 Adina Wise1,2
Adina Wise1,2 Roberto A. Ortega1,2
Roberto A. Ortega1,2 Deborah Raymond1,2
Deborah Raymond1,2 Alessandra Cervera3,4,5Emma Thorn3,4,5Katherine Leaver1,2
Alessandra Cervera3,4,5Emma Thorn3,4,5Katherine Leaver1,2 David S. Russell6
David S. Russell6 Susan B. Bressman1,2
Susan B. Bressman1,2 John F. Crary3,4,5*
John F. Crary3,4,5* Rachel Saunders-Pullman1,2*
Rachel Saunders-Pullman1,2*- 1Department of Neurology, Mount Sinai Beth Israel Medical Center, New York, NY, United States
- 2Department of Neurology, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 3Departments of Pathology, Neuroscience, and Artificial Intelligence & Human Health, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 4Neuropathology Brain Bank & Research CoRE, Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 5Ronald M. Loeb Center for Alzheimer’s Disease, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 6Institute for Neurodegenerative Disorders, New Haven, CT, United States
Background: LRRK2 variants have been associated with immune dysregulation as well as immune-related disorders such as IBD. A possible relationship between multiple sclerosis (MS) and LRRK2 PD has also been suggested. Further, neuropathologic studies of homozygous LRRK2 G2019S carriers with Parkinson’s disease (PD) are rare, and there are no systematic reports of clinical features in those cases.
Methods: We investigated the co-occurrence of PD and MS in our research cohort and report on two cases of MS in LRRK2 PD as well as neuropathological findings for one.
Results: MS preceded PD in 1.4% (2/138) of participants with LRRK2 G2019S variants, and in none (0/638) with idiopathic PD (p = 0.03). One case with MS and PD was a LRRK2 G2019S homozygous carrier, and neuropathology showed evidence of substantia nigra pars compacta degeneration and pallor without Lewy deposition, as well as multiple white matter lesions consistent with MS-related demyelination.
Discussion: The increased prevalence of MS in LRRK2 PD further supports an important role for immune function for LRRK2 PD. This co-occurrence, while rare, suggests that MS may be an expression of the LRRK2 G2019S variant that includes both MS and PD, with MS predating features diagnostic of PD. The neuropathology suggests that the MS-related effects occurred independent of synuclein deposition. Importantly, and in addition, the neuropathological results not only support the MS diagnosis, but provide further evidence that Lewy body pathology may be absent even in homozygote LRRK2 carriers.
Introduction
An immune role for LRRK2 in mediating PD has been suggested (1), and the question of increased frequency of concomitant multiple sclerosis and LRRK2 G2019S-related PD (LRRK2 PD) has been raised (2). Homozygous LRRK2-related Parkinson disease (PD) has been reported and does not appear motorically worse than heterozygous LRRK2 PD (3), however, descriptions of non-motor features and neuropathology in these cases are limited (4).
Methods
Participants were part of genetic studies of PD at Mount Sinai Beth Israel/Mount Sinai, and screening for LRRK2 G2019S and eleven GBA variants was performed as previously reported (5). Twelve individuals with both LRRK2 G2019S and GBA1 variants were excluded from the analysis. History of multiple sclerosis was ascertained by patient report and prevalence of MS was compared between LRRK2 PD and non-LRRK2 PD groups. The non-LRRK2 group was comprised of 142 individuals with GBA PD and 496 individuals with idiopathic PD. In the case with neuropathology, olfaction was measured by University of Pennsylvania Smell Identification Test (UPSIT), striatal dopamine transporter density assessment using iodine-123–labeled 2-β-carboxymethoxy-3-β-(4-iodophenyl)tropane single photon emission-computed tomography (β-CIT SPECT), and transcranial sonography (6, 7) was performed. The Mount Sinai IRB approved both the overall studies (AAAE0748 and AAAF3108), and the neuropathological protocols; next-of-kin provided consent for brain donation. Histopathological workup consisted of a comprehensive neuroanatomical sampling assessed according to standard protocols as previously described (8). Routine (hematoxylin & eosin, Luxol fast blue, and Bielschowsky silver) and a battery of immunohistochemical (IHC) stains for phospho-tau (AT8, 1:1000, Invitrogen), β-amyloid (4G8, 1:8000, BioLegend), TAR DNA-binding protein 43 (TDP-43, 1:5000, Proteintech), α-synuclein (LB509, 1:3000, Abcam), and neurofilament (SMI-32, 1:1000, Biolegend).
