 Vanessa I. Romero
Vanessa I. Romero Samantha Sáenz1
Samantha Sáenz1 Kazuyoshi Hosomichi
Kazuyoshi Hosomichi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol. , 15 April 2024
Sec. Neurogenetics
Volume 15 - 2024 | https://doi.org/10.3389/fneur.2024.1376643
This article is part of the Research Topic Consanguinity and Rare Genetic Neurological Diseases View all 9 articles
Epilepsy, characterized by recurrent seizures, impacts 70–80% of patients, leading to cognitive deficits. The intricate relationship between seizure control and cognitive impairment remains complex. Epileptic encephalopathy (EE), an intensified form often rooted in genetic factors, is detectable through next-generation sequencing, aiding in precise diagnoses, family counseling, and potential treatment strategies. We present a case involving two sisters with refractory generalized seizures evolving into dysarthria, dysphagia, ataxia, and cognitive decline. Despite normal physical exams, abnormal electroencephalogram results consistent with epilepsy were noted. Whole Exome Sequencing identified heterozygous variants in the alanyl-tRNA synthetase (AARS) and Calcium Voltage-Gated Channel Subunit Alpha 1 (CACNA1A) genes. The AARS variant (c.C2083T, p.R695*) was maternal, while the CACNA1A variant (c.G7400C, p.R2467P) was paternal. Patients A and B exhibited a unique blend of neurological and psychiatric conditions, distinct from common disorders that begin adolescence, like Juvenile Myoclonic Epilepsy. Whole Exome Sequencing uncovered an AARS gene and CACNA1A gene, linked to various autosomal dominant phenotypes. Presence in both parents, coupled with familial reports of migraines and seizures, provides insight into accelerated symptom progression. This study underscores the importance of genetic testing in decoding complex phenotypes and emphasizes the value of documenting family history for anticipating related symptoms and future health risks.
Epilepsy, characterized by recurrent seizures, is a prevalent neurological disorder that induces irreversible brain damage through frequent and recurrent seizures, leading to cognitive changes (1, 2). Cognitive impairment, observed in 70–80% of epilepsy patients, manifests as memory loss, cognitive slowing, and attention deficits, significantly impacting daily functions (3). Multiple factors contribute to cognitive dysfunction, including organic brain lesions, uncontrolled seizures, drug treatment, and individual mental abilities (2, 4). Conflicting findings exist regarding the relationship between seizure control and cognitive impairment, emphasizing the complex interplay between epilepsy and cognitive function across diverse populations and contexts (1, 5).
Epileptic encephalopathy (EE) represents a severe form of epilepsy in which epileptic activity exacerbates cognitive and behavioral impairments beyond the anticipated effects of the underlying pathology alone (6). Manifesting across various age groups, EE is commonly associated with genetic etiologies, often identified through next-generation sequencing (NGS) techniques (7). One significant genetic aspect involves disruptions in voltage-gated channels, particularly voltage-gated sodium channels (VGSCs) such as SCN1A, SCN2A, and SCN8A, implicated in epilepsies of varying severity (8–10). Potassium channelopathies, including KCNQ2 and KCNQ3 mutations, and disruptions in inwardly rectifying potassium (Kir) channels (e.g., KCNB1) also contribute to EE, affecting the frequency and duration of action potentials (11, 12). Calcium channelopathies, particularly mutations in CACNA1A, have been linked to various neurological phenotypes, including epilepsy, hemiplegic migraine, and episodic ataxia (13–17). Understanding the intricate relationship between genetic factors and channelopathies in epileptic encephalopathy is crucial for precise diagnosis, management, and genetic counseling.
Our patients presented with unexplained neurological signs and symptoms, for which we performed next-generation sequencing. Genetic testing, such as Whole Exome Sequencing (WES), is increasingly utilized in different specialties, including neurological and neurogenic conditions, particularly in phenotypes with broad variability and overlap of potential genetic causes (18). WES facilitates accurate diagnosis for these patients, enabling family counseling, recurrence prevention, and treatment, when available (19). Additionally, next-generation sequencing allows the association of novel phenotypes with previously described genes, as observed in our patients (18).
Patient A, a 20-year-old Hispanic female, was previously in good health. At the age of 15, she experienced the onset of generalized myoclonic-tonic–clonic seizures (HP: 0032795) that proved refractory to medical treatment. One year after the onset of epilepsy, she began to manifest symptoms of dysarthria (HP: 0001260), dysphagia (HP: 0002015), and ataxia (HP: 0001251). By the age of 18, she exhibited a progressive cognitive decline (HP: 0001268), losing the ability to speak, walk, and eat. During the physical examination, no dysmorphic features were observed. Her language skills were characterized by mutism (HP: 0002300). She demonstrated alexithymia (HP: 0031433), with delayed spoken language comprehension (HP: 5200240). Muscular strength was assessed at 4/5 in all four extremities using the Medical Research Council (MRC) Scale, accompanied by areflexia (HP: 0001284), erratic myoclonus (HP: 0025357), and gait ataxia (HP: 0002066). The electroencephalogram (Figure 1) revealed generalized slowness in the background activity, along with abundant multifocal epileptic activity and generalized activity. Cerebral magnetic resonance image did not show structural lesion. At the time of our visit, the patient was highly dependent and required assistance with her daily living activities like bathing, dressing, toileting, transferring, continence and feeding (KATZ ADL of 0/6).

