
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 11 March 2024
Sec. Dementia and Neurodegenerative Diseases
Volume 15 - 2024 | https://doi.org/10.3389/fneur.2024.1320663
 Spyros Papapetropoulos1
Spyros Papapetropoulos1 Jeffrey M. Gelfand2
Jeffrey M. Gelfand2 Takuya Konno3
Takuya Konno3 Takeshi Ikeuchi3
Takeshi Ikeuchi3 Angela Pontius1
Angela Pontius1 Andreas Meier1*Farid Foroutan4
Andreas Meier1*Farid Foroutan4 Zbigniew K. Wszolek5
Zbigniew K. Wszolek5Introduction: Because adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) is a rare, rapidly progressive, debilitating, and ultimately fatal neurodegenerative disease, a rapid and accurate diagnosis is critical. This analysis examined the frequency of initial misdiagnosis of ALSP via comprehensive review of peer-reviewed published cases.
Methods: Data were extracted from a MEDLINE search via PubMed (January 1, 1980, through March 22, 2022) from eligible published case reports/series for patients with an ALSP diagnosis that had been confirmed by testing for the colony-stimulating factor-1 receptor gene (CSF1R) mutation. Patient demographics, clinical symptoms, brain imaging, and initial diagnosis data were summarized descriptively. Categorical data for patient demographics, symptoms, and brain imaging were stratified by initial diagnosis category to test for differences in initial diagnosis based on each variable.
Results: Data were extracted from a cohort of 291 patients with ALSP from 93 published case reports and case series. Mean (standard deviation) age of symptom onset was 43.2 (11.6) years. A family history of ALSP was observed in 59.1% of patients. Cognitive impairment (47.1%) and behavioral and psychiatric abnormalities (26.8%) were the most frequently reported initial symptoms. Of 291 total cases, an accurate initial diagnosis of ALSP was made in 72 cases (24.7%) and the most frequent initial misdiagnosis categories were frontotemporal dementia (28 [9.6%]) and multiple sclerosis (21 [7.2%]). Of the 219 cases (75.3%) that were initially mis- or undiagnosed, 206 cases (94.1%) were later confirmed as ALSP by immunohistology, imaging, and/or genetic testing; for the remaining 13 cases, no final diagnosis was reported. Initial diagnosis category varied based on age, family history, geographic region, mode of inheritance, and presenting symptoms of pyramidal or extrapyramidal motor dysfunction, behavioral and psychiatric abnormalities, cognitive impairment, and speech difficulty. Brain imaging abnormalities were common, and initial diagnosis category was significantly associated with white matter hyperintensities, white matter calcifications, and ventricular enlargement.
Discussion: In this literature analysis, ALSP was frequently misdiagnosed. Improving awareness of this condition and distinguishing it from other conditions with overlapping presenting symptoms is important for timely management of a rapidly progressive disease such as ALSP.
Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) is a rare, hereditary, autosomal dominant neurodegenerative disorder with typical onset between 40 and 50 years of age (mean age, 43 years [range: 18–78 years]) (1, 2). Loss-of-function mutations in the colony-stimulating factor-1 receptor gene (CSF1R) cause the morphologic abnormalities that are characteristic of ALSP, including distended neuronal axons, pigmented glial cells, and demyelination of cerebral white matter (3, 4). The treatment of ALSP remains an unmet medical need, as no symptomatic or disease-modifying therapies are currently approved to reverse, delay, or stop the progression of this disabling disorder (3, 4). The natural course of ALSP is marked by rapidly progressive and debilitating cognitive impairment, moderate to severe motor dysfunction, and neuropsychiatric complications, leading to impaired quality of life and death within approximately 6–8 years from symptom onset (1, 4, 5).
Symptom onset and progression among patients with ALSP is variable, even within the same family, and differential diagnosis of ALSP can be challenging due to the overlapping presentation of symptoms and radiologic features that can mimic other neurodegenerative and white matter diseases (inflammatory, vascular, or genetic) (4–6). A previously conducted comprehensive review of the literature examined disorders with clinical symptoms that overlap with ALSP (3), highlighting that the differential diagnosis of ALSP should include Alzheimer’s disease, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), atypical Parkinson’s disease, other leukodystrophies, frontotemporal dementia, progressive multiple sclerosis, and vascular dementia (3–5).
Validated diagnostic criteria for ALSP that were developed through a retrospective case study combine specific core features, exclusionary findings, and supportive findings to generate the diagnosis of ALSP as definite, probable, or possible (7); however, independent analyses have suggested that these criteria demonstrate poor specificity (8, 9), which may be because they were designed to avoid missing atypical ALSP cases (8). Genetic testing for confirmation of a CSF1R mutation is required for a definitive diagnosis, but limited accessibility to genetic testing may delay diagnosis. Another diagnostic model for ALSP has been evaluated, but its applicability is limited by a restrictive neuroimaging and neuropathologic approach (10).
Because no regulatory-approved disease-modifying therapies are currently available for ALSP (3), rapid and accurate diagnosis of ALSP is critical for initiating clinical care as well as for the design of future studies to test experimental and interventional therapies. Thus, the objective of this retrospective literature analysis was to leverage a comprehensive review of published cases from the peer-reviewed literature (3) to examine the frequency of misdiagnosis among a cohort of patients with ALSP. Discussion of therapies that are commonly used to temporarily relieve the motor, mood, and behavior symptoms of ALSP as well as experimental approaches, such as hematopoietic stem cell transplantation and others, are outside the scope of this current work but are covered in detail elsewhere in the literature (3, 4, 11).
A cohort of patients with ALSP was collated from individual case reports and series identified by a literature search. Limited findings from this analysis have been reported elsewhere (3), but this is the first report of the full methodology.
ALSP case series and case reports were identified via a PubMed search using the following search terms: “adult-onset leukodystrophy with neuroaxonal spheroids and pigmented glia,” “adult-onset leukoencephalopathy with axonal spheroids and pigmented glia,” “ALSP,” “hereditary diffuse leukoencephalopathy with spheroids,” “HDLS,” “pigmentary orthochromatic leukodystrophy,” “POLD,” and “CSF1R-related leukoencephalopathy.” Search results were limited to English-language articles published between January 1, 1980, and March 22, 2022. Abstracts of the identified articles were manually curated for eligible publications that clearly delineated clinical details in case reports and case series, after which the remaining publications were reviewed in detail by 4 reviewers (employees of and consultants to Vigil Neuroscience, Inc.) to confirm eligibility.
Articles eligible for inclusion in this analysis were case series and case reports with clearly delineated clinical details of adults aged ≥ 18 years (living or deceased) with a diagnosis of ALSP, pigmentary orthochromatic leukodystrophy, or hereditary diffuse leukoencephalopathy with spheroids (HDLS) confirmed by genetic testing for the CSF1R mutation, brain imaging, and/or brain histopathology. Publications that did not have adequate clinical data, included data from non-case studies, or that confirmed the presence of mutation in the alanyl-tRNA synthetase gene (AARS) via genetic testing were excluded. Among the excluded cases were members from the Swedish family in which HDLS was originally identified, since genetic testing has recently shown the likely etiological cause in affected members of this family to be a genetic variant of AARS, rather than CSF1R (12).
Individual patient data were extracted from a cohort of 291 patients with ALSP from 93 published case reports and series. The reviewers entered the data into a master spreadsheet under the appropriate demographic and clinical characteristic headings. Prior to statistical analysis, all extracted data were independently reviewed and examined for accuracy. Disputes regarding data accuracy were resolved through consultation and consensus among reviewers. Data from the same patient in multiple case reports were extracted and entered only once into the master spreadsheet to avoid duplication.
Categorical patient information collected from each case report included the following: demographics (sex, family history, geographic region), CSF1R mutation and its corresponding exon position (intron, within exons 18–21, outside of exons 18–21) and protein region (signal peptide, immunoglobulin-like domain, juxtamembrane domain, tyrosine kinase domain 1, kinase insertion domain, tyrosine kinase domain 2, carboxyl-terminal domain), initial diagnosis category (ALSP, frontotemporal dementia, multiple sclerosis, cardiovascular disease familial leukoencephalopathy, adult-onset leukodystrophy, Alzheimer’s disease, nonspecific neurodegeneration or dementia; Supplementary Table 1), clinical symptoms (cognitive impairment, pyramidal motor abnormalities, extrapyramidal motor abnormalities, behavioral and psychiatric dysfunction, speech dysfunction) at presentation and during progression, abnormal brain imaging (atrophy, corpus callosum abnormalities, ventricular enlargement, white matter hyperintensities, white matter calcification), concurrent medications, and clinical outcome assessment scores. Ages of onset and death, disease duration if alive, and survival time (years) were collected as numerical (continuous) variables. Concurrent medications and clinical outcome assessments were not analyzed due to a paucity of data.
Analyses were descriptive for continuous data (mean, standard deviation [SD], median, range) and categorical data (frequency, percentage). Categorical data for demographics, symptoms, and brain imaging were also stratified by initial diagnosis category. An analysis of variance model was used to evaluate whether initial diagnosis category varied based on the frequency distribution between categorical variables for patient demographics, exon position of CSF1R mutation, protein region of CSF1R mutation, symptoms, or brain imaging. Fisher’s exact test or Chi-square testing was used to test for statistical significance at an α level of 0.05 and marginal significance at a level of 0.10.
