 Katarzyna Radomska1*
Katarzyna Radomska1* Zofia Leszczyńska1
Zofia Leszczyńska1 Rafal Becht2
Rafal Becht2 Monika Zaborek - Łyczba3Anna Rzepakowska4Jakub Lubiński1
Monika Zaborek - Łyczba3Anna Rzepakowska4Jakub Lubiński1 Marcin Szymański3
Marcin Szymański3- 1Department of Otolaryngology, Pomeranian Medical University, Szczecin, Poland
- 2Department of Oncology, Pomeranian Medical University, Szczecin, Poland
- 3Department of Otolaryngology, Medical University of Lublin, Lublin, Poland
- 4Department of Otolaryngology, Medical University of Warsaw, Warsaw, Poland
Paragangliomas are rare tumors originating from the paraventricular bodies of the autonomic nervous system located in the adrenal glands, chest, abdomen, pelvis and head and neck. Tumors of this type account for 0.5% of head and neck cancers, 0.03% of all cancers and their incidence is estimated at 1–30/100,000 per year. Head and Neck Paragangliomas (HNPGL) are localized in carotid body, tympanic cavity or jugular foramen. It is established that HNPGL may be associated with mutations of the SDH complex, with SDHD being the most prevalent. However, SDHB, SDHC and SDHAF are also potential causes. The aforementioned mutations are influenced by various risk factors, including young age, a positive family history of paraganglioma, the presence of metastases and gender The purpose of this study is to summarize the results of genetic testing performed on patients with head and neck paraganglioma and to create an up-to-date genetic diagnosis algorithm for patients with HNPGL based on previous studies published in the literature that can be used in daily practice. Several papers observed that among SDHD mutation carriers, most or all of those studied had HNPGL, and SDHB mutations were more frequently found in the presence of metastasis. Based on the results, it was concluded that there is no basis for genetic testing for VHL in patients without a positive family history. In each algorithm proposed by different authors, proposals for rational genetic diagnosis were analyzed based on the studies cited by the author and the analyses included in our paper. For the analysis of the treatment algorithms, the following were included: Martin, Mannelli, Neumann, Gupta. Subsequently, publications related to the genetic diagnosis of HNPGL were analyzed to verify the proposed algorithms in light of the latest genetic studies and to establish an updated diagnostic management scheme.
Introduction
Paragangliomas are rare tumors originating from the paraventricular bodies of the autonomic nervous system (1) located in the adrenal glands, chest, abdomen, pelvis and head and neck. Tumors of this type account for 0.5% of head and neck cancers, 0.03% of all cancers (2), and their incidence is estimated at 1–30/100,000 per year (3). Paragangliomas can originate from the paraganglial bodies of the sympathetic and parasympathetic nervous system, with head and neck paragangliomas (HNPGLs) originating from parasympathetic bodies and are not catecholamine-secreting tumors (1, 2, 4–8). Sympathetic chain PGLs more commonly secrete catecholamines compared to other PGLs, though all HNPGLs may potentially possess this feature (9). A recent study reports that only 3.7% of all HNPGLs lead to elevated normetanephrine levels. Interestingly, this study also found higher levels of methoxytyramine, the O-methylated metabolite of dopamine in benign HNPGLs, which contrasts the prior suggestion of an increase in methoxytyramine and dopamine in metastatic PGLs (10). The tumors are most often benign, but 10–15% may metastasize to bone, liver, lung or lymph nodes (1, 11–13). The current World Health Organization classification divides these tumors into non-metastatic and metastatic (13–15) and recommends using this term instead of “malignant tumors.” They can occur as sporadic, multiple or as part of genetically determined syndromes (3, 7, 16–18).
Genetic testing in patients with paragangliomas is complementary to diagnosis and allows management to be targeted to detect potential multiple tumors or metastases. In addition, tests performed in family members can lead to the diagnosis of paraganglioma at an early stage, while they are still asymptomatic, and implement appropriate management. It is established that HNPGL may be associated with mutations of the SDH complex, with SDHD being the most prevalent. However, SDHB, SDHC and SDHAF are also potential causes. The aforementioned mutations are influenced by various risk factors, including young age, a positive family history of paraganglioma, the presence of metastases and gender (1, 3, 5, 12).
To date, no such work has been conducted for the Polish population, and in determining the indications for diagnosis it is necessary to rely on the results of studies for groups of patients from other countries. Nevertheless, at present, the extension of diagnostics in patients with HNPGL by genetic testing should be an essential part of the treatment process. To this end, bearing in mind also the potential costs of testing, it is worth using a diagnostic algorithm.
Aim of the study
The purpose of this study is to summarize the results of genetic testing performed on patients with head and neck paraganglioma and to create an up-to-date genetic diagnosis algorithm for patients with HNPGL based on previous studies published in the literature that can be used in daily practice.
Methodics
Databases from Medline, Embase, Scopus, Google Scholar from 2000 to X 2023 were searched. Inclusion criteria included papers describing genetic findings in patients with head and neck paragangliomas. Original papers, review papers, and case reports and family studies describing a genetic background or family history of HNPGL were included in the review. Search words used—head and neck paraganglioma (HNPGL) genetic spectrum, HNPGL genetic classification, hereditary HNPGL, SDH mutations in HNPGL, genetic overview of HNPGL, genetic testing in HNPGL. We searched for all results related to the genetic background of HNPGL as well as included review papers that proposed algorithms for genetic diagnosis. Papers examining paragangliomas outside the head and neck region were excluded from the analysis. Also, analyses investigating the molecular mechanisms of mutation effects on PGL development, without clinical analysis, were not included in this paper. Due to the rarity of HNPGL, case reports and family line studies were included in the paper.
Data were downloaded on genetic test results and their correlation with clinical data. The genetic diagnostic algorithms proposed so far were analyzed. Algorithms related to diagnostic imaging or proposed treatment were not included in the study.
The selection of publications for the study is shown in Figure 1.
