 Mariane Vicente
Mariane Vicente Kwaku Addo-Osafo
Kwaku Addo-Osafo Keith Vossel
Keith Vossel- Mary S. Easton Center for Alzheimer’s Research and Care, Department of Neurology, David Geffen School of Medicine at University of California, Los Angeles, CA, United States
Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB) stand as the prevailing sources of neurodegenerative dementia, impacting over 55 million individuals across the globe. Patients with AD and DLB exhibit a higher prevalence of epileptic activity compared to those with other forms of dementia. Seizures can accompany AD and DLB in early stages, and the associated epileptic activity can contribute to cognitive symptoms and exacerbate cognitive decline. Aberrant neuronal activity in AD and DLB may be caused by several mechanisms that are not yet understood. Hyperexcitability could be a biomarker for early detection of AD or DLB before the onset of dementia. In this review, we compare and contrast mechanisms of network hyperexcitability in AD and DLB. We examine the contributions of genetic risk factors, Ca2+ dysregulation, glutamate, AMPA and NMDA receptors, mTOR, pathological amyloid beta, tau and α-synuclein, altered microglial and astrocytic activity, and impaired inhibitory interneuron function. By gaining a deeper understanding of the molecular mechanisms that cause neuronal hyperexcitability, we might uncover therapeutic approaches to effectively ease symptoms and slow down the advancement of AD and DLB.
Introduction
Hyperexcitability can be defined as an increased likelihood of firing at the level of the neuron from certain stimuli and/or due to decreased firing thresholds (1). This heightened excitability can clinically present itself as epilepsy. As per the official definition by the International League Against Epilepsy (2), “Epilepsy is characterized by repeated spontaneous bursts of neuronal hyperactivity and high synchronization in the brain.” Epilepsy has emerged as a significant global health issue, impacting approximately 70 million individuals worldwide (3–6). Hyperactivity occurs in neuronal populations or brain regions when the frequency of activity is above normal rates. Brain activity is normally regulated with precise timing and regional specificity, however, high synchronization or hypersynchrony denotes an increase in neuronal coordination and cellular firing (7, 8). While epilepsy can manifest in any stage of life, it is notably more common among individuals aged 65 years and older, reaching a prevalence of 5.7% in the Cardiovascular Health Study (9). Increasingly, there is a growing recognition that late-onset epilepsy, starting after age 55, is often not an isolated condition but is frequently linked to neurodegenerative diseases like Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB) (10–12).
AD constitutes 60–70% of all dementia cases and is characterized by a gradual decline in memory and other cognitive functions. At present, there are more than 57 million people globally living with dementia, and this figure is predicted to double every two decades, reaching 74.7 million by 2030 (Alzheimer’s Disease International). The buildup of extracellular clusters of amyloid beta (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) consisting of hyperphosphorylated tau protein in the cortical and limbic regions of the human brain signifies the disease’s pathological features (13–17). The accumulation of Aβ plaques and NFTs is connected with notable loss of neurons and synapses, along with neuroinflammation (18). In this context, there is a growing number of studies showing that patients with AD exhibit epilepsy, which may be a harbinger or indicator of the disease (11, 19–24). The prevalence of epilepsy in patients with AD is around 10 to 22% (21, 25, 26), while epileptiform activity, with varying characteristics, can be detected in patients with AD and with or without diagnosed epilepsy (23, 27–34). Seizures can begin in preclinical or clinical stages of AD (20, 23, 35, 36). The preponderance of seizures in AD lacks motor characteristics, rendering their diagnosis complex and potentially leading to an underreporting of seizures (23, 36, 37). Some studies suggest seizures can increase the production and deposition of Aβ and hyperphosphorylated tau in the brain and cause a decline in cognition in patients with AD (24, 38–41). Late-onset epilepsy increases risk of AD by around three-fold (12, 42). Notably, AD predisposes patients to develop epilepsy and late-onset epilepsy predisposes patients to develop AD highlighting the bidirectionality between diseases (11, 19).
DLB ranks as the second most frequent neurodegenerative dementia among individuals above the age of 65 (43–45). Clinical criteria encompass cognitive fluctuations, visual hallucinations, rapid eye movement sleep behavior disorder, and parkinsonism (45, 46). The neuropathology of DLB is marked by neuronal Lewy bodies and Lewy neurites, consisting of aggregates of α-synuclein that impact the brainstem along with extensive limbic and neocortical areas (47). This pathology also involves the loss of midbrain dopamine cells and cholinergic neurons in ventral forebrain nuclei, nucleus basalis of Meynert (48, 49). Furthermore, Aβ plaques and NFTs are present in a majority of DLB cases (50, 51). Analogous to AD, individuals with DLB also experience seizures (52). Marawar et al. (53) demonstrated a higher occurrence of seizures in DLB compared to the general population, with a rate of 3.8% in pathologically confirmed DLB across the United States. Meanwhile, Beagle et al. (52) identified a cumulative probability of 14.7% for DLB patients to develop seizures and a 5.1% prevalence of new-onset seizures in a population from the Memory Aging Center at the University of California, San Francisco, while other studies observed a 2–3% seizure prevalence rate in cohorts from Italy, United States, and Sweden (53–55).
In spite of the presence of antiseizure medications, roughly a third of individuals with epilepsy are unable to manage their seizures or develop resistance to the impact of these medications (56–59). This underscores the urgent need to create novel and inventive treatment approaches for epilepsy. Beyond that, therapeutic interventions targeting the molecular mechanisms of neuronal hyperexcitability have promise for treating disorders linked to increased excitability, such as AD and DLB. For example, Vossel et al. (60) showed that low doses of levetiracetam can improve spatial memory and executive function in AD patients with detectable epileptic activity. Levetiracetam also improved attention, oral fluency, and overall cognition in AD patients in a case–control study (61). Also, the clinical trial HOPE4MCI (NCT03486938) uses low dose levetiracetam which has been shown to decrease hippocampal hyperexcitability and attenuate cognitive decline by improving task related memory performance in amnestic mild cognitive impairment (62, 63). These studies show that levetiracetam can improve diverse cognitive functions in various stages of AD, reflecting multiple cortical regions that exhibit hyperexcitability in the disease. As a potential marker of neurodegeneration and pathology progression in AD and DLB, the early detection of cortical hyperexcitability and its mechanistic understanding is instrumental. Hyperexcitability may begin or be a result of neuropathology and may arise due to a number of different factors at varying time points in AD and DLB. Though hyperexcitability has been previously explored in the context of AD (1, 24, 31, 60, 64, 65), the role and mechanisms of hyperexcitability in DLB (66–69), as well as its similarities and differences with AD requires more research. In this review, we explore shared and distinct molecular mechanisms associated with hyperexcitability in AD and DLB, encompassing factors such as genetic risk factors, Ca2 and glutamate contributions, cholinergic pathways, AMPA and NMDA receptors, mTOR, pathological Aβ, tau and α-synuclein, genetic risk factors, altered microglial and astrocytic activity, and impaired inhibitory interneuron function (Figure 1).
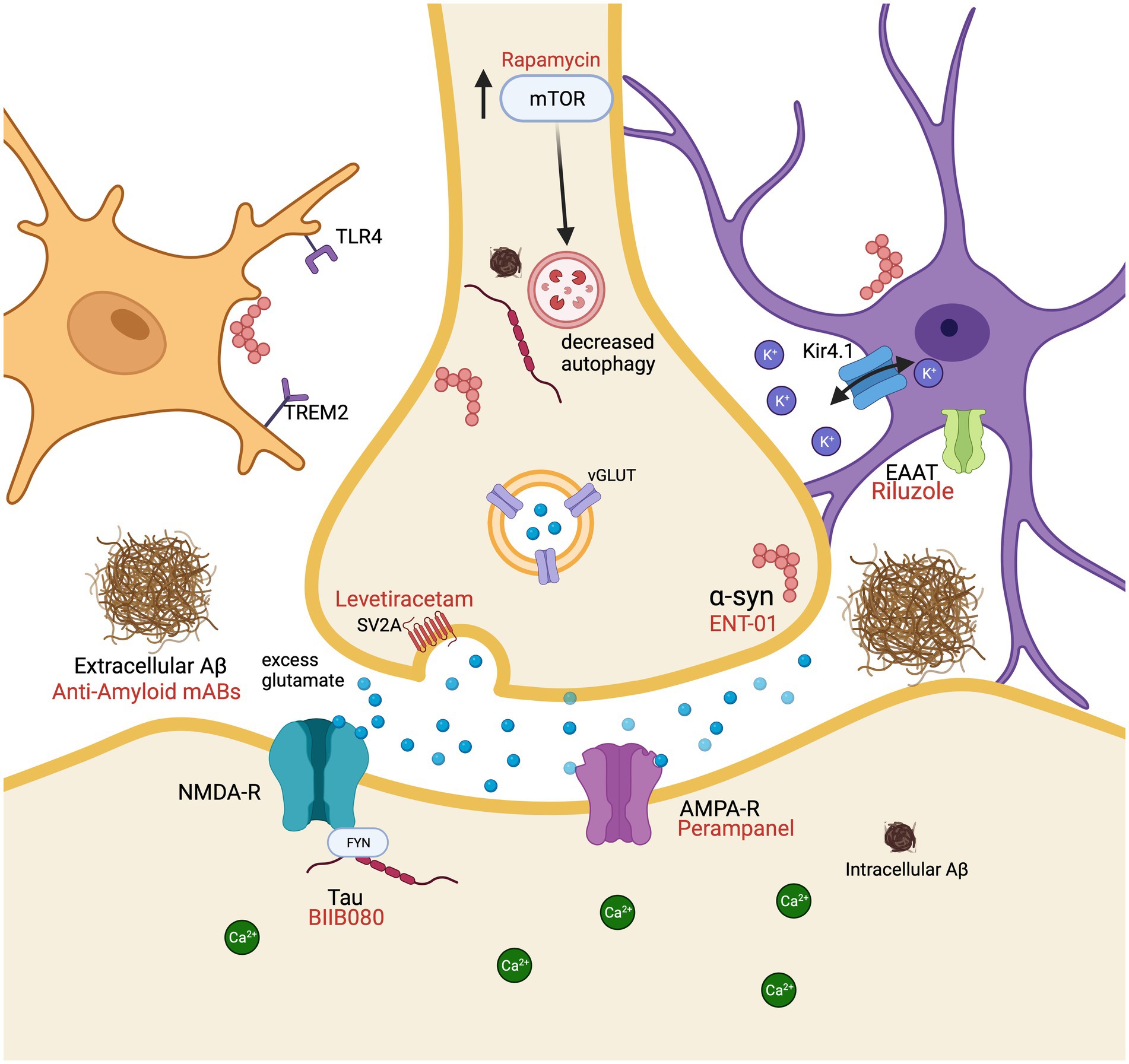
Figure 1. Molecular mechanisms resulting in cellular hyperexcitability associated with Alzheimer’s disease and dementia with Lewy bodies in a glutamatergic neuron surrounded by a microglial cell (peach) and an astrocyte (purple). Pharmacological interventions (red) are displayed by their receptor or protein of action. Aβ, amyloid beta; AMPA-R, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; EAAT, excitatory amino acid transporters; mAbs, monoclonal antibodies; NMDA-R, N-methyl-D-aspartate receptor; TLR4, toll like receptor 4; TREM2, triggering receptor expressed on myeloid cells 2; mTOR, mechanistic target of rapamycin; SV2A, synaptic vesicle glycoprotein 2A; vGLUT, vesicular glutamate transporters. Created with BioRender.com.
Genetic risk factors
APOE
The apolipoprotein E (APOE) ε4 allele is implicated in cerebrovascular, mental, and neurological disorders but stands as the primary genetic susceptibility factor for AD, and also increases the severity of neuropathology in DLB (70–75). In the context of hyperexcitability, APOE ε4 (APOE4) has not been associated with early-onset epilepsy, within 12 months of age, (76), but APOE4 has been linked to an increased risk of late-onset epilepsy, starting after age 60, and there exists an allele dose dependence on the incidence of late-onset epilepsy of 2.87, 4.13, and 7.05 per 1,000 person-years for 0, 1, and 2 APOE ε4 alleles, respectively (42, 77). These results persisted when participants with strokes were censored, suggesting that APOE4 confers epilepsy risk through mechanisms beyond its effects on cerebrovascular disease (42). A meta-analysis demonstrated that individuals carrying the APOE4 allele and experiencing temporal lobe epilepsy exhibit seizure onset nearly 4 years earlier than those without the allele (78). Another investigation revealed that individuals with temporal lobe epilepsy and APOE4 have an elevated risk of experiencing verbal learning deficits, particularly among those with a longer epilepsy duration (79). Similarly, mice expressing the human APOE4 allele develop a seizure phenotype that is either absent or less pronounced in mice expressing human APOE2 or APOE3 (80).
The exact mechanisms by which APOE4 promotes heightened neural excitability remain to be fully elucidated. APOE is involved in cholesterol metabolism and transportation, stabilization and solubilization of lipoproteins, and maintaining lipid homeostasis. Additionally, it plays a role in synaptic plasticity, signal transduction, and immunomodulation (81–83). In vitro studies utilizing human induced pluripotent stem cell-derived neurons expressing APOE4 demonstrate increased excitability compared to APOE3 isogenic controls. This heightened excitability might be attributed to an elevated expression of synaptic proteins like synaptophysin and PSD-95, the upregulation of genes involved in neuronal differentiation, and alterations in cholesterol metabolism (84). Due to the critical importance of APOE in shaping neuronal structure, establishing synapses, and regulating ion channels, changes in cholesterol and lipid concentrations can significantly impact neural excitability (85). For example, in rat hippocampal neurons, changed cholesterol levels differentially affect fast transient currents and delay rectifying currents modulating hyperexcitability (86). A clinical demonstration of importance is evident in Niemann-Pick type C (NPC) disease. In NPC, dysregulation of cholesterol transport and accumulation, can result in an AD-like phenotype, including cortical neurodegeneration, tau hyperphosphorylation, Aβ deposition, and hyperexcitability (87, 88). Vivas et al. (88) has shown that decreased transport of cholesterol from lysosomes disrupts ion channel activity and ultimately results in neuronal hyperexcitability. This mechanism is mediated by the reduction in phosphatidylinositol 4,5-bisphosphate in the plasma membrane resulting in a decrease in KCNQ2/3 current and increased excitability (88). Finally, microglia and astrocytes harboring APOE4 exhibit slowed uptake of extracellular Aβ (84). Consequently, elevated Aβ levels can also lead to increased neural activity. The plethora of physiological functions APOE is involved in results in numerous pathways by which APOE can contribute to hyperexcitability and targeted with therapeutics in AD and DLB (Table 1).