Figure 1. (A) Electroencephalogram of patient A with a bipolar montage showed generalized slowness in the background activity, along with abundant multifocal epileptic activity and generalized activity. (B) Electroencephalogram of patient B. Electroencephalogram showing reported generalized slowness of background activity with abundant multifocal epileptic activity, predominantly affecting the posterior regions, consistent with a non-convulsive status epilepticus. (C) Electroencephalogram in normal conditions showed generalized slowness in the background activity, along with abundant multifocal epileptic activity and generalized activity.
Patient B, the sister of Patient A, is a 17-year-old Hispanic female who was also previously in good health. She presented to the hospital with a one-year history of generalized myoclonic-tonic–clonic seizures primarily occurring during sleep, persisting daily despite pharmacological treatment. Alongside epilepsy, she exhibited psychiatric symptoms characterized by delusional perception (HP: 5200424), auditory hallucination (HP: 0008765), emotional lability (HP: 0000712), and insomnia (HP: 0100785), coupled with progressive cognitive decline that led to her abrupt discontinuation of school attendance. During the physical examination, no dysmorphic features were observed, but she displayed confusion (HP: 0001289), bradyphrenia (HP: 0031843), and delayed spoken language comprehension (HP: 5200240). Also, gait ataxia (HP: 0002066) was observed. Magnetic resonance imaging of the brain revealed no abnormalities. The electroencephalogram (Figure 1) reported generalized slowness of background activity with abundant multifocal epileptic activity, predominantly affecting the posterior regions, consistent with a non-convulsive status epilepticus (Supplementary Figure 1). At the time of our visit, the patient was dependent and required assistance with her daily living activities like bathing, dressing, transferring, continence and feeding (KATZ ADL of 1/6).
Chromosome microarray analysis for Patient B confirmed the absence of copy number changes or copy-neutral regions. The patient’s father, who reported a history of headaches and memory impairment (HP: 0002354) during adolescence, has since resolved those symptoms and has not experienced any seizures. The patient’s mother is in overall good health. Additionally, two female and two male cousins from the patient’s father’s side have presented with seizures but without any other accompanying neurological symptoms (Figure 2). We conducted a Whole Exome Sequencing (WES) analysis on the peripheral blood samples obtained from both the sisters and their parents to ascertain the genetic condition. The parents were not consanguineous but were born and live in the same rural area.

Figure 2. Family pedigree. The father had migraine and memory impairment during adolescence. Relatives from the father side have seizures.
We used a capture of target regions using probes, followed by next-generation sequencing with Illumina technology. The total number of reads was 41,660,733, whereas that of the mapped reads was 41,603,016 (99.86%). We aligned the raw data using the Burrows–Wheeler Aligner software, sorted and merged the data using the Picard tools software, and identified the nucleotide variants (SNV) and insertions or deletions (indel) using GATK. The GRCh37 version of the human genome was taken as reference.
After performing WES, we identified 1,529 variants causing non-synonymous, stopgain, and frameshift changes; selected those with minor allele frequency (<0.01) in various databases; and detected two variants. The first one a nonsense heterozygote C > T (c.C2083T) variant of uncertain significance (db SNP rs761043713) in 15th exon of alanyl-tRNA synthetase (AARS), resulting in a arginine to a premature stop codon change at position 695 (p.R695*). The nonsense variant has a frequency of 0.0000323 in The Genome Aggregation Database (gnomAD version V2.1.1). The variant was heterozygous in the sisters and the mother, not in the father. The second variant is a missense heterozygote G > C (c.G7400C) variant of uncertain significance in 47 exon of Calcium Voltage-Gated Channel Subunit Alpha 1 (CACNA1A) gene, resulting in a change from arginine to proline (p.R2467P). The nonsense variant has a frequency of 0.00002368 in The Genome Aggregation Database (gnomAD version V2.1.1). The variant was heterozygous in the sisters and the father, not in the mother. Algorithm developed to predict the effect of missense changes on protein structure and function result in the following: PolyPhen-2: “Damaging” (0.981). The amino acid residue arginine is found in multiple mammalian species, suggesting that this missense negatively affects the function of the protein (Supplementary Figure 2).
Modeling the CACNA1A mutant isoform using Swiss Model and AlphaFold revealed an unchanged globular protein architecture with no discernible modifications in secondary motifs like beta turns and alpha helices (20, 21). Analysis showed the R2467P mutation alters the final loop, forming an intrinsically disordered region (IDR). This mutation changes residue 2,467 from a polar amino acid (arginine) to a non-polar proline with an amide lateral chain, impacting electron interaction, as seen in Figures 3A–E. Proline’s association with alpha helix formation suggests its role in maintaining IDR folding (22–24). Disorder in this region plays a crucial role in communication, scaffolding, and self-activation/inhibition (25–28). Disruption in CACNA1A’s communication within the extensive calcium equilibrium network may lead to neural electrophysiological perturbations, contributing to conductivity-related pathologies like seizures and epilepsy (29).