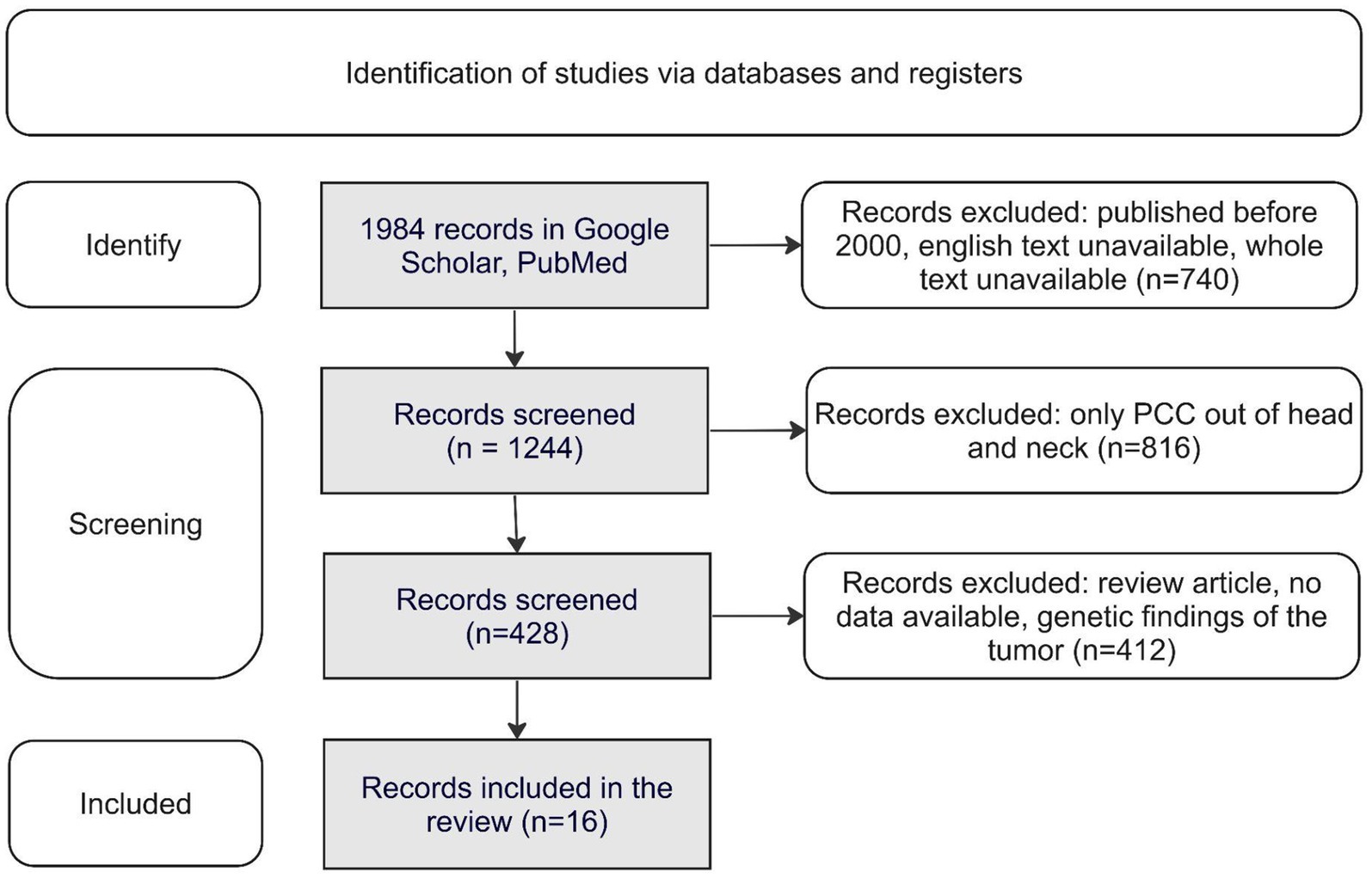
Figure 1. Identification of studies via databases and registers. PCC, Pheochromocytoma.
Results
The results of genetic testing for HNPGL are shown in Table 1, and papers with repeated patient groups are omitted. The most common mutations found were in the SDH complex (2, 22, 25, 27, 28), with mutations within SDHD most common for HNPGL, and SDHB, SDHC and SDHAF2 mutations less common. The papers showed that the main risk factors for mutations were young age, a positive family history of PCC/PGL, multifocality (12, 20, 24), metastatic tumors (lymph nodes, bone, liver, lung) and gender (22). However, there remained a group of patients without risk factors who also had genetic mutations (17).
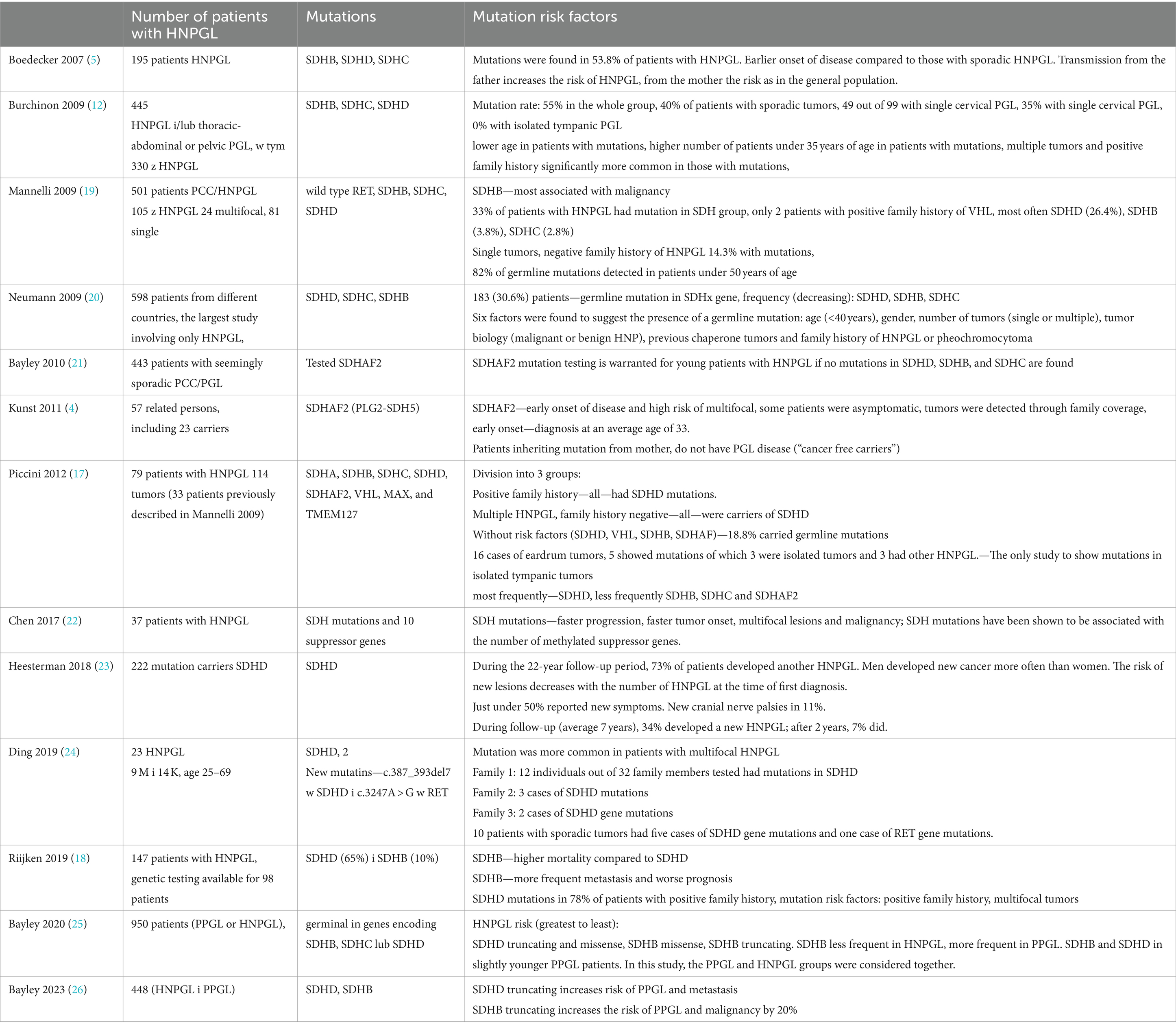
Table 1. Genetic testing in patients with HNPGL in order of publication years.
