 Nan Lin
Nan Lin Xi-Yan Gao2*
Xi-Yan Gao2* Xiao Li
Xiao Li Wen-Ming Chu
Wen-Ming Chu- 1College of Acupuncture and Tuina of Henan University of Chinese Medicine, Zhengzhou, Henan, China
- 2The Third Affiliated Hospital of Henan University of Chinese Medicine, Zhengzhou, Henan, China
The hallmark pathological features of Alzheimer’s disease (AD) consist of senile plaques, which are formed by extracellular β-amyloid (Aβ) deposition, and neurofibrillary tangles, which are formed by the hyperphosphorylation of intra-neuronal tau proteins. With the increase in clinical studies, the in vivo imbalance of iron homeostasis and the dysfunction of synaptic plasticity have been confirmed to be involved in AD pathogenesis. All of these mechanisms are constituted by the abnormal accumulation of misfolded or conformationally altered protein aggregates, which in turn drive AD progression. Proteostatic imbalance has emerged as a key mechanism in the pathogenesis of AD. Ubiquitination modification is a major pathway for maintaining protein homeostasis, and protein degradation is primarily carried out by the ubiquitin-proteasome system (UPS). In this review, we provide an overview of the ubiquitination modification processes and related protein ubiquitination degradation pathways in AD, focusing on the microtubule-associated protein Tau, amyloid precursor protein (APP), divalent metal transporter protein 1 (DMT1), and α-amino-3-hyroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors. We also discuss recent advances in ubiquitination-based targeted therapy for AD, with the aim of contributing new ideas to the development of novel therapeutic interventions for AD.
1 Introduction
AD is a progressive neurodegenerative disorder with clinical manifestations of memory loss and cognitive dysfunction. It is the most common form of dementia and with a prevalence, which severely affects patients’ quality of life (1). Recent data from Scheltens et al. in 2021 suggests that the prevalence of dementia will double in Europe and triple globally by 2050. this estimation will be three times higher based on the biological (rather than clinical) definition of AD (2). Understanding the pathogenesis of AD remains a challenge, with the formation of senile plaques due to extracellular β-amyloid (Aβ) deposition and neurofibrillary tangles resulting from hyperphosphorylation of intra-neuronal tau proteins being key hallmarks (3). The misfolding of proteins leads to the aggregation of Aβ and tau into toxic fibrillar structures, impairing their normal functionality (4). Protein homeostasis is maintained through the critical pathway of ubiquitination, with the ubiquitin-proteasome system (UPS) responsible for degrading 80–90% of proteins in the nucleus and cytoplasm. This system is vital for the timely degradation of short-lived, damaged, and misfolded proteins (5). In addition, it has been found that both iron death and synaptic dysfunction are closely associated with the development of AD (6, 7). Thus, the Ferroptosis pathway plays a key role in AD progression, as well as ubiquitination modifications of molecular proteins associated with synaptic plasticity. Notably, ubiquitination modifications of iron-related pathways and synaptic plasticity molecules are implicated in AD advancement. This paper aims to explore the intricate mechanisms of ubiquitination modification in Alzheimer’s disease, offering valuable insights into potential targets for future clinical interventions in AD treatment.
2 Ubiquitination
Ubiquitination is a vital post-translational modification mediated by ubiquitin (Ub), it is essential for cell division, differentiation, protein quality control, gene expression, DNA repair, protein transport, and signal transduction (8). This intricate process of transferring Ub to a substrate protein by three enzymes undergoes a cascade reaction, which consists of a three-step reaction: a. Activation: Ubiquitin-activating enzymes (E1s) are the initial enzymes necessary for the binding of ubiquitin to substrate proteins. These enzymes, devoid of effects on the specificity of target proteins, form a high-energy thioester by facilitating bond between their structural cysteine (Cys) residue and the lysine (Lys) residue at the C-terminus of ubiquitin in the presence of ATP energy, thereby activating ubiquitin. b. Binding: The activated Ub is then transferred to ubiquitin-conjugating enzymes (E2s), binding to the Cys residue of E2s through a thioester bond. All E2s share a conserved core structural domain of approximately 150 amino acid residues, featuring a central Cys residue that dictates their enzymatic activity. This dynamic process involves the E2 shuttling between E1 and E3, as E1 and E3 are linked to the E2 by the same motif, forming an essential part of the reaction cycle. c. Ligations: Activated ubiquitin is either attached directly to the protein substrate via the E2 or is attached to the substrate by ubiquitin-protein ligases (E3). E3 transfer ubiquitin to the target protein through the formation of an amino-isopeptide bond between the carboxyl terminus of ubiquitin and the ε-amino group of the target protein’s Lys residue (9) (Figure 1). Among these, E3 plays a key role in the ubiquitination pathway by connecting E2 to the specific substrate, transferring the activated ubiquitin chain to the lysine residue of the specific substrate, and degrading the protein purposely by recognizing the multimeric ubiquitin chain. The specific recognition of various substrates by the complex and diverse E3 family members enables the ubiquitination pathway to demonstrate a high level of selectivity in protein degradation.
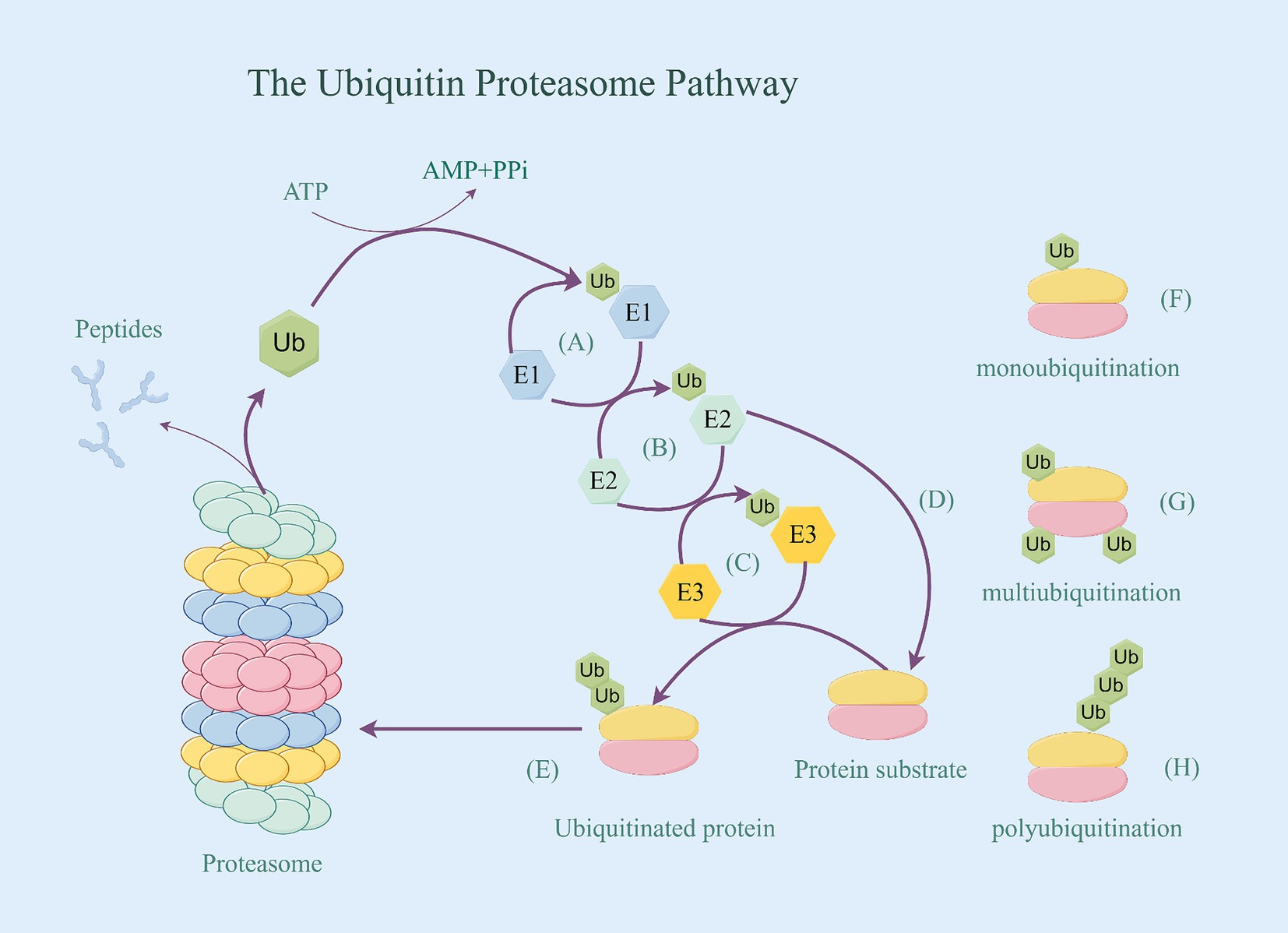
Figure 1. The ubiquitin proteasome pathway. (A) Activation: E1’s structural cysteine (Cys) residue and the lysine (Lys) residue at the C-terminus of ubiquitin in the presence of ATP energy bond together to form a high-energy thioester, thereby activating ubiquitin. (B) Binding: The activated Ub is then transferred to E2, binding to the Cys residue of E2 through a thioester bond. (C) Ligations: E3 transfer the activated ubiquitin to the target protein. (D) Activated ubiquitin is attached directly to the protein substrate via the E2. (E) Proteins tagged with ubiquitin be identified by the proteasome, thereby being degraded. (F) Target protein’s monoubiquitination. (G) Target protein’s multiubiquitination. (H) Target protein’s polyubiquitination.
