 Vincenzo Marcelli1,2*
Vincenzo Marcelli1,2* Beatrice Giannoni3
Beatrice Giannoni3 Giampiero Volpe4
Giampiero Volpe4 Mario Faralli5,6
Mario Faralli5,6 Anna Rita Fetoni2
Anna Rita Fetoni2 Vito E. Pettorossi6
Vito E. Pettorossi6- 1Audiology and Vestibology Unit, Department of ENT, Ospedale del Mare, ASL Napoli 1 Centro, Napoli, Italy
- 2Department of Neuroscience, Reproductive Science and Dentistry, Section of Audiology, University of Naples ‘’Federico II’’, Napoli, Italy
- 3Department of Neuroscience, Psychology, Drug’s Area and Child’s Health, University of Florence, Florence, Italy
- 4Department of Neurology, Ospedale San Luca di Vallo della Lucania, ASL Salerno, Salerno, Italy
- 5Department of ENT, University of Perugia, Perugia, Italy
- 6Department of Medicine and Surgery, University of Perugia, Perugia, Italy
Downbeat nystagmus (DBN) is a neuro-otological finding frequently encountered by clinicians dealing with patients with vertigo. Since DBN is a finding that should be understood because of central vestibular dysfunction, it is necessary to know how to frame it promptly to suggest the correct diagnostic-therapeutic pathway to the patient. As knowledge of its pathophysiology has progressed, the importance of this clinical sign has been increasingly understood. At the same time, clinical diagnostic knowledge has increased, and it has been recognized that this sign may occur sporadically or in association with others within defined clinical syndromes. Thus, in many cases, different therapeutic solutions have become possible. In our work, we have attempted to systematize current knowledge about the origin of this finding, the clinical presentation and current treatment options, to provide an overview that can be used at different levels, from the general practitioner to the specialist neurologist or neurotologist.
Introduction
Downbeat nystagmus (DBN) is the most common form of acquired central nystagmus (1) and is therefore a very frequent finding in daily clinical practice in both the primary care and outpatient settings, from the first to third level of complexity.
Since the initial descriptions, there have been many acquisitions that have led to an ever-greater understanding of the pathophysiological mechanisms underlying DBN, and there have been many implications that these findings have had in both the clinical and therapeutic fields. Despite the large number of publications and some recent reviews, a comprehensive view of the clinical and experimental evidence together with a perspective of pathophysiological interpretation for the different aspects of DBN seems necessary and useful also for clinical purposes. In this review, we trace the steps from the first descriptions of vertical nystagmus to the current “downbeat nystagmus syndrome” (DBNS). Thereafter, we will describe what is known about the clinical characteristics of DBN, we will make an overall overview of the various pathophysiological mechanisms that underlie it, and we will describe the central nervous structures of the central nervous structures whose alterations underlie the signs and symptoms.
Ultimately, our intention was to provide a unified view of this syndrome in terms of etiology, of clinical and nystagmus features, associated oto-neurological signs, and genetic aspects, useful for the identification of the disorder and, sometimes, for the rapid establishment of the most appropriate diagnostic and therapeutic procedure.
First descriptions of vertical nystagmus
Vertical nystagmus received little attention in the early medical literature. For instance, Boehm in 1857 (2) and Fuchs in 1908 (3) did not even discuss vertical nystagmus in their papers. In 1921, Wilbrand and Saenger (4) started to note that vertical nystagmus could be produced in experimental animals by cutting the anterior semicircular canal (SCC) in the labyrinth and found it in some patients with severe bilateral visual loss, drug toxicity (barbiturates and quinine), multiple sclerosis, spasmus nutans, lesions of the pons and medulla (tuberculoma and meningioma), and encephalitis lethargica. In 1950, Berens and McAlpine (5) stated: “Bilateral vertical or mixed movements of nystagmus are rare…. Unfortunately, the direction of the nystagmus has no diagnostic value, and the main clinical problem is the differential diagnosis of whether it is peripheral or central.” On the contrary, Walsh (6) reported in his 1947 review that purely vertical nystagmus was “central or cerebellar,” describing it in two patients with Arnold-Chiari malformation and in one subject affected by cerebellar ataxia, bilateral pyramidal tract involvement and hydrocephalus. However, Walsh does not describe the characteristics of the nystagmus. Although all these authors recognized vertical nystagmus as a sign of central nervous system lesion, they specifically never described its direction.
First descriptions of downbeat nystagmus, its causes and the effects of eccentric gaze and head-body position
Cogan and Barrows (7) described a vertical nystagmus in nine patients with platybasia or Arnold-Chiari malformation; such a sign was present in the primary gaze position in eight subjects of his series. For the first time, the authors described the direction of the nystagmus; two patients had DBN. In one patient, DBN was present in central gaze and decreased during reading (convergence). In the other, DBN was present only in lateral gaze. All but one of the nine patients had symptoms and signs due to compression of the brainstem and/or cerebellum. Cogan (8) published the first study to systematically describe the characteristics, causes and localization of DBN. In particular, the effects of eccentric gaze and head or body position were evaluated. The most common symptoms associated with DBN were found to be oscillopsia and gait difficulty due to ataxia or corticospinal tract impairment. The radiological and surgical findings led Cogan to suggest that lesions causing DBN may be localized in the cerebellum or in the lower brainstem. After this historical paper numerous authors (9–14) confirmed that cerebellar lesions such as Arnold-Chiari malformations, hereditary and sporadic cerebellar degenerations, and other acquired cerebellar degenerations (alcoholic, paraneoplastic and anoxic) can cause DBN. It is important to note that almost all researchers have also already described some peculiarities of DBN, such as the increase in the velocity of its slow component in lateral gaze and in certain positions of the head.
Experimental studies in animals have also confirmed that some lesions usually confined to the caudal cerebellum can produce DBN (15–20).
Evidence of downbeat nystagmus syndrome with nystagmus, perceptual and balance alteration
The association of DBN with oscillopsia, impaired environmental motion perception, mislocation of objects in the direction of the slow phase of nystagmus and an imbalance that causes the body to oscillate predominantly in the antero-posterior direction, constitute a true syndrome called “Downbeat Nystagmus Syndrome” (DBNS) characterized by a tonic vestibular imbalance of neural circuits in the sagittal plane due to damage to central neural pathways.
Characteristics of downbeat nystagmus (DBN)
DBN is usually clearly visible in the primary gaze position, but it can be present, alternatively, or simultaneously with this state, even in the eccentric position of gaze. The most important feature of DBN is its strong gravitational dependence. Convergence and lateral gaze can increase its slow phase velocity (SPV) or even cause nystagmus. As DBN most often follows Alexander’s law (21–23) downward gaze makes the nystagmus more apparent. DBN is not inhibited by visual fixation (24–26).
Other conditions such as hyperventilation, head shaking, and visual fixation can affect the quality of DBN and sometimes change its direction. Very rarely, DBN occurs in combination with internuclear ophthalmoplegia. In this case it is disconjugate, with a vertical component in one eye and a torsional component in the other (27).
The slow phase of nystagmus is usually linear but may show either a negative or positive exponential trend in the presence of brainstem neural integrator dysfunction.
Because DBN is caused by an imbalance of the reflex in the vertical plane, the eye movement has a torsional component in lateral gaze (Table 1).

Table 1. Cerebellar and extracerebellar sites of lesion and the mechanisms underlying them.
As mentioned above, the DBN can be observed in primary (1) or eccentric eye position (2).
1) Nystagmus that is present in primary gaze position.
Regarding the nystagmus present in the primary gaze position, we can recognize it in two different components:
a) A gravity-independent (GI) direction-fixed, chin-directed DBN;
b) a gravity-dependent (GD) direction-changing vertical nystagmus.
The effect that gravity has on vertical drift and VOR through stimulation of otolith receptors is well documented (28). In fact, even normal subjects show vertical drift/nystagmus in the dark which is systematically modulated by both the position of the eyes in the orbit and by the orientation of the head in space (29, 30) In the presence of a cerebellar lesion, this directional influence of gravity is greatly increased (31–33) and electrophysiological studies demonstrated that both the flocculus (34, 35) and the nodulus (36) modulate otolith-ocular reflexes. The role of the cerebellum in controlling the modulation exerted by gravity on the DBN has been clarified by the administration of 3,4-diaminopyridine (3,4-DAP), a drug which acts on the Purkinje cells. Indeed, it has been demonstrated that the drug affects the cerebellar gravity-dependence of nystagmus in different sagittal head positions but does not alter nystagmus in the standing position (37). The modulation of the GD component in the sagittal plane could be due to disinhibition of the canalar reflex in the same plane, modulated by the macular input. As a result of this modulation, in the prone position, as in head flexion, the GD component is directed upwards, whereas in the supine position, as in head extension, the GD component is directed downwards. Due to the presence of these two components, the direction and SPV of the nystagmus are determined by the combination of a fixed GI component (always directed upwards) and a variable vertically directed GD component. In the prone position, the SPV of the nystagmus has the highest values: the GI component directed upwards is indeed added to the GD component directed in the same direction, which has the maximum intensity in this position. On the other hand, the SPV is lowest in the supine position: the GD component shows its maximum downward drift, counteracting the upward drift of the GI component. The opposite direction of the vertical drift caused by the two components is therefore responsible for a very low SPV in the supine position. In the upright position, the GD component is close to zero, so the vertical drift of the eyes in this position is mainly due to the GI component. In the head hanging position, the SPV increased again compared to the supine position because the downward GD component decreased, probably due to the different position of the head and/or the compression force on neural structures, as in Arnold-Chiari disease.
2) DBN that is present only in eccentric gaze on vertical plane.
In such a condition, an orbital-position-dependent effect is present that is likely due to a damage of the vertical neural integrator (VNI) is present. Then, when looking downwards, the result will be a slow centripetal drift of the eyes, which in turn will produce a retinal slip which will generate a rapid eye movement to recover gaze eccentricity, ultimately producing or enhancing the DBN. In this case, the slow phase of the DBN is not linear but either negative or positive exponential.
Although DBN in primary gaze is a sign of CNS dysfunction, it is not rarely possible to observe a positional down beat nystagmus (pDBN), in the presence of which a differential diagnosis with anterior canal BPPV or apogeotropic variant of PSC BPPV is necessary (38–42).
Pathophysiology for DBN
To understand how DBN can develop, it is necessary to consider the existence of an innate asymmetry for the vestibulo-oculomotor vertical reflexes in favor of upward eye movements both in the peripheral apparatus and in the central circuits. Such an imbalance is probably designed to counteract the gravitational forces that naturally oppose upward eye, head, and body movement.
Evidence for vertical reflex asymmetry
The first evidence for an upward reflex preponderance comes from the observation that in cats, dogs, monkeys and humans, in which DB has been shown to be more difficult to suppress than up beat nystagmus (43–46), there is a clear tendency for upward ocular drift in darkness, which increases when the head is tilted away from the upright position due to an otolith-driven central mechanism (47). On the other hand, it has been shown (48, 49) that optokinetic nystagmus and optokinetic afternystagmus as well as nystagmus induced by vertical rotation predominate in the downward direction. In fact, the time constant for DBN is about 15 s, which is like that of horizontal nystagmus, whereas for upbeat nystagmus it is about 8 s. This intrinsic time constant asymmetry is also modulated by the otolith end organs, which suppress the velocity storage mechanism for downward directed nystagmus in the upright position and enhance it during downward low frequencies head movements (50). Further confirming the asymmetry in the sagittal plane and the role of the macula, in the upright subject, is that the forward pitch sensation during the surround movement that produced the downward OKN is stronger than the backward pitch sensation associated with upward OKN, and this effect is significantly enhanced in the side lateral position (51).
Peripheral asymmetry of the vertical vestibulo-ocular reflex
At the peripheral level, the preponderance of upward eye movements is supported by the anatomo-physiological properties of the vertical SCCs (34). Indeed, the anterior SCCs are more aligned with the sagittal plane than the posterior canals. Therefore, the ampullofugal stimulus, which is excitatory for the vertical SCCs, excites the cupula of the anterior SCCs better during downward head rotation than the cupula of the posterior SCCs during upward head rotation. Thus, the vertical head rotation signal is naturally asymmetric, manifesting a preponderance during downward rotations (Figure 1). This state of asymmetry must be subject to central inhibitory control. Moreover, the antigravitational effects must be modulated according to the action of gravity and therefore according to the different positions of the head in space (Figure 2). Consequently, if these central inhibitory control areas are damaged, a nystagmus with a slow upward phase and a fast downward phase (DBN) can be induced.
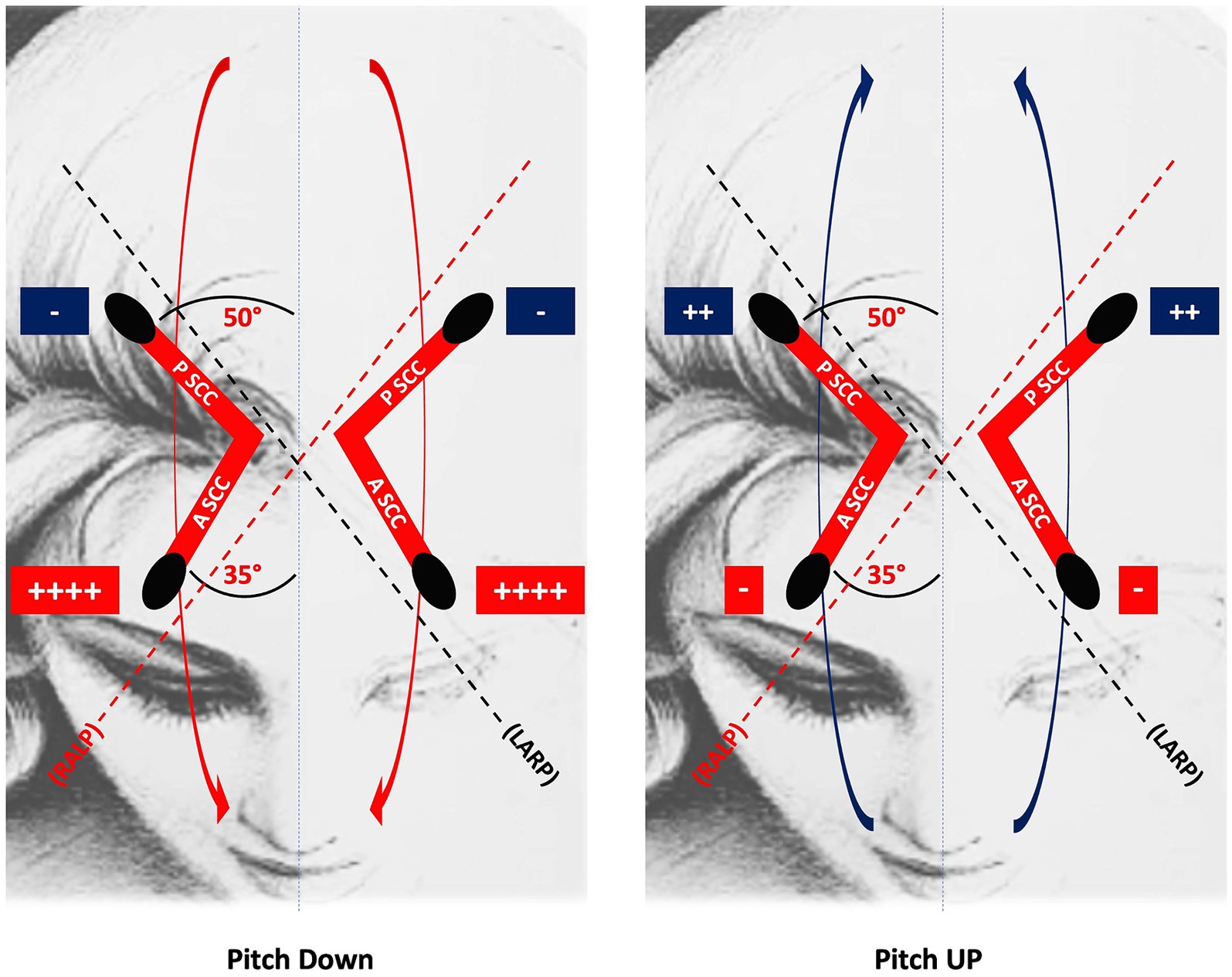
Figure 1. The upward eye response to downward head rotation is greater than the downward eye response to upward head rotation because the anterior SCC is more aligned with the sagittal plane than the posterior SCC: thus, the ampullofugal, excitatory response of the anterior SCC cupula to downward head rotation is greater than the homologous response of the posterior SCC cupula to upward head rotation. RALP, right anterior, left posterior; LARP, left anterior, right posterior; PSCC, posterior semicircular canal; ASCC, anterior semicircular canal.