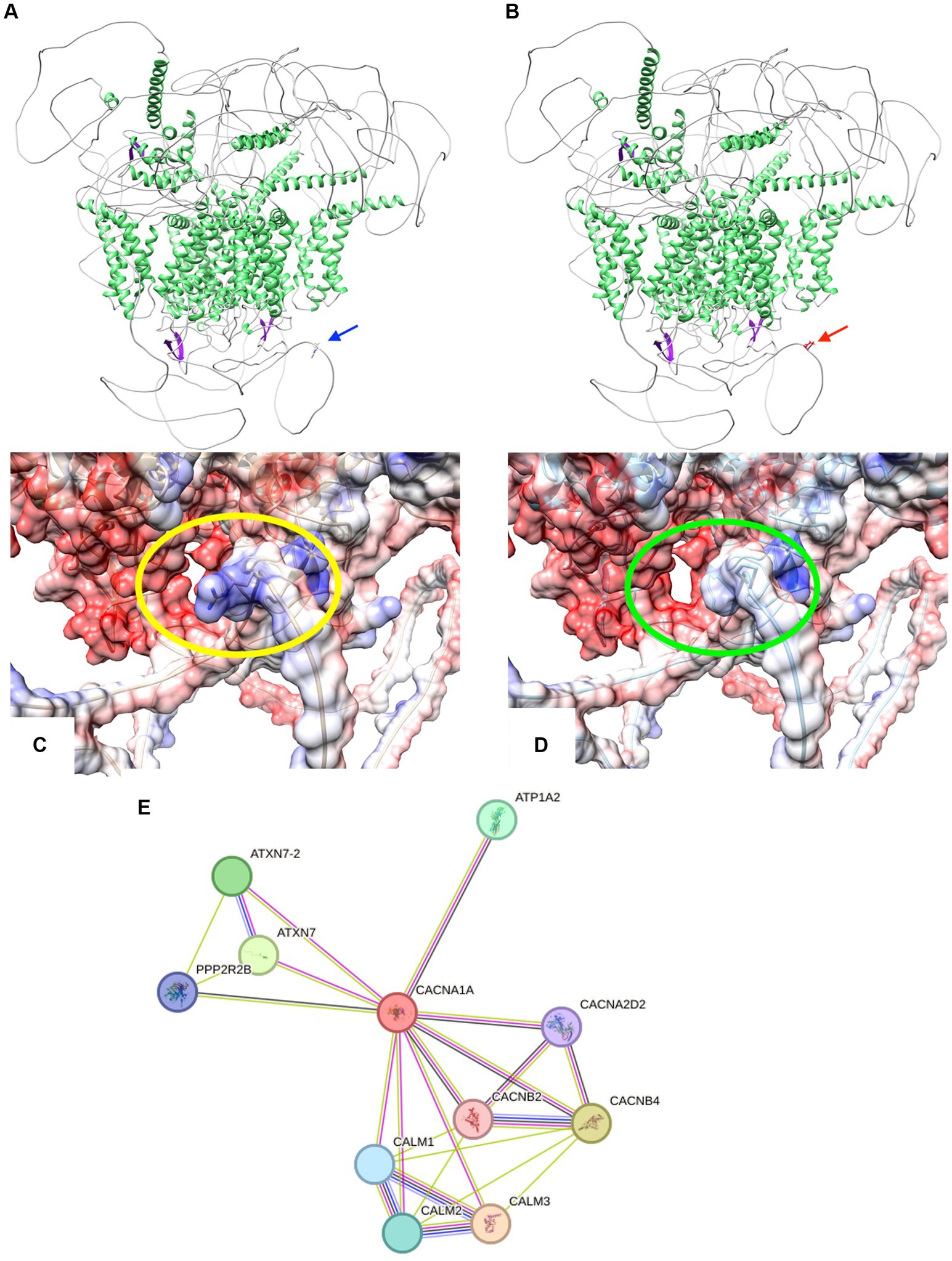
Figure 3. (A,B) Cartoon representations illustrate the protein structures of both CACNA1A wild-type (wt) and the R2467P mutant. The structures are color-coded by secondary elements: white represents loops, green indicates helices, and purple denotes sheets. On the left (blue arrow) is the wt structure, featuring the polar residue arginine. On the right (red arrow) is the R2467P mutant, characterized by the non-polar residue proline. (C,D) Surface representations depict the protein structures of both CACNA1A wild-type (wt) and the R2467P mutant. The structures are color-coded based on electrostatic values: red represents negative values (−10 min), white corresponds to 0, and blue signifies positive values (10 max). On the left (within the yellow oval) is the wt structure, featuring the polar residue arginine. On the right (within the green oval) is the R2467P mutant, characterized by the non-polar residue proline. Note the discernible modification in the electrostatic surface of this Intrinsically Disordered Region (IDR) caused by the mutation. (E) STRING network analysis of CACNA1A reveals a comprehensive interaction network involving proteins associated with its interaction. The connections are highlighted by co-expression (black), text mining (green), protein homology (purple), and experimentally determined interactions (pink).
