 Rui Gao
Rui Gao Lihua Gu
Lihua Gu Wenchao Zuo
Wenchao Zuo Pan Wang
Pan Wang- Department of Neurology, Tianjin Huanhu Hospital, Nankai University, Tianjin, China
Background: MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes) is a common subtype of mitochondrial encephalomyopathy. However, few studies have explored the relationship between biochemical markers and prognosis. This study aimed to explore the relationship between clinical and biochemical markers and prognosis of patients with MELAS.
Methods: This was a retrospective single-center study. A total of 39 MELAS patients were followed for an average of 7.3 ± 4.7 (range 1–21 years). All patients underwent detailed demographic registration, neurological examinations, biochemical and mitochondrial DNA analyses, muscle biopsy. Throughout the follow-up period, the modified Rankin Scale (mRS) scores, recurrent strokes rates, and mortality were tracked.
Results: All patients initially presented with stroke-like episodes. Of the 39 subjects who were followed, 8 died, primarily due to acute stroke-like episodes and status epilepticus. Univariate analysis showed a higher risk of mortality in patients with severe lactate elevation compared to those with normal and mildly elevated levels (OR = 5.714, 95% CI 1.086–30.071, p = 0.040). While the absence of anemia was associated with a lower risk of death compared to those with anemia (OR = 0.175, 95% CI 0.033–0.921, p = 0.040). In multivariate analysis, severe lactate elevation (OR = 7.279, 95% CI 1.102–48.086, p = 0.039) and anemia (OR = 0.137, 95% CI 0.021–0.908, p = 0.039) were identified as independent predictors of mortality. MRS scores were categorized as follows: 41% of patients scored 0 to 2, 38.5% scored 3 to 5, and 20.5% had a score of 6 or had died. There was a positive correlation between lactic acid levels and MRS scores (r = 0.460, p = 0.003). In contrast, hemoglobin levels were negatively correlated with MRS scores (r = −0.375, p = 0.015). Furthermore, a positive correlation was observed between MRS scores and the frequency of stroke-like episodes (r = 0.280, p = 0.042).
Conclusion: Our study found that the majority of patients with MELAS had poor clinical outcomes. Anemia and significantly increased lactate levels were identified as indicators of poor prognosis in MELAS. Early intervention may lead to improvements in clinical outcomes.
1 Introduction
Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Episodes (MELAS) is a prevalent maternally inherited mitochondrial disorder. It is a multi-system disorder with a wide range of manifestations, characterized by recurrent stroke-like episodes, progressive intellectual decline, and dementia (1). MELAS is associated with high morbidity and mortality rates. Among patients with the m.3243A > G mutation, earlier central nervous system involvement is often associated with a more severe progression of the disease (2). However, research on the risk factors for mortality in MELAS is limited, primarily focusing on clinical symptoms, with even fewer studies investigating biochemical indicators. For instance, an 8-year follow-up study on clinical indicators and their prognostic implications in a Chinese cohort (3), a 5-year follow-up study in a Japanese cohort (4), and a study examining baseline clinical characteristics, mutation load in urine and blood, and major adverse events in a French cohort (5) have been conducted. We conducted a retrospective follow-up study with a follow-up period of up to 21 years on a cohort of MELAS patients to examine the relationships between clinical and biochemical parameters, cerebrospinal fluid lactate levels, and prognosis.
2 Methods
2.1 Subjects
In this retrospective, single-center study, the cohort included symptomatic MELAS patients treated at Tianjin Huanhu Hospital between January 2003 and January 2023. All participants met the diagnostic criteria established in previously published studies (6–8). All patients underwent full-length mitochondrial DNA (mtDNA) sequencing and/or relevant nuclear gene testing, as well as muscle biopsy.
2.2 Clinical and biochemical data collection
The onset of MELAS was defined by the presence of initial symptoms or events, such as seizures, stroke-like episodes, or severe headaches, which were associated with neuroimaging abnormalities (4). Patient characteristics, including gender, age at onset, prodromal symptoms, and clinical manifestations, were recorded. Biochemical data collected at the onset of the disease included cerebrospinal fluid lactate, venous blood lactate, creatine kinase (CK), CK-MB, creatinine, cholesterol, hemoglobin, triglycerides, uric acid, fasting blood glucose, and glycated hemoglobin. Mild hyperlactatemia was defined as a blood lactate level greater than the upper limit of the normal range (>2 mmol/L), while severe hyperlactatemia was defined as a blood lactate level greater than twice the upper limit of the normal range (>4 mmol/L). Similarly, mild elevation in cerebrospinal fluid (CSF) lactate was defined as a level exceeding the upper normal limit (>2.2 mmol/L), and severe elevation was defined as a level exceeding two times the upper normal limit (>4.4 mmol/L). The levels of CK, hemoglobin, creatinine, and uric acid were adjusted according to reference values specific to age and gender. All biochemical markers were collected during fasting and resting states to minimize variability. Cerebrospinal fluid was obtained via lumbar puncture, performed under sterile conditions.
2.3 Cohort follow-up
The follow-up period concluded in January 2024. The follow-up duration ranged from 1 to 21 years, with medical histories obtained through face-to-face visits or telephone interviews. Using the mRS, patients’ disability status was assessed, and mortality data were collected (9). Importantly, none of the patients were lost to follow-up throughout the study period.
