 Bin Ren1†
Bin Ren1† Xiaoyan Wu
Xiaoyan Wu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol. , 21 March 2024
Sec. Epilepsy
Volume 15 - 2024 | https://doi.org/10.3389/fneur.2024.1359287
This article is part of the Research Topic Case Reports in Epilepsy: volume III - 2023 View all 6 articles
The SYN1 gene encodes synapsin I, variants within the SYN1 gene are linked to X-linked neurodevelopmental disorders with high clinical heterogeneity, with reflex epilepsies (REs) being a representative clinical manifestation. This report analyzes a Chinese pedigree affected by seizures associated with SYN1 variants and explores the genotype–phenotype correlation. The proband, a 9-year-old boy, experienced seizures triggered by bathing at the age of 3, followed by recurrent absence seizures, behavioral issues, and learning difficulties. His elder brother exhibited a distinct clinical phenotype, experiencing sudden seizures during sleep at the age of 16, accompanied by hippocampal sclerosis. Whole exome sequencing (WES) confirmed a pathogenic SYN1 variant, c.1647_1650dup (p. Ser551Argfs*134), inherited in an X-linked manner from their mother. Notably, this variant displayed diverse clinical phenotypes in the two brothers and one previously reported case in the literature. Retrospective examination of SYN1 variants revealed an association between truncating variants and the pathogenicity of REs, and non-truncating variants are more related to developmental delay/intellectual disability (DD/ID). In summary, this study contributes to understanding complex neurodevelopmental disorders associated with SYN1, highlighting the clinical heterogeneity of gene variants and emphasizing the necessity for comprehensive genetic analysis in elucidating the pathogenic mechanisms of such diseases.
Reflex epilepsies (REs) are characterized by recurrent seizures consistently triggered by specific stimuli, such as light, music, hot water, and bathing (1). Limited research has been conducted on the genetic underpinnings of REs, with SYN1 being one of the few identified causative genes in humans (2, 3). SYN1 encodes synapsin I, a neuronal phosphoprotein that regulates axonogenesis and synaptogenesis (4). SYN1 gene variants have been implicated in two X-linked disorders: epilepsy with variable learning disabilities and behavior disorders (EPILX, [MIM:300491]) and intellectual developmental disorder 50 (XLID50, [MIM:300115]). EPILX manifests as epileptic seizures, learning difficulties, autism spectrum disorders, aggressive behavior, behavioral abnormalities, or variable developmental delays (2, 5–7). In contrast, XLID50 is characterized by varying degrees of intellectual disability, autistic features, and mild brain malformations, typically without seizures (6, 8–10).
The disease phenotype associated with SYN1 variants is highly heterogeneous, with individuals within the same family carrying identical genetic variants displaying a broad spectrum of clinical manifestations and varying degrees of severity (5, 6, 11). REs, a distinctive clinical hallmark, are frequently observed in individuals with SYN1 variants. Seizures are mostly triggered by water exposure (bathing, showering, swimming), but other stimuli like tooth brushing, rubbing with a towel, fever, and so on have been reported (4–7, 12, 13). Though recent reports suggest a tendency for seizures, especially REs to be more prevalent in individuals with truncating variants, while non-truncating variants are associated with developmental disorder (6), definitive genotype–phenotype are yet to be established and require intensive investigation.
In this context, our study aims to contribute valuable insights into the genotype–phenotype correlations of SYN1-related disorders through a Chinese pedigree with two male patients and literature Review. In this family, two brothers were identified with SYN1 truncating variants, and displayed different clinical features. Additionally, our study involves a comprehensive review of the pathogenicity of truncating and non-truncating variants from existing literature, laying the foundation for future in-depth investigations into SYN1-related disorders.
A 9-year-old boy (proband, II-1) and his elder brother (II-2) with epilepsy were admitted at Affiliated Hospital of Guilin Medical College. Whole exome sequencing (WES) was performed on both patients. The parents of the siblings were also enrolled in this investigation to elucidate the genetic inheritance pattern. This study was approved by the Institutional Research Ethics Committee of the Affiliated Hospital of Guilin Medical College (Ethical Approval Number:2022QTLL) and written informed consent was obtained from the parents of the pedigree.
Genomic DNA was extracted from whole blood samples for library preparation using the MagPure Tissue&Blood DNA LQ Kit (Magen). The T345V1 Exome Research Panel probes (iGeneTech, Shanghai, China) were utilized to capture the exon region following the manufacturer’s recommendations. The raw data was sequenced on NovaSeq6000 platforms (Illumina). Reads were mapped to the hg19 reference genome using BWA MEM (v0.7.17). PCR and optical duplicate marking were conducted using GATK (v4.1.4.0), and local realignment around indels and base quality score recalibration were carried out by GATK (v3.8.1). Finally, variants were identified using GATK HaplotypeCaller 1 (v3.8.1) and filtered by bcftools with the following criteria (v1.9): mapping quality≥40; depth ≥ 4.
We prioritized the rare variants with a minor allele frequency < 0.005 in Genome Aggregation Database (gnomAD),1 and Exome Aggregation Consortium. We retained potentially pathogenic variants, including frameshift, nonsense, canonical splice site, initiation codon, in-frame, and missense variants. Variants were further filtered based on epilepsy-associated gene model and possible inheritance models. Candidate variants were confirmed and co-segregation studies were performed using Sanger sequencing. Candidate variants were analyzed according to the American College of Medical Genetics and Genomics (ACMG) guidelines (14).