Based on the criteria outlined in the joint consensus of the ACMG (American College of Medical Genetics) and AMP (American Association of Molecular Pathology) for classifying pathogenic variants, we propose reclassifying this variant as a pathogenic Variant. It aligns with characteristics demonstrating supporting pathogenicity (PS1) as CACNA1A is associated with autosomal dominant Developmental and epileptic encephalopathy (OMIM: 617106), Episodic ataxia (OMIM: 108500), Migraine, familial hemiplegic with progressive cerebellar ataxia (OMIM: 141500), Spinocerebellar ataxia (OMIM: 183086) and Lennox–Gastaut syndrome (OMIM: 615369); (PS2) as the mother has one of the variants and the father has the other variant; its low frequency in control databases (PS4), and moderate evidence of pathogenicity (PM4) based on the conservation of the nucleotide.
Common neurological disorders of this age like generalized idiopathic seizures including juvenile myoclonic epilepsy and tonic–clonic seizures on awakening lack of cognitive and psychiatric alterations.
Both previous healthy adolescent patients presented with a progressive neurological disorder characterized by refractory epilepsy, cognitive decline, and ataxia, raising concerns beyond the common neurological disorder of this age, like generalized idiopathic seizures including juvenile myoclonic epilepsy and tonic–clonic seizures on awakening lack of cognitive and psychiatric alterations. Consequently, our investigation delved into mitochondrial encephalopathy, autoimmune encephalitis, and POLG-related disorders, with detailed characteristics outlined in Table 1. Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes syndrome (MELAS), a rare disorder commencing in childhood, primarily affecting the nervous system and muscles, manifest seizures as the predominant early symptoms, accompanied by stroke-like episodes featuring hemiparesis, altered consciousness, vision and hearing loss, loss of motor skills, and intellectual disability (30). Myoclonus Epilepsy associated with Ragged-red fibers (MERRF syndrome) is marked by myoclonic seizures, myopathy, ataxia, and dementia (30). Autoimmune encephalitis encompasses conditions where the immune system erroneously attacks healthy brain cells, causing brain inflammation with neurologic and/or psychiatric symptoms characterizing this condition (31). Finally, POLG-related disorders encompass various syndromes linked to a mutation in the nuclear POLG gene, representing the most prevalent single-gene cause of mitochondrial disease (32). Descriptions of six conditions falling under POLG-related disorders range from severe encephalopathy to ophthalmologic symptoms (32). This comprehensive exploration provides a foundation for understanding the unique neurologic and psychiatric features observed in our adolescent patients.
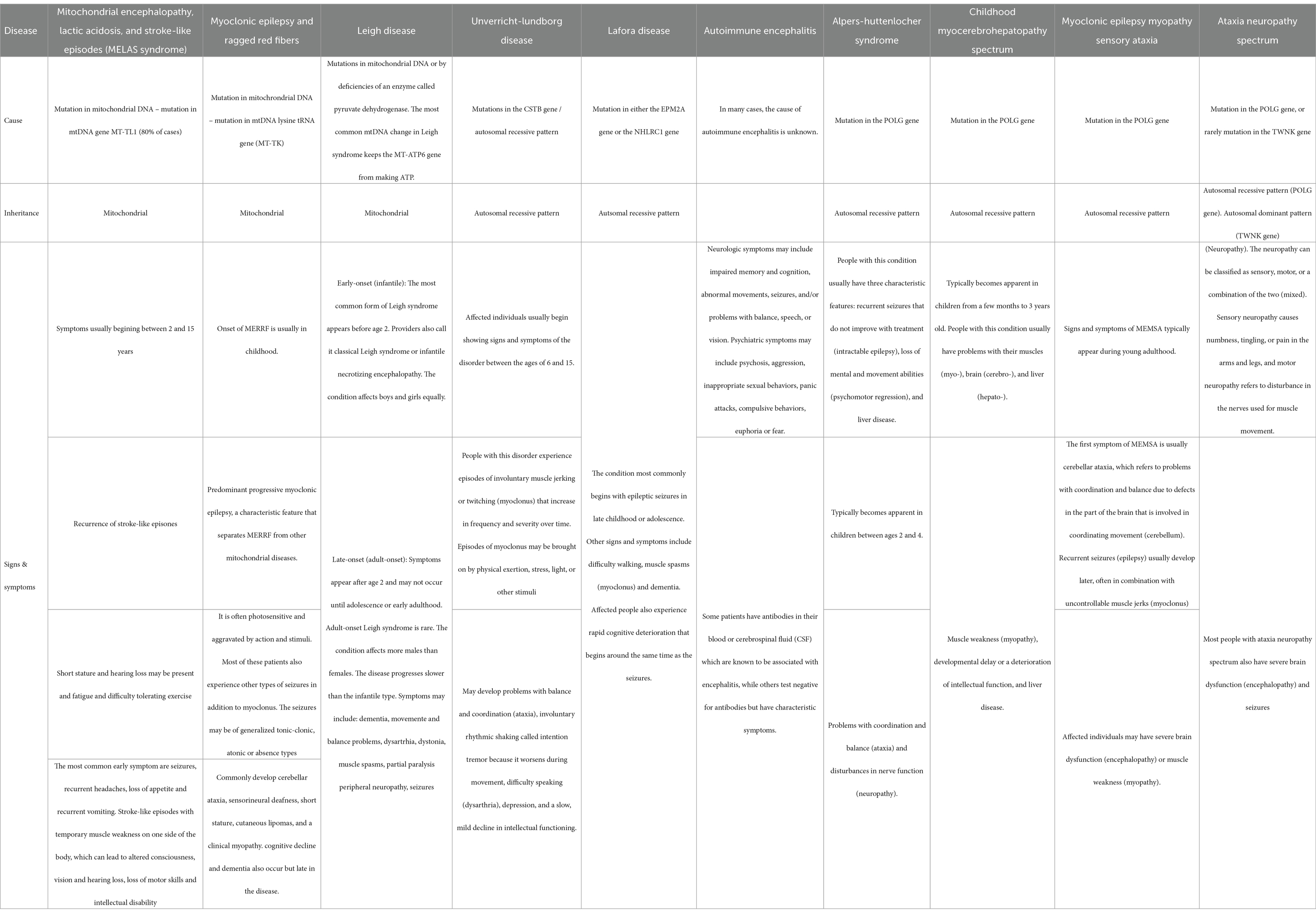
Table 1. Differential diagnosis comparison.