Data for 291 patients with a confirmed diagnosis of ALSP were extracted from 93 eligible published case reports (1, 2, 11, 13–102). Of these, only 14 cases from 7 articles (39, 58, 64, 79, 85, 86, 94) were characterized prior to the identification of CSF1R mutation as the genetic basis of ALSP (published in 2011) (72).
Overall, the mean (SD) age of ALSP onset was 43.2 (11.6) years, cases were approximately evenly divided between females (48.1%) and males (42.6%), and a family history of ALSP was reported for more than half (59.1%) (Table 1). Approximately one-third of cases in this analysis were from Asia (36.1%), Europe (32.6%), and North America (24.4%). CSF1R mutations were most often located within exons 18–21 (55.7%) compared with elsewhere (outside of exons 18–21, 24.1%; intronic, 7.9%) and most mapped to either tyrosine kinase domain 2 (67.7%) or domain 1 (15.1%) of the CSF1R protein. The most frequently reported initial symptoms were cognitive impairment (47.1%) and behavioral and psychiatric abnormalities (26.8%), followed by extrapyramidal motor symptoms (16.2%), pyramidal motor dysfunction (11.7%), and speech difficulty (11.3%).
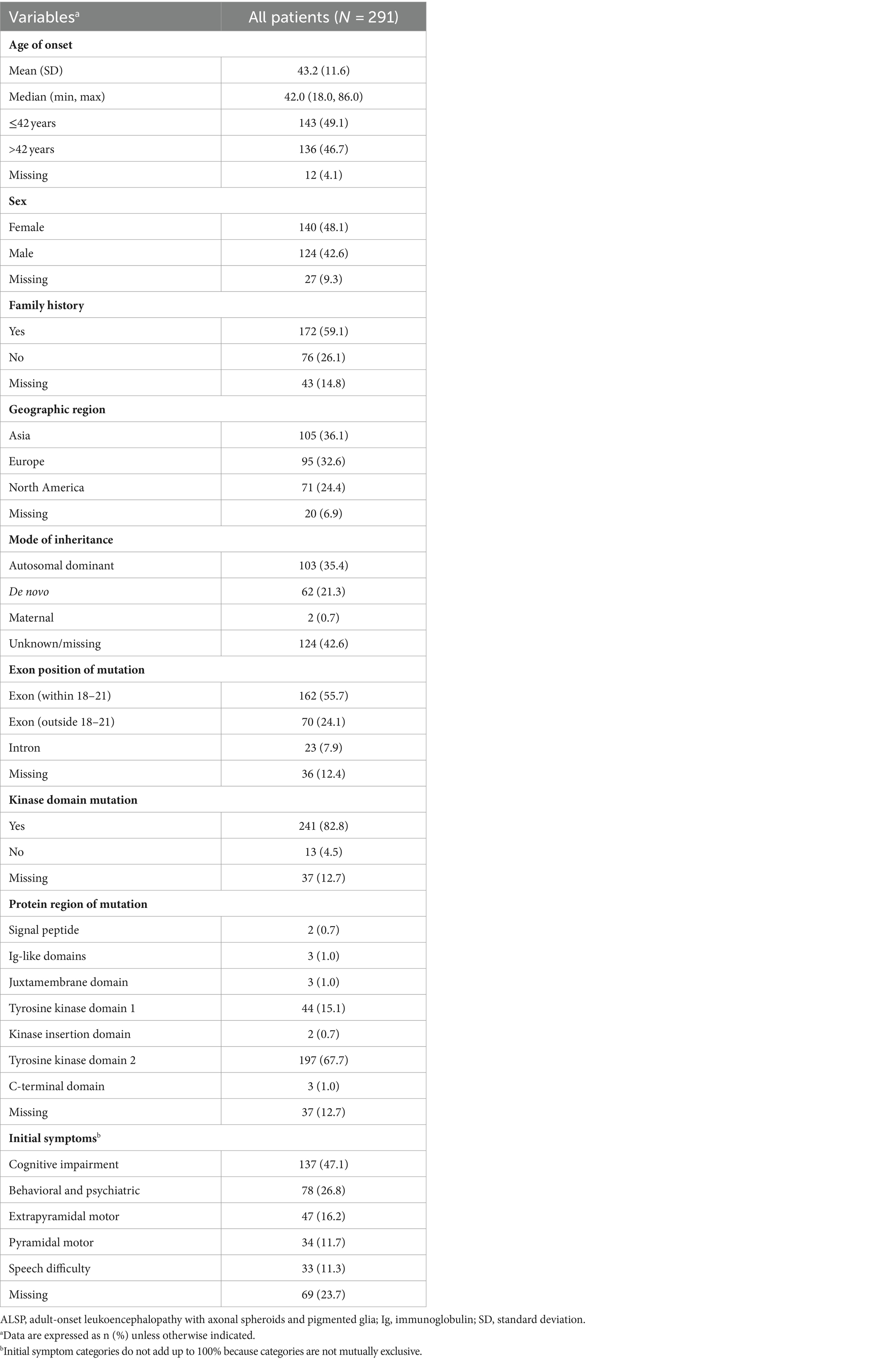
Table 1. Demographics and initial symptoms of patients with ALSP.
Table 2 shows the distribution of initial diagnoses within this cohort. Within the cohort of 291 patients, 77 cases (26.5%) did not report an initial diagnosis and only 72 cases (24.7%) received an accurate initial diagnosis of ALSP. Initial misdiagnosis with several other neurodegenerative and white matter diseases was common, including frontotemporal dementia (9.6%), multiple sclerosis (7.2%), cerebrovascular disease (3.1%), familial leukoencephalopathy (2.7%), Alzheimer’s disease (2.4%), adult-onset leukodystrophy (1.7%), and other miscellaneous disorders (7.2%). In addition, 14.8% of cases were diagnosed nonspecifically with multiple disorders or using broader phenotypic terms (e.g., “leukodystrophy,” “leukoencephalopathy,” or “dementia”). Of the 219 cases (75.3%) that were initially mis- or undiagnosed (Figure 1), 206 cases (94.1%) were later confirmed as ALSP by immunohistology, imaging, and/or genetic testing, whereas a final diagnosis was not reported for 13 cases (5.9%). Of the 206 ALSP-confirmed cases, only 13 cases were confirmed without genetic testing, 11 of which were reported prior to the introduction of the genetic test.
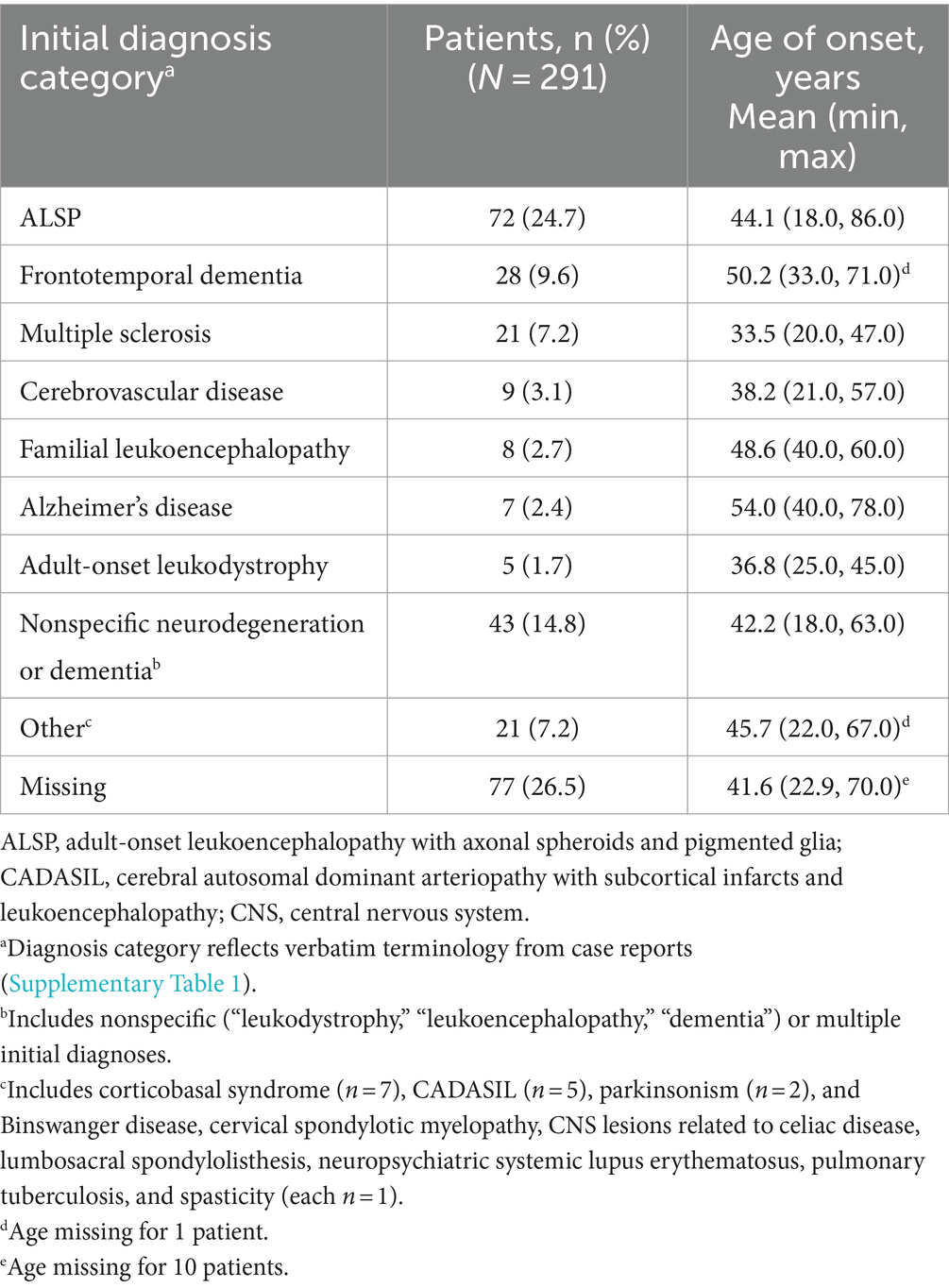
Table 2. Initial diagnosis categories of patients.
