Liechti-Gallati, S, Pongratz, DE, Müller-Höcker, J, et al. A systematic mutation screen of 10 nuclear and 25 mitochondrial candidate genes in 21 patients with cytochrome c oxidase (COX) deficiency shows tRNA (Ser)(UCN) mutations in a subgroup with syndromal encephalopathy. J Med Genet. (1998) 35:895–900. doi: 10.1136/jmg.35.11.895
102. Debray, F-G, Seneca, S, Gonce, M, Vancampenhaut, K, Bianchi, E, Boemer, F, et al. Mitochondrial encephalomyopathy with cytochrome c oxidase deficiency caused by a novel mutation in the MTCO1 gene. Mitochondrion. (2014) 17:101–5. doi: 10.1016/j.mito.2014.06.003
103. Wang, W, Sun, Y, Lin, Y, Xu, X, Zhao, D, Ji, K, et al. A novel nonsense variant in MT-CO3 causes MELAS syndrome. Neuromuscul Disord. (2021) 31:558–65. doi: 10.1016/j.nmd.2021.02.020
104. Pillai, NR, AlDhaheri, NS, Ghosh, R, Lim, J, Streff, H, Nayak, A, et al. Biallelic variants in COX4I1 associated with a novel phenotype resembling Leigh syndrome with developmental regression, intellectual disability, and seizures. Am J Med Genet A. (2019) 179:2138–43. doi: 10.1002/ajmg.a.61288
105. Martinelli, D, Catteruccia, M, Piemonte, F, Pastore, A, Tozzi, G, Dionisi-Vici, C, et al. EPI-743 reverses the progression of the pediatric mitochondrial disease—genetically defined Leigh syndrome. Mol Genet Metab. (2012) 107:383–8. doi: 10.1016/j.ymgme.2012.09.007
106. Rak, M, Bénit, P, Chrétien, D, Bouchereau, J, Schiff, M, El-Khoury, R, et al. Mitochondrial cytochrome c oxidase deficiency. Clin Sci. (2016) 130:393–407. doi: 10.1042/CS20150707
107. Ling, Q, Rioux, M, Hu, Y, Lee, M, and Gray, SJ. Adeno-associated viral vector serotype 9-based gene replacement therapy for SURF1-related Leigh syndrome. Mol Ther Methods Clin Dev. (2021) 23:158–68. doi: 10.1016/j.omtm.2021.09.001
108. Zech, M, Kopajtich, R, Steinbrücker, K, Bris, C, Gueguen, N, Feichtinger, RG, et al. Variants in mitochondrial ATP synthase cause variable neurologic phenotypes. Ann Neurol. (2022) 91:225–37. doi: 10.1002/ana.26293
109. Jonckheere, AI, Smeitink, JA, and Rodenburg, RJ. Mitochondrial ATP synthase: architecture, function and pathology. J Inherit Metab Dis. (2012) 35:211–25. doi: 10.1007/s10545-011-9382-9
110. Stendel, C, Neuhofer, C, Floride, E, Yuqing, S, Ganetzky, RD, Park, J, et al. Delineating MT-ATP6-associated disease: from isolated neuropathy to early onset neurodegeneration. Neurology Genetics. (2020) 6:e393. doi: 10.1212/NXG.0000000000000393
111. Licchetta, L, Ferri, L, La Morgia, C, Zenesini, C, Caporali, L, Lucia Valentino, M, et al. Epilepsy in MT-ATP6-related mils/NARP: correlation of elettroclinical features with heteroplasmy. Ann Clin Trans Neurol. (2021) 8:704–10. doi: 10.1002/acn3.51259
112. Mäkelä-Bengs, P, Suomalainen, A, Majander, A, Rapola, J, Kalimo, H, Nuutila, A, et al. Correlation between the clinical symptoms and the proportion of mitochondrial DNA carrying the 8993 point mutation in the NARP syndrome. Pediatr Res. (1995) 37:634–9. doi: 10.1203/00006450-199505000-00014
113. Knight, KM, Shelkowitz, E, Larson, AA, Mirsky, DM, Wang, Y, Chen, T, et al. The mitochondrial DNA variant m. 9032T> C in MT-ATP6 encoding p.(Leu169Pro) causes a complex mitochondrial neurological syndrome. Mitochondrion. (2020) 55:8–13. doi: 10.1016/j.mito.2020.08.009