Several papers observed that among SDHD mutation carriers, most or all of those studied had HNPGL (8, 12, 18, 19, 22, 29, 30), and SDHB mutations were more frequently found in the presence of metastasis (12, 18, 19, 22). Based on the results, it was concluded that there is no basis for genetic testing for VHL in patients without a positive family history (12, 31).
In each algorithm proposed by different authors, proposals for rational genetic diagnosis were analyzed based on the studies cited by the author and the analyses included in our paper. For the analysis of the treatment algorithms, the following were included: Martin (32), Mannelli (19), Neumann (20), and Gupta (31). Subsequently, publications related to the genetic diagnosis of HNPGL were analyzed to verify the proposed algorithms in light of the latest genetic studies and to establish an updated diagnostic management scheme.
The first of these algorithms, proposed by Martin et al. (32), deals with the diagnosis of paragangliomas in general, without dividing them into HNPGL or other tumors, and only proposes genetic diagnosis with SDHD, SDHB and SDHC testing in one aspect, without considering possible risk factors. With the development of genetic testing, new and different schemes have emerged, making diagnosis more targeted, depending on the primary location and the presence of additional factors (19) (Figure 2).
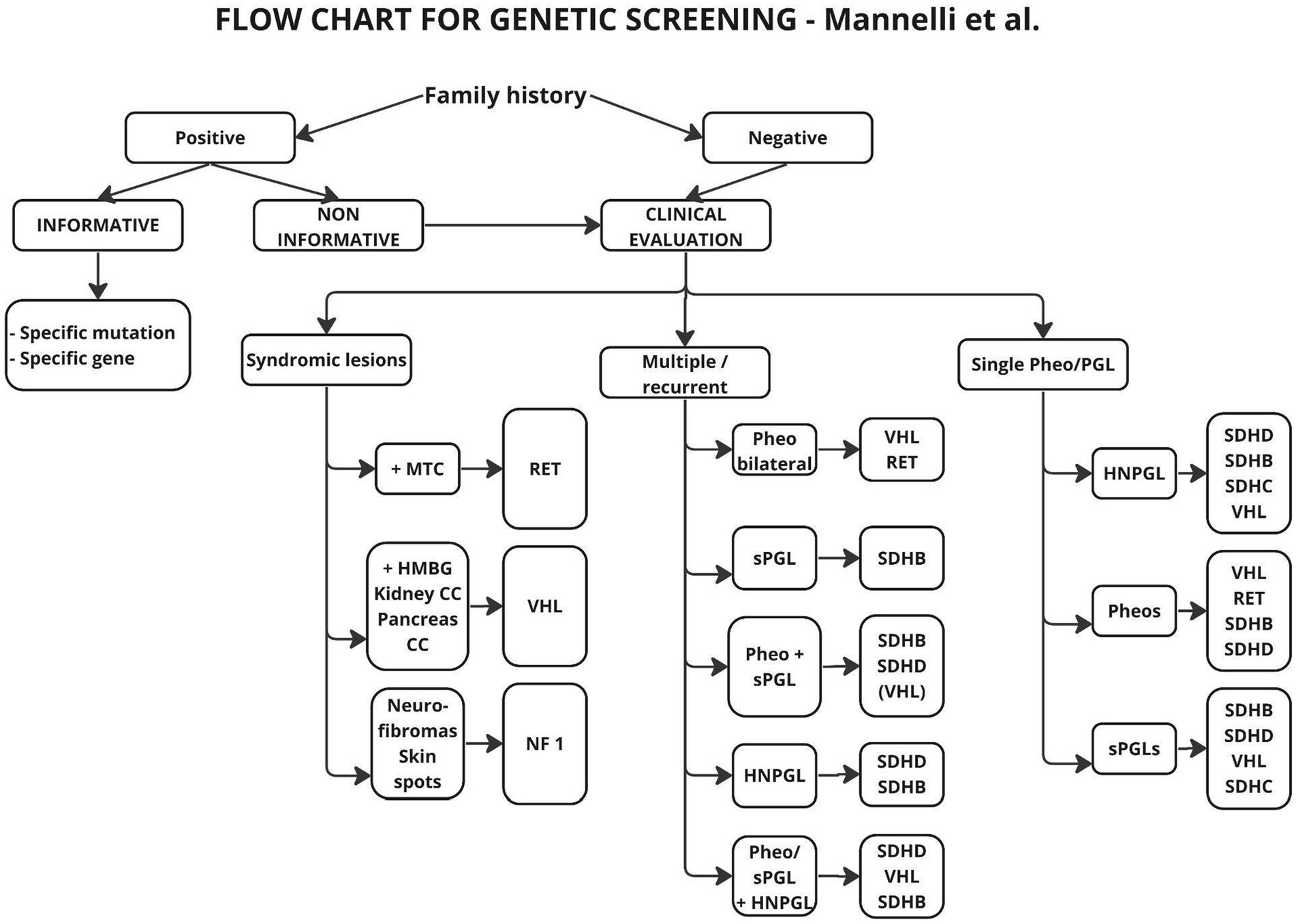
Figure 2. Flow chart suggested for genetic analysis in patients affected by Pheos or PGLs. The genes reported in the boxes are those more likely to be found mutated according to clinical picture. MTC, Medullary thyroid carcinoma; HMGB, hemangioblastomas; CC, cancer/cysts.