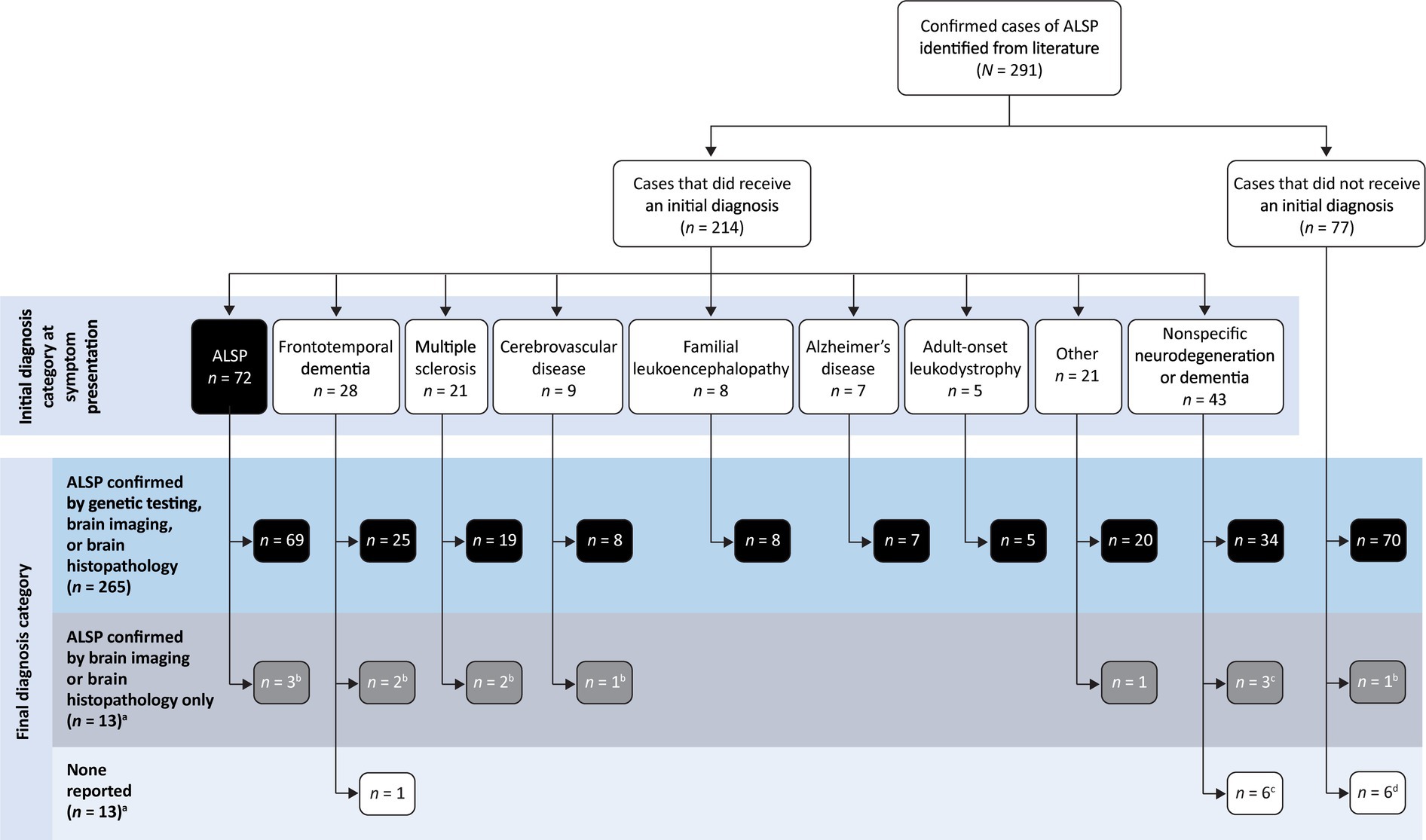
Figure 1. Summary of initial and final diagnosis categories. a14 cases from 7 publications were published prior to the identification of CSF1R mutation as the genetic basis of ALSP. bAll, c2, or d1 cases published prior to the identification of CSF1R mutation as the genetic basis of ALSP.
Analysis of the influence of initial diagnosis category on demographic and disease characteristics is shown in Table 3. Initial diagnosis categories varied significantly based on age (< 42 vs. ≥ 42; p < 0.001), with initial diagnoses categorized as Alzheimer’s disease and frontotemporal dementia tending toward older ages (mean age ≥ 50 years) and as multiple sclerosis tending toward younger ages (mean age < 40 years). The initial diagnosis category also significantly differed based on having a family history of ALSP (p = 0.046); higher proportions of patients with a family history of ALSP received an initial diagnosis within the categories of familial leukoencephalopathy (87.5%) and cerebrovascular disease (77.8%) compared with multiple sclerosis (47.6%) and adult-onset leukoencephalopathy (20.0%). Initial diagnosis category was also significantly associated with geographical region (p < 0.001), mode of inheritance (p < 0.001), and presenting symptoms of pyramidal (p = 0.027) or extrapyramidal (p = 0.020) motor dysfunction, behavioral and psychiatric abnormalities (p = 0.047), cognitive impairment (p = 0.007), and speech difficulty (p = 0.003). Among the 34 cases with initial diagnoses categorized as Alzheimer’s disease or frontotemporal dementia, none were reported to have pyramidal motor symptoms and only 1 case had extrapyramidal motor dysfunction, compared with higher proportions of patients with motor symptoms (9.5–30.2%) across the other initial diagnosis categories. Behavioral and psychiatric abnormalities and cognitive impairment were each observed in only 1 patient with an initial diagnosis in the cerebrovascular disease category compared with higher proportions of patients across the other initial diagnosis categories (behavioral and psychiatric abnormalities, 12.5–52.4%; cognitive impairment, 41.9–87.5%). Initial diagnosis did not vary based on sex or location of CSF1R mutation (exon number, kinase domain, or protein region).
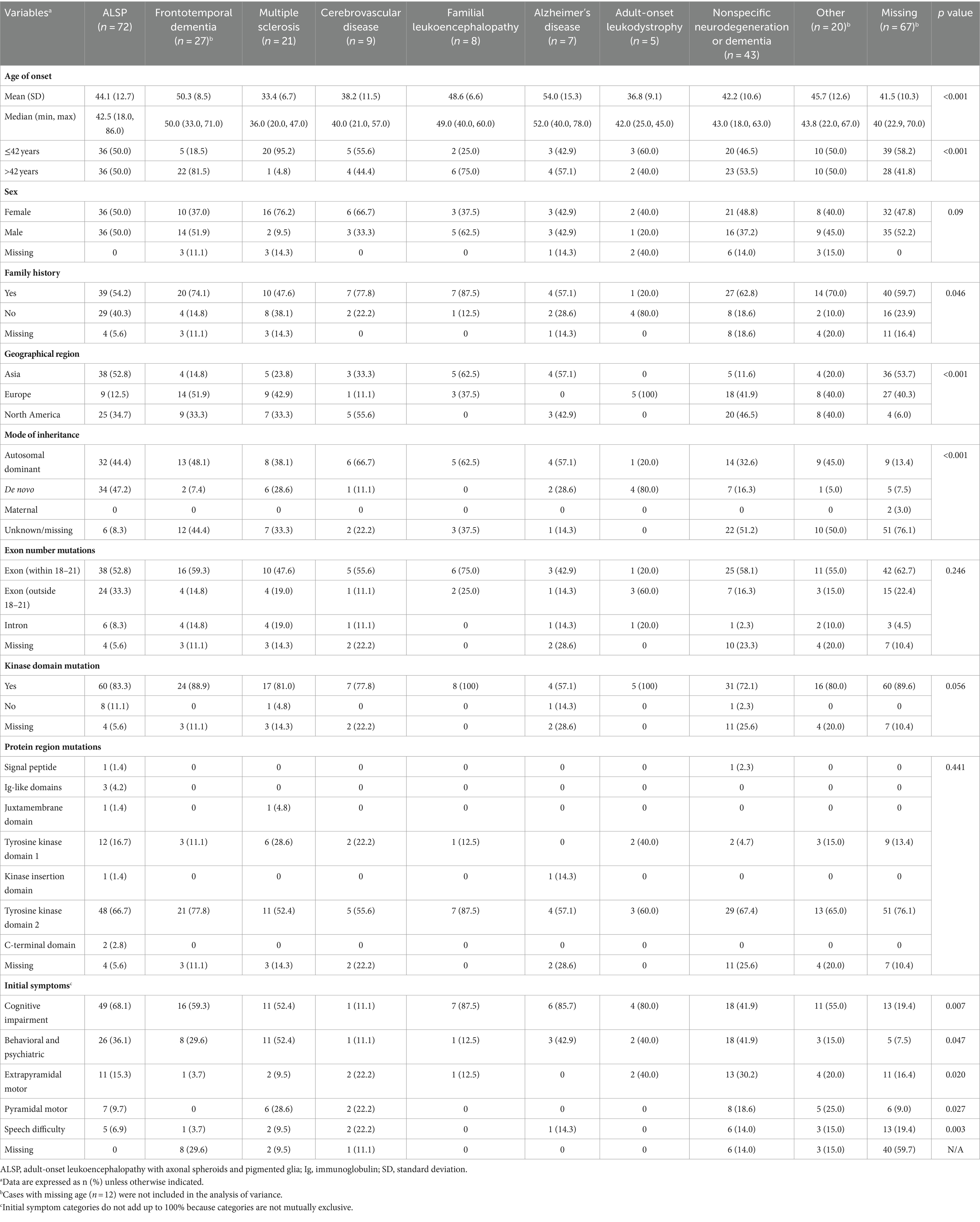
Table 3. Demographics analyzed by initial diagnosis category.