The process of attaching a single ubiquitin to an intracellular protein is known as monoubiquitination. When multiple Ub monomers simultaneously attach multiple different lysine residues in a target protein, it is referred to as multiubiquitination. Additionally, multiple ubiquitin monomers can form a polyubiquitinated chain on a target protein through different Lys residues (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, and Lys63), or Met1 residues can be interlinked to the target protein to create a polyubiquitinated chain (such as K48chain,K63chain), known as polyubiquitination (10) (Figure 1). The polyubiquitinated Ub residues determine the structure of the Ub chain. For example, the M1 chain and the K63 chain adopt an “open” conformation similar to a linear chain, while the K48 chain has a compact “forked” globular conformation (11). All these different ubiquitination structures and linkages and their combinations form a highly complex “ubiquitin code” that determines the future fate of target proteins (12). Overall, the functions of different forms of ubiquitination can be summarized in three aspects: a. Alter the stability of target proteins, causing them to be degraded by UPS; b. Alter the functional activity of target proteins; and c. Shift the localization of target proteins.
Most protein clearance in cells relies on ubiquitin, as proteins must be covalently modified with Ub before entering the proteasome for degradation. Proteins tagged with polyubiquitin can be identified by the proteasome. Research from Maniv et al. shows that dysregulation of ubiquitination may contribute to the development of AD (13). Ubiquitination modification is a dynamic and reversible process. E1/2/3 enzymes positively catalyze the ubiquitination reaction, while deubiquitinating enzymes (DUBs) reverse the process of ubiquitination. DUBs have been found to promote AD by cleaving ubiquitin from ubiquitination-modified substrate proteins, causing the substrate proteins to evade degradation by the proteasome (14). Research has found that ubiquitin levels were significantly elevated in the AD brain as measured by immunoassay. Ubiquitin-positive pathological protein aggregates in AD such as Aβ peptide, whose levels show a significant increase (15). Label-free mass spectrometry (MS)-based proteomic analysis has showed an 80% increase in ubiquitination levels in AD, with a total of 800 ubiquitination sites (15). Furthermore, the ubiquitination cascade has emerged as an attractive target for therapeutic intervention in AD (16).
3 Ubiquitination modification of AD-related proteins
3.1 Ubiquitination of Tau
Tau proteins are microtubule-associated proteins (MAPs) predominantly located in the axons of neurons within the central nervous system. Their primary functions include promoting and maintaining microtubule protein formation, stability, and regulating axonal transport (17). Anomalies in the aggregation of tau proteins closely correlate with neuronal loss and cognitive dysfunction. Excessive phosphorylation causes conformational changes in tau leading to tau protein aggregation, which is a toxic form of tau protein (18). Tau polymerization and degradation are regulated by a variety of post-translational modifications, among which ubiquitination modifications are critical for Tau to enter the degradation system. Phosphorylated Tau can be degraded by ubiquitination into the proteasome and lysosome (19, 20). The process of ubiquitination modification can be reversed by deubiquitinases (DUBs), including ubiquitin-specific protease 10 (USP10), a highly conserved deubiquitinating enzyme expressed widely in the brain, which promotes the aggregation of tau (21). In AD, the microtubule-associated protein tau has the highest number of ubiquitination sites per protein among ubiquitinated proteins (22), highlighting the essential role of the ubiquitination modification process carried out by tau via the Ubiquitin-Proteasome System (UPS) in AD.
The E3 CHIP plays a role in regulating the ubiquitin-dependent degradation of Tau in vivo. CHIP is part of the RING/U-box-type family, This family make up the largest E3 family that contains the RING or U-box catalytic domain (23), they do not bind ubiquitin directly but mediate the transfer of ubiquitin from bound E2 (E2-Ub) to the target substrate (24). CHIP acts as a quality regulator for the cellular proteome and is specifically designed to target misfolded proteins for degradation (25). On the one hand, CHIP can directly bind to Tau and promote its ubiquitination in vivo and in vitro (26). CHIP promotes the degradation of phosphorylated tau via UPS by binding to Hsc70/Hsp70 or Hsp90 chaperones (27). More precisely, the lysine clamp found in the CHIP tetrapeptide repeat sequence (TPR) structural domain, formed by the odd-numbered helices, attracts Hsp70 and Hsp90 family members by selectively binding to their C-terminal (I/M) EEVD motifs. This process completes the ubiquitination modification (28). On the other hand, CHIP has the capability to attach to the E2 enzyme Ubc13, which is responsible for catalyzing the ubiquitination of multiple lysine residues in the four repeat regions of Tau, even in the absence of Hsp70 (29). These lysine residues are largely present in Tau filaments from the brains of AD patients (30) (Figure 2). Tau protein serves as a substrate for CHIP by binding to it through various interaction sites, with a predominant presence in the N-terminal and C-terminal regions (specifically at residues 46–57 and 413–428). Additionally, there are some interactions in the region which is rich with proline and part of the N2 structural domain. The multisite recognition pattern is facilitated by the remarkable flexibility of Tau, as it is a long and intrinsically disordered polypeptide capable of exploring a large conformational space. This allows it to provide multiple individual contact points for interactions with protein chaperones (31).
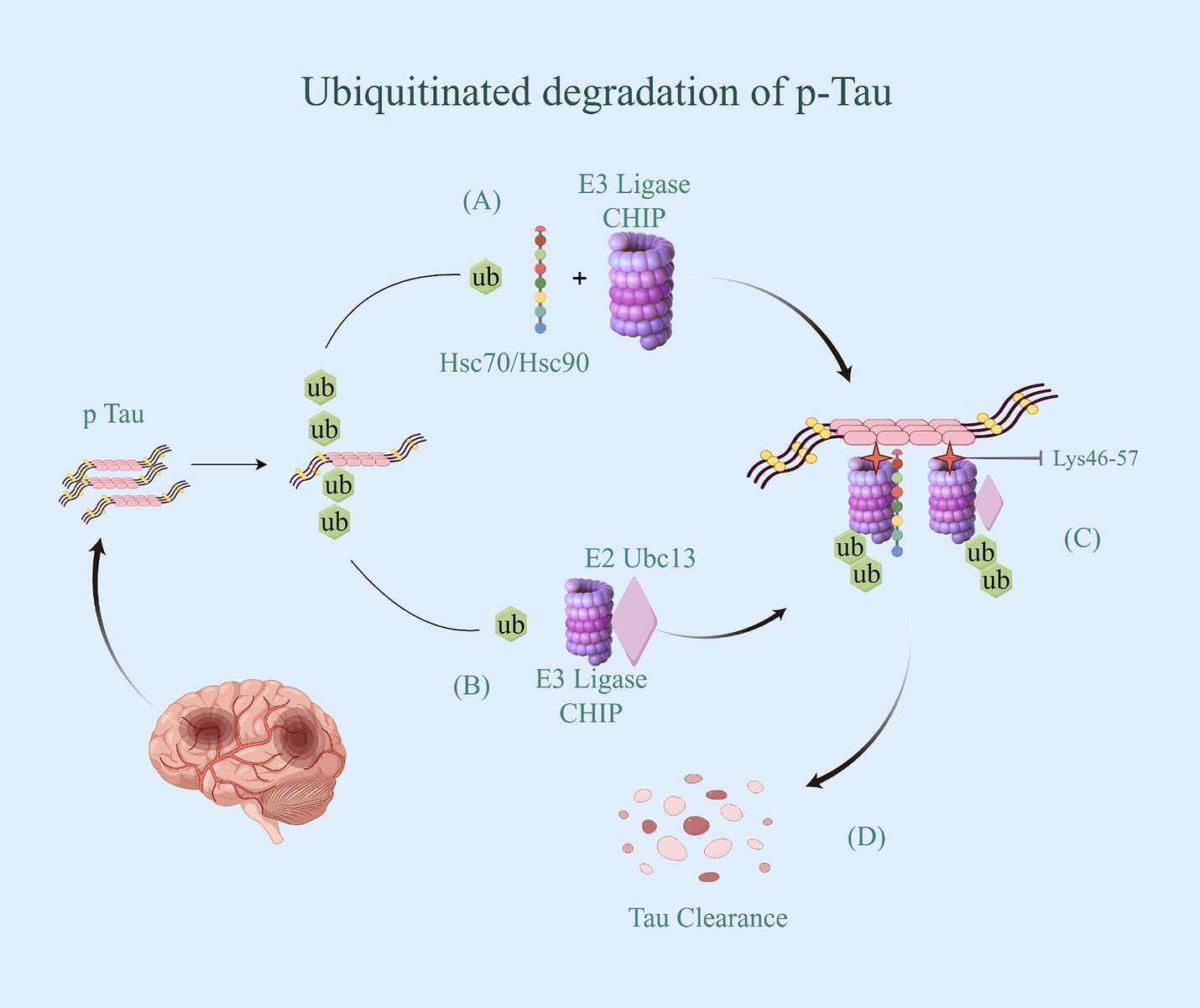
Figure 2. Ubiquitinated degradation of p-Tau. (A) CHIP binding to Hsc70/Hsp70 or Hsp90 chaperones transfer the activated Ub. (B) CHIP attach to the E2 enzyme Ubc13, and bond the activated Ub. (C) CHIP with Ub specifically bind to p-Tua’s Lysine residues 46–57 in N-terminal and C-terminal regions, and promote p-Tua’s ubiquitination in vivo and in vitro. (D) p-Tua with ubiquitin be identified by the proteasome, thereby being degraded.