In the above scheme, a positive family history and multifocal tumor were taken as risk factors for mutations, but the age of the patients was not taken into account. In the description included in the paper, it is suggested in the aspect of age, to take 50 years as a cut-off point and above this age to abandon genetic testing, since only 5% of patients in this group showed mutations. Other authors (20, 31) suggest 40 years or even 35 years as the cutoff age (12, 20) (Figure 3).
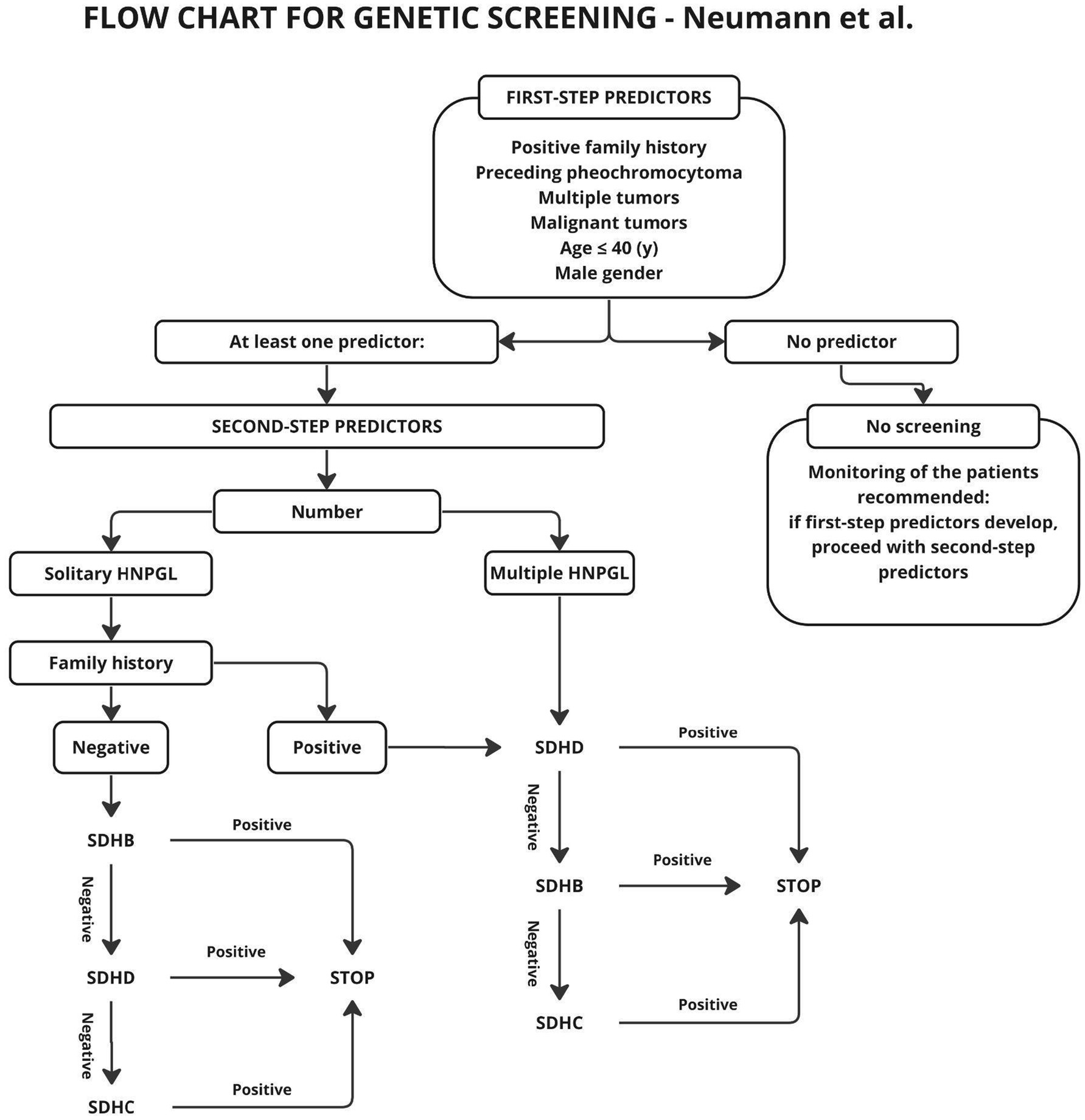
Figure 3. General algorithm for mutation screening of HNP patients. Only those with a first-step predictor being positive enter the main algorithm where second-step predictors are applied.
In the scheme proposed by Neumann et al. (20), genetic testing was to be performed only in patients with risk factors such as positive family history, previous pheochromocytoma, multiple tumors, malignant tumors, age under 40 years or gender. As shown in later studies, also patients without the aforementioned risk factors may carry genetic mutations, and genetic diagnosis should be performed for this group as well (31, 33). The publication of subsequent studies resulted in the algorithm described by Gupta et al. (31), shown in Figure 4.