Table 4 presents brain imaging abnormalities of patients stratified by all initial diagnosis categories. Initial diagnosis category varied significantly based on white matter hyperintensities, white matter calcification, and ventricular enlargement. No brain imaging abnormalities were reported for any cases with an initial diagnosis category of familial leukoencephalopathy, and those initially diagnosed with ALSP had the highest (corpus callosum irregularities, 44.4% vs. 0–40.0%) or second highest (white matter hyperintensities, 91.7% vs. 0–100%; white matter calcification, 38.9% vs. 0–44.4%; ventricular enlargement, 27.8% vs. 0–28.6%) proportions of brain imaging irregularities compared with the other initial diagnosis categories.
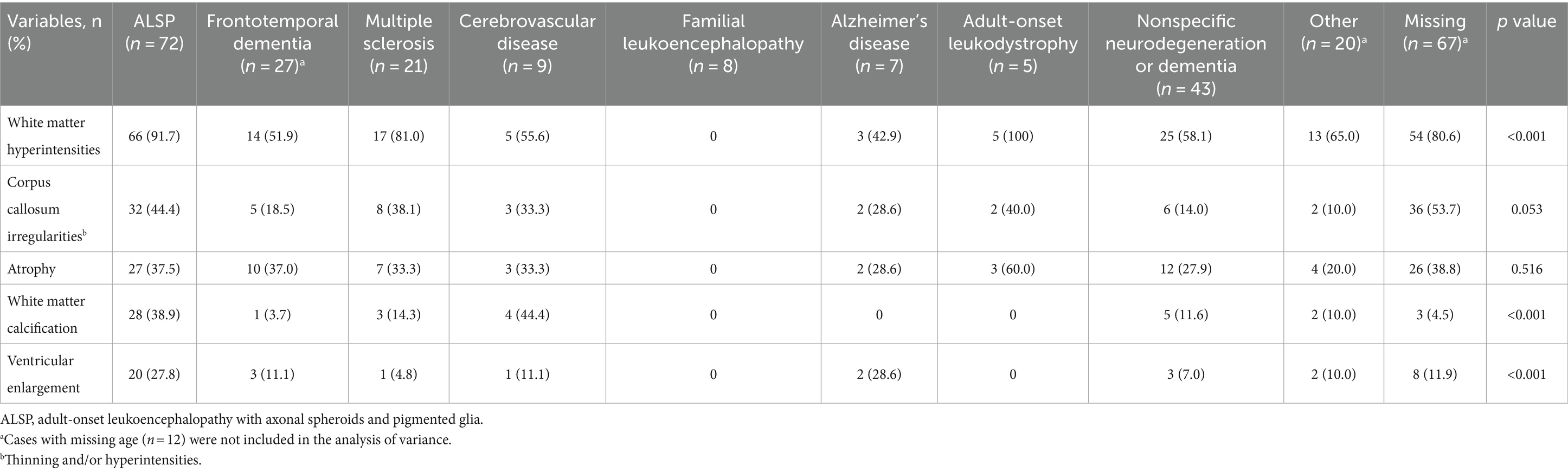
Table 4. Brain imaging abnormalities analyzed by initial diagnosis category.
To our knowledge, the findings of this analysis of published cases represent the largest case series to date of patients with ALSP. Initial misdiagnosis of ALSP was common, with an accurate initial diagnosis achieved in only 24.7% of patients with ALSP. This is likely due in part to symptoms that overlap with other disorders, including early-onset Alzheimer’s disease (executive functioning, memory, language, and personality changes), frontotemporal dementia (problems with social behavior, personality, and language), familial leukoencephalopathy (behavioral or cognitive decline), multiple sclerosis (cognitive problems, language problems, or motor neuron impairment), and cerebrovascular diseases (speech difficulty, confusion, and memory derangement) (5, 103–107).
A rapid, accurate diagnosis of ALSP is critical in providing supportive symptom management and potentially allowing for early therapeutic intervention. Definitive diagnosis of ALSP can be confirmed with genetic testing for pathogenic CSF1R mutations in the clinical context of characteristic symptoms (cognitive impairment, moderate to severe motor dysfunction, and neuropsychiatric symptoms), typical brain imaging abnormalities, and in some cases with a supportive family history (as a subset of patients have de novo patterns of inheritance) (3, 7). Although genetic testing is crucial for a definitive diagnosis of ALSP, use in clinical practice may be limited due to cost and availability (7).
Examination of categorical data for patients stratified by initial diagnosis category suggested that age, family history, geographic region, and mode of inheritance were associated with the selection of initial diagnosis. Similarly, initial diagnoses appear to have been influenced by presenting symptoms such as pyramidal or extrapyramidal motor dysfunction, behavioral and psychiatric abnormalities, cognitive impairment, and speech difficulty. Stratification of brain imaging abnormalities by initial diagnosis category also demonstrated statistically significant associations between initial diagnosis category and the presence of white matter hyperintensities, ventricular enlargement, white matter calcification, and corpus callosum irregularities. Abnormal diffusion on brain MRI, which can persist for extended periods of time, has also been observed in patients with ALSP in recent literature (1, 7, 11, 20), but the presence of diffusion abnormalities was not systematically examined in this analysis.
These data are consistent with current evidence indicating the importance of brain imaging in accurately diagnosing ALSP (2–4). The diagnostic criteria developed for ALSP combine brain imaging with presenting characteristics such as patient age, symptoms (cognitive, psychiatric, and motor dysfunction), and inheritance patterns (autosomal dominant or sporadic) (7). Retrospective application of these criteria to more than 150 patients with ALSP and CSF1R-positive mutations, CSF1R-negative leukoencephalopathies, or CADASIL showed a sensitivity of 99% to accurately detect probable or possible cases and an adequate specificity of 88% to exclude non-ALSP cases (7).
This retrospective literature analysis is subject to several limitations. The development of this cohort and associated individual patient data was based entirely upon published case reports and case series, which are contingent upon the reporting standards associated with original patient medical records; thus, potential errors or omissions in the clinical descriptions may have resulted in inaccurate, incomplete, or missing clinical assessments of some symptoms of ALSP. Further, the geographic locations of the authors and clinics providing each case report exhibited considerable variation, which may have generated heterogeneity in interpretation and reporting of clinical manifestations.
For this cohort, the initial diagnosis information extracted from the primary sources constituted an unavoidably heterogeneous dataset, with some imprecision in reporting in the published literature, that necessitated application of discrete categories for interpretation; this analysis is limited by the subjective nature and potential for overlap inherent in such categorization. This analysis also reflects the state of the ALSP literature over this time period. Initial diagnosis data were missing in 26.5% of cases, and the available data for another 14.8% of cases necessitated a judgment be made as to whether nonspecific or multiple initial diagnoses (i.e., “leukodystrophy” or “Alzheimer’s disease, atypical CADASIL, or atypical multiple sclerosis”) should be categorized as true misdiagnoses or as broad tentative phenotypic diagnoses that should be considered a correct initial diagnosis that was given with the intent for later revision. Furthermore, a handful of cases were reported prior to the introduction of genetic testing for a CSF1R mutation that is now necessary to confirm an ALSP diagnosis, which may also have added an additional, albeit small, element of uncertainty.
An analysis of the literature often includes measures to assess the risk of reporting bias arising from study design, conduct, or analyses; however, the individual patient-level data that comprise this dataset are derived from case series and case reports. Therefore, assessments of bias in study design, conduct, or analyses are not applicable to this cohort. Because the sampling procedure for this cohort was limited to cases published in peer-reviewed journals, these results are inherently subject to publication bias. Not all clinical cases are necessarily or systematically published in the peer-reviewed literature, and published case reports may sometimes tend to highlight unique or “interesting” clinical features. For these reasons, a cohort based entirely of published case reports may not represent the clinical experience of all patients living with ALSP, and the estimates obtained from this cohort may not be fully generalizable to all patients living with this disease. Absolute rates of ALSP misdiagnosis could differ from that reported in this analysis, and future comparisons of this cohort to new patient cases and/or clinical trial data may be necessary for broader generalizability.