Sanger sequencing was performed on the two brothers and their parents to confirm the candidate variant identified in SYN1 through WES analysis. The primer design was accomplished using Primer Premier 5 and synthesized by MapBioo (Shanghai, China). The forward and reverse primer sequences were as follows: Forward: 5′-TTCCAAGTCCCACCTCAGCG-3′; Reverse: 5′-TTGGCGGAGCCGGGCCAGA-3′. Amplified products underwent sequencing utilizing an ABI 3730 DNA sequencer (Applied Biosystems).
The proband (II-1), initially manifested loss of consciousness at the age of 3, which was triggered by bathing in a bathtub filled with lukewarm water (approximately 37°C). The episodes included loss of consciousness, perioral cyanosis and hypotonia, and lasted for approximately 1 min before consciousness was regained. Subsequently, several seizures occurred specifically during bathing. To mitigate this, bathing in the bathtub was discontinued. The patient’s mother adapted by using a warm wet towel to cleanse him, a measure that successfully averted any subsequent seizure activity. Notably, after a hiatus of 4–5 months, the child was able to return to his regular routine of bathing without experiencing any further episodes of seizures. Commencing at age 6, he exhibited recurring absence epilepsy episodes, spontaneously resolving within seconds, but occurring as frequently as daily, particularly during homework sessions. Interictal electroencephalography (EEG) disclosed 2–3 Hz sharp slow wave discharges, notably in the left occipital and middle/posterior temporal regions during sleep, along with sharp wave discharges in the right anterior temporal lobe on another EEG test. The patient reported experiencing episodes of staring and the ictal EEG showed high amplitude spike-and-wave activity and sharp-and-slow wave activity across multiple leads, with both long and short bursts occurring. Magnetic resonance spectroscopy (MRS) analysis showed metabolic abnormalities in the hippocampal head on both sides, the left body, and the right tail, suggesting neuronal loss and gliosis. Additionally, the proband displayed abnormal behavioral and psychiatric features, including uncontrolled temper (especially during homework), attention deficit hyperactivity disorder (ADHD), and learning difficulties. The patient initially received treatment with 125 mg of magnesium valproate sustained release tablets twice a day, which reduced the frequency of absence seizures. After 2 weeks, the dosage was adjusted to 250 mg twice a day. Following this adjustment, the frequency of absence seizures during homework periods further diminished from 4 to 5 times daily to 1–2 times daily. As the seizures had minimal impact on the patient’s daily life, it was decided to maintain the current treatment plan without any immediate changes.
Reflex epilepsies, such as hot water epilepsy and bathing epilepsy, are characterized by seizures triggered by specific stimuli, particularly involving water during bathing. Hot water epilepsy, a well-known variety that is frequent in South India, manifests a few key clinical characteristics: (1) triggering of seizures by pouring hot water, with temperatures ranging from 38°C to 55°C, over the head; (2) the absence of neurodevelopmental problems or detectable abnormalities in neuro-imaging for most affected children; (3) generally favourable outcome for the majority of children diagnosed with hot water epilepsy (15–19). But a less well-known infantile variety, primarily documented in France, exhibits several distinct clinical features: (1) Seizures commence before the age of 1 year, triggered by immersion in hot water at approximately 37.5°C; (2) Similar to the variant prevalent in South India, these children typically do not exhibit neurodevelopmental problems or abnormalities in neuro-imaging; (3) The prognosis for these children is also generally positive; (4) EEGs often reveal a focal onset of seizures, predominantly in the temporal region; (5) No other type of seizure was reported (20). The phenotypes of our patient do not align with the characteristics of the aforementioned diseases. Bathing epilepsy, in contrast, is characterized by seizures that can be triggered by immersion in water at temperatures close to or below the normal body temperature, and some patients may experience neurological symptoms (2, 18, 21, 22). The diagnosis of bathing epilepsy is supported by the proband’s clinical presentation, which more closely meets the diagnostic criteria for this condition.
His elder brother (II-2), now aged 16, exhibited normal developmental history and intelligence until experiencing a sudden epileptic seizure during sleep for 3 times in 1 month at the age of 16, characterized by loss of consciousness and teeth clenching, with no identifiable trigger. Ambulatory EEG revealed dispersed epileptic discharges during sleep, predominantly in the left occipital region, without any clinical events occurring during monitoring. MRS demonstrated bilateral hippocampal sclerosis and abnormal neurometabolic profiles indicative of neuronal loss and gliosis. Antiepileptic therapy with magnesium valproate sustained release tablets (250 mg twice daily) successfully prevented further seizure for 2-year follow-up.
The EEG of the two brothers show some notable similarities. Both individuals display epileptiform discharges, which are more prominent on the left side and tend to occur during sleep. Interestingly, the brother exhibits a broader distribution of epileptiform discharges compared to the proband. The consistency across their EEG and MRS results strengthens the likelihood of a shared etiology behind their neurological presentations.
The two brothers were the only children in their family. No family history of neurological disorders, behavioral issues, or febrile seizures was reported. Both parents were healthy and nonconsanguineous.
Whole Exome Sequencing identified a frameshift variant in SYN1 [NM_006950.3:c.1647_1650dup (p.Ser551Argfs*134)] in both brothers (Figure 1). Sanger sequencing confirmed maternal inheritance of the variant (Figure 1). The X-linked pattern of segregation was observed, with affected siblings being hemizygous, unaffected mother being heterozygous and father being wild-type (father; PP1). The variant c.1647_1650dup (p.Ser551Argfs*134) is predicted to cause a frameshift that could potentially result in nonsense-mediated mRNA decay (PVS1) (23). The variant was absent in gnomAD v4.0.0 databases (PM2_Supporting). The variant was classified as conflicting classifications of pathogenicity in ClinVar with “Pathogenic” and “Uncertain significance”(Variation ID:1994692). Based on the above evidence, this variant can be classified as pathogenic (PVS1 + PM2_Supporting+PP1) according to ACMG guidelines (14, 23, 24).