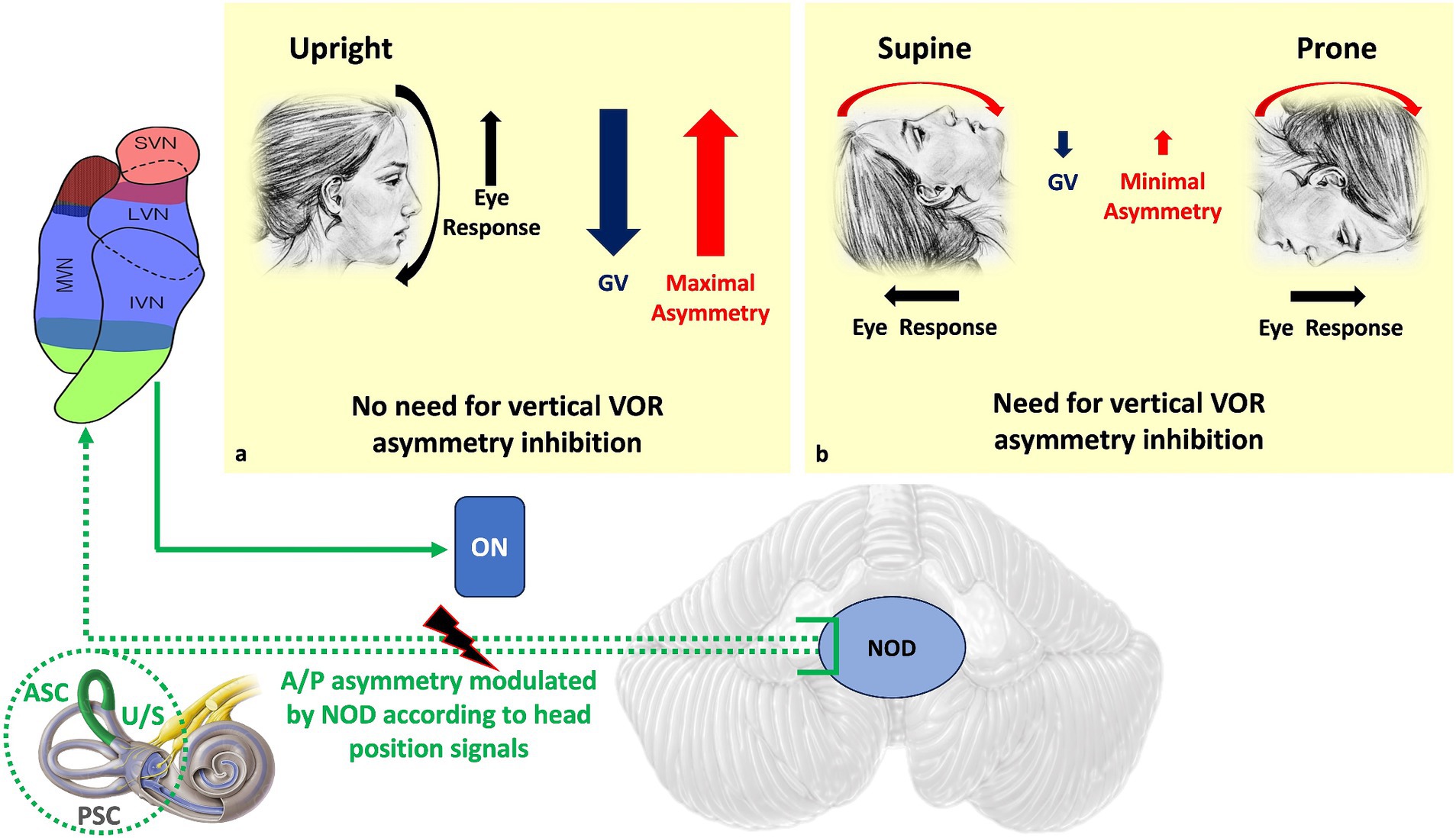
Figure 2. Dashed green is the NOD circuit that modulates the A/P asymmetry of the vertical VOR and upward slow phase due to different gravitational vector effects on the macula depending on head position. In the upright position (a), the preponderance of upward eye responses is needed to counteract the effect of gravity, so peripheral asymmetry should not be inhibited. In the supine and prone positions (b), where gravity no longer opposes movement, the natural asymmetry of the VOR is no longer necessary. Therefore, the asymmetry of the vertical VOR should be inhibited. Direct NOD lesion in humans produces positional DBN. ASC, Anterior semicircular canal; PSC, Posterior semicircular canal; U/S, Utricle/Sacculus; ON, Oculomotor nuclei; A/P, Anterior/Posterior; NOD, Nodulus/Uvula; GV, gravitational vector.
Asymmetry of the centers controlling the vertical reflex
Regarding the circuits that control vertical asymmetry, it has been shown that some cerebellar and extra-cerebellar structures are dedicated to controlling the activity of VOR responses in the vertical plane. These areas have been identified in the flocculus/paraflocculus, nodulus, vermis and some brainstem areas.
1. Role of the Flocculus/Paraflocculus.
a. Cerebellar Purkinje cells of the rostral and caudal zones of the flocculus (F/PF). These cells are involved in the inhibitory control of upward vertical rotational VOR, projecting to superior vestibular nucleus (52–55). In fact, their lesion results in relative hyperactivity of the anterior SCCs circuit (14, 34, 55–57), which, in turn, generates a DBN. Such a hyperactivity is confirmed by the finding of an increased upward VOR gain when performing a downward head impulse test (14, 58–60). Since the F/PF is involved in both the generation and in control of smooth pursuit and the control of ocular eccentricity, a lesion involving these structures will also result in impaired vertical smooth pursuit and gaze-evoked nystagmus (GEN). Downbeat nystagmus seen with flocculus/paraflocculus lesions cannot be suppressed with visual fixation.
b. Cerebellar flocculus/paraflocculus (F/PF) and the dorsal larger cell Y-group (Yd) of the Y-group nucleus. The Y-group nucleus of the vestibular nuclear complex is a well-defined nucleus in the brainstem. A subpopulation of the Y-group nucleus, the dorsal larger Y group (Yd), contains a large population of excitatory neurons that target up gaze motoneurons in the contralateral oculomotor nucleus and GABAergic inhibitory neurons that target motoneurons in the ipsilateral oculomotor and trochlear nuclei (58–61). The Y-group nucleus receives a disynaptic excitatory input from the ipsilateral anterior and posterior SCCs via neurons in the superior vestibular nucleus and caudal medial vestibular nucleus (MVN), and a strong inhibitory input from Purkinje cells of the F/PF (62–65). A lesion of the Purkinje cells results in disinhibition of the Y-group cells, which leads to upward sliding of the eyes, and induces DBN.
2. Role of the Nodulus/Uvula and otolith.
It has been shown that almost half of the Purkinje cells sensitive to vertical SCCs activation in the nodulus/uvula (NOD) are modulated by otolith signals (28, 36, 56, 58, 66, 67), that signals from SCCs and otolith receptors reach specific areas of the NU where they are integrated (68), and that the same signals often converge on a single Purkinje cell. It is also interesting to note that the afferent signals from anterior and posterior SCCs project in different areas. In this way, the two circuits have the possibility of separate modulation for movements in both the frontal and sagittal plane, to influence differently the preponderance of the peripheral vestibular signals. Such convergence of inputs therefore allows the control of the inhibitory SCCs to be modulated by head position signals.
In particular, the natural preponderance of responses inherent to the circuitry of the anterior SCCs can be modulated during downward head movement depending on how the force of gravity opposes the eye and head responses (Figure 2).
The role of the nodule/uvula is further supported by the observation of increased intensity of the DBN following neurosurgical aspiration of these structures in monkeys (69) and the loss of orientation of the horizontal afternystagmus, which also indicates the orientational role of the nodule in ocular responses (see “Associated oto-neurological signs” section).
In addition, it has been shown that the velocity of the slow phase of head-down nystagmus is no longer inhibited after nodule removal (66, 70, 71) and that direct NOD lesion in humans produces positional DBN (72).
In contrast to the downbeat nystagmus seen in flocculus/paraflocculus lesions, the nystagmus due to nodulus/uvula lesions can be suppressed by visual fixation, is only seen in the dark, and the slow phase velocity is not increased in lateral gaze or decreased in up gaze. These findings suggest that downbeat nystagmus in nodulus/uvula lesions is unlikely to be related to an abnormal vertical ocular motor integrator, but rather to a central vestibular imbalance, possibly in the otolith-ocular pathways that mediate the vertical translational VOR (69).
In this way, direct damage to the otolith-ocular circuit can also cause DBN (58). In humans, DBN has been shown to be induced by static head tilt and linear acceleration independent of the canal planes and corresponding to the static and dynamic components of otolith function (73, 74). Changes in static head orientation in the sagittal plane are known to influence DBN through otolith modulation (30, 75). The role of otolith inputs is also demonstrated during oscillation about the upright position: the gain of the upward VOR increases and the gain of the downward VOR decreases, with a degree of VOR asymmetry greater than would be expected from a simple summation of spontaneous nystagmus with normal SCCs reflexes (58).
In addition, mutant mice with calcium P/Q channelopathy, which lose many of the critical electrical properties of the cerebellar Purkinje cell dendrite, show hyperactivity of the otolith-ocular reflexes (76) and static ocular hyperdeviation, which is the murine equivalent of the upward velocity bias in DBN (77). As mentioned above, the imbalance of the macular-canal pathways would potentially explain the modulation of nystagmus by the gravitational vectors in most, if not all, patients, and damage to these structures would therefore produce a disinhibition of the macular-canal interaction (21, 37, 58–60, 72, 78–83). The role of macular input is further demonstrated by the change in DBN SPV induced by convergence, a reflex that is modulated by the macula.
3. Role of cerebellar areas implied in vertical smooth pursuit: flocculus/paraflocculus, vermis Nodulus/Uvula
a. Flocculus/paraflocculus. A central imbalance in vertical smooth pursuit (SP) tone has been considered as a possible origin of DBN due to impairment of Purkinje cells of the F/PF and its complex pathway (medial and superior VN, dorsolateral pontine nuclei, ocular motor dorsal vermis, fastigial nucleus, nucleus reticularis tegmenti pontis, prepositus nuclei). In particular, the just reported bias of Purkinje cells to discharge with downward movements could explain the asymmetric vertical pursuit in patients with downbeat nystagmus. A lesion involving such cells would result in upward eye gliding and consequent DBN.
b. Vermis. In addition to the Purkinje cells of the flocculus, there is another Purkinje population in the dorsal vermis (lobules VI, VII) that plays a critical role in vertical SP eye movements, firing more effectively during downward than upward smooth pursuit, hence the term “downward directed floccular cells” (60, 78, 84, 85). Therefore, also a lesion involving such cells would result in upward eye gliding and consequent DBN.
c. Nodulus/uvula. A critical role for the NOD in vertical pursuit, particularly in sustained downward pursuit, has been demonstrated, and in monkeys lesion of the NOD increased spontaneous upward eye drift in the dark. This finding confirms a role for the NOD in eye fixation and in the generation of downbeat nystagmus (83).
4. Role of extra-cerebellar areas: vertical neural integrator, floor of the fourth ventricle, medulla oblongata, pons, lower motor neurons
a) The neural integrator for vertical eye movements is a complex neural network whose structures are largely located in the midbrain and project indirectly, from the interstitial nucleus of Cajal [via the paramedian tract nucleus (PMT)], or directly, via the PMT, to the FL/PFL. The VNI receives velocity commands from all the conjugate eye movements systems and converts them into a position command suitable for maintaining vertical gaze eccentricity once it has been reached if this is required. Damage to these structures can lead to DBN, as has been shown in non-human primates with lesions of the PMT (86), which determined a reduced FL/PFL input, a reduced PC output and, finally, a hypofunction of the FL/PFL. A lesion of this neural network can also alter the integrator time constant and the intrinsic orientation coordinate system, and thus still result in DBN as the eyes are unable to maintain eye position correctly and rapid reset phases are triggered to regain downward position (21, 55, 81, 86). In addition, the VNI may become “leaky” or “unstable,” characteristically modifying the DBN intensity:
a. a “leaky” NI determines a velocity decreasing slow-phase waveforms and nystagmus intensity which increases looking in the direction of the fast phase.
b. an “unstable” NI determines a velocity increasing slow-phase waveforms and nystagmus intensity which increases looking in the direction of the slow phase.
b) The floor of the fourth ventricle contains the vertical VOR pathways starting from the posterior SCCs, whose input reaches the medial and lateral vestibular nuclei. Injury of this pathway will result in the loss of the tonic activity of the posterior SCCs, resulting in upward ocular drift and DBN (87). The possible involvement of the medial longitudinal fasciculus, traveling in a craniocaudal direction near the midline within the tegmentum of the midbrain and dorsal pons immediately ventral to the cerebral aqueduct and fourth ventricle, justifies the possible associated internuclear ophthalmoplegia.
c) The medulla oblongata contains the paramedian tract neuron (PMT) cells, which exert a physiological tonic excitation on floccular Purkinje cells. Injury of these cells reduces the activity of floccular Purkinje cells, which in turn reduces the inhibition of secondary vestibular nucleus neurons that receive input from the anterior SCCs. This disinhibition induces upward ocular drift and thus the production of a DBN (88, 89).
d) The Pons contains the nucleus hypoglossi prepositus (NPH), that is the basic structure of horizontal neural integrator (HNI). An injury to the HNI may also result in a DBN. Experimental animal study and case reports have shown that NPH dysfunction can also results in vertical ocular deviations, suggesting some overlap between horizontal and vertical gaze control (90). Of course, the proximity between the NPH and MVN, approximately 0.9–1.1 mm apart in felines (91, 92), does not exclude that the lesion may also affect the VMN, in addition to that of the neural integrator, which would be the true cause of DBN.
e) Lower motor neurons. Finally, it must also be remembered the existence of an association between DBN and the lower motor neuron disease, whose etiology remains unknown (93, 94).