2.4 Statistical analysis
The Mann–Whitney U test was utilized to detect differences between continuous variables, while Fisher’s exact test was used for categorical variables. The relationships between mRS scores, lactate levels, hemoglobin levels, and age were assessed using Kendall’s tau-b correlation coefficient. Univariate and multivariate analyses were performed using a binary logistic regression model. To evaluate the potential influence of follow-up duration on the results, a sensitivity analysis was conducted. This included follow-up time categorized into intervals (0–4 years, 4–8 years, 8–12 years, 12–16 years, 16–20 years, and over 20 years) in the multivariable logistic regression analysis. Data analysis was performed using SPSS version 29.0. A p-value of less than 0.05 was regarded as statistically significant.
3 Results
3.1 Demographic and pathological findings of MELAS in the cohort study
A total of 39 patients were enrolled in the study. Of these, approximately 64% of patients were male and 36% were female. The mean age at the onset of the disease was 33.5 ± 10.2 years. A positive family history was noted in 74.4% of the patients. Pathogenic mutations of mtDNA were identified in 39 patients, including m.3243A > G (n = 30) and m.3136A > G (n = 1). The remaining 8 patients met the Bernier criteria (7), and were definitively diagnosed through muscle biopsies. Muscle biopsies were performed on all 39 patients. Ragged-red fibers (RRF) were observed in 29 patients (74.4%), and SDH strongly reactive blood vessels (SSV) were observed in 28 patients (71.8%). The mean time interval from the first episode to the last visit at our institute or death was 7.5 ± 5.2 years. The average number of stroke-like episodes during the follow-up period was 3.2 ± 1.5 (ranging from 1 to 6). Among the 36 patients who were followed up, 8 died. The mean age of the patients who died was 37.8 ± 12.1 years. The time from onset to death was 4.1 ± 2.5 years. Six patients died from stroke-like episodes, with status epilepticus being the clinical manifestation prior to death. The remaining deaths were attributed to cardiac insufficiency (1) and pulmonary infection (1; Table 1).
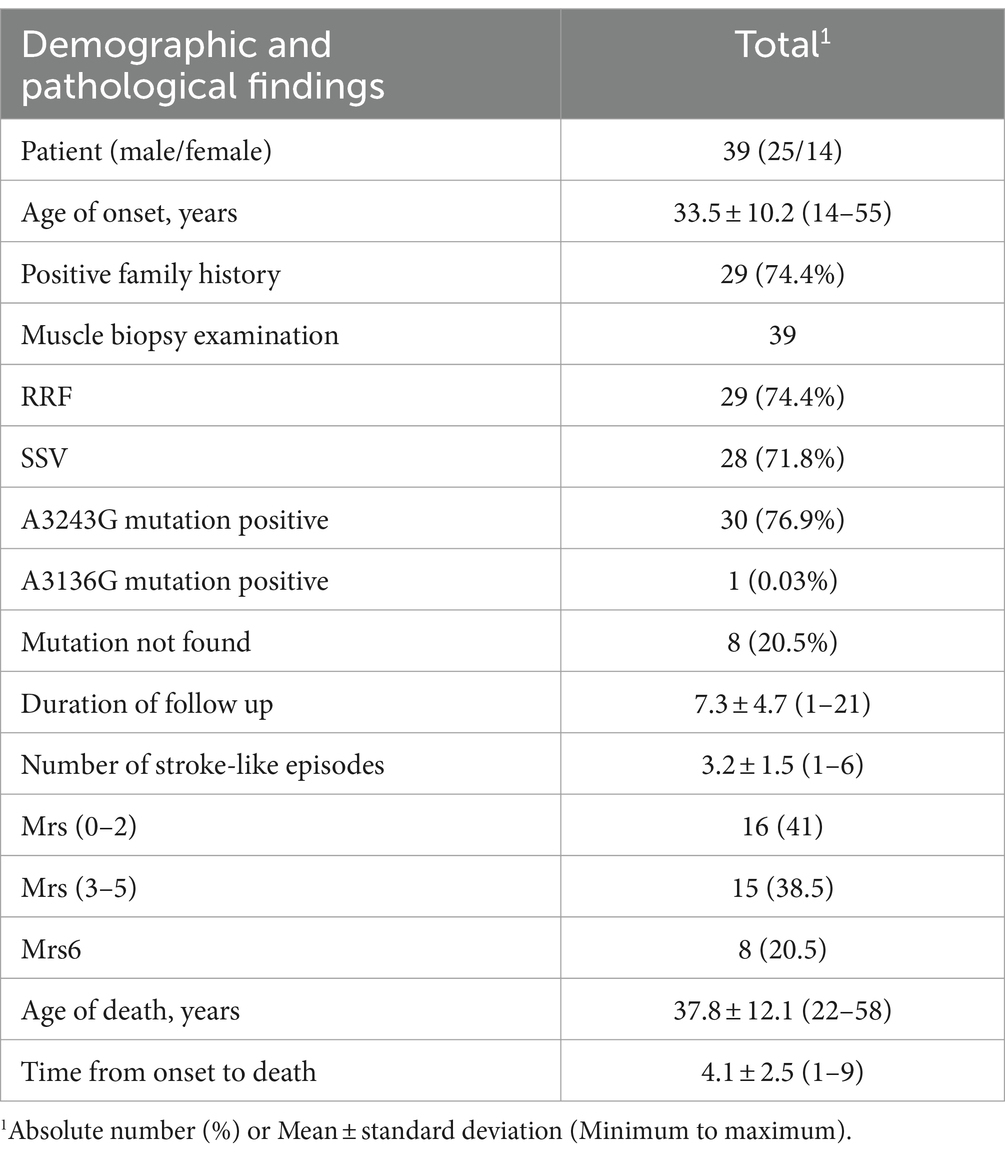
Table 1. Demographic and pathological characteristics in MELAS cases.