CHIP, serving as a co-chaperone and an E3 ubiquitin ligase, is responsible for the ubiquitination and degradation of proteins. It plays a significant role in the development of AD (32). CHIP controls protein folding homeostasis that determines whether to refold or dephosphorylate pathologic aggregates in neural cells. If CHIP is not able to function properly, the degradation process will be severely compromised and accumulation of proteins will occur because of overburdened proteasomal and lysosomal systems (33). It has been reported that CHIP-positive tau inclusions were detected in AD (34). With the increase of research reports in recent years, it has been found that in addition to CHIP, E3 ubiquitin ligases such as Parkin (35), TRAF6 (36), and Hrd1 (37) also have the capacity to mediate the degradation of phosphorylated Tau and complete ubiquitination modification through the UPS pathway.
3.2 Ubiquitination of APP
APP is a type I single-channel transmembrane protein that undergoes processing and sorting by the endoplasmic reticulum and Golgi/trans-Golgi network before being secreted to the cytoplasmic membrane. Upon reaching the cytoplasmic membrane, it can be cleaved by α-secretase, leading to the production of the neuroprotective sAPPα fragment (38). Aβ acts as a product of a series of enzyme digestion pathways. Its over-production/aggregation is the main pathological feature of AD. The post-translational modification of APP plays a key regulatory role in the production and degradation of Aβ (39). Abnormalities in the ubiquitin modification process of APP have been identified as central to the pathological changes induced by Aβ deposition in AD (40).
The ubiquitin-dependent degradation of APP is primarily facilitated by the E3 ligases HRD1 and FBL2 through the UPS pathway (41, 42). HRD1 is an E3 ligase located in the endoplasmic reticulum that is upregulated during endoplasmic reticulum stress and is known to prevent endoplasmic reticulum stress-induced apoptosis (43). Study about loss of HRD1-mediated protein degradation have shown that the protein levels of HRD1 are significantly decreased in the cerebral cortex of Alzheimer’s disease patients, leading to the accumulation of APP (41). HRD1 acts as an E3 ligase for APP, binding specifically to APP at the proline-rich region of HRD1. This interaction facilitates the ubiquitination and subsequent proteasome-dependent degradation of APP, ultimately reducing the production of Aβ (41). However, the exact ubiquitin site of HRD1 binding to APP remains unclear, warranting further research to predict and validate this binding. F-box and leucine rich repeat protein2 (FBL2) belongs to the family of F-box proteins (44). It contains an F-box domain and also has 11 leucine-rich repeat regions for interaction with specific substrates. The expression of FBL2 which is the component of the complex E3 ubiquitin ligase Skp1-Cullin1-F-box (SCF) is reduced in the brains of AD patients (45). Binding FBL2 to APP and promoting its ubiquitination can reduce Aβ production. In this process, FBL2 facilitates the ubiquitination of both intracellular and cell surface APP. The ubiquitinated intracellular APP is then degraded by the proteasome, while the ubiquitinated cell surface APP does not undergo endocytosis. It subsequently reduces the amount of APP protein in lipid rafts and β-secretase APP cleavage (42). APP, as a substrate protein, binds specifically to FBL2 mainly through site lysine 651. What’s more, FBL2 can regulate APP metabolism by promoting ubiquitination at this site.
3.3 Ubiquitination of the Ferroptosis-related protein DMT1
Ferroptosis is an iron-dependent, novel mode of programmed cell death distinct from apoptosis, cell necrosis, and cell autophagy (46). The development of AD is closely linked to Ferroptosis (47), the iron accumulation has been shown to accelerate age-related plaque deposition and the production of neurogenic fiber tangles (48, 49). Abnormalities in proteins associated with iron uptake, storage, and export can cause an imbalance in iron homeostasis and induce cellular iron death. Ubiquitination is one of the most important posttranslational modifications for proteasomal degradation of target proteins mediated by specific ligases, and it is involved in Ferroptosis, and protein degradation by regulating protein stability (50). The transmembrane transport of iron ions is crucial for regulating cellular iron balance. Divalent metal transporter protein 1 (DMT1), a widely distributed transmembrane metal ion transporter, is responsible for the uptake of various divalent metal ions, including ferric ions (51). It was also found that DMT1 exhibits high expression levels in AD brains (52), and a pathological increase in DMT1 levels induces neurofibrillary tangles (53). DMT1 has 12 transmembrane segments and is expressed in neurons, allowing for the doping of metals from the extracellular environment and/or recycling of endosomes (54). Once Fe2+ are translocated into the cytoplasm via DMT1, they are metabolically utilized to maintain iron homeostasis. The degradation of DMT1 is primarily regulated by ubiquitination modifications, and this process is crucial for preventing iron accumulation and neuronal apoptosis (55). Therefore, the ubiquitinated degradation of DMT1 is also significant in the context of AD.
The regulation of iron ion channels and transport proteins involves ubiquitination, mediated by the Nedd4 family of E3 ubiquitin ligases. These ligases typically interact with their substrates through WW structural domains, binding to specific motifs in the target proteins (56). However, not all potential targets of these E3 ligases contain these binding motifs. As a result, auxiliary proteins may facilitate the interaction between the Nedd4 family members and their targets (57). For instance, Nedd4-2 is a ubiquitin ligase for DMT1 polyubiquitination under metal-induced stress (54). Additionally, its interacting protein Ndfip1, also known as Nedd4WW structural domain binding protein 5 (N4WBP5) (58), has the ability to facilitate the ubiquitinated degradation of various target proteins, including DMT1 (59). Ndfip1 serves as a bridging protein that facilitates the recruitment of Nedd4-2 and binds to DMT1 to complete the ubiquitination and subsequent degradation of DMT1. It has been observed that reduced levels of Ndfip1 in the AD brain are linked to elevated DMT1 levels (60). Throughout the ubiquitination modification process, Ndfip1 plays a crucial role in targeting E3 ligase ubiquitinated proteins as an auxiliary articulating protein, forming a complex by specifically binding Nedd4-2 and DMT1 (54). This complex accomplishes the ubiquitination degradation of DMT1, resulting in the downregulation of DMT1 expression and activity, ultimately reducing intracellular iron accumulation (Figure 3). However, the precise ubiquitin site of Ndfip1/Nedd4-2-mediated DMT1 degradation remains unknown, and future studies are needed to predict and verify this ubiquitin site through experimental design.
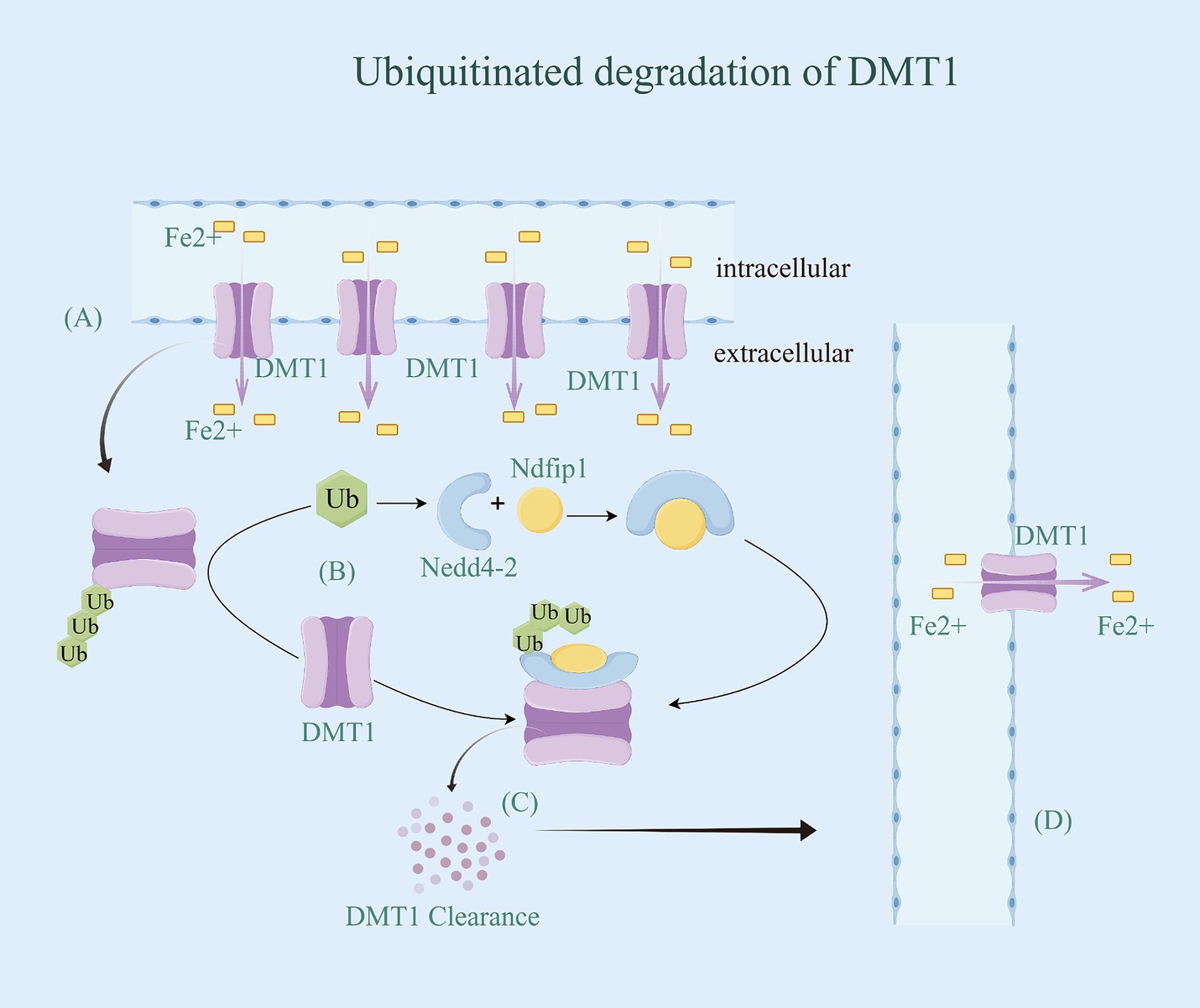
Figure 3. Ubiquitinated degradation of DMTl. (A) DMT1 has 12 transmembrane segments and is expressed in neurons, allowing for the doping of Fe2+ from the extracellular environment and/or recycling of endosomes. Once Fe2+ are translocated into the cytoplasm via DMT1, they are metabolically utilized to maintain iron homeostasis. (B) Ndfip1 serves as a bridging protein that facilitates the recruitment of Nedd4-2 with Ub and binds to DMT1 to complete the ubiquitination and subsequent degradation of DMT1. (C) DMT1 with ubiquitin be identified by the proteasome, thereby being degraded. (D) With the downregulation of DMT1 expression and activity, intracellular iron accumulation be reduced.