In conclusion, despite the application of diagnostic criteria to distinguish ALSP from other disorders that present similarly and the development of a genetic test for the CSF1R mutation, diagnosis of ALSP remains challenging. Previous studies have estimated a prevalence of at least 10,000 cases of ALSP in the United States (61, 108, 109); however, only a small number of patients with confirmed ALSP have been currently identified (3, 109), which may be due, at least in part, to the high rate of initial misdiagnosis. Therefore, increased awareness of ALSP and further investigation and characterization into its presenting symptoms are needed to improve diagnostic accuracy of this debilitating disorder.
This review of published ALSP case reports was registered in the Research Registry Database under unique identification number 1251.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
This study was exempt from requiring ethics approval or written informed consent because it is a retrospective analysis of previously collected and published data.
SP: Conceptualization, Methodology, Data curation, Writing – review & editing, Investigation. JG: Writing – review & editing, Investigation. TK: Writing – review & editing, Investigation. TI: Writing – review & editing, Investigation. AP: Writing – review & editing, Data curation. AM: Writing – review & editing, Data curation, Investigation. FF: Formal analysis, Writing – review & editing, Investigation. ZW: Writing – review & editing, Investigation, Data curation.
The authors declare financial support was received for the research, writing, and/or publication of this article. This study was supported by Vigil Neuroscience, Inc.
Leslie Leahy, PhD, a publication consultant to Vigil Neuroscience, Inc., assisted with organization and review of the manuscript. Medical writing and editorial support were provided by Morgan C. Hill, PhD, CMPP, and Melissa Austin of Apollo Medical Communications, part of Helios Global Group, and funded by Vigil Neuroscience, Inc.
SP and AP are former full-time employees of Vigil Neuroscience, Inc. JG receives research support to his institution from Vigil Neuroscience, Inc. for clinical trials, from Genentech/Roche for clinical trials, and personal fees for consulting from Arialys. TK and TI are funded by AMED JP23dk0207060, a public grant from the Japanese government to support research on ALSP. FF is a consultant for Vigil Neuroscience, Inc. ZW is partially supported by the NIH/NIA and NIH/NINDS (1U19AG063911, FAIN: U19AG063911), Mayo Clinic Center for Regenerative Medicine, gifts from the Donald G. and Jodi P. Heeringa Family, the Haworth Family Professorship in Neurodegenerative Diseases fund, and the Albertson Parkinson’s Research Foundation; he serves as PI or Co-PI on Biohaven Pharmaceuticals, Inc. (BHV4157-206) and Vigil Neuroscience, Inc. (VGL101-01.002, VGL101-01.201, PET tracer development protocol, CSF1R biomarker and repository project, and ultra-high field MRI in the diagnosis and management of CSF1R-related adult-onset leukoencephalopathy with axonal spheroids and pigmented glia) projects/grants; he serves as Co-PI of the Mayo Clinic APDA Center for Advanced Research, as an external advisory board member for Vigil Neuroscience, Inc., and as a consultant on neurodegenerative medical research for Eli Lilly and Company. AM is a full-time employee of Vigil Neuroscience, Inc.
This study received funding from Vigil Neuroscience, Inc. The funder had the following involvement with the study: study design; data collection, analysis, and interpretation; and drafting, revising, and submitting this article for publication.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2024.1320663/full#supplementary-material
1. Konno, T, Yoshida, K, Mizuno, T, Kawarai, T, Tada, M, Nozaki, H, et al. Clinical and genetic characterization of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation. Eur J Neurol. (2017) 24:37–45. doi: 10.1111/ene.13125
2. Sundal, C, Van Gerpen, JA, Nicholson, AM, Wider, C, Shuster, EA, Aasly, J, et al. MRI characteristics and scoring in HDLS due to CSF1R gene mutations. Neurology. (2012) 79:566–74. doi: 10.1212/WNL.0b013e318263575a
3. Papapetropoulos, S, Pontius, A, Finger, E, Karrenbauer, V, Lynch, DS, Brennan, M, et al. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: review of clinical manifestations as foundations for therapeutic development. Front Neurol. (2022) 12:788168. doi: 10.3389/fneur.2021.788168
4. Sundal, C, and Wszolek, ZK. CSF1R-related adult-onset leukoencephalopathy with axonal spheroids and pigmented glia In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al., editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (2017). 1993–2023.
5. NORD. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. (2021). Available at: https://rarediseases.org/rare-diseases/adult-onset-leukoencephalopathy-with-axonal-spheroids-and-pigmented-glia/ (Accessed January 23, 2024)
6. Lynch, DS, Wade, C, Paiva, ARB, John, N, Kinsella, JA, Merwick, A, et al. Practical approach to the diagnosis of adult-onset leukodystrophies: an updated guide in the genomic era. J Neurol Neurosurg Psychiatry. (2019) 90:543–54. doi: 10.1136/jnnp-2018-319481
7. Konno, T, Yoshida, K, Mizuta, I, Mizuno, T, Kawarai, T, Tada, M, et al. Diagnostic criteria for adult-onset leukoencephalopathy with axonal spheroids and pigmented glia due to CSF1R mutation. Eur J Neurol. (2018) 25:142–7. doi: 10.1111/ene.13464
8. Kondo, Y, Matsushima, A, Nagasaki, S, Nakamura, K, Sekijima, Y, and Yoshida, K. Factors predictive of the presence of a CSF1R mutation in patients with leukoencephalopathy. Eur J Neurol. (2020) 27:369–75. doi: 10.1111/ene.14086
9. Ayrignac, X, Carra-Dalliere, C, Codjia, P, Mouzat, K, Castelnovo, G, Ellie, E, et al. Evaluation of CSF1R-related adult onset leukoencephalopathy with axonal spheroids and pigmented glia diagnostic criteria. Eur J Neurol. (2022) 29:329–34. doi: 10.1111/ene.15115
10. Mao, C, Zhou, L, Zhou, L, Yang, Y, Niu, J, Li, J, et al. Biopsy histopathology in the diagnosis of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP). Neurol Sci. (2020) 41:403–9. doi: 10.1007/s10072-019-04116-7
11. Konno, T, Kasanuki, K, Ikeuchi, T, Dickson, DW, and Wszolek, ZK. CSF1R-related leukoencephalopathy: a major player in primary microgliopathies. Neurology. (2018) 91:1092–104. doi: 10.1212/WNL.0000000000006642
12. Sundal, C, Carmona, S, Yhr, M, Almstrom, O, Ljungberg, M, Hardy, J, et al. An AARS variant as the likely cause of Swedish type hereditary diffuse leukoencephalopathy with spheroids. Acta Neuropathol Commun. (2019) 7:188. doi: 10.1186/s40478-019-0843-y
13. Adams, SJ, Kirk, A, and Auer, RN. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP): integrating the literature on hereditary diffuse leukoencephalopathy with spheroids (HDLS) and pigmentary orthochromatic leukodystrophy (POLD). J Clin Neurosci. (2018) 48:42–9. doi: 10.1016/j.jocn.2017.10.060
14. Ahmed, R, Guerreiro, R, Rohrer, JD, Guven, G, Rossor, MN, Hardy, J, et al. A novel A781V mutation in the CSF1R gene causes hereditary diffuse leucoencephalopathy with axonal spheroids. J Neurol Sci. (2013) 332:141–4. doi: 10.1016/j.jns.2013.06.007
15. Alturkustani, M, Keith, J, Hazrati, LN, Rademakers, R, and Ang, LC. Pathologic staging of white matter lesions in adult-onset leukoencephalopathy/leukodystrophy with axonal spheroids. J Neuropathol Exp Neurol. (2015) 74:233–40. doi: 10.1097/NEN.0000000000000168
16. Arshad, F, Vengalil, S, Maskomani, S, Kamath, SD, Kulanthaivelu, K, Mundlamuri, RC, et al. Novel CSF1R variant in adult-onset leukoencephalopathy masquerading as frontotemporal dementia: a follow-up study. Neurocase. (2021) 27:484–9. doi: 10.1080/13554794.2021.2022704
17. Ayrignac, X, Carra-Dalliere, C, Menjot de Champfleur, N, Denier, C, Aubourg, P, Bellesme, C, et al. Adult-onset genetic leukoencephalopathies: a MRI pattern-based approach in a comprehensive study of 154 patients. Brain. (2015) 138:284–92. doi: 10.1093/brain/awu353
18. Bai, Y, Lu, L, Cui, Y, Li, J, Liu, Y, Liu, L, et al. Analysis of clinical and neuroimaging features in a Chinese family with hereditary diffuse leukoencephalopathy with neuroaxonal spheroids. Chin J Neurol. (2018) 51:877–81. doi: 10.3760/cma.j.issn.1006-7876.2018.11.004
19. Battisti, C, Di Donato, I, Bianchi, S, Monti, L, Formichi, P, Rufa, A, et al. Hereditary diffuse leukoencephalopathy with axonal spheroids: three patients with stroke-like presentation carrying new mutations in the CSF1R gene. J Neurol. (2014) 261:768–72. doi: 10.1007/s00415-014-7257-3
20. Bender, B, Klose, U, Lindig, T, Biskup, S, Nägele, T, Schöls, L, et al. Imaging features in conventional MRI, spectroscopy and diffusion weighted images of hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS). J Neurol. (2014) 261:2351–9. doi: 10.1007/s00415-014-7509-2