3.2 Clinical manifestations before onset and onset
All the patients in our study exhibited stroke-like episodes. Prior to the onset of the condition, the most frequently observed symptoms included hearing loss, general fatigue, and short stature, which were reported by the patients or noted during assessments. At the onset of the stroke-like episodes, the predominant symptoms observed were headache and seizures. Additional clinical symptoms observed both before and at onset are listed in Table 2.
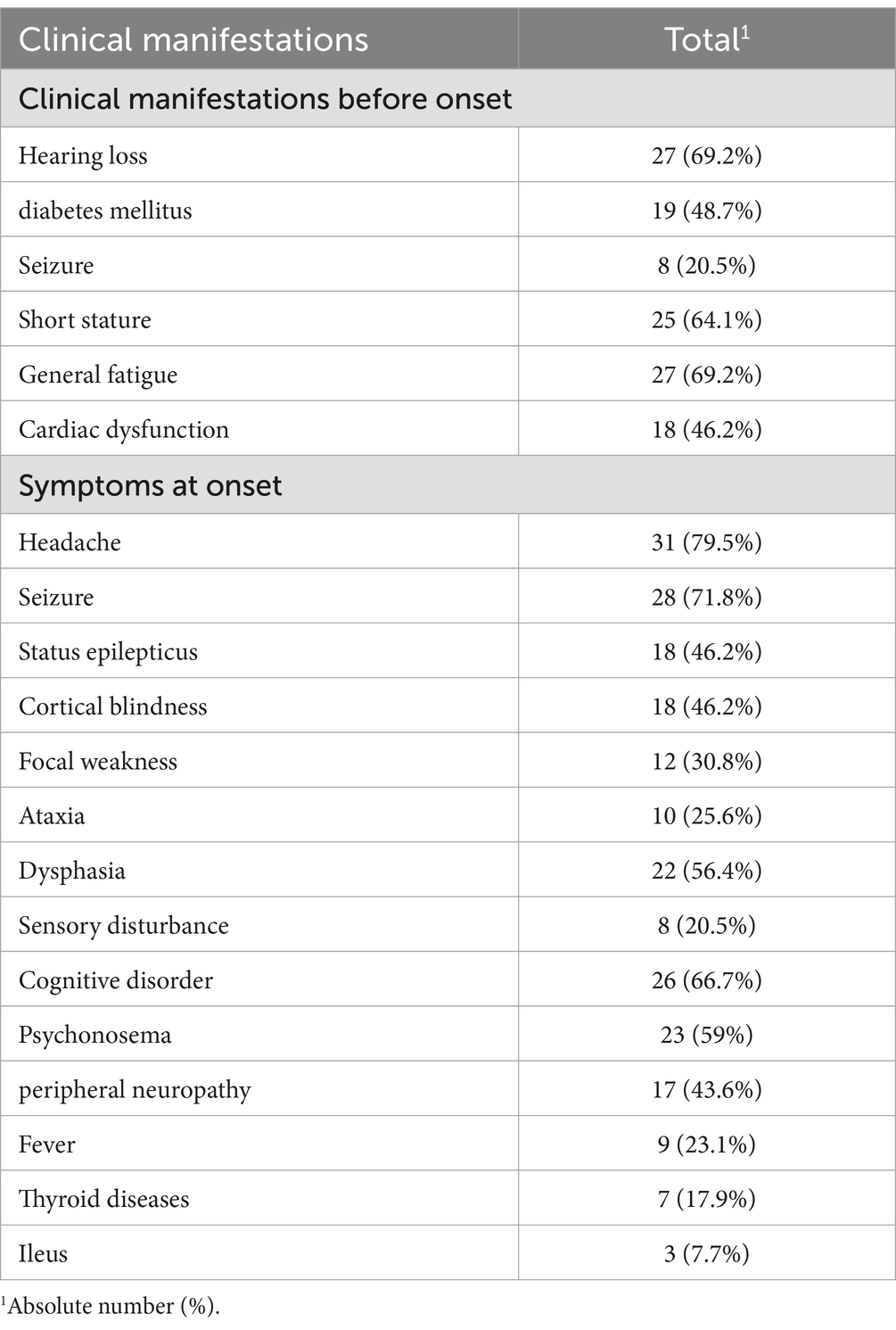
Table 2. Clinical symptoms before and at disease onset.
3.3 Cerebrospinal fluid and blood parameters
In our study, the elevation of lactate levels in blood and cerebrospinal fluid (CSF) was observed as follows: mild elevations in blood lactate levels were seen in 69.2% of patients, while severe elevations were noted in 30.8% of patients. For CSF lactate testing, mildly elevated levels were observed in 94.9% of patients, and severe elevations were noted in 38.5%. Blood hemoglobin tests indicated anemia, defined as hemoglobin levels below the normal range, in 30.8% of patients. A small proportion of patients exhibited abnormally elevated levels of blood CK, CK-MB, creatinine, cholesterol, triglycerides, and uric acid. The proportion of patients with abnormally elevated fasting blood glucose levels showed a roughly equal distribution compared to those without such elevations, and a similar pattern was observed for glycated hemoglobin levels (Table 3).