Our WES analysis revealed a mutation in the AARS gene, encoding alanyl-tRNA synthetase critical for attaching alanine to its cognate tRNA in protein translation’s initial step (33). As a member of the highly conserved class II tRNA synthases family, alanyl-tRNA synthetase interprets the RNA code, attaching specific amino acids to tRNAs with cognate trinucleotide anticodons (34). Enzymes comprise a catalytic domain interacting with the tRNA’s amino acid acceptor-Tpsi C helix and a second domain interacting with the rest of the tRNA structure (34). Disease-causing mutations often affect the aminoacylation and editing domains (33). Four phenotypes linked to AARS gene mutations include Hereditary Diffuse Leukoencephalopathy with Spheroids (autosomal dominant), characterized by neurodegeneration and psychiatric symptoms (33–36); axonal Charcot–Marie–Tooth disease type 2 (autosomal dominant), a common inherited neuropathy (37–40); Developmental and Epileptic Encephalopathy, an autosomal recessive neurologic disorder (41, 42); and Nonphotosensitive Trichothiodystrophy, an autosomal recessive disorder with characteristic features (43, 44). Despite sharing the gene mutation, our patients do not fit these phenotypes. These healthy females exhibited typical motor and neurological development until age 15, displaying progressive symptoms from seizures to motor and cognitive alterations. Patient B presented additional psychiatric symptoms. In the ClinVar database, we identified 27 nonsense variants of the AARS gene: 16 classified as pathogenic, three as likely pathogenic, nine as variants of uncertain significance, and one with conflicting classifications. These nonsense variants have been associated with conditions such as Charcot–Marie–Tooth disease type 2, Developmental and epileptic encephalopathy type 29, axonal type 2N Charcot–Marie–Tooth disease, and various inborn genetic diseases. The variant most like ours was categorized as pathogenic for Charcot–Marie–Tooth disease type 2, resulting in a p.Y690* mutation (NC_000016.10: 70258139: A > C).
Our patients harbor a second mutation in the CACNA1A gene, responsible for encoding the transmembrane pore-forming subunit of the P/Q-type or CaV2.1 voltage-gated calcium channel (45). These voltage-dependent Ca (2+) channels play a crucial role in facilitating Ca (2+) ion entry into excitable cells and participate in various Ca (2+)-dependent processes, including muscle contraction, hormone or neurotransmitter release, and gene expression (46). Mutations in this gene were associated with cerebellar ataxia, hemiplegic migraine and epileptic encephalopathy, demonstrating a broad phenotypic spectrum with autosomal dominant inheritance (13–17). The pathogenic variant in CACNA1A (c.C835T, p.R279C) has been documented to result in diverse manifestations among carriers, ranging from severe epileptic encephalopathy to episodic ataxia and cerebellar ataxia with intellectual deficiency, highlighting significant incomplete penetrance (14). Functional studies conducted in mouse thalamic relay neurons shed light on the impact of different CACNA1A variants, underscoring the pivotal role of calcium channels in synaptic dysfunction and clinical manifestations (47).
Both of our patients present with a mutation in the AARS gene, which is also presented in their mom, but they are the first of their mother’s family side with symptoms suggesting incomplete penetrance and an autosomal dominant inheritance. Additionally, both patients present with a mutation in the CACNA1A gene, which is also present in their father. The latter presented with migraines during his teenage years, and four other family members of their father’s family side have reported seizures. Comprehensive whole exome sequencing was performed, thoroughly examining all genes associated with the clinical symptoms and additional genes as specified previously. This led us to identify AARS and CACNA1A as the only genes likely to be implicated in the etiology of the condition. We speculate that having these two mutations could explain the fast progression of symptoms and deterioration.