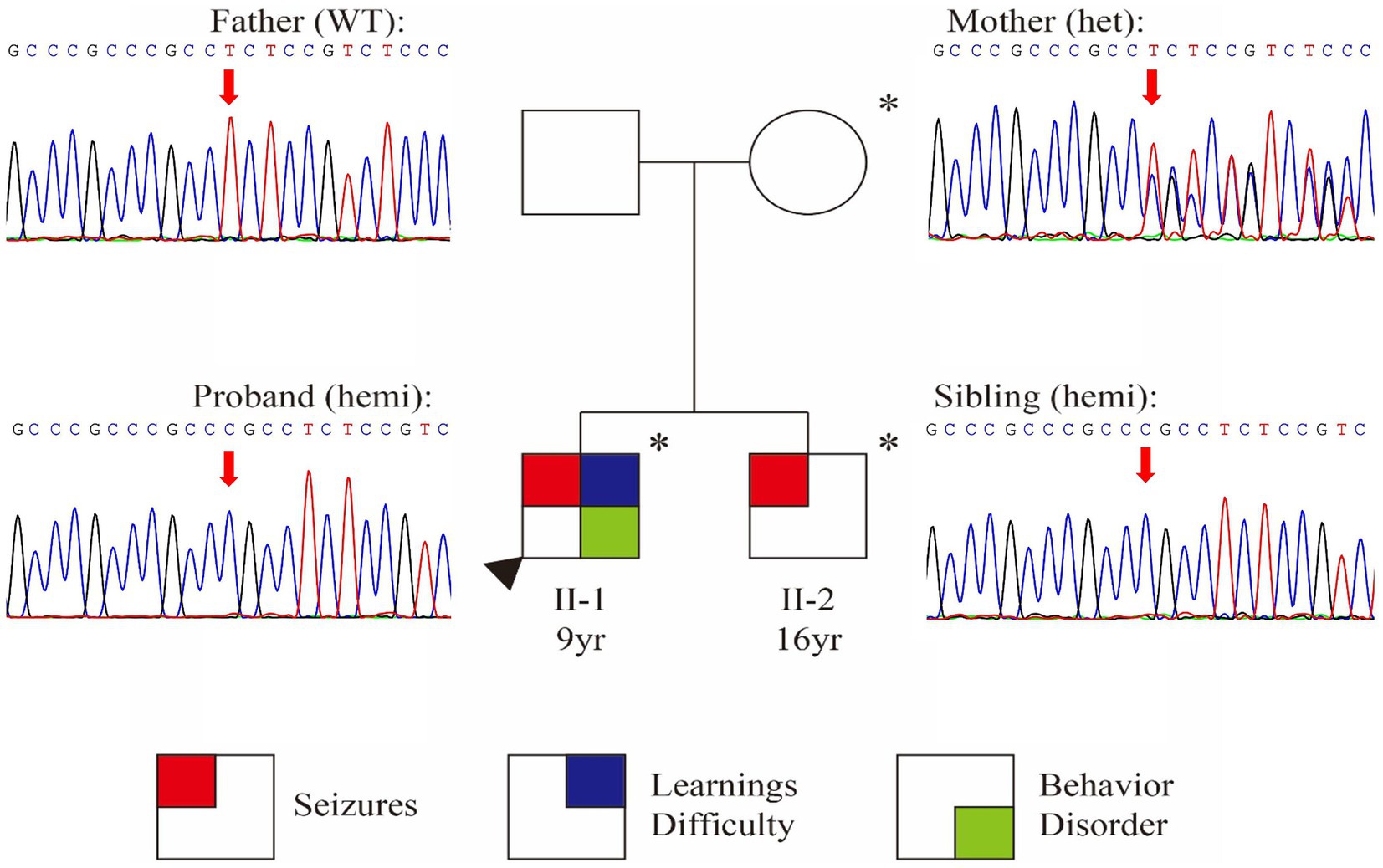
Figure 1. Identification of a frameshift variant in SYN1. Pedigree chart and Sanger confirmation of the family. Individuals with the SYN1 variant are represented with an asterisk (*) symbol.
The c.1647_1650dup (p.Ser551Argfs*134) variant was previously reported in a boy with seizures, global developmental delay, mild intellectual disability, autism spectrum disorder, motor stereotypies, attention-deficit/hyperactivity disorders, who experienced reflex seizures after bathing, showering, fingernail clipping (2). Including our patients, c.1647_1650dup has been reported in three patients with detailed records available, but the clinical phenotypes were highly variable, even the two brothers in the same family. All the three cases demonstrated a favorable response to antiepileptic drugs. In the case reported by Accogli, the affected individual achieved seizure freedom following treatment with Oxcarbazepine, Sulthiame, Valproate, and Lamotrigine (2).
In addition, this variant has been submitted three times in ClinVar database, with each submission coming from patients identified through clinical testing. The patient submitted by Invitae was diagnosed as “epilepsy, X-linked 1, with variable learning disabilities and behavior disorders,” while the patients submitted by Baylor Genetics were diagnosed with “intellectual disability, X-linked 50” and “Epilepsy, X-linked 1, with variable learning disabilities and behavior disorders.” These phenotypes align with those related to SYN1 disease. However, without specific clinical information, it’s unclear whether they experience reflex seizures or how they respond to anti-epileptic drugs, which inhibits further analysis of clinical variations.