3.4 Ubiquitination of AMPARs
Synapses are central for information transfer between neurons. Synaptic plasticity, the biological foundation of learning and memory, governs the structure and function of synapses (61). Structural plasticity is marked by changes in synapse number and morphology, while functional plasticity balances Long-term potentiation (LTP) and Long-term depression (LTD) activities (62). Impairment of synaptic functional plasticity correlates strongly with memory impairment in early AD (63, 64). AD is characterized by progressive and irreversible memory impairment, and associated with inhibition of LTP and enhancement of LTD in the hippocampus (65, 66). The establishment of memory and cognitive abilities is closely linked to synaptic plasticity. Impaired synaptic plasticity has been identified as one of the key causative factors and early pathological features of Alzheimer’s disease (AD) (67). It has been shown that synaptic adaptation and stability are ultimately regulated by synaptic proteins, including removal, addition, and post-translational modifications (68). Ubiquitin-dependent protein degradation is a critical step in memory consolidation (69). In recent years, a role for protein degradation by the ubiquitin–proteasome pathway in synaptic plasticity has been discovered (70, 71). Dong et al. reported that the proteasome plays a facilitatory role in the maintenance part of L-LTP (72). Their data indicate Proteasome inhibition leads to postsynaptic changes such as stabilization of newly synthesized proteins in dendrites, and causes presynaptic changes in the hippocampus such as modulation of transmitter release. These all show that UPS mediated degradation may have broad implications for synaptic plasticity under physiological conditions, as well as synaptic dysfunction in AD with which abnormal protein degradation is associated (73). Post-synaptic density AMPA receptors (AMPARs) play a crucial role in supporting synaptic transmission and plasticity (74). Increasing the number of AMPARs has been shown to enhance synaptic transmission efficiency (75). Impaired AMPAR function is associated with cognitive deficits in the early stages of AD, and its excitatory damage can lead to neurodegeneration in the late stages of the disease (76). Furthermore, post-translational ubiquitination of AMPAR has emerged as a significant factor in AD, regulating the surface expression of these receptors and playing a key role in the disease.
AMPARs, the primary mediators of excitatory synaptic transmission in the brain, consist of two dimers of a combination of four subunits (GluA1-GluA4). Additionally, different subunits are capable of conferring specific physiological properties to the AMPARs channel functions (77). Each subunit has a variable C-terminus, which is a key factor in the transport of AMPARs. Among them, the GluA1 and GluA2 subunits play important roles in cytosis and internalization (78). Ubiquitination of both subunits is critical for regulating the surface and synaptic expression of AMPARs (79). It has been shown that promoting GluA1 expression improves synaptic transmission efficacy, and its hyperubiquitination leads to amyloid-β (Aβ)-induced downregulation of surface AMPAR expression and inhibition of excitatory synaptic transmission (80). The activity-dependent ubiquitination of GluA1 occurs at the C-terminal Lys-868 residue and is primarily mediated by the E3 ligases Nedd4-1, Nedd4-2, and RNF220 (81–83). Their co-localization and binding to AMPARs trigger GluA1 ubiquitination and promote the internalized degradation of AMPARs. GluA2 is also an important subunit that determines the function of AMPAR, and its expression in tetramers correlates with the rate of inactivation, single-channel conductance, and Ca2+ permeability of AMPARs (84). The process of GluA2 internalization, where it is taken back into the synapse from the postsynaptic membrane, weakens synaptic transmission when increased (75). Consequently, an overactivation of receptors for AMPARs containing the GluA2 subunit results in dysfunction and potential death of neurons in AD. The RING-type E3 ubiquitin ligase, RNF167, is a transmembrane protein that resides in endosomes and lysosomes and is involved in regulating the endolysosomal pathway. GluA2, as one of the substrates of RNF167, is ubiquitinated by first binding to the E2 coupling enzyme UBE2N, and then the RING structural domain of RNF167 binds to the coupling enzyme between them, completing the ubiquitin degradation of GluA2 (85), and enhancing synaptic transmission efficacy. Additionally, the ubiquitination site is mainly located on the Lys-870/Lys-882 residues at the C-terminal end of the GluA2 subunit (79) (Figure 4). Notably, ubiquitination of the GluA1 subunit inhibits synaptic transmission, whereas ubiquitination of the GluA2 subunit enhances synaptic transmission. This is due to the different physiological functions of the different subunits in the transport of AMPARs.
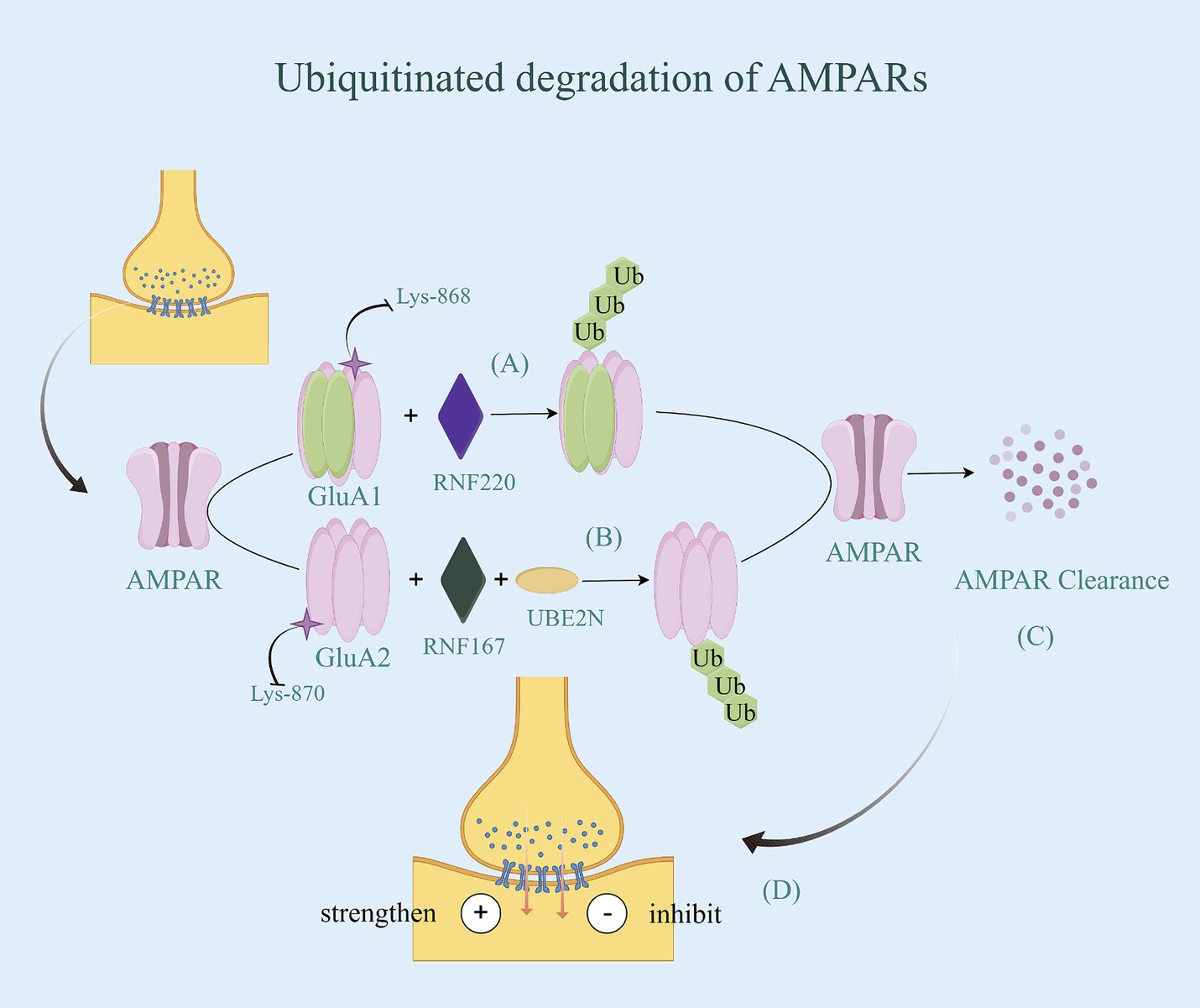
Figure 4. Ubiquitinated degradation of AMPARs. (A) The activity-dependent ubiquitination of GluA1 occurs at the C-terminal Lys-868 residue and is primarily mediated by the E3 RNF220. Their co-localization and binding to AMPARs trigger GluA1 ubiquitination and promote the internalized degradation of AMPARs. (B) GluA2, as one of the substrates of RNF167, is ubiquitinated by first binding to the E2 coupling enzyme UBE2N, and then the RING structural domain of RNF167 binds to the coupling enzyme between them, completing the ubiquitin degradation of GluA2, and enhancing synaptic transmission efficacy. Additionally, the ubiquitination site is mainly located on the Lys-870/Lys-882 residues at the C-terminal end of the GluA2 subunit. (C) AMPARs with ubiquitin be identified by the proteasome, thereby being degraded. (D) Due to the different physiological functions of the different subunits in the transport of AMPARs, ubiquitination of the GluA1 subunit inhibits synaptic transmission, whereas ubiquitination of the GluA2 subunit enhances synaptic transmission.