Two crucial lessons emerge from these cases. Firstly, the significance and efficacy of genetic testing for diverse phenotypes become evident. In our patients, the implementation of Whole Exome Sequencing (WES) proved essential for achieving a definitive diagnosis. Unfortunately, in Latin American countries like Ecuador, access to genetic specialists and genetic testing is limited, underscoring the necessity for forming collaborative partnerships with specialists from other countries (48). This collaborative approach aims to promote universal, equitable, and accessible healthcare for all individuals. Secondly, the family history of our patients underscores the importance of meticulous family history documentation and the utilization of pedigrees. These tools aid in identifying other family members exhibiting related symptoms, deciphering the pattern of transmission, and identifying potential health problems that an individual may be at increased risk for in the future (38). Thus, emphasizing the significance of comprehensive family history assessment contributes to a more accurate understanding of genetic conditions and facilitates proactive health management.
Our study unravels a complex interplay of genetic factors contributing to a novel phenotype in two adolescent patients with a dual mutation in the AARS and CACNA1A genes. The distinct progression of symptoms from seizures to motor and cognitive alterations, including psychiatric symptoms in Patient B, challenges the conventional understanding of AARS-related phenotypes. The CACNA1A mutation further adds to the complexity, showcasing a broad phenotypic spectrum with significant incomplete penetrance. The familial inheritance of these mutations correlates with an accelerated symptom progression, emphasizing their potential role in the clinical manifestation. We underscore the essential role of genetic testing, especially Whole Exome Sequencing, in diagnosing diverse phenotypes, highlighting the need for international collaborations to improve healthcare accessibility, and emphasizes the significance of detailed family history documentation for a comprehensive and collaborative approach to genetic research and healthcare, ensuring universal access and proactive management for individuals and families with complex genetic conditions.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
The studies involving humans were approved by CEISH Universidad San Francisco de Quito. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
VR: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. SS: Data curation, Investigation, Visualization, Writing – original draft, Writing – review & editing. BA-A: Formal analysis, Validation, Visualization, Writing – original draft, Writing – review & editing. DD: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. KH: Data curation, Formal analysis, Funding acquisition, Methodology, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Academic Articles Publication Fund of Universidad San Francisco de Quito USFQ.
The authors are indebted to the patients and the parents for their willingness to participate in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2024.1376643/full#supplementary-material
1. Özer Çelik, A, Kurt, P, Yener, G, Alkin, T, Öztura, İ, and Baklan, B. Comparison of cognitive impairment between patients having epilepsy and psychogenic nonepileptic seizures. Nöro Psikiyatri Arş. (2015) 52:163–8. doi: 10.5152/npa.2015.7290
2. Helmstaedter, Christoph. The impact of epilepsy on cognitive function. (2014). Available: https://jnnp.bmj.com/content/84/9/e1.6 (Accessed December 13, 2023)
3. Elger, CE, Helmstaedter, C, and Kurthen, M. Chronic epilepsy and cognition. Lancet Neurol. (2004) 3:663–72. doi: 10.1016/S1474-4422(04)00906-8
4. Lado, FA, Laureta, EC, and Moshé, SL. Seizure-induced hippocampal damage in the mature and immature brain. Epileptic Disord Int Epilepsy J Videotape. (2002) 4:83–97. doi: 10.1684/j.1950-6945.2002.tb00479.x
5. Witt, J-A, and Helmstaedter, C. Should cognition be screened in new-onset epilepsies? A study in 247 untreated patients. J Neurol. (2012) 259:1727–31. doi: 10.1007/s00415-012-6526-2
6. Berg, AT, Berkovic, SF, Brodie, MJ, Buchhalter, J, Cross, JH, van Emde Boas, W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE commission on classification and terminology, 2005-2009. Epilepsia. (2010) 51:676–85. doi: 10.1111/j.1528-1167.2010.02522.x
7. Bartolini, E. Inherited developmental and epileptic encephalopathies. Neurol Int. (2021) 13:555–68. doi: 10.3390/neurolint13040055
8. Cheah, CS, Westenbroek, RE, Roden, WH, Kalume, F, Oakley, JC, Jansen, LA, et al. Correlations in timing of sodium channel expression, epilepsy, and sudden death in Dravet syndrome. Channels Austin Tex. (2013) 7:468–72. doi: 10.4161/chan.26023
9. Liao, Y, Deprez, L, Maljevic, S, Pitsch, J, Claes, L, Hristova, D, et al. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain J Neurol. (2010) 133:1403–14. doi: 10.1093/brain/awq057