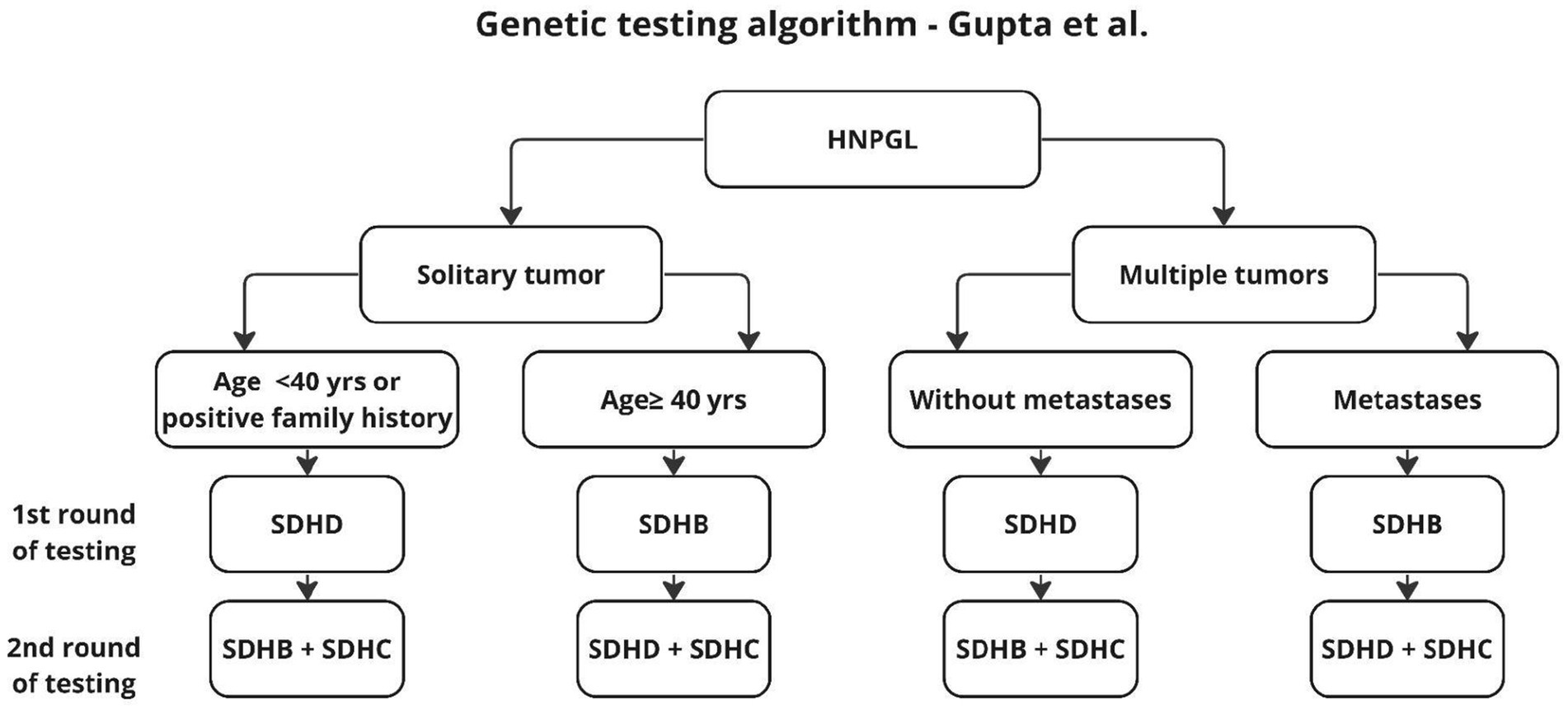
Figure 4. Algorithm according to Gupta et al. (31).
The algorithm proposed by Gupta applies only to HNPGL, dates back to 2019, and proposes two stages of testing to reduce diagnostic costs. In the first stage, the authors recommend genetic testing according to the risk factors found and an algorithm for patients without such factors, with the firąst stage additionally distinguishing age groups, unlike previous authors who proposed genetic testing for the entire SDHx group (19, 20, 32). This approach seems reasonable because, as shown in earlier work, 2 mutations are not found in the same patient (20). However, in light of the results of other studies, it seems reasonable to supplement the above algorithm with SDHAF2 testing, in patients younger than 40 years and in multifocal tumors (4, 21) (Figure 5).
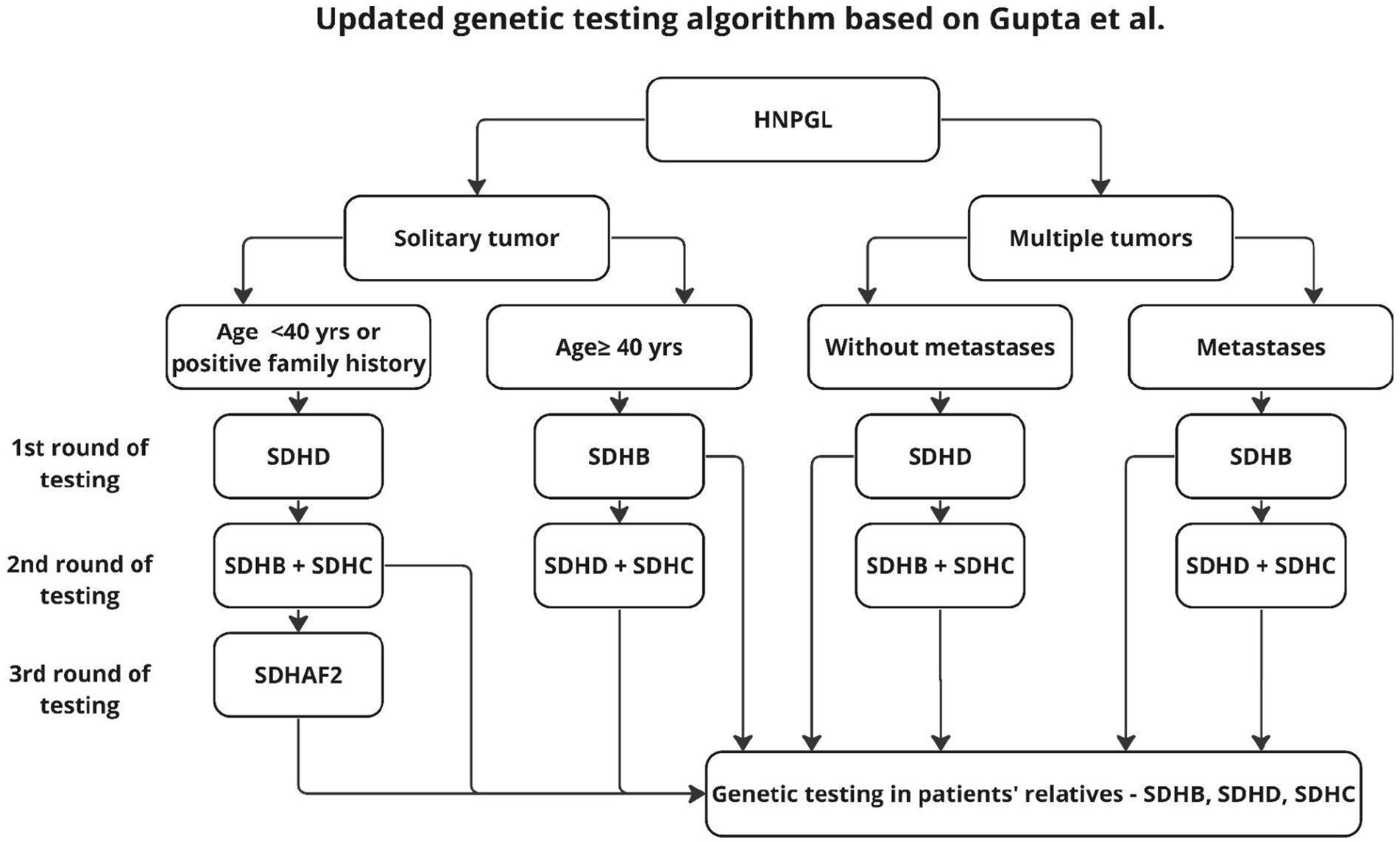
Figure 5. Updated algorithm based on Gupta’s proposal.