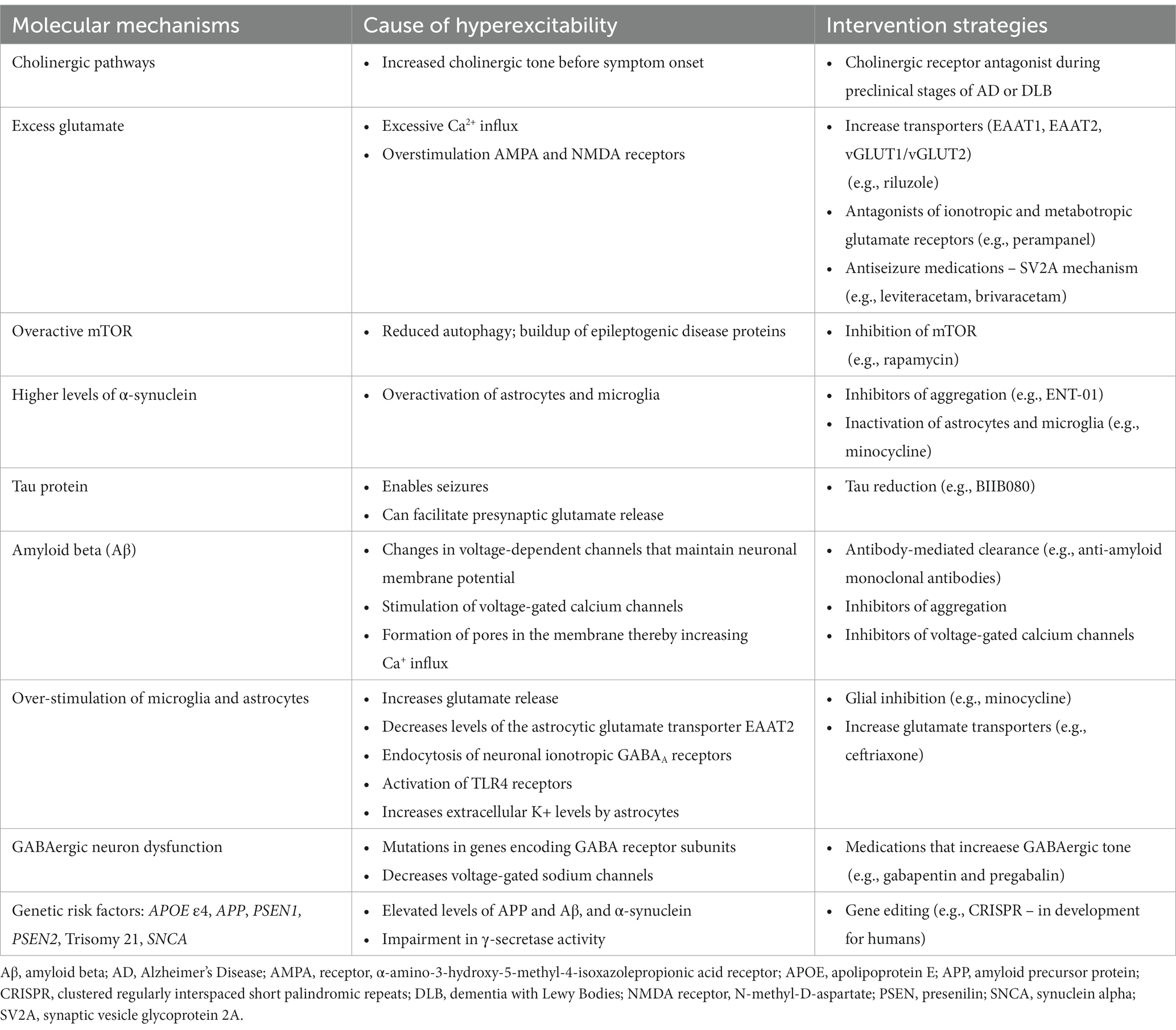
Table 1. Summary of molecular mechanisms cause hyperexcitability and intervention strategies associated with Alzheimer’s disease and dementia with Lewy Bodies.
APP, PSEN1, and PSEN2
Early-onset familial AD, which constitutes less than 1% of cases, can be triggered by highly penetrant mutations in genes encoding amyloid precursor protein (APP) on chromosome 21, presenilin 1 (PSEN1) on chromosome 14, and presenilin 2 (PSEN2) on chromosome 1 (89–91). Among the roughly 35 distinct APP mutations associated with AD pathogenesis are gene locus duplications and point mutations in the coding region, leading to amino acid substitutions. Duplication of the entire gene or locus results in elevated APP and Aβ levels, favoring the formation of Aβ plaques (92, 93). PSEN1 and PSEN2 are not only involved in γ-secretase but also in cleaving other type I integral proteins like the Notch receptor (94). Likewise, mutations in PSEN1 and PSEN2 hinder γ-secretase activity, causing an imbalance in the Aβ1–42 to Aβ1–40 ratio due to Aβ1–42 overproduction or Aβ1–40 underproduction, or a combination thereof. The Aβ1–42 to Aβ1–40 ratio is significant because an increase in this ratio increases the aggregation and neurotoxicity of the Aβ protein while a decrease in the ratio can decrease deposition (95–98). APP, PSEN1, and PSEN2 mutations contribute to neural excitability by activating the mentioned mechanisms via elevated Aβ levels and amyloid plaque formation. It is important to note that early-onset AD is not only related to APP, PSEN1, and PSEN2. Alterations in these three genes only account for 5–10% of early-onset AD with remaining genes and risk factors still to be discovered and studied (89, 99–101). Beyond these known genetic variants causing AD, individuals with Down syndrome possess an extra copy of chromosome 21, housing APP, and face an elevated risk of early-onset AD and seizures (102, 103). Estimates indicate that more than 50% of people with Down syndrome will develop Alzheimer’s with symptoms emerging in their 50s and 60s (104, 105).
SNCA
Mutations in the SNCA gene, which encodes α-synuclein, lead to parkinsonian disorders, notably including DLB (106–108). Among the numerous mutations, A30P, E46K, G51D, and duplications and triplications of the SNCA gene, of specific interest is the A53T point mutation (106, 109, 110). Recent investigations have uncovered that mice expressing human α-synuclein with the A53T mutation manifest a phenotype akin to the human condition (110). They exhibit deficits in long-term potentiation and learning and memory. Furthermore, these mice display a left shift in electroencephalography (EEG) spectral power, mirroring the EEG slowing observed in patients with DLB (110–112). The EEG slowing and shift in spectral power to more delta signifies network dysfunction, a loss of cholinergic neurons, and symptoms of DLB (66, 68). Similarly, Morris et al. (66) demonstrated that neuronal overexpression of wild-type α-synuclein in transgenic mice (Thy1-SYN line 61) also leads to EEG slowing. Both of these models experience seizures and present molecular alterations in the hippocampus that suggest abnormal network excitability, including a depletion of calbindin in the dentate gyrus. These collective findings suggest that higher levels or dysfunction of α-synuclein may contribute to the neuronal hyperactivity found in DLB.
Degeneration of cholinergic pathways
Acetylcholine is an ester of acetic acid and choline that is released by cholinergic neurons (113, 114). Acetylcholine plays a crucial role as one of the neurotransmitters implicated in cognitive functions like memory and executive function. In both DLB and AD, deficiencies in cholinergic activity are observable (115, 116). These deficiencies manifest as reduced acetylcholine levels and irregularities in the expression of nicotinic and muscarinic receptors. Notably, the extent of cholinergic deficits tends to be more pronounced in DLB when compared to AD, even though DLB typically exhibits less brain volume loss (49, 117). The decline of cholinergic neurons projecting to the cortex contributes to a deceleration of cortical oscillations as seen on EEG, resulting in a shift of spectral power from higher frequency bands (alpha, beta, gamma) to lower ones (delta, theta) (118, 119). DLB patients experience a more significant loss of cholinergic neurons, displaying more pronounced EEG slowing (49). Additionally, DLB patients demonstrate greater clinical improvement with the usage of common acetylcholinesterase inhibitors such as donepezil, rivastigmine, and galantamine compared to AD patients (120, 121). It is unknown whether neurodegeneration of cholinergic neurons contributes to hyperexcitability. However, animal models suggest that early changes in cholinergic tone could contribute to epilepsy in preclinical stages of AD. Interictal spikes have been observed during the rapid eye movement stage of sleep in Tg2576 mice expressing human amyloid precursor protein (APP) at a very young age (5 weeks old), long before the deposition of Aβ (122). After administration of muscarinic cholinergic receptor antagonist, atropine, the investigators observed a reduction in interictal spikes, suggesting that there may be a phase of high cholinergic tone, contributing to epilepsy, prior to reductions in acetylcholine (122). In contrast, donepezil, a cholinesterase inhibitor had no significant effect on interictal spikes (122). Another study using the APPswe/PS1dE9 mouse model presenting with spike–wave discharges (SWDs), showed that donepezil does not have a significant effect on epileptic activity whereas atropine decreases SWDs and results in EEG slowing (123). This information suggests that before the degeneration of cholinergic neurons in AD and DLB, there could be a phase of increased cholinergic tone that contributes to an increase in neuronal activity and epilepsy.
Glutamate
Glutamate, among the most extensively studied neurotransmitters within the central nervous system, is a non-essential amino acid synthesized within neurons and glial cells using glucose and α-ketoglutarate. It is ubiquitously distributed throughout the brain (124). Glutamate holds significance in cognitive functions like memory and learning, playing a pivotal role in neuronal excitability by expediting swift synaptic activity in neurons—a process regulated by astrocytes and other glial cells (125). The distribution of glutamate across distinct brain compartments is orchestrated by specific transporters and enzymes accountable for its metabolism. Surplus glutamate is eliminated by glial cells through excitatory amino acid transporters (EAAT1, EAAT2) (126). Notably, reduced expression levels of EAAT1 and EAAT2 have been observed in cases of epilepsy (127, 128), while mutations in the SLC1A3 and SLC1A2 genes that encodes EAAT1 and EAAT2, can result in episodic ataxia 6, characterized by symptoms of epilepsy, long lasting ataxia attacks and headaches, and epileptic encephalopathies, respectively (129, 130).
Inside astrocytes, glutamate undergoes a transformation into glutamine, subsequently being released and taken up by the neuronal presynaptic compartment. There, it is converted back into glutamate, which then accumulates within synaptic vesicles via vesicular glutamate transporters (vGLUT1/vGLUT2). Although astrocytes are commonly discussed collectively, they are an extremely diverse cell population. A recently described subpopulation of astrocytes specifically mediates the release of glutamate (131). Ultimately, this intricate process facilitates highly efficient neurotransmission within tri or tetrapartite synapses (132, 133).
An imbalance in the expression of vGLUT1 was observed in post-mortem human brain samples at the advanced stages of both AD and DLB (134). Similarly, Liraz et al. (135) discovered reduced levels of vGLUT in the hippocampal neurons of APOE4 mice. Previous research studies have pointed to a decline in the capacity and protein expression of glutamate transporters, as well as a specific loss of vGLUT in AD patients (136–138). A postmortem study showed increases in EAAT1 levels in a subset of pyramidal neurons exhibiting degeneration in the AD brain (139), whereas another postmortem study and in vitro assay showed impaired function of EAAT2 in the AD brain (140). Pharmacological administration of riluzole increases glutamate transporter expression, and in the P301L mouse model reverses glutamate related alterations and associated cognitive decline (141). Consequently, elevated levels of glutamate contribute to excitotoxicity and neuronal cell death (142). These findings collectively suggest that as the disease advances, the transporters responsible for glutamate reuptake become less effective, potentially leading to increased neuronal excitability.
Glutamate toxicity primarily arises from an excessive influx of Ca2+ (143, 144). Dubinsky (145) demonstrated that hippocampal neurons exposed to toxic levels of glutamate maintained elevated Ca2+ levels for around 1 h before returning to baseline levels. As calcium signaling governs a spectrum of cellular processes, the outcome of Ca2+ overload entails the activation of catabolic enzymes like calpain I (146), phospholipases, and the release of arachidonic acid (147). This cascade results in an escalation of reactive oxygen and nitrogen species and the eventual collapse of neuronal cells through cytoskeletal degradation and membrane deterioration. Clinically, this associates with the progressive decline in cognition and memory, as well as brain atrophy in AD patients (148, 149). This is further evident in epilepsy where the seizures ultimately cause excitotoxicity by starting the aforementioned cascade and leading to neuronal cell death and loss (150, 151).
Upon release from synaptic vesicles, glutamate initiates the activation of diverse ionotropic (AMPA, kainate, NMDA) and metabotropic (mGluR1 and mGluR5 in group I, mGluR2 and mGluR3 in group II, and mGluR4,6,8 in group III) glutamate receptors, primarily located in the postsynaptic region (152). The overstimulation of these receptors contributes to the generation of free radicals, possibly as a result of the continued calcium influx, inducing oxidative stress and subsequently disrupting mitochondrial functions (152, 153). This mitochondrial dysfunction plays a role in initiating and advancing epilepsy by triggering sequences of apoptosis (154).