10. Larsen, J, Carvill, GL, Gardella, E, Kluger, G, Schmiedel, G, Barisic, N, et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology. (2015) 84:480–9. doi: 10.1212/WNL.0000000000001211
11. Dedek, K, Fusco, L, Teloy, N, and Steinlein, OK. Neonatal convulsions and epileptic encephalopathy in an Italian family with a missense mutation in the fifth transmembrane region of KCNQ2. Epilepsy Res. (2003) 54:21–7. doi: 10.1016/s0920-1211(03)00037-8
12. Miceli, F, Striano, P, Soldovieri, MV, Fontana, A, Nardello, R, Robbiano, A, et al. A novel KCNQ3 mutation in familial epilepsy with focal seizures and intellectual disability. Epilepsia. (2015) 56:e15–20. doi: 10.1111/epi.12887
13. Barros, J, Damásio, J, Tuna, A, Alves, I, Silveira, I, Pereira-Monteiro, J, et al. Cerebellar ataxia, hemiplegic migraine, and related phenotypes due to a CACNA1A missense mutation: 12-year follow-up of a large Portuguese family. JAMA Neurol. (2013) 70:235–40. doi: 10.1001/jamaneurol.2013.591
14. Angelini, C, van Gils, J, Bigourdan, A, Jouk, PS, Lacombe, D, Menegon, P, et al. Major intra-familial phenotypic heterogeneity and incomplete penetrance due to a CACNA1A pathogenic variant. Eur J Med Genet. (2019) 62:103530. doi: 10.1016/j.ejmg.2018.08.011
15. Di Stefano, V, Rispoli, MG, Pellegrino, N, Graziosi, A, Rotondo, E, Napoli, C, et al. Diagnostic and therapeutic aspects of hemiplegic migraine. J Neurol Neurosurg Psychiatry. (2020) 91:764–71. doi: 10.1136/jnnp-2020-322850
16. Indelicato, E, and Boesch, S. From genotype to phenotype: expanding the clinical Spectrum of CACNA1A variants in the era of next generation sequencing. Front Neurol. (2021) 12:639994. doi: 10.3389/fneur.2021.639994
17. Le Roux, M, Barth, M, Gueden, S, de Cepoy, PD, Aeby, A, Vilain, C, et al. CACNA1A-associated epilepsy: Electroclinical findings and treatment response on seizures in 18 patients. Eur J Paediatr Neurol EJPN Off J Eur Paediatr Neurol Soc. (2021) 33:75–85. doi: 10.1016/j.ejpn.2021.05.010
18. Johnson, NE. Whole-exome sequencing in neurologic practice: reducing the diagnostic odyssey. Neurol Genet. (2015) 1:e37. doi: 10.1212/NXG.0000000000000037
19. Muthaffar, OY. The utility of whole exome sequencing in diagnosing pediatric neurological disorders. Balk J Med Genet BJMG. (2020) 23:17–24. doi: 10.2478/bjmg-2020-0028
20. Waterhouse, A, Bertoni, M, Bienert, S, Studer, G, Tauriello, G, Gumienny, R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. (2018) 46:W296–303. doi: 10.1093/nar/gky427
21. Jumper, J, Evans, R, Pritzel, A, Green, T, Figurnov, M, Ronneberger, O, et al. Highly accurate protein structure prediction with alpha fold. Nature. (2021) 596:583–9. doi: 10.1038/s41586-021-03819-2
22. Yun, RH, Anderson, A, and Hermans, J. Proline in alpha-helix: stability and conformation studied by dynamics simulation. Proteins. (1991) 10:219–28. doi: 10.1002/prot.340100306
23. Woolfson, DN, and Williams, DH. The influence of proline residues on alpha-helical structure. FEBS Lett. (1990) 277:185–8. doi: 10.1016/0014-5793(90)80839-b
24. Barlow, DJ, and Thornton, JM. Helix geometry in proteins. J Mol Biol. (1988) 201:601–19. doi: 10.1016/0022-2836(88)90641-9
25. Kessel, A., and Ben-Tal, N. Introduction to proteins | structure, function, and motion. 2nd. Chapman and Hall/CRC: New York. (2024).
26. van der Lee, R, Buljan, M, Lang, B, Weatheritt, RJ, Daughdrill, GW, Dunker, AK, et al. Classification of intrinsically disordered regions and proteins. Chem Rev. (2014) 114:6589–631. doi: 10.1021/cr400525m
27. Uversky, VN. Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J. (2015) 282:1182–9. doi: 10.1111/febs.13202
28. Holehouse, AS, and Kragelund, BB. The molecular basis for cellular function of intrinsically disordered protein regions. Nat Rev Mol Cell Biol. (2023) 25:187–211. doi: 10.1038/s41580-023-00673-0
29. Szklarczyk, D, Gable, AL, Lyon, D, Junge, A, Wyder, S, Huerta-Cepas, J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. (2019) 47:D607–13. doi: 10.1093/nar/gky1131
30. Scaglia, F. MELAS syndrome – symptoms, causes, treatment | NORD. (2011). Available at: https://rarediseases.org/rare-diseases/melas-syndrome/ (Accessed December 13, 2023).
31. Autoimmune encephalitis – about the disease – genetic and rare diseases information center. (2023). Available at: https://rarediseases.info.nih.gov/diseases/11979/autoimmune-encephalitis (Accessed December 13, 2023)
32. Rahman, S, and Copeland, WC. POLG-related disorders and their neurological manifestations. Nat Rev Neurol. (2019) 15:40–52. doi: 10.1038/s41582-018-0101-0
33. Sundal, C, Carmona, S, Yhr, M, Almström, O, Ljungberg, M, Hardy, J, et al. An AARS variant as the likely cause of Swedish type hereditary diffuse leukoencephalopathy with spheroids. Acta Neuropathol Commun. (2019) 7:188. doi: 10.1186/s40478-019-0843-y
34. Turner, AJ, and Hick, PE. Inhibition of aldehyde reductase by acidic metabolites of the biogenic amines. Biochem Pharmacol. (1975) 24:1731–3. doi: 10.1016/0006-2952(75)90016-7
35. Lin, W-L, Wszolek, ZK, and Dickson, DW. Hereditary diffuse leukoencephalopathy with spheroids: ultrastructural and immunoelectron microscopic studies. Int J Clin Exp Pathol. (2010) 3:665–74.