114. Ganetzky, RD, Stendel, C, McCormick, EM, Zolkipli-Cunningham, Z, Goldstein, AC, Klopstock, T, et al. MT-ATP6 mitochondrial disease variants: phenotypic and biochemical features analysis in 218 published cases and cohort of 14 new cases. Hum Mutat. (2019) 40:499–515. doi: 10.1002/humu.23723
115. Morava, E, Rodenburg, RJ, Hol, F, De Vries, M, Janssen, A, Van Den Heuvel, L, et al. Clinical and biochemical characteristics in patients with a high mutant load of the mitochondrial T8993G/C mutations. Am J Med Genet A. (2006) 140:863–8. doi: 10.1002/ajmg.a.31194
116. Gurses, C, Azakli, H, Alptekin, A, Cakiris, A, Abaci, N, Arikan, M, et al. Mitochondrial DNA profiling via genomic analysis in mesial temporal lobe epilepsy patients with hippocampal sclerosis. Gene. (2014) 538:323–7. doi: 10.1016/j.gene.2014.01.030
117. Mkaouar-Rebai, E, Kammoun, F, Chamkha, I, Kammoun, N, Hsairi, I, Triki, C, et al. A de novo mutation in the adenosine triphosphatase (ATPase) 8 gene in a patient with mitochondrial disorder. J Child Neurol. (2010) 25:770–5. doi: 10.1177/0883073809344351
118. De Meirleir, L, Seneca, S, Lissens, W, De Clercq, I, Eyskens, F, Gerlo, E, et al. Respiratory chain complex V deficiency due to a mutation in the assembly gene ATP12. J Med Genet. (2004) 41:120–4. doi: 10.1136/jmg.2003.012047
119. Torraco, A, Verrigni, D, Rizza, T, Meschini, MC, Vazquez-Memije, ME, Martinelli, D, et al. TMEM70: a mutational hot spot in nuclear ATP synthase deficiency with a pivotal role in complex V biogenesis. Neurogenetics. (2012) 13:375–86. doi: 10.1007/s10048-012-0343-8
120. Sgarbi, G, Casalena, GA, Baracca, A, Lenaz, G, DiMauro, S, and Solaini, G. Human NARP mitochondrial mutation metabolism corrected with α-ketoglutarate/aspartate: a potential new therapy. Arch Neurol. (2009) 66:951–7. doi: 10.1001/archneurol.2009.134
121. Mattiazzi, M, Vijayvergiya, C, Gajewski, CD, DeVivo, DC, Lenaz, G, Wiedmann, M, et al. The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum Mol Genet. (2004) 13:869–79. doi: 10.1093/hmg/ddh103
122. Alcázar-Fabra, M, Trevisson, E, and Brea-Calvo, G. Clinical syndromes associated with coenzyme Q10 deficiency. Essays Biochem. (2018) 62:377–98. doi: 10.1042/EBC20170107
123. Desbats, MA, Lunardi, G, Doimo, M, Trevisson, E, and Salviati, L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J Inherit Metab Dis. (2015) 38:145–56. doi: 10.1007/s10545-014-9749-9
124. Alcázar-Fabra, M, Rodríguez-Sánchez, F, Trevisson, E, and Brea-Calvo, G. Primary coenzyme Q deficiencies: A literature review and online platform of clinical features to uncover genotype-phenotype correlations. Free Radic Biol Med. (2021) 167:141–80. doi: 10.1016/j.freeradbiomed.2021.02.046
125. Laugwitz, L, Seibt, A, Herebian, D, Peralta, S, Kienzle, I, Buchert, R, et al. Human COQ4 deficiency: delineating the clinical, metabolic and neuroimaging phenotypes. J Med Genet. (2022) 59:878–87. doi: 10.1136/jmedgenet-2021-107729
126. Brea-Calvo, G, Haack, TB, Karall, D, Ohtake, A, Invernizzi, F, Carrozzo, R, et al. COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am J Hum Genet. (2015) 96:309–17. doi: 10.1016/j.ajhg.2014.12.023
127. Lu, M, Zhou, Y, Wang, Z, Xia, Z, Ren, J, and Guo, Q. Clinical phenotype, in silico and biomedical analyses, and intervention for an east Asian population-specific c. 370G> A (p. G124S) COQ4 mutation in a Chinese family with CoQ10 deficiency-associated Leigh syndrome. J Hum Genet. (2019) 64:297–304. doi: 10.1038/s10038-019-0563-y