Results
MS was present and preceded PD in 1.4% (2/138) of participants with LRRK2 G2019S variants. This was increased relative to the non-LRRK2 PD group in which no participants had an MS diagnosis that preceded the onset of PD (p = 0.03). In the subset of women, 2.9% (2/69) of LRRK2 PD participants had preceding diagnoses of MS (p = 0.05).
In the case with neuropathology, the participant received the diagnosis of MS in her mid-30’s when she developed left leg numbness, and this was attributed to a cervical cord lesion. During her 40’s, she experienced relapses and remissions with MRIs consistent with demyelinating plaques. Notable exacerbations included left arm weakness with loss of dexterity and, later, weakness of the left leg. Cervical spine MRI at age 67 showed abnormal signal intensity without cord expansion. Head CT at age 75 showed a hypodensity in the right frontal centrum semiovale, extending into the right periventricular region, with a similar, less prominent focus in the left centrum semiovale and subcortical white matter. She declined disease modifying treatment for MS. At age 67, she developed left arm tremor, and at age 68, she was diagnosed with PD. β-CIT SPECT revealed marked, symmetrically reduced uptake indicating significant loss of striatal dopamine neuronal innervation (Figure 1D). At age 72, examination demonstrated right hand rest tremor and bilateral action/postural tremors. In addition, there was bilateral rigidity and bradykinesia, both worse on the left. She had moderately stooped posture, required assistance with walking and tended to lose balance spontaneously. MoCA score was 21/30 (normosmic for age and sex, and her performance placed her in the best latent class, which was associated with the least severe motor decline) (9). Nigral transcranial sonography was within normal range (0.144 cm2 and 0.121 cm2) (6, 7). At age 78, she developed endometroid ovarian cancer and she died at age 80 from sepsis.
On gross neuropathological examination, there were numerous, chronic, white matter lesions bilaterally, often seen at the gray-white matter junction and periventricularly, as well as throughout the frontal, parietal, temporal and occipital lobes, consistent with demyelinating lesions (Figure 1A). In the cervical spinal cord, histological examination showed pallor and reactive gliosis in the lateral funiculus, with intact axonal neurofilament staining consistent with demyelination (Figures 1B,C). In the midbrain, there was severe pallor evident grossly; microscopically, there was loss of pigmented neurons in the substantia nigra without Lewy bodies, which was confirmed by immunohistochemistry targeting α-synuclein (Figures 1E,F,H), however mild (1+) Lewy bodies were found in the medulla and olfactory bulb (Figures 1G,M,N). Rare α-synuclein positive Lewy neurites of undetermined significance were noted in the white matter of the brainstem (Figure 1O). Additionally, the cingulate gyrus, amygdala, frontal cortex, and hippocampus were all negative for α-synuclein (Figures 1I–L).