Recent research highlights NMDA receptors (NMDARs) as contributors to neuronal hyperexcitability, suggesting that abnormal activation of these receptors, particularly through Ca2+ influx, is implicated in hyperexcitability (155, 156). NMDARs possess a significantly higher permeability for calcium ions compared to other ionotropic glutamate receptors (iGluRs), thus facilitating hyperactivity through calcium influx (155, 156). Memantine, an NMDAR antagonist, has been found to reduce Ca2+ influx and improve cognition and behavior in moderate-to-advanced AD (157). On the other hand, direct links between AMPA receptors and epilepsy in AD and DLB are more limited. Elevated levels of AMPA receptors have been observed in the brains of various epilepsy types, in humans and animal models (158, 159), and there is evidence of changes in receptor function through increased levels of AMPA and NMDA receptor subunits in human and mouse epileptic brains (160, 161).
Studies such as that by Teravski et al. (110) involving A53T α-synuclein-expressing neurons have indicated postsynaptic dysfunction, including reduced amplitude of miniature postsynaptic currents and a lower ratio of AMPA to NMDA receptor currents. Such changes coincide with the development of epileptic activity in this model. If the loss of AMPA receptors occurs in GABAergic inhibitory neurons, this could enhance the activity of neurons receiving their projections, potentially leading to neural network hyperactivity in DLB. Further exploration of the roles of NMDA and AMPA receptors in AD and DLB could yield valuable insights into potential treatments for epilepsy associated with these diseases.
Overactivation of mTOR Pathway
mTOR, mechanistic target of rapamycin, is a highly conserved serine/threonine protein kinase that forms two distinct complexes, mTORC1 and mTORC2. External triggers including energy, oxygen, DNA damage, and amino acids activate the mTOR complexes, and they are implicated in a breadth of physiological functions including cell survival, growth, proliferation, metabolism, protein synthesis and signaling (162). In the brain, mTOR expression is widespread, affecting many neuronal and glial cell types playing a role in axonal development, synaptic plasticity, and neuronal excitability (163). Another crucial role function of mTOR signaling is autophagy, the process of degrading and recycling components of dysfunctional cells and proteins (164). Recently however, the role of autophagy has been expanded and shown to affect neuronal excitability (165). An ATG5 deficient mouse model shows impairment and decreases in protein kinase A (PKA) signaling from the lack of PKA subunit turnover (165). In addition to increased excitatory neurotransmission, alterations in synapses and disruption in AMPA receptor function, seizures also present as a common phenotype in these mice. This further highlights mTOR’s myriad functions and its contribution to hyperexcitability and warrants further investigation in the context of neurodegeneration.
Overactivation and dysregulation of mTOR can result in severe pathological changes. mTOR association with hyperexcitability and seizures can be attenuated pharmacologically (125, 166, 167). mTOR hyperactivation is observed in Tuberous Sclerosis Complex (TSC) which presents with epileptic seizures and autism-like traits (162). Loss of the TSC gene in mouse models results in seizures and epilepsy that can be attenuated with the mTOR inhibitor, rapamycin (168). mTOR overactivation can also be activated by seizures evidenced by an increase in phospho-S6 expression in a kainic acid seizure mouse model (169). Inhibition of mTOR and restoration of the excitatory imbalance causing seizures and epilepsy may provide additional benefit for AD and DLB where hyperexcitability may participate in a positive feedback loop (40).
Pertaining to AD and DLB, postmortem examinations revealed increased mTOR activation in AD, DLB, and Parkinson’s disease and associations with deficits in autophagy (170–173). Seizures have been shown to both activate mTOR and worsen AD pathology and cognitive deficits. Rapamycin administration can attenuate cognitive deficits in AD models through an increase in autophagy and/or decrease in hyperexcitability, further linking overactivation of mTOR activity and its contribution to AD (174, 175). Within the context of hyperexcitability, mTOR in DLB may be underappreciated and understudied. Since autophagy deficits have been implicated in DLB, hyperexcitability may be a mechanistic link with mTOR. A better understanding of these mechanisms and connection to hyperexcitability in AD and DLB may allow for more targeted therapeutics beyond rapamycin in lessening the overactivation of mTOR and burden of its wide-ranging effects.
Proteinopathies
Alpha-synuclein
Alpha-synuclein is a protein composed of 140 amino acids. It was initially identified in association with synaptic vesicles within the presynaptic nerve terminal and has demonstrated interactions with membranes (176, 177). This protein modulates synaptic transmission, influences the density of synaptic vesicles, and contributes to neuronal plasticity (178, 179).
Beyond its synaptic functions, extracellular alpha-synuclein has a pivotal impact on neuroinflammation, neurotoxicity, and the propagation of pathological changes (180). It is transported into the extracellular space following active secretion or release from dying neurons. The exact mechanism behind the secretion of alpha-synuclein is unknown. However, research by Paillusson et al. (181) indicated that enteric neurons can release it via conventional endoplasmic reticulum/Golgi-dependent exocytosis, which is driven by neuronal activity.
Clinical and experimental studies demonstrate that α-synuclein expression participates in epilepsy (182–185). Tweedy et al. (186) demonstrated hippocampal network hyperexcitability in young transgenic mice expressing human mutant alpha-synuclein. Yang et al. (185) identified anomalous accumulations of this protein in hippocampal samples taken from individuals with mesial temporal lobe epilepsy (MTLE). These deposits were correlated with the loss of neuronal cells and reactive gliosis, indicating a potential link between the presence of the protein and the pathological changes seen in MTLE. Another clinical study in children with epilepsy showed that higher levels of serum α-synuclein correlated with disease severity (182). In the same way that α-synuclein levels are associated with seizures in epilepsy patients, it may also be associated with epileptic events in AD and DLB. A mechanism by which α-synuclein contributes to epilepsy could be activation of astrocytes and microglia, enhancing glial proinflammatory activity cytokines, nitric oxide, and reactive oxygen species (184, 187). More investigations are warranted to determine whether lowering α-synuclein levels or inhibiting its aggregation in the brain modulates epilepsy.
Tau protein
The microtubule-associated protein tau predominantly resides within axons, where it plays a vital role in assembling microtubules. Tau can also be located in various neuronal compartments, such as somatodendritic regions and nuclei, and it is even detectible within glial cells (188, 189). In cases of pathology, tau undergoes hyperphosphorylation within neurons, diminishing its affinity for tubulin. This leads to the aggregation of tau into neuropil filaments or NFTs, giving rise to tauopathies (190–192).
Brain aggregates of hyperphosphorylated tau have been noted in patients with epilepsy as well as various models of epilepsy (40, 68, 193). This indicates that the abnormal aggregation of phosphorylated tau might play a role in the pathogenesis of epilepsy. Additionally, the tau protein seems to contribute to the development of epilepsy in the context of AD and DLB. Referencing Hwang et al. (40), endogenous tau acts as an enabler of hyperexcitability and seizures and, within the context of epilepsy and AD, a complex balance may occur in an attempt to decrease hyperexcitability. Total tau is reduced after 2 months in a status epilepticus (SE) model of epilepsy (194). After 4 months, tau levels return to normal while phosphorylation at tau sites S202/T205 is reduced by about 50%. This may highlight how tau changes in response to hyperexcitability over time in an attempt to reach homeostasis.
Numerous investigations have demonstrated that genetically altering tau or diminishing tau levels can result in an increase or decrease in seizures and epileptic activity across different animal models. Ablating both tau and Fyn in a mouse model shows robust neuroprotection from pentylenetetrazol, including increased seizure latency, reduced seizure stage, and reduced gliosis (195). Roberson et al. (196) demonstrated that reducing normal tau prevents the occurrence of spontaneous epileptiform activity across multiple lines of transgenic mice expressing human APP. Conversely, transgenic mice that overexpressed wild-type human tau or tau with an A152T mutation exhibit epileptiform activity and heightened susceptibility to seizures (197). The A152T tau mutation induces more pronounced network hyperexcitability compared to wild-type tau (197). In vitro studies using the rTg4510 mouse model, which features mutant (P301L) human tau, revealed increased neuronal excitability in the cortex’s layer 3 even before the formation of NFTs. In the CA1 region of the hippocampus, pyramidal neurons display heightened firing, while inhibitory interneurons exhibit reduced activity, indicating a breakdown in inhibitory synaptic transmission (198, 199).
Recent investigations involving mice expressing human α-synuclein with the A53T mutation highlighted that endogenous tau contributes to hyperexcitability and that epileptic activity diminishes in the absence of tau (68). Delving deeper into the pathways influenced by tau, Decker et al. (200) demonstrated that hyperphosphorylated tau could stimulate presynaptic glutamate release, resulting in hyperexcitability. The toxicity of glutamate has been linked to tau-mediated neuronal cell death and behavioral deficits in drosophila (201).
The observation that physiological endogenous tau levels in adult mice impact seizure susceptibility suggests that similar relationships might exist in humans, potentially influencing the risk of developing seizures. This supports the notion that reducing tau could contribute to preventing seizures (202) and offers an opportunity for pharmacological intervention targeting tau.
Amyloid beta (Aβ)
Amyloid precursor protein (APP) is a transmembrane protein encompassing a sizable extracellular domain and a smaller intracellular segment. Amyloid-beta (Aβ) peptides stem from the proteolytic cleavage of APP, sequentially catalyzed by β-secretase and γ-secretase. In pathological conditions, Aβ peptides amass into dense fibrillary plaques. Aβ has been demonstrated to incite network dysregulation, culminating in heightened synchronicity and seizures. This increased neuronal activity, in turn, exacerbates neurodegeneration (203). Recent investigations indicate that Aβ possesses epileptogenic properties and can significantly influence the trajectory of cognitive decline (12, 204). Ovsepian and O’Leary (205) proposed that seizures might foster the deposition of Aβ plaques. This epileptogenic potential of Aβ was validated in the APP/PS1 model, where neurons exhibiting epileptic discharges were found to colocalize with Aβ plaques (206). Evidence also suggests that Aβ could have epileptogenic effects even during pre-plaque stages. Hyperactivity among hippocampal neurons during the initial phases of Aβ pathology, when Aβ fibrils remain soluble, has been observed in APP/PS1 mice (122, 207). APP/PS1 mice also present with an increase in soluble and insoluble Aβ1-42 and an increase in seizure susceptibility with corneal kindling (208). Aβ oligomers, being synaptotoxic, might trigger epileptic discharges prior to plaque deposition (209). Exposure to Aβ oligomers can also lead to spontaneous neuronal firing in hippocampal neurons (210).
Studies indicate that Aβ-triggered neuronal epileptic activity is tied to alterations in voltage-dependent channels that regulate the neuronal membrane potential. In a drosophila model expressing human Aβ42, Ping et al. (211) demonstrated that fewer Kv4 channels in neurons promote hyperexcitability, while Kv2 and Kv3 channels remained unaffected. Other research has shown that Aβ can perturb calcium homeostasis by either stimulating voltage-gated calcium channels or creating membrane pores, thereby augmenting calcium influx (212). Additionally, Aβ can influence glutamate release. Talantova et al. (213) illustrated that Aβ interacts with α7 nicotinic acetylcholine receptors, leading to the release of astrocytic glutamate, which subsequently activates extrasynaptic NMDA receptors on neurons. Similarly, Zott et al. (214) employed Aβ-amyloidosis models to reveal that hyperactivity is initiated by dampening glutamate reuptake. Soluble Aβ oligomers hinder the uptake of glutamate and intensify extrasynaptic NMDAR activation. Thus, Aβ can trigger a sequence of molecular events culminating in neural hyperexcitability.
Glia and neuroinflammation
Glial cells are brain defense cells comprising microglia, astrocytes, and oligodendrocytes. When stimulated, microglia and reactive astrocytes release modulators to facilitate the recovery of the tissue from damage (215, 216). However, the continuous stimulation of the glial network causes a cascade of molecular events leading to neuroinflammation (215, 217). Investigators have previously proposed that neuroinflammation stimulates heightened neuronal activity and seizures, and the disruption of glial immunoinflammatory function is considered a factor that could predispose to or play a role in the emergence of seizures (218, 219). Therefore, inflammatory mediators and epileptic seizures form a vicious positive feedback loop, reinforcing each other (220). This vicious cycle can be found in diseases with neuroinflammatory conditions such as AD and DLB and to be responsible for epileptogenesis.
Elevated concentrations of pro-inflammatory cytokines, notably interleukin-1β (IL-1β), IL-6, and tumor necrosis factor-α (TNF-α), have been linked to epileptic seizures (221). The increased concentration of pro-inflammatory mediators can participate in hyperexcitability by increasing glutamate release via decreasing levels of the astrocytic glutamate transporter EAAT2 (excitatory amino acid transporter 2) (222, 223). Before and after seizures, there is an increase in the levels of pro-inflammatory cytokines and the expression of their receptors in both glial cells and neurons (221). In epileptic and AD patients, TNFα levels are elevated in the brain (224, 225), and TNFα increases the sensitivity of AMPA and NMDA glutamatergic receptors in the postsynaptic neuron, leading to excitotoxicity (223, 226). TNFα also induces endocytosis of neuronal ionotropic GABAA receptors, so that neurotransmission becomes more excitatory, leading to epilepsy (227). Furthermore, Xiaoqin et al. (85) found that the intracerebroventricular injection of IL-1β in rats leads to a reduction in cortical and hippocampal GABA concentration, while simultaneously increasing glutamate release. This alteration in neurotransmitter balance enhances the brain’s vulnerability to seizures.