Discussion
Over the past few years, one can observe changes related to the approach to genetic diagnosis in paragangliomas. Undoubtedly, this is related to the rapid expansion of knowledge about different types of mutations and their impact on tumor biology. Not insignificant is the fact of developing diagnostic methods and improving accessibility to such tests. Based on the data collected from the literature, it can be noted that initially genetic testing was recommended for patients with HNPGL only in groups with risk factors, i.e., patients with young age, positive family history of pheochromocytoma, in multiple, metastatic tumors (formerly called malignant). As research has evolved, some recommendations have evolved or new ones have emerged.
Young age for different authors did not always mean the same metric age. Depending on the author, it ranged from 35 to 50 years of age (12, 19, 20, 22, 34). Burnichon et al. determined a young age for patients <35 years old, with SDHD, SDHB and SDHC mutations (12). Mannelli et al. did not recommend genetic testing in patients >50 years of age, justifying this with the observation that 82% of patients with mutations were younger than 50 (19). After Piccini extended the study in 2012, age was lowered to 45 years—as a risk factor for mutations (17). In a group of 598 patients with HNPGL described by Neumann et al. (20), mutations were found significantly more often for a population aged <40 years.
In the following years, studies were conducted on additional groups of genes which allowed the detection of SDHAF2 mutations (21) in young patients and recommended these tests if previous tests for SDHD, SDHB and SDHC are negative. These observations were confirmed in Kunst’s study, where patients with SDHAF2 mutation were found to have PGL in patients up to 33 years of age after further diagnostic imaging. The study by Zhu et al. 2015 (34)—studied 23 patients with HNPGL and a negative family history. Showed mutations in 8 patients, of which 7 occurred in patients younger than 45 years of age. When this group of patients was included in the larger Chen study (22), <40 years of age was considered young.
In terms of positive family history, the findings are consistent and confirm a higher frequency of mutations in these groups (12, 17–20, 24). There is little variation in mutation frequency from 78% (18) to 100% (17) for this group of patients.
In multiple, multifocal tumors, mutations are found with a frequency ranging from 38.8% (19), to as high as 100% if HNPGL coexists with PCC (19). In the works of many authors, mutations in the SDH complex—SDHD, SDHB, SDHC, SDHAF2—have been confirmed to be more frequent in multiple tumors (4, 12, 16, 20). When multiple tumors are found, they may be part of genetic syndromes associated with paraganglioma formation (1, 7, 13, 33, 35, 36). In contrast to multiple tumors, mutations in SDHB have been found in this group of patients when the ability to give metastasis to other organs such as lymph nodes, bone, liver (1, 11, 12). Due to the more malignant clinical course in such cases, Zhu et al. (34) authors suggested including family members in genetic and clinical care in situations where SDHB mutations were found. In the genetic diagnostic algorithm, Gupta (31) proposes separating the group of patients with multiple tumors into those with and without metastasis, and if metastatic features are found, perform first-line testing for SDHB. In light of the presented study results, it seems reasonable to include such a procedure in the genetic diagnostic algorithm. When planning diagnosis of family members can furthermore take into account the gender of the patients, which is important in determining the mutation carrier. Transmission of the mutation from the father increases the risk of behavior, while in the case of carrier in the mother, there is no need for screening in children (4, 16, 23).
Some controversy surrounds genetic testing in patients with isolated eardrum paraganglioma (Fisch A type). In a 2009 Burnichon study, 330 patients were studied with tumors in the tympanic cavity and none of them had mutations typical of HNPGL (12). However, the paper did not specify the number of patients with this type of paraganglioma. Based on the results, it was concluded that patients with isolated eardrum HNPGL did not require genetic testing. In subsequent studies, this group of patients was excluded from genetic diagnosis (12). In 2012, Piccini et al. (17) included these patients in their study and obtained positive mutation results in 6/16 patients, of which 3 patients with type A paraganglioma were accompanied by multiple lesions and 3 had isolated tumors of the tympanic cavity. Also, isolated eardrum HNPGL were diagnosed in a study by Heesterman (23), where patients with SDHD mutations were observed. Therefore, it seems reasonable to include patients with tympanic PGLs in future genetic studies, not only in situations where they are part of multifocal tumors.
In light of the findings presented regarding the necessity of genetic testing in HNPGL in patients with risk factors. There remains, however, a group of patients with isolated tumors not burdened by such factors. Previous publications have shown mutations in this group of patients as well, such as in the work of Mannelli (19) where 14.3% of patients with a negative family history and isolated tumors were found to have mutations. This study was extended several years later by Piccini (17) where mutations were found in 18.8% of patients without risk factors. Also among the patients studied by Chen et al. (22) were patients with SDHD mutations without risk factors. Therefore, a modern diagnostic algorithm should include all patients with HNPGL.
In the analyses presented so far, studies for the Polish population have been included in a multicenter study by Neumann et al. (20), who included 23 patients from Poland. However, the results were analyzed collectively for the entire group of 598 patients with HNPGL, so it is difficult to draw conclusions for our country’s population on this basis. In 2008, a description of 2 cases of familial HNPGL in Polish patients was presented (37). In 2015, a paper describing 14 patients with multiple head and neck chaperones was also published based on the Polish group (38). In none of these papers, however, genetic testing was performed, so it is reasonable to initiate genetic testing also in the Polish population in order for the management algorithm to be based not only on data from the literature but also on the results of the study.