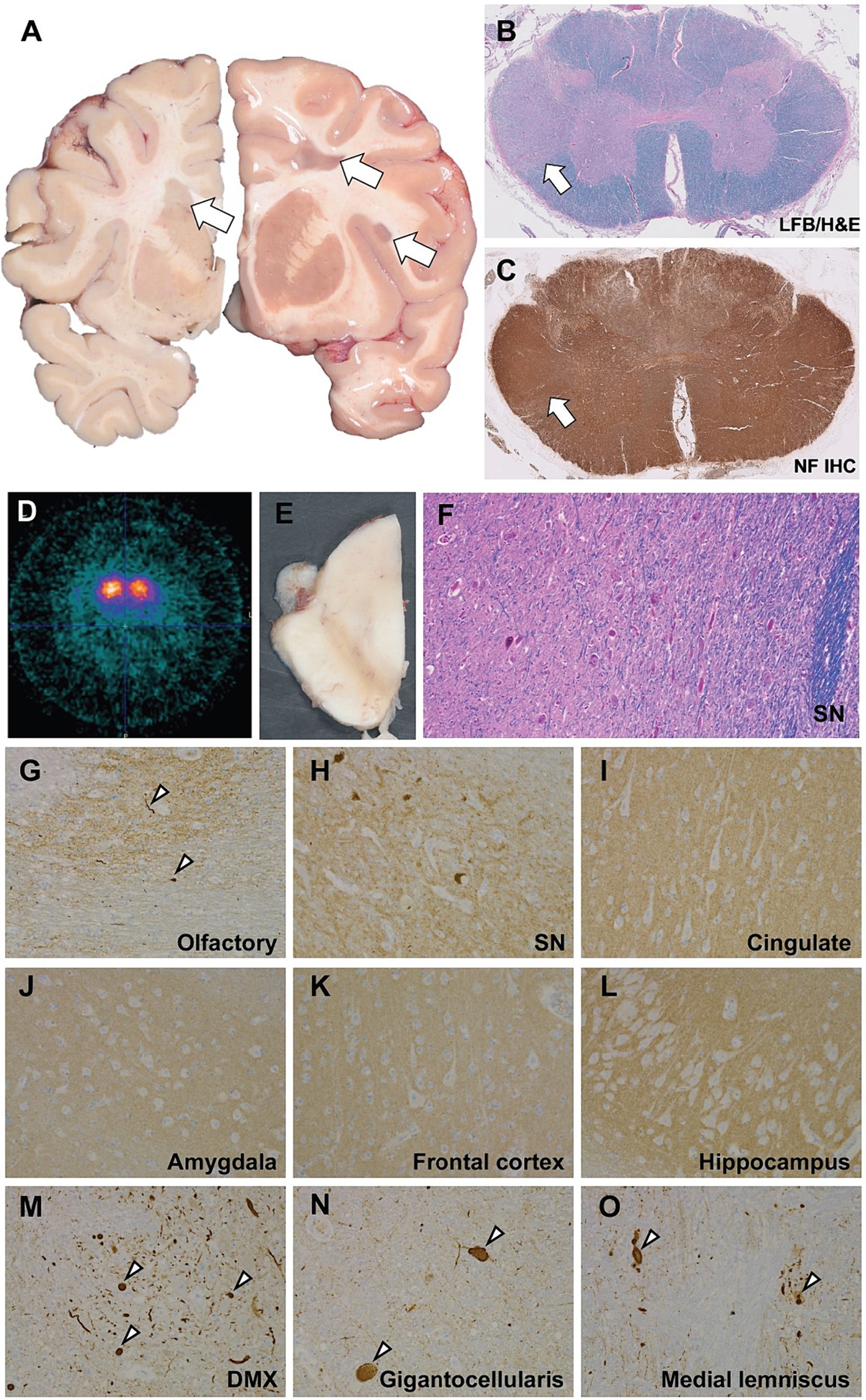
Figure 1. Neuropathological and neuroimaging evaluation demonstrating multiple sclerosis and brainstem degeneration in the absence of nigral Lewy bodies. (A) Gross coronal sections showing scattered white matter plaques (white arrows). (B) Luxol fast blue counterstained hematoxyoin & eosin stained (LH&E) section from the cervical spinal cord showing myelin pallor in the lateral funiculus. (C) Immunohistochemistry (IHC) using antisera targeting neurofilament shows intact axons consistent with a demyelinating process. (D) b-CIT SPECT revealed marked, symmetrically reduced uptake indicating significant loss of striatal dopamine neuronal innervation. (E) Gross hemisection of the midbrain demonstrating marked pallor in the substantia nigra. (F) High power (20x) photomicrograph of an LH&E-stained section through the midbrain showing marked loss of pigmented dopaminergic neurons and reactive gliosis. Immunohistochemistry to α-synuclein shows mild Lewy pathology (white arrowheads) in the olfactory bulb (G), negative staining in the substantia nigra (H), cingulate gyrus (I), amygdala (J), frontal cortex (K), hippocampus (L), and positive staining in the medulla, including the dorsal motor nucleus of the vagus (DMX; M), nucleus gigantocellularis (N), and medial lemniscus (O).