The spectrum of SYN1 gene variants is extensive, encompassing truncating variants such as frameshift, nonsense, splice-site, and start-loss variants, alongside non-truncating variants, including missense alterations, in-frame deletion, and in-frame insertion (Figure 2). Syn1 knockout mice have been observed to display an epileptic phenotype (25, 26), which is similar to the phenotype seen in human patients. Functional studies have also confirmed that truncating variants of SYN1 can impair synaptic function (9, 27). According to ClinGen dosage sensitivity curation standard operating procedures (23), the evidence supporting haploinsufficiency in SYN1-related X-linked complex neurodevelopmental disorder is substantial, establishing loss of function as the pathogenic mechanism for SYN1-related disorders. SYN1 truncating variants were reported in 62 patients (Table 1), with a total of 25 different variants. All 25 variants were loss of function variants, classified as likely pathogenic or pathogenic (Figure 2). Truncating variants introduce a premature termination codon, leading to nonsense-mediated mRNA decay or generating a truncated protein that lacks phosphorylation sites (27–29). These alterations disrupt synaptic vesicle transport, increasing network excitability and firing/bursting activity (9, 27, 30).
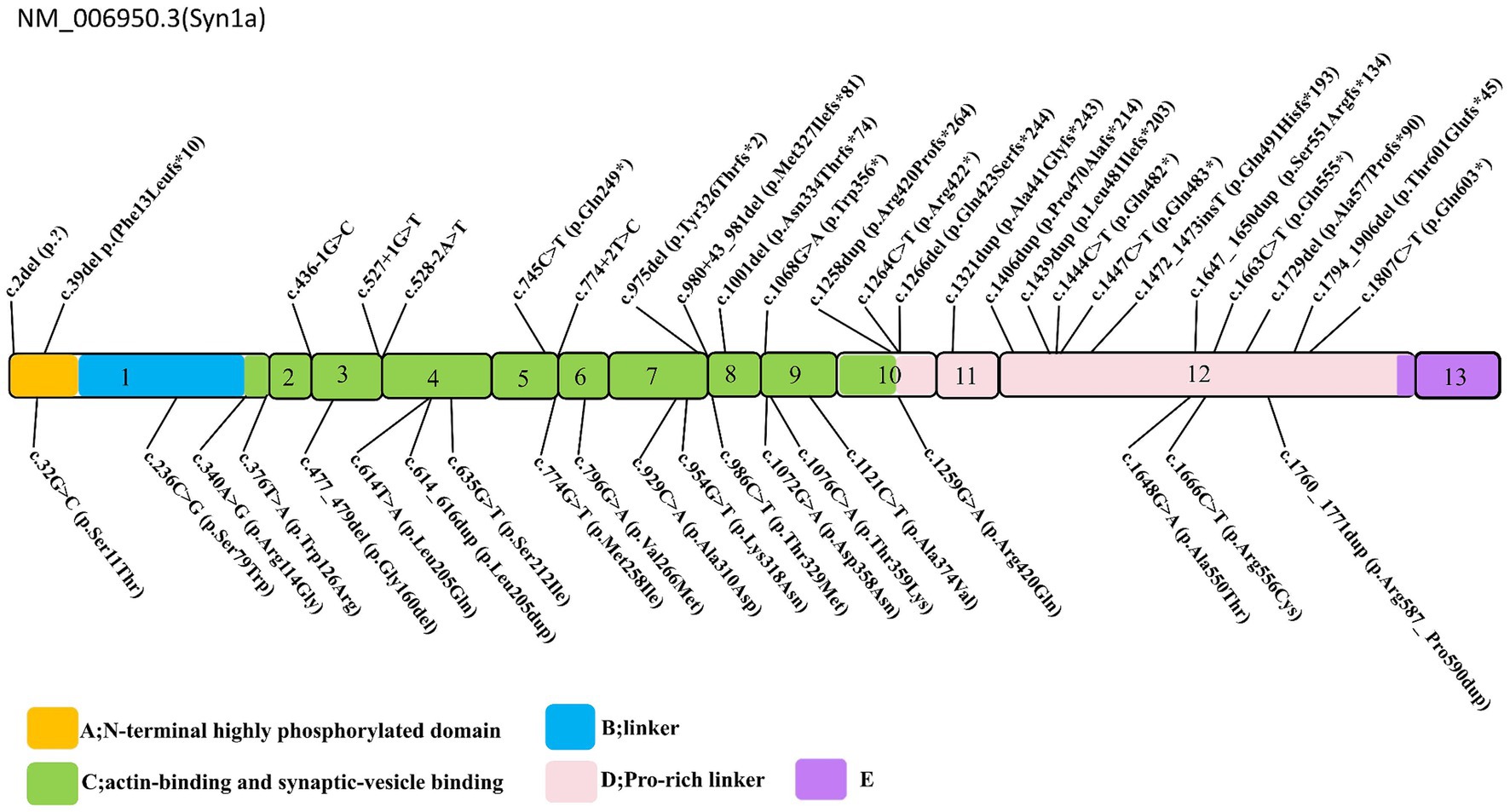
Figure 2. Schematic representation of the SYN1 gene. Exons 1–13 are depicted, truncating variants depicted above, non-truncating variants depicted below, the variants Thr567Ala, Ala51Gly, and Gly240Arg are removed. The conserved A (N-terminal highly phosphorylated domain), B (linker), and C (functional domain) domains are depicted in yellow, blue, green, respectively; the D, E domains in pink, and purple.