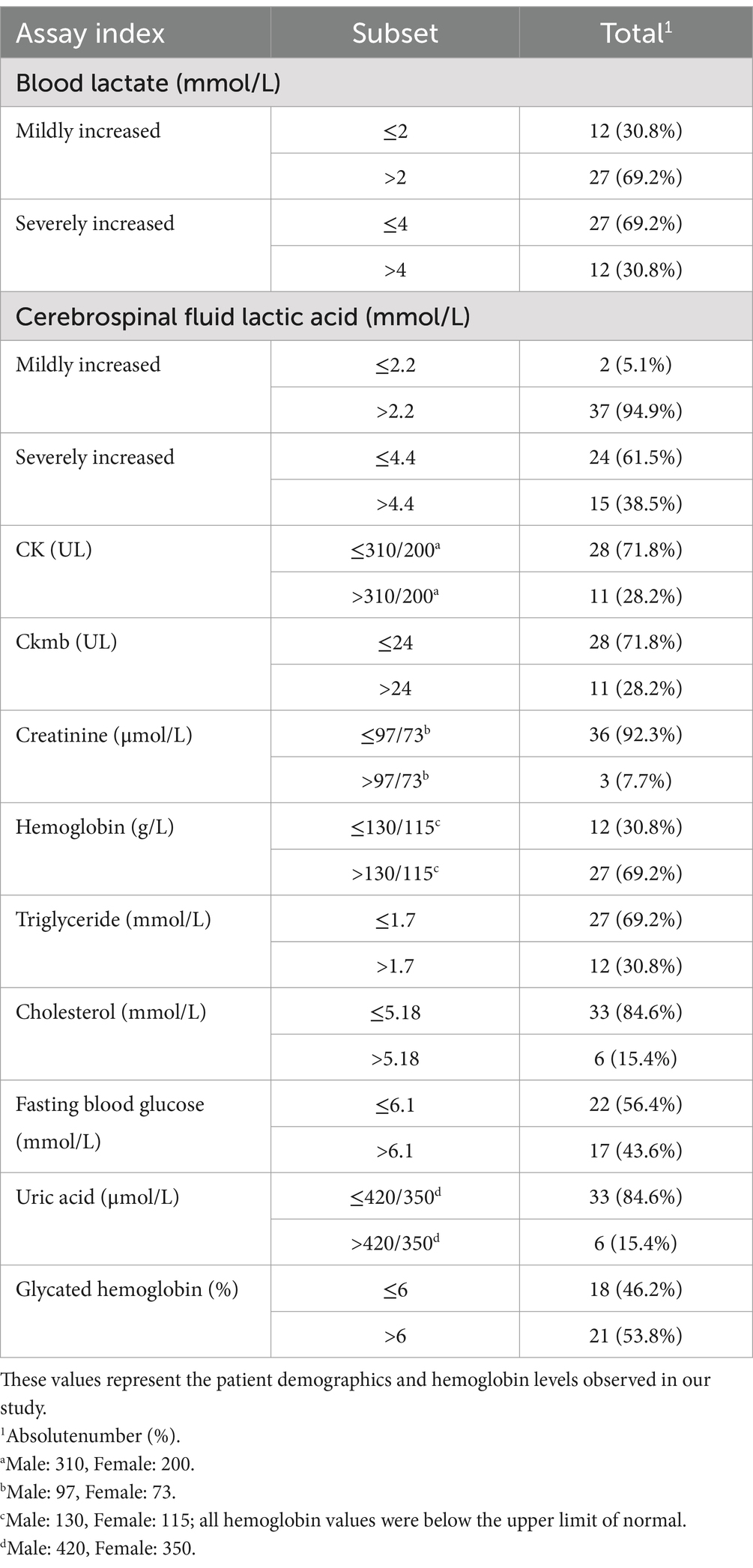
Table 3. Biochemical markers in cerebrospinal fluid and blood.
3.4 Risk factors for mortality
In the univariate analysis, patients with severe hyperlactatemia had a higher risk of mortality compared to those with normal and mildly elevated lactate levels (OR = 5.714, 95% CI 1.086–30.071, p = 0.040). Patients without anemia had a lower risk of death compared to those with anemia (OR = 0.175, 95% CI 0.033–0.921, p = 0.040). Multifactorial analysis, incorporating single positive indicators along with gender and age, showed that patients with severe hyperlactatemia had a higher risk of mortality compared to those with normal and mildly elevated lactate levels (OR = 7.279, 95% CI 1.102–48.086, p = 0.039). Additionally, patients without anemia had a lower risk of death compared to those with anemia (OR = 0.137, 95% CI 0.021–0.908, p = 0.039; Supplementary Table S1). We further evaluated the impact of follow-up duration on the results, and the sensitivity analysis did not alter the findings of the primary analysis (Supplementary Table S2).
3.5 mRS scores
The average mRS score of the patients in our study was 3.18 ± 1.57. We divided the MRS scores into three levels:41% of patients had an mRS score of 0–2,38.5% had a score of 3–5, and 20.5% either died or had an mRS score of 6. Lactate levels exhibited a positive correlation with mRS scores (r = 0.460, p = 0.003, Figure 1), and hemoglobin levels were negatively correlated with mRS scores (r = −0.375, p = 0.015, Figure 2). Additionally, we discovered a positive correlation between mRS scores and the frequency of stroke-like episodes (r = 0.280, p = 0.042). However, we did not find a correlation between mRS scores and the age of onset (r = −0.114, p = 0.374).