128. Chung, WK, Martin, K, Jalas, C, Braddock, SR, Juusola, J, Monaghan, KG, et al. Mutations in COQ4, an essential component of coenzyme Q biosynthesis, cause lethal neonatal mitochondrial encephalomyopathy. J Med Genet. (2015) 52:627–35. doi: 10.1136/jmedgenet-2015-103140
129. Traschütz, A, Schirinzi, T, Laugwitz, L, Murray, NH, Bingman, CA, Reich, S, et al. Clinico-genetic, imaging and molecular delineation of COQ8A-ataxia: a multicenter study of 59 patients. Ann Neurol. (2020) 88:251–63. doi: 10.1002/ana.25751
130. Arntsen, V, Sand, T, Hikmat, O, Samsonsen, C, Bindoff, LA, and Brodtkorb, E. A characteristic occipital epileptiform EEG pattern in ADCK3-related mitochondrial disease. Epileptic Disord. (2021) 23:281–90. doi: 10.1684/epd.2021.1269
131. Horvath, R, Czermin, B, Gulati, S, Demuth, S, Houge, G, Pyle, A, et al. Adult-onset cerebellar ataxia due to mutations in CABC1/ADCK3. J Neurol Neurosurg Psychiatry. (2012) 83:174–8. doi: 10.1136/jnnp-2011-301258
132. Mignot, C, Apartis, E, Durr, A, Marques Lourenço, C, Charles, P, Devos, D, et al. Phenotypic variability in ARCA2 and identification of a core ataxic phenotype with slow progression. Orphanet J Rare Dis. (2013) 8:1–10. doi: 10.1186/1750-1172-8-173
133. Uccella, S, Pisciotta, L, Severino, M, Bertini, E, Giacomini, T, Zanni, G, et al. Photoparoxysmal response in ADCK3 autosomal recessive ataxia: a case report and literature review. Epileptic Disord. (2021) 23:153–60. doi: 10.1684/epd.2021.1243
134. Quinzii, C, Naini, A, Salviati, L, Trevisson, E, Navas, P, DiMauro, S, et al. A mutation in Para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet. (2006) 78:345–9. doi: 10.1086/500092
135. Helbig, KL, Farwell Hagman, KD, Shinde, DN, Mroske, C, Powis, Z, Li, S, et al. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med. (2016) 18:898–905. doi: 10.1038/gim.2015.186
136. Herebian, D, Seibt, A, Smits, SH, Rodenburg, RJ, Mayatepek, E, and Distelmaier, F. 4-Hydroxybenzoic acid restores CoQ10 biosynthesis in human COQ 2 deficiency. Annals of clinical and translational neurol. (2017) 4:902–8. doi: 10.1002/acn3.486
137. Doimo, M, Trevisson, E, Airik, R, Bergdoll, M, Santos-Ocaña, C, Hildebrandt, F, et al. Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. (2014) 1842:1–6. doi: 10.1016/j.bbadis.2013.10.007
138. Falk, MJ, Polyak, E, Zhang, Z, Peng, M, King, R, Maltzman, JS, et al. Probucol ameliorates renal and metabolic sequelae of primary CoQ deficiency in Pdss2 mutant mice. EMBO Mol Med. (2011) 3:410–27. doi: 10.1002/emmm.201100149
139. Santucci, R, Sinibaldi, F, Cozza, P, Polticelli, F, and Fiorucci, L. Cytochrome c: an extreme multifunctional protein with a key role in cell fate. Int J Biol Macromol. (2019) 136:1237–46. doi: 10.1016/j.ijbiomac.2019.06.180
140. Zhao, S, Aviles, ER Jr, and Fujikawa, DG. Nuclear translocation of mitochondrial cytochrome c, lysosomal cathepsins B and D, and three other death-promoting proteins within the first 60 minutes of generalized seizures. J Neurosci Res. (2010) 88:1727–37. doi: 10.1002/jnr.22338