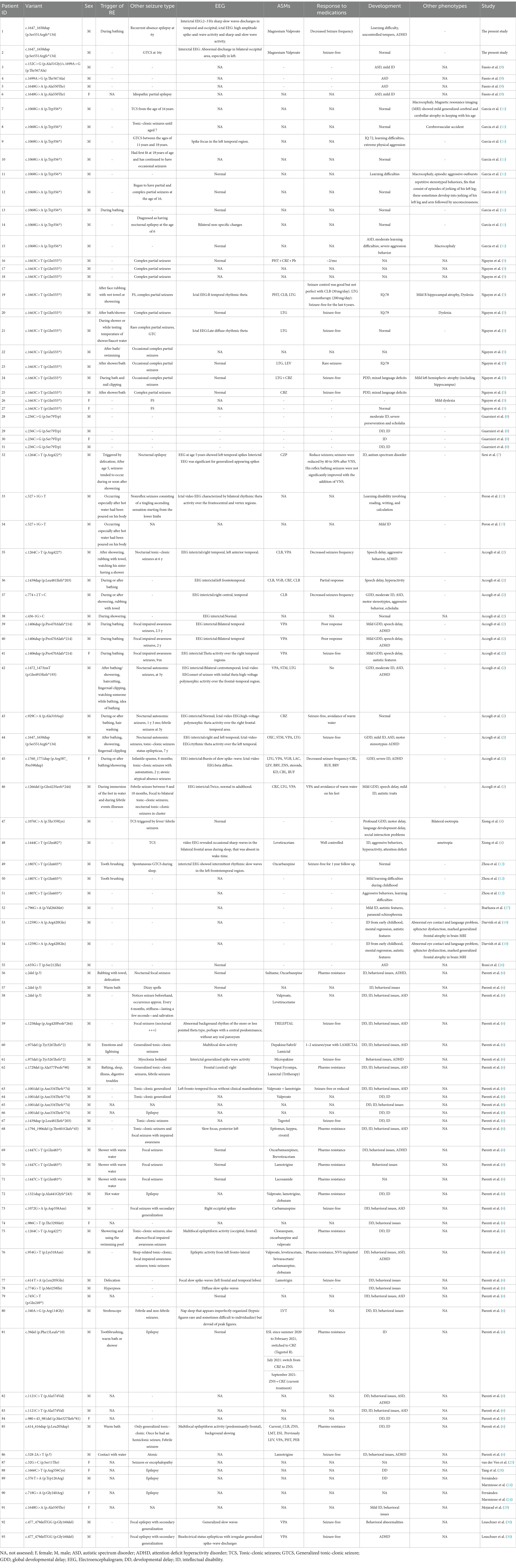
Table 1. Clinical and genetic characteristics of patients with SYN1-related disorders in literature and the study.
A meticulous re-evaluation of the pathogenicity of 23 reported non-truncating variants in 31 patients unveiled that most reported missense variants could be appropriately categorized as Variants of Uncertain Significance (VUS) by the ACMG guidelines (14, 23, 24) (Tables 1, 2). According to ClinGen recommendations for PP3/BP4 criteria updated in 2022, we relied on the REVEL score to provide supporting, moderate, and strong levels of evidence for pathogenicity or benignity (31). Functional studies indicated that SYN1 missense variants, such as Ser79Trp, Arg420Gln, Ala550Thr, and Thr567Ala, may exert an impact on neuronal development and nerve terminal targeting. These variants were associated with the disruption of synaptic vesicle pools and an increased frequency of excitatory spontaneous release events (8–10). However, it is noteworthy that the Thr567Ala variant was detected in 755 hemizygous and 48 homozygous healthy carriers in gnomAD v4.0.0. The high allele frequency implies a proclivity toward benign polymorphism rather than a pathogenic variant. Ala51Gly was also noted with high allele frequency in gnomAD v4.0.0 with 670 hemizygous and 42 homozygous healthy carriers, whereas Gly240Arg shows an incongruent pattern of inheritance (32). The three variants (Thr567Ala, Ala51Gly, Gly240Arg) tend to be benign. Uncertainty persists regarding the functional implications of other non-truncating variants, considering the limited scope of in vitro experiments conducted thus far. The question of whether SYN1 missense variants significantly contribute to SYN1-related disease remains elusive in light of these experimental constraints.