4 Conclusion and discussion
Ubiquitin-dependent protein degradation is essential for neuronal health, where protein remodeling underlies specific brain processes such as memory and learning as well as synaptic plasticity (86). This paper not only explores the process of ubiquitination modification but also provides an overview of the ubiquitin degradation of proteins associated with Alzheimer’s disease (AD), particularly focusing on the significant role of E3 ubiquitin ligases in this process (Table 1). Considering the complexity and diversity of ubiquitination modifications, the discussion here is limited to the regulation of protein degradation. In fact, the signaling pathways involved in ubiquitination also play a crucial role. For instance, in the NF-κB pathway, the IκBα protein is degraded through ubiquitination modification, lifting the inhibition of NF-κB and enabling it to enter the nucleus to carry out transcriptional activities, regulating inflammatory and immune responses (87). Additionally, more E3 ubiquitin ligases have been implicated in AD, such as mitochondrial ubiquitin ligase (MITOL/MARCH5), situated in the outer membrane of mitochondria, and contributing to the maintenance of mitochondrial homeostasis by clearing protein aggregates that have accumulated on mitochondria (88). MITOL deficiency has been shown to disrupt mitochondrial dynamics, leading to mitochondrial damage and worsening cognitive decline in the APP/PS1 mouse model. However, further investigation is required to understand the detailed pathways of action and ubiquitin sites.
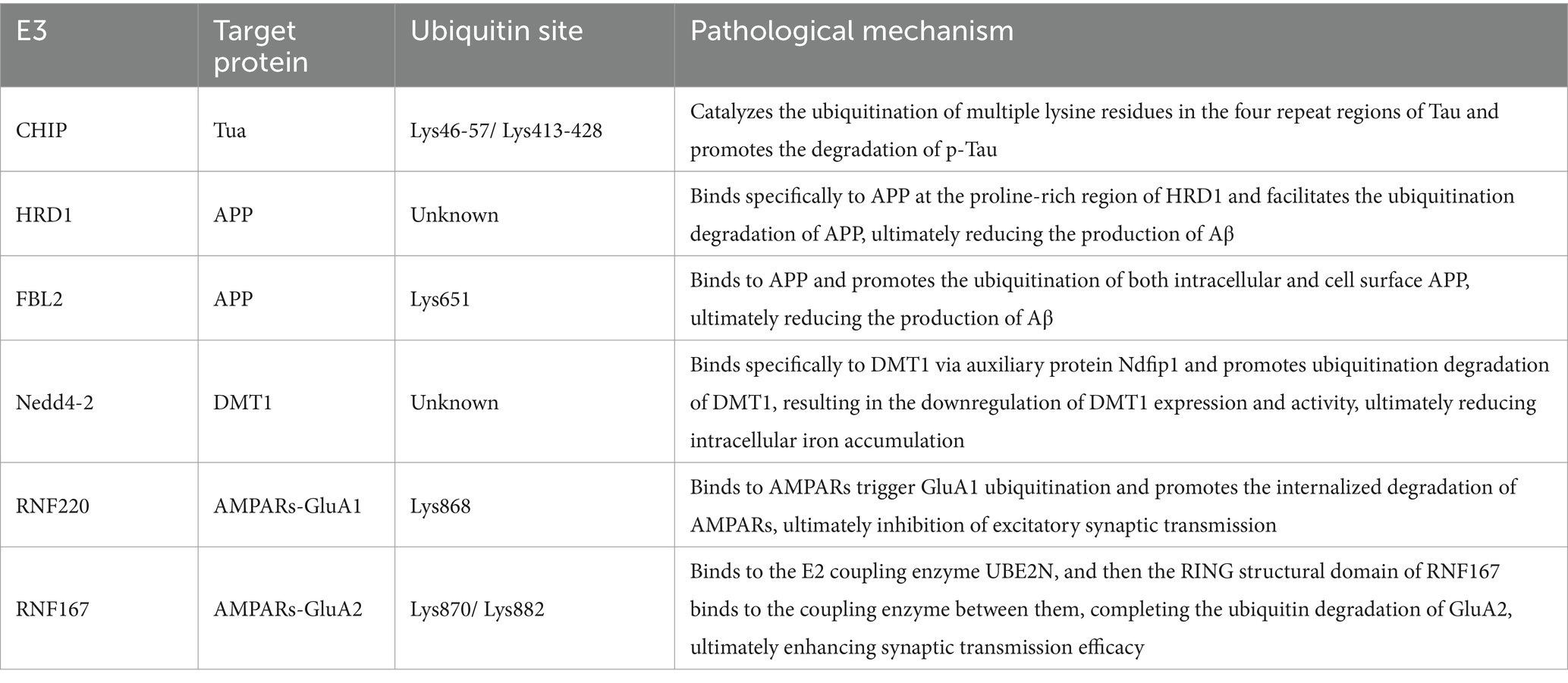
Table 1. Pathological mechanism of E3 involved in ubiquitination of AD-related proteins.
AD is characterized as a protein-related disorder, with disturbances in protein homeostasis playing a crucial role. Compared to other neurodegenerative diseases such as Parkinson’s, which relies primarily on ubiquitin-dependent mitochondrial autophagy to accomplish protein degradation due to its defective mitochondrial function (89), AD relies primarily on UPS. UPS is an important post-translational modification for protein degradation and control of homeostasis. The enzymes involved in UPS, such as E1, E2, E3 ligases, and DUB, regulate disease-induced protein aggregation and degradation by controlling the degree of ubiquitination (90). During the ubiquitination degradation process of AD-associated proteins, E3, acting as a bridge to transfer activated Ub from E2 to the substrate proteins, is specifically recognized by the proteasome for degradation, which in turn delays the development of AD. On the other hand, DUBs remove Ub from substrate proteins by cleaving the peptide or isopeptide bond between Ub and substrate, rescuing the substrate proteins from the ubiquitination degradation pathway and preventing them from being degraded. This in turn leads to the accumulation of AD-associated neurotoxicity proteins and facilitates the development of AD. Extensive research has been dedicated to identifying novel targets for AD treatment. Given the complex mechanisms and diverse proteins involved, this paper highlights the significance of four major protein degradation pathways in AD pathogenesis, emphasizing the potential of targeting key components in these pathways for therapeutic interventions. With the increase in clinical studies, modulators based on ubiquitination-targeted therapy for AD have emerged. For instance, the modulator geniposide increases HRD1 expression, promoting the phosphorylation of inositol requiring enzyme 1 alpha (IRE1α), resulting in accelerated degradation of amyloid precursor protein (APP) (91). Another modulator, X-box binding protein (XBP-1), indirectly decreases the expression and activity of β-site APP cleaving enzyme (BACE1) by up-regulating HRD1, reducing amyloid-beta (Aβ) production (92). The small molecule agonists Sulforaphane, Anisomycin and Peptidoglycan (PGN), which promote CHIP expression, inhibit the progression of AD by promoting the expression of CHIP in the cerebral cortex and hippocampus, leading to ubiquitinated degradation of phosphorylated Tau proteins, and may be targeted drugs for the treatment of neurological disorders (93). Additionally, the involvement of deubiquitinating enzymes (DUBs), including ubiquitin-specific protease14 (USP14), in AD development has been identified. USP14, a proteasome-associated ubiquitin-specific protease, plays a critical role in neurodegenerative diseases, inflammatory responses, tumorigenesis and other aspects (94). Studies have shown that USP14 inhibits ubiquitination of hyperphosphorylated Tau proteins and induces further exacerbation of AD. The small molecule inhibitor of USP14, IU1-47, has been shown to be targetable for the treatment of AD by accelerating the degradation of hyperphosphorylated Tau proteins through inhibition of USP14 activity (95). Lee et al. conducted preclinical study to test whether the small molecule inhibitor of USP14, IU1, could inhibit the trimming of ubiquitin chains by the proteasome and whether IU1 could enhance proteasome function in cells (96). The result shows IU1 enhanced proteasome function and was helpful for Tua degradation. Furthermore, it could potentially be used to eliminate misfolded proteins more effectively. However, the efficacy of the IU1 series of compounds for the treatment of AD by targeting USP14 requires further clinical trials studies.