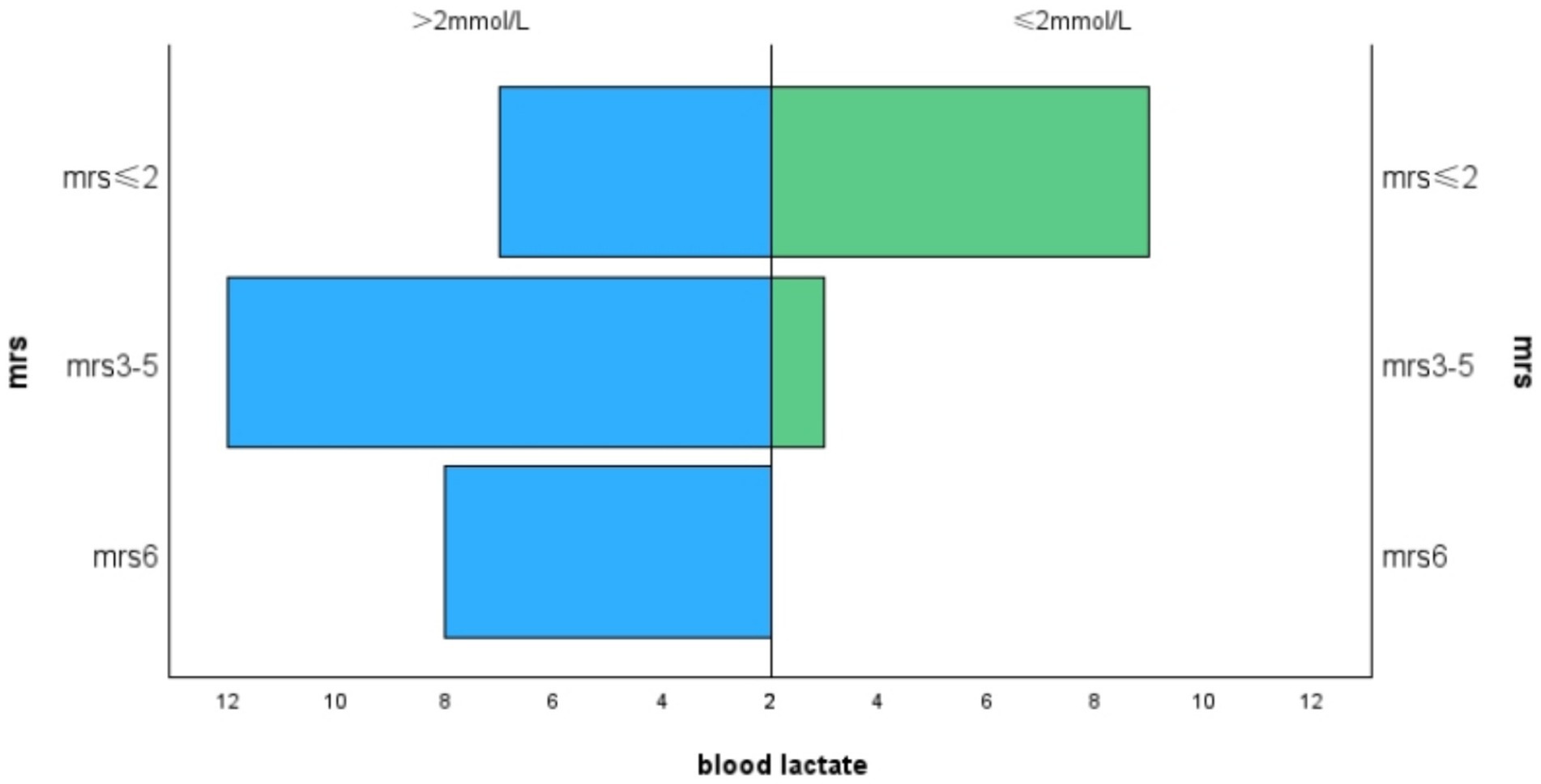
Figure 1. Correlation between MRS and blood lactate levels.
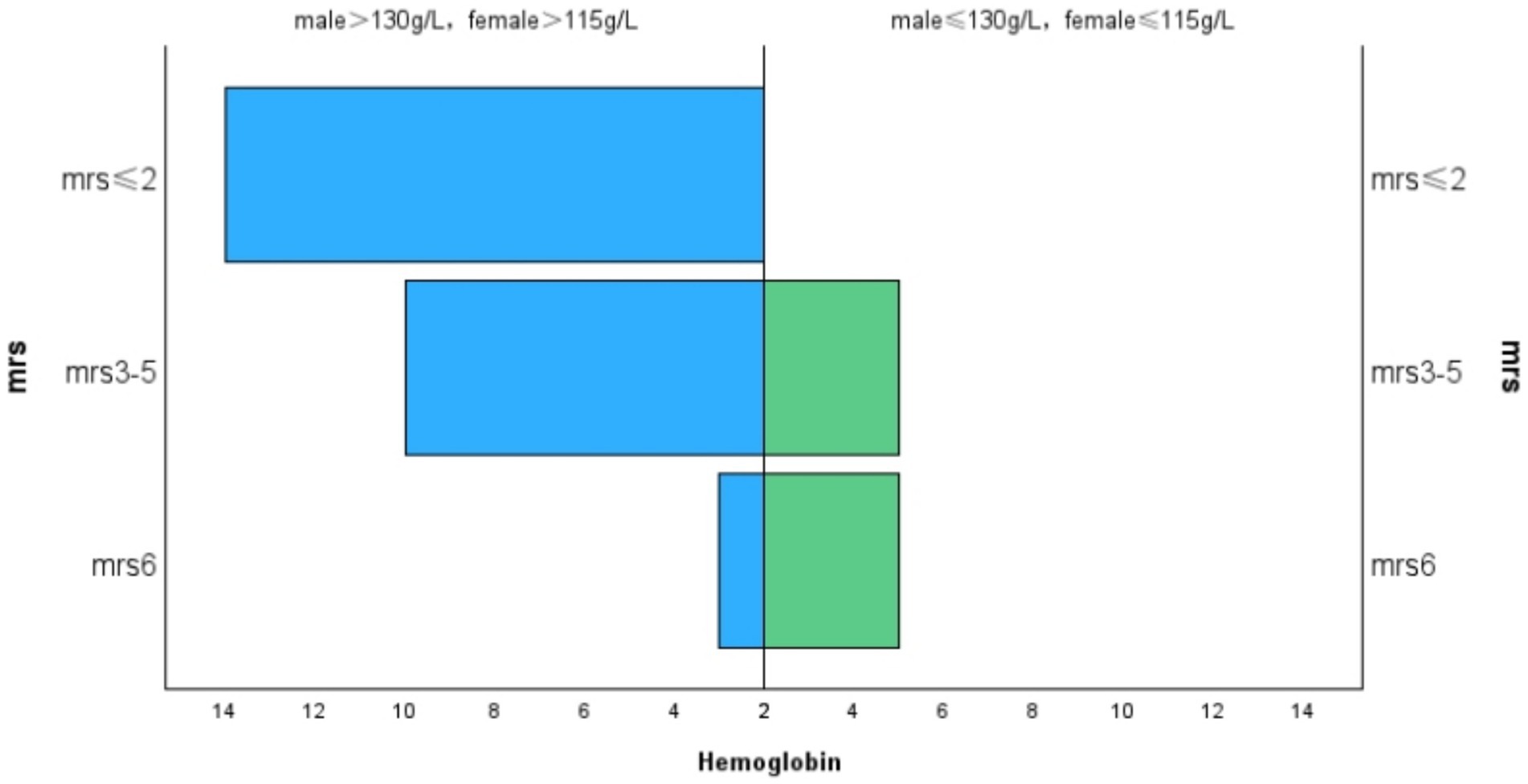
Figure 2. Correlation between MRS and hemoglobin.