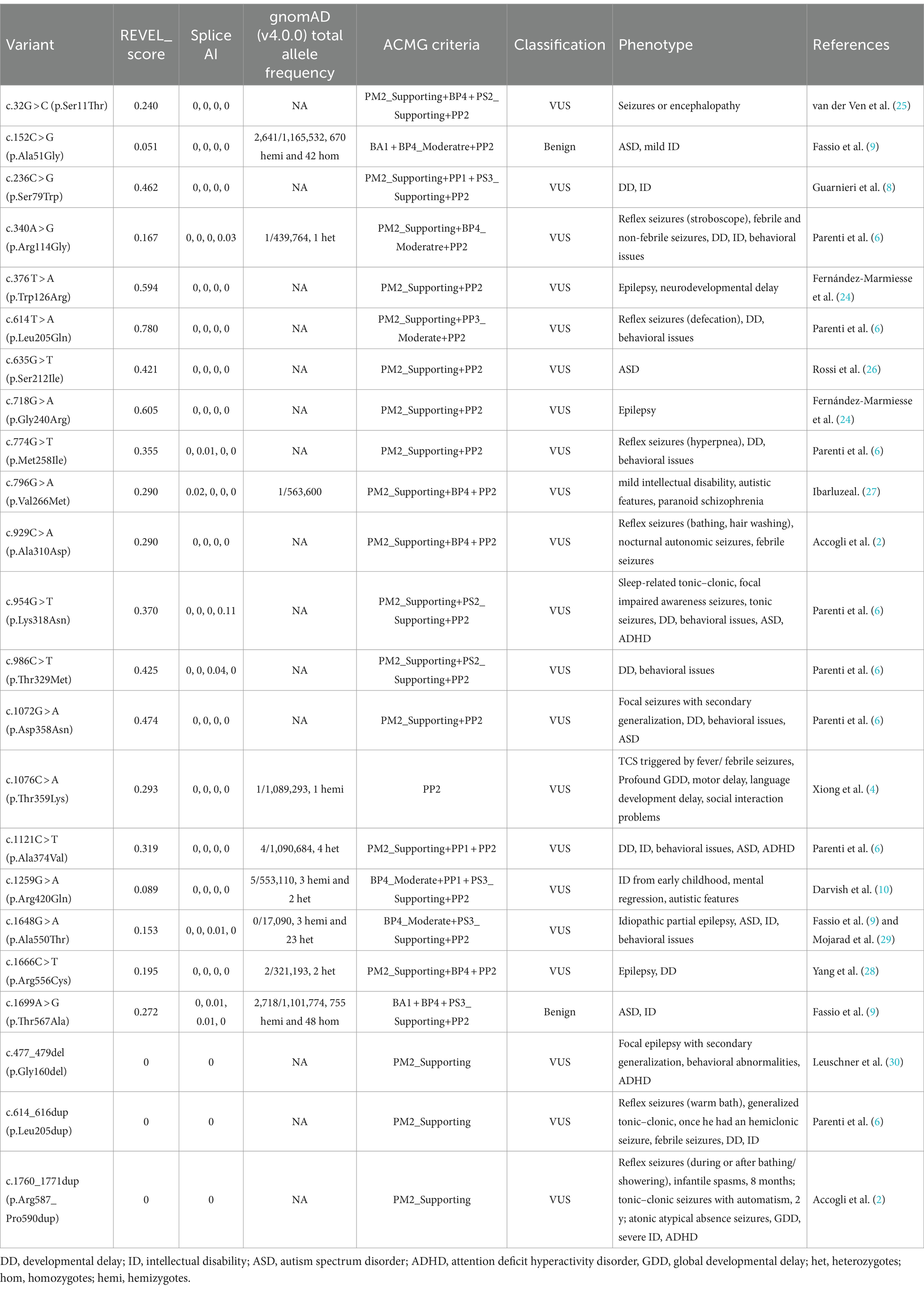
Table 2. The types and classification of reported SYN1 gene non-truncating variants.
This report elucidated the identification of a maternally inherited SYN1 pathogenic variant in two brothers. Notably, the SYN1 variant exhibited clinical heterogeneity in both brothers and one previously reported case in the literature (2). A comprehensive review of the literature was undertaken to delineate the prevalence of key clinical features associated with SYN1-related disorders, resulting in the compilation of data from a total of 93 documented patients with SYN1-associated diseases (Table 1). Among those with SYN1 truncating variants, 56/62(90.3%) patients reported seizures, and a substantial number of experienced diverse seizure types. In contrast, the non-truncating variant cohort reported only 16/31(51.6%) instances of seizures. These data strongly support the notion that patients with SYN1 truncating variants are more prone to seizures.
Specifically, in epilepsy patients with SYN1 truncating variants, 35/56(62.5%) patients reported reflex seizures triggered by activities such as bathing, showering, toothbrushing, rubbing with a towel, fever, fingernail clipping, falling asleep, watching others showering or bathing, and gastrointestinal discomfort (Table 1). In contrast, the non-truncating was 6/16(37.5%) reported instances of seizures, triggered by bathing/showering, defecation, hyperpnea, stroboscope, and hair washing (Table 1). It seems that truncating variants of the SYN1 gene may tend to manifest REs.
We also analyzed the frequency of spontaneous seizures (non-REs) in epilepsy patients across two groups. The occurrence of non-REs was 21/56(37.5%) in the truncating group and 10/16(62.5%) in the non-truncating group (Table 1). This result further corroborates that reflex seizures may be the primary manifestation in patients with truncating variants. This observation underscores the importance of identifying specific SYN1 variants to understand unique clinical trajectories, emphasizing the heterogeneity of SYN1-related neurodevelopmental disorders. It should also be noted that, although there is a difference between the truncating and non-truncating groups, epilepsy in general is common in patients with SYN1 variants, with a notable tendency to exhibit reflex epilepsy.
Furthermore, the evaluation of the developmental delay/intellectual disability (DD/ID) phenotype revealed intriguing patterns. A larger proportion of individuals with non-truncating variants 23/31(74%) exhibited DD/ID compared to those with truncating variants 31/62(50%; Table 1). The findings align with previous research (6) but extend our understanding by emphasizing the nuanced relationship between genetic variants and clinical outcomes. However, a deeper understanding of the mechanisms contributing to phenotypic diversity in SYN1-related disorders necessitates further investigation.
To date, a total of 81 males and 12 females have been reported with putative SYN1-related disorders. Females with SYN1 variants have exhibited a broad clinical heterogeneity and incomplete penetrance. These female patients have displayed a wide range of clinical manifestations, from simple febrile seizures to severe epileptic encephalopathy, with some individuals presenting DD/ID without seizures (Table 1). Interestingly, epilepsy triggered by bathing was observed in two female patients (Table 1). No differences in variant types were found when compared to male patients, with both truncating and non-truncating variants reported (Table 1). Moreover, two female patients had reported de novo variants and two others had reported paternal inherited variants, while somatic mosaicism in the blood was identified in the unaffected father of one of the female individuals (6).