The microscopic examination of the brain yielded various regions that were p-tau (AT8) positive including rare structures in the white matter of the substantia nigra, mild (1+) neurofibrillary tangles (NFTs) in the striatum, putamen, nucleus basalis of Meynert, amygdala, hypothalamus, and upper pons. Additionally, there were severe (3+) NFTs present in the inner and outer layers of the entorhinal area, and rare granular p-tau positive clusters and threads in the inferior frontal cortex. These findings are consistent with a Braak stage of III (B2).
Phospho-TDP-43 proteinopathy was negative in the olfactory bulb, midbrain at level of red nucleus, and all levels of the spinal cord including the cauda equina.
The second participant with LRRK2 PD was diagnosed with MS at age 45 after the onset of horizontal diplopia. At age 48 she experienced bilateral lower extremity weakness and was treated with interferon beta-1a but discontinued due to side effects. She was switched to glatiramer acetate and stayed on this medication for at least 8 years, but did not take it consistently. Clinical features in her case were also consistent with a relapsing–remitting phenotype. At age 66, she developed stooped posture and difficulty with gait. She was diagnosed with PD and started on carbidopa-levodopa with improvement. MRI at age 75 demonstrated elongated, flame-shaped T2 hyperintense lesions perpendicular to the lateral ventricle wall as well as peri-colossally and at the parietal-occipital junction. Age-related atrophy of the bilateral cerebral hemispheres was also noted.
Discussion
The higher rate of MS preceding PD, albeit an uncommon occurrence, supports that MS may be part of the phenotypic spectrum of LRRK2 GS019S carriage, and provides further support for immune relation in LRRK2. The numerous, white matter lesions seen on neuropathology in one of the participants with MS and LRRK2 PD, and on MRI in the other, together with nigral cell loss in the first case, substantiate a chronic, demyelinating disorder separate from, and preceding, PD. The development of MS prior to PD in individuals with the G2019S variant is not altogether surprising as LRRK2 plays an important role in a variety of immune responses and is found within monocytes, macrophages, B lymphocytes and other immune cells [Reviewed by Zhang et al. (10)]. Several studies have shown that cells with this gain of function mutation (increased kinase activity) have exaggerated pro-inflammatory responses, including heightened release of cytokines (11, 12). Correspondingly, associations between LRRK2 carrier status and other immune conditions, including Inflammatory Bowel Disease (IBD)/Crohn’s Disease, have been raised (13–15). Genome wide associations have found that LRRK2 G2019S mutations are a risk factor for both Crohn’s disease (CD) and PD, with inflammatory processes proposed as common mechanistic pathways (15). Given these established links, it is worth considering that impairments in LRRK2 G2019S may create the conditions whereby immune dysregulation underlies a subtype of neurological disease with features of both MS and PD.
Increased prevalence of autoimmune diseases relative to other PD groups was found in a Norwegian sample of 100 LRRK2 carriers, and included three individuals (3%) diagnosed with MS (2). In roughly the same geographic location, overall MS prevalence among the local population has been estimated at 0.16%, suggesting that LRRK2 carrier status may indeed confer an increased risk of developing MS (16). While our sample prevalence was smaller (1.4%), these results are highly suggestive of an association between MS and LRRK2 PD that is not attributable to chance.
The presented neuropathological findings suggest that homozygous LRRK2 mutation carriers do not have greater PD severity than heterozygotes. These results also highlight the absence of Lewy body pathology in a significant subset (17). Though a putaminal demyelinating lesion was seen on neuropathological examination, it is unlikely that this unilateral lesion was the cause of this participant’s bilateral PD symptoms. In addition, most MS patients who have basal ganglia or midbrain lesions do not develop signs of PD and parkinsonism is quite rare in MS patients overall (18).
Among the over 30 cases of homozygous LRRK2 mutations reported to date, most are biallelic G2019S variants (3, 4, 19–22). The phenotype is similar in individuals carrying either one or two LRRK2 G2019S mutations and, in general, both homozygous and heterozygous mutations are associated with low rates of cognitive impairment and psychiatric co-morbidity. PD in LRRK2 homozygous carriers is not thought to be more penetrant than in heterozygous carriers and there is no evidence to suggest more severe clinical manifestations (3, 19–22). In line with these observations, our homozygous participant presented with symptoms that were, overall, consistent with relatively mild and slowly progressive Parkinson disease.