Another potential mechanism underlying seizure activation is the engagement of TLR4 receptors (228). TLR4 acts as the primary receptor for the proinflammatory mediator High Mobility Group Box 1 (HMGB1). Activation of TLR4 via HMGB1 sets off seizures by initiating a Ca2+ influx subsequent to the phosphorylation of the NR2B subunit of the NMDAR. In support of this, Maroso et al. (229) revealed an elevation in TLR4 expression in hippocampal samples from individuals with drug-resistant temporal lobe epilepsy compared to control subjects. Furthermore, inflammation triggers the release of reactive oxygen species and reactive nitrogen species, thereby heightening susceptibility to seizures and intensifying the inflammatory milieu in the brain (230). This inflammatory environment gives rise to mediators like pro-inflammatory cytokines, transforming growth factor-β, and prostaglandin E2 (231). These mediators stimulate astrocytes and impact glutamate release, culminating in hyperexcitability.
Triggering receptor expressed on myeloid cells 2 (TREM2) are primarily expressed by microglia and play a role in the immune response. TREM2 expression in the brain has been found to be increased in AD and thought to provide an adaptive response to AD pathology, while reduction in TREM2 and mutant variants increases susceptibility to hyperexcitability and epileptic activity (232, 233). In regards to DLB, results are mixed as to whether soluble TREM2 is increased, and more research is needed to determine TREM2’s influence on DLB-related hyperexcitability (234–236).
In addition to releasing inflammatory mediators, glial cells, particularly astrocytes, play a role in maintaining ion balance by clearing extracellular potassium (K+) during neuronal repolarization. Wang et al. (237) demonstrated that the onset of seizures is linked to elevated extracellular K+ levels due to astrocytic activity. Notably, the protein expression of the astrocytic potassium channel Kir4.1 is diminished in both a mouse model of AD and in the brains of AD patients (238).
Astrocytes often undergo reactive changes, termed reactive astrocytosis, characterized by increased astrocyte size and number. These changes are frequently observed alongside neuronal loss and synaptic reorganization (239). Reactive astrocytosis is present in conditions such as epilepsy, AD, and DLB, and it might contribute to neural hyperexcitability by influencing the function of astrocytic membrane K+ channels (240–242). In light of these insights, understanding the underlying mechanisms of inflammation in the development of epilepsy could pave the way for the discovery of promising antiseizure medications.
GABAergic dysfunction
Gamma-aminobutyric acid (GABA) serves as the primary inhibitory neurotransmitter within the central nervous system. It is synthesized by the enzyme glutamic acid decarboxylase (240). This neurotransmitter is primarily found in interneurons that establish synapses on cell bodies and nearby axon segments. Released into the synaptic cleft, GABA exerts its influence through activation of GABAA and GABAB receptors. GABAA receptors function as ligand-gated ion channels, promptly inducing inhibition by enhancing chloride influx into cells. In the context of AD, studies have shown moderate reductions in GABAA receptors within the brain (243, 244). GABAB receptors, on the other hand, are G protein-coupled ion channels that augment extracellular potassium transport while concurrently decreasing calcium influx. Mutations in genes encoding GABA receptor subunits have been linked to a range of epileptic disorders (245).
Investigations point to a potential mechanism involving GABAergic dysfunction contributing to hyperexcitability by influencing voltage-gated sodium channels. Studies by Verret et al. (246) and Hamm et al. (247) demonstrated variable decreases in Nav1.1 and Nav1.6 within hippocampus and somatosensory cortex mouse models of AD. These channels enhance gamma oscillations during exploration, which can help suppress epileptiform discharges. Consequently, Nav1.1 and Nav1.6 hold potential as targets for addressing epileptic seizures in the context of AD.
Conclusion
In conclusion, hyperexcitability in AD and DLB arises from a combination of multiple factors working together to disrupt regulation of neuronal excitability. In this context, we observe that genetic predispositions, initial elevations in cholinergic activity, excessive calcium influx causing glutamate toxicity, heightened NMDA and AMPA receptor sensitivity, overactivation of mTOR, disruptions in calcium homeostasis due to Aβ, tau, and α-synuclein, hyperstimulation of microglia and astrocytes, and GABA dysfunction collectively contribute to the promotion of hyperexcitability in AD and DLB. By understanding the specific dysfunctions within these pathways, it becomes possible to develop targeted therapeutic strategies aimed at restoring proper neuronal excitability. Such interventions hold the potential to alleviate the symptoms associated with these neurodegenerative disorders, offering hope for improved treatments and better quality of life for affected individuals.
Author contributions
MV: Writing – review & editing. KA-O: Writing – review & editing. KV: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by NIH grants R01 NS033310 (KV), R01 AG058820 (KV), R01 AG075955 (KV), R56 AG074473 (KV), and UH2 AG083254 (KV).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Targa Dias Anastacio, H, Matosin, N, and Ooi, L. Neuronal hyperexcitability in Alzheimer's disease: what are the drivers behind this aberrant phenotype? Transl Psychiatry. (2022) 12:257. doi: 10.1038/s41398-022-02024-7
2. Scheffer, IE, Berkovic, S, Capovilla, G, Connolly, MB, French, J, Guilhoto, L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. (2017) 58:512–21. doi: 10.1111/epi.13709
3. Collaborators, GBDN. Global, regional, and national burden of neurological disorders, 1990-2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. (2019) 18:459–80. doi: 10.1016/S1474-4422(18)30499-X
4. Falco-Walter, J. Epilepsy-definition, classification, pathophysiology, and epidemiology. Semin Neurol. (2020) 40:617–23. doi: 10.1055/s-0040-1718719
5. Fiest, KM, Sauro, KM, Wiebe, S, Patten, SB, Kwon, CS, Dykeman, J, et al. Prevalence and incidence of epilepsy: A systematic review and meta-analysis of international studies. Neurology. (2017) 88:296–303. doi: 10.1212/WNL.0000000000003509
6. Thijs, RD, Surges, R, O'brien, TJ, and Sander, JW. Epilepsy in adults. Lancet. (2019) 393:689–701. doi: 10.1016/S0140-6736(18)32596-0
7. Jiruska, P, De Curtis, M, Jefferys, JG, Schevon, CA, Schiff, SJ, and Schindler, K. Synchronization and desynchronization in epilepsy: controversies and hypotheses. J Physiol. (2013) 591:787–97. doi: 10.1113/jphysiol.2012.239590
8. Margineanu, DG. Epileptic hypersynchrony revisited. Neuroreport. (2010) 21:963–7. doi: 10.1097/WNR.0b013e32833ed111
9. Choi, H, Pack, A, Elkind, MS, Longstreth, WT Jr, Ton, TG, and Onchiri, F. Predictors of incident epilepsy in older adults: the cardiovascular health study. Neurology. (2017) 88:870–7. doi: 10.1212/WNL.0000000000003662
10. Costa, C, Romoli, M, Liguori, C, Farotti, L, Eusebi, P, Bedetti, C, et al. Alzheimer's disease and late-onset epilepsy of unknown origin: two faces of beta amyloid pathology. Neurobiol Aging. (2019) 73:61–7. doi: 10.1016/j.neurobiolaging.2018.09.006
11. Johnson, EL, Krauss, GL, Kucharska-Newton, A, Albert, MS, Brandt, J, Walker, KA, et al. Dementia in late-onset epilepsy: the atherosclerosis risk in communities study. Neurology. (2020) 95:e3248–56. doi: 10.1212/WNL.0000000000011080
12. Keret, O, Hoang, TD, Xia, F, Rosen, HJ, and Yaffe, K. Association of Late-Onset Unprovoked Seizures of unknown etiology with the risk of developing dementia in older veterans. JAMA Neurol. (2020) 77:710–5. doi: 10.1001/jamaneurol.2020.0187
13. Deture, MA, and Dickson, DW. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener. (2019) 14:32. doi: 10.1186/s13024-019-0333-5
14. Fan, L, Mao, C, Hu, X, Zhang, S, Yang, Z, Hu, Z, et al. New insights into the pathogenesis of Alzheimer's disease. Front Neurol. (2019) 10:1312. doi: 10.3389/fneur.2019.01312
15. Kinney, JW, Bemiller, SM, Murtishaw, AS, Leisgang, AM, Salazar, AM, and Lamb, BT. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement. (2018) 4:575–90. doi: 10.1016/j.trci.2018.06.014
16. Perl, DP. Neuropathology of Alzheimer's disease. Mt Sinai J Med. (2010) 77:32–42. doi: 10.1002/msj.20157
17. Soria Lopez, JA, Gonzalez, HM, and Leger, GC. Alzheimer's disease. Handb Clin Neurol. (2019) 167:231–55. doi: 10.1016/B978-0-12-804766-8.00013-3
18. Mangalmurti, A, and Lukens, JR. How neurons die in Alzheimer's disease: implications for neuroinflammation. Curr Opin Neurobiol. (2022) 75:102575. doi: 10.1016/j.conb.2022.102575
19. Dun, C, Zhang, Y, Yin, J, Su, B, Peng, X, and Liu, L. Bi-directional associations of epilepsy with dementia and Alzheimer's disease: a systematic review and meta-analysis of longitudinal studies. Age Ageing. (2022) 51:afac010. doi: 10.1093/ageing/afac010
20. Fang, Y, Si, X, Wang, J, Wang, Z, Chen, Y, Liu, Y, et al. Alzheimer disease and epilepsy: A Mendelian randomization study. Neurology. (2023) 101:e399–409. doi: 10.1212/WNL.0000000000207423
21. Friedman, D, Honig, LS, and Scarmeas, N. Seizures and epilepsy in Alzheimer's disease. CNS Neurosci Ther. (2012) 18:285–94. doi: 10.1111/j.1755-5949.2011.00251.x
22. Giorgi, FS, Saccaro, LF, Busceti, CL, Biagioni, F, and Fornai, F. Epilepsy and Alzheimer's disease: potential mechanisms for an association. Brain Res Bull. (2020) 160:107–20. doi: 10.1016/j.brainresbull.2020.04.009
23. Vossel, KA, Beagle, AJ, Rabinovici, GD, Shu, H, Lee, SE, Naasan, G, et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. (2013) 70:1158–66. doi: 10.1001/jamaneurol.2013.136
24. Vossel, KA, Tartaglia, MC, Nygaard, HB, Zeman, AZ, and Miller, BL. Epileptic activity in Alzheimer's disease: causes and clinical relevance. Lancet Neurol. (2017) 16:311–22. doi: 10.1016/S1474-4422(17)30044-3
25. Mendez, M, and Lim, G. Seizures in elderly patients with dementia: epidemiology and management. Drugs Aging. (2003) 20:791–803. doi: 10.2165/00002512-200320110-00001
26. Miranda, DDC, and Brucki, SMD. Epilepsy in patients with Alzheimer's disease: A systematic review. Dement Neuropsychol. (2014) 8:66–71. doi: 10.1590/S1980-57642014DN81000010
27. Ciliento, R, Gjini, K, Dabbs, K, Hermann, B, Riedner, B, Jones, S, et al. Prevalence and localization of nocturnal epileptiform discharges in mild cognitive impairment. Brain Commun. (2023) 5:fcad302. doi: 10.1093/braincomms/fcad302
28. Devulder, A, Macea, J, Kalkanis, A, De Winter, FL, Vandenbulcke, M, Vandenberghe, R, et al. Subclinical epileptiform activity and sleep disturbances in Alzheimer's disease. Brain Behav. (2023) 13:e3306. doi: 10.1002/brb3.3306
29. Horvath, AA, Papp, A, Zsuffa, J, Szucs, A, Luckl, J, Radai, F, et al. Subclinical epileptiform activity accelerates the progression of Alzheimer's disease: A long-term EEG study. Clin Neurophysiol. (2021) 132:1982–9. doi: 10.1016/j.clinph.2021.03.050
30. Lam, AD, Deck, G, Goldman, A, Eskandar, EN, Noebels, J, and Cole, AJ. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer's disease. Nat Med. (2017) 23:678–80. doi: 10.1038/nm.4330
31. Lam, AD, Sarkis, RA, Pellerin, KR, Jing, J, Dworetzky, BA, Hoch, DB, et al. Association of epileptiform abnormalities and seizures in Alzheimer disease. Neurology. (2020) 95:e2259–70. doi: 10.1212/wnl.0000000000010612
32. Musaeus, CS, Frederiksen, KS, Andersen, BB, Hogh, P, Kidmose, P, Fabricius, M, et al. Detection of subclinical epileptiform discharges in Alzheimer's disease using long-term outpatient EEG monitoring. Neurobiol Dis. (2023) 183:106149. doi: 10.1016/j.nbd.2023.106149
33. Vossel, KA, Ranasinghe, KG, Beagle, AJ, Mizuiri, D, Honma, SM, Dowling, AF, et al. Incidence and impact of subclinical epileptiform activity in Alzheimer's disease. Ann Neurol. (2016) 80:858–70. doi: 10.1002/ana.24794
34. Yeh, WC, Hsu, CY, Li, KY, Chien, CF, Huang, LC, and Yang, YH. Association between subclinical epileptiform discharge and the severity of cognitive decline in Alzheimer's disease: A longitudinal cohort study. J Alzheimers Dis. (2022) 90:305–12. doi: 10.3233/JAD-220567
35. Amatniek, JC, Hauser, WA, Delcastillo-Castaneda, C, Jacobs, DM, Marder, K, Bell, K, et al. Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia. (2006) 47:867–72. doi: 10.1111/j.1528-1167.2006.00554.x
36. Sarkis, RA, Dickerson, BC, Cole, AJ, and Chemali, ZN. Clinical and neurophysiologic characteristics of unprovoked seizures in patients diagnosed with dementia. J Neuropsychiatry Clin Neurosci. (2016) 28:56–61. doi: 10.1176/appi.neuropsych.15060143
37. Horvath, A, Szucs, A, Hidasi, Z, Csukly, G, Barcs, G, and Kamondi, A. Prevalence, semiology, and risk factors of epilepsy in Alzheimer's disease: An ambulatory EEG study. J Alzheimers Dis. (2018) 63:1045–54. doi: 10.3233/JAD-170925
38. Canet, G, Zub, E, Zussy, C, Hernandez, C, Blaquiere, M, Garcia, V, et al. Seizure activity triggers tau hyperphosphorylation and amyloidogenic pathways. Epilepsia. (2022) 63:919–35. doi: 10.1111/epi.17186
39. Dolev, I, Fogel, H, Milshtein, H, Berdichevsky, Y, Lipstein, N, Brose, N, et al. Spike bursts increase amyloid-beta 40/42 ratio by inducing a presenilin-1 conformational change. Nat Neurosci. (2013) 16:587–95. doi: 10.1038/nn.3376