This report delineates a familial presentation of seizures associated with a pathogenic variant in the SYN1 gene, thereby broadening the phenotype spectrum of SYN1 gene-related epilepsy disorders. Through a comprehensive review of existing literature, a discernible pattern emerges wherein truncating variants of the SYN1 gene tend to manifest in seizure-related symptoms, whereas non-truncating variants are more frequently associated with DD/ID. This discovery may aid in anticipating the phenotypes linked with SYN1-related conditions.
Furthermore, upon re-evaluating reported SYN1 non-truncating variants, a substantial portion of these variants are presently classified as VUS. Consequently, exercising caution is recommended when interpreting SYN1 missense variants in clinical diagnostics. It is imperative to conduct additional functional studies to definitively ascertain the pathogenicity of these variants.
In summary, our findings contribute valuable insights into the role of the SYN1 gene in epilepsy disorders, underscoring the necessity for additional research on its genetic variations. These results bear implications for enhancing diagnostic accuracy and providing more informed guidance for patients and their families affected by SYN1-related conditions.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding authors.
The studies involving humans were approved by the Medical Ethics Committee of Affiliated Hospital of Guilin Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
BR: Conceptualization, Data curation, Formal analysis, Methodology, Project administration, Resources, Supervision, Writing – review & editing. XW: Data curation, Formal analysis, Investigation, Writing – original draft, Writing – review & editing. YZ: Investigation, Writing – original draft, Writing – review & editing, Formal analysis. LC: Formal analysis, Investigation, Methodology, Project administration, Writing – review & editing, Conceptualization. JJ: Investigation, Methodology, Project administration, Resources, Writing – original draft, Conceptualization.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Project of Guangxi Natural Science Foundation (grant no. 2022GXNSFAA103020).
BR, XW, YZ, and LC were employed by the company Shanghai Nyuen Biotechnology Co., Ltd.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Okudan, ZV, and Özkara, Ç. Reflex epilepsy: triggers and management strategies. Neuropsychiatr Dis Treat. (2018) 14:327–37. doi: 10.2147/Ndt.S107669
2. Accogli, A, Wiegand, G, Scala, M, Cerminara, C, Iacomino, M, Riva, A, et al. Clinical and genetic features in patients with reflex bathing epilepsy. Neurology. (2021) 97:E577–86. doi: 10.1212/Wnl.0000000000012298
3. Italiano, D, Striano, P, Russo, E, Leo, A, Spina, E, Zara, F, et al. Genetics of reflex seizures and epilepsies in humans and animals. Epilepsy Res. (2016) 121:47–54. doi: 10.1016/J.Eplepsyres.2016.01.010
4. Xiong, J, Duan, H, Chen, S, Kessi, M, He, F, Deng, X, et al. Familial Syn1 variants related neurodevelopmental disorders in Asian pediatric patients. BMC Med Genet. (2021) 14:182. doi: 10.1186/S12920-021-01028-4
5. Nguyen, DK, Rouleau, I, Sénéchal, G, Ansaldo, AI, Gravel, M, Benfenati, F, et al. X-linked focal epilepsy with reflex bathing seizures: characterization of a distinct epileptic syndrome. Epilepsia. (2015) 56:1098–108. doi: 10.1111/Epi.13042
6. Parenti, I, Leitão, E, Kuechler, A, Villard, L, Goizet, C, Courdier, C, et al. The different clinical facets of Syn1-related neurodevelopmental disorders. Front Cell Dev Biol. (2022) 10:1019715. doi: 10.3389/Fcell.2022.1019715
7. Sirsi, D, Armstrong, D, Munoz-Bibiloni, J, Redondo, B, and Jy, P. Syn1 gene mutation in a child with focal epilepsy and reflex bathing seizures. J Pediatr Epilepsy. (2017) 6:119–24. doi: 10.1055/s-0037-1599193
8. Guarnieri, FC, Pozzi, D, Raimondi, A, Fesce, R, Valente, MM, Delvecchio, VS, et al. A novel Syn1 missense mutation in non-syndromic X-linked intellectual disability affects synaptic vesicle life cycle, clustering and mobility. Hum Mol Genet. (2017) 26:4699–714. doi: 10.1093/Hmg/Ddx352
9. Fassio, A, Patry, L, Congia, S, Onofri, F, Piton, A, Gauthier, J, et al. Syn1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum Mol Genet. (2011) 20:2297–307. doi: 10.1093/Hmg/Ddr122
10. Darvish, H, Lj, A, Tafakhori, A, Mesias, R, Ahmadifard, A, Sanchez, E, et al. Phenotypic and genotypic characterization of families with complex intellectual disability identified pathogenic genetic variations in known and novel disease genes. Sci Rep. (2020) 10:968. doi: 10.1038/S41598-020-57929-4
11. Garcia, CC, Blair, HJ, Seager, M, Coulthard, A, Tennant, S, Buddles, M, et al. Identification of a mutation in Synapsin I, a synaptic vesicle protein, in a family with epilepsy. J Med Genet. (2004) 41:183–6. doi: 10.1136/Jmg.2003.013680
12. Zhou, Q, Wang, J, Xia, L, Li, R, Zhang, Q, and Pan, S. Syn1 mutation causes X-linked Toothbrushing epilepsy in a Chinese family. Front Neurol. (2021) 12:736977. doi: 10.3389/Fneur.2021.736977
13. Peron, A, Baratang, NV, Canevini, MP, Campeau, PM, and Vignoli, A. Hot water epilepsy and Syn1 variants. Epilepsia. (2018) 59:2162–3. doi: 10.1111/Epi.14572
14. Richards, S, Aziz, N, Bale, S, Bick, D, Das, S, Gastier-Foster, J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/Gim.2015.30
15. Sharma, A, and Sankhyan, N. Hot-water epilepsy in children. Indian J Pediatr. (2021) 88:857–8. doi: 10.1007/S12098-021-03840-3
16. Satishchandra, P. Hot-water epilepsy. Epilepsia. (2003) 44:29–32. doi: 10.1046/J.1528-1157.44.S.1.14.X
17. Yalçin Ad, Toydemir He, Forta H. Hot Water Epilepsy. Clinical and electroencephalographic features of 25 cases. Epilepsy Behav. (2006) 9:89–94. doi: 10.1016/J.Yebeh.2006.03.013
18. Mosquera-Gorostidi, A, Azcona-Ganuza, G, Me, Y-P, García De Gurtubay, I, and Aguilera-Albesa, S. Ictal video-electroencephalography findings in bathing seizures: two new cases and review of the literature. Pediatr Neurol. (2019) 99:76–81. doi: 10.1016/J.Pediatrneurol.2019.04.017
19. Meghana, A, Sinha, S, Tn, S, Dk, S, and Satishchandra, P. Hot water epilepsy clinical profile and treatment--a prospective study. Epilepsy Res. (2012) 102:160–6. doi: 10.1016/J.Eplepsyres.2012.05.011
20. Plouin, P, and Vigevano, VF. Reflex Seizures In Infancy. In: Wolf P, Inoue Y, Zifkin B, and Eurotext JL, editors. Reflex epilepsies: progress in understanding. Montrouge: John Libbey Eurotext (2004) 115–122.
