 Yang Liu1*
Yang Liu1* Youqi Chen2
Youqi Chen2 Ming Gao2
Ming Gao2 Jia Luo2Yanan Wang2Yihan Wang3
Jia Luo2Yanan Wang2Yihan Wang3 Yu Gao4
Yu Gao4 Laiyu Yang3Jingning Wang2Ningxin Wang2
Laiyu Yang3Jingning Wang2Ningxin Wang2- 1Department of Endocrinology, Affiliated Hospital of Jilin Medical University, Jilin, China
- 2Bethune First Hospital of Jilin University, Changchun, China
- 3Bethune Third Hospital of Jilin University, Changchun, China
- 4Clinical College, The Second Affiliated Hospital of Harbin Medical University, Harbin, China
Background: Earlier observational studies have demonstrated a correlation between glioma and the risk of neurodegenerative diseases (NDs), but the causality and direction of their associations remain unclear. The objective of this study was to ascertain the causal link between glioma and NDs using Mendelian randomization (MR) methodology.
Methods: Genome-wide association study (GWAS) data were used in a two-sample bi-directional MR analysis. From the largest meta-analysis GWAS, encompassing 18,169 controls and 12,488 cases, summary statistics data on gliomas was extracted. Summarized statistics for NDs, including Alzheimer’s disease (AD), multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS) and Parkinson’s disease (PD) were obtained from the GWAS of European ancestry. Inverse variance weighted (IVW) method was elected as the core MR approach with weighted median (WM) method and MR-Egger method as complementary methods. In addition, sensitivity analyses were performed. A Bonferroni correction was used to correct the results.
Results: Genetically predicted glioma had been related to decreased risk of AD. Specifically, for all glioma (IVW: OR = 0.93, 95% CI = 0.90–0.96, p = 4.88 × 10−6) and glioblastoma (GBM) (IVW: OR = 0.93, 95% CI = 0.91–0.95, p = 5.11 × 10−9). We also found that genetically predicted all glioma has a suggestive causative association with MS (IVW: OR = 0.90, 95% CI = 0.81–1.00, p = 0.045). There was no evidence of causal association between glioma and ALS or PD. According to the results of reverse MR analysis, no discernible causal connection of NDs was found on glioma. Sensitivity analyses validated the robustness of the above associations.
Conclusion: We report evidence in support of potential causal associations of different glioma subtypes with AD and MS. More studies are required to uncover the underlying mechanisms of these findings.
Introduction
NDs are major health challenges that have drawn tremendous focus in the previous few years. NDs are an assembly of heterogeneous neurological disorders that involve progressive neuronal damage and death, such as AD, PD, Huntington’s disease, MS and ALS (1). These illnesses significantly diminish patients’ quality of life and frequently result in mortality (2). The etiology of NDs is complex and not well understood. Currently, treatments for NDs have limited effectiveness, with no existing cures available (3). And the prognosis for individuals afflicted with NDs is typically bleak (4). Further research is imperative to enhance the understanding of the underlying causes of NDs and to advance the development of more efficacious treatments.
Gliomas are primary brain tumors that account for approximately 80% of all malignant brain tumors (5). Their 5-year survival rate is less than 20%, indicating a terrible prognosis and imposing a significant health burden on patients (6). Gliomas are divided into different histologic subtypes, the most common and aggressive of which is GBM, which has a 5-year survival rate of only 6.8% (7). The cost of treating individuals with glioma is anticipated to be considerable. Besides, the etiology of glioma is poorly understood.
The relationship between NDs and glioma has been studied to some extent, yet the results remain uncertain. In recent years, increasing prospective studies have supported the correlation between glioma and NDs (8). For instance, several clinical findings imply that glioma growth and progression may be initiated by pre-existing MS or may be facilitated by it (9). Besides, there are more similarities between NDs and brain cancer than previously thought. In addition to having comparable epidemiological and molecular characteristics, these illnesses are associated with some risk factors, such as aging and inflammation (10). Notably, many observational studies may have limitations due to sample numbers and potential confounding variables (11). Therefore, it’s uncertain how NDs and the risk of glioma causally relate.
Utilizing genetic variants as instrumental variables (IVs) to infer the causal relationships between exposures and diseases, MR is a potent approach (12). By exploiting the random assignment of genes during meiosis, MR can circumvent some of the biases inherent in observational studies, such as confusion and inverse causality (13).
In the current research, we implemented a two-sample MR framework to explore the potential role of different glioma subtypes (all glioma, GBM, non-GBM) in the development of four NDs, including AD, MS, ALS, and PD. In addition, we conducted bidirectional MR using gliomas as outcomes to test the direction of association.
Methods
Fundamental MR principles
To explore the causal impact of the risk factor with a desired result, we employed genetic variants as IVs (Figure 1). The study followed three fundamental principles, including: (1) associated with exposure, (2) not associated with confounders of the association between exposure and outcome, and (3) only associated with outcome via their association with exposure (14).
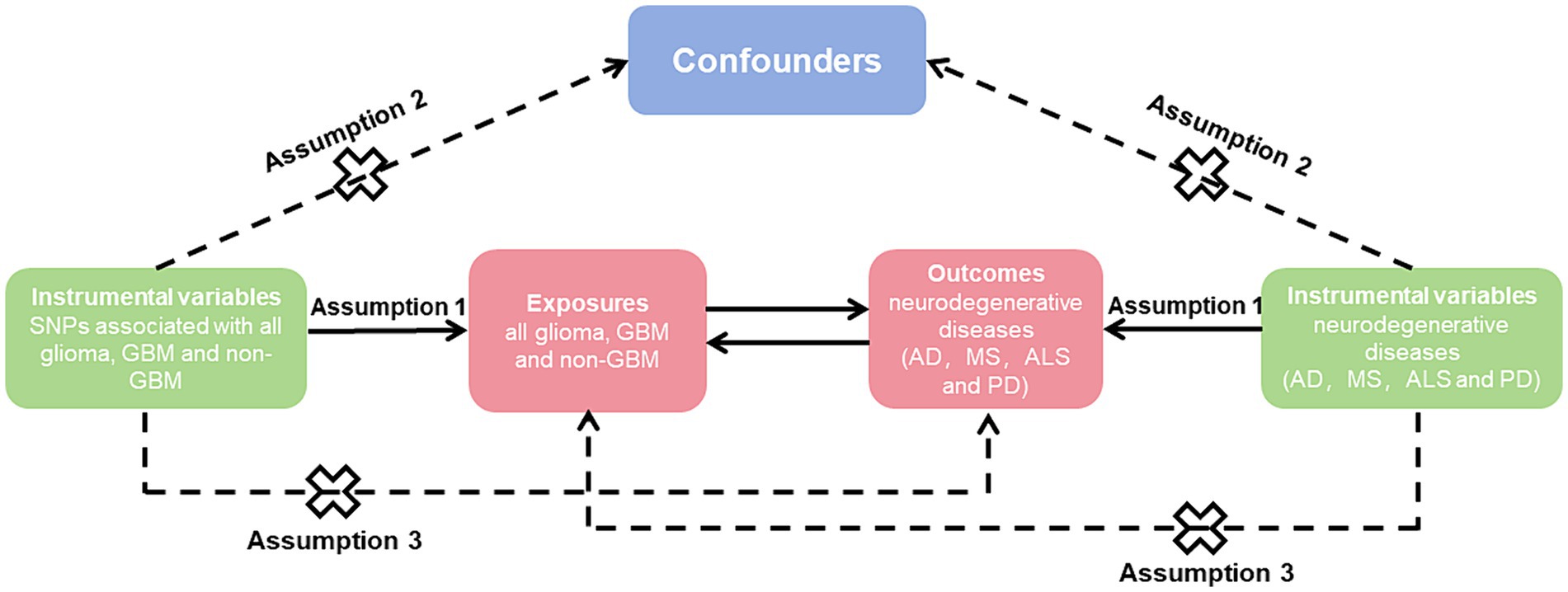
Figure 1. Mendelian randomization causal diagram with three assumptions. (1) Instrumental variables (IVs) must be associated with exposure (p < 5 × 10−8); (2) IVs are not associated with confounders of the association between exposure and outcome; (3) IVs have no direct effect on the outcome, except through exposure. SNP, single nucleotide polymorphisms; GBM, glioblastoma; AD, Alzheimer’s disease; MS, multiple sclerosis; ALS, amyotrophic lateral sclerosis; PD, Parkinson’s disease.
NDs and glioma GWAS dataset
The two-sample MR studies assumed that the separate samples were used for the exposure and outcome. Therefore, the GWAS summarized data of NDs that had a significant overlap with glioma phenotypes were eliminated. The GWAS summarized data for glioma were obtained from the strongest meta-analysis GWAS, which included 6,183 individuals with glioblastoma multiforme (GBM), 5,820 individuals with non-GBM, and 18,169 European ancestry controls from eight unique GWAS databases (15). The International Multiple Sclerosis Genetics Consortium (IMSGC) provided general information for MS which involves 9,772 cases and 17,376 controls of European descent (16). Summary statistics of PD, AD and ALS were derived from different GWAS. Summary statistics of PD contains 294 cases and 456,054 controls of European ancestry (17). Summary statistics of AD comprise 21,982 late-onset AD individuals with European ancestry, 53,042 European ancestry individuals with a family history of AD, and 397,844 controls with European ancestry (18). Summary statistics of ALS covers 12,577 cases and 23,475 controls (19).
MR analysis
To generate IVs, statistically significant threshold [p < 5 × 10−8; linkage disequilibrium (LD) r2 < 0.001, LD distance > 10,000 kb] was set (20). For PD, there was no IV’s p-value less than 5 × 10−8, so we relaxed it to 5 × 10−6 (21). We used the IVW approach as the principal analytical approach. The IVW method is a statistical technique used in meta-analysis to combine the results of multiple studies (22). It is based on the principle that studies with larger sample sizes and lower variance should be given more weight in the analysis. We also made reference to the outcomes of other types of models, such as MR-Egger (23) and weighted median (24). A minimum of 50% of the single nucleotide polymorphisms (SNPs) are assumed to be legitimate using the weighted median technique, which provides consistent causal estimates under this assumption. A random effects model may be applied because of the significant heterogeneity among the analyses (Figure 2) (25). Bonferroni correction method was used to adjust the significance level of hypothesis testing (26). If the IVW and weighted median approaches yield consistent results for the direction as well as the magnitude of the causal effects, the presence of causality is indicated (27) and the p values of Bonferroni correcting are less than 2.08E−3 (0.05/24). p values less than 0.05 but greater than 2.08E−3 are interpreted as suggestive of a causal relationship.
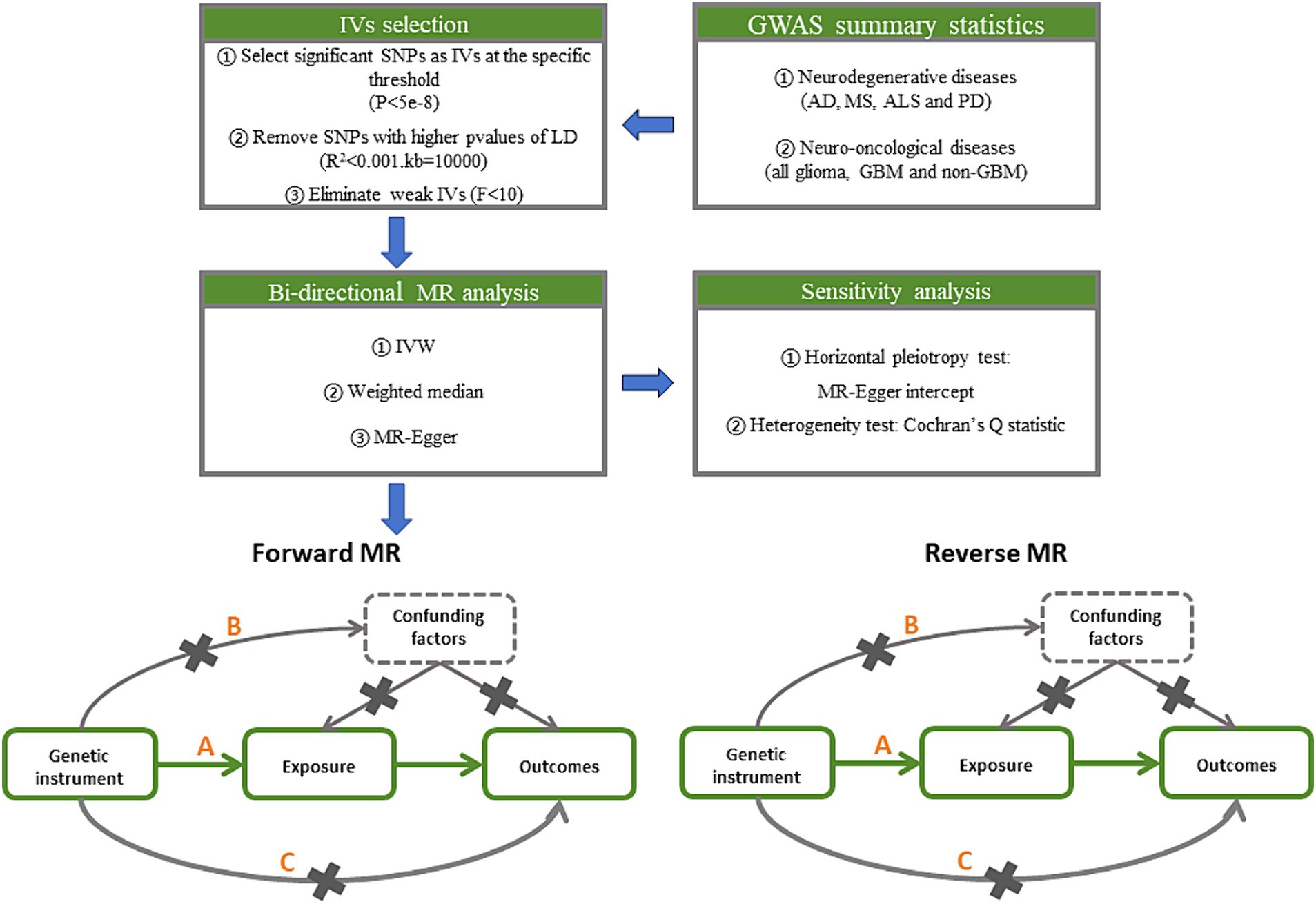
Figure 2. Study design pipeline of the two-sample bi-directional Mendelian randomization analysis. GWAS, Genome-wide association study; AD, Alzheimer’s disease; MS, multiple sclerosis; ALS, amyotrophic lateral sclerosis; PD, Parkinson’s disease; GBM, glioblastoma; IV, instrumental variables; SNP, single nucleotide polymorphisms; LD, linkage disequilibrium; IVW, inverse variance weighted.
Sensitivity analyses
Additionally, in MR studies, horizontal pleiotropy and outlier SNPs can be detected using MR-Egger intercept (28). There are no horizontal pleiotropy effects present if the p value of the MR-Egger intercept is higher than 0.05. The heterogeneity between SNPs can be determined using Cochran Q statistics, which will guarantee the validity of the MR analysis (29). The strength of genetic instruments can be measured using F-statistics, which is calculated using the sample size, number of SNPs, and R-squared value (30). A low F-statistics value (less than 10) may indicate weak instrument bias (31). The analysis can be performed using the “Two Sample MR” package in R software (32).
Results
Table 1 displays the enrolled GWAS studies’ summary data. To sum up, 5 GWAS studies (4 GWAS of neurodegenerative diseases and 1 GWAS of glioma) were enrolled in this MR study.
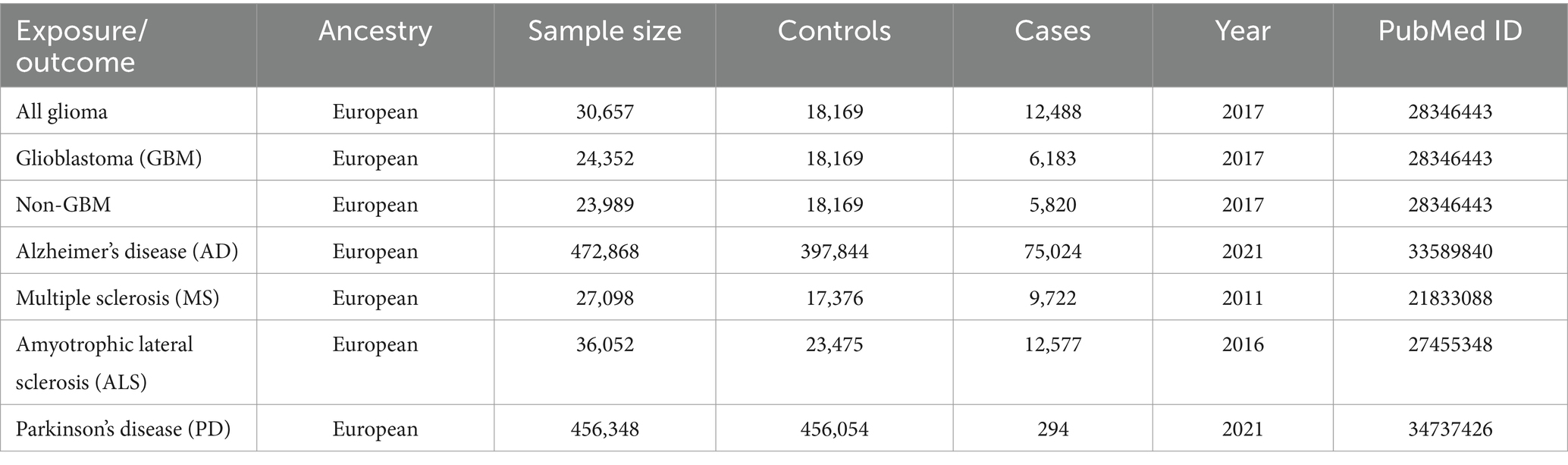
Table 1. A brief description of each GWAS summary statistics.
Causal association of glioma on NDs
There were 12–18 SNPs used for MR assessments in the initial analyzing. All SNPs were considered robust since their F statistics were all greater than the threshold of 10 (Supplementary Table 1). The results of the MR evaluation and the sensitivity analysis of causality of glioma on NDs are displayed in Tables 2A, 2B, 2C and Supplementary Tables 2, 3.

Table 2A. Mendelian randomization estimates, heterogeneity test and pleiotropy test of all glioma on neurodegenerative diseases.
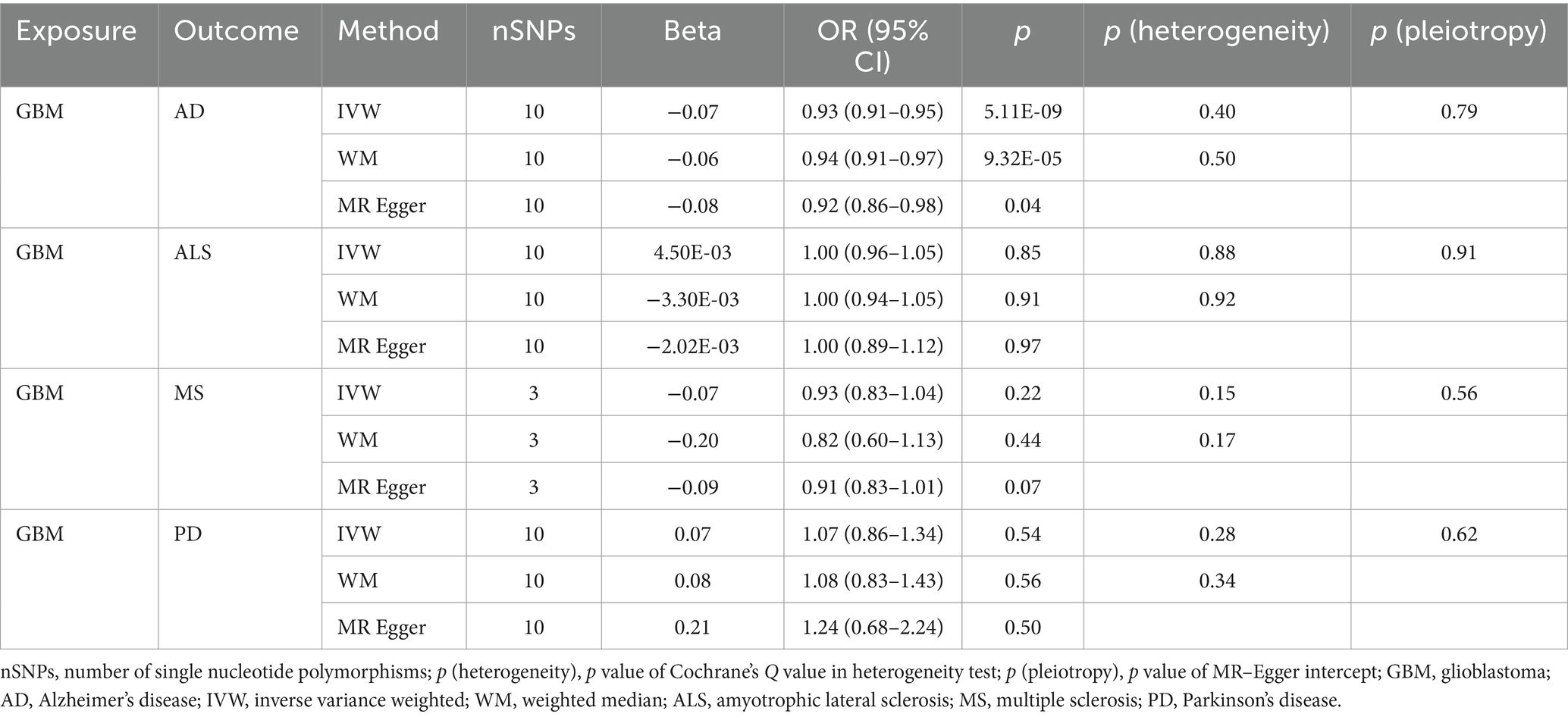
Table 2B. Mendelian randomization estimates, heterogeneity test and pleiotropy test of GBM on neurodegenerative diseases.
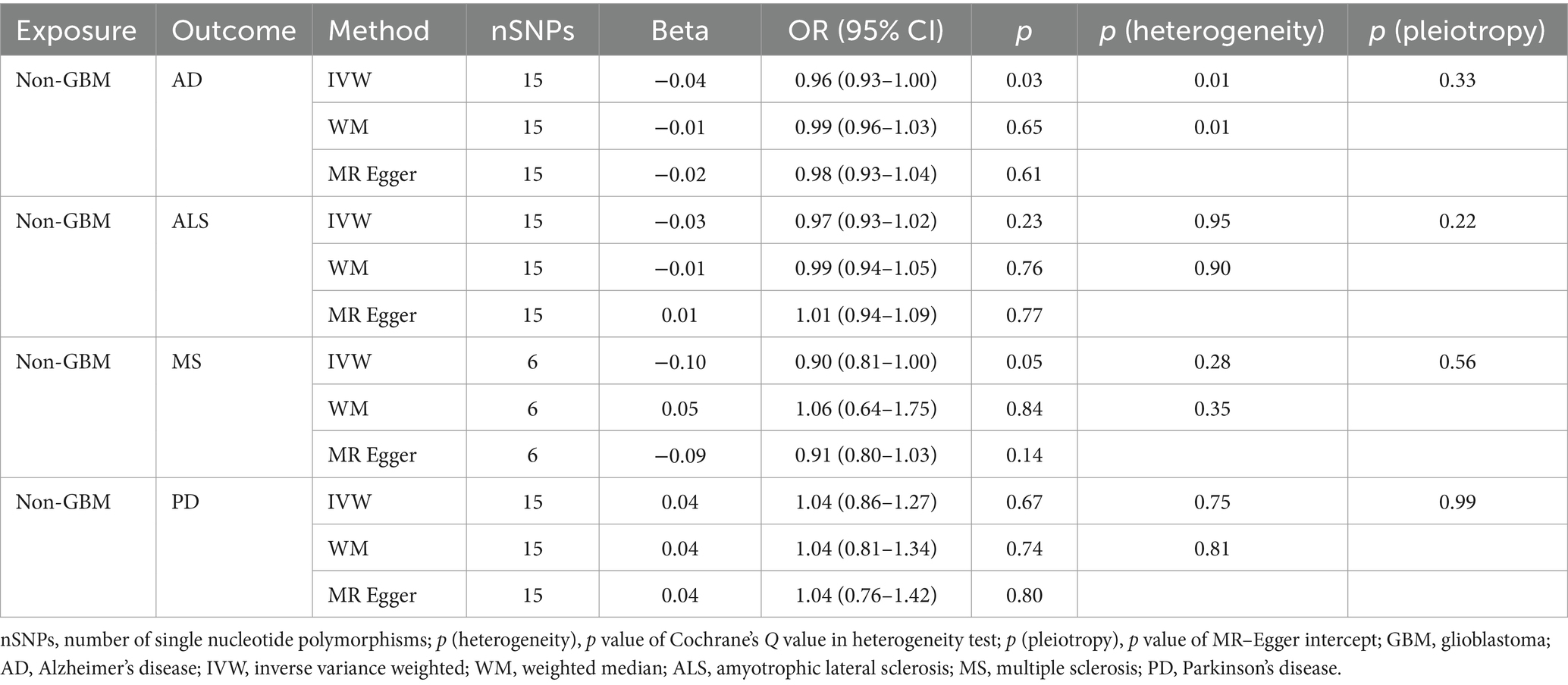
Table 2C. Mendelian randomization estimates, heterogeneity test and pleiotropy test of non-GBM on neurodegenerative diseases.
Our research revealed a strong causal link between AD and genetically predicted gliomas. Specifically, for all glioma (IVW: OR = 0.93, 95% CI = 0.90–0.96, p = 4.88 × 10−6; WM: OR = 0.93, 95% CI = 0.90–0.97, p = 3.30 × 10−4) and GBM (IVW: OR = 0.93, 95% CI = 0.91–0.95, p = 5.11 × 10−9; WM: OR = 0.94, 95% CI = 0.91–0.97, p = 9.32 × 10−5). It is noteworthy that a suggestive causative association between genetically predicted all glioma and MS was discovered (padj > 0.05 and IVW p < 0.05) and genetically predicted non-GBM appeared suggestively to be causally related to AD (padj > 0.05 and IVW p < 0.05). Furthermore, a link between glioma and the likelihood of developing ALS and PD was not found (IVW p > 0.05) (Tables 2A, 2B, 2C).
Causal association of NDs on glioma
We designated the SNPs linked to NDs as exposure IVs to evaluate the link of causality between NDs and glioma. Four to forty SNPs were utilized in the MR calculations. All SNPs were robust since their F values were much higher than the cutoff of 10 (Supplementary Table 4). A summary is provided in Tables 3A, 3B and 3C and Supplementary Tables 5, 6 of the results obtained from the MR analysis and sensitivity analysis exploring the causative association between NDs and gliomas.
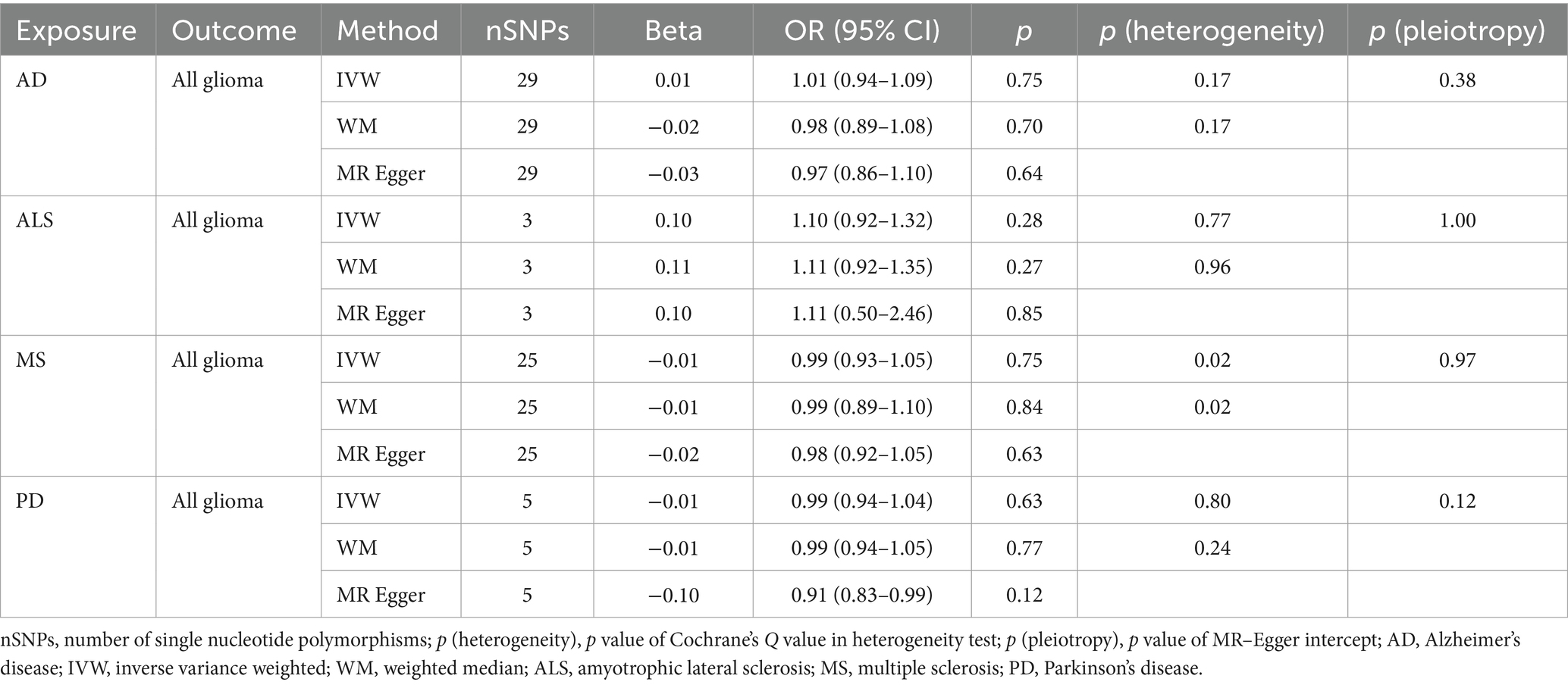
Table 3A. Mendelian randomization estimates, heterogeneity test and pleiotropy test of neurodegenerative diseases on all glioma.
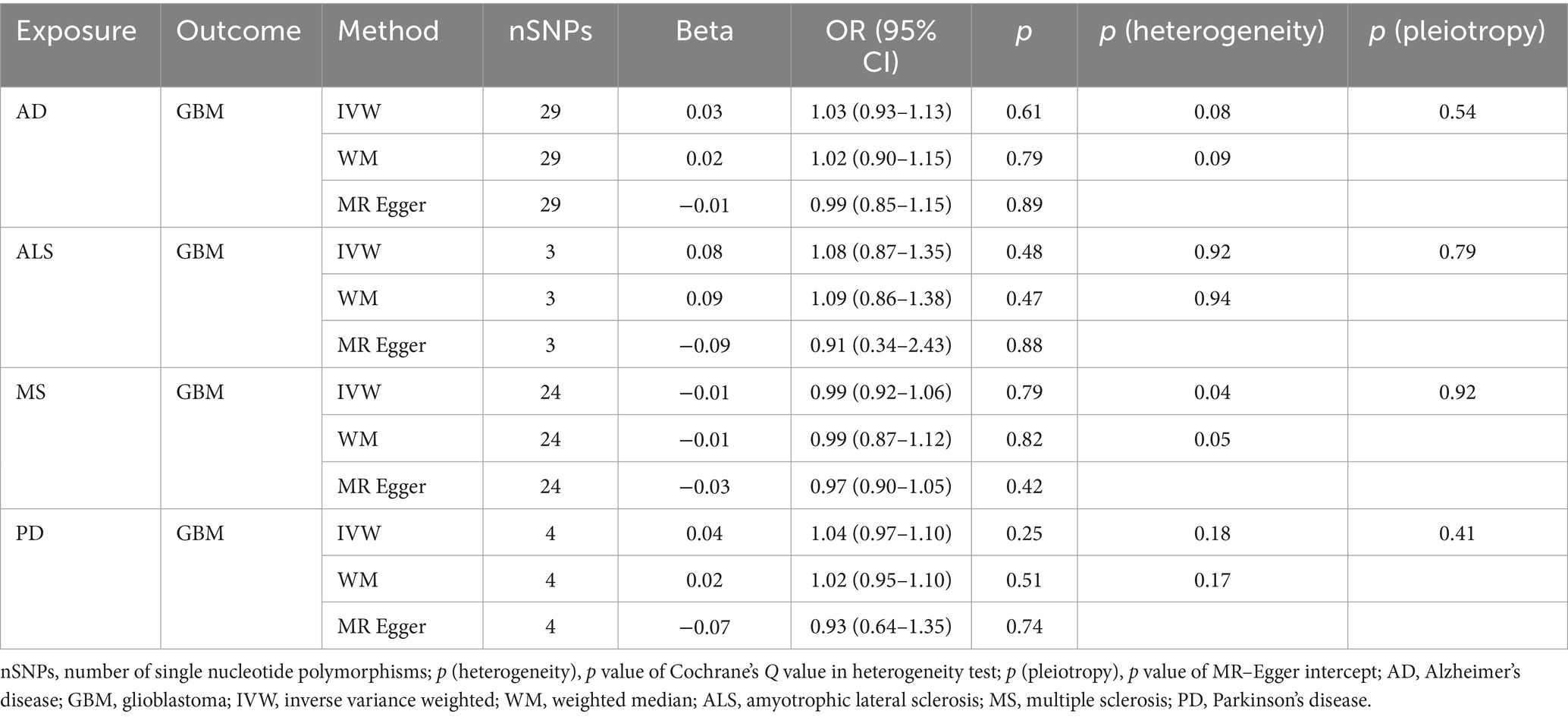
Table 3B. Mendelian randomization estimates, heterogeneity test and pleiotropy test of neurodegenerative diseases on GBM.
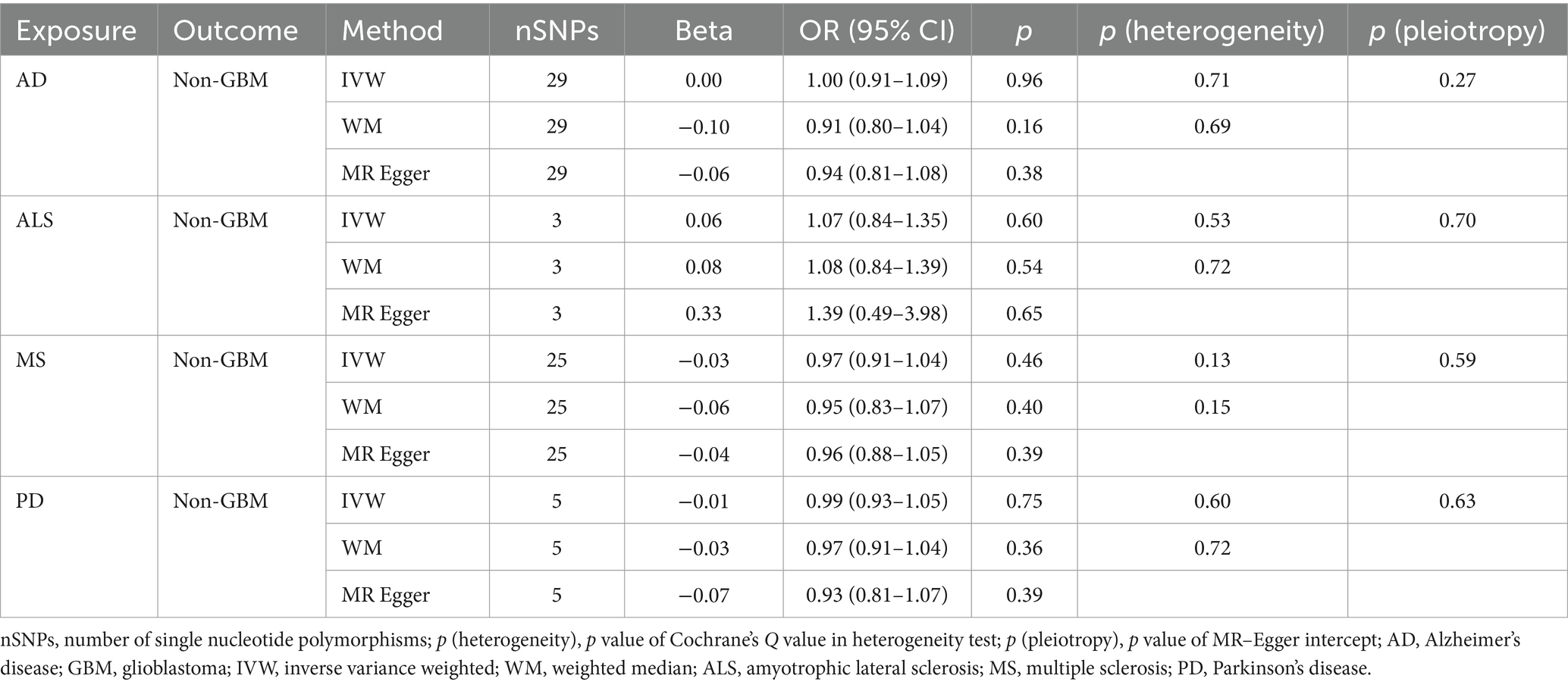
Table 3C. Mendelian randomization estimates, heterogeneity test and pleiotropy test of neurodegenerative diseases on non-GBM.
NDs with the risk of GBM, non-GBM, and all glioma did not show any causal relationships (p > 0.05) (Tables 3A, 3B and 3C). Pleiotropy analyses showed that our MR results had no horizontal pleiotropy (Supplementary Table 6).
Discussion
NDs are a group of neurological disorders that affect the lives of millions of people worldwide (33). Despite extensive research, the etiology of NDs remains unclear (34). There are several observational studies exploring the causal relationship between NDs and gliomas (8). Due to small sample sizes and inherent biases, establishing causality is difficult (35). We used MR analysis in the present study to investigate the causality and direction of association between different subtypes of glioma and AD, MS, ALS and PD. In this comprehensive analysis of gliomas with risk of NDs, we observed that genetically predicted all glioma and GBM has significant causality with lower risk of AD. Our results also indicate some evidence in favor of a potentially causative link between all glioma and MS, although the association was not survived correction for multiple testing.
Although some research has indicated that the risk of glioma changes possibly during the AD progress, it is unclear what biological mechanism glioma may use to defend against AD (36). A variety of NDs can impact the central nervous system (CNS). Among NDs, AD is the most prevalent (37). Recognizing a wide range of potential threats to the CNS, microglia, which are the main instinctive immune cells in the brain, can quickly and powerfully activate both the inflammatory and immune systems to defend the brain (38). Risk variations of AD that are connected to the microglia of the elderly brain have contributed to an important function for microglia in contemporary AD research, such as TREM2, CD33, INPP5D, HLA-DQA1, and ATXN7L (39). Various studies suggest that inadequate lipid processing and microglial phagocytosis of ab plaques may be at least partially responsible for the disease, even though the exact role of microglia in the process is still unknown (40). In addition, microglia or macrophages can make up to 30–50% of the cells in gliomas (41). Within and surrounding glioma tissue, macrophages and microglial cells proliferate and take on an amoeboid appearance. Microglia can be attracted to glioma cells by the secretion of scatter factor, hepatocyte growth factor and so on (42). It’s possible that glioma survivors’ cells, such as microglia, may have developed a unique phenotype during the illness and therapy that inhibits the progression of AD. Additionally, gliomas are associated with some of the same risk genes for ADs, one example is TREM2 (43). According to GWAS, TREM2 may be essential to the pathophysiology of AD (44). TREM2 may have a variety of roles in microglial processes related to AD brain homeostasis (45). TREM2 can function alone or in conjunction with additional molecules, such as apolipoprotein E (APOE), to affect microglial functions in disorders caused by amyloid and tau, as well as inflammation and metabolism (45–47). Moreover, it was discovered that excessive TREM2 expression in malignant brain tumors is linked to a worse prognosis, while low TREM2 expression is linked to a higher chance of survival (48). It is conceivable that competing genotypes of the same gene are linked to both AD and brain cancer, explaining the seemingly paradoxical results of AD and glioma sharing overexpressed genes but having negatively correlated prevalence. Our research indicates that glioma risk may have a protective effect against AD development and validates its involvement in AD etiology.
While glioma and MS co-occurring is extremely uncommon, reports of such cases have been documented dating back to 1949 (49), which raises the possibility of underlying mechanisms between glioma and MS, perhaps resulting from the environment or heredity. However, whether there exists a causal association and the effect direction remains unknown. Consideration between glioma and MS may be given to the possible involvement of DNA methylation. Glioma growth and progression are significantly influenced by epigenetic changes, which are regarded as a marker (50). DNA methylation, including hypermethylation, hypomethylation across the genome, and hypomethylation specific to certain genes, has been implicated in majority of studies on epigenetic changes in GBM thus far (51). In MS patients, several brain regions have mismethylated genes having a particular profile or low methylation, like the cytosine in the promoter of the myelin enzyme peptidylarginine deiminase-2 in MS-normal-appearing white matter (52). Furthermore, Sahm et al. identified differentially methylated regions (DMRs) involving immune-related genes, such as human leukocyte antigen (HLA) and interleukin regions, by comparative analysis of genome-wide DNA methylation patterns in gliomas occurring in patients with and without MS (53). Additionally, from an immunological point of view, gliomas and MS represent opposing paradigmatic states inside the CNS. Significant immunological abnormalities (54), including as CD4 lymphopenia (55), elevated regulatory T cell percentages in peripheral blood, and changes in cytokine profiles from Th1 to Th2 (56), are present in glioma patients. Whereas overactive immune reactions cause MS. MS is the most prevalent immune-mediated brain illness with radiological features. It is distinguished by axonal damage, severe demyelination, lesion formation in the brain and spinal cord, opening of the blood–brain barrier (BBB), and infiltration of inflammatory immune cells (57). Interestingly, the majority of the recently discovered MS risk genes are immune system-related (58). Meanwhile, germline and somatic immune system changes have been proposed as potential contributors to the pathophysiology of adult glioma in epidemiological investigations (59). Therefore, immune impairment in glioma patients may be a potential mechanism for reducing MS.
Similar to NDs in terms of age range and tissue type, CNS tumors also develop. But there is few epidemiological data about the correlation between NDs and this kind of tumor (60). According to some research, there is a positive correlation between a better prognosis for gliomas and genes linked to both ALS and PD (61). Among these genes, there is the highest association between high Tau/MAPT expression and many markers of longer life in patients with gliomas. Although tau protein has been shown to express in glial cells and gliomas, it controls microtubule dynamics and stability in neurons (62). However, regarding the control of Tau/MAPT transcription in tumors, not much is known (62). In addition, CNS tumors is characterized by a high rate of morbidity and mortality. The majority of these neoplasms develop infrequently, and a number of risk factors, including concurrent illnesses like Parkinson’s disease and exposure to electromagnetic fields or ionizing radiation, have been linked to their formation (63). Most juvenile recessive autosomal cases of PD are caused by Parkin. Parkin and p53’s balance is upset in both PD and brain tumors (64). The significance of the functional interaction between Parkin and p53 is noteworthy, and the pathogenic mutations that disrupt it are probably responsible for the genesis of both PD and gliomas. But it’s still unknown how PD affects the growth of glioma. There was no evidence in our investigation of a causal link between NDs and gliomas. To validate that, more investigation is required.
Current studies showed there is a possible correlation existing between NDs and treatment linked to gliomas. For instance, methylene blue (MB), a medication that has been around for a century, has ability to accept electrons from NADH and transfer them to cytochrome C, offering a different route for electron transfer (65). In glioma treatment, MB reduces glioma proliferation in cells, stops the glioma cell cycle at S-phase, and reverses the Warburg effect by increasing mitochondrial phosphorylation by oxidation (66). A clinical Phase II trial evaluated the effects of MB therapy on cognitive impairment in 332 presumably AD patients were presented (67). Furthermore, several studies have shown that MB protects neurons and astrocytes from a variety of stressors in vitro and in rat models of AD and PD (68, 69). Our results suggest that glioma is associated with AD and MS. However, the glioma data used in our MR analysis did not include information on whether patients with glioma received treatment. Further research is necessary to determine how therapies in glioma patients affects NDs.
Our study has several advantages. Firstly, a two-sample bi-directional MR approach was used to draw causal conclusions between glioma and NDs risk while controlling for confounders and reverse causality. Secondly, in comparison to previous research, ours demonstrated robust validity and generalizability since it used data on glioma from the biggest GWAS dataset, comprising 12,488 cases and 18,020 controls, as well as data on NDs from an impartial large-scale GWAS dataset. Thirdly, we incorporated fresh elements. That have not been studied before in previous MR research, such as MS and ALS.
However, it is important to take into account this study’s shortcomings as well. Firstly, due to the lack of data on gender or age stratification, our analysis may be influenced by these factors. Secondly, our study was limited to using European-ancestry whole-genome association data, which may result in limited applicability of our findings to other populations. Nevertheless, our goal is to include all populations in our analysis as much as feasible.
Conclusion
In summary, our findings showed a genetic correlation between glioma and NDs. Meanwhile, the risk of AD and MS may be lowered by glioma. These results contribute to our understanding of the function of glioma in NDs and will enable the development of therapeutic medications for glioma complications in upcoming clinical trials. In addition, our research sheds further light on the development of NDs and justifies more study to identify the precise pathways underlying their pathophysiology.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
YL: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. YC: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization. MG: Conceptualization, Writing – review & editing. JL: Methodology, Writing – review & editing. YaW: Methodology, Writing – review & editing. YiW: Validation, Writing – review & editing. YG: Investigation, Writing – review & editing. LY: Methodology, Writing – review & editing. JW: Writing – review & editing. NW: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2024.1413015/full#supplementary-material
References
1. Dugger, BN, and Dickson, DW. Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol. (2017) 9:a028035. doi: 10.1101/cshperspect.a028035
2. Rehman, MU, Sehar, N, Dar, NJ, Khan, A, Arafah, A, Rashid, S, et al. Mitochondrial dysfunctions, oxidative stress and neuroinflammation as therapeutic targets for neurodegenerative diseases: an update on current advances and impediments. Neurosci Biobehav Rev. (2023) 144:104961. doi: 10.1016/j.neubiorev.2022.104961
3. Kim, J, and Mook-Jung, I. Special issue on neurodegenerative diseases and their therapeutic approaches. Exp Mol Med. (2015) 47:e146. doi: 10.1038/emm.2015.13
4. Feldman, EL, Goutman, SA, Petri, S, Mazzini, L, Savelieff, MG, Shaw, PJ, et al. Amyotrophic lateral sclerosis. Lancet. (2022) 400:1363–80. doi: 10.1016/s0140-6736(22)01272-7
5. Wagner, MW, Jabehdar Maralani, P, Bennett, J, Nobre, L, Lim-Fat, MJ, Dirks, P, et al. Brain tumor imaging in adolescents and young adults: 2021 WHO updates for molecular-based tumor types. Radiology. (2024) 310:e230777. doi: 10.1148/radiol.230777
6. Nakagawa, K, Aoki, Y, Fujimaki, T, Tago, M, Terahara, A, Karasawa, K, et al. High-dose conformal radiotherapy influenced the pattern of failure but did not improve survival in glioblastoma multiforme. Int J Radiat Oncol Biol Phys. (1998) 40:1141–9. doi: 10.1016/s0360-3016(97)00911-5
7. Srivastava, R, Dodda, M, Zou, H, Li, X, and Hu, B. Tumor niches: perspectives for targeted therapies in glioblastoma. Antioxid Redox Signal. (2023) 39:904–22. doi: 10.1089/ars.2022.0187
8. Cuevas-Diaz Duran, R, Wang, CY, Zheng, H, Deneen, B, and Wu, JQ. Brain region-specific gene signatures revealed by distinct astrocyte subpopulations unveil links to glioma and neurodegenerative diseases. eNeuro. (2019) 6:ENEURO.0288–18.2019. doi: 10.1523/eneuro.0288-18.2019
9. Hemminki, K, Liu, X, Forsti, A, Ji, J, Sundquist, J, and Sundquist, K. Subsequent brain tumors in patients with autoimmune disease. Neuro Oncol. (2013) 15:1142–50. doi: 10.1093/neuonc/not070
10. Schwartz, L, Peres, S, Jolicoeur, M, and da Veiga Moreira, J. Cancer and Alzheimer’s disease: intracellular pH scales the metabolic disorders. Biogerontology. (2020) 21:683–94. doi: 10.1007/s10522-020-09888-6
11. D'Onofrio, BM, Sjölander, A, Lahey, BB, Lichtenstein, P, and Öberg, AS. Accounting for confounding in observational studies. Annu Rev Clin Psychol. (2020) 16:25–48. doi: 10.1146/annurev-clinpsy-032816-045030
12. Burgess, S. Sample size and power calculations in Mendelian randomization with a single instrumental variable and a binary outcome. Int J Epidemiol. (2014) 43:922–9. doi: 10.1093/ije/dyu005
13. Skrivankova, VW, Richmond, RC, Woolf, BAR, Yarmolinsky, J, Davies, NM, Swanson, SA, et al. Strengthening the reporting of observational studies in epidemiology using Mendelian randomization: the STROBE-MR statement. JAMA. (2021) 326:1614–21. doi: 10.1001/jama.2021.18236
14. Smith, GD. Mendelian randomization for strengthening causal inference in observational studies: application to gene × environment interactions. Perspect Psychol Sci. (2010) 5:527–45. doi: 10.1177/1745691610383505
15. Melin, BS, Barnholtz-Sloan, JS, Wrensch, MR, Johansen, C, Il'yasova, D, Kinnersley, B, et al. Genome-wide association study of glioma subtypes identifies specific differences in genetic susceptibility to glioblastoma and non-glioblastoma tumors. Nat Genet. (2017) 49:789–94. doi: 10.1038/ng.3823
16. Sawcer, S, Hellenthal, G, Pirinen, M, Spencer, CC, Patsopoulos, NA, Moutsianas, L, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. (2011) 476:214–9. doi: 10.1038/nature10251
17. Jiang, L, Zheng, Z, Fang, H, and Yang, J. A generalized linear mixed model association tool for biobank-scale data. Nat Genet. (2021) 53:1616–21. doi: 10.1038/s41588-021-00954-4
18. Schwartzentruber, J, Cooper, S, Liu, JZ, Barrio-Hernandez, I, Bello, E, Kumasaka, N, et al. Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat Genet. (2021) 53:392–402. doi: 10.1038/s41588-020-00776-w
19. van Rheenen, W, Shatunov, A, Dekker, AM, McLaughlin, RL, Diekstra, FP, Pulit, SL, et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet. (2016) 48:1043–8. doi: 10.1038/ng.3622
20. Lu, C, Chen, Q, Tao, H, Xu, L, Li, J, Wang, C, et al. The causal effect of inflammatory bowel disease on diffuse large B-cell lymphoma: two-sample Mendelian randomization study. Front Immunol. (2023) 14:1171446. doi: 10.3389/fimmu.2023.1171446
21. Kwok, MK, and Schooling, CM. Herpes simplex virus and Alzheimer’s disease: a Mendelian randomization study. Neurobiol Aging. (2021) 99:101.e11–3. doi: 10.1016/j.neurobiolaging.2020.09.025
22. Yang, Q, Borges, MC, Sanderson, E, Magnus, MC, Kilpi, F, Collings, PJ, et al. Associations between insomnia and pregnancy and perinatal outcomes: evidence from Mendelian randomization and multivariable regression analyses. PLoS Med. (2022) 19:e1004090. doi: 10.1371/journal.pmed.1004090
23. Burgess, S, and Thompson, SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. (2017) 32:377–89. doi: 10.1007/s10654-017-0255-x
24. Bowden, J, Davey Smith, G, Haycock, PC, and Burgess, S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. (2016) 40:304–14. doi: 10.1002/gepi.21965
25. Barili, F, Parolari, A, Kappetein, PA, and Freemantle, N. Statistical primer: heterogeneity, random- or fixed-effects model analyses? Interact Cardiovasc Thorac Surg. (2018) 27:317–21. doi: 10.1093/icvts/ivy163
26. Sedgwick, P. Multiple hypothesis testing and Bonferroni’s correction. BMJ. (2014) 349:g6284. doi: 10.1136/bmj.g6284
27. Xiao, G, He, Q, Liu, L, Zhang, T, Zhou, M, Li, X, et al. Causality of genetically determined metabolites on anxiety disorders: a two-sample Mendelian randomization study. J Transl Med. (2022) 20:475. doi: 10.1186/s12967-022-03691-2
28. Deng, Y, and Wong, MCS. Association between rheumatoid arthritis and osteoporosis in Japanese populations: a Mendelian randomization study. Arthritis Rheumatol. (2023) 75:1334–43. doi: 10.1002/art.42502
29. Peterson, MD, Rhea, MR, Sen, A, and Gordon, PM. Resistance exercise for muscular strength in older adults: a meta-analysis. Ageing Res Rev. (2010) 9:226–37. doi: 10.1016/j.arr.2010.03.004
30. Weir, BS, and Hill, WG. Estimating F-statistics. Annu Rev Genet. (2002) 36:721–50. doi: 10.1146/annurev.genet.36.050802.093940
31. Rodrigues, KT, Mekahli, D, Tavares, MF, and Van Schepdael, A. Development and validation of a CE-MS method for the targeted assessment of amino acids in urine. Electrophoresis. (2016) 37:1039–47. doi: 10.1002/elps.201500534
32. Yang, M, Wan, X, Zheng, H, Xu, K, Xie, J, Yu, H, et al. No evidence of a genetic causal relationship between ankylosing spondylitis and gut microbiota: a two-sample Mendelian randomization study. Nutrients. (2023) 15:1057. doi: 10.3390/nu15041057
33. Wang, W, Gopal, S, Pocock, R, and Xiao, Z. Glycan mimetics from natural products: new therapeutic opportunities for neurodegenerative disease. Molecules. (2019) 24:4604. doi: 10.3390/molecules24244604
34. Makdissi, S, Parsons, BD, and Di Cara, F. Towards early detection of neurodegenerative diseases: a gut feeling. Front Cell Dev Biol. (2023) 11:1087091. doi: 10.3389/fcell.2023.1087091
35. Zuflacht, JP, Shao, Y, Kronish, IM, Edmondson, D, Elkind, MSV, Kamel, H, et al. Psychiatric hospitalization increases short-term risk of stroke. Stroke. (2017) 48:1795–801. doi: 10.1161/strokeaha.116.016371
36. Lehrer, S. Glioma and Alzheimer’s disease. J Alzheimers Dis Rep. (2018) 2:213–8. doi: 10.3233/adr-180084
37. Castaneda, A, Ferraz, R, Vieira, M, Cardoso, I, Vasconcelos, V, and Martins, R. Bridging cyanobacteria to neurodegenerative diseases: a new potential source of bioactive compounds against Alzheimer’s disease. Mar Drugs. (2021) 19:343. doi: 10.3390/md19060343
38. Spiteri, AG, Wishart, CL, Pamphlett, R, Locatelli, G, and King, NJC. Microglia and monocytes in inflammatory CNS disease: integrating phenotype and function. Acta Neuropathol. (2022) 143:179–224. doi: 10.1007/s00401-021-02384-2
39. Olah, M, Patrick, E, Villani, AC, Xu, J, White, CC, Ryan, KJ, et al. A transcriptomic atlas of aged human microglia. Nat Commun. (2018) 9:539. doi: 10.1038/s41467-018-02926-5
40. Shi, Y, and Holtzman, DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. (2018) 18:759–72. doi: 10.1038/s41577-018-0051-1
41. Morantz, RA, Wood, GW, Foster, M, Clark, M, and Gollahon, K. Macrophages in experimental and human brain tumors. Part 2: studies of the macrophage content of human brain tumors. J Neurosurg. (1979) 50:305–11. doi: 10.3171/jns.1979.50.3.0305
42. Badie, B, Schartner, J, Klaver, J, and Vorpahl, J. In vitro modulation of microglia motility by glioma cells is mediated by hepatocyte growth factor/scatter factor. Neurosurgery. (1999) 44:1077–82; discussion 1082–3. doi: 10.1097/00006123-199905000-00075
43. Abbott, A. Is ‘friendly fire’ in the brain provoking Alzheimer’s disease? Nature. (2018) 556:426–8. doi: 10.1038/d41586-018-04930-7
44. Karch, CM, and Goate, AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. (2015) 77:43–51. doi: 10.1016/j.biopsych.2014.05.006
45. Yeh, FL, Wang, Y, Tom, I, Gonzalez, LC, and Sheng, M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. (2016) 91:328–40. doi: 10.1016/j.neuron.2016.06.015
46. Zheng, H, Liu, CC, Atagi, Y, Chen, XF, Jia, L, Yang, L, et al. Opposing roles of the triggering receptor expressed on myeloid cells 2 and triggering receptor expressed on myeloid cells-like transcript 2 in microglia activation. Neurobiol Aging. (2016) 42:132–41. doi: 10.1016/j.neurobiolaging.2016.03.004
47. Schmid, CD, Sautkulis, LN, Danielson, PE, Cooper, J, Hasel, KW, Hilbush, BS, et al. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J Neurochem. (2002) 83:1309–20. doi: 10.1046/j.1471-4159.2002.01243.x
48. Wang, XQ, Tao, BB, Li, B, Wang, XH, Zhang, WC, Wan, L, et al. Overexpression of TREM2 enhances glioma cell proliferation and invasion: a therapeutic target in human glioma. Oncotarget. (2016) 7:2354–66. doi: 10.18632/oncotarget.6221
49. Munch-Petersen, CJ. A case of disseminated sclerosis and glioma of the brain in the same patient. Acta Psychiatr Neurol. (1949) 24:599–605. doi: 10.1111/j.1600-0447.1949.tb07342.x
50. Chaligne, R, Gaiti, F, Silverbush, D, Schiffman, JS, Weisman, HR, Kluegel, L, et al. Epigenetic encoding, heritability and plasticity of glioma transcriptional cell states. Nat Genet. (2021) 53:1469–79. doi: 10.1038/s41588-021-00927-7
51. Uddin, MS, Mamun, AA, Alghamdi, BS, Tewari, D, Jeandet, P, Sarwar, MS, et al. Epigenetics of glioblastoma multiforme: from molecular mechanisms to therapeutic approaches. Semin Cancer Biol. (2022) 83:100–20. doi: 10.1016/j.semcancer.2020.12.015
52. Mastronardi, FG, Noor, A, Wood, DD, Paton, T, and Moscarello, MA. Peptidyl argininedeiminase 2 CpG island in multiple sclerosis white matter is hypomethylated. J Neurosci Res. (2007) 85:2006–16. doi: 10.1002/jnr.21329
53. Sahm, K, Kessler, T, Eisele, P, Ratliff, M, Sperk, E, König, L, et al. Concurrent gliomas in patients with multiple sclerosis. Commun Med. (2023) 3:186. doi: 10.1038/s43856-023-00381-y
54. Elliott, L, Brooks, W, and Roszman, T. Role of interleukin-2 (IL-2) and IL-2 receptor expression in the proliferative defect observed in mitogen-stimulated lymphocytes from patients with gliomas. J Natl Cancer Inst. (1987) 78:919–22.
55. Brooks, WH, Netsky, MG, Normansell, DE, and Horwitz, DA. Depressed cell-mediated immunity in patients with primary intracranial tumors. Characterization of a humoral immunosuppressive factor. J Exp Med. (1972) 136:1631–47. doi: 10.1084/jem.136.6.1631
56. Brooks, WH, Roszman, TL, and Rogers, AS. Impairment of rosette-forming T lymphocytes in patients with primary intracranial tumors. Cancer. (1976) 37:1869–73. doi: 10.1002/1097-0142(197604)37:4<1869::aid-cncr2820370435>3.0.co;2-q
57. Lassmann, H. Classification of demyelinating diseases at the interface between etiology and pathogenesis. Curr Opin Neurol. (2001) 14:253–8. doi: 10.1097/00019052-200106000-00001
58. International Multiple Sclerosis Genetics Consortium. Network-based multiple sclerosis pathway analysis with GWAS data from 15,000 cases and 30,000 controls. Am J Hum Genet. (2013) 92:854–65. doi: 10.1016/j.ajhg.2013.04.019
59. Botella, A, Delvaux, M, Bueno, L, and Frexinos, J. Intracellular pathways triggered by galanin to induce contraction of pig ileum smooth muscle cells. J Physiol. (1992) 458:475–86. doi: 10.1113/jphysiol.1992.sp019428
60. Driver, JA, Beiser, A, Au, R, Kreger, BE, Splansky, GL, Kurth, T, et al. Inverse association between cancer and Alzheimer’s disease: results from the Framingham heart study. BMJ. (2012) 344:e1442. doi: 10.1136/bmj.e1442
61. Gargini, R, Segura-Collar, B, and Sánchez-Gómez, P. Novel functions of the neurodegenerative-related gene tau in cancer. Front Aging Neurosci. (2019) 11:231. doi: 10.3389/fnagi.2019.00231
62. Avila, J, Jiménez, JS, Sayas, CL, Bolós, M, Zabala, JC, Rivas, G, et al. Tau structures. Front Aging Neurosci. (2016) 8:262. doi: 10.3389/fnagi.2016.00262
63. Alegría-Loyola, MA, Galnares-Olalde, JA, and Mercado, M. Tumors of the central nervous system. Rev Med Inst Mex Seguro Soc. (2017) 55:330–40.
64. Checler, F, and Alves da Costa, C. Interplay between Parkin and p53 governs a physiological homeostasis that is disrupted in Parkinson’s disease and cerebral cancer. Neurodegener Dis. (2014) 13:118–21. doi: 10.1159/000354075
65. Atamna, H, Nguyen, A, Schultz, C, Boyle, K, Newberry, J, Kato, H, et al. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. (2008) 22:703–12. doi: 10.1096/fj.07-9610com
66. Poteet, E, Choudhury, GR, Winters, A, Li, W, Ryou, MG, Liu, R, et al. Reversing the Warburg effect as a treatment for glioblastoma. J Biol Chem. (2013) 288:9153–64. doi: 10.1074/jbc.M112.440354
67. Gura, T. Hope in Alzheimer’s fight emerges from unexpected places. Nat Med. (2008) 14:894. doi: 10.1038/nm0908-894
68. Rojas, JC, Bruchey, AK, and Gonzalez-Lima, F. Neurometabolic mechanisms for memory enhancement and neuroprotection of methylene blue. Prog Neurobiol. (2012) 96:32–45. doi: 10.1016/j.pneurobio.2011.10.007
Keywords: Mendelian randomization, neurodegenerative diseases, glioma, genetics, Alzheimer’s disease, multiple sclerosis
Citation: Liu Y, Chen Y, Gao M, Luo J, Wang Y, Wang Y, Gao Y, Yang L, Wang J and Wang N (2024) Association between glioma and neurodegenerative diseases risk: a two-sample bi-directional Mendelian randomization analysis. Front. Neurol. 15:1413015. doi: 10.3389/fneur.2024.1413015
Edited by:
Maria Semitekolou, Biomedical Research Foundation of the Academy of Athens (BRFAA), GreeceReviewed by:
Kulvinder Kochar Kaur, Kulvinder Kaur Centre for Human Reproduction, IndiaMubashir Mintoo, University of Kansas, United States
Copyright © 2024 Liu, Chen, Gao, Luo, Wang, Wang, Gao, Yang, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Liu, MTE0Mjk5OTAxMEBxcS5jb20=