Conclusion
A genetic diagnosis of paraganglioma should be an integral part of the diagnostic process. The use of this diagnostic tool enables the early detection of pathological lesions and the initiation of treatment in family members of patients, thereby reducing the risk of complications. Furthermore, the rationalization of genetic diagnostics has an impact on the cost and efficiency of the process, which may lead to the applicability of diagnostic algorithms in clinical practice.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
KR: Conceptualization, Investigation, Methodology, Writing – original draft, Supervision. ZL: Data curation, Formal analysis, Visualization, Writing – original draft. RB: Supervision, Writing – review & editing. MZ-Ł: Project administration, Writing – review & editing. AR: Software, Writing – review & editing. JL: Writing – review & editing, Project administration, Software. MS: Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CC, Cancer/cysts; HMBG, Hemangioblastomas; HNPGL, Head and neck paraganglioma; MAX, MAX mutation; MTC, Medullary thyroid carcinoma; PCC(s)/Pheo(s), Pheochromocytoma(s); PGL(s), Paraganglioma(s); PPGL, Pheochromocytoma-paraganglioma (syndrome); RET, “Rearranged during transformation” proto-oncogene; SDH, Succinate dehydrogenase; SDHA, Succinate dehydrogenase complex flavoprotein subunit A; SDHAF2, Succinate dehydrogenase complex assembly factor 2; SDHB, Succinate dehydrogenase complex iron sulfur subunit B; SDHC, Succinate dehydrogenase complex subunit C; SDHD, Succinate dehydrogenase complex subunit D; sPGL(s), Secreting paraganglioma(s); TMEM 127, Transmembrane 127 protein mutation; VHL, Von Hippel–Lindau (gene/disease)
References
1. Majewska, A, Budny, B, Ziemnicka, K, Ruchała, M, and Wierzbicka, M. Head and neck paragangliomas—a genetic overview. Int J Mol Sci. (2020) 21:1–11. doi: 10.3390/ijms21207669
2. Polanowski, P, Kotecka-Blicharz, A, Chmielik, E, Ole, K, Wygoda, A, Rutkowski, T, et al. Paragangliomas of the head and neck region. Nowotwory. (2018) 68:132–9. doi: 10.5603/NJO.2018.0021
3. Guha, A, Musil, Z, Vicha, A, Zelinka, T, Pacak, K, Astl, J, et al. A systematic review on the genetic analysis of paragangliomas: primarily focused on head and neck paragangliomas. Neoplasma. (2019) 66:671–80. doi: 10.4149/neo_2018_181208N933
4. Kunst, HPM, Rutten, MH, de Mönnink, JP, Hoefsloot, LH, Timmers, HJLM, Marres, HAM, et al. SDHAF2 (PGL2-SDH5) and hereditary head and neck paraganglioma. Clin Cancer Res. (2011) 17:247–54. doi: 10.1158/1078-0432.CCR-10-0420
5. Boedeker, CC, Neumann, HPH, Maier, W, Bausch, B, Schipper, J, and Ridder, GJ. Malignant head and neck paragangliomas in SDHB mutation carriers. Otolaryngol Head Neck Surg. (2007) 137:126–9. doi: 10.1016/j.otohns.2007.01.015
6. Rijken, JA, de Vos, B, Leemans, CR, Zwezerijnen, GJCB, de Graaf, P, Hensen, EF, et al. Management of Multiple Secreting Paragangliomas in a succinate dehydrogenase subunit D (SDHD) variant carrier. Ann Thorac Surg. (2020) 109:e87–90. doi: 10.1016/j.athoracsur.2019.05.037
7. Myssiorek, D, Ferlito, A, Silver, CE, Rodrigo, JP, Baysal, BE, Fagan, JJ, et al. Screening for familial paragangliomas. Oral Oncol. (2008) 44:532–7. doi: 10.1016/j.oraloncology.2007.06.010
8. Hussain, I, Husain, Q, Baredes, S, Eloy, JA, Jyung, RW, and Liu, JK. Molecular genetics of paragangliomas of the skull base and head and neck region: implications for medical and surgical management. A review. J Neurosurg. (2014) 120:321–30. doi: 10.3171/2013.10.JNS13659
9. Smith, JD, Ellsperman, SE, Basura, GJ, and Else, T. Re-evaluating the prevalence and factors characteristic of catecholamine secreting head and neck paragangliomas. Endocrinol, Diabetes and Metabolism. (2021) 4:e00256. doi: 10.1002/edm2.256
10. Richter, S, Qiu, B, Ghering, M, Kunath, C, Constantinescu, G, Luths, C, et al. Head/neck paragangliomas: focus on tumor location, mutational status and plasma methoxytyramine. Endocr Relat Cancer. (2022) 29:213–24. doi: 10.1530/ERC-21-0359
11. Watts, D, Jaykar, MT, Bechmann, N, and Wielockx, B. Hypoxia signaling pathway: a central mediator in endocrine tumors. Front Endocrinol. (2023) 13. doi: 10.3389/fendo.2022.1103075
12. Burnichon, N, Rohmer, V, Amar, L, Herman, P, Leboulleux, S, Darrouzet, V, et al. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab. (2009) 94:2817–27. doi: 10.1210/jc.2008-2504
13. Lin, EP, Chin, BB, Fishbein, L, Moritani, T, Montoya, SP, Ellika, S, et al. Head and neck Paragangliomas: an update on the molecular classification, state-of-the-art imaging, and management recommendations. Radiol Imag Cancer. (2022) 4:e210088. doi: 10.1148/rycan.210088
14. Koopman, K, Gaal, J, and de Krijger, RR. Pheochromocytomas and paragangliomas: new developments with regard to classification, genetics, and cell of origin. Cancer. (2019) 11:1070. doi: 10.3390/cancers11081070
15. Williams, MD, and Tischler, AS. Update from the 4th edition of the World Health Organization classification of head and neck Tumours: Paragangliomas. Head Neck Pathol. (2017) 11:88–95. doi: 10.1007/s12105-017-0786-1
16. Boedeker, CC, Hensen, EF, Neumann, HPH, Maier, W, van Nederveen, FH, Suárez, C, et al. Genetics of hereditary head and neck paragangliomas. Head Neck. (2014) 36:907–16. doi: 10.1002/hed.23436
17. Piccini, V, Rapizzi, E, Bacca, A, di Trapani, G, Pulli, R, Giachè, V, et al. Head and neck paragangliomas: genetic spectrum and clinical variability in 79 consecutive patients. Endocr Relat Cancer. (2012) 19:149–55. doi: 10.1530/ERC-11-0369