40. Hwang, K, Vaknalli, RN, Addo-Osafo, K, Vicente, M, and Vossel, K. Tauopathy and epilepsy comorbidities and underlying mechanisms. Front Aging Neurosci. (2022) 14:903973. doi: 10.3389/fnagi.2022.903973
41. Joutsa, J, Rinne, JO, Hermann, B, Karrasch, M, Anttinen, A, Shinnar, S, et al. Association between childhood-onset epilepsy and amyloid burden 5 decades later. JAMA Neurol. (2017) 74:583–90. doi: 10.1001/jamaneurol.2016.6091
42. Johnson, EL, Krauss, GL, Lee, AK, Schneider, ALC, Dearborn, JL, Kucharska-Newton, AM, et al. Association between midlife risk factors and late-onset epilepsy: results from the atherosclerosis risk in communities study. JAMA Neurol. (2018) 75:1375–82. doi: 10.1001/jamaneurol.2018.1935
43. Hogan, DB, Fiest, KM, Roberts, JI, Maxwell, CJ, Dykeman, J, Pringsheim, T, et al. The prevalence and incidence of dementia with Lewy bodies: a systematic review. Can J Neurol Sci. (2016) 43:S83–95. doi: 10.1017/cjn.2016.2
44. Kane, JPM, Surendranathan, A, Bentley, A, Barker, SAH, Taylor, JP, Thomas, AJ, et al. Clinical prevalence of Lewy body dementia. Alzheimers Res Ther. (2018) 10:19. doi: 10.1186/s13195-018-0350-6
45. Mckeith, IG, Boeve, BF, Dickson, DW, Halliday, G, Taylor, JP, Weintraub, D, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB consortium. Neurology. (2017) 89:88–100. doi: 10.1212/WNL.0000000000004058
46. Yamada, M, Komatsu, J, Nakamura, K, Sakai, K, Samuraki-Yokohama, M, Nakajima, K, et al. Diagnostic criteria for dementia with Lewy bodies: updates and future directions. J Mov Disord. (2020) 13:1–10. doi: 10.14802/jmd.19052
47. Goedert, M, Spillantini, MG, Del Tredici, K, and Braak, H. 100 years of Lewy pathology. Nat Rev Neurol. (2013) 9:13–24. doi: 10.1038/nrneurol.2012.242
48. Harding, AJ, and Halliday, GM. Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol. (2001) 102:355–63. doi: 10.1007/s004010100390
49. Tiraboschi, P, Hansen, LA, Alford, M, Sabbagh, MN, Schoos, B, Masliah, E, et al. Cholinergic dysfunction in diseases with Lewy bodies. Neurology. (2000) 54:407–11. doi: 10.1212/WNL.54.2.407
50. Mattila, PM, Roytta, M, Torikka, H, Dickson, DW, and Rinne, JO. Cortical Lewy bodies and Alzheimer-type changes in patients with Parkinson's disease. Acta Neuropathol. (1998) 95:576–82. doi: 10.1007/s004010050843
51. Schneider, JA, Arvanitakis, Z, Bang, W, and Bennett, DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. (2007) 69:2197–204. doi: 10.1212/01.wnl.0000271090.28148.24
52. Beagle, AJ, Darwish, SM, Ranasinghe, KG, La, AL, Karageorgiou, E, and Vossel, KA. Relative incidence of seizures and myoclonus in Alzheimer's disease, dementia with Lewy bodies, and frontotemporal dementia. J Alzheimers Dis. (2017) 60:211–23. doi: 10.3233/JAD-170031
53. Marawar, R, Wakim, N, Albin, RL, and Dodge, H. Seizure occurrence and related mortality in dementia with Lewy bodies. Epilepsy Behav. (2020) 111:107311. doi: 10.1016/j.yebeh.2020.107311
54. Arnaldi, D, Donniaquio, A, Mattioli, P, Massa, F, Grazzini, M, Meli, R, et al. Epilepsy in neurodegenerative dementias: A clinical, epidemiological, and EEG study. J Alzheimers Dis. (2020) 74:865–74. doi: 10.3233/JAD-191315
55. Zelano, J, Brigo, F, and Garcia-Patek, S. Increased risk of epilepsy in patients registered in the Swedish dementia registry. Eur J Neurol. (2020) 27:129–35. doi: 10.1111/ene.14043
57. Engel, J Jr. Approaches to refractory epilepsy. Ann Indian Acad Neurol. (2014) 17:S12–7. doi: 10.4103/0972-2327.128644
58. Kwan, P, Schachter, SC, and Brodie, MJ. Drug-resistant epilepsy. N Engl J Med. (2011) 365:919–26. doi: 10.1056/NEJMra1004418
59. Stafstrom, CE, and Carmant, L. Seizures and epilepsy: an overview for neuroscientists. Cold Spring Harb Perspect Med. (2015) 5:a022426. doi: 10.1101/cshperspect.a022426
60. Vossel, K, Ranasinghe, KG, Beagle, AJ, La, A, Ah Pook, K, Castro, M, et al. Effect of Levetiracetam on cognition in patients with Alzheimer disease with and without epileptiform activity: A randomized clinical trial. JAMA Neurol. (2021) 78:1345–54. doi: 10.1001/jamaneurol.2021.3310
61. Cumbo, E, and Ligori, LD. Levetiracetam, lamotrigine, and phenobarbital in patients with epileptic seizures and Alzheimer's disease. Epilepsy Behav. (2010) 17:461–6. doi: 10.1016/j.yebeh.2010.01.015
62. Bakker, A, Krauss, GL, Albert, MS, Speck, CL, Jones, LR, Stark, CE, et al. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. (2012) 74:467–74. doi: 10.1016/j.neuron.2012.03.023
63. Rosenzweig-Lipson, S, Barton, R, Gallagher, M, and Mohs, R. HOPE4MCI trial: first trial targeting reduction of hippocampal overactivity to treat mild cognitive impairment due to Alzheimer’s disease with AGB101. Alzheimers Dement. (2021) 17:e057813. doi: 10.1002/alz.057813
64. Ranasinghe, KG, Kudo, K, Hinkley, L, Beagle, A, Lerner, H, Mizuiri, D, et al. Neuronal synchrony abnormalities associated with subclinical epileptiform activity in early-onset Alzheimer's disease. Brain. (2022) 145:744–53. doi: 10.1093/brain/awab442
65. Toniolo, S, Sen, A, and Husain, M. Modulation of brain Hyperexcitability: potential new therapeutic approaches in Alzheimer's disease. Int J Mol Sci. (2020) 21:9318. doi: 10.3390/ijms21239318
66. Morris, M, Sanchez, PE, Verret, L, Beagle, AJ, Guo, W, Dubal, D, et al. Network dysfunction in alpha-synuclein transgenic mice and human Lewy body dementia. Ann Clin Transl Neurol. (2015) 2:1012–28. doi: 10.1002/acn3.257
67. Musaeus, CS, Kjaer, TW, Cacic Hribljan, M, Andersen, BB, Hogh, P, Kidmose, P, et al. Subclinical epileptiform activity in dementia with Lewy bodies. Mov Disord. (2023) 38:1861–70. doi: 10.1002/mds.29531
68. Peters, ST, Fahrenkopf, A, Choquette, JM, Vermilyea, SC, Lee, MK, and Vossel, K. Ablating tau reduces Hyperexcitability and moderates electroencephalographic slowing in transgenic mice expressing A53T human alpha-Synuclein. Front Neurol. (2020) 11:563. doi: 10.3389/fneur.2020.00563
69. Zhao, B, Shen, LX, Ou, YN, Ma, YH, Dong, Q, Tan, L, et al. Risk of seizures and subclinical epileptiform activity in patients with dementia: A systematic review and meta-analysis. Ageing Res Rev. (2021) 72:101478. doi: 10.1016/j.arr.2021.101478
70. Borroni, B, Grassi, M, Costanzi, C, Archetti, S, Caimi, L, and Padovani, A. APOE genotype and cholesterol levels in lewy body dementia and Alzheimer disease: investigating genotype-phenotype effect on disease risk. Am J Geriatr Psychiatry. (2006) 14:1022–31. doi: 10.1097/01.JGP.0000225088.29353.08
71. Dickson, DW, Heckman, MG, Murray, ME, Soto, AI, Walton, RL, Diehl, NN, et al. APOE epsilon4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology. (2018) 91:e1182–95. doi: 10.1212/WNL.0000000000006212
72. Koutsodendris, N, Nelson, MR, Rao, A, and Huang, Y. Apolipoprotein E and Alzheimer's disease: findings, hypotheses, and potential mechanisms. Annu Rev Pathol. (2022) 17:73–99. doi: 10.1146/annurev-pathmechdis-030421-112756
73. Lumsden, AL, Mulugeta, A, Zhou, A, and Hypponen, E. Apolipoprotein E (APOE) genotype-associated disease risks: a phenome-wide, registry-based, case-control study utilising the UK biobank. EBioMedicine. (2020) 59:102954. doi: 10.1016/j.ebiom.2020.102954
74. Saunders, AM, Strittmatter, WJ, Schmechel, D, George-Hyslop, PH, Pericak-Vance, MA, Joo, SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. (1993) 43:1467–72. doi: 10.1212/WNL.43.8.1467
75. Sun, YY, Wang, Z, and Huang, HC. Roles of ApoE4 on the pathogenesis in Alzheimer's disease and the potential therapeutic approaches. Cell Mol Neurobiol. (2023) 43:3115–36. doi: 10.1007/s10571-023-01365-1
76. Blumcke, I, Brockhaus, A, Scheiwe, C, Rollbrocker, B, Wolf, HK, Elger, CE, et al. The apolipoprotein E epsilon 4 allele is not associated with early onset temporal lobe epilepsy. Neuroreport. (1997) 8:1235–7. doi: 10.1097/00001756-199703240-00035
77. Liang, Y, Zhou, Z, Wang, H, Cheng, X, Zhong, S, and Zhao, C. Association of apolipoprotein E genotypes with epilepsy risk: A systematic review and meta-analysis. Epilepsy Behav. (2019) 98:27–35. doi: 10.1016/j.yebeh.2019.06.015
78. Kauffman, MA, Consalvo, D, Moron, DG, Lereis, VP, and Kochen, S. ApoE epsilon4 genotype and the age at onset of temporal lobe epilepsy: a case-control study and meta-analysis. Epilepsy Res. (2010) 90:234–9. doi: 10.1016/j.eplepsyres.2010.05.007
79. Gambardella, A, Aguglia, U, Chifari, R, Labate, A, Manna, I, Serra, P, et al. ApoE epsilon4 allele and disease duration affect verbal learning in mild temporal lobe epilepsy. Epilepsia. (2005) 46:110–7. doi: 10.1111/j.0013-9580.2005.15804.x
80. Hunter, JM, Cirrito, JR, Restivo, JL, Kinley, RD, Sullivan, PM, Holtzman, DM, et al. Emergence of a seizure phenotype in aged apolipoprotein epsilon 4 targeted replacement mice. Brain Res. (2012) 1467:120–32. doi: 10.1016/j.brainres.2012.05.048
81. Chernick, D, Ortiz-Valle, S, Jeong, A, Qu, W, and Li, L. Peripheral versus central nervous system APOE in Alzheimer's disease: interplay across the blood-brain barrier. Neurosci Lett. (2019) 708:134306. doi: 10.1016/j.neulet.2019.134306
82. Eichner, JE, Dunn, ST, Perveen, G, Thompson, DM, Stewart, KE, and Stroehla, BC. Apolipoprotein E polymorphism and cardiovascular disease: a HuGE review. Am J Epidemiol. (2002) 155:487–95. doi: 10.1093/aje/155.6.487
83. Martins, IJ, Hone, E, Foster, JK, Sunram-Lea, SI, Gnjec, A, Fuller, SJ, et al. Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer's disease and cardiovascular disease. Mol Psychiatry. (2006) 11:721–36. doi: 10.1038/sj.mp.4001854
84. Lin, YT, Seo, J, Gao, F, Feldman, HM, Wen, HL, Penney, J, et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer's disease phenotypes in human iPSC-derived brain cell types. Neuron. (2018) 98:1294. doi: 10.1016/j.neuron.2018.06.011
85. Xiaoqin, Z, Zhengli, L, Changgeng, Z, Xiaojing, W, and Li, L. Changes in behavior and amino acid neurotransmitters in the brain of rats with seizure induced by IL-1beta or IL-6. J Huazhong Univ Sci Technolog Med Sci. (2005) 25:236–9. doi: 10.1007/BF02828129
86. Guo, J, Chi, S, Xu, H, Jin, G, and Qi, Z. Effects of cholesterol levels on the excitability of rat hippocampal neurons. Mol Membr Biol. (2008) 25:216–23. doi: 10.1080/09687680701805541
87. Malnar, M, Hecimovic, S, Mattsson, N, and Zetterberg, H. Bidirectional links between Alzheimer's disease and Niemann-pick type C disease. Neurobiol Dis. (2014) 72:37–47. doi: 10.1016/j.nbd.2014.05.033
88. Vivas, O, Tiscione, SA, Dixon, RE, Ory, DS, and Dickson, EJ. Niemann-pick type C disease reveals a link between lysosomal cholesterol and PtdIns (4, 5) P (2) that regulates neuronal excitability. Cell Rep. (2019) 27:2636–2648 e 2634. doi: 10.1016/j.celrep.2019.04.099
89. Dai, MH, Zheng, H, Zeng, LD, and Zhang, Y. The genes associated with early-onset Alzheimer's disease. Oncotarget. (2018) 9:15132–43. doi: 10.18632/oncotarget.23738
90. Kim, DH, Yeo, SH, Park, JM, Choi, JY, Lee, TH, Park, SY, et al. Genetic markers for diagnosis and pathogenesis of Alzheimer's disease. Gene. (2014) 545:185–93. doi: 10.1016/j.gene.2014.05.031
91. Lanoiselee, HM, Nicolas, G, Wallon, D, Rovelet-Lecrux, A, Lacour, M, Rousseau, S, et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. (2017) 14:e1002270. doi: 10.1371/journal.pmed.1002270
92. Ovchinnikov, DA, Korn, O, Virshup, I, Wells, CA, and Wolvetang, EJ. The impact of APP on Alzheimer-like pathogenesis and gene expression in down syndrome iPSC-derived neurons. Stem Cell Rep. (2018) 11:32–42. doi: 10.1016/j.stemcr.2018.05.004
93. Sleegers, K, Brouwers, N, Gijselinck, I, Theuns, J, Goossens, D, Wauters, J, et al. APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain. (2006) 129:2977–83. doi: 10.1093/brain/awl203
94. De Strooper, B, Iwatsubo, T, and Wolfe, MS. Presenilins and gamma-secretase: structure, function, and role in Alzheimer disease. Cold Spring Harb Perspect Med. (2012) 2:a006304. doi: 10.1101/cshperspect.a006304
95. Gu, L, and Guo, Z. Alzheimer's Abeta42 and Abeta40 peptides form interlaced amyloid fibrils. J Neurochem. (2013) 126:305–11. doi: 10.1111/jnc.12202
96. Kim, J, Onstead, L, Randle, S, Price, R, Smithson, L, Zwizinski, C, et al. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. (2007) 27:627–33. doi: 10.1523/JNEUROSCI.4849-06.2007
97. Kuperstein, I, Broersen, K, Benilova, I, Rozenski, J, Jonckheere, W, Debulpaep, M, et al. Neurotoxicity of Alzheimer's disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J. (2010) 29:3408–20. doi: 10.1038/emboj.2010.211
98. Pauwels, K, Williams, TL, Morris, KL, Jonckheere, W, Vandersteen, A, Kelly, G, et al. Structural basis for increased toxicity of pathological abeta42: abeta 40 ratios in Alzheimer disease. J Biol Chem. (2012) 287:5650–60. doi: 10.1074/jbc.M111.264473
99. Brouwers, N, Sleegers, K, and Van Broeckhoven, C. Molecular genetics of Alzheimer's disease: an update. Ann Med. (2008) 40:562–83. doi: 10.1080/07853890802186905
100. Mol, MO, van der Lee, SJ, Hulsman, M, Pijnenburg, YAL, and Scheltens, PNetherlands Brain Bank, et al. Mapping the genetic landscape of early-onset Alzheimer's disease in a cohort of 36 families. Alzheimers Res Ther. (2022) 14:77. doi: 10.1186/s13195-022-01018-3
101. Wingo, TS, Lah, JJ, Levey, AI, and Cutler, DJ. Autosomal recessive causes likely in early-onset Alzheimer disease. Arch Neurol. (2012) 69:59–64. doi: 10.1001/archneurol.2011.221