21. Chen, J, Luo, S, Li, N, Li, H, Han, J, and Ling, L. A novel missense mutation of the CSF1R gene causes incurable CSF1R-related leukoencephalopathy: case report and review of literature. Int J Gen Med. (2020) 13:1613–20. doi: 10.2147/IJGM.S286421
22. Cheng, X, Shen, W, Zou, H, Shen, L, Gu, X, Huang, D, et al. Analysis of CSF1R gene mutation in a Chinese family with hereditary diffuse leukoencephalopathy with neuroaxonal spheroids. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2015) 32:208–12. doi: 10.3760/cma.j.issn.1003-9406.2015.02.012
23. Chu, M, Wang, DX, Cui, Y, Kong, Y, Liu, L, Xie, KX, et al. Three novel mutations in Chinese patients with CSF1R-related leukoencephalopathy. Ann Transl Med. (2021) 9:1072. doi: 10.21037/atm-21-217
24. Codjia, P, Ayrignac, X, Mochel, F, Mouzat, K, Carra-Dalliere, C, Castelnovo, G, et al. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: an MRI study of 16 French cases. AJNR Am J Neuroradiol. (2018) 39:1657–61. doi: 10.3174/ajnr.A5744
25. Daida, K, Nishioka, K, Li, Y, Nakajima, S, Tanaka, R, and Hattori, N. CSF1R mutation p.G589R and the distribution pattern of brain calcification. Intern Med. (2017) 56:2507–12. doi: 10.2169/internalmedicine.8462-16
26. Di Donato, I, Stabile, C, Bianchi, S, Taglia, I, Mignarri, A, Salvatore, S, et al. A novel CSF1R mutation in a patient with clinical and neuroradiological features of hereditary diffuse leukoencephalopathy with axonal spheroids. J Alzheimers Dis. (2015) 47:319–22. doi: 10.3233/JAD-150097
27. Du, Q, Chen, H, Shi, Z, Zhang, Y, Wang, J, and Zhou, H. A novel mutation in the CSF1R gene causes hereditary diffuse leukoencephalopathy with axonal spheroids. Neurol Sci. (2019) 40:1287–90. doi: 10.1007/s10072-018-3693-7
28. Eichler, FS, Li, J, Guo, Y, Caruso, PA, Bjonnes, AC, Pan, J, et al. CSF1R mosaicism in a family with hereditary diffuse leukoencephalopathy with spheroids. Brain. (2016) 139:1666–72. doi: 10.1093/brain/aww066
29. Foulds, N, Pengelly, RJ, Hammans, SR, Nicoll, JA, Ellison, DW, Ditchfield, A, et al. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia caused by a novel R782G mutation in CSF1R. Sci Rep. (2015) 5:10042. doi: 10.1038/srep10042
30. Fujioka, S, Broderick, DF, Sundal, C, Baker, MC, Rademakers, R, and Wszolek, ZK. An adult-onset leukoencephalopathy with axonal spheroids and pigmented glia accompanied by brain calcifications: a case report and a literature review of brain calcifications disorders. J Neurol. (2013) 260:2665–8. doi: 10.1007/s00415-013-7093-x
31. Gelfand, JM, Greenfield, AL, Barkovich, M, Mendelsohn, BA, Van Haren, K, Hess, CP, et al. Allogeneic HSCT for adult-onset leukoencephalopathy with spheroids and pigmented glia. Brain. (2020) 143:503–11. doi: 10.1093/brain/awz390
32. Gore, E, Manley, A, Dees, D, Appleby, BS, and Lerner, AJ. A young-onset frontal dementia with dramatic calcifications due to a novel CSF1R mutation. Neurocase. (2016) 22:257–62. doi: 10.1080/13554794.2016.1175635
33. Granberg, T, Hashim, F, Andersen, O, Sundal, C, and Karrenbauer, VD. Hereditary diffuse leukoencephalopathy with spheroids - a volumetric and radiological comparison with multiple sclerosis patients and healthy controls. Eur J Neurol. (2016) 23:817–22. doi: 10.1111/ene.12948
34. Guerreiro, R, Kara, E, Le Ber, I, Bras, J, Rohrer, JD, Taipa, R, et al. Genetic analysis of inherited leukodystrophies: genotype-phenotype correlations in the CSF1R gene. JAMA Neurol. (2013) 70:875–82. doi: 10.1001/jamaneurol.2013.698
35. Ho, VM, Hovsepian, DA, and Shieh, PB. Myelopathy in a patient with leukodystrophy due to CSF1R mutation. Neurol Genet. (2019) 5:e376. doi: 10.1212/NXG.0000000000000376
36. Hoffmann, S, Murrell, J, Harms, L, Miller, K, Meisel, A, Brosch, T, et al. Enlarging the nosological spectrum of hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS). Brain Pathol. (2014) 24:452–8. doi: 10.1111/bpa.12120
37. Huang, H, Cao, L, and Chen, H. Dynamic analysis of CSF1R-related leukoencephalopathy on magnetic resonance imaging: a case report. BMC Neurol. (2021) 21:156. doi: 10.1186/s12883-021-02182-z
38. Inui, T, Kawarai, T, Fujita, K, Kawamura, K, Mitsui, T, Orlacchio, A, et al. A new CSF1R mutation presenting with an extensive white matter lesion mimicking primary progressive multiple sclerosis. J Neurol Sci. (2013) 334:192–5. doi: 10.1016/j.jns.2013.08.020
39. Itoh, K, Shiga, K, Shimizu, K, Muranishi, M, Nakagawa, M, and Fushiki, S. Autosomal dominant leukodystrophy with axonal spheroids and pigmented glia: clinical and neuropathological characteristics. Acta Neuropathol. (2006) 111:39–45. doi: 10.1007/s00401-005-1113-6
40. Jin, C, Washimi, Y, Yoshida, K, Hashizume, Y, and Yazawa, I. Characterization of spheroids in hereditary diffuse leukoencephalopathy with axonal spheroids. J Neurol Sci. (2015) 352:74–8. doi: 10.1016/j.jns.2015.03.033
41. Karle, KN, Biskup, S, Schüle, R, Schweitzer, KJ, Krüger, R, Bauer, P, et al. De novo mutations in hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS). Neurology. (2013) 81:2039–44. doi: 10.1212/01.wnl.0000436945.01023.ac
42. Kawakami, I, Iseki, E, Kasanuki, K, Minegishi, M, Sato, K, Hino, H, et al. A family with hereditary diffuse leukoencephalopathy with spheroids caused by a novel c.2442+2T>C mutation in the CSF1R gene. J Neurol Sci. (2016) 367:349–55. doi: 10.1016/j.jns.2016.06.013
43. Kim, EJ, Shin, JH, Lee, JH, Kim, JH, Na, DL, Suh, YL, et al. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia linked CSF1R mutation: report of four Korean cases. J Neurol Sci. (2015) 349:232–8. doi: 10.1016/j.jns.2014.12.021
44. Kim, M, Lee, H, Cho, HJ, Young Chun, S, Shin, JH, Kim, EJ, et al. Pathologic correlation of paramagnetic white matter lesions in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. J Neuropathol Exp Neurol. (2017) 76:924–8. doi: 10.1093/jnen/nlx086
45. Kinoshita, M, Yoshida, K, Oyanagi, K, Hashimoto, T, and Ikeda, S. Hereditary diffuse leukoencephalopathy with axonal spheroids caused by R782H mutation in CSF1R: case report. J Neurol Sci. (2012) 318:115–8. doi: 10.1016/j.jns.2012.03.012
46. Kitani-Morii, F, Kasai, T, Tomonaga, K, Saito, K, Mizuta, I, Yoshioka, A, et al. Hereditary diffuse leukoencephalopathy with spheroids characterized by spastic hemiplegia preceding mental impairment. Intern Med. (2014) 53:1377–80. doi: 10.2169/internalmedicine.53.1932
47. Kleinfeld, K, Mobley, B, Hedera, P, Wegner, A, Sriram, S, and Pawate, S. Adult-onset leukoencephalopathy with neuroaxonal spheroids and pigmented glia: report of five cases and a new mutation. J Neurol. (2013) 260:558–71. doi: 10.1007/s00415-012-6680-6
48. Koga, S, Tipton, PW, Wierenga, KJ, Dickson, DW, and Wszolek, ZK. Neuropathological findings of CSF1R-related leukoencephalopathy after long-term immunosuppressive therapy. Mov Disord. (2022) 37:439–40. doi: 10.1002/mds.28919
49. Kondo, Y, Kinoshita, M, Fukushima, K, Yoshida, K, and Ikeda, S. Early involvement of the corpus callosum in a patient with hereditary diffuse leukoencephalopathy with spheroids carrying the de novo K793T mutation of CSF1R. Intern Med. (2013) 52:503–6. doi: 10.2169/internalmedicine.52.8879
50. Konno, T, Broderick, DF, Mezaki, N, Isami, A, Kaneda, D, Tashiro, Y, et al. Diagnostic value of brain calcifications in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. AJNR Am J Neuroradiol. (2017) 38:77–83. doi: 10.3174/ajnr.A4938
51. Konno, T, Tada, M, Tada, M, Koyama, A, Nozaki, H, Harigaya, Y, et al. Haploinsufficiency of CSF-1R and clinicopathologic characterization in patients with HDLS. Neurology. (2014) 82:139–48. doi: 10.1212/WNL.0000000000000046
52. Kortvelyessy, P, Krageloh-Mann, I, Mawrin, C, Heinze, HJ, Bittner, D, Wieland, I, et al. Hereditary diffuse leukoencephalopathy with spheroids (HDLS) with a novel CSF1R mutation and spinal cord involvement. J Neurol Sci. (2015) 358:515–7. doi: 10.1016/j.jns.2015.09.370
53. Kraya, T, Quandt, D, Pfirrmann, T, Kindermann, A, Lampe, L, Schroeter, ML, et al. Functional characterization of a novel CSF1R mutation causing hereditary diffuse leukoencephalopathy with spheroids. Mol Genet Genomic Med. (2019) 7:e00595. doi: 10.1002/mgg3.595
54. La Piana, R, Webber, A, Guiot, MC, Del Pilar, CM, and Brais, B. A novel mutation in the CSF1R gene causes a variable leukoencephalopathy with spheroids. Neurogenetics. (2014) 15:289–94. doi: 10.1007/s10048-014-0413-1
55. Labauge, P, Berger, E, Magnin, E, Rumbach, L, Mine, M, and Renard, D. A new form of leukoencephalopathy with calcifications and cysts with nonrecessive inheritance and absence of gadolinium enhancement. Eur Neurol. (2012) 67:151–3. doi: 10.1159/000331937
56. Lan, MY, Liu, JS, Chang, CC, Chen, YF, Su, CS, Peng, CH, et al. Clinicopathologic and genetic studies of 2 patients with hereditary diffuse leukoencephalopathy with axonal spheroids. Alzheimer Dis Assoc Disord. (2016) 30:73–6. doi: 10.1097/WAD.0000000000000067
57. Leng, C, Lu, L, Wang, G, Zhang, Y, Xu, Y, Lin, X, et al. A novel dominant-negative mutation of the CSF1R gene causes adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. Am J Transl Res. (2019) 11:6093–101.