Lewy bodies, long-considered to be distinctive features of PD-associated neurodegeneration, are absent in a significant portion of LRRK2 cases. Among 37 autopsy LRRK2 PD cases, only 17 had Lewy bodies with Lewy body pathology found more frequently in G2019S carriers than in other LRRK2 variants (17). Though motor features were similar among LRRK2 PD cases with and without Lewy body pathology, non-motor features, including orthostatic hypotension, anxiety and cognitive impairment, were more frequent in the Lewy body group (17). Of interest, there was no nigral hyperechogenicity on transcranial sonography.
While olfaction has not been consistently reported in PD autopsy cases, absence of olfactory loss, and its potential association with isolated nigral degeneration, is of great interest. We have previously reported on two clusters of olfactory performance in G2019S LRRK2 PD: in the worse-performing cluster, PD age of onset was earlier and motor decline was faster (9). We, therefore, hypothesized that the cluster with better olfaction may show isolated nigral degeneration. While larger clinical-autopsy studies are necessary to further assess this correlation, these results raise the question as to whether excellent olfaction may be a clinical surrogate for absence of synuclein pathology. This is supported by recent CSF synuclein seeding assay studies demonstrating lower frequency of SSA abnormalities in normosmic LRRK2 PD (23).
Our participant’s significant bilateral nigral degeneration, without substantial Lewy Body pathology, extends the LRRK2 G2019S neuropathologic phenotype to suggest that the absence of Lewy body deposition may occur even in biallelic G2019S carriers. A prior report of two homozygous R1441 mutation carriers also showed prominent nigral degeneration without alpha-synuclein pathology (24). In a separate neuropathological analysis of 10 LRRK2 G2019S carriers (two homozygous and eight heterozygous), only half of the examined samples had alpha-synuclein pathology; unfortunately, the homozygous cases were not separately delineated (4).
Late in life this participant developed endometrial cancer. A possible association between LRRK2 variants and non-skin cancers has also been reported, though the mechanisms of the link remains incompletely understood (25).
This study bolsters a hypothesized link between immunologic disease and LRRK2 PD, and raises important questions about shared immune pathophysiology contributing to disease development. It also highlights the potential for personalized approaches through the lens of the pathologic heterogeneity of LRRK2 PD. In particular, evidence (or lack thereof) of microscopic Lewy body pathology, through synuclein or surrogates for pathology, may guide choice of anti-synuclein agents. Thus, our results emphasize the importance of continuing to work toward a clear delineation of PD subtypes, as therapeutic trials focused on groups with shared causality are far more likely to succeed. In addition, neuropathological findings support that homozygous LRRK2 mutations are not associated with worse brain pathology than heterozygote LRRK2 mutations, though we cannot exclude the possibility that, in this case, LRRK2 homozygosity may have contributed to the development of MS. Our findings suggest the need to investigate concurrent immune and inflammatory diseases in Parkinson’s disease, and especially in LRRK2 PD.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the deceased patient’s family for publication of the details of their medical case and any accompanying images.
Author contributions
AW: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. RO: Writing – original draft, Writing – review & editing. DeR: Writing – original draft, Writing – review & editing. AC: Writing – original draft, Writing – review & editing. ET: Writing – original draft, Writing – review & editing. KL: Writing – original draft, Writing – review & editing. DaR: Writing – original draft, Writing – review & editing. SB: Writing – original draft, Writing – review & editing. JC: Writing – original draft, Writing – review & editing. RS-P: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was funded by the Empire Clinical Research Investigator Program, National Institute of Neurological Disorders and Stroke (NINDS) (U01NS107016-01A1), Michael J Fox Foundation for Parkinson’s Research, Parkinson’s Disease Foundation (JC), National Institute of Aging (RF1NS095252), National Institute of Neurological Disorders and Stroke (R01AG054008, R01NS086736).