102. Rahman, MM, and Fatema, K. Seizures in down syndrome: An update. Mymensingh Med J. (2019) 28:712–5.
103. Salehi, A, Ashford, JW, and Mufson, EJ. The link between Alzheimer's disease and down syndrome. A historical perspective. Curr Alzheimer Res. (2016) 13:2–6. doi: 10.2174/1567205012999151021102914
104. Fortea, J, Zaman, SH, Hartley, S, Rafii, MS, Head, E, and Carmona-Iragui, M. Alzheimer's disease associated with down syndrome: a genetic form of dementia. Lancet Neurol. (2021) 20:930–42. doi: 10.1016/S1474-4422(21)00245-3
105. Lott, IT, and Head, E. Dementia in down syndrome: unique insights for Alzheimer disease research. Nat Rev Neurol. (2019) 15:135–47. doi: 10.1038/s41582-018-0132-6
106. Kruger, R, Kuhn, W, Muller, T, Woitalla, D, Graeber, M, Kosel, S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. (1998) 18:106–8. doi: 10.1038/ng0298-106
107. Polymeropoulos, MH, Lavedan, C, Leroy, E, Ide, SE, Dehejia, A, Dutra, A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. (1997) 276:2045–7. doi: 10.1126/science.276.5321.2045
108. Zarranz, JJ, Alegre, J, Gomez-Esteban, JC, Lezcano, E, Ros, R, Ampuero, I, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. (2004) 55:164–73. doi: 10.1002/ana.10795
109. Mehra, S, Sahay, S, and Maji, SK. Alpha-Synuclein misfolding and aggregation: implications in Parkinson's disease pathogenesis. Biochim Biophys Acta Proteins Proteom. (2019) 1867:890–908. doi: 10.1016/j.bbapap.2019.03.001
110. Teravskis, PJ, Covelo, A, Miller, EC, Singh, B, Martell-Martinez, HA, Benneyworth, MA, et al. A53T mutant alpha-Synuclein induces tau-dependent postsynaptic impairment independently of neurodegenerative changes. J Neurosci. (2018) 38:9754–67. doi: 10.1523/jneurosci.0344-18.2018
111. Duda, JE, Giasson, BI, Mabon, ME, Miller, DC, Golbe, LI, Lee, VM, et al. Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol. (2002) 104:7–11. doi: 10.1007/s00401-002-0563-3
112. Lee, MK, Stirling, W, Xu, Y, Xu, X, Qui, D, Mandir, AS, et al. Human α-synuclein-harboring familial Parkinson's disease-linked ala-53 → Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A. (2002) 99:8968–73. doi: 10.1073/pnas.132197599
113. Gigout, S, Wierschke, S, Lehmann, TN, Horn, P, Dehnicke, C, and Deisz, RA. Muscarinic acetylcholine receptor-mediated effects in slices from human epileptogenic cortex. Neuroscience. (2012) 223:399–411. doi: 10.1016/j.neuroscience.2012.07.044
114. Picciotto, MR, Higley, MJ, and Mineur, YS. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron. (2012) 76:116–29. doi: 10.1016/j.neuron.2012.08.036
115. Chen, ZR, Huang, JB, Yang, SL, and Hong, FF. Role of cholinergic signaling in Alzheimer's disease. Molecules. (2022) 27:1816. doi: 10.3390/molecules27061816
116. Duda, JE. Pathology and neurotransmitter abnormalities of dementia with Lewy bodies. Dement Geriatr Cogn Disord. (2004) 17:3–14. doi: 10.1159/000074677
117. Barber, R, Ballard, C, Mckeith, IG, Gholkar, A, and O'brien, JT. MRI volumetric study of dementia with Lewy bodies: a comparison with AD and vascular dementia. Neurology. (2000) 54:1304–9. doi: 10.1212/WNL.54.6.1304
118. Andersson, M, Hansson, O, Minthon, L, Rosen, I, and Londos, E. Electroencephalogram variability in dementia with lewy bodies, Alzheimer's disease and controls. Dement Geriatr Cogn Disord. (2008) 26:284–90. doi: 10.1159/000160962
119. Bonanni, L, Thomas, A, Tiraboschi, P, Perfetti, B, Varanese, S, and Onofrj, M. EEG comparisons in early Alzheimer's disease, dementia with Lewy bodies and Parkinson's disease with dementia patients with a 2-year follow-up. Brain. (2008) 131:690–705. doi: 10.1093/brain/awm322
120. Lam, B, Hollingdrake, E, Kennedy, JL, Black, SE, and Masellis, M. Cholinesterase inhibitors in Alzheimer's disease and Lewy body spectrum disorders: the emerging pharmacogenetic story. Hum Genomics. (2009) 4:91–106. doi: 10.1186/1479-7364-4-2-91
121. Simard, M, and Van Reekum, R. The acetylcholinesterase inhibitors for treatment of cognitive and behavioral symptoms in dementia with Lewy bodies. J Neuropsychiatry Clin Neurosci. (2004) 16:409–25. doi: 10.1176/jnp.16.4.409
122. Kam, K, Duffy, AM, Moretto, J, Lafrancois, JJ, and Scharfman, HE. Interictal spikes during sleep are an early defect in the Tg2576 mouse model of beta-amyloid neuropathology. Sci Rep. (2016) 6:20119. doi: 10.1038/srep20119
123. Jin, N, Ziyatdinova, S, Gureviciene, I, and Tanila, H. Response of spike-wave discharges in aged APP/PS1 Alzheimer model mice to antiepileptic, metabolic and cholinergic drugs. Sci Rep. (2020) 10:11851. doi: 10.1038/s41598-020-68845-y
124. Schousboe, A, Scafidi, S, Bak, LK, Waagepetersen, HS, and Mckenna, MC. Glutamate metabolism in the brain focusing on astrocytes. Adv Neurobiol. (2014) 11:13–30. doi: 10.1007/978-3-319-08894-5_2
125. Albrecht, J, and Zielinska, M. Mechanisms of excessive extracellular glutamate accumulation in temporal lobe epilepsy. Neurochem Res. (2017) 42:1724–34. doi: 10.1007/s11064-016-2105-8
126. Czapski, GA, and Strosznajder, JB. Glutamate and GABA in microglia-neuron cross-talk in Alzheimer's disease. Int J Mol Sci. (2021) 22:11677. doi: 10.3390/ijms222111677
127. Sarac, S, Afzal, S, Broholm, H, Madsen, FF, Ploug, T, and Laursen, H. Excitatory amino acid transporters EAAT-1 and EAAT-2 in temporal lobe and hippocampus in intractable temporal lobe epilepsy. APMIS. (2009) 117:291–301. doi: 10.1111/j.1600-0463.2009.02443.x
128. Soni, N, Reddy, BV, and Kumar, P. GLT-1 transporter: an effective pharmacological target for various neurological disorders. Pharmacol Biochem Behav. (2014) 127:70–81. doi: 10.1016/j.pbb.2014.10.001
129. Chivukula, AS, Suslova, M, Kortzak, D, Kovermann, P, and Fahlke, C. Functional consequences of SLC1A3 mutations associated with episodic ataxia 6. Hum Mutat. (2020) 41:1892–905. doi: 10.1002/humu.24089
130. Kovermann, P, Kolobkova, Y, Franzen, A, and Fahlke, C. Mutations associated with epileptic encephalopathy modify EAAT2 anion channel function. Epilepsia. (2022) 63:388–401. doi: 10.1111/epi.17154
131. De Ceglia, R, Ledonne, A, Litvin, DG, Lind, BL, Carriero, G, Latagliata, EC, et al. Specialized astrocytes mediate glutamatergic gliotransmission in the CNS. Nature. (2023) 622:120–9. doi: 10.1038/s41586-023-06502-w
132. Mahmoud, S, Gharagozloo, M, Simard, C, and Gris, D. Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cell. (2019) 8:184. doi: 10.3390/cells8020184
133. Robinson, MB. The family of sodium-dependent glutamate transporters: a focus on the GLT-1/EAAT2 subtype. Neurochem Int. (1998) 33:479–91. doi: 10.1016/S0197-0186(98)00055-2
134. Garcia-Esparcia, P, Diaz-Lucena, D, Ainciburu, M, Torrejon-Escribano, B, Carmona, M, Llorens, F, et al. Glutamate transporter GLT1 expression in Alzheimer disease and dementia with Lewy bodies. Front Aging Neurosci. (2018) 10:122. doi: 10.3389/fnagi.2018.00122
135. Liraz, O, Boehm-Cagan, A, and Michaelson, DM. ApoE4 induces Abeta42, tau, and neuronal pathology in the hippocampus of young targeted replacement apoE4 mice. Mol Neurodegener. (2013) 8:16.