4 Discussion
MELAS syndrome is characterized by high mortality and morbidity. Previous research demonstrated that the mortality rate was higher in MELAS patients who had focal seizures or stroke-like episodes (10). These findings suggest that identifying risk factors for mortality or disability may help predict the clinical course of the disease and facilitate early clinical intervention. We conducted a multivariate analysis to identify clinical and biochemical factors associated with poor prognosis in MELAS.
The average mRS score of the patients in this study was 3.18 ± 1.57, which is consistent with previous studies reporting an mRS score of 3.3 ± 1.8 (3), indicating a poor prognosis for the majority of patients. Among the 8 deceased MELAS patients, the average age at death was 37.8 ± 12.1 years, which was higher than the 23.3 ± 11.9 years reported in a study involving Chinese patients (3), but similar to the 34.5 ± 19 years observed in another study of American patients (10).
The main causes of death in this study were stroke-like episodes and status epilepticus, which aligns with findings from previous research. Zhang et al. reported that most deaths among MELAS patients resulted from stroke-like episodes, which frequently presented with clinical manifestations such as status epilepticus and metabolic acidosis (3). Moreover, Fayssoil et al. observed that the majority of deaths among their MELAS patients were attributed to stroke-like episodes and status epilepticus, while other causes included heart failure and bowel obstruction (5). These studies underscore the critical importance of early identification and treatment of stroke-like episodes and status epilepticus.
One patient in our cohort succumbed to heart failure. Heart failuer has been reported as the cause of death for 71% of adult patients with the m.3243A > G mutation (11). Given the high rate of left ventricular involvement, it is recommended that patients with MELAS syndrome undergo regular cardiac examinations (12). One patient in our cohort died from a lung infection, which aligns with prior studies reporting mortality due to complications such as dyspnea and pulmonary edema (12). Kaufmann et al. reported that intestinal obstruction is another significant cause of mortality among MELAS patients (10), highlighting the need for comprehensive care that includes gastrointestinal assessment. These observations underscore the necessity of addressing not only neurological symptoms in MELAS patients but also monitoring for potential damage to vital organs, including the heart, lungs, and gastrointestinal system.
According to a prognostic study conducted by Zhang et al. on Chinese MELAS patients, a negative correlation was observed between mRS scores and age of onset, indicating that an earlier onset of the disease is associated with more severe outcomes. Additionally, stroke-like episodes served as independent predictors of death (3). Conversely, a clinical investigation in Taiwan on MELAS patients carrying the m.3243A > G mutation identified seizures and status epilepticus as the most significant predictors of unfavorable prognosis, while age of onset and visceral organ damage did not significantly impact outcomes (12). In a study of 43 adult patients in France, Fayssoil et al. identified a urine mutation load of ≥45%, along with left ventricular hypertrophy and seizures, as significant factors associated with hospitalization or death (5). In this study, we found a positive correlation between mRS scores and the frequency of stroke like episodes. This finding suggests that increased frequency of central nervous system involvement may correlate with more severe clinical manifestations in MELAS patients.
The multivariate analysis indicated that significant elevations in lactate levels were associated with an increased risk of death. Elevated lactate levels correlated with higher mRS scores, reflecting greater disability. The majority of patients with MELAS syndrome exhibited elevated lactate levels in both blood and cerebrospinal fluid, as supported by previous studies (13). Mitochondrial dysfunction led to increased anaerobic glycolysis in muscle tissues, resulting in elevated blood lactate levels due to insufficient oxidative phosphorylation (14). Previous studies have identified serum lactate as an independent risk predictor for stroke-like episodes in carriers of the m.3243A > G mutation, suggesting a potential association with poor prognosis (15). Thus far, the most significant biomarker indicating the severity of MELAS disease is the lactate level measured in the ventricles through brain magnetic resonance spectroscopy (16), suggesting that elevated CSF lactate correlates with worsening disease severity (10). In individuals with the m.3243A > G mutation, the levels of heteroplasmy in skeletal muscle and blood have been positively associated with disease severity (17). Given the small sample size of this study, further investigation with larger cohorts is needed to ascertain whether severely elevated blood lactate consistently correlates with poor prognosis and mortality in MELAS patients.