Although ubiquitination events are considered a promising target for AD therapy, the identification of highly potent and specific E3 and DUB modulators has not been successful due to the complexity of the enzyme cascade involved in ubiquitin-binding. E3 and DUB have a synergistic or exclusive role in functionality. Therefore, more in-depth research is needed to explore the mechanism of E3 and DUB action in AD, as well as the balance between ubiquitination and deubiquitination. In recent years, the treatment of AD by proteolysistargeting chimera (PROTAC) is also very promising. However, there are still few ligands for E3 ligase, which has a certain degree of limitation on the treatment of AD. Whether there are new E3s and related target proteins or known E3s and target proteins that can regulate the development of AD through novel mechanisms needs future studies. Furthermore, whether they can be used clinically as therapeutic targets for AD also requires in-depth study with the aim of providing new approaches for the future treatment of AD.
Author contributions
NL: Writing – original draft, Writing – review & editing. X-YG: Conceptualization, Writing – review & editing. XL: Resources, Writing – review & editing. W-MC: Visualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This review article was supported by grants from the Henan Province’s Scientific Research Program for the Creation of “Double First Class” of Traditional Chinese Medicine (HSRP-DFCTCM-2023-3-14) and the Postdoctoral Fellowship Program of CPSF (GZC20240442).
Acknowledgments
The figures were created by Figdraw, for which we would like to express our gratitude.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dubois, B, von Arnim, C, Burnie, N, Bozeat, S, and Cummings, J. Biomarkers in Alzheimer's disease: role in early and differential diagnosis and recognition of atypical variants. Alzheimers Res Ther. (2023) 15:175. doi: 10.1186/s13195-023-01314-6
2. Scheltens, P, De Strooper, B, Kivipelto, M, Holstege, H, Chételat, G, Teunissen, CE, et al. Alzheimer's disease. Lancet. (2021) 397:1577–90. doi: 10.1016/S0140-6736(20)32205-4
3. Khan, S, Barve, KH, and Kumar, MS. Recent advancements in pathogenesis, diagnostics and treatment of Alzheimer's disease. Curr Neuropharmacol. (2020) 18:1106–25. doi: 10.2174/1570159X18666200528142429
4. Abiose, O, Rutledge, J, Moran-Losada, P, Belloy, ME, Wilson, EN, He, Z, et al. Post-translational modifications linked to preclinical Alzheimer's disease-related pathological and cognitive changes. Alzheimers Dement. (2024) 20:1851–67. doi: 10.1002/alz.13576
5. Fernández-Cruz, I, and Reynaud, E. Proteasome subunits involved in neurodegenerative diseases. Arch Med Res. (2021) 52:1–14. doi: 10.1016/j.arcmed.2020.09.007
6. Bao, WD, Pang, P, Zhou, XT, Hu, F, Xiong, W, Chen, K, et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer's disease. Cell Death Differ. (2021) 28:1548–62. doi: 10.1038/s41418-020-00685-9
7. Ju, Y, and Tam, KY. Pathological mechanisms and therapeutic strategies for Alzheimer's disease. Neural Regen Res. (2022) 17:543–9. doi: 10.4103/1673-5374.320970
8. French, ME, Koehler, CF, and Hunter, T. Emerging functions of branched ubiquitin chains. Cell Discov. (2021) 7:6. doi: 10.1038/s41421-020-00237-y
9. Yu, J, Qin, B, and Lou, Z. Ubiquitin and ubiquitin-like molecules in DNA double strand break repair. Cell Biosci. (2020) 10:13. doi: 10.1186/s13578-020-0380-1
10. Komander, D, and Rape, M. The ubiquitin code. Annu Rev Biochem. (2012) 81:203–29. doi: 10.1146/annurev-biochem-060310-170328
11. Swatek, KN, and Komander, D. Ubiquitin modifications. Cell Res. (2016) 26:399–422. doi: 10.1038/cr.2016.39
12. Komander, D. The emerging complexity of protein ubiquitination. Biochem Soc Trans. (2009) 37:937–53. doi: 10.1042/BST0370937
13. Maniv, I, Sarji, M, Bdarneh, A, Feldman, A, Ankawa, R, Koren, E, et al. Altered ubiquitin signaling induces Alzheimer's disease-like hallmarks in a three-dimensional human neural cell culture model. Nat Commun. (2023) 14:5922. doi: 10.1038/s41467-023-41545-7
14. Zhu, F, Zheng, GL, and Tian, XJ. Progress of autophagy-related deubiquitinating enzymes and their small molecule inhibitors. Adv Biochem Biophys. (2020) 47:210–23. doi: 10.16476/j.pibb.2019.0301
15. Abreha, MH, Dammer, EB, Ping, L, Zhang, T, Duong, DM, Gearing, M, et al. Quantitative analysis of the brain Ubiquitylome in Alzheimer's disease. Proteomics. (2018) 18:e1800108. doi: 10.1002/pmic.201800108
16. De Silva, A, and Page, RC. Ubiquitination detection techniques. Exp Biol Med (Maywood). (2023) 248:1333–46. doi: 10.1177/15353702231191186
17. Wegmann, S, Biernat, J, and Mandelkow, E. A current view on tau protein phosphorylation in Alzheimer's disease. Curr Opin Neurobiol. (2021) 69:131–8. doi: 10.1016/j.conb.2021.03.003
18. LaFerla, FM, Green, KN, and Oddo, S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci. (2007) 8:499–509. doi: 10.1038/nrn2168
19. Bierer, LM, Hof, PR, Purohit, DP, Carlin, L, Schmeidler, J, Davis, KL, et al. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer's disease. Arch Neurol. (1995) 52:81–8. doi: 10.1001/archneur.1995.00540250089017
20. Kang, J, and Müller-Hill, B. Differential splicing of Alzheimer's disease amyloid a4 precursor RNA in rat tissues: PreA4(695) mRNA is predominantly produced in rat and human brain. Biochem Biophys Res Commun. (1990) 166:1192–200. doi: 10.1016/0006-291x(90)90992-v
21. Wei, Z. Deubiquitinating enzyme USP10 promotes tau aggregation involved in Alzheimer's disease pathogenesis and its mechanisms. China: Huazhong University of Science and Technology (2022).