136. Kirvell, SL, Esiri, M, and Francis, PT. Down-regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer's disease. J Neurochem. (2006) 98:939–50. doi: 10.1111/j.1471-4159.2006.03935.x
137. Takahashi, K, Foster, JB, and Lin, CL. Glutamate transporter EAAT2: regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell Mol Life Sci. (2015) 72:3489–506. doi: 10.1007/s00018-015-1937-8
138. Takahashi, K, Kong, Q, Lin, Y, Stouffer, N, Schulte, DA, Lai, L, et al. Restored glial glutamate transporter EAAT2 function as a potential therapeutic approach for Alzheimer's disease. J Exp Med. (2015) 212:319–32. doi: 10.1084/jem.20140413
139. Scott, HL, Pow, DV, Tannenberg, AE, and Dodd, PR. Aberrant expression of the glutamate transporter excitatory amino acid transporter 1 (EAAT1) in Alzheimer's disease. J Neurosci. (2002) 22:RC206. doi: 10.1523/JNEUROSCI.22-03-j0004.2002
140. Scott, HA, Gebhardt, FM, Mitrovic, AD, Vandenberg, RJ, and Dodd, PR. Glutamate transporter variants reduce glutamate uptake in Alzheimer's disease. Neurobiol Aging. (2011) 32:553.e1–553.e11. doi: 10.1016/j.neurobiolaging.2010.03.008
141. Hunsberger, HC, Weitzner, DS, Rudy, CC, Hickman, JE, Libell, EM, Speer, RR, et al. Riluzole rescues glutamate alterations, cognitive deficits, and tau pathology associated with P301L tau expression. J Neurochem. (2015) 135:381–94. doi: 10.1111/jnc.13230
142. Alijanpour, S, Miryounesi, M, and Ghafouri-Fard, S. The role of excitatory amino acid transporter 2 (EAAT2) in epilepsy and other neurological disorders. Metab Brain Dis. (2023) 38:1–16. doi: 10.1007/s11011-022-01091-5
143. Mattson, MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci. (2008) 1144:97–112. doi: 10.1196/annals.1418.005
144. Tymianski, M, Charlton, MP, Carlen, PL, and Tator, CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. (1993) 13:2085–104. doi: 10.1523/JNEUROSCI.13-05-02085.1993
145. Dubinsky, JM. Intracellular calcium levels during the period of delayed excitotoxicity. J Neurosci. (1993) 13:623–31. doi: 10.1523/JNEUROSCI.13-02-00623.1993
146. Siman, R, Noszek, JC, and Kegerise, C. Calpain I activation is specifically related to excitatory amino acid induction of hippocampal damage. J Neurosci. (1989) 9:1579–90. doi: 10.1523/JNEUROSCI.09-05-01579.1989
147. Lazarewicz, JW, Wroblewski, JT, and Costa, E. N-methyl-D-aspartate-sensitive glutamate receptors induce calcium-mediated arachidonic acid release in primary cultures of cerebellar granule cells. J Neurochem. (1990) 55:1875–81. doi: 10.1111/j.1471-4159.1990.tb05771.x
148. Danysz, W, Parsons, CG, Mobius, HJ, Stoffler, A, and Quack, G. Neuroprotective and symptomatological action of memantine relevant for alzheimer’s disease — a unified glutamatergic hypothesis on the mechanism of action. Neurotox Res. (2000) 2:85–97. doi: 10.1007/BF03033787
149. Wenk, GL. Neuropathologic changes in Alzheimer's disease: potential targets for treatment. J Clin Psychiatry. (2006) 67:3–7.
150. Barker-Haliski, M, and White, HS. Glutamatergic mechanisms associated with seizures and epilepsy. Cold Spring Harb Perspect Med. (2015) 5:a022863. doi: 10.1101/cshperspect.a022863
151. Green, JL, Dos Santos, WF, and Fontana, ACK. Role of glutamate excitotoxicity and glutamate transporter EAAT2 in epilepsy: opportunities for novel therapeutics development. Biochem Pharmacol. (2021) 193:114786. doi: 10.1016/j.bcp.2021.114786
152. Hanada, T. Ionotropic glutamate receptors in epilepsy: A review focusing on AMPA and NMDA receptors. Biomol Ther. (2020) 10:464. doi: 10.3390/biom10030464
153. Srivastava, A, Das, B, Yao, AY, and Yan, R. Metabotropic glutamate receptors in Alzheimer's disease synaptic dysfunction: therapeutic opportunities and Hope for the future. J Alzheimers Dis. (2020) 78:1345–61. doi: 10.3233/jad-201146
154. Mendez-Armenta, M, Nava-Ruiz, C, Juarez-Rebollar, D, Rodriguez-Martinez, E, and Gomez, PY. Oxidative stress associated with neuronal apoptosis in experimental models of epilepsy. Oxidative Med Cell Longev. (2014) 2014:293689:1–12. doi: 10.1155/2014/293689
155. Hanson, JE, Ma, K, Elstrott, J, Weber, M, Saillet, S, Khan, AS, et al. GluN2A NMDA receptor enhancement improves brain oscillations, synchrony, and cognitive functions in Dravet syndrome and Alzheimer's disease models. Cell Rep. (2020) 30:e384:381–396.e4. doi: 10.1016/j.celrep.2019.12.030
156. Lerdkrai, C, Asavapanumas, N, Brawek, B, Kovalchuk, Y, Mojtahedi, N, Olmedillas Del Moral, M, et al. Intracellular ca(2+) stores control in vivo neuronal hyperactivity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. (2018) 115:E1279–88. doi: 10.1073/pnas.1714409115
157. Matsunaga, S, Kishi, T, and Iwata, N. Memantine monotherapy for Alzheimer's disease: a systematic review and meta-analysis. PLoS One. (2015) 10:e0123289. doi: 10.1371/journal.pone.0123289
158. Brines, ML, Sundaresan, S, Spencer, DD, and De Lanerolle, NC. Quantitative autoradiographic analysis of ionotropic glutamate receptor subtypes in human temporal lobe epilepsy: up-regulation in reorganized epileptogenic hippocampus. Eur J Neurosci. (1997) 9:2035–44. doi: 10.1111/j.1460-9568.1997.tb01371.x
159. Graebenitz, S, Kedo, O, Speckmann, EJ, Gorji, A, Panneck, H, Hans, V, et al. Interictal-like network activity and receptor expression in the epileptic human lateral amygdala. Brain. (2011) 134:2929–47. doi: 10.1093/brain/awr202
160. Mathern, GW, Pretorius, JK, Leite, JP, Kornblum, HI, Mendoza, D, Lozada, A, et al. Hippocampal AMPA and NMDA mRNA levels and subunit immunoreactivity in human temporal lobe epilepsy patients and a rodent model of chronic mesial limbic epilepsy. Epilepsy Res. (1998) 32:154–71. doi: 10.1016/S0920-1211(98)00048-5
161. Ying, Z, Babb, TL, Comair, YG, Bushey, M, and Touhalisky, K. Increased densities of AMPA Glu R1 subunit proteins and presynaptic mossy fiber sprouting in the fascia dentata of human hippocampal epilepsy. Brain Res. (1998) 798:239–46. doi: 10.1016/S0006-8993(98)00421-1
162. Saxton, RA, and Sabatini, DM. mTOR signaling in growth, metabolism, and disease. Cell. (2017) 169:361–71. doi: 10.1016/j.cell.2017.03.035
163. Bockaert, J, and Marin, P. mTOR in brain physiology and pathologies. Physiol Rev. (2015) 95:1157–87. doi: 10.1152/physrev.00038.2014
164. Stavoe, AKH, and Holzbaur, ELF. Autophagy in neurons. Annu Rev Cell Dev Biol. (2019) 35:477–500. doi: 10.1146/annurev-cellbio-100818-125242
165. Overhoff, M, Tellkamp, F, Hess, S, Tolve, M, Tutas, J, Faerfers, M, et al. Autophagy regulates neuronal excitability by controlling cAMP/protein kinase A signaling at the synapse. EMBO J. (2022) 41:e110963. doi: 10.15252/embj.2022110963
166. Butler, CR, Boychuk, JA, and Smith, BN. Effects of rapamycin treatment on neurogenesis and synaptic reorganization in the dentate gyrus after controlled cortical impact injury in mice. Front Syst Neurosci. (2015) 9:163. doi: 10.3389/fnsys.2015.00163
167. Citraro, R, Leo, A, Constanti, A, Russo, E, and De Sarro, G. mTOR pathway inhibition as a new therapeutic strategy in epilepsy and epileptogenesis. Pharmacol Res. (2016) 107:333–43. doi: 10.1016/j.phrs.2016.03.039
168. Zeng, LH, Xu, L, Gutmann, DH, and Wong, M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. (2008) 63:444–53. doi: 10.1002/ana.21331
169. Zeng, LH, Rensing, NR, and Wong, M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. (2009) 29:6964–72. doi: 10.1523/JNEUROSCI.0066-09.2009
170. An, WL, Cowburn, RF, Li, L, Braak, H, Alafuzoff, I, Iqbal, K, et al. Up-regulation of phosphorylated/activated p 70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. Am J Pathol. (2003) 163:591–607. doi: 10.1016/S0002-9440(10)63687-5
171. Crews, L, Spencer, B, Desplats, P, Patrick, C, Paulino, A, Rockenstein, E, et al. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One. (2010) 5:e9313. doi: 10.1371/journal.pone.0009313
172. Hou, X, Watzlawik, JO, Fiesel, FC, and Springer, W. Autophagy in Parkinson's disease. J Mol Biol. (2020) 432:2651–72. doi: 10.1016/j.jmb.2020.01.037
173. Li, X, Alafuzoff, I, Soininen, H, Winblad, B, and Pei, JJ. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer's disease brain. FEBS J. (2005) 272:4211–20. doi: 10.1111/j.1742-4658.2005.04833.x
174. Gourmaud, S, Stewart, DA, Irwin, DJ, Roberts, N, Barbour, AJ, Eberwine, G, et al. The role of mTORC1 activation in seizure-induced exacerbation of Alzheimer's disease. Brain. (2022) 145:324–39. doi: 10.1093/brain/awab268
175. Spilman, P, Podlutskaya, N, Hart, MJ, Debnath, J, Gorostiza, O, Bredesen, D, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One. (2010) 5:e9979. doi: 10.1371/journal.pone.0009979
176. Fortin, DL, Troyer, MD, Nakamura, K, Kubo, S, Anthony, MD, and Edwards, RH. Lipid rafts mediate the synaptic localization of alpha-synuclein. J Neurosci. (2004) 24:6715–23. doi: 10.1523/JNEUROSCI.1594-04.2004
177. Maroteaux, L, Campanelli, JT, and Scheller, RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. (1988) 8:2804–15. doi: 10.1523/JNEUROSCI.08-08-02804.1988
178. Cabin, DE, Shimazu, K, Murphy, D, Cole, NB, Gottschalk, W, Mcilwain, KL, et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J Neurosci. (2002) 22:8797–807. doi: 10.1523/JNEUROSCI.22-20-08797.2002
179. Cheng, F, Vivacqua, G, and Yu, S. The role of alpha-synuclein in neurotransmission and synaptic plasticity. J Chem Neuroanat. (2011) 42:242–8. doi: 10.1016/j.jchemneu.2010.12.001
180. Lee, HJ, Bae, EJ, and Lee, SJ. Extracellular α-synuclein—a novel and crucial factor in Lewy body diseases. Nat Rev Neurol. (2014) 10:92–8. doi: 10.1038/nrneurol.2013.275
181. Paillusson, S, Clairembault, T, Biraud, M, Neunlist, M, and Derkinderen, P. Activity-dependent secretion of alpha-synuclein by enteric neurons. J Neurochem. (2013) 125:512–7. doi: 10.1111/jnc.12131
182. Choi, J, Kim, SY, Kim, H, Lim, BC, Hwang, H, Chae, JH, et al. Serum alpha-synuclein and IL-1beta are increased and correlated with measures of disease severity in children with epilepsy: potential prognostic biomarkers? BMC Neurol. (2020) 20:85. doi: 10.1186/s12883-020-01662-y
183. Hussein, AM, Eldosoky, M, El-Shafey, M, El-Mesery, M, Ali, AN, Abbas, KM, et al. Effects of metformin on apoptosis and alpha-synuclein in a rat model of pentylenetetrazole-induced epilepsy. Can J Physiol Pharmacol. (2019) 97:37–46. doi: 10.1139/cjpp-2018-0266
184. Li, A, Choi, YS, Dziema, H, Cao, R, Cho, HY, Jung, YJ, et al. Proteomic profiling of the epileptic dentate gyrus. Brain Pathol. (2010) 20:1077–89. doi: 10.1111/j.1750-3639.2010.00414.x
185. Yang, JW, Czech, T, Felizardo, M, Baumgartner, C, and Lubec, G. Aberrant expression of cytoskeleton proteins in hippocampus from patients with mesial temporal lobe epilepsy. Amino Acids. (2006) 30:477–93. doi: 10.1007/s00726-005-0281-y
186. Tweedy, C, Kindred, N, Curry, J, Williams, C, Taylor, JP, Atkinson, P, et al. Hippocampal network hyperexcitability in young transgenic mice expressing human mutant alpha-synuclein. Neurobiol Dis. (2021) 149:105226. doi: 10.1016/j.nbd.2020.105226
187. Marques, O, and Outeiro, TF. Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis. (2012) 3:e350. doi: 10.1038/cddis.2012.94
188. Gotz, J, Probst, A, Spillantini, MG, Schafer, T, Jakes, R, Burki, K, et al. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. (1995) 14:1304–13. doi: 10.1002/j.1460-2075.1995.tb07116.x
189. Papasozomenos, SC, and Binder, LI. Phosphorylation determines two distinct species of tau in the central nervous system. Cell Motil Cytoskeleton. (1987) 8:210–26. doi: 10.1002/cm.970080303
190. Kovacs, GG. Tauopathies. Handb Clin Neurol. (2017) 145:355–68. doi: 10.1016/B978-0-12-802395-2.00025-0
191. Meng, JX, Zhang, Y, Saman, D, Haider, AM, De, S, Sang, JC, et al. Hyperphosphorylated tau self-assembles into amorphous aggregates eliciting TLR4-dependent responses. Nat Commun. (2022) 13:2692. doi: 10.1038/s41467-022-30461-x
192. Xia, Y, Prokop, S, Gorion, KM, Kim, JD, Sorrentino, ZA, Bell, BM, et al. Tau Ser 208 phosphorylation promotes aggregation and reveals neuropathologic diversity in Alzheimer's disease and other tauopathies. Acta Neuropathol Commun. (2020) 8:88. doi: 10.1186/s40478-020-00967-w