Hematological abnormalities, including anemia and various forms of cytopenia, have been reported in approximately 10 to 30% of patients with mitochondrial diseases (18). Defects in the mitochondrial respiratory chain impair the differentiation capacity of hematopoietic stem cells, resulting in cytopenias (18, 19). Our study found that 30.8% of patients exhibited anemia, a slightly higher proportion than expected, potentially influenced by patient selection criteria, though dietary or other factors affecting hemoglobin levels could not be ruled out. Research on POLG-related mitochondrial diseases has also demonstrated that anemia is significantly associated with worse prognosis (20).
Currently, several small cohort studies have investigated potential treatments for MELAS. A study involving two MELAS patients indicated that taurine administration resulted in no further seizures or stroke-like episodes, accompanied by a reduction in serum lactate and pyruvate levels (21). An investigation demonstrated that after 48 weeks of sodium pyruvate treatment in patients with mitochondrial disease, blood lactate concentration and lateral ventricular lactate levels significantly declined from baseline, showing a clear trend of improvement in the Newcastle Mitochondrial Disease Adult Scale (NMDAS) partial scores (22). Arginine administration was found to lower blood lactate concentrations in MELAS patients (13) and to reduce the frequency of stroke-like episode recurrences (23). Recent studies have indicated that the mammalian target of rapamycin inhibitor rapamycin promotes erythroid differentiation in pluripotent stem cells and significantly improves the anemic phenotype in patients with Mitochondrial Leukoencephalopathy, Lactic Acidosis, and Stroke-like episodes (24).
This study has several limitations. First, as a retrospective cohort study, it was inherently vulnerable to recall bias. Secondly, selection bias may have been introduced, as younger patients could have sought treatment at pediatric hospitals, despite our institution being a specialized neurology center. Additionally,. our multivariate analysis primarily focused on clinical variables and biochemical markers, without thoroughly investigating the relationships between clinical phenotypes, mtDNA heteroplasmy, or emerging biomarkers such as fibroblast growth factor 21 (FGF-21) and growth differentiation factor 15 (GDF-15) (25), both of which have been linked to mitochondrial disease severity (26). Future studies should investigate these biomarkers, along with genetic mutations, to gain a more comprehensive understanding of their prognostic significance. Although standard protocols for biochemical data collection were followed, the venous lactate levels might have been artificially elevated as a result of patient restlessness during the sampling process (27).
In conclusion, our study demonstrated that severe elevations in venous lactate levels and the presence of anemia are associated with increased mortality and adverse outcomes in patients with MELAS. Early identification of these indicators could aid in assessing patient prognosis and facilitate graded management, potentially leading to improved outcomes.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by the Human Participants Ethics Committee of Tianjin Huanhu Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
RG: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. LG: Investigation, Methodology, Writing – review & editing. WZ: Data curation, Formal analysis, Writing – review & editing. PW: Writing – review & editing, Conceptualization, Investigation.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Natural Science Foundation of China (81901101), Science and Technology Project of Tianjin Municipal Health Committee (TJWJ2022MS032), and Tianjin Key Medical Discipline (Specialty) Construction (TJYXZDXK-052B).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2024.1491283/full#supplementary-material
References
1. Kaufmann, P, Engelstad, K, Wei, Y, Kulikova, R, Oskoui, M, Battista, V, et al. Protean phenotypic features of the A3243G mitochondrial DNA mutation. Arch Neurol. (2009) 66:85–91. doi: 10.1001/archneurol.2008.526
2. DiMauro, S, Schon, E, Carelli, V, and Hirano, M. The clinical maze of mitochondrial neurology. Nat Rev Neurol. (2013) 9:429–44. doi: 10.1038/nrneurol.2013.126
3. Zhe, Z, Danhua, Z, Xiao, Z, Hui, X, Xinhua, B, Yun, Y, et al. Survival analysis of a cohort of Chinese patients with mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) based on clinical features. J Neurol Sci. (2018) 385:151–5. doi: 10.1016/j.jns.2017.12.033
4. Shuichi, Y, Nataliya, P, Junko, N, Koju, K, Noriko, K, Toyojiro, M, et al. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. (2011) 1820:619–24. doi: 10.1016/j.bbagen.2011.03.015
5. Fayssoil, A, Laforêt, P, Bougouin, W, Jardel, C, Lombès, A, Bécane, HM, et al. Prediction of long‐term prognosis by heteroplasmy levels of the m.3243A>G mutation in patients with themitochondrial encephalomyopathy, lactic acidosis and stroke‐like episodessyndrome (2017) 24:255–61. doi: 10.1111/ene.13176