22. Li, L, Jiang, Y, Wang, JZ, Liu, R, and Wang, X. Tau ubiquitination in Alzheimer's disease. Front Neurol. (2021) 12:786353. doi: 10.3389/fneur.2021.786353
23. Sampson, C, Wang, Q, Otkur, W, Zhao, H, Lu, Y, Liu, X, et al. The roles of E3 ubiquitin ligases in cancer progression and targeted therapy. Clin Transl Med. (2023) 13:e1204. doi: 10.1002/ctm2.1204
24. Metzger, MB, Pruneda, JN, Klevit, RE, and Weissman, AM. RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim Biophys Acta. (2014) 1843:47–60. doi: 10.1016/j.bbamcr.2013.05.026
25. Hatakeyama, S, Matsumoto, M, Kamura, T, Murayama, M, Chui, DH, Planel, E, et al. U-box protein carboxyl terminus of Hsc70-interacting protein (CHIP) mediates poly-ubiquitylation preferentially on four-repeat tau and is involved in neurodegeneration of tauopathy. J Neurochem. (2004) 91:299–307. doi: 10.1111/j.1471-4159.2004.02713.x
26. Kim, JH, Lee, J, Choi, WH, Park, S, Park, SH, Lee, JH, et al. CHIP-mediated hyperubiquitylation of tau promotes its self-assembly into the insoluble tau filaments. Chem Sci. (2021) 12:5599–610. doi: 10.1039/d1sc00586c
27. Joshi, V, Amanullah, A, Upadhyay, A, Mishra, R, Kumar, A, and Mishra, A. A decade of boon or burden: what has the CHIP ever done for cellular protein quality control mechanism implicated in neurodegeneration and aging? Front Mol Neurosci. (2016) 9:93. doi: 10.3389/fnmol.2016.00093
28. Wang, L, Liu, YT, Hao, R, Chen, L, Chang, Z, Wang, HR, et al. Molecular mechanism of the negative regulation of Smad1/5 protein by carboxyl terminus of Hsc70-interacting protein (CHIP). J Biol Chem. (2011) 286:15883–94. doi: 10.1074/jbc.M110.201814
29. Munari, F, Barracchia, CG, Franchin, C, Parolini, F, Capaldi, S, Romeo, A, et al. Semisynthetic and enzyme-mediated conjugate preparations illuminate the ubiquitination-dependent aggregation of tau protein. Angew Chem Int Ed Engl. (2020) 59:6607–11. doi: 10.1002/anie.201916756
30. Wesseling, H, Mair, W, Kumar, M, Schlaffner, CN, Tang, S, Beerepoot, P, et al. Tau PTM profiles identify patient heterogeneity and stages of Alzheimer's disease. Cell. (2020) 183:1699–1713.e13. doi: 10.1016/j.cell.2020.10.029
31. Munari, F, Mollica, L, Valente, C, Parolini, F, Kachoie, EA, Arrigoni, G, et al. Structural basis for chaperone-independent ubiquitination of tau protein by its E3 ligase CHIP. Angew Chem Int Ed Engl. (2022) 61:e202112374. doi: 10.1002/anie.202112374
32. Wang, T, Wang, W, Wang, Q, Xie, R, Landay, A, and Chen, D. The E3 ubiquitin ligase CHIP in normal cell function and in disease conditions. Ann N Y Acad Sci. (2020) 1460:3–10. doi: 10.1111/nyas.14206
33. Dickey, CA, Patterson, C, Dickson, D, and Petrucelli, L. Brain CHIP: removing the culprits in neurodegenerative disease. Trends Mol Med. (2007) 13:32–8. doi: 10.1016/j.molmed.2006.11.003
34. Petrucelli, L, Dickson, D, Kehoe, K, Taylor, J, Snyder, H, Grover, A, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. (2004) 13:703–14. doi: 10.1093/hmg/ddh083
35. Mazanetz, MP, and Fischer, PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov. (2007) 6:464–79. doi: 10.1038/nrd2111
36. Oddo, S, Vasilevko, V, Caccamo, A, Kitazawa, M, Cribbs, DH, and LaFerla, FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. (2006) 281:39413–23. doi: 10.1074/jbc.M608485200
37. Li, Q. Ubiquitin ligase Hrd1-mediated tau protein degradation. China: Anhui Medical University (2008).
38. Zhang, YW, Thompson, R, Zhang, H, and Xu, H. APP processing in Alzheimer's disease. Mol Brain. (2011) 4:3. doi: 10.1186/1756-6606-4-3
39. Dawkins, E, and Small, DH. Insights into the physiological function of the β-amyloid precursor protein: beyond Alzheimer's disease. J Neurochem. (2014) 129:756–69. doi: 10.1111/jnc.12675
40. Tsatsanis, A, Dickens, S, Kwok, J, Wong, BX, and Duce, JA. Post translational modulation of β-amyloid precursor protein trafficking to the cell surface alters neuronal Iron homeostasis. Neurochem Res. (2019) 44:1367–74. doi: 10.1007/s11064-019-02747-y
41. Kaneko, M, Koike, H, Saito, R, Kitamura, Y, Okuma, Y, and Nomura, Y. Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-beta generation. J Neurosci. (2010) 30:3924–32. doi: 10.1523/JNEUROSCI.2422-09.2010
42. Watanabe, T, Hikichi, Y, Willuweit, A, Shintani, Y, and Horiguchi, T. FBL2 regulates amyloid precursor protein (APP) metabolism by promoting ubiquitination-dependent APP degradation and inhibition of APP endocytosis. J Neurosci. (2012) 32:3352–65. doi: 10.1523/JNEUROSCI.5659-11.2012
43. Kaneko, M, Ishiguro, M, Niinuma, Y, Uesugi, M, and Nomura, Y. Human HRD1 protects against ER stress-induced apoptosis through ER-associated degradation. FEBS Lett. (2002) 532:147–52. doi: 10.1016/s0014-5793(02)03660-8
44. Ilyin, GP, Rialland, M, Glaise, D, and Guguen-Guillouzo, C. Identification of a novel Skp2-like mammalian protein containing F-box and leucine-rich repeats. FEBS Lett. (1999) 459:75–9. doi: 10.1016/s0014-5793(99)01211-9
45. Blalock, EM, Geddes, JW, Chen, KC, Porter, NM, Markesbery, WR, and Landfield, PW. Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci USA. (2004) 101:2173–8. doi: 10.1073/pnas.0308512100
46. Dixon, SJ, Lemberg, KM, Lamprecht, MR, Skouta, R, Zaitsev, EM, Gleason, CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
47. Wang, F, Wang, J, Shen, Y, Li, H, Rausch, WD, and Huang, X. Iron Dyshomeostasis and Ferroptosis: a new Alzheimer's disease hypothesis? Front Aging Neurosci. (2022) 14:830569. doi: 10.3389/fnagi.2022.830569
48. Kim, AC, Lim, S, and Kim, YK. Metal ion effects on Aβ and tau aggregation. Int J Mol Sci. (2018) 19:128. doi: 10.3390/ijms19010128
49. Becerril-Ortega, J, Bordji, K, Fréret, T, Rush, T, and Buisson, A. Iron overload accelerates neuronal amyloid-β production and cognitive impairment in transgenic mice model of Alzheimer's disease. Neurobiol Aging. (2014) 35:2288–301. doi: 10.1016/j.neurobiolaging.2014.04.019
50. Wang, X, Wang, Y, Li, Z, Qin, J, and Wang, P. Regulation of Ferroptosis pathway by ubiquitination. Front Cell Dev Biol. (2021) 9:699304. doi: 10.3389/fcell.2021.699304
51. Nevo, Y, and Nelson, N. The mutation F227I increases the coupling of metal ion transport in DCT1. J Biol Chem. (2004) 279:53056–61. doi: 10.1074/jbc.M408398200
52. Zheng, W, Xin, N, Chi, ZH, Zhao, BL, Zhang, J, Li, JY, et al. Divalent metal transporter 1 is involved in amyloid precursor protein processing and Abeta generation. FASEB J. (2009) 23:4207–17. doi: 10.1096/fj.09-135749
53. Xie, L, Zheng, W, Xin, N, Xie, JW, Wang, T, and Wang, ZY. Ebselen inhibits iron-induced tau phosphorylation by attenuating DMT1 up-regulation and cellular iron uptake. Neurochem Int. (2012) 61:334–40. doi: 10.1016/j.neuint.2012.05.016
54. Howitt, J, Putz, U, Lackovic, J, Doan, A, Dorstyn, L, Cheng, H, et al. Divalent metal transporter 1 (DMT1) regulation by Ndfip1 prevents metal toxicity in human neurons. Proc Natl Acad Sci USA. (2009) 106:15489–94. doi: 10.1073/pnas.0904880106
55. Lu, L, Jifu, C, Xia, J, and Wang, J. E3 ligases and DUBs target ferroptosis: a potential therapeutic strategy for neurodegenerative diseases. Biomed Pharmacother. (2024) 175:116753. doi: 10.1016/j.biopha.2024.116753
56. Scheffner, M, Nuber, U, and Huibregtse, JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. (1995) 373:81–3. doi: 10.1038/373081a0
57. Putz, U, Howitt, J, Lackovic, J, Foot, N, Kumar, S, Silke, J, et al. Nedd4 family-interacting protein 1 (Ndfip1) is required for the exosomal secretion of Nedd4 family proteins. J Biol Chem. (2008) 283:32621–7. doi: 10.1074/jbc.M804120200
58. Low, LH, Chow, YL, Li, Y, Goh, CP, Putz, U, Silke, J, et al. Nedd4 family interacting protein 1 (Ndfip1) is required for ubiquitination and nuclear trafficking of BRCA1-associated ATM activator 1 (BRAT1) during the DNA damage response. J Biol Chem. (2015) 290:7141–50. doi: 10.1074/jbc.M114.613687
59. Foot, NJ, Dalton, HE, Shearwin-Whyatt, LM, Dorstyn, L, Tan, SS, Yang, B, et al. Regulation of the divalent metal ion transporter DMT1 and iron homeostasis by a ubiquitin-dependent mechanism involving Ndfips and WWP2. Blood. (2008) 112:4268–75. doi: 10.1182/blood-2008-04-150953