193. Thom, M, Liu, JY, Thompson, P, Phadke, R, Narkiewicz, M, Martinian, L, et al. Neurofibrillary tangle pathology and Braak staging in chronic epilepsy in relation to traumatic brain injury and hippocampal sclerosis: a post-mortem study. Brain. (2011) 134:2969–81. doi: 10.1093/brain/awr209
194. Concepcion, FA, Ekstrom, NA, Khan, MN, Estes, OO, and Poolos, NP. Progressive dysregulation of tau phosphorylation in an animal model of temporal lobe epilepsy. Neuroscience. (2023) 522:42–56. doi: 10.1016/j.neuroscience.2023.04.020
195. Putra, M, Puttachary, S, Liu, G, Lee, G, and Thippeswamy, T. Fyn-tau ablation modifies PTZ-induced seizures and post-seizure hallmarks of early Epileptogenesis. Front Cell Neurosci. (2020) 14:592374. doi: 10.3389/fncel.2020.592374
196. Roberson, ED, Halabisky, B, Yoo, JW, Yao, J, Chin, J, Yan, F, et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. (2011) 31:700–11. doi: 10.1523/JNEUROSCI.4152-10.2011
197. Maeda, S, Djukic, B, Taneja, P, Yu, GQ, Lo, I, Davis, A, et al. Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep. (2016) 17:530–51. doi: 10.15252/embr.201541438
198. Crimins, JL, Rocher, AB, and Luebke, JI. Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathol. (2012) 124:777–95. doi: 10.1007/s00401-012-1038-9
199. Witton, J, Staniaszek, LE, Bartsch, U, Randall, AD, Jones, MW, and Brown, JT. Disrupted hippocampal sharp-wave ripple-associated spike dynamics in a transgenic mouse model of dementia. J Physiol. (2016) 594:4615–30. doi: 10.1113/jphysiol.2014.282889
200. Decker, JM, Kruger, L, Sydow, A, Dennissen, FJ, Siskova, Z, Mandelkow, E, et al. The tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep. (2016) 17:552–69. doi: 10.15252/embr.201541439
201. Kilian, JG, Hsu, HW, Mata, K, Wolf, FW, and Kitazawa, M. Astrocyte transport of glutamate and neuronal activity reciprocally modulate tau pathology in Drosophila. Neuroscience. (2017) 348:191–200. doi: 10.1016/j.neuroscience.2017.02.011
202. Devos, SL, Goncharoff, DK, Chen, G, Kebodeaux, CS, Yamada, K, Stewart, FR, et al. Antisense reduction of tau in adult mice protects against seizures. J Neurosci. (2013) 33:12887–97. doi: 10.1523/JNEUROSCI.2107-13.2013
203. Palop, JJ, and Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci. (2016) 17:777–92. doi: 10.1038/nrn.2016.141
204. Costa, C, Romoli, M, and Calabresi, P. Late onset epilepsy and Alzheimer's disease: exploring the dual pathogenic role of amyloid-beta. Brain. (2018) 141:e60. doi: 10.1093/brain/awy162
205. Ovsepian, SV, and O'leary, VB. Neuronal activity and amyloid plaque pathology: an update. J Alzheimers Dis. (2016) 49:13–9. doi: 10.3233/JAD-150544
206. Busche, MA, Eichhoff, G, Adelsberger, H, Abramowski, D, Wiederhold, KH, Haass, C, et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. (2008) 321:1686–9. doi: 10.1126/science.1162844
207. Minkeviciene, R, Rheims, S, Dobszay, MB, Zilberter, M, Hartikainen, J, Fulop, L, et al. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. (2009) 29:3453–62. doi: 10.1523/JNEUROSCI.5215-08.2009
208. Vande Vyver, M, Barker-Haliski, M, Aourz, N, Nagels, G, Bjerke, M, Engelborghs, S, et al. Higher susceptibility to 6 Hz corneal kindling and lower responsiveness to antiseizure drugs in mouse models of Alzheimer's disease. Epilepsia. (2022) 63:2703–15. doi: 10.1111/epi.17355
209. Ziyatdinova, S, Ronnback, A, Gurevicius, K, Miszczuk, D, Graff, C, Winblad, B, et al. Increased epileptiform EEG activity and decreased seizure threshold in Arctic APP transgenic mouse model of Alzheimer's disease. Curr Alzheimer Res. (2016) 13:817–30. doi: 10.2174/1567205013666160129095508
210. Shankar, GM, Li, S, Mehta, TH, Garcia-Munoz, A, Shepardson, NE, Smith, I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. (2008) 14:837–42. doi: 10.1038/nm1782
211. Ping, Y, Hahm, ET, Waro, G, Song, Q, Vo-Ba, DA, Licursi, A, et al. Linking abeta42-induced hyperexcitability to neurodegeneration, learning and motor deficits, and a shorter lifespan in an Alzheimer's model. PLoS Genet. (2015) 11:e1005025. doi: 10.1371/journal.pgen.1005025
212. Ho, R, Ortiz, D, and Shea, TB. Amyloid-beta promotes calcium influx and neurodegeneration via stimulation of L voltage-sensitive calcium channels rather than NMDA channels in cultured neurons. J Alzheimers Dis. (2001) 3:479–83. doi: 10.3233/JAD-2001-3507
213. Talantova, M, Sanz-Blasco, S, Zhang, X, Xia, P, Akhtar, MW, Okamoto, S, et al. Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A. (2013) 110:E2518–27. doi: 10.1073/pnas.1306832110
214. Zott, B, Simon, MM, Hong, W, Unger, F, Chen-Engerer, HJ, Frosch, MP, et al. A vicious cycle of beta amyloid-dependent neuronal hyperactivation. Science. (2019) 365:559–65. doi: 10.1126/science.aay0198
215. Glass, CK, Saijo, K, Winner, B, Marchetto, MC, and Gage, FH. Mechanisms underlying inflammation in neurodegeneration. Cell. (2010) 140:918–34. doi: 10.1016/j.cell.2010.02.016
216. Patani, R, Hardingham, GE, and Liddelow, SA. Functional roles of reactive astrocytes in neuroinflammation and neurodegeneration. Nat Rev Neurol. (2023) 19:395–409. doi: 10.1038/s41582-023-00822-1
217. Bernaus, A, Blanco, S, and Sevilla, A. Glia crosstalk in Neuroinflammatory diseases. Front Cell Neurosci. (2020) 14:209. doi: 10.3389/fncel.2020.00209
218. Devinsky, O, Vezzani, A, Najjar, S, De Lanerolle, NC, and Rogawski, MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci. (2013) 36:174–84. doi: 10.1016/j.tins.2012.11.008
219. Pracucci, E, Pillai, V, Lamers, D, Parra, R, and Landi, S. Neuroinflammation: A signature or a cause of epilepsy? Int J Mol Sci. (2021) 22:6981. doi: 10.3390/ijms22136981
220. Sanz, P, and Garcia-Gimeno, MA. Reactive glia inflammatory signaling pathways and epilepsy. Int J Mol Sci. (2020) 21:4096. doi: 10.3390/ijms21114096
221. De Vries, EE, Van Den Munckhof, B, Braun, KP, Van Royen-Kerkhof, A, De Jager, W, and Jansen, FE. Inflammatory mediators in human epilepsy: A systematic review and meta-analysis. Neurosci Biobehav Rev. (2016) 63:177–90. doi: 10.1016/j.neubiorev.2016.02.007
222. Bedner, P, and Steinhauser, C. TNFalpha-driven astrocyte purinergic signaling during Epileptogenesis. Trends Mol Med. (2019) 25:70–2. doi: 10.1016/j.molmed.2018.12.001
223. Clark, IA, and Vissel, B. Excess cerebral TNF causing glutamate excitotoxicity rationalizes treatment of neurodegenerative diseases and neurogenic pain by anti-TNF agents. J Neuroinflammation. (2016) 13:236. doi: 10.1186/s12974-016-0708-2
224. Kaur, D, Sharma, V, and Deshmukh, R. Activation of microglia and astrocytes: a roadway to neuroinflammation and Alzheimer's disease. Inflammopharmacology. (2019) 27:663–77. doi: 10.1007/s10787-019-00580-x
225. Vargas-Sanchez, K, Mogilevskaya, M, Rodriguez-Perez, J, Rubiano, MG, Javela, JJ, and Gonzalez-Reyes, RE. Astroglial role in the pathophysiology of status epilepticus: an overview. Oncotarget. (2018) 9:26954–76. doi: 10.18632/oncotarget.25485
226. Beattie, EC, Stellwagen, D, Morishita, W, Bresnahan, JC, Ha, BK, Von Zastrow, M, et al. Control of synaptic strength by glial TNFalpha. Science. (2002) 295:2282–5. doi: 10.1126/science.1067859
227. Henstridge, CM, Tzioras, M, and Paolicelli, RC. Glial contribution to excitatory and inhibitory synapse loss in neurodegeneration. Front Cell Neurosci. (2019) 13:63. doi: 10.3389/fncel.2019.00063
228. Balosso, S, Liu, J, Bianchi, ME, and Vezzani, A. Disulfide-containing high mobility group box-1 promotes N-methyl-D-aspartate receptor function and excitotoxicity by activating toll-like receptor 4-dependent signaling in hippocampal neurons. Antioxid Redox Signal. (2014) 21:1726–40. doi: 10.1089/ars.2013.5349
229. Maroso, M, Balosso, S, Ravizza, T, Liu, J, Aronica, E, Iyer, AM, et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. (2010) 16:413–9. doi: 10.1038/nm.2127
230. Terrone, G, Balosso, S, Pauletti, A, Ravizza, T, and Vezzani, A. Inflammation and reactive oxygen species as disease modifiers in epilepsy. Neuropharmacology. (2020) 167:107742. doi: 10.1016/j.neuropharm.2019.107742
231. Terrone, G, Salamone, A, and Vezzani, A. Inflammation and epilepsy: preclinical findings and potential clinical translation. Curr Pharm Des. (2017) 23:5569–76. doi: 10.2174/1381612823666170926113754
232. Das, M, Mao, W, Shao, E, Tamhankar, S, Yu, GQ, Yu, X, et al. Interdependence of neural network dysfunction and microglial alterations in Alzheimer's disease-related models. iScience. (2021) 24:103245. doi: 10.1016/j.isci.2021.103245
233. Das, M, Mao, W, Voskobiynyk, Y, Necula, D, Lew, I, Petersen, C, et al. Alzheimer risk-increasing TREM2 variant causes aberrant cortical synapse density and promotes network hyperexcitability in mouse models. Neurobiol Dis. (2023) 186:106263. doi: 10.1016/j.nbd.2023.106263
234. Morenas-Rodriguez, E, Alcolea, D, Suarez-Calvet, M, Munoz-Llahuna, L, Vilaplana, E, Sala, I, et al. Different pattern of CSF glial markers between dementia with Lewy bodies and Alzheimer's disease. Sci Rep. (2019) 9:7803. doi: 10.1038/s41598-019-44173-8
235. Wilson, EN, Swarovski, MS, Linortner, P, Shahid, M, Zuckerman, AJ, Wang, Q, et al. Soluble TREM2 is elevated in Parkinson's disease subgroups with increased CSF tau. Brain. (2020) 143:932–43. doi: 10.1093/brain/awaa021
236. Zhou, W., Zhou, Y., and Li, J. (2023). Association between cerebrospinal fluid soluble TREM2, Alzheimer's Disease and Other Neurodegenerative Diseases. J Clin Med 12:3589. doi: 10.3390/jcm12103589
237. Wang, F, Qi, X, Zhang, J, and Huang, JH. Astrocytic modulation of potassium under seizures. Neural Regen Res. (2020) 15:980–987.
238. Wilcock, DM, Vitek, MP, and Colton, CA. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer's disease. Neuroscience. (2009) 159:1055–69. doi: 10.1016/j.neuroscience.2009.01.023
239. Shapiro, LA, Wang, L, and Ribak, CE. Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia. (2008) 49:33–41. doi: 10.1111/j.1528-1167.2008.01491.x
240. Cohen-Gadol, AA, Pan, JW, Kim, JH, Spencer, DD, and Hetherington, HH. Mesial temporal lobe epilepsy: a proton magnetic resonance spectroscopy study and a histopathological analysis. J Neurosurg. (2004) 101:613–20. doi: 10.3171/jns.2004.101.4.0613
241. De Sousa, RAL. Reactive gliosis in Alzheimer's disease: a crucial role for cognitive impairment and memory loss. Metab Brain Dis. (2022) 37:851–7. doi: 10.1007/s11011-022-00953-2
242. Lopez-Valdes, HE, and Martinez-Coria, H. The role of Neuroinflammation in age-related dementias. Rev Investig Clin. (2016) 68:40–8.
243. Govindpani, K, Turner, C, Waldvogel, HJ, Faull, RLM, and Kwakowsky, A. Impaired expression of GABA signaling components in the Alzheimer's disease middle temporal gyrus. Int J Mol Sci. (2020) 21:8704. doi: 10.3390/ijms21228704
244. Limon, A, Reyes-Ruiz, JM, and Miledi, R. Loss of functional GABA(A) receptors in the Alzheimer diseased brain. Proc Natl Acad Sci U S A. (2012) 109:10071–6. doi: 10.1073/pnas.1204606109
245. Fu, X, Wang, YJ, Kang, JQ, and Mu, TW. GABA(A) receptor variants in epilepsy In: SJ Czuczwar, editor. epilepsy. Brisbane, AU: (2022)
246. Verret, L, Mann, EO, Hang, GB, Barth, AM, Cobos, I, Ho, K, et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. (2012) 149:708–21. doi: 10.1016/j.cell.2012.02.046
Keywords: Alzheimer’s disease, dementia with Lewy bodies, epilepsy, network hyperexcitability, epileptic activity
Citation: Vicente M, Addo-Osafo K and Vossel K (2024) Latest advances in mechanisms of epileptic activity in Alzheimer’s disease and dementia with Lewy Bodies. Front. Neurol. 15:1277613. doi: 10.3389/fneur.2024.1277613
Edited by:
Helen E. Scharfman, Nathan Kline Institute for Psychiatric Research, United StatesReviewed by:
Melissa Barker-Haliski, University of Washington, United StatesHolly Hunsberger, Rosalind Franklin University of Medicine and Science, United States
Copyright © 2024 Vicente, Addo-Osafo and Vossel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keith Vossel, a3Zvc3NlbEBtZWRuZXQudWNsYS5lZHU=