141. Luan, G, Zhao, Y, Zhai, F, Chen, Y, and Li, T. Ketogenic diet reduces Smac/diablo and cytochrome c release and attenuates neuronal death in a mouse model of limbic epilepsy. Brain Res Bull. (2012) 89:79–85. doi: 10.1016/j.brainresbull.2012.07.002
142. Lou, X, Zhou, Y, Liu, Z, Xie, Y, Zhang, L, Zhao, S, et al. De novo frameshift variant in MT-ND1 causes a mitochondrial complex I deficiency associated with MELAS syndrome. Gene. (2023) 860:147229. doi: 10.1016/j.gene.2023.147229
143. Ng, YS, Lax, NZ, Maddison, P, Alston, CL, Blakely, EL, Hepplewhite, PD, et al. MT-ND5 mutation exhibits highly variable neurological manifestations at low mutant load. EBioMedicine. (2018) 30:86–93. doi: 10.1016/j.ebiom.2018.02.010
144. Mayr, JA, Bodamer, O, Haack, TB, Zimmermann, FA, Madignier, F, Prokisch, H, et al. Heterozygous mutation in the X chromosomal NDUFA1 gene in a girl with complex I deficiency. Mol Genet Metab. (2011) 103:358–61. doi: 10.1016/j.ymgme.2011.04.010
145. Hoefs, SJ, Dieteren, CE, Distelmaier, F, Janssen, RJ, Epplen, A, Swarts, HG, et al. NDUFA2 complex I mutation leads to Leigh disease. Am J Hum Genet. (2008) 82:1306–15. doi: 10.1016/j.ajhg.2008.05.007
146. Nesti, C, Ticci, C, Rubegni, A, Doccini, S, Scaturro, G, Vetro, A, et al. Additive effect of DNAJC30 and NDUFA9 mutations causing Leigh syndrome. J Neurol. (2023) 270:3266–9. doi: 10.1007/s00415-023-11673-7
147. Schuelke, M, Smeitink, J, Mariman, E, Loeffen, J, Plecko, B, Trijbels, F, et al. Mutant NDUFV1 subunit of mitochondrial complex I causes leukodystrophy and myoclonic epilepsy. Nat Genet. (1999) 21:260–1. doi: 10.1038/6772
148. Ortigoza-Escobar, JD, Oyarzabal, A, Montero, R, Artuch, R, Jou, C, Jiménez, C, et al. Ndufs4 related Leigh syndrome: A case report and review of the literature. Mitochondrion. (2016) 28:73–8. doi: 10.1016/j.mito.2016.04.001
149. Varlamov, DA, Kudin, AP, Vielhaber, S, Schröder, R, Sassen, R, Becker, A, et al. Metabolic consequences of a novel missense mutation of the mtDNA CO I gene. Hum Mol Genet. (2002) 11:1797–805. doi: 10.1093/hmg/11.16.1797
150. Jennions, E, Olsson-Engman, M, Visuttijai, K, Wiksell, Å, Fluriach Dominguez, N, Kollberg, G, et al. A novel homozygous pathogenic missense variant in COX6B1: further delineation of the phenotype. Am J Med Genet A. (2024) 194:e63783. doi: 10.1002/ajmg.a.63783
151. Hallmann, K, Kudin, AP, Zsurka, G, Kornblum, C, Reimann, J, Stüve, B, et al. Loss of the smallest subunit of cytochrome c oxidase, COX8A, causes Leigh-like syndrome and epilepsy. Brain. (2016) 139:338–45. doi: 10.1093/brain/awv357
152. Pitceathly, RD, Rahman, S, Wedatilake, Y, Polke, JM, Cirak, S, Foley, AR, et al. NDUFA4 mutations underlie dysfunction of a cytochrome c oxidase subunit linked to human neurological disease. Cell Rep. (2013) 3:1795–805. doi: 10.1016/j.celrep.2013.05.005
153. van der Ven, AT, Cabrera-Orefice, A, Wente, I, Feichtinger, RG, Tsiakas, K, Weiss, D, et al. Expanding the phenotypic and biochemical spectrum of NDUFAF3-related mitochondrial disease. Mol Genet Metab. (2023) 140:107675. doi: 10.1016/j.ymgme.2023.107675
154. Bianciardi, L, Imperatore, V, Fernandez-Vizarra, E, Lopomo, A, Falabella, M, Furini, S, et al. Exome sequencing coupled with mRNA analysis identifies NDUFAF6 as a Leigh gene. Mol Genet Metab. (2016) 119:214–22. doi: 10.1016/j.ymgme.2016.09.001