Acknowledgments
We sincerely thank all the participants and their family members for their invaluable contributions to this research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cook, DA, Kannarkat, GT, Cintron, AF, Butkovich, LM, Fraser, KB, Chang, J, et al. LRRK2 levels in immune cells are increased in Parkinson's disease. NPJ Parkinson's Dis. (2017) 3:11. doi: 10.1038/s41531-017-0010-8
2. Aasly, JO . Inflammatory diseases among Norwegian LRRK2 mutation carriers. A 15-years follow-up of a cohort. Front Neurosci. (2021) 15:634666. doi: 10.3389/fnins.2021.634666
3. Ishihara, L, Warren, L, Gibson, R, Amouri, R, Lesage, S, Dürr, A, et al. Clinical features of Parkinson disease patients with homozygous leucine-rich repeat kinase 2 G2019S mutations. Arch Neurol. (2006) 63:1250–4. doi: 10.1001/archneur.63.9.1250
4. Henderson, MX, Sengupta, M, Trojanowski, JQ, and Lee, VMY. Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol Commun. (2019) 7:183. doi: 10.1186/s40478-019-0836-x
5. Miltenberger-Miltenyi, G, Ortega, RA, Domingo, A, Yadav, R, Nishiyama, A, Raymond, D, et al. Genetic risk variants in New Yorkers of Puerto Rican and Dominican Republic heritage with Parkinson’s disease. npj Parkinson’s Disease (2023) 9:160.
6. Brüggemann, N, Hagenah, J, Stanley, K, Klein, C, Wang, C, Raymond, D, et al. Substantia nigra hyperechogenicity with LRRK2 G2019S mutations. Mov Disord. (2011) 26:885–8. doi: 10.1002/mds.23644
7. Pullman, M, Ortega, R, Glickman, A, Deik, A, Raymond, D, Marder, K, et al. Increased substantia Nigra echogenicity in LRRK2 family members without mutations. Mov Disord. (2018) 33:1504–5. doi: 10.1002/mds.27443
8. Ditzel, RM Jr, Walker, RH, Nirenberg, MJ, Tetlow, AM, Farrell, K, Lind-Watson, KJ, et al. An autopsy series of seven cases of VPS13A disease (chorea-Acanthocytosis). Mov Disord. (2023) 38:2163–72. doi: 10.1002/mds.29589
9. Saunders-Pullman, R, Ortega, RA, Wang, C, Raymond, D, Elango, S, Leaver, K, et al. Association of Olfactory Performance with Motor Decline and age at onset in people with Parkinson disease and the LRRK2 G2019S variant. Neurology. (2022) 99:e814–23. doi: 10.1212/WNL.0000000000200737
10. Zhang, M, Li, C, Ren, J, Wang, H, Yi, F, Wu, J, et al. The double-faceted role of leucine-rich repeat kinase 2 in the Immunopathogenesis of Parkinson's disease. Front Aging Neurosci. (2022) 14:909303. doi: 10.3389/fnagi.2022.909303
11. Ahmadi Rastegar, D, Hughes, LP, Perera, G, Keshiya, S, Zhong, S, Gao, J, et al. Effect of LRRK2 protein and activity on stimulated cytokines in human monocytes and macrophages. NPJ Parkinsons Dis. (2022) 8:34. doi: 10.1038/s41531-022-00297-9
12. Moehle, MS, Daher, JPL, Hull, TD, Boddu, R, Abdelmotilib, HA, Mobley, J, et al. The G2019S LRRK2 mutation increases myeloid cell chemotactic responses and enhances LRRK2 binding to actin-regulatory proteins. Hum Mol Genet. (2015) 24:4250–67. doi: 10.1093/hmg/ddv157
13. Peter, I, and Strober, W. Immunological features of LRRK2 function and its role in the gut-brain Axis governing Parkinson’s disease. J Parkinsons Dis. (2023) 13:279–96. doi: 10.3233/JPD-230021
14. Hui, KY, Fernandez-Hernandez, H, Hu, J, Schaffner, A, Pankratz, N, Hsu, NY, et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn's disease and Parkinson's disease. Sci Transl Med. (2018) 10:eaai7795. doi: 10.1126/scitranslmed.aai7795