18. Rijken, JA, de Vos, B, van Hest, LP, Dreijerink, KMA, den Heijer, M, Wisselink, W, et al. Evolving management strategies in head and neck paragangliomas: a single-Centre experience with 147 patients over a 60-year period. Clin Otolaryngol. (2019) 44:836–41. doi: 10.1111/coa.13380
19. Mannelli, M, Castellano, M, Schiavi, F, Filetti, S, Giacchè, M, Mori, L, et al. Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab. (2009) 94:1541–7. doi: 10.1210/jc.2008-2419
20. Neumann, HPH, Erlic, Z, Boedeker, CC, Rybicki, LA, Robledo, M, Hermsen, M, et al. Clinical predictors for germline mutations in head and neck paraganglioma patients: cost reduction strategy in genetic diagnostic process as fail-out. Cancer Res. (2009) 69:3650–6. doi: 10.1158/0008-5472.CAN-08-4057
21. Bayley, J-P, Cascon, A, Lourdes Sampietro, M, Gaal, J, and Korpershoek, E. Articles SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. (2010) 11:366–72. doi: 10.1016/S1470-2045(10)70007-3
22. Chen, H, Zhu, W, Li, X, Xue, L, Wang, Z, and Wu, H. Genetic and epigenetic patterns in patients with the head-and-neck paragangliomas associate with differential clinical characteristics. J Cancer Res Clin Oncol. (2017) 143:953–60. doi: 10.1007/s00432-017-2355-0
23. Heesterman, BL, Lisa De Pont, MH, Andel Van Der Mey, GL, Bayley, J-P, Eleonora Corssmit, PM, Frederik Hes, J, et al. Clinical progression and metachronous paragangliomas in a large cohort of SDHD germline variant carriers. Eur J Hum Genet. (2018) 26:1339–47. doi: 10.1038/s41431-018-0116-4
24. Ding, Y, Feng, Y, Wells, M, Huang, Z, and Chen, X. SDHx gene detection and clinical phenotypic analysis of multiple paraganglioma in the head and neck. Laryngoscope. (2019) 129:E67–71. doi: 10.1002/lary.27509
25. Bayley, JP, Bausch, B, Rijken, JA, van Hulsteijn, LT, Jansen, JC, Ascher, D, et al. Variant type is associated with disease characteristics in SDHB, SDHC and SDHD-linked phaeochromocytoma-paraganglioma. J Med Genet. (2020) 57:96–103. doi: 10.1136/jmedgenet-2019-106214
26. Bayley, JP, Bausch, B, Jansen, JC, Hensen, EF, van der Tuin, K, Corssmit, EP, et al. SDHB variant type impacts phenotype and malignancy in pheochromocytoma-paraganglioma. J Med Genet. (2023) 60:25–32. doi: 10.1136/jmedgenet-2020-107656
27. Mete, O, and Wenig, BM. Update from the 5th edition of the World Health Organization classification of head and neck tumors: overview of the 2022 WHO classification of head and neck neuroendocrine neoplasms. Head Neck Pathol. (2022) 16:123–42. doi: 10.1007/s12105-022-01435-8
28. Nosé, V, and Lazar, AJ. Update from the 5th edition of the World Health Organization classification of head and neck tumors: familial tumor syndromes. Head Neck Pathol. (2022) 16:143–57. doi: 10.1007/s12105-022-01414-z
29. Burnichon, N, Abermil, N, Buffet, A, Favier, J, and Gimenez-Roqueplo, AP. The genetics of paragangliomas. Eur Ann Otorhinolaryngol Head Neck Dis. (2012) 129:315–8. doi: 10.1016/j.anorl.2012.04.007
30. Sandow, L, Thawani, R, Kim, MS, and Heinrich, MC. Paraganglioma of the head and neck: a review. Endocr Pract. (2023) 29:141–7. doi: 10.1016/j.eprac.2022.10.002
31. Gupta, N, Strome, SE, and Hatten, KM. Is routine genetic testing warranted in head and neck paragangliomas? Laryngoscope. (2019) 129:1491–3. doi: 10.1002/lary.27656
32. Martin, TPC, Irving, RM, and Maher, ER. The genetics of paragangliomas: a review. Clin Otolaryngol. (2007) 32:7–11. doi: 10.1111/j.1365-2273.2007.01378.x
33. Capatina, C, Ntali, G, Karavitaki, N, and Grossman, AB. The management of head-and-neck paragangliomas. Endocr Relat Cancer. (2013) 20:223. doi: 10.1530/ERC-13-0223
34. Zhu, WD, Wang, ZY, Chai, YC, Wang, XW, Chen, DY, and Wu, H. Germline mutations and genotype-phenotype associations in head and neck paraganglioma patients with negative family history in China. Eur J Med Genet. (2015) 58:433–8. doi: 10.1016/j.ejmg.2015.05.008
35. Boedeker, CC, Erlic, Z, Richard, S, Kontny, U, Gimenez-Roqueplo, AP, Cascon, A, et al. Head and neck paragangliomas in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. J Clin Endocrinol Metab. (2009) 94:1938–44. doi: 10.1210/jc.2009-0354
36. Offergeld, C, Brase, C, Yaremchuk, S, Mader, I, Rischke, HC, Gläsker, S, et al. Head and neck paragangliomas: clinical and molecular genetic classification. Clinics. (2012) 67:19–28. doi: 10.6061/clinics/2012(Sup01)05
37. Szyfter, W, Wierzbicka, M, and Gawȩcki, W. Multiple and familial paragangliomas of the head and neck review of literature and report of two cases. Otolaryngol Pol. (2008) 62:530–5. doi: 10.1016/s0030-6657(08)70308-1
Keywords: head and neck, tumor, paraganglioma, pheochromoacytoma, geneti algorithm, genetic diagnosis
Citation: Radomska K, Leszczyńska Z, Becht R, Zaborek - Łyczba M, Rzepakowska A, Lubiński J and Szymański M (2024) Algorithm of genetic diagnosis for patients with head and neck paraganglioma—update. Front. Neurol. 15:1437027. doi: 10.3389/fneur.2024.1437027
Edited by:
Wiesław Konopka, Polish Mother’s Memorial Hospital Research Institute, PolandReviewed by:
Jozef Mierzwinski, Children’s Hospital of Bydgoszcz, PolandHanna Klimza, Poznan University of Medical Sciences, Poland
Copyright © 2024 Radomska, Leszczyńska, Becht, Zaborek - Łyczba, Rzepakowska, Lubiński and Szymański. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katarzyna Radomska, a2F0YXJ6eW5hLmFtZXJuaWtAcHVtLmVkdS5wbA==