155. Barbato, A, Gori, G, Sacchini, M, Pochiero, F, Bargiacchi, S, Traficante, G, et al. Endocrinological features and epileptic encephalopathy in COX deficiency due to SCO1 mutations: case series and review of literature. Endocr. Connect. (2024) 13:e240221. doi: 10.1530/EC-24-0221
156. Mansour, H, Sabbagh, S, Bizzari, S, El-Hayek, S, Chouery, E, Gambarini, A, et al. The Lebanese allele in the PET100 gene: report on two new families with cytochrome c oxidase deficiency. J Pediatr Genet. (2019) 8:172–8. doi: 10.1055/s-0039-1685172
157. Wei, X, Du, M, Li, D, Wen, S, Xie, J, Li, Y, et al. Mutations in FASTKD2 are associated with mitochondrial disease with multi-OXPHOS deficiency. Hum Mutat. (2020) 41:961–72. doi: 10.1002/humu.23985
158. Shah, R, and Balasubramaniam, S. Clinical phenotype of FASTKD2 mutation. J Pediatr Neurosci. (2021) 16:319–22. doi: 10.4103/jpn.JPN_199_20
159. Magner, M, Dvorakova, V, Tesarova, M, Mazurova, S, Hansikova, H, Zahorec, M, et al. TMEM70 deficiency: long-term outcome of 48 patients. J Inherit Metab Dis. (2015) 38:417–26. doi: 10.1007/s10545-014-9774-8
160. Bonnefont, JP, Bastin, J, Laforêt, P, Aubey, F, Mogenet, A, Romano, S, et al. Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin Pharmacol Ther. (2010) 88:101–8. doi: 10.1038/clpt.2010.55
161. Pastore, A, Petrillo, S, Tozzi, G, Carrozzo, R, Martinelli, D, Dionisi-Vici, C, et al. Glutathione: a redox signature in monitoring EPI-743 therapy in children with mitochondrial encephalomyopathies. Mol Genet Metab. (2013) 109:208–14. doi: 10.1016/j.ymgme.2013.03.011
162. Mizuguchi, Y, Hatakeyama, H, Sueoka, K, Tanaka, M, and Goto, YI. Low dose resveratrol ameliorates mitochondrial respiratory dysfunction and enhances cellular reprogramming. Mitochondrion. (2017) 34:43–8. doi: 10.1016/j.mito.2016.12.006
163. Madsen, KL, Buch, AE, Cohen, BH, Falk, MJ, Goldsberry, A, Goldstein, A, et al. Safety and efficacy of omaveloxolone in patients with mitochondrial myopathy: MOTOR trial. Neurology. (2020) 94:e687–98. doi: 10.1212/WNL.0000000000008861
164. Khan, NA, Auranen, M, Paetau, I, Pirinen, E, Euro, L, Forsström, S, et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol Med. (2014) 6:721–31. doi: 10.1002/emmm.201403943
165. Lee, CF, Caudal, A, Abell, L, Nagana Gowda, GA, and Tian, R. Targeting NAD(+) metabolism as interventions for mitochondrial disease. Sci Rep. (2019) 9:3073. doi: 10.1038/s41598-019-39419-4
166. van de Weijer, T, Phielix, E, Bilet, L, Williams, EG, Ropelle, ER, Bierwagen, A, et al. Evidence for a direct effect of the NAD+ precursor acipimox on muscle mitochondrial function in humans. Diabetes. (2015) 64:1193–201. doi: 10.2337/db14-0667
167. Pirinen, E, Auranen, M, Khan, NA, Brilhante, V, Urho, N, Pessia, A, et al. Niacin cures systemic NAD(+) deficiency and improves muscle performance in adult-onset mitochondrial myopathy. Cell Metab. (2020) 31:1078–1090.e5. doi: 10.1016/j.cmet.2020.04.008
168. Katsyuba, E, Mottis, A, Zietak, M, De Franco, F, van der Velpen, V, Gariani, K, et al. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature. (2018) 563:354–9. doi: 10.1038/s41586-018-0645-6
169. Patgiri, A, Skinner, OS, Miyazaki, Y, Schleifer, G, Marutani, E, Shah, H, et al. An engineered enzyme that targets circulating lactate to alleviate intracellular NADH:NAD(+) imbalance. Nat Biotechnol. (2020) 38:309–13. doi: 10.1038/s41587-019-0377-7