15. Tsafaras, G, and Baekelandt, V. The role of LRRK2 in the periphery: link with Parkinson's disease and inflammatory diseases. Neurobiol Dis. (2022) 172:105806. doi: 10.1016/j.nbd.2022.105806
16. Dahl, OP, Aarseth, JH, Myhr, KM, Nyland, H, and Midgard, R. Multiple sclerosis in Nord-Trøndelag County, Norway: a prevalence and incidence study. Acta Neurol Scand. (2004) 109:378–84. doi: 10.1111/j.1600-0404.2004.00244.x
17. Kalia, LV, Lang, AE, Hazrati, LN, Fujioka, S, Wszolek, ZK, Dickson, DW, et al. Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol. (2015) 72:100–5. doi: 10.1001/jamaneurol.2014.2704
18. Ghosh, R, Roy, D, Dubey, S, das, S, and Benito-León, J. Movement disorders in multiple sclerosis: an update. Tremor Other Hyperkinet Mov. (2022) 12:14. doi: 10.5334/tohm.671
19. du Toit, N, van Coller, R, Anderson, DG, Carr, J, and Bardien, S. Frequency of the LRRK2 G2019S mutation in south African patients with Parkinson's disease. Neurogenetics. (2019) 20:215–8. doi: 10.1007/s10048-019-00588-z
20. Ishihara, L, Gibson, RA, Warren, L, Amouri, R, Lyons, K, Wielinski, C, et al. Screening for LRRK2 G2019S and clinical comparison of Tunisian and north American Caucasian Parkinson's disease families. Mov Disord. (2007) 22:55–61. doi: 10.1002/mds.21180
21. Nichols, WC, Pankratz, N, Hernandez, D, Paisán-Ruíz, C, Jain, S, Halter, CA, et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson's disease. Lancet. (2005) 365:410–2. doi: 10.1016/S0140-6736(05)17828-3
22. Lesage, S, Dürr, A, Tazir, M, Lohmann, E, Leutenegger, AL, Janin, S, et al. LRRK2 G2019S as a cause of Parkinson's disease in north African Arabs. N Engl J Med. (2006) 354:422–3. doi: 10.1056/NEJMc055540
23. Siderowf, A, Concha-Marambio, L, Lafontant, DE, Farris, CM, Ma, Y, Urenia, PA, et al. Assessment of heterogeneity among participants in the Parkinson's progression markers initiative cohort using α-synuclein seed amplification: a cross-sectional study. Lancet Neurol. (2023) 22:407–17. doi: 10.1016/S1474-4422(23)00109-6
24. Takanashi, M, Funayama, M, Matsuura, E, Yoshino, H, Li, Y, Tsuyama, S, et al. Isolated nigral degeneration without pathological protein aggregation in autopsied brains with LRRK2 p.R1441H homozygous and heterozygous mutations. Acta Neuropathol Commun. (2018) 6:105. doi: 10.1186/s40478-018-0617-y
Keywords: multiple sclerosis, Parkinson’s disease, LRRK2, genetic, genetic risk, Parkinson’s genes, immunology & inflammation, autoimmune diseases
Citation: Wise A, Ortega RA, Raymond D, Cervera A, Thorn E, Leaver K, Russell DS, Bressman SB, Crary JF and Saunders-Pullman R (2024) Multiple sclerosis in LRRK2 G2019S Parkinson’s disease and isolated nigral degeneration in a homozygous variant carrier. Front. Neurol. 15:1450654. doi: 10.3389/fneur.2024.1450654
Edited by:
Emilia Mabel Gatto, Sanatorio de la Trinidad Mitre, ArgentinaReviewed by:
Alberto J. Espay, University of Cincinnati, United StatesMartin Emiliano Cesarini, INEBA Institute of Neurosciences Buenos Aires, Argentina
Copyright © 2024 Wise, Ortega, Raymond, Cervera, Thorn, Leaver, Russell, Bressman, Crary and Saunders-Pullman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rachel Saunders-Pullman, UmFjaGVsLnNhdW5kZXJzLXB1bGxtYW5AbW91bnRzaW5haS5vcmc=; John F. Crary, am9obi5jcmFyeUBtb3VudHNpbmFpLm9yZw==