21. Braun, M, Et, P, Simpkins, A, Kozlik, S, and Curtis, C. Novel bathing epilepsy in a patient with 2q22.3q23.2 deletion. Seizure. (2021) 91:1–4. doi: 10.1016/J.Seizure.2021.05.007
22. Franzoni, E, Gentile, V, Grosso, S, Brunetto, D, Dm, C, and Balestri, P. Bathing epilepsy: report of two Caucasian cases. Epileptic Disord. (2010) 12:88–90. doi: 10.1684/Epd.2010.0295
23. Abou Tayoun, AN, Pesaran, T, DiStefano, MT, Oza, A, Rehm, HL, Biesecker, LG, et al. Recommendations for interpreting the loss of function Pvs1 Acmg/amp variant criterion. Hum Mutat. (2018) 39:1517–24. doi: 10.1002/Humu.23626
24. Nykamp, K, Anderson, M, Powers, M, Garcia, J, Herrera, B, Yy, H, et al. Sherloc: a comprehensive refinement of the Acmg-amp variant classification criteria. Genet Med. (2017) 19:1105–17. doi: 10.1038/Gim.2017.37
25. Rosahl, TW, Spillane, D, Missler, M, Herz, J, Selig, DK, Wolff, JR, et al. Essential functions of Synapsins I and ii in synaptic vesicle regulation. Nature. (1995) 375:488–93. doi: 10.1038/375488a0
26. Li, L, Chin, LS, Shupliakov, O, Brodin, L, Sihra, TS, Hvalby, O, et al. Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in Synapsin I-deficient mice. Proc Natl Acad Sci USA. (1995) 92:9235–9. doi: 10.1073/Pnas.92.20.9235
27. Giannandrea, M, Fc, G, Gehring, N, Monzani, E, Benfenati, F, Ae, K, et al. Nonsense-mediated Mrna decay and loss-of-function of the protein underlie the X-linked epilepsy associated with the W356× mutation in Synapsin I. PLoS One. (2013) 8:e67724. doi: 10.1371/journal.pone.0067724
28. Lewis, BP, Green, RE, and Brenner, SE. Evidence for the widespread coupling of alternative splicing and nonsense-mediated Mrna decay in humans. Proc Natl Acad Sci USA. (2003) 100:189–92. doi: 10.1073/Pnas.0136770100
29. Chang, YF, Imam, JS, and Wilkinson, MF. The nonsense-mediated decay Rna surveillance pathway. Annu Rev Biochem. (2007) 76:51–74. doi: 10.1146/Annurev.Biochem.76.050106.093909
30. Lignani, G, Raimondi, A, Ferrea, E, Rocchi, A, Paonessa, F, Cesca, F, et al. Epileptogenic Q555x Syn1 mutant triggers imbalances in release dynamics and short-term plasticity. Hum Mol Genet. (2013) 22:2186–99. doi: 10.1093/Hmg/Ddt071
31. Pejaver, V, Byrne, AB, Feng, BJ, Pagel, KA, Mooney, SD, Karchin, R, et al. Calibration of computational tools for missense variant pathogenicity classification and Clingen recommendations for Pp3/Bp4 criteria. Am J Hum Genet. (2022) 109:2163–77. doi: 10.1016/J.Ajhg.2022.10.013
Keywords: SYN1, reflex epilepsy, bathing epilepsy, genotype–phenotype correlation, clinical heterogeneity
Citation: Ren B, Wu X, Zhou Y, Chen L and Jiang J (2024) SYN1 variant causes X-linked neurodevelopmental disorders: a case report of variable clinical phenotypes in siblings. Front. Neurol. 15:1359287. doi: 10.3389/fneur.2024.1359287
Edited by:
Fernando Cendes, State University of Campinas, BrazilReviewed by:
Ilaria Meloni, University of Siena, ItalyCopyright © 2024 Ren, Wu, Zhou, Chen and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lijuan Chen, Y2hlbmxpanVhbkBuaHdhLWdyb3VwLmNvbQ==; Jingzi Jiang, amlhbmdqaW5nemlAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.