Effects of ocular convergence and ocular deviation on DBN
Ocular convergence and ocular lateral deviation have been reported to induce or facilitate DBN (1, 57, 95). A possible explanation for these effects is the presence of a cross-coupling mechanism between the circuits of convergence and lateral gaze deviation and vertical eye movements. It is likely that lesions producing DBN can also destroy these separate circuits. Regarding convergence, it is well known that the vertical otolith-ocular reflex should depend on the distance between the target and the observer, with an increase in the reflex gain in the presence of a near object to be observed (96). Thus, the reflex is facilitated along with the activation of vergence. In DBN patients, the two interrelated mechanisms of vergence and gain enhancement are cross-coupled. Therefore, vergence could enhance any vestibular reflex, but if there is an imbalance in the vertical circuits, vergence could reveal or induce DBN. A similar cross-coupling may be responsible for the occurrence of DBN with lateral eye deviation. The lateral deviation of the eye could be interpreted by the central nervous system because of a lateral displacement of the head, activating the translational macular reflex in the vertical plane, making the eyes sliding upwards (68, 73, 97, 98). Because of the damage of the center controlling the otolithic networks, cross coupling between horizontal and vertical otolithic reflexes may occur, inducing increased excitability in the vertical reflex. Therefore, in presence of an imbalance in the vertical eye movement control, this control excitability would facilitate the DBN (57).
Figure 3 summarizes the possible cerebellar and extracerebellar sites of lesion and the mechanisms underlying them.
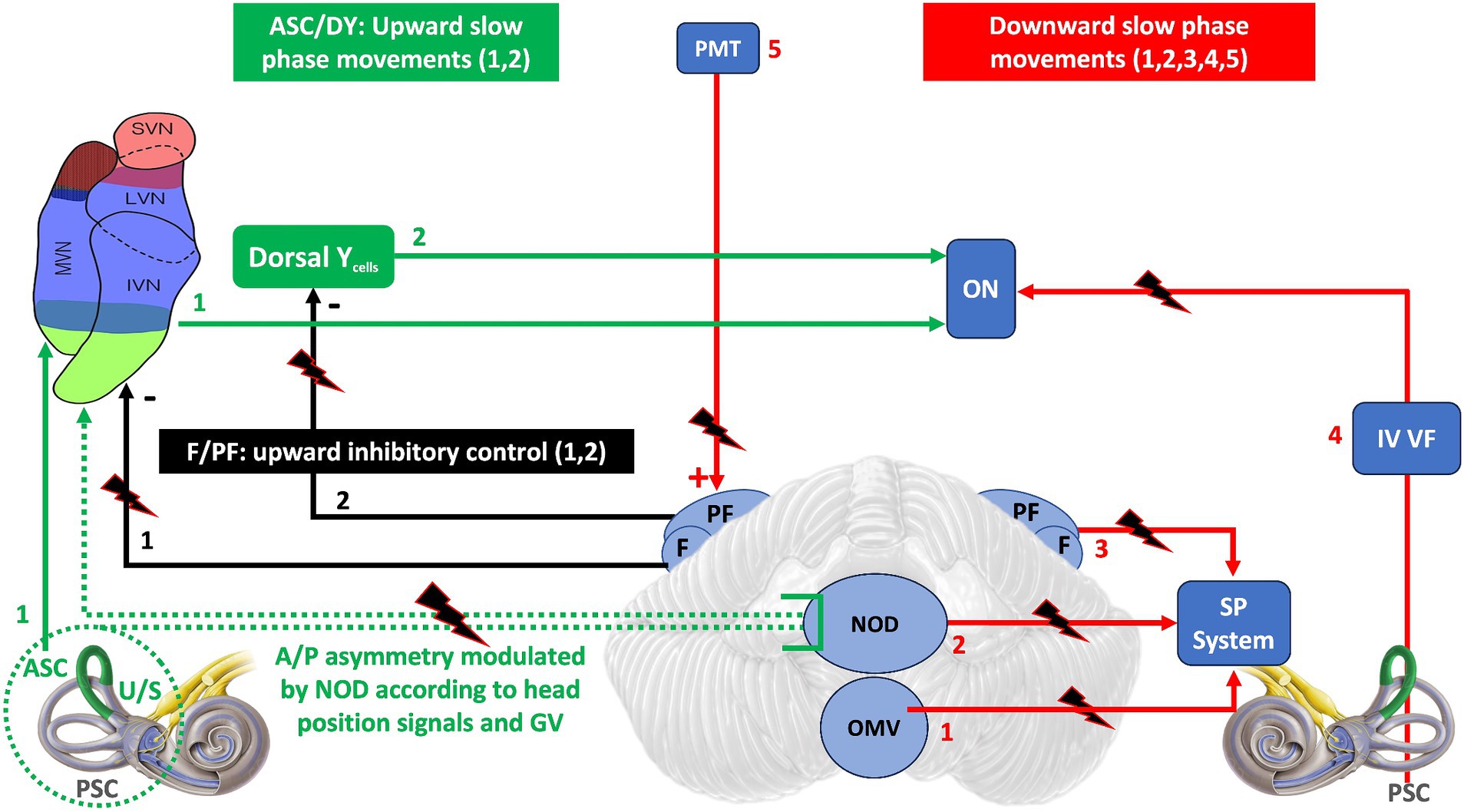
Figure 3. The diagram is a schematic representation of the neural areas involved in the control of vertical eye movements and the possible locations of lesions that may result in DBN. In green are the areas directly responsible for generating an upward slow phase: ASC and dorsal Y. In black are the FL/PF inhibitory efferents to the vestibular nuclei that modulate the upward slow phase. In red are the areas destined to generate a downward slow phase: 1,2,3,4 directly. 5 (PMT) indirectly, because by exciting the F/PF, it favors the inhibition of the upward slow phase and thus the generation of a downward slow phase. Dashed green as in Figure 2. ASC, Anterior semicircular canal; PSC, Posterior semicircular canal; U/S, Utricle/Sacculus; A/P, Anterior/Posterior; GV, Gravitational vector; F/FP, Flocculus/Paraflocculus; DY, Dorsal Y cells; PMT, paramedian tract nucleus; ON, Oculomotor nuclei; OMV, Oculomotor vermis; NOD, Nodulus/Uvula; SP, Smooth Pursuit; IV VF, Floor of VI ventricle.
Circadian modulation of DBN
The DBN decreases spontaneously during the day when the subject remains in an upright position and in darkness. Indeed, the slow phase velocity (SPV) decreases by about 50% during the day and remains constant in the afternoon (99). On the other hand, the SPV measured in the upright position is lower after resting in the upright position than after resting in the supine or prone position. This effect does not depend on fixation, eye position or body position. Several hypotheses have been proposed to account for the possible role of otolith signals in stabilizing vertical circuits of the VOR during prolonged upright rest (100, 101). Other influences can be considered, such as the role of extraocular proprioception (102), efference copy (103, 104), sleep inertia, which is a transient state of reduced arousal that occurs immediately after awakening from sleep (105–107) and has a negative influence on Purkinje cell function. Finally, the effect of resting for 2 h in darkness was also considered, which significantly lowered the post-rest DBN, in contrast to the effect of resting in light (108).
Symptoms of downbeat nystagmus syndrome
The symptomatology of Downbeat Nystagmus Syndrome (DBNS) is essentially characterized by oscillopsia and motion illusion, diplopia, internuclear ophthalmoplegia (INO) and/or skew deviation, sagittal imbalance, and increased risk of falls.
Oscillopsia (109) is the most disabling symptom. It is caused by the shifting of images on the retina because of the involuntary slow phase of nystagmus. In the absence of an efferent copy, which is generated only in the presence of a voluntary movement, the pathological displacement of the eyes creates an illusion of movement of the visual scene. The illusion is less intense compared to the magnitude of the slow-phase eye movements, presumably due to a reduction in the sensitivity of visual motion perception (110), which is useful to mitigate the annoyance of illusory oscillations. Indeed, the estimated ratio between the illusory motion and the slow-phase angular velocity (SPAV) is 0.371. However, this perceptual reduction also reduces the visual feedback necessary for motion control, thus altering responses to movement and altering foot trajectory and positioning during walking, ultimately resulting in additional imbalance (111, 112).
On the other hand, the same motion illusion due to oscillopsia generates a compensatory vestibulo-spinal reflex (VSR) that contributes to the imbalance. In fact, under physiological conditions, flexion of the head and/or trunk activates the vertical VOR, responsible for an upward slow phase, and the vestibulo-collic and vestibulo-spinal reflexes, responsible for an increase in posterior spinal muscle tone necessary to balance the flexion.
Under pathological conditions, the tonic asymmetry generated not by the subject’s movement but by the central vestibular pathways causes an upward eye movement and generates an antero-pulsion misperception characterized by an illusory head and/or body flexion. The resulting postural compensation leads to an increase in posterior spinal muscle tone to compensate for the illusory flexion, which is characterized by retropulsion and postural oscillations in the antero-posterior direction. These effects are not dependent on vision, as they have been demonstrated even with the eyes closed (113). In conclusion, the postural oscillations present with eyes open are a direct result of several mechanisms induced by the lesion and/or an indirect consequence of the illusory perception of body displacement.
In addition, is typical of the DBS a peculiar gait disturbance characterized by a reduced step speed, a shorter swing phase, and a longer double-support stance phase (114) due to impaired sensorimotor integration caused by the vestibular-cerebellar lesion. Notably, DBN frequency and postural imbalance were significantly reduced during walking compared to standing (p < 0.001), and the frequency of nystagmus during walking was further modulated in a manner that depended on the specific phase of the gait cycle (p = 0.015) (115). The attenuation of DBN symptoms may result directly from the locomotor activity itself, reflecting selective central and/or peripheral suppression of sensory feedback during locomotion. Theoretical approaches (116, 117), clinical observations (118, 119), and experimental evidence (120, 121) suggest the presence of a predictive feedforward regulation of gaze and postural stability during locomotion. Thus, efference copies of the locomotor command could also counteract the destabilizing vestibulo-cerebellar drive that has been suggested to cause central oculomotor and postural impairments in DBN patients.
Diplopia, internuclear ophthalmoplegia (INO) and/or skew deviation (14, 122), when present, are due to concomitant involvement of the MLF (27, 55).
Interestingly, patients with DBS may have worse symptoms in the morning and instead improve during the rest of the day (99), may improve after resting in an upright position (123) and after being left in the dark (108).
Associated oto-neurological signs
Due to the presence of spontaneous vertical nystagmus, smooth pursuit and optokinetic reflex in the vertical plane are altered, configuring a “false” (extrinsic) alteration of these reflexes. On the other hand, the alteration of smooth pursuit in the vertical plane is greater than would be expected from spontaneous nystagmus alone and from the deficit gain of smooth pursuit and the optokinetic reflex. Such a finding would seem to indicate a more widespread and “real” alteration in the central vestibular pathways.
In addition, due to these extensive changes, gaze-evoked and rebound nystagmus in the horizontal plane can often be observed. These findings are probably due to the close topographical and functional relationship between the cerebellar pathways for vertical movements and those for horizontal movements, especially those originating from the flocculus and vermis (108). In these cases, smooth pursuit may be apparently normal when the target is moving upward, the slow phase of nystagmus being in favor of smooth eye movement.
In some patients with DBS, unilateral or bilateral deficit vestibulopathy, especially at high frequencies, may be observed.
In addition to DBN, it is not uncommon to observe cross-coupled (perverted) nystagmus during caloric stimulation. The latter can be attributed to a disruption of the nodulus-uvula complex, the structure physiologically responsible for conjugating the plane of the reflex response with the plane in which the stimulus occurs (32).
Finally, the association of DBN with polyneuropathy and mono- or bilateral deficit vestibulopathy, especially at high frequencies, is not uncommon. The latter association suggests a common pathogenesis (1, 124, 125) probably involving calcium channel abnormalities, particularly in Purkinje cells.
Etiology
Approximately 40% of DBS have an unknown cause and are considered idiopathic primary forms. In the remaining cases, secondary forms, a sporadic (i.e., vascular, malformative, degenerative, neoplastic, infectious, paraneoplastic, toxic, deficient, and iatrogenic) or genetic cause can be recognized. Various sites and structures may be involved in these secondary forms:
1. Cerebellum: flocculus/paraflocculus (FL/PFL), vermis, cerebellar hemispheres.
2. Brainstem: PMT neurons, pontine ventral tegmental tract (VTT).
3. Cervicomedullary junction (CMJ).
4. Foramen magnum region, and atlanto-occipital joint.
The most frequent causes of acquired forms of DBS are:
1. Vascular pathologies such as:
a) ischemic and hemorrhagic stroke (1, 88, 122, 126–129);
b) arterial malformations as: dolichoectasia of the vertebrobasilar circulation (130–132);
c) compression of the vertebral artery (133).
2. Inflammatory and autoimmune diseases such as:
a) optic neuropathy (134);
b) demyelinating pathologies (89, 122, 135–137);
c) autoimmune thyroiditis and Hashimoto’s encephalopathy (138);
e) anti-GAD antibodies associated with DBN, idiopathic cerebellar ataxia, stiff-person syndrome, epilepsy and limbic encephalitis (143);
f) anti-NMDA receptor encephalitis (144).
3. Infectious diseases such as:
a) cephalic tetanus, which inhibits GABAergic Purkinje cells (145);
b) herpes encephalitis affecting brainstem or flocculus (146, 147);
c) West Nile virus encephalitis affecting the CMJ (148);
d) chronic human T-lymphotropic virus- 1 (HTLV-1) infection causing degeneration of cerebellar vermis (149).