58. Letournel, F, Etcharry-Bouyx, F, Verny, C, Barthelaix, A, and Dubas, F. Two clinicopathological cases of a dominantly inherited, adult onset orthochromatic leucodystrophy. J Neurol Neurosurg Psychiatry. (2003) 74:671–3. doi: 10.1136/jnnp.74.5.671
59. Li, S, Zhu, Y, and Yao, M. Spinal cord involvement in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. JAMA Neurol. (2020) 77:1169–70. doi: 10.1001/jamaneurol.2020.2204
60. Lopes, C, Duarte, S, Santos, E, Reis, C, and Pinto, M. Intrafamilial heterogeneity in hereditary diffuse leukoencephalopathy with axonal spheroids. Neurol Clin Pract. (2019) 9:500–2. doi: 10.1212/CPJ.0000000000000647
61. Lynch, DS, Jaunmuktane, Z, Sheerin, UM, Phadke, R, Brandner, S, Milonas, I, et al. Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series. J Neurol Neurosurg Psychiatry. (2016) 87:512–9. doi: 10.1136/jnnp-2015-310788
62. Lynch, DS, Brandao, R, de Paiva, A, Zhang, WJ, Bugiardini, E, Freua, F, et al. Clinical and genetic characterization of leukoencephalopathies in adults. Brain. (2017) 140:1204–11. doi: 10.1093/brain/awx045
63. Makary, MS, Awan, U, Kisanuki, YY, and Slone, HW. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: clinical and imaging characteristics. Neuroradiol J. (2019) 32:139–42. doi: 10.1177/1971400918822136
64. Marotti, JD, Tobias, S, Fratkin, JD, Powers, JM, and Rhodes, CH. Adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia: report of a family, historical perspective, and review of the literature. Acta Neuropathol. (2004) 107:481–8. doi: 10.1007/s00401-004-0847-x
65. Meyer-Ohlendorf, M, Braczynski, A, Al-Qaisi, O, Gessler, F, Biskup, S, Weise, L, et al. Comprehensive diagnostics in a case of hereditary diffuse leukodystrophy with spheroids. BMC Neurol. (2015) 15:103. doi: 10.1186/s12883-015-0368-3
66. Miura, T, Mezaki, N, Konno, T, Iwasaki, A, Hara, N, Miura, M, et al. Identification and functional characterization of novel mutations including frameshift mutation in exon 4 of CSF1R in patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. J Neurol. (2018) 265:2415–24. doi: 10.1007/s00415-018-9017-2
67. Mochel, F, Delorme, C, Czernecki, V, Froger, J, Cormier, F, Ellie, E, et al. Haematopoietic stem cell transplantation in CSF1R-related adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. J Neurol Neurosurg Psychiatry. (2019) 90:1375–6. doi: 10.1136/jnnp-2019-320701
68. Nicholson, AM, Baker, MC, Finch, NA, Rutherford, NJ, Wider, C, Graff-Radford, NR, et al. CSF1R mutations link POLD and HDLS as a single disease entity. Neurology. (2013) 80:1033–40. doi: 10.1212/WNL.0b013e31828726a7
69. Okamoto, M, Takeshita, J, Takahashi, K, Tanaka, A, Yoshida, K, and Kuriyama, M. Retraction: adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: a case presented brain calcification and corpus callosum atrophy from over 10 years before the onset of dementia. Rinsho Shinkeigaku. (2017) 57:521–6. doi: 10.5692/clinicalneurol.cn-001072
70. Oyanagi, K, Kinoshita, M, Suzuki-Kouyama, E, Inoue, T, Nakahara, A, Tokiwai, M, et al. Adult onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) and Nasu-Hakola disease: lesion staging and dynamic changes of axons and microglial subsets. Brain Pathol. (2017) 27:748–69. doi: 10.1111/bpa.12443
71. Prieto-Morin, C, Ayrignac, X, Ellie, E, Tournier-Lasserve, E, and Labauge, P. CSF1R-related leukoencephalopathy mimicking primary progressive multiple sclerosis. J Neurol. (2016) 263:1864–5. doi: 10.1007/s00415-016-8197-x
72. Rademakers, R, Baker, M, Nicholson, AM, Rutherford, NJ, Finch, N, Soto-Ortolaza, A, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet. (2011) 44:200–5. doi: 10.1038/ng.1027
73. Riku, Y, Ando, T, Goto, Y, Mano, K, Iwasaki, Y, Sobue, G, et al. Early pathologic changes in hereditary diffuse leukoencephalopathy with spheroids. J Neuropathol Exp Neurol. (2014) 73:1183–90. doi: 10.1097/NEN.0000000000000139
74. Robinson, JL, Suh, E, Wood, EM, Lee, EB, Coslett, HB, Raible, K, et al. Common neuropathological features underlie distinct clinical presentations in three siblings with hereditary diffuse leukoencephalopathy with spheroids caused by CSF1R p.Arg782His. Acta Neuropathol Commun. (2015) 3:42. doi: 10.1186/s40478-015-0219-x
75. Rosenstein, I, Andersen, O, Victor, D, Englund, E, Granberg, T, Hedberg-Oldfors, C, et al. Four Swedish cases of CSF1R-related leukoencephalopathy: visualization of clinical phenotypes. Acta Neurol Scand. (2022) 145:599–609. doi: 10.1111/ane.13589
76. Saitoh, BY, Yamasaki, R, Hayashi, S, Yoshimura, S, Tateishi, T, Ohyagi, Y, et al. A case of hereditary diffuse leukoencephalopathy with axonal spheroids caused by a de novo mutation in CSF1R masquerading as primary progressive multiple sclerosis. Mult Scler. (2013) 19:1367–70. doi: 10.1177/1352458513489854
77. Saitoh, BY, Yamasaki, R, Hiwatashi, A, Matsushita, T, Hayashi, S, Mitsunaga, Y, et al. Discriminative clinical and neuroimaging features of motor-predominant hereditary diffuse leukoencephalopathy with axonal spheroids and primary progressive multiple sclerosis: a preliminary cross-sectional study. Mult Scler Relat Disord. (2019) 31:22–31. doi: 10.1016/j.msard.2019.03.008
78. Schuberth, M, Levin, J, Sawalhe, D, Schwarzkopf, R, von Baumgarten, L, Ertl-Wagner, B, et al. Hereditary diffuse leukencephalopathy with spheroids: a microgliopathy due to CSF1 receptor impairment. Nervenarzt. (2014) 85:465–70. doi: 10.1007/s00115-014-4052-4
79. Shannon, P, Wherrett, JR, and Nag, S. A rare form of adult onset leukodystrophy: orthochromatic leukodystrophy with pigmented glia. Can J Neurol Sci. (1997) 24:146–50. doi: 10.1017/s0317167100021491
80. Shu, Y, Long, L, Liao, S, Yang, J, Li, J, Qiu, W, et al. Involvement of the optic nerve in mutated CSF1R-induced hereditary diffuse leukoencephalopathy with axonal spheroids. BMC Neurol. (2016) 16:171. doi: 10.1186/s12883-016-0694-0
81. Sohn, EH, Lee, J, Lee, AY, and Shin, JH. A case of CSF1R-related leukoencephalopathy: serial neuroimaging and neuropsychological tests. Neurocase. (2021) 27:415–8. doi: 10.1080/13554794.2021.1981947
82. Stoiloudis, P, Parissis, D, Smyrni, N, Stardeli, T, Afrantou, T, Konstantinopoulou, E, et al. Hereditary diffuse leukoencephalopathy with spheroids mimicking primary progressive aphasia: report of a Greek case. Neurol Sci. (2021) 42:3431–3. doi: 10.1007/s10072-021-05257-4
83. Sundal, C, Baker, M, Karrenbauer, V, Gustavsen, M, Bedri, S, Glaser, A, et al. Hereditary diffuse leukoencephalopathy with spheroids with phenotype of primary progressive multiple sclerosis. Eur J Neurol. (2015) 22:328–33. doi: 10.1111/ene.12572