60. Tian, J, Zheng, W, Li, XL, Cui, YH, and Wang, ZY. Lower expression of Ndfip1 is associated with Alzheimer disease pathogenesis through decreasing DMT1 degradation and increasing Iron influx. Front Aging Neurosci. (2018) 10:165. doi: 10.3389/fnagi.2018.00165
61. Hong, M, Kim, M, Kim, TW, Park, SS, Kim, MK, Park, YH, et al. Treadmill exercise improves motor function and short-term memory by enhancing synaptic plasticity and neurogenesis in Photothrombotic stroke mice. Int Neurourol J. (2020) 24:S28–38. doi: 10.5213/inj.2040158.079
62. Ganguly, K, and Poo, MM. Activity-dependent neural plasticity from bench to bedside. Neuron. (2013) 80:729–41. doi: 10.1016/j.neuron.2013.10.028
63. Li, W, Kong, LH, Wang, H, Shen, F, Wang, YW, Zhou, H, et al. High-frequency electroacupuncture evidently reinforces hippocampal synaptic transmission in Alzheimer's disease rats. Neural Regen Res. (2016) 11:801–6. doi: 10.4103/1673-5374.182708
64. Shankar, GM, and Walsh, DM. Alzheimer's disease: synaptic dysfunction and Abeta. Mol Neurodegener. (2009) 4:48. doi: 10.1186/1750-1326-4-48
65. Larson, J, Lynch, G, Games, D, and Seubert, P. Alterations in synaptic transmission and long-term potentiation in hippocampal slices from young and aged PDAPP mice. Brain Res. (1999) 840:23–35. doi: 10.1016/s0006-8993(99)01698-4
66. Gureviciene, I, Ikonen, S, Gurevicius, K, Sarkaki, A, van Groen, T, Pussinen, R, et al. Normal induction but accelerated decay of LTP in APP + PS1 transgenic mice. Neurobiol Dis. (2004) 15:188–95. doi: 10.1016/j.nbd.2003.11.011
67. Zhang, Y, Zhang, J, Wang, Y, and Yao, J. Global trends and prospects about synaptic plasticity in Alzheimer's disease: a bibliometric analysis. Front Aging Neurosci. (2023) 15:1234719. doi: 10.3389/fnagi.2023.1234719
68. Mabb, AM, and Ehlers, MD. Ubiquitination in postsynaptic function and plasticity. Annu Rev Cell Dev Biol. (2010) 26:179–210. doi: 10.1146/annurev-cellbio-100109-104129
69. Artinian, J, McGauran, AM, De Jaeger, X, Mouledous, L, Frances, B, and Roullet, P. Protein degradation, as with protein synthesis, is required during not only long-term spatial memory consolidation but also reconsolidation. Eur J Neurosci. (2008) 27:3009–19. doi: 10.1111/j.1460-9568.2008.06262.x
70. Hegde, AN, and DiAntonio, A. Ubiquitin and the synapse. Nat Rev Neurosci. (2002) 3:854–61. doi: 10.1038/nrn961
71. Hegde, AN. Proteolysis, synaptic plasticity and memory. Neurobiol Learn Mem. (2017) 138:98–110. doi: 10.1016/j.nlm.2016.09.003
72. Dong, C, Upadhya, SC, Ding, L, Smith, TK, and Hegde, AN. Proteasome inhibition enhances the induction and impairs the maintenance of late-phase long-term potentiation. Learn Mem. (2008) 15:335–47. doi: 10.1101/lm.984508
73. Gong, B, Cao, Z, Zheng, P, Vitolo, OV, Liu, S, Staniszewski, A, et al. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell. (2006) 126:775–88. doi: 10.1016/j.cell.2006.06.046
74. Wang, C, Yu, Y, Xie, K, and Wang, G. Progress of AMPA receptors in Alzheimer's disease. J Tianjin Med Univ. (2020) 26:292–5.
75. Twomey, EC, Yelshanskaya, MV, Grassucci, RA, Frank, J, and Sobolevsky, AI. Elucidation of AMPA receptor-stargazin complexes by cryo-electron microscopy. Science. (2016) 353:83–6. doi: 10.1126/science.aaf8411
76. Parameshwaran, K, Dhanasekaran, M, and Suppiramaniam, V. Amyloid beta peptides and glutamatergic synaptic dysregulation. Exp Neurol. (2008) 210:7–13. doi: 10.1016/j.expneurol.2007.10.008
77. Keifer, J, and Zheng, Z. AMPA receptor trafficking and learning. Eur J Neurosci. (2010) 32:269–77. doi: 10.1111/j.1460-9568.2010.07339.x
78. Cheng, RZ, Wang, K, Sun, WM, Chun, XJ, Yan, ND, Zhang, W, et al. Effects of the kidney enhancing and turbidity-reducing formula on internalization in Alzheimer's disease model mice. World TCM. (2023) 18:1679–83. doi: 10.3969/j.issn.1673-7202.2023.12.006
79. Widagdo, J, Chai, YJ, Ridder, MC, Chau, YQ, Johnson, RC, Sah, P, et al. Activity-dependent ubiquitination of GluA1 and GluA2 regulates AMPA receptor intracellular sorting and degradation. Cell Rep. (2015) 10:783–95. doi: 10.1016/j.celrep.2015.01.015
80. Zhang, Y, Guo, O, Huo, Y, Wang, G, and Man, HY. Amyloid-β induces AMPA receptor ubiquitination and degradation in primary neurons and human brains of Alzheimer's disease. J Alzheimers Dis. (2018) 62:1789–801. doi: 10.3233/JAD-170879
81. Schwarz, LA, Hall, BJ, and Patrick, GN. Activity-dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. J Neurosci. (2010) 30:16718–29. doi: 10.1523/JNEUROSCI.3686-10.2010
82. Zhu, J, Lee, KY, Jewett, KA, Man, HY, Chung, HJ, and Tsai, NP. Epilepsy-associated gene Nedd4-2 mediates neuronal activity and seizure susceptibility through AMPA receptors. PLoS Genet. (2017) 13:e1006634. doi: 10.1371/journal.pgen.1006634
83. Ma, P, Wan, LP, Li, Y, He, CH, Song, NN, Zhao, S, et al. RNF220 is an E3 ubiquitin ligase for AMPA receptors to regulate synaptic transmission. Sci Adv. (2022) 8:eabq4736. doi: 10.1126/sciadv.abq4736
84. Zhao, ZH, and Guo, J. Ampa receptors and Alzheimer's disease. Chem Life. (2010) 30:202–6. doi: 10.13488/j.smhx.2010.02.032
85. Ghilarducci, K, Cabana, VC, Desroches, C, Chabi, K, Bourgault, S, Cappadocia, L, et al. Functional interaction of ubiquitin ligase RNF167 with UBE2D1 and UBE2N promotes ubiquitination of AMPA receptor. FEBS J. (2021) 288:4849–68. doi: 10.1111/febs.15796
86. Dörrbaum, AR, Kochen, L, Langer, JD, and Schuman, EM. Local and global influences on protein turnover in neurons and glia. eLife. (2018) 7:7. doi: 10.7554/eLife.34202
87. Chen, J, and Chen, ZJ. Regulation of NF-κB by ubiquitination. Curr Opin Immunol. (2013) 25:4–12. doi: 10.1016/j.coi.2012.12.005
88. Park, YJ, Dodantenna, N, Kim, Y, Kim, TH, Lee, HS, Yoo, YS, et al. MARCH5-dependent NLRP3 ubiquitination is required for mitochondrial NLRP3-NEK7 complex formation and NLRP3 inflammasome activation. EMBO J. (2023) 42:e113481. doi: 10.15252/embj.2023113481
89. Junqueira, SC, Centeno, E, Wilkinson, KA, and Cimarosti, H. Post-translational modifications of Parkinson's disease-related proteins: phosphorylation, SUMOylation and ubiquitination. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:2001–7. doi: 10.1016/j.bbadis.2018.10.025
90. Do, HA, and Baek, KH. Cellular functions regulated by deubiquitinating enzymes in neurodegenerative diseases. Ageing Res Rev. (2021) 69:101367. doi: 10.1016/j.arr.2021.101367
91. Cui, H, Deng, M, Zhang, Y, Yin, F, and Liu, J. Geniposide increases unfolded protein response-mediating HRD1 expression to accelerate APP degradation in primary cortical neurons. Neurochem Res. (2018) 43:669–80. doi: 10.1007/s11064-018-2469-z
92. Gerakis, Y, Dunys, J, Bauer, C, and Checler, F. Aβ42 oligomers modulate β-secretase through an XBP-1s-dependent pathway involving HRD1. Sci Rep. (2016) 6:37436. doi: 10.1038/srep37436
93. Zhang, S, Hu, ZW, Mao, CY, Shi, CH, and Xu, YM. CHIP as a therapeutic target for neurological diseases. Cell Death Dis. (2020) 11:727. doi: 10.1038/s41419-020-02953-5
94. Massa, F, Tammaro, R, Prado, MA, Cesana, M, Lee, BH, Finley, D, et al. The deubiquitinating enzyme Usp14 controls ciliogenesis and hedgehog signaling. Hum Mol Genet. (2019) 28:764–77. doi: 10.1093/hmg/ddy380
95. Wang, F, Ning, S, Yu, B, and Wang, Y. USP14: structure, function, and target inhibition. Front Pharmacol. (2021) 12:801328. doi: 10.3389/fphar.2021.801328
Keywords: Alzheimer’s disease, ubiquitination, protein degradation, Tua, APP, DMT1, AMPARs
Citation: Lin N, Gao X-Y, Li X and Chu W-M (2024) Involvement of ubiquitination in Alzheimer’s disease. Front. Neurol. 15:1459678. doi: 10.3389/fneur.2024.1459678
Edited by:
Mayur Doke, University of Miami Health System, United StatesReviewed by:
Isabella Altilio, University of Miami, United StatesSumedha Kapre, Texas A&M University, United States
Copyright © 2024 Lin, Gao, Li and Chu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xi-Yan Gao, Z2FveGl5YW5AeWVhaC5uZXQ=