170. Johnson, SC, Yanos, ME, Kayser, EB, Quintana, A, Sangesland, M, Castanza, A, et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science. (2013) 342:1524–8. doi: 10.1126/science.1244360
171. Andreux, PA, Blanco-Bose, W, Ryu, D, Burdet, F, Ibberson, M, Aebischer, P, et al. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab. (2019) 1:595–603. doi: 10.1038/s42255-019-0073-4
172. Klopstock, T, Yu-Wai-Man, P, Dimitriadis, K, Rouleau, J, Heck, S, Bailie, M, et al. A randomized placebo-controlled trial of idebenone in Leber's hereditary optic neuropathy. Brain. (2011) 134:2677–86. doi: 10.1093/brain/awr170
173. Zhao, W, Xu, Z, Cao, J, Fu, Q, Wu, Y, Zhang, X, et al. Elamipretide (SS-31) improves mitochondrial dysfunction, synaptic and memory impairment induced by lipopolysaccharide in mice. J Neuroinflammation. (2019) 16:230. doi: 10.1186/s12974-019-1627-9
174. Domínguez-González, C, Madruga-Garrido, M, Mavillard, F, Garone, C, Aguirre-Rodríguez, FJ, Donati, MA, et al. Deoxynucleoside therapy for thymidine kinase 2-deficient myopathy. Ann Neurol. (2019) 86:293–303. doi: 10.1002/ana.25506
175. Saada, A. Insights into deoxyribonucleoside therapy for mitochondrial TK2 deficient mtDNA depletion. EBioMedicine. (2019) 47:14–5. doi: 10.1016/j.ebiom.2019.08.005
176. Russell, S, Bennett, J, Wellman, JA, Chung, DC, Yu, ZF, Tillman, A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. (2017) 390:849–60. doi: 10.1016/S0140-6736(17)31868-8
177. Venkatesh, A, Li, Z, Christiansen, A, Lim, KH, Kach, J, Hufnagel, R, et al. Antisense oligonucleotide mediated increase of OPA1 expression using TANGO technology for the treatment of autosomal dominant optic atrophy. Invest Ophthalmol Vis Sci. (2020) 61:2755.
178. Falabella, M, Minczuk, M, Hanna, MG, Viscomi, C, and Pitceathly, RDS. Gene therapy for primary mitochondrial diseases: experimental advances and clinical challenges. Nat Rev Neurol. (2022) 18:689–98. doi: 10.1038/s41582-022-00715-9
179. Gorman, GS, McFarland, R, Stewart, J, Feeney, C, and Turnbull, DM. Mitochondrial donation: from test tube to clinic. Lancet. (2018) 392:1191–2. doi: 10.1016/S0140-6736(18)31868-3
180. Mok, BY, de Moraes, MH, Zeng, J, Bosch, DE, Kotrys, AV, Raguram, A, et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. (2020) 583:631–7. doi: 10.1038/s41586-020-2477-4
181. Adashi, EY, Rubenstein, DS, Mossman, JA, Schon, EA, and Cohen, IG. Mitochondrial disease: replace or edit? Science. (2021) 373:1200–1. doi: 10.1126/science.abg0491
182. Jeppesen, TD, Schwartz, M, Olsen, DB, Wibrand, F, Krag, T, Dunø, M, et al. Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain. (2006) 129:3402–12. doi: 10.1093/brain/awl149
Keywords: epilepsy, mitochondrial complex, coenzyme Q, cytochrome C, genes
Citation: Zhang X, Zhang B, Tao Z and Liang J (2025) Mitochondrial disease and epilepsy in children. Front. Neurol. 15:1499876. doi: 10.3389/fneur.2024.1499876
Edited by:
Hua-Jun Feng, Massachusetts General Hospital, Harvard Medical School, United StatesReviewed by:
Raffaele Falsaperla, Unità di Pediatria e Emergenza Pediatrica, Policlinico San Marco, ItalyDeepa Krishnakumar, Cambridge University Hospitals NHS Foundation Trust, United Kingdom
Copyright © 2025 Zhang, Zhang, Tao and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianmin Liang, bGlhbmdqbUBqbHUuZWR1LmNu