84. Sundal, C, Fujioka, S, Van Gerpen, JA, Wider, C, Nicholson, AM, Baker, M, et al. Parkinsonian features in hereditary diffuse leukoencephalopathy with spheroids (HDLS) and CSF1R mutations. Parkinsonism Relat Disord. (2013) 19:869–77. doi: 10.1016/j.parkreldis.2013.05.013
85. Sundal, C, Lash, J, Aasly, J, Øygarden, S, Roeber, S, Kretzschman, H, et al. Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS): a misdiagnosed disease entity. J Neurol Sci. (2012) 314:130–7. doi: 10.1016/j.jns.2011.10.006
86. Tan, K, Brewer, J, Rowe, DB, Jenkins, B, Powers, JM, and Buckland, ME. Adult onset leucodystrophy with neuroaxonal spheroids and pigmented glia (ALSP): report of a new kindred. Neuropathol Appl Neurobiol. (2012) 38:95–100. doi: 10.1111/j.1365-2990.2011.01173.x
87. Taylor, RG, Alyamany, B, Pandey, S, Kertesz, A, Ang, LC, and Finger, E. Looking glass syndromes: two sides of the same gene. Can J Neurol Sci. (2019) 46:115–20. doi: 10.1017/cjn.2018.354
88. Terasawa, Y, Osaki, Y, Kawarai, T, Sugimoto, T, Orlacchio, A, Abe, T, et al. Increasing and persistent DWI changes in a patient with hereditary diffuse leukoencephalopathy with spheroids. J Neurol Sci. (2013) 335:213–5. doi: 10.1016/j.jns.2013.08.027
89. Tian, WT, Zhan, FX, Liu, Q, Luan, XH, Zhang, C, Shang, L, et al. Clinicopathologic characterization and abnormal autophagy of CSF1R-related leukoencephalopathy. Transl Neurodegener. (2019) 8:32. doi: 10.1186/s40035-019-0171-y
90. Tipton, PW, Stanley, ER, Chitu, V, and Wszolek, ZK. Is pre-symptomatic immunosuppression protective in CSF1R-related leukoencephalopathy? Mov Disord. (2021) 36:852–6. doi: 10.1002/mds.28515
91. Tsai, MC, and Sung, YF. Handwriting impairment in a case of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia caused by a novel mutation in the CSF1R gene. Neurol Sci. (2022) 43:2109–10. doi: 10.1007/s10072-021-05819-6
92. Tsai, PC, Fuh, JL, Yang, CC, Chang, A, Lien, LM, Wang, PN, et al. Clinical and genetic characterization of adult-onset leukoencephalopathy caused by CSF1R mutations. Ann Clin Transl Neurol. (2021) 8:2121–31. doi: 10.1002/acn3.51467
93. Ueda, S, Yamashita, H, Hikiami, R, Sawamoto, N, Yoshida, K, and Takahashi, R. A novel A792D mutation in the CSF1R gene causes hereditary diffuse leukoencephalopathy with axonal spheroids characterized by slow progression. eNeurologicalSci. (2015) 1:7–9. doi: 10.1016/j.ensci.2015.07.001
94. Van Gerpen, JA, Wider, C, Broderick, DF, Dickson, DW, Brown, LA, and Wszolek, ZK. Insights into the dynamics of hereditary diffuse leukoencephalopathy with axonal spheroids. Neurology. (2008) 71:925–9. doi: 10.1212/01.wnl.0000325916.30701.21
95. Wang, M, and Zhang, X. A novel CSF-1R mutation in a family with hereditary diffuse leukoencephalopathy with axonal spheroids misdiagnosed as hydrocephalus. Neurogenetics. (2019) 20:155–60. doi: 10.1007/s10048-019-00579-0
96. Wu, L, Liu, J, Sha, L, Wang, X, Li, J, Dong, J, et al. Sporadic cases with novel mutations and pedigree in hereditary leukoencephalopathy with axonal spheroids. J Alzheimers Dis. (2017) 56:893–8. doi: 10.3233/JAD-161193
97. Wu, X, Sun, C, Wang, X, Liu, Y, Wu, W, and Jia, G. Identification of a de novo splicing mutation in the CSF1R gene in a Chinese patient with hereditary diffuse leukoencephalopathy with spheroids. Neurol Sci. (2022) 43:3265–72. doi: 10.1007/s10072-021-05755-5
98. Yang, X, Huang, P, Tan, Y, and Xiao, Q. A novel splicing mutation in the CSF1R gene in a family with hereditary diffuse leukoencephalopathy with axonal spheroids. Front Genet. (2019) 10:491. doi: 10.3389/fgene.2019.00491
99. Zhan, FX, Zhu, ZY, Liu, Q, Zhou, HY, Luan, XH, Huang, XJ, et al. Altered structural and functional connectivity in CSF1R-related leukoencephalopathy. Brain Imaging Behav. (2021) 15:1655–66. doi: 10.1007/s11682-020-00360-0
100. Zheng, S, and Wang, J. A case of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) with a high antinuclear antibody titer. Neurol Sci. (2021) 42:5387–9. doi: 10.1007/s10072-021-05631-2
101. Zhuang, LP, Liu, CY, Li, YX, Huang, HP, and Zou, ZY. Clinical features and genetic characteristics of hereditary diffuse leukoencephalopathy with spheroids due to CSF1R mutation: a case report and literature review. Ann Transl Med. (2020) 8:11. doi: 10.21037/atm.2019.12.17
102. Zur-Wyrozumska, K, Kaczmarska, P, and Mensah-Glanowska, P. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with an A792D mutation in the CSF1R gene in a polish patient. Neurol Neurochir Pol. (2021) 55:322–4. doi: 10.5603/PJNNS.a2021.0012
103. Alzheimer’s Association. 2023 Alzheimer’s disease: facts and figures. (2023). Available at: https://www.alz.org/media/documents/alzheimers-facts-and-figures.pdf (Accessed January 19, 2024)
104. NIH National Institute on Aging. What are frontotemporal disorders? Causes, symptoms, and treatment. (2021). Available at: https://www.nia.nih.gov/health/frontotemporal-disorders/what-are-frontotemporal-disorders-causes-symptoms-and-treatment#:~:text=Frontotemporal%20disorders%20(FTD)%2C%20sometimes,work%2C%20or%20difficulty%20with%20walking (Accessed January 19, 2024)
105. Vanderver, A. Genetic leukoencephalopathies in adults. Continuum. (2016) 22:916–42. doi: 10.1212/CON.0000000000000338
106. National Multiple Sclerosis Society. MS signs & symptoms. (2023). Available at: https://www.nationalmssociety.org/Symptoms-Diagnosis/MS-Symptoms (Accessed January 19, 2024)
107. NIH National Institute on Aging. Vascular dementia: causes, symptoms, and treatments. (2021). Available at: https://www.nia.nih.gov/health/vascular-dementia/vascular-dementia-causes-symptoms-and-treatments (Accessed January 19, 2024)
108. Ahmed, RM, Murphy, E, Davagnanam, I, Parton, M, Schott, JM, Mummery, CJ, et al. A practical approach to diagnosing adult onset leukodystrophies. J Neurol Neurosurg Psychiatry. (2014) 85:770–81. doi: 10.1136/jnnp-2013-305888
Keywords: adult-onset leukoencephalopathy with axonal spheroids and pigmented glia, ALSP, CSF1R-related leukoencephalopathy, diagnosis, misdiagnosis
Citation: Papapetropoulos S, Gelfand JM, Konno T, Ikeuchi T, Pontius A, Meier A, Foroutan F and Wszolek ZK (2024) Clinical presentation and diagnosis of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: a literature analysis of case studies. Front. Neurol. 15:1320663. doi: 10.3389/fneur.2024.1320663
Edited by:
Giovanni Rizzo, IRCCS Institute of Neurological Sciences of Bologna (ISNB), ItalyReviewed by:
Ettore Salsano, IRCCS Carlo Besta Neurological Institute Foundation, ItalyCopyright © 2024 Papapetropoulos, Gelfand, Konno, Ikeuchi, Pontius, Meier, Foroutan and Wszolek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas Meier, YW1laWVyQHZpZ2lsbmV1cm8uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.