36. Sundal, C, Ekholm, S, Nordborg, C, Jönsson, L, Börjesson-Hanson, A, Lindén, T, et al. Update of the original HDLS kindred: divergent clinical courses. Acta Neurol Scand. (2012) 126:67–75. doi: 10.1111/j.1600-0404.2011.01624.x
37. Latour, P, Thauvin-Robinet, C, Baudelet-Méry, C, Soichot, P, Cusin, V, Faivre, L, et al. A major determinant for binding and Aminoacylation of tRNAAla in cytoplasmic Alanyl-tRNA Synthetase is mutated in dominant axonal Charcot-Marie-tooth disease. Am J Hum Genet. (2010) 86:77–82. doi: 10.1016/j.ajhg.2009.12.005
38. Lin, K-P, Soong, BW, Yang, CC, Huang, LW, Chang, MH, Lee, IH, et al. The mutational spectrum in a cohort of Charcot-Marie-tooth disease type 2 among the Han Chinese in Taiwan. PLoS One. (2011) 6:e29393. doi: 10.1371/journal.pone.0029393
39. McLaughlin, HM, Sakaguchi, R, Giblin, W, NIH Intramural Sequencing CenterWilson, TE, Biesecker, L, et al. A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-tooth disease type 2N (CMT2N). Hum Mutat. (2012) 33:244–53. doi: 10.1002/humu.21635
40. Weterman, MAJ, Kuo, M, Kenter, SB, Gordillo, S, Karjosukarso, DW, Takase, R, et al. Hypermorphic and hypomorphic AARS alleles in patients with CMT2N expand clinical and molecular heterogeneities. Hum Mol Genet. (2018) 27:4036–50. doi: 10.1093/hmg/ddy290
41. Nakayama, T, Wu, J, Galvin-Parton, P, Weiss, J, Andriola, MR, Hill, RS, et al. Deficient activity of alanyl-tRNA synthetase underlies an autosomal recessive syndrome of progressive microcephaly, hypomyelination, and epileptic encephalopathy. Hum Mutat. (2017) 38:1348–54. doi: 10.1002/humu.23250
42. Simons, C, Griffin, LB, Helman, G, Golas, G, Pizzino, A, Bloom, M, et al. Loss-of-function Alanyl-tRNA Synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am J Hum Genet. (2015) 96:675–81. doi: 10.1016/j.ajhg.2015.02.012
43. Botta, E, Theil, AF, Raams, A, Caligiuri, G, Giachetti, S, Bione, S, et al. Protein instability associated with AARS1 and MARS1 mutations causes trichothiodystrophy. Hum Mol Genet. 30:1711–20. doi: 10.1093/hmg/ddab123
44. King, MD, Gummer, CL, and Stephenson, JB. Trichothiodystrophy-neurotrichocutaneous syndrome of Pollitt: a report of two unrelated cases. J Med Genet. (1984) 21:286–9. doi: 10.1136/jmg.21.4.286
45. Kordasiewicz, HB, Thompson, RM, Clark, HB, and Gomez, CM. C-termini of P/Q-type Ca2+ channel alpha 1A subunits translocate to nuclei and promote polyglutamine-mediated toxicity. Hum Mol Genet. (2006) 15:1587–99. doi: 10.1093/hmg/ddl080
46. Diriong, S, Lory, P, Williams, ME, Ellis, SB, Harpold, MM, and Taviaux, S. Chromosomal localization of the human genes for alpha 1A, alpha 1B, and alpha 1E voltage-dependent Ca2+ channel subunits. Genomics. (1995) 30:605–9. doi: 10.1006/geno.1995.1284
47. Zhang, Y, Mori, M, Burgess, DL, and Noebels, JL. Mutations in high-voltage-activated calcium channel genes stimulate low-voltage-activated currents in mouse thalamic relay neurons. J Neurosci. (2002) 22:6362–71. doi: 10.1523/JNEUROSCI.22-15-06362.2002
Keywords: AARS, CACNA1A, case review, epilepsy, progressive cognitive decline, epileptic encephalopathy
Citation: Romero VI, Sáenz S, Arias-Almeida B, DiCapua D and Hosomichi K (2024) AARS and CACNA1A mutations: diagnostic insights into a case report of uncommon epileptic encephalopathy phenotypes in two siblings. Front. Neurol. 15:1376643. doi: 10.3389/fneur.2024.1376643
Edited by:
Suzanne Lesage, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Alessandro Capuano, Azienda Sanitaria Locale di Viterbo, ItalyCopyright © 2024 Romero, Sáenz, Arias-Almeida, DiCapua and Hosomichi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vanessa I. Romero, dnJvbWVyb0B1c2ZxLmVkdS5lYw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.