6. Hirano, M, and Pavlakis, SG. Topical review: mitochondrial myopathy, encephalopathy, lactic acidosis, and Strokelike episodes (MELAS): current concepts. Child Neurol Open. (1994) 9:4–13. doi: 10.1177/088307389400900102
7. Bernier, FP, Boneh, A, Dennett, X, Chow, CW, Cleary, MA, and Thorburn, DR. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. (2002) 59:1406–11. doi: 10.1212/01.WNL.0000033795.17156.00
8. Sumit, P, Amy, G, Mary Kay, K, Fernando, S, Gregory, ME, Russell, S, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the mitochondrial medicine society. Genet Med. (2014) 17:689–701. doi: 10.1038/gim.2014.177
9. Sandra, B, Urs, F, Edith, W, Heinrich, PM, Eugen, B, Sarah, B, et al. Acute ischemic stroke in children versus young adults. Ann Neurol. (2011) 70:245–54. doi: 10.1002/ana.22427
10. Kaufmann, P, Engelstad, K, Wei, Y, Kulikova, R, Oskoui, M, Sproule, DM, et al. Natural history of MELAS associated with mitochondrial DNA m.3243A>G genotype. Neurology. (2011) 77:1965–71. doi: 10.1212/WNL.0b013e31823a0c7f
11. Majamaa-Voltti, KAM, Winqvist, S, Remes, AM, Tolonen, U, Pyhtinen, J, Uimonen, S, et al. A 3-year clinical follow-up of adult patients with 3243A>G in mitochondrial DNA (2006) 66:1470–5. doi: 10.1212/01.wnl.0000216136.61640.79
12. Liu, C, Chang, C, Kuo, H, Ro, L, Liou, C, Wei, Y, et al. Prognosis of symptomatic patients with the A3243G mutation of mitochondrial DNA. J Formos Med Assoc. (2012) 111:489–94. doi: 10.1016/j.jfma.2011.06.014
13. Ayman, WE-H, Adekunle, MA, Jeremy, J, and Fernando, SJMGM. MELAS syndrome: clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab. (2015) 116:4–12. doi: 10.1016/j.ymgme.2015.06.004
14. Hao, C, Qian, H, Hafiz Khuram, R, Thitsavanh, C, Sandeep, S, Pabitra, R, et al. An analysis of the clinical and imaging features of mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Somatosens Mot Res. (2020) 37:45–9. doi: 10.1080/08990220.2020.1720636
15. Yi Shiau, N, Nichola, ZL, Alasdair, PB, Daniel, E, Mark, RB, Tuomo, P, et al. Forecasting stroke-like episodes and outcomes in mitochondrial disease. Brain. (2021) 145:542–54. doi: 10.1093/brain/awab353
16. Rohit, S, Bryn, R, Kristin, E, Owen, SS, Erin, S, Ronald, GH, et al. Circulating markers of NADH-reductive stress correlate with mitochondrial disease severity. J Clin Invest. (2021) 131:e136055. doi: 10.1172/JCI136055
17. John, PG, Sarah, JP, Yi Shiau, N, Charlotte, LA, Emma, LB, Steven, AH, et al. mtDNA heteroplasmy level and copy number indicate disease burden in m.3243A>G mitochondrial disease. EMBO Mol Med. (2018) 10:e8262. doi: 10.15252/emmm.201708262
18. Arthavan, S, Juliana, T, and Bindu, PS. Hematologic manifestations in primary mitochondrial diseases. J Pediatr Hematol Oncol. (2024) 46:e338–47. doi: 10.1097/MPH.0000000000002890
19. Elena, A, Samuel, EW, Lauren, PD, Benjamin, JT, Sébastien, M, Paul, TS, et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat Cell Biol. (2017) 19:614–25. doi: 10.1038/ncb3529
20. Omar, H, Charalampos, T, Claus, K, Magnhild, R, Chantal, MET, Eylert, B, et al. The presence of anaemia negatively influences survival in patients with POLG disease. J Inherit Metab Dis. (2017) 40:861–6. doi: 10.1007/s10545-017-0084-9
21. Mitsue, R, Yutaka, O, Alexander, MW, Kiyomi, N, Harumi, I, Naomi, K, et al. Taurine ameliorates impaired the mitochondrial function and prevents stroke-like episodes in patients with MELAS. Intern Med. (2012) 51:3351–7. doi: 10.2169/internalmedicine.51.7529
22. Yasutoshi, K, Nataliya, P, Eisuke, I, Kazutaka, N, and Masashi, T. Biomarkers and clinical rating scales for sodium pyruvate therapy in patients with mitochondrial disease. Mitochondrion. (2019) 48:11–5. doi: 10.1016/j.mito.2019.02.001
23. Yasutoshi, K, Nataliya, P, Eisuke, I, Hidefumi, N, Akiko, I, Yasuhiro, S, et al. Therapeutic regimen of l-arginine for MELAS: 9-year, prospective, multicenter, clinical research. J Neurol. (2018) 265:2861–74. doi: 10.1007/s00415-018-9057-7
24. Bichen, W, Deyang, S, Shuang, Y, Yu, L, Haoyuan, L, Mutian, C, et al. Mitochondrial tRNA pseudouridylation governs erythropoiesis. Blood. (2024) 144:657–71. doi: 10.1182/blood.2023022004
25. Kristin, NV, Omar, H, Hanne Linda, N, Christian, AV, and Laurence, AB. Serum biomarkers in primary mitochondrial disorders. Brain Commun. (2021) 3:fcaa222. doi: 10.1093/braincomms/fcaa222
26. Shuichi, Y, Yasunori, F, Akiko, I, Yoshihiro, F, Hajime, A, Tatsuyuki, K, et al. Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann Neurol. (2015) 78:814–23. doi: 10.1002/ana.24506
Keywords: MELAS, prognosis, clinical markers, biochemical markers, risk factor
Citation: Gao R, Gu L, Zuo W and Wang P (2024) Long-term prognostic factors and outcomes in mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes: a clinical and biochemical marker analysis. Front. Neurol. 15:1491283. doi: 10.3389/fneur.2024.1491283
Edited by:
Massimiliano Filosto, University of Brescia, ItalyReviewed by:
Christina Liang, Royal North Shore Hospital, AustraliaChiara La Morgia, IRCCS Institute of Neurological Sciences of Bologna (ISNB), Italy
Helin Zheng, Children‘s Hospital of Chongqing Medical University, China
Copyright © 2024 Gao, Gu, Zuo and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pan Wang, d3Bhb2ZlaWVyQDE2My5jb20=