4. Degenerative pathologies such as:
a) multiple system atrophy (38);
b) amyotrophic lateral sclerosis (150);
c) motor neuronopathy (151).
5. Malformative conditions such as:
a) Arnold Chiari disease (7, 152–155) (reported in 4–7% of patients with type I Arnold Chiari malformation);
b) syringobulbia (14, 122, 156, 157);
c) platybasia (8, 122, 158, 159);
d) “foramen magnum syndrome” (160).
6. Paraneoplastic syndromes include small cell lung cancer (with anti-CV2/CRMP5, anti-Hu, anti-Zic1/4 antibodies), ovarian and breast cancer (with anti-Yo antibodies), testicular neoplasms (with anti-Ma antibodies), thymoma (with anti-CV2/CRMP5 antibodies) and Hodgkin’s lymphoma (with anti-Tr antibodies) (161); anti-Yo and anti-Tr antibodies syndromes involving cerebellar degeneration (162–165), anti-Ma antibodies encephalitis (166, 167), anti-HU antibodies brainstem encephalitis (168).
7. Toxic conditions that may be attributed to toluene exposure and, above all, to ethanol chronic abuse (169, 170), the latter resulting in vitamin B1 deficiency and Wernicke’s encephalopathy (171, 172). Interestingly, also in non-chronic alcoholics forms, an excessive alcohol intake can cause reversible, short-lived DBN (173, 174).
8. Deficiency-type cerebellar disorders, which include vitamin B12 and Magnesium deficiency, a modulator of excitatory Ca2++ NMDA channels in cerebellar granule cells, which is also often associated with a lack in vit B1 (175–180).
9. Iatrogenic form due to:
a) antiepileptic drugs such as phenytoin (181), carbamazepine (even in therapeutic range) (182), lamotrigine (183), pregabalin (184). These drugs inhibit voltage-gated sodium channels Nav1.1 and 1.6, which are heavily expressed in Purkinje cells (185);
b) antiarrhythmic agents, such amiodarone (186);
c) intravenous opioids morphine, fentanyl, pethidine and piritramide (187–190), as well as intrathecal morphine (191). These drugs may generate a DBN by virtue of the agonism of m-opioid receptors in the cerebellum inhibiting the inhibitory cerebellar projections to the superior vestibular nucleus;
d) gastric acid suppressants such as ranitidine (192);
e) lithium toxicity is only rarely a cause of DBN, and no clear mechanism has been elucidated (193–197), such as quietiapine fumarate and estazolam (198).
10. Other possible causes as head trauma, intracranial hypertension, hydrocephalus (199); vestibular migraine (199–201) and tumors, also including metastasis (202–205).
11. Complex hereditary neurodegenerative disease such as:
a) Spinocerebellar ataxia type 6 (SCA6) (206–209);
b) Episodic ataxia type 2 (EA2) (210–213) and familial hemiplegic migraine type 1 (FHM1) (214–216), both caused by mutations in the CACNA1A gene, which encodes for voltage-gated calcium channel widely expressed in Purkinje and granule cells;
c) Idiopathic cerebellar degeneration such sporadic adult-onset ataxia and multisystem atrophy (1);
d) Familial episodic ataxia (213, 217, 218);
e) Ataxia- telangiectasia (219);
f) Mitochondrial Encephalopathy with Lactic Acidosis and Stroke-like episodes (MELAS) (220).
Tran et al. (221) have recently proposed the following useful etiological classification:
1. DBN because of structurally evident pathology;
2. DBN associated with specified non-structurally evident causes;
3. Idiopathic DBN, which includes patients with:
a) “pure” DBN, without other signs or symptoms except for the oculomotor disorders associated with floccular disorders (i.e., impaired smooth pursuit, optokinetic reflex and/or lack of visual suppression of the VOR);
b) “cerebellar” DBN (i.e., DBN associated with cerebellar ataxia or dysarthria);
c) “syndromic” form (i.e., DBN associated with floccular signs/symptoms, gait abnormalities, limb ataxia, dysarthria, dysmetria, dysdiadochokinesis, tremor, hypotonia, and so forth).
In the “syndromic” form, other authors also include at least two of the following: bilateral vestibulopathy, cerebellar signs and/or polyneuropathy, the latter reflecting multisystem neurodegeneration or channelopathy (1).
Genetic aspects
To explain the genesis of DBN, some genetic aspects that can alter the functioning of cerebellar cells, and in particular Purkinje cells, have also been studied.
For example, adult murine CACNA1A mutants, in which P/Q channels present in cerebellar Purkinje cells and responsible for calcium currents are particularly reduced, show an upward displacement of the eye globes of the eye and a reduced high-frequency VOR gain. This finding supports that a channelopathy may be the underlying cause in some cases of idiopathic downbeat nystagmus. The efficacy of a potassium channel blocker, such as 4-aminopyridine, in reducing downbeat nystagmus and improving horizontal and vertical VOR gain would support this hypothesis. The appearance of the signs only in adult mice would also suggest that a genetic polymorphism in the CACNA1A gene predisposes to the DBS, which would occur because of age-related degenerative phenomena (222–224).
Other interesting information comes from chromosome 5q14.1, which contains the genes DHFR (essential for folate metabolism) and MSH3 (involved in cell cycle regulation, apoptosis, and genome stability). Disruption of these genes is responsible for cerebellar disorders (225–227).
Finally, it is important to remember the role of the fibroblast growth factor 14 (FGF14) gene, located on chromosome 13, and highly expressed in Purkinje cells, which represent the only output of the cerebellar cortex. FGF14 modulates the intrinsic excitability of cerebellar Purkinje neurons (228) positively regulates voltage-gated KCNQ2/3 potassium channels (Kv7.2/7.3), voltage-gated sodium channels (which have nine well-characterized isoforms: NaV1.1-NaV1.9) (229) and presynaptic Ca2+ voltage-gated channels. A distinctive feature of Purkinje cells is the robust expression of Nav1.6-encoded sodium channels, which underlie the “resurgent” sodium current that is critical for maintaining the characteristic high-frequency burst of these cells. More than 80% of Fgf14−/− Purkinje cells are quiescent and do not repeatedly activate in response to excitatory stimuli. Immunohistochemistry revealed reduced expression of Nav1.6 protein in Purkinje Fgf14−/− cells. These observations suggest that FGF14 is necessary for normal expression of Nav1.6 in Purkinje neurons and that its absence, by altering the expression of Nav1.6 channels, impairs spontaneous and tonic repetitive activation of Purkinje cells. The recent identification of an association between a variation in intron 1 of the fibroblast growth factor 14 (FGF14) gene and idiopathic DBN in a genome-wide association study (GWAS) (225) and of a dominantly inherited (GAA)≥250 repeat expansion in intron 1 of FGF14 as the cause of spinocerebellar ataxia 27B (SCA27B)/GAA-FGF14 ataxia (230, 231), a late-onset slowly progressive cerebellar syndrome that is frequently associated with DBN (230, 232) provided evidence that FGF14 (AAA) repeat expansions, including (GAA)200–249 and (GAA)<200, are a monogenic cause of idiopatic DBN syndrome (230, 232–235). The frequency of FGF14 (GAA)≥250 expansions in DBN syndromes is 56% (42/75) for DBN plus additional isolated cerebellar ocular motor signs, 72% (23/32) for DBN plus cerebellar ataxia, 30% (17/56) for DBN plus cerebellar ocular motor signs and/or ataxia and extracerebellar features, which included bilateral vestibulopathy (BVP) and/or polyneuropathy, and 0% (0/7) for pure DBN (235). The unexpected high frequency of FGF14 (GAA) ≥250 repeat expansions in patients with a predominantly sporadic non-ataxic DBN presentation may be due to hte fact that DBN is a milder phenotypic presentation of GAA FGF14 disease, in which overt cerebellar ataxia and other multisystemic involvement can be absent or, in some cases, develop later in the disease course. The identification of additional floccular/parafloccular cerebellar ocular motor signs in all (GAA)≥250-FGF14 patients with DBN, which may present in isolation early in the disease course, up to 8 years prior to the development of gait impairment, indicates that the basic dysfunction arises from this cerebellar region and that pure DBN is an uncommon manifestation in GAA-FGF14 disease. Finally, DBN plus additional isolated cerebellar ocular motor signs but without overt ataxia, despite long disease duration (up to 16 years), raises the possibility that GAA-FGF14 disease may remain limited to the cerebellar ocular motor system without broader cerebellar involvement in a subset of patients. For these reasons, genetic testing for FGF14 GAA repeat expansion should become part of the diagnostic work-up of patients with idiopathic DBN (235). Another interesting aspect, in these patients bilateral vestibulopathy (BVP) has a low frequency occurrence of (only 10% of cases), is a late feature in GAA-FGF14 disease compared to cerebellar ataxia, developing on average more than 10 years after disease onset, is significantly less frequent in (GAA)≥250-FGF14 compared to (GAA)<200-FGF14 DBN syndromes, and tends to remain relatively mild in GAA-FGF14-related DBN syndromes despite prolonged disease duration. Moreover, impairment of the visual fixation suppression of the VOR is most strongly associated with and predictive of (GAA)≥250-FGF14 DBN and (29) (GAA)200–249-FGF14 than (GAA)<200-FGF14 patients (235).
What has just been reported justifies the symptomatic benefit of 4-aminopyridine, a blocker of mainly Kv1 (A-type) potassium channels, which is the drug of choice for the treatment of downbeat nystagmus because it acts precisely by increasing the excitability of CPs43 and restoring the neuronal firing precision of Purkinje neurons. The efficacy of 4 aminopyridine is in fact demonstrated in a mouse model of ataxia that mimics SCA6 (57, 236), in episodic ataxia type 2 (EA2) and in GAA-FGF14 ataxia (SCA27B), which are also characterized by DBN (232). Particularly, response to 4-AP represents a strong predictor of (GAA)≥250-FGF14 and (GAA)200–249-FGF14 status in patients with idiopathic DBN (positive predictive value, 89%).
Conclusion
An important finding most often indicative of central vestibular dysfunction is the DBN whose presence is often included in a currently well-characterized clinical syndrome.
The diagnostic significance of this finding has only recently been widely understood as research into its origin and pathophysiology has progressed. We now know in sufficient detail the neural structures responsible for the delicate control of eye movements in the vertical plane and can therefore focus our attention on these structures when confronted with a DBN.
Nowadays therefore, DBN is to be considered one of those highly localizing pathologic findings, and its recognition should first lead to detailed neuro-radiologic studies to identify the most common structural chronic or acute causes of this sign.
However, DBN can also hide subtle causes that can only be identified through specific hematologic and clinical studies, which must be known because, even in these special cases, a state of diagnostic and therapeutic urgency can be realized.
Sometimes, to correctly define a DBN and its syndrome, it is also necessary to think about performing specific genetic studies aimed at detecting diseases of a neuro-degenerative nature.
Thus, DBN and DBNS therapy today can in some cases be causal, i.e., aimed at eliminating the triggering cause (as can be the case especially with toxic, metabolic, deficiency, autoimmune forms), but when this is not possible, it can also be symptomatic, i.e., aimed at reducing the effects that the presence of this sign can cause.
The aim of our work was to provide a comprehensive and as complete as possible picture of DBN and to give a possible explanation of the different functional aspects observable in the presence of DBN. Therefore, this paper may be a useful tool for the specialist who finds himself dealing with these patients in any evaluation context, but also for the clinician who finds himself dealing with such a sign and pathology even in a non-specialist context, such as in a first aid environment.
Author contributions
VM: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. BG: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. GV: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. MF: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. AF: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. VP: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2024.1394859/full#supplementary-material
SUPPLEMENTARY VIDEO S1 | AS, a 36-year-old female, presented for observation complaining of mild gait ataxia and imbalance aggravated by exertion, brief episodes of vertigo, difficulty observing moving objects, and intention tremor for approximately 6 months. Rare episodes of diplopia had also been present for several years. Neuro-otologic evaluation revealed a spontaneous downbeat nystagmus of low amplitude and frequency, not inhibited by fixation, accentuated by eccentric lateral gaze but especially by the prone position. The latter finding was particularly suggestive. The subsequent neurological examination revealed hyperreflexia and extensor plantar responses. Molecular genetic testing performed approximately 2 months later revealed spinocerebellar ataxia type 6. The patient's mother is also affected by SCA6.
SUPPLEMENTARY VIDEO S2 | AC, a 76-year-old female, presented for evaluation of positional vertigo, which had started about 15 days earlier and was associated with imbalance, but more importantly with uncontrollable vomiting. For these reasons, the patient had already undergone a cranial MRI, which was normal. At the time of the visit, the neuro-otological evaluation was negative, so she was advised to return in case of a new acute phase. Three days later, the patient returned for evaluation of very severe imbalance and tremor. Neuro-otologic evaluation revealed intense spontaneous downbeat nystagmus. A biochemical blood test revealed a decrease in serum magnesium (Mg) level (0.2 mg/dL; normal range 1.7–2.5), resulting in a deficiency-type cerebellar disorder. Mg supplementation (400 mg/day, orally) was initiated, with rapid improvement of the imbalance and tremor. Fifteen days later, the patient was asymptomatic, and the blood magnesium level was 2.05.
References
1. Wagner, JN, Glaser, M, Brandt, T, and Strupp, M. Downbeat nystagmus: aetiology and comorbidity in 117 patients. J Neurol Neurosurg Psychiatry. (2008) 79:672–7. doi: 10.1136/jnnp.2007.126284
3. Fuchs, E In: A Duane, editor. Fuchs’ text-book of ophthalmology. 3rd ed. Philadelphia: JB Lippincott (1908)
4. Wilbrand, H, and Saenger, A. Die Pathologie der Bahnen und Centren der Augenmuskeln In: JF Bergmann, editor. Die Neurologie des Auges, vol. VIII. München und Wiesbaden (1922).
5. Berens, C, and McAlpine, PT. Motor anomalies of the eye In: C Berens, editor. The eye and its diseases. 2nd ed. Philadelphia: WB Saunders (1950). 765–6.
7. Cogan, DG, and Barrows, LJ. Platybasia and the Arnold-Chiari malformation. AMA Arch Ophthalmol. (1954) 52:13–29. doi: 10.1001/archopht.1954.00920050015002
8. Cogan, DG. Down-beat nystagmus. Arch Ophthalmol. (1968) 80:757–68. doi: 10.1001/archopht.1968.00980050759015
9. Yee, RD. Eye movement recording as a clinical tool. Ophthalmology. (1983) 90:211–22. doi: 10.1016/s0161-6420(83)34571-1
10. Zee, DS, Friendlich, AR, and Robinson, DA. The mechanism of downbeat nystagmus. Arch Neurol. (1974) 30:227–37. doi: 10.1001/archneur.1974.00490330035006
11. Zee, DS, Yee, RD, Cogan, DG, Robinson, DA, and Engel, WK. Ocular motor abnormalities in hereditary cerebellar ataxia. Brain. (1976) 99:207–34. doi: 10.1093/brain/99.2.207
12. Yee, RD, Baloh, RW, Honrubia, V, Lau, CG, and Jenkins, HA. Slow build-up of optokinetic nystagmus associated with downbeat nystagmus. Invest Ophthalmol Vis Sci. (1979) 18:622–9
13. Baloh, RW, and Spooner, JW. Downbeat nystagmus: a type of central vestibular nystagmus. Neurology. (1981) 31:304–10. doi: 10.1212/wnl.31.3.304
14. Halmagyi, GM, Rudge, P, Gresty, MA, and Sanders, MD. Downbeating nystagmus. A review of 62 cases. Arch Neurol. (1983) 40:777–84. doi: 10.1001/archneur.40.13.777
15. Spiegel, EA, and Scala, NP. Vertical nystagmus following lesions of the cerebellar vermis. Arch Ophthalmol. (1941) 26:661–9. doi: 10.1001/archopht.1941.00870160137012
16. Allen, G, and Fernandez, C. Experimental observations in postural nystagmus. I. Extensive lesions in the posterior vermis of the cerebellum. Acta Otolaryngol. (1960) 51:2–14. doi: 10.3109/00016486009124460
17. Fernandez, C, Alzate, R, and Lindsay, JR. Experimental observations on postural nystagmus. II lesions of the nodulus. Ann Otol Rhinol Laryngol. (1960) 69:94–114. doi: 10.1177/000348946006900108
18. Fernandez, C, and Frederickson, JM. Experimental cerebellar lesions and their effect on vestibular function. Acta Otolaryngol Suppl. (1964) 58:52–62. doi: 10.3109/00016486409134628
19. Takemori, S, and Suzuki, M. Cerebellar contribution to oculomotor function. ORL J Otorhinolaryngol Relat Spec. (1977) 39:209–17. doi: 10.1159/000275359
20. Zee, DS, Yamazaki, A, Butler, PH, and Gücer, G. Effects of ablation of flocculus and paraflocculus on eye movements in primates. J Neurophysiol. (1981) 46:878–99. doi: 10.1152/jn.1981.46.4.878
21. Zee, DS, Leigh, RJ, and Mathieu-Millaire, F. Cerebellar control of ocular gaze stability. Ann Neurol. (1980) 7:37–40. doi: 10.1002/ana.410070108
22. Alexander, G. Die Ohrenkrankheiten im Kindesalter In: M Pfaundler and A Schlossmann, editors. Handbuch der Kinderheilkunde. Leibzig: Vogel (1912). 84–96.
23. Robinson, DA, Zee, DS, Hain, TC, Holmes, A, and Rosenberg, LF. Alexander's law: its behavior and origin in the human vestibulo-ocular reflex. Ann Neurol. (1984) 16:714–22. doi: 10.1002/ana.410160614
24. Fukuda, J, Highstein, SM, and Ito, M. Cerebellar inhibitory control of the vestibulo-ocular reflex investigated in rabbit 3rd nucleus. Exp Brain Res. (1972) 14:511–26. doi: 10.1007/BF00236593
25. Cohen, B, and Takemori, S. Visual inhibition of nystagmus by the flocculus. Trans Am Neurol Assoc. (1973) 98:52–6
26. Takemori, S, and Cohen, B. Loss of visual suppression of vestibular nystagmus after flocculus lesions. Brain Res. (1974) 72:213–24. doi: 10.1016/0006-8993(74)90860-9
27. Nozaki, S, Mukuno, K, and Ishikawa, S. Internuclear ophthalmoplegia associated with ipsilateral downbeat nystagmus and contralateral incyclorotatory nystagmus. Ophthalmologica. (1983) 187:210–6. doi: 10.1159/000309328
28. Barmack, NH, and Pettorossi, VE. The otolithic origin of the vertical vestibuloocular reflex following bilateral blockage of the vertical semicircular canals in the rabbit. J Neurosci. (1988) 8:2827–35. doi: 10.1523/JNEUROSCI.08-08-02827.1988
29. Goltz, HC, Irving, EL, Steinbach, MJ, and Eizenman, M. Vertical eye position control in darkness: orbital position and body orientation interact to modulate drift velocity. Vis Res. (1997) 37:789–98. doi: 10.1016/s0042-6989(96)00217-9
30. Bisdorff, AR, Sancovic, S, Debatisse, D, Bentley, C, Gresty, MA, and Bronstein, AM. Positional nystagmus in the dark in normal subjects. Neuro-Ophthalmology. (2000) 24:283–90. doi: 10.1076/0165-8107(200008)2411-VFT283
31. Angelaki, DE, and Hess, BJ. Inertial representation of angular motion in the vestibular system of rhesus monkeys. II. Otolith-controlled transformation that depends on an intact cerebellar nodulus. J Neurophysiol. (1995) 73:1729–51. doi: 10.1152/jn.1995.73.5.1729
32. Wearne, S, Raphan, T, and Cohen, B. Control of spatial orientation of the angular vestibuloocular reflex by the nodulus and uvula. J Neurophysiol. (1998) 79:2690–715. doi: 10.1152/jn.1998.79.5.2690
33. Wiest, G, Deecke, L, Trattnig, S, and Mueller, C. Abolished tilt suppression of the vestibulo-ocular reflex caused by a selective uvulo-nodular lesion. Neurology. (1999) 52:417–9. doi: 10.1212/wnl.52.2.417
34. Böhmer, A, and Straumann, D. Pathomechanism of mammalian downbeat nystagmus due to cerebellar lesion: a simple hypothesis. Neurosci Lett. (1998) 250:127–30. doi: 10.1016/s0304-3940(98)00450-9
35. Snyder, LH, and King, WM. Behavior and physiology of the macaque vestibulo-ocular reflex response to sudden off-axis rotation: computing eye translation. Brain Res Bull. (1996) 40:293–301. doi: 10.1016/0361-9230(96)00118-9
36. Precht, W, Volkind, R, Maeda, M, and Giretti, ML. The effects of stimulating the cerebellar nodulus in the cat on the responses of vestibular neurons. Neuroscience. (1976) 1:301–12. doi: 10.1016/0306-4522(76)90057-9
37. Helmchen, C, Sprenger, A, Rambold, H, Sander, T, Kömpf, D, and Straumann, D. Effect of 3,4-diaminopyridine on the gravity dependence of ocular drift in downbeat nystagmus. Neurology. (2004) 63:752–3. doi: 10.1212/01.wnl.0000136226.52639.35
38. Bertholon, P, Bronstein, AM, Davies, RA, Rudge, P, and Thilo, KV. Positional down beating nystagmus in 50 patients: cerebellar disorders and possible anterior semicircular canalithiasis. J Neurol Neurosurg Psychiatry. (2002) 72:366–72. doi: 10.1136/jnnp.72.3.366
39. Heidenreich, KD, Kerber, KA, Carender, WJ, Basura, GJ, and Telian, SA. Persistent positional nystagmus: a case of superior Semicircular Canal benign paroxysmal positional Vertigo? Laryngoscope. (2011) 121:1818–20. doi: 10.1002/lary.21848
40. Vannucchi, P, Pecci, R, and Giannoni, B. Posterior semicircular canal benign paroxysmal positional vertigo presenting with torsional downbeating nystagmus: an apogeotropic variant. Int J Otolaryngol. (2012) 2012:413603. doi: 10.1155/2012/413603
41. Vannucchi, P, Pecci, R, Giannoni, B, Di Giustino, F, Santimone, R, and Mengucci, A. Apogeotropic posterior Semicircular Canal benign paroxysmal positional Vertigo: some clinical and therapeutic considerations. Audiol Res. (2015) 5:130. doi: 10.4081/audiores.2015.130
42. Giannoni, B, Marcelli, V, Verdolin, I, Checcucci, C, Pollastri, F, and Pecci, R. Congruous torsional down beating nystagmus in the third position of the Semont's maneuver in patients treated for Canalithiasis of posterior Semicircular Canal benign paroxysmal positional Vertigo: its significance and prognostic value. Front Neurol. (2020) 11:949. doi: 10.3389/fneur.2020.00949
43. Darlot, C, López-Barneo, J, and Tracey, D. Asymmetries of vertical vestibular nystagmus in the cat. Exp Brain Res. (1981) 41-41:420–6. doi: 10.1007/BF00238901
44. Baloh, RW, Richman, L, Yee, RD, and Honrubia, V. The dynamics of vertical eye movements in normal human subjects. Aviat Space Environ Med. (1983) 54:32–8
45. Benson, AJ, and Guedry, FE Jr. Comparison of tracking-task performance and nystagmus during sinusoidal oscillation in yaw and pitch. Aerosp Med. (1971) 42:593–601
46. Barnes, GR, Benson, AJ, and Prior, AR. Visual-vestibular interaction in the control of eye movement. Aviat Space Environ Med. (1978) 49:557–64
47. Rude, SA, and Baker, JF. Otolith orientation and downbeat nystagmus in the normal cat. Exp Brain Res. (1996) 111:144–8. doi: 10.1007/BF00229564
48. Matsuo, V, and Cohen, B. Vertical optokinetic nystagmus and vestibular nystagmus in the monkey: up-down asymmetry and effects of gravity. Exp Brain Res. (1984) 53:197–216. doi: 10.1007/BF00238150
49. Takahashi, M, Sakurai, S, and Kanzaki, J. Horizontal and vertical optokinetic nystagmus in man. ORL J Otorhinolaryngol Relat Spec. (1978) 40:43–52. doi: 10.1159/000275385
50. Igarashi, M, Takahashi, M, Kubo, T, Levy, JK, and Homick, JL. Effect of macular ablation on vertical optokinetic nystagmus in the squirrel monkey. ORL J Otorhinolaryngol Relat Spec. (1979) 40:312–8. doi: 10.1159/000275422
51. Young, LR, Oman, CM, and Dichgans, JM. Influence of head orientation on visually induced pitch and roll sensation. Aviat Space Environ Med. (1975) 46:264–8
52. Miles, FA, Fuller, JH, Braitman, DJ, and Dow, BM. Long-term adaptive changes in primate vestibuloocular reflex. III. Electrophysiological observations in flocculus of normal monkeys. J Neurophysiol. (1980) 43:1437–76. doi: 10.1152/jn.1980.43.5.1437
53. Chubb, MC, and Fuchs, AF. Contribution of y group of vestibular nuclei and dentate nucleus of cerebellum to generation of vertical smooth eye movements. J Neurophysiol. (1982) 48:75–99. doi: 10.1152/jn.1982.48.1.75
54. Fukushima, K, Fukushima, J, Kaneko, CR, and Fuchs, AF. Vertical Purkinje cells of the monkey floccular lobe: simple-spike activity during pursuit and passive whole body rotation. J Neurophysiol. (1999) 82:787–803. doi: 10.1152/jn.1999.82.2.787
55. Pierrot-Deseilligny, C, and Milea, D. Vertical nystagmus: clinical facts and hypotheses. Brain. (2005) 128:1237–46. doi: 10.1093/brain/awh532
56. Ito, M, Nisimaru, N, and Yamamoto, M. Specific patterns of neuronal connections involved in the control of the rabbit’s vestibulo-ocular reflexes by the cerebellar flocculus. J Physiol. (1977) 265:833–54. doi: 10.1113/jphysiol.1977.sp011747
57. Leigh, RJ, and Zee, DS. The neurology of eye movements. 5th ed. New York, NY: Oxford University Press (2015).
58. Gresty, M, Barratt, H, Rudge, P, and Page, N. Analysis of downbeat nystagmus. Otolithic vs semicircular canal influences. Arch Neurol. (1986) 43:52–5. doi: 10.1001/archneur.1986.00520010048021
59. Walker, MF, and Zee, DS. Asymmetry of the pitch vestibulo-ocular reflex in patients with cerebellar disease. Ann N Y Acad Sci. (2005) 1039:349–58. doi: 10.1196/annals.1325.033
60. Stone, LS, and Lisberger, SG. Visual responses of Purkinje cells in the cerebellar flocculus during smooth-pursuit eye movements in monkeys. I Simple spikes J Neurophysiol. (1990) 63:1241–61. doi: 10.1152/jn.1990.63.5.1241
61. Gacek, RR. Location of brain stem neurons projecting to the oculomotor nucleus in the cat. Exp Neurol. (1977) 57:725–49. doi: 10.1016/0014-4886(77)90105-4
62. Nagao, S, Kitamura, T, Nakamura, N, Hiramatsu, T, and Yamada, J. Differences of the primate flocculus and ventral paraflocculus in the mossy and climbing fiber input organization. J Comp Neurol. (1997) 382:480–98. doi: 10.1002/(sici)1096-9861(19970616)382:4<480::aid-cne5>3.0.co;2-z
63. Blanks, RH, Precht, W, and Torigoe, Y. Afferent projections to the cerebellar flocculus in the pigmented rat demonstrated by retrograde transport of horseradish peroxidase. Exp Brain Res. (1983) 52:293–306. doi: 10.1007/BF00236639
64. Partsalis, AM, Zhang, Y, and Highstein, SM. Dorsal Y group in the squirrel monkey. II. Contribution of the cerebellar flocculus to neuronal responses in normal and adapted animals. J Neurophysiol. (1995) 73:632–50. doi: 10.1152/jn.1995.73.2.632
65. Langer, T, Fuchs, AF, Chubb, MC, Scudder, CA, and Lisberger, SG. Floccular efferents in the rhesus macaque as revealed by autoradiography and horseradish peroxidase. J Comp Neurol. (1985) 235:26–37. doi: 10.1002/cne.902350103
66. Barmack, NH, and Pettorossi, VE. Adaptive balance in posterior cerebellum. Front Neurol. (2021) 12:635259. doi: 10.3389/fneur.2021.635259
67. Wylie, DR, De Zeeuw, CI, Di Giorgi, PL, and Simpson, JI. Projections of individual Purkinje cells of identified zones in the ventral nodulus to the vestibular and cerebellar nuclei in the rabbit. J Comp Neurol. (1994) 349:448–63. doi: 10.1002/cne.903490309
68. Walker, MF, Tian, J, Shan, X, Tamargo, RJ, Ying, H, and Zee, DS. The cerebellar nodulus/uvula integrates otolith signals for the translational vestibulo-ocular reflex. PLoS One. (2010) 5:e13981. doi: 10.1371/journal.pone.0013981
69. Walker, MF, Tian, J, Shan, X, Ying, H, Tamargo, RJ, and Zee, DS. Enhancement of the bias component of downbeat nystagmus after lesions of the nodulus and uvula. Ann N Y Acad Sci. (2009) 1164:482–5. doi: 10.1111/j.1749-6632.2008.03717.x
70. Barmack, NH, Errico, P, Ferraresi, A, Fushiki, H, Pettorossi, VE, and Yakhnitsa, V. Cerebellar nodulectomy impairs spatial memory of vestibular and optokinetic stimulation in rabbits. J Neurophysiol. (2002) 87:962–75. doi: 10.1152/jn.00528.2001
71. Pettorossi, VE, Grassi, S, Errico, P, and Barmack, NH. Role of cerebellar nodulus and uvula on the vestibular quick phase spatial constancy. Acta Otolaryngol Suppl. (2001) 121:155–9. doi: 10.1080/000164801750388342
72. Kronenbuerger, M, Olivi, A, and Zee, DS. Pearls & oy-sters: positional vertigo and vertical nystagmus in medulloblastoma: a picture is worth a thousand words. Neurology. (2018) 90:e352–4. doi: 10.1212/WNL.0000000000004866
73. Chambers, BR, Ell, JJ, and Gresty, MA. A case of downbeat nystagmus influenced by otolith stimulation. Ann Neurol. (1983) 13:204–7. doi: 10.1002/ana.410130218
74. Lowenstein, O, and Saunders, RD. Otolith-controlled responses from the first-order neurons of the labyrinth of the bullfrog (Rana catesbeiana) to changes in linear acceleration. Proc R Soc Lond B Biol Sci. (1975) 191:475–505. doi: 10.1098/rspb.1975.0140
75. Kim, JI, Somers, JT, Stahl, JS, Bhidayasiri, R, and Leigh, RJ. Vertical nystagmus in normal subjects: effects of head position, nicotine and scopolamine. J Vestib Res. (2000) 10:291–300. doi: 10.3233/VES-2000-10606
76. Stahl, JS, Thumser, ZC, and Oommen, BS. The ataxic mouse as a model for studying downbeat nystagmus. J Vestib Res. (2012) 22:221–41. doi: 10.3233/VES-120463
77. Stahl, JS, James, RA, Oommen, BS, Hoebeek, FE, and De Zeeuw, CI. Eye movements of the murine P/Q calcium channel mutant tottering, and the impact of aging. J Neurophysiol. (2006) 95:1588–607. doi: 10.1152/jn.00318.2005
78. Glasauer, S, Stephan, T, Kalla, R, Marti, S, and Straumann, D. Up-down asymmetry of cerebellar activation during vertical pursuit eye movements. Cerebellum. (2009) 8:385–8. doi: 10.1007/s12311-009-0109-5
79. Dieterich, M, and Brandt, T. Functional brain imaging of peripheral and central vestibular disorders. Brain. (2008) 131:2538–52. doi: 10.1093/brain/awn042
80. Bense, S, Best, C, Buchholz, HG, Wiener, V, Schreckenberger, M, Bartenstein, P, et al. 18F-fluorodeoxyglucose hypometabolism in cerebellar tonsil and flocculus in downbeat nystagmus. Neuroreport. (2006) 17:599–603. doi: 10.1097/00001756-200604240-00009
81. Glasauer, S, Hoshi, M, Kempermann, U, Eggert, T, and Büttner, U. Three-dimensional eye position and slow phase velocity in humans with downbeat nystagmus. J Neurophysiol. (2003) 89:338–54. doi: 10.1152/jn.00297.2002
82. Halmagyi, GM, and Leigh, RJ. Upbeat about downbeat nystagmus. Neurology. (2004) 63:606–7. doi: 10.1212/01.wnl.0000138374.62591.f3
83. Walker, MF, Tian, J, Shan, X, Tamargo, RJ, Ying, H, and Zee, DS. Lesions of the cerebellar nodulus and uvula impair downward pursuit. J Neurophysiol. (2008) 100:1813–23. doi: 10.1152/jn.01193.2007
84. Shinmei, Y, Yamanobe, T, Fukushima, J, and Fukushima, K. Purkinje cells of the cerebellar dorsal vermis: simple-spike activity during pursuit and passive whole-body rotation. J Neurophysiol. (2002) 87:1836–49. doi: 10.1152/jn.00150.2001
85. Hüfner, K, Stephan, T, Kalla, R, Deutschländer, A, Wagner, J, Holtmannspötter, M, et al. Structural and functional MRIs disclose cerebellar pathologies in idiopathic downbeat nystagmus. Neurology. (2007) 69:1128–35. doi: 10.1212/01.wnl.0000276953.00969.48
86. Nakamagoe, K, Iwamoto, Y, and Yoshida, K. Evidence for brainstem structures participating in oculomotor integration. Science. (2000) 288:857–9. doi: 10.1126/science.288.5467.857
87. de Jong, JM, Cohen, B, Matsuo, V, and Uemura, T. Midsagittal pontomedullary brain stem section: effects on ocular adduction and nystagmus. Exp Neurol. (1980) 68:420–42. doi: 10.1016/0014-4886(80)90098-9
88. Nakamagoe, K, Shimizu, K, Koganezawa, T, and Tamaoka, A. Downbeat nystagmus due to a paramedian medullary lesion. J Clin Neurosci. (2012) 19:1597–9. doi: 10.1016/j.jocn.2012.03.017
89. Nakamagoe, K, Fujizuka, N, Koganezawa, T, Yamaguchi, T, and Tamaoka, A. Downbeat nystagmus associated with damage to the medial longitudinal fasciculus of the pons: a vestibular balance control mechanism via the lower brainstem paramedian tract neurons. J Neurol Sci. (2013) 328:98–101. doi: 10.1016/j.jns.2013.02.017
90. Sanchez, K, and Rowe, FJ. Role of neural integrators in oculomotor systems: a systematic narrative literature review. Acta Ophthalmol. (2018) 96:e111–8. doi: 10.1111/aos.13307
91. Mettens, P, Godaux, E, Cheron, G, and Galiana, HL. Effect of muscimol microinjections into the prepositus hypoglossi and the medial vestibular nuclei on cat eye movements. J Neurophysiol. (1994) 72:785–802. doi: 10.1152/jn.1994.72.2.785
92. Arnold, DB, Robinson, DA, and Leigh, RJ. Nystagmus induced by pharmacological inactivation of the brainstem ocular motor integrator in monkey. Vis Res. (1999) 39:4286–95. doi: 10.1016/s0042-6989(99)00142-x
93. Anagnostou, E, Papadimas, G, Rentzos, M, and Zambelis, T. Downbeat nystagmus and lower motor neuron disease: 14 years follow-up. J Neurol. (2018) 265:714–6. doi: 10.1007/s00415-018-8775-1
94. Delva, A, Thakore, N, Pioro, EP, Poesen, K, Saunders-Pullman, R, Meijer, IA, et al. Finger extension weakness and downbeat nystagmus motor neuron disease syndrome: a novel motor neuron disorder? Muscle Nerve. (2017) 56:1164–8. doi: 10.1002/mus.25669
95. Carl, JR, Yee, RD, and Baloh, RW. Convergence and gaze effects on vertical nystagmus. Invest Ophthalmol Vis Sci (ARVO Suppl). (1982) 22:265.
96. Crane, BT, and Demer, JL. Human horizontal Vestibulo-ocular reflex initiation: effects of acceleration, target distance, and unilateral deafferentation. J Neurophysiol. (1998) 80:1151–66. doi: 10.1152/jn.1998.80.3.1151
97. Patel, VR, and Zee, DS. The cerebellum in eye movement control: nystagmus, coordinate frames and disconjugacy. Eye. (2015) 29:191–5. doi: 10.1038/eye.2014.271
98. Kattah, JC, McClelland, C, and Zee, DS. Vertical nystagmus in Wernicke’s encephalopathy: pathogenesis and role of central processing of information from the otoliths. J Neurol. (2019) 266:139–45. doi: 10.1007/s00415-019-09326-9
99. Spiegel, R, Rettinger, N, Kalla, R, Lehnen, N, Straumann, D, Brandt, T, et al. The intensity of downbeat nystagmus during daytime. Ann N Y Acad Sci. (2009) 1164:293–9. doi: 10.1111/j.1749-6632.2009.03865.x
100. Marti, S, Palla, A, and Straumann, D. Gravity dependence of ocular drift in patients with cerebellar downbeat nystagmus. Ann Neurol. (2002) 52:712–21. doi: 10.1002/ana.10370
101. Eron, JN, Cohen, B, Raphan, T, and Yakushin, SB. Adaptation of orientation vectors of otolith-related central vestibular neurons to gravity. J Neurophysiol. (2008) 100:1686–90. doi: 10.1152/jn.90289.2008
102. Büttner-Ennever, JA, and Horn, AK. The neuroanatomical basis of oculomotor disorders: the dual motor control of extraocular muscles and its possible role in proprioception. Curr Opin Neurol. (2002) 15:35–43. doi: 10.1097/00019052-200202000-00007
103. Glasauer, S, Schneider, E, Jahn, K, Strupp, M, and Brandt, T. How the eyes move the body. Neurology. (2005) 65:1291–3. doi: 10.1212/01.wnl.0000175132.01370.fc
104. Klier, EM, Hess, BJ, and Angelaki, DE. Human visuospatial updating after passive translations in three-dimensional space. J Neurophysiol. (2008) 99:1799–809. doi: 10.1152/jn.01091.2007
105. Ferrara, M, and De Gennaro, L. The sleep inertia phenomenon during the sleep-wake transition: theoretical and operational issues. Aviat Space Environ Med. (2000) 71:843–8
106. Jewett, ME, Wyatt, JK, Ritz-De Cecco, A, Khalsa, SB, Dijk, DJ, and Czeisler, CA. Time course of sleep inertia dissipation in human performance and alertness. J Sleep Res. (1999) 8:1–8. doi: 10.1111/j.1365-2869.1999.00128.x
107. Tassi, P, and Muzet, A. Sleep inertia. Sleep Med Rev. (2000) 4:341–53. doi: 10.1053/smrv.2000.0098
108. Spiegel, R, Claassen, J, Teufel, J, Bardins, S, Schneider, E, Lehrer Rettinger, N, et al. Resting in darkness improves downbeat nystagmus: evidence from an observational study. Ann N Y Acad Sci. (2016) 1375:66–73. doi: 10.1111/nyas.13172
109. Büchele, W, Brandt, T, and Degner, D. Ataxia and oscillopsia in downbeat-nystagmus vertigo syndrome. Adv Otorhinolaryngol. (1983) 30:291–7. doi: 10.1159/000407661
110. Dieterich, M, and Brandt, TH. Impaired motion perception in congenital nystagmus and acquired ocular motor palsy. Clin Vis Sci. (1987) 1:337–45.
111. Wuehr, M, Schniepp, R, Pradhan, C, Ilmberger, J, Strupp, M, Brandt, T, et al. Differential effects of absent visual feedback control on gait variability during different locomotion speeds. Exp Brain Res. (2013) 224:287–94. doi: 10.1007/s00221-012-3310-6
112. Hallemans, A, Ortibus, E, Meire, F, and Aerts, P. Low vision affects dynamic stability of gait. Gait Posture. (2010) 32:547–51. doi: 10.1016/j.gaitpost.2010.07.018
114. Stolze, H, Klebe, S, Petersen, G, Raethjen, J, Wenzelburger, R, Witt, K, et al. Typical features of cerebellar ataxic gait. J Neurol Neurosurg Psychiatry. (2002) 73:310–2. doi: 10.1136/jnnp.73.3.310
115. Dietrich, H, Pradhan, C, Heidger, F, Schniepp, R, and Wuehr, M. Downbeat nystagmus becomes attenuated during walking compared to standing. J Neurol. (2022) 269:6222–7. doi: 10.1007/s00415-022-11106-x
116. Chagnaud, BP, Simmers, J, and Straka, H. Predictability of visual perturbation during locomotion: implications for corrective efference copy signaling. Biol Cybern. (2012) 106:669–79. doi: 10.1007/s00422-012-0528-0
117. MacNeilage, PR, and Glasauer, S. Quantification of head movement predictability and implications for suppression of vestibular input during locomotion. Front Comput Neurosci. (2017) 11:47. doi: 10.3389/fncom.2017.00047
118. Brandt, T, Strupp, M, and Benson, J. You are better off running than walking with acute vestibulopathy. Lancet. (1999) 354:746. doi: 10.1016/S0140-6736(99)03179-7
119. Jahn, K, Strupp, M, and Brandt, T. Both actual and imagined locomotion suppress spontaneous vestibular nystagmus. Neuroreport. (2002) 13:2125–8. doi: 10.1097/00001756-200211150-00027
120. Dietrich, H, Heidger, F, Schniepp, R, MacNeilage, PR, Glasauer, S, and Wuehr, M. Head motion predictability explains activity-dependent suppression of vestibular balance control. Sci Rep. (2020) 10:668. doi: 10.1038/s41598-019-57400-z
121. Dietrich, H, and Wuehr, M. Selective suppression of the vestibulo-ocular reflex during human locomotion. J Neurol. (2019) 266:101–7. doi: 10.1007/s00415-019-09352-7
122. Yee, RD. Downbeat nystagmus: characteristics and localization of lesions. Trans Am Ophthalmol Soc. (1989) 87:984–1032
123. Spiegel, R, Kalla, R, Rettinger, N, Schneider, E, Straumann, D, Marti, S, et al. Head position during resting modifies spontaneous daytime decrease of downbeat nystagmus. Neurology. (2010) 75:1928–32. doi: 10.1212/WNL.0b013e3181feb22f
124. Zingler, VC, Cnyrim, C, Jahn, K, Weintz, E, Fernbacher, J, Frenzel, C, et al. Causative factors and epidemiology of bilateral vestibulopathy in 255 patients. Ann Neurol. (2007) 61:524–32. doi: 10.1002/ana.21105
125. Mold, JW, Vesely, SK, Keyl, BA, Schenk, JB, and Roberts, M. The prevalence, predictors, and consequences of peripheral sensory neuropathy in older patients. J Am Board Fam Pract. (2004) 17:309–18. doi: 10.3122/jabfm.17.5.309
126. Park, H, Kim, HJ, Cha, MJ, Lee, JY, Koh, IS, and Nam, HS. A case of cerebellar infarction caused by acute subclavian thrombus following minor trauma. Yonsei Med J. (2013) 54:1538–41. doi: 10.3349/ymj.2013.54.6.1538
127. Huh, YE, and Kim, JS. Patterns of spontaneous and head-shaking nystagmus in cerebellar infarction: imaging correlations. Brain. (2011) 134:3662–71. doi: 10.1093/brain/awr269
128. Rousseaux, M, Dupard, T, Lesoin, F, Barbaste, P, and Hache, JC. Upbeat and downbeat nystagmus occurring successively in a patient with posterior medullary haemorrhage. J Neurol Neurosurg Psychiatry. (1991) 54:367–9. doi: 10.1136/jnnp.54.4.367
129. Wagner, J, Lehnen, N, Glasauer, S, Rettinger, N, Büttner, U, Brandt, T, et al. Downbeat nystagmus caused by a paramedian ponto-medullary lesion. J Neurol. (2009) 256:1572–4. doi: 10.1007/s00415-009-5153-z
130. Jacobson, DM, and Corbett, JJ. Downbeat nystagmus associated with dolichoectasia of the vertebrobasilar artery. Arch Neurol. (1989) 46:1005–8. doi: 10.1001/archneur.1989.00520450075023
131. Himi, T, Kataura, A, Tokuda, S, Sumi, Y, Kamiyama, K, and Shitamichi, M. Downbeat nystagmus with compression of the medulla oblongata by the dolichoectatic vertebral arteries. Am J Otol. (1995) 16:377–81
132. Krespi, Y, Verstichel, P, Masson, C, and Cambier, J. Nystagmus battant vers le bas et dolichoectasie artérielle vertébrobasilaire [downbeat nystagmus and vertebrobasilar arterial dolichoectasia]. Rev Neurol (Paris). (1995) 151:196–7
133. Rosengart, A, Hedges, TR 3rd, Teal, PA, DeWitt, LD, Wu, JK, Wolpert, S, et al. Intermittent downbeat nystagmus due to vertebral artery compression. Neurology. (1993) 43:216–8. doi: 10.1212/wnl.43.1_part_1.216
134. Malik, S, Furlan, AJ, Sweeney, PJ, Kosmorsky, GS, and Wong, M. Optic neuropathy: a rare paraneoplastic syndrome. J Clin Neuroophthalmol. (1992) 12:137–41
135. Graves, J, and Balcer, LJ. Eye disorders in patients with multiple sclerosis: natural history and management. Clin Ophthalmol. (2010) 4:1409–22. doi: 10.2147/OPTH.S6383
136. Masucci, EF, and Kurtzke, JF. Downbeat nystagmus secondary to multiple sclerosis. Ann Ophthalmol. (1988) 20:347–8
137. Anagnostou, E, Mandellos, D, Limbitaki, G, Papadimitriou, A, and Anastasopoulos, D. Positional nystagmus and vertigo due to a solitary brachium conjunctivum plaque. J Neurol Neurosurg Psychiatry. (2006) 77:790–2. doi: 10.1136/jnnp.2005.084624
138. Selim, M, and Drachman, DA. Ataxia associated with Hashimoto's disease: progressive non-familial adult onset cerebellar degeneration with autoimmune thyroiditis. J Neurol Neurosurg Psychiatry. (2001) 71:81–7. doi: 10.1136/jnnp.71.1.81
139. Hadjivassiliou, M, Boscolo, S, Davies-Jones, GA, Grünewald, RA, Not, T, Sanders, DS, et al. The humoral response in the pathogenesis of gluten ataxia. Neurology. (2002) 58:1221–6. doi: 10.1212/wnl.58.8.1221
140. Habek, M, Hojsak, I, Barun, B, and Brinar, VV. Downbeat nystagmus, ataxia and spastic tetraparesis due to coeliac disease. Neurol Sci. (2011) 32:911–4. doi: 10.1007/s10072-011-0506-7
141. Hadjivassiliou, M, Grünewald, R, Sharrack, B, Sanders, D, Lobo, A, Williamson, C, et al. Gluten ataxia in perspective: epidemiology, genetic susceptibility and clinical characteristics. Brain. (2003) 126:685–91. doi: 10.1093/brain/awg050
142. Hadjivassiliou, M, Martindale, J, Shanmugarajah, P, Grünewald, RA, Sarrigiannis, PG, Beauchamp, N, et al. Causes of progressive cerebellar ataxia: prospective evaluation of 1500 patients. J Neurol Neurosurg Psychiatry. (2017) 88:301–9. doi: 10.1136/jnnp-2016-314863
143. Antonini, G, Nemni, R, Giubilei, F, Gragnani, F, Ceschin, V, Morino, S, et al. Autoantibodies to glutamic acid decarboxylase in downbeat nystagmus. J Neurol Neurosurg Psychiatry. (2003) 74:998–9. doi: 10.1136/jnnp.74.7.998
144. Tsuyusaki, Y, Sakakibara, R, Kishi, M, Tateno, F, and Yoshida, T. Downbeat nystagmus as the initial manifestation of anti-NMDAR encephalitis. Neurol Sci. (2014) 35:125–6. doi: 10.1007/s10072-013-1511-9
145. Orwitz, JI, Galetta, SL, and Teener, JW. Bilateral trochlear nerve palsy and downbeat nystagmus in a patient with cephalic tetanus. Neurology. (1997) 49:894–5. doi: 10.1212/wnl.49.3.894
146. Hirst, LW, Clark, AW, Wolinsky, JS, Zee, DS, Kaizer, H, Miller, NR, et al. Downbeat nystagmus. A case report of herpetic brain stem encephalitis. J Clin Neuroophthalmol. (1983) 3:245–9
147. Shaikh, AG, Termsarasab, P, Riley, DE, and Katirji, B. The floccular syndrome in herpes simplex type 1 encephalitis. J Neurol Sci. (2013) 325:154–5. doi: 10.1016/j.jns.2012.11.002
148. Prasad, S, Brown, MJ, and Galetta, SL. Transient downbeat nystagmus from West Nile virus encephalomyelitis. Neurology. (2006) 66:1599–600. doi: 10.1212/01.wnl.0000216267.34612.ad
149. Castillo, LC, Gracia, F, Román, GC, Levine, P, Reeves, WC, and Kaplan, J. Spinocerebellar syndrome in patients infected with human T-lymphotropic virus types I and II (HTLV-I/HTLV-II): report of 3 cases from Panama. Acta Neurol Scand. (2000) 101:405–12. doi: 10.1034/j.1600-0404.2000.80180.x
150. Donaghy, C, Thurtell, MJ, Pioro, EP, Gibson, JM, and Leigh, RJ. Eye movements in amyotrophic lateral sclerosis and its mimics: a review with illustrative cases. J Neurol Neurosurg Psychiatry. (2011) 82:110–6. doi: 10.1136/jnnp.2010.212407
151. Thakore, NJ, Pioro, EP, Rucker, JC, and Leigh, RJ. Motor neuronopathy with dropped hands and downbeat nystagmus: a distinctive disorder? A case report. BMC Neurol. (2006) 6:3. doi: 10.1186/1471-2377-6-3
152. Dones, J, De Jesús, O, Colen, CB, Toledo, MM, and Delgado, M. Clinical outcomes in patients with Chiari I malformation: a review of 27 cases. Surg Neurol. (2003) 60:142–7. doi: 10.1016/s0090-3019(03)00131-9
153. Goodwin, D, and Halvorson, AR. Chiari I malformation presenting as downbeat nystagmus: clinical presentation, diagnosis, and management. Optometry. (2012) 83:80–6
154. Shaikh, AG, and Ghasia, FF. Neuro-ophthalmology of type 1 Chiari malformation. Expert Rev Ophthalmol. (2015) 10:351–7. doi: 10.1586/17469899.2015.1057505
155. Kumar, A, Patni, AH, and Charbel, F. The Chiari I malformation and the neurotologist. Otol Neurotol. (2002) 23:727–35. doi: 10.1097/00129492-200209000-00021
156. Wall, M. Brainstem syndromes In: W Bradley, R Daroff, GM Fenichel, and J Jankovic, editors. Neurology in clinical practice: principles of diagnosis and management. 4th ed. Philadelphia: Butterworth-Heinemann (Elsevier) (2004) p. 19991–5.
157. Nógues, M, López, L, and Meli, F. Neuro-ophthalmologic complications of syringobulbia. Curr Neurol Neurosci Rep. (2010) 10:459–66. doi: 10.1007/s11910-010-0139-x
158. Yeow, YK, and Tjia, TL. The localizing value of downbeat nystagmus. Singapore Med J. (1989) 30:273–6
159. Cai, CX, Siringo, FS, Odel, JG, Lignelli-Dipple, A, Lanzman, BA, Gindin, T, et al. Downbeat nystagmus secondary to familial hemophagocytic lymphohistiocytosis. J Neuroophthalmol. (2014) 34:57–60. doi: 10.1097/WNO.0000000000000064
160. Yasuoka, S, and Takakura, K. Proposal for the definition of "foramen magnum syndrome"--foramen magnum tumor and abnormalities. No To Shinkei. (1983) 35:1001–7
161. Bussière, M, Al-Khotani, A, Steckley, JL, Nicolle, M, and Nicolle, D. Paraneoplastic downbeat nystagmus. Can J Ophthalmol. (2008) 43:243–5. doi: 10.3129/i08-026
162. Peterson, K, Rosenblum, MK, Kotanides, H, and Posner, JB. Paraneoplastic cerebellar degeneration. I. A clinical analysis of 55 anti-Yo antibody-positive patients. Neurology. (1992) 42:1931–7. doi: 10.1212/wnl.42.10.1931
163. Hammack, J, Kotanides, H, Rosenblum, MK, and Posner, JB. Paraneoplastic cerebellar degeneration. II. Clinical and immunologic findings in 21 patients with Hodgkin's disease. Neurology. (1992) 42:1938–43. doi: 10.1212/wnl.42.10.1938
164. Ogawa, E, Sakakibara, R, Kawashima, K, Yoshida, T, Kishi, M, Tateno, F, et al. VGCC antibody-positive paraneoplastic cerebellar degeneration presenting with positioning vertigo. Neurol Sci. (2011) 32:1209–12. doi: 10.1007/s10072-011-0648-7
165. Rosenfeld, MR, and Dalmau, J. Central nervous system paraneoplastic disease. Handb Clin Neurol. (2012) 105:853–64. doi: 10.1016/B978-0-444-53502-3.00027-6
166. Dalmau, J, Graus, F, Villarejo, A, Posner, JB, Blumenthal, D, Thiessen, B, et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain. (2004) 127:1831–44. doi: 10.1093/brain/awh203
167. Yamamoto, T, and Tsuji, S. Anti-Ma2-associated encephalitis and paraneoplastic limbic encephalitis. Brain Nerve. (2010) 62:838–51
168. Saiz, A, Bruna, J, Stourac, P, Vigliani, MC, Giometto, B, Grisold, W, et al. Anti-Hu-associated brainstem encephalitis. J Neurol Neurosurg Psychiatry. (2009) 80:404–7. doi: 10.1136/jnnp.2008.158246
169. Zasorin, NL, and Baloh, RW. Downbeat nystagmus with alcoholic cerebellar degeneration. Arch Neurol. (1984) 41:1301–2. doi: 10.1001/archneur.1984.04050230087028
170. Fetter, M, Haslwanter, T, Bork, M, and Dichgans, J. New insights into positional alcohol nystagmus using three-dimensional eye-movement analysis. Ann Neurol. (1999) 45:216–23. doi: 10.1002/1531-8249(199902)45:2<216::aid-ana12>3.0.co;2-f
171. Ota, Y, Capizzano, AA, Moritani, T, Naganawa, S, Kurokawa, R, and Srinivasan, A. Comprehensive review of Wernicke encephalopathy: pathophysiology, clinical symptoms and imaging findings. Jpn J Radiol. (2020) 38:809–20. doi: 10.1007/s11604-020-00989-3
172. Ramulu, P, Moghekar, A, Chaudhry, V, Zee, DS, and Reich, SG. Wernicke's encephalopathy. Neurology. (2002) 59:846. doi: 10.1212/wnl.59.6.846
173. Rosenberg, ML. Reversible downbeat nystagmus secondary to excessive alcohol intake. J Clin Neuroophthalmol. (1987) 7:23–5
174. Zuccoli, G, Santa Cruz, D, Bertolini, M, Rovira, A, Gallucci, M, Carollo, C, et al. MR imaging findings in 56 patients with Wernicke encephalopathy: nonalcoholics may differ from alcoholics. AJNR Am J Neuroradiol. (2009) 30:171–6. doi: 10.3174/ajnr.A1280
175. Mayfrank, L, and Thoden, U. Downbeat nystagmus indicates cerebellar or brain-stem lesions in vitamin B12 deficiency. J Neurol. (1986) 233:145–8. doi: 10.1007/BF00314420
176. Akdal, G, Yener, GG, Ada, E, and Halmagyi, GM. Eye movement disorders in vitamin B12 deficiency: two new cases and a review of the literature. Eur J Neurol. (2007) 14:1170–2. doi: 10.1111/j.1468-1331.2007.01824.x
177. Saul, RF, and Selhorst, JB. Downbeat nystagmus with magnesium depletion. Arch Neurol. (1981) 38:650–2. doi: 10.1001/archneur.1981.00510100078014
178. Sedehizadeh, S, Keogh, M, and Wills, AJ. Reversible hypomagnesaemia-induced subacute cerebellar syndrome. Biol Trace Elem Res. (2011) 142:127–9. doi: 10.1007/s12011-010-8757-3
179. Du Pasquier, R, Vingerhoets, F, Safran, AB, and Landis, T. Periodic downbeat nystagmus. Neurology. (1998) 51:1478–80. doi: 10.1212/wnl.51.5.1478
180. Viola, P, Marcelli, V, Sculco, D, Pisani, D, Caglioti, A, Ricciardiello, F, et al. Vestibular disorders after kidney transplantation: focus on the pathophysiological mechanisms underlying the vertical nystagmus associated with tacrolimus-related hypomagnesemia. Int J Environ Res Public Health. (2022) 19:2260. doi: 10.3390/ijerph19042260
181. Berger, JR, and Kovacs, AG. Downbeat nystagmus with phenytoin. J Clin Neuroophthalmol. (1982) 2:209–11
182. Chrousos, GA, Cowdry, R, Schuelein, M, Abdul-Rahim, AS, Matsuo, V, and Currie, JN. Two cases of downbeat nystagmus and oscillopsia associated with carbamazepine. Am J Ophthalmol. (1987) 103:221–4. doi: 10.1016/s0002-9394(14)74231-x
183. Alkawi, A, Kattah, JC, and Wyman, K. Downbeat nystagmus as a result of lamotrigine toxicity. Epilepsy Res. (2005) 63:85–8. doi: 10.1016/j.eplepsyres.2004.11.004
184. Choi, JY, Park, YM, Woo, YS, Kim, SU, Jung, JM, and Kwon, DY. Perverted head-shaking and positional downbeat nystagmus in pregabalin intoxication. J Neurol Sci. (2014) 337:243–4. doi: 10.1016/j.jns.2013.12.007
185. Wu, D, and Thijs, RD. Anticonvulsant-induced downbeat nystagmus in epilepsy. Epilepsy Behav Case Rep. (2015) 4:74–5. doi: 10.1016/j.ebcr.2015.07.003
186. Arbusow, V, Strupp, M, and Brandt, T. Amiodarone-induced severe prolonged head-positional vertigo and vomiting. Neurology. (1998) 51:917. doi: 10.1212/wnl.51.3.917
187. Rottach, KG, Wohlgemuth, WA, Dzaja, AE, Eggert, T, and Straube, A. Effects of intravenous opioids on eye movements in humans: possible mechanisms. J Neurol. (2002) 249:1200–5. doi: 10.1007/s00415-002-0806-1
188. Henderson, RD, and Wijdicks, EF. Downbeat nystagmus associated with intravenous patient-controlled administration of morphine. Anesth Analg. (2000) 91:691–2. doi: 10.1213/00000539-200009000-00036
189. Kaut, O, and Kornblum, C. Transient downbeat nystagmus after intravenous administration of the opioid piritramide. J Neuroophthalmol. (2010) 30:164. doi: 10.1097/WNO.0b013e3181c25477
190. Paciuc-Beja, M, and Mendieta, G. Downbeat nystagmus induced by sedation in LASIK. Case Rep Ophthalmol Med. (2012) 2012:171679. doi: 10.1155/2012/171679
191. Korff, C, Peter, M, and Burkhard, PR. Downbeat nystagmus as a manifestation of intrathecal morphine toxicity. Eur J Anaesthesiol. (2007) 24:201–2. doi: 10.1017/S0265021506001724
192. Butragueño Laiseca, L, Toledo Del Castillo, B, Rodríguez Jimenez, C, Manrique-Rodríguez, S, and Pérez, MJ. Downbeat nystagmus due to ranitidine in a pediatric patient. Eur J Paediatr Neurol. (2017) 21:682–4. doi: 10.1016/j.ejpn.2017.01.015
193. Corbett, JJ, Jacobson, DM, Thompson, HS, Hart, MN, and Albert, DW. Downbeating nystagmus and other ocular motor defects caused by lithium toxicity. Neurology. (1989) 39:481–7. doi: 10.1212/wnl.39.4.481
194. Halmagyi, GM, Lessell, I, Curthoys, IS, Lessell, S, and Hoyt, WF. Lithium-induced downbeat nystagmus. Am J Ophthalmol. (1989) 107:664–70. doi: 10.1016/0002-9394(89)90265-1
195. Jørgensen, JS, Landschoff Lassen, L, and Wegener, M. Lithium-induced downbeat nystagmus and horizontal gaze palsy. Open Ophthalmol J. (2016) 10:126–8. doi: 10.2174/1874364101610010126
196. Schein, F, Manoli, P, and Cathébras, P. Lithium-induced downbeat nystagmus. Am J Ophthalmol Case Rep. (2017) 7:74–5. doi: 10.1016/j.ajoc.2017.06.012
197. Hong, H, and Lyu, IJ. A case of skew deviation and downbeat nystagmus induced by Lithium. BMC Ophthalmol. (2019) 19:257. doi: 10.1186/s12886-019-1270-3
198. Zhang, S, Lang, Y, Wang, W, Wu, Y, Yan, S, Zhang, T, et al. Analysis of etiology and clinical features of spontaneous downbeat nystagmus: a retrospective study. Front Neurol. (2024) 15:1326879. doi: 10.3389/fneur.2024.1326879
199. Phadke, JG, Hern, JE, and Blaiklock, CT. Downbeat nystagmus--a false localising sign due to communicating hydrocephalus. J Neurol Neurosurg Psychiatry. (1981) 44:459. doi: 10.1136/jnnp.44.5.459
200. Lempert, T, Neuhauser, H, and Daroff, RB. Vertigo as a symptom of migraine. Ann N Y Acad Sci. (2009) 1164:242–51. doi: 10.1111/j.1749-6632.2009.03852.x
201. Strupp, M, Versino, M, and Brandt, T. Vestibular migraine. Handb Clin Neurol. (2010) 97:755–71. doi: 10.1016/S0072-9752(10)97062-0
202. Weiner, HL, Freed, D, Woo, HH, Rezai, AR, Kim, R, and Epstein, FJ. Intra-axial tumors of the cervicomedullary junction: surgical results and long-term outcome. Pediatr Neurosurg. (1997) 27:12–8. doi: 10.1159/000121219
203. Nair, AP, Mehrotra, A, Das, KK, Srivastava, AK, Sahu, RN, and Kumar, R. Clinico-radiological profile and nuances in the management of cervicomedullary junction intramedullary tumors. Asian J Neurosurg. (2014) 9:21–8. doi: 10.4103/1793-5482.131060
204. Shih, RY, and Koeller, KK. Intramedullary masses of the spinal cord: radiologic-pathologic correlation. Radiographics. (2020) 40:1125–45. doi: 10.1148/rg.2020190196
205. Lv, J, Liu, B, Quan, X, Li, C, Dong, L, and Liu, M. Intramedullary spinal cord metastasis in malignancies: an institutional analysis and review. Onco Targets Ther. (2019) 12:4741–53. doi: 10.2147/OTT.S193235
206. Harada, H, Tamaoka, A, Watanabe, M, Ishikawa, K, and Shoji, S. Downbeat nystagmus in two siblings with spinocerebellar ataxia type 6 (SCA 6). J Neurol Sci. (1998) 160:161–3. doi: 10.1016/s0022-510x(98)00250-0
207. Jen, JC, Yue, Q, Karrim, J, Nelson, SF, and Baloh, RW. Spinocerebellar ataxia type 6 with positional vertigo and acetazolamide responsive episodic ataxia. J Neurol Neurosurg Psychiatry. (1998) 65:565–8. doi: 10.1136/jnnp.65.4.565
208. Yabe, I, Sasaki, H, Takeichi, N, Takei, A, Hamada, T, Fukushima, K, et al. Positional vertigo and macroscopic downbeat positioning nystagmus in spinocerebellar ataxia type 6 (SCA6). J Neurol. (2003) 250:440–3. doi: 10.1007/s00415-003-1020-5
209. Ueno, T, Nishizawa, H, Suzuki, C, Nunomura, JI, and Tomiyama, M. Downbeat nystagmus as an initial clinical sign in spinocerebellar ataxia type 6. Neurol Sci. (2017) 38:1543–5. doi: 10.1007/s10072-017-2973-y
210. Choi, KD, and Choi, JH. Episodic ataxias: clinical and genetic features. J Mov Disord. (2016) 9:129–35. doi: 10.14802/jmd.16028
211. Strupp, M, Zwergal, A, and Brandt, T. Episodic ataxia type 2. Neurotherapeutics. (2007) 4:267–73. doi: 10.1016/j.nurt.2007.01.014
212. Jen, J, Kim, GW, and Baloh, RW. Clinical spectrum of episodic ataxia type 2. Neurology. (2004) 62:17–22. doi: 10.1212/01.wnl.0000101675.61074.50
213. Strupp, M, Kalla, R, Claassen, J, Adrion, C, Mansmann, U, Klopstock, T, et al. A randomized trial of 4-aminopyridine in EA2 and related familial episodic ataxias. Neurology. (2011) 77:269–75. doi: 10.1212/WNL.0b013e318225ab07
214. Yabe, I, Kitagawa, M, Suzuki, Y, Fujiwara, K, Wada, T, Tsubuku, T, et al. Downbeat positioning nystagmus is a common clinical feature despite variable phenotypes in an FHM1 family. J Neurol. (2008) 255:1541–4. doi: 10.1007/s00415-008-0970-z
215. Suzuki, M, Fujiwara, K, Tsubuku, T, Yabe, I, Sasaki, H, and Fukuda, S. Time course of downbeat positioning nystagmus in familial hemiplegic migraine type 1 treated with acetazolamide. J Neurol Sci. (2016) 368:206–8. doi: 10.1016/j.jns.2016.07.020
216. Elliott, MA, Peroutka, SJ, Welch, S, and May, EF. Familial hemiplegic migraine, nystagmus, and cerebellar atrophy. Ann Neurol. (1996) 39:100–6. doi: 10.1002/ana.410390115
217. Baloh, RW. Episodic ataxias 1 and 2. Handb Clin Neurol. (2012) 103:595–602. doi: 10.1016/B978-0-444-51892-7.00042-5
218. Cha, YH, Lee, H, Jen, JC, Kattah, JC, Nelson, SF, and Baloh, RW. Episodic vertical oscillopsia with progressive gait ataxia: clinical description of a new episodic syndrome and evidence of linkage to chromosome 13q. J Neurol Neurosurg Psychiatry. (2007) 78:1273–5. doi: 10.1136/jnnp.2006.111138
219. Shaikh, AG, Marti, S, Tarnutzer, AA, Palla, A, Crawford, TO, Straumann, D, et al. Gaze fixation deficits and their implication in ataxia-telangiectasia. J Neurol Neurosurg Psychiatry. (2009) 80:858–64. doi: 10.1136/jnnp.2008.170522
220. Shinmei, Y, Kase, M, Suzuki, Y, Nitta, T, Chin, S, Yoshida, K, et al. Ocular motor disorders in mitochondrial encephalopathy with lactic acid and stroke-like episodes with the 3271 (T-C) point mutation in mitochondrial DNA. J Neuroophthalmol. (2007) 27:22–8. doi: 10.1097/WNO.0b013e3180334cb0
221. Tran, TM, Lee, MS, and McClelland, CM. Downbeat nystagmus: a clinical review of diagnosis and management. Curr Opin Ophthalmol. (2021) 32:504–14. doi: 10.1097/ICU.0000000000000802
222. Stahl, JS. Eye movements of the murine P/Q calcium channel mutant rocker, and the impact of aging. J Neurophysiol. (2004) 91:2066–78. doi: 10.1152/jn.01068.2003
223. Stahl, JS, and James, RA. Neural integrator function in murine CACNA1A mutants. Ann N Y Acad Sci. (2005) 1039:580–2. doi: 10.1196/annals.1325.072
224. Kalla, R, Glasauer, S, Schautzer, F, Lehnen, N, Büttner, U, Strupp, M, et al. 4-aminopyridine improves downbeat nystagmus, smooth pursuit, and VOR gain. Neurology. (2004) 62:1228–9. doi: 10.1212/01.wnl.0000118287.68294.e5
225. Strupp, M, Maul, S, Konte, B, Hartmann, AM, Giegling, I, Wollenteit, S, et al. A variation in FGF14 is associated with downbeat nystagmus in a genome-wide association study. Cerebellum. (2020) 19:348. doi: 10.1007/s12311-020-01113-x
226. Pablo, J, and Pitt, GS. FGF14 is a regulator of KCNQ2/3 channels. Proc Natl Acad Sci USA. (2016) 114:154–9. doi: 10.1073/pnas.1610158114
227. Shaikh, AG, and Manto, M. Genome-wide association study points new direction for downbeat nystagmus research. Cerebellum. (2020) 19:345–7. doi: 10.1007/s12311-020-01128-4
228. Shakkottai, VG, Xiao, M, Xu, L, Wong, M, Nerbonne, JM, Ornitz, DM, et al. FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol Dis. (2009) 33:81–8. doi: 10.1016/j.nbd.2008.09.019
229. Laezza, F, Lampert, A, Kozel, MA, Gerber, BR, Rush, AM, Nerbonne, JM, et al. FGF14 N-terminal splice variants differentially modulate Nav1.2 and Nav1.6-encoded sodium channels. Mol Cell Neurosci. (2009) 42:90–101. doi: 10.1016/j.mcn.2009.05.007
230. Pellerin, D, Danzi, MC, Wilke, C, Renaud, M, Fazal, S, Dicaire, MJ, et al. Deep Intronic FGF14 GAA repeat expansion in late-onset cerebellar Ataxia. N Engl J Med. (2023) 388:128–41. doi: 10.1056/NEJMoa2207406
231. Rafehi, H, Read, J, Szmulewicz, DJ, Davies, KC, Snell, P, Fearnley, LG, et al. An intronic GAA repeat expansion in FGF14 causes the autosomal-dominant adult-onset ataxia SCA27B/ATX-FGF14. Am J Hum Genet. (2023) 110:1018. doi: 10.1016/j.ajhg.2023.05.005
232. Wilke, C, Pellerin, D, Mengel, D, Traschütz, A, Danzi, MC, Dicaire, MJ, et al. GAA-FGF14 ataxia (SCA27B): phenotypic profile, natural history progression and 4-aminopyridine treatment response. Brain. (2023) 146:4144–57. doi: 10.1093/brain/awad157
233. Pellerin, D, Wilke, C, Traschütz, A, Nagy, S, Currò, R, Dicaire, MJ, et al. Intronic FGF14 GAA repeat expansions are a common cause of ataxia syndromes with neuropathy and bilateral vestibulopathy. J Neurol Neurosurg Psychiatry. (2024) 95:175–9. doi: 10.1136/jnnp-2023-331490
234. Hengel, H, Pellerin, D, Wilke, C, Fleszar, Z, Brais, B, Haack, T, et al. As frequent as Polyglutamine spinocerebellar ataxias: SCA27B in a large German autosomal dominant Ataxia cohort. Mov Disord. (2023) 38:1557–8. doi: 10.1002/mds.29559
235. Pellerin, D, Heindl, F, Wilke, C, Danzi, MC, Traschütz, A, Ashton, C, et al. Intronic FGF14 GAA repeat expansions are a common cause of downbeat nystagmus syndromes: frequency, phenotypic profile, and 4-aminopyridine treatment response. EBioMedicine. (2024) 102:105076. doi: 10.1101/2023.07.30.23293380
Keywords: downbeat nystagmus, downbeat nystagmus syndrome, vertical reflex asymmetry, neural integrator, central neurological disorders
Citation: Marcelli V, Giannoni B, Volpe G, Faralli M, Fetoni AR and Pettorossi VE (2024) Downbeat nystagmus: a clinical and pathophysiological review. Front. Neurol. 15:1394859. doi: 10.3389/fneur.2024.1394859
Edited by:
Sergio Carmona, INEBA Institute of Neurosciences Buenos Aires, ArgentinaReviewed by:
Alessandra Rufa, University of Siena, ItalySeung-Han Lee, Chonnam National University, Republic of Korea
Copyright © 2024 Marcelli, Giannoni, Volpe, Faralli, Fetoni and Pettorossi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincenzo Marcelli, dmltYXJAb25